 Bruno Saleme1,2
Bruno Saleme1,2 Gopinath Sutendra1,2,3*
Gopinath Sutendra1,2,3*- 1Department of Medicine, University of Alberta, Edmonton, AB, Canada
- 2Mazankowski Alberta Heart Institute, University of Alberta, Edmonton, AB, Canada
- 3Cancer Research Institute of Northern Alberta, University of Alberta, Edmonton, AB, Canada
Chemotherapy-induced cardiotoxicity (CIC) is an emerging clinical problem with significant healthcare costs and no preventative therapies (1, 2). Identifying selective therapeutic targets in CIC is difficult, in part, because the mechanisms of drug toxicity vary between chemotherapeutics. For example, cardiotoxicity can be acute or chronic, transient or permanent, and can affect myocardial contractility, cardiomyocyte conduction or the myocardial vascular system (3). Thus, candidate CIC therapies would need to target many features involved in cardiac dysfunction, and additionally should not prevent chemotherapy-mediated tumor regression. Although most would agree that investing in new therapies that specifically target the tumor, while not affecting other normal tissues, including the heart would be ideal, this approach is currently impractical, as even the most selective cancer therapies have been associated with cardiotoxicity (1). For example, Bcr-Abl kinase is a specific gene fusion that causes chronic myeologenous leukemia (CML) (4), and although Bcr-Abl kinase inhibitors, including imatinib mesylate are effective in treating CML (5), they are also associated with cardiotoxicity in pre-clinical animal studies and patients (6), suggesting that alternative adjuvant therapies that can prevent, limit or improve CIC need to be developed. The most commonly used preventative therapy for CIC is dexrazoxane (7), and although dexrazoxane has shown some benefit in preventing CIC (7), it has also been associated with prevention of chemotherapy-induced tumor regression (8), and increased incidence in the development of certain types of cancer in pediatric patients (9, 10). In addition, current treatment guidelines for patients diagnosed with CIC often result in discontinuation of the chemotherapy (regardless of the tumor responsiveness) and initiation into standard heart failure treatment regimes (which include β-blockers and angiotensin inhibitors) (11). In both options, for either prevention or treatment of CIC, the myocardium appears to have precedence over the tumor, with patients receiving suboptimal care for their cancer. Rather than separating our treatment regime to focus either on heart failure or cancer, an ideal approach would look for common pathways identified in both tissues, with the aim to limit or improve chemotherapy-induced heart failure, but not prevent (or even enhance) chemotherapy-induced tumor regression. In this opinion article, we will discuss metabolic pathways that appear to be induced in both the failing heart and tumor, suggesting that metabolic therapies could provide an alternative approach for treating CIC, without hindering or potentially even improving chemotherapy-induced tumor regression.
In recent years several metabolic pathways have been identified in the failing myocardium, resulting in the emergence of metabolic therapies that appear to be beneficial against several forms of heart failure in both animals and patients (12, 13). The myocardium is the most energetically demanding organ of our body, and predominantly utilizes long-chain fatty acids and glucose as the primary substrates to generate adenosine triphosphate (ATP), which is required for myocardial contractility (14). In normal conditions, glucose is metabolized to pyruvate in the cytoplasm by glycolysis (GLY), generating ~2 ATP (14). Pyruvate can be further metabolized in the mitochondria to acetyl-CoA, the substrate for the Krebs' cycle, in a process termed glucose oxidation (GO), and this requires the pyruvate dehydrogenase complex (PDC) (14). Alternatively, long-chain fatty acids can also be metabolized in the mitochondria to generate acetyl-CoA via fatty acid β-oxidation (FAO) (14). The reducing equivalents NADH and FADH2 produced from the Krebs' cycle can enter the electron transport chain (ETC) to produce ~32 ATP (14). The normal myocardium generates the majority of its ATP (~60–90%) from mitochondrial fatty acid β-oxidation (FAO) and glucose oxidation (GO), with cytoplasmic GLY providing a minimal alternative energy-producing pathway (14, 15). Several studies have reported that a transition from a normal to failing myocardium is associated with a switch in energy metabolism from mitochondrial GO to cytoplasmic GLY (13–15). Furthermore, GLY appears to be uncoupled from GO, in part, because PDC is actively inhibited by pyruvate dehydrogenase kinase (PDK) (14). The increase in GLY (and uncoupling to GO) results in an increase in the production of lactate and protons (H+) in the cytoplasm. This buildup of H+ eventually results in a decrease in cardiac efficiency, since the cardiomyocytes utilize a large amount of ATP to restore ion homeostasis, at the expense of ATP-dependent contractility (14). Thus, this shift in energy metabolism impairs cardiac contractility and conductance.
A prominent metabolic transcription factor that has been shown to be important in the switch in energy metabolism from GO to GLY is hypoxia-inducible factor 1α (HIF1α) (16). HIF1α is a transcription factor that is induced in the failing myocardium, and is associated with increased expression of glucose transporters, glycolytic enzymes, and PDK (17–19). Thus, HIF1α can directly increase GLY (via increasing glucose uptake into the cell and increasing the levels of glycolytic enzymes) and inhibit GO (via the induction of PDK), resulting in decreased cardiac efficiency. Several studies have shown that coupling GLY with GO can improve cardiac function in several heart failure models. For example, inhibition of PDK with the small molecule compound dicholoracetate (DCA) improves cardiac function in both ischemic and afterload-induced heart failure models (20–24). Furthermore, inhibition of FAO with Ranolazine or Trimetazidine, which subsequently increase GO [via the Randle cycle; (25)], improves cardiac function in multiple preclinical heart failure models and in patients (26–29). Therefore, increasing GO (either directly with PDK inhibitors or indirectly with FAO inhibitors) appears to reverse the metabolic remodeling observed in the failing heart and improve cardiac efficiency and function. A recent study has implicated a similar metabolic remodeling in sunitinib-induced heart failure [i.e., increased GLY (30)], suggesting that therapeutically increasing GO in CIC would be beneficial in this form of heart failure as well. In addition, several chemotherapeutics, including anthracyclines or tyrosine kinase inhibitors are associated with cardiac metabolic dysfunction (30–38), providing further evidence that metabolic therapies could be beneficial against a variety of cardiotoxic chemotherapy agents.
Intriguingly, a similar metabolic remodeling has also been identified in cancer progression (39, 40). In 1927 Otto Warburg observed that most cancer cells utilized aerobic GLY, and this was associated with decreased mitochondrial respiration (41). It is now well described that cancer cells have a similar uncoupling of GLY with GO to the failing myocardium, however, unlike the failing myocardium, this metabolic profile provides cancer cells with a survival advantage (39, 42). For example, the increase in GLY in cancer results in an increase in other glycolytic branching pathways, including the pentose phosphate pathway or serine biosynthetic pathway, which generates nucleotides or amino acids, respectively, both required for cell proliferation (40, 43). Alternatively, the decrease in mitochondrial GO provides cancer cells with apoptosis resistance (19, 39, 44). The inhibition of PDC (and GO) in cancer cells is associated with an increase in the mitochondrial membrane potential, which subsequently increases the threshold for activation of the mitochondrial permeability transition pore and thus, mitochondrial dependent apoptosis (19, 39, 44). Similar to the failing myocardium, HIF1α is also induced in cancer cells and is associated with an increase in the expression of glucose transporters, glycolytic enzymes and PDK (resulting in suppressed mitochondrial GO). Inhibition of PDK (and increasing GO), with DCA in cancer cells results in decreased proliferation and enhanced mitochondrial-dependent apoptosis, resulting in decreased tumor growth in several pre-clinical animal models (19, 44–47), and in a small clinical trial in glioblastoma patients (48). Alternatively, other compounds that also increase GO, including the pyruvate kinase activator TEPP-46, has shown benefit against tumor progression (49). Taken together, these studies provide strong evidence that therapeutically increasing GO is a valid approach for decreasing tumor progression. In addition, our group had shown that increasing GO with DCA was sufficient to decrease HIF1α activity (44), providing a strong positive feedback loop that would potentiate the increase in the GO/GLY ratio, in cancer.
Recent evidence has also implicated HIF1α with the reductive glutamine pathway in cancer (50–52). The reductive glutamine pathway is associated with decreased GO, and provides cancer cells with sufficient mitochondrial substrates (i.e., citrate) to sustain lipogenesis, a critical requirement for proliferating cancer cells (50). Similarly the reductive glutamine pathway has also been implicated in right-sided heart failure, as well (53), and inhibition of this pathway has been shown to be beneficial against both heart failure and cancer progression (51–53). Intriguingly, enhancing GO has been shown to inhibit the reductive glutamine pathway (51, 53), suggesting that metabolic therapies which increase GO could have alternative benefits against heart failure and cancer progression, in addition to altering energy metabolism.
In conclusion, a similar metabolic profile (i.e., uncoupling of GLY with GO) appears to be prominent in both heart failure and cancer (see Figure 1). Therapeutically increasing GO in either the failing myocardium or tumor results in improved cardiac function or tumor regression, respectively, suggesting that a similar metabolic therapy could be beneficial in CIC. Although intriguing, much work is required to address if metabolic therapies could be advantageous against this emerging and prominent clinical condition.
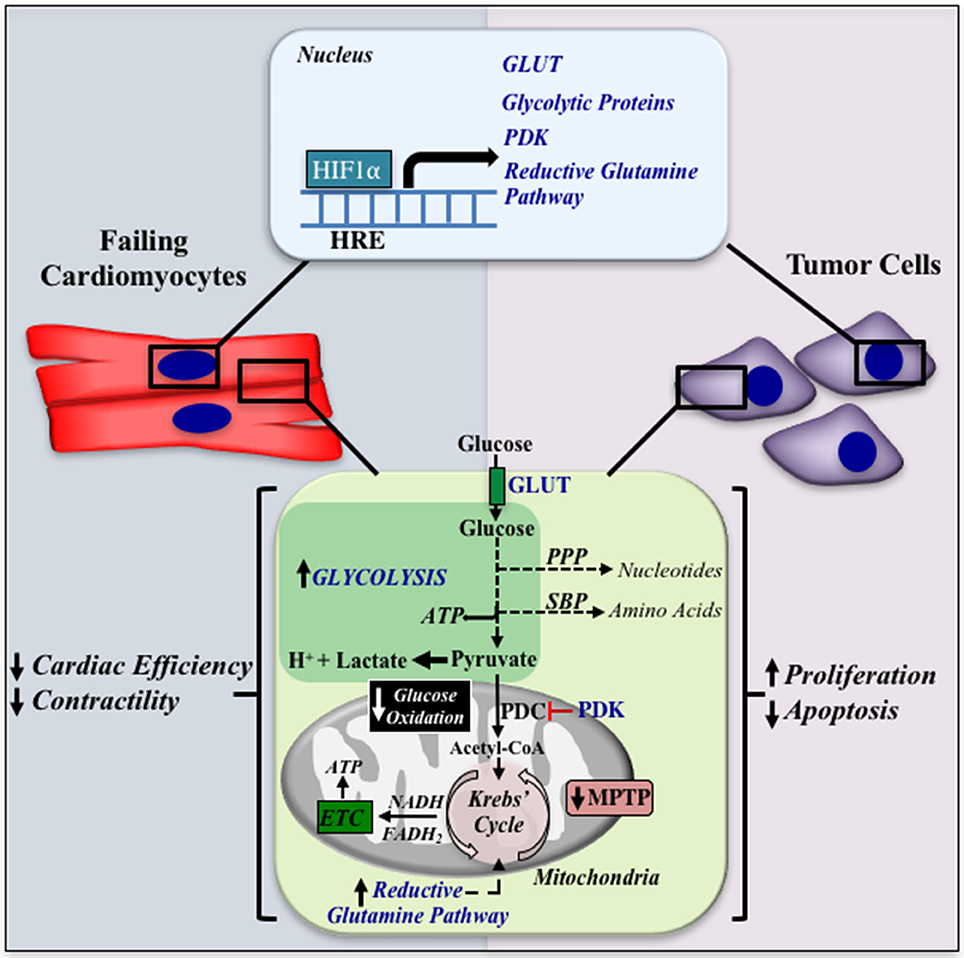
Figure 1. A similar metabolic remodeling in heart failure and cancer. HIF1α is induced in both the failing cardiomyocyte and cancer cells, and can increase the expression of glucose transporters, glycolytic enzymes, pyruvate dehydrogenase kinase (PDK), and the reductive glutamine pathway (shown in blue font). The increase in glycolysis is accompanied with the shuttling of glycolytic intermediates into the PPP and SBP, increasing nucleotide and amino acid synthesis, respectively. Furthermore, uncoupling of glycolysis with glucose oxidation results in an increase in lactate and H+ production in the cytoplasm. Inhibition of glucose oxidation is associated with closure of the MPTP, leading to apoptosis resistance. These metabolic alterations result in decreased cardiac efficiency and contractility in the heart and increased proliferation and apoptosis resistance in the tumor. HRE, Hypoxia Response Element; GLUT, Glucose Transporter; PPP, Pentose Phosphate Pathway; SBP, Serine Biosynthetic Pathway; PDK, Pyruvate Dehydrogenase Kinase; PDC, Pyruvate Dehydrogenase Complex; ETC: Electron Transport Chain; MPTP, Mitochondrial Permeability Transition Pore.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
GS is supported by an Alberta Innovates Translational Health Chair in Cardio-Oncology and an Alberta and National New Investigator Award from Heart and Stroke Foundation of Canada, along with grants from the Canadian Institutes of Health Research and Mazankowski Alberta Heart Institute/Heart and Stroke Foundation of Alberta.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SG and handling editor declared their shared affiliation.
References
1. Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. (2010) 102:14–25. doi: 10.1093/jnci/djp440
2. Nolan MT, Plana JC, Thavendiranathan P, Shaw L, Si L, Marwick TH. Cost-effectiveness of strain-targeted cardioprotection for prevention of chemotherapy-induced cardiotoxicity. Int J Cardiol. (2016) 212:336–45. doi: 10.1016/j.ijcard.2016.02.137
3. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. (2015) 12:620. doi: 10.1038/nrcardio.2015.133
4. Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. (2001) 344:1084–86. doi: 10.1056/NEJM200104053441409
5. Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. (2006) 355:2408–17. doi: 10.1056/NEJMoa062867
6. Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. (2006) 12:908–16. doi: 10.1038/nm1446
7. Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. (2004) 351:145–53. doi: 10.1056/NEJMoa035153
8. Sehested M, Jensen PB, Sorensen BS, Holm B, Friche E, Demant EJ. Antagonistic effect of the cardioprotector (+)-1,2-bis(3,5-dioxopiperazinyl-1-yl)propane (ICRF-187) on DNA breaks and cytotoxicity induced by the topoisomerase II directed drugs daunorubicin and etoposide (VP-16). Biochem Pharmacol. (1993) 46:389–93. doi: 10.1016/0006-2952(93)90514-W
9. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. (2007) 25:493–500. doi: 10.1200/JCO.2005.02.3879
10. Salzer WL, Devidas M, Carroll WL, Winick N, Pullen J, Hunger SP, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia (2010) 24:355–70. doi: 10.1038/leu.2009.261
11. Virani SA, Dent S, Brezden-Masley C, Clarke B, Davis MK, Jassal DS, et al. Canadian cardiovascular society guidelines for evaluation and management of cardiovascular complications of cancer therapy. Can J Cardiol. (2016) 32:831–41. doi: 10.1016/j.cjca.2016.02.078
12. Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, Mochizuki S. Optimization of cardiac metabolism in heart failure. Curr Pharm Des. (2011) 17:3846–53. doi: 10.2174/138161211798357773
13. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
14. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
15. Sankaralingam S, Lopaschuk GD. Cardiac energy metabolic alterations in pressure overload–induced left and right heart failure (2013 Grover Conference Series). Pulm Circ. (2015) 5:15–28. doi: 10.1086/679608
16. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. (2011) 10:671–84. doi: 10.1038/nrd3504
17. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
18. Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med. (2013) 91:1315–27. doi: 10.1007/s00109-013-1059-4
19. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. (2007) 11:37–51. doi: 10.1016/j.ccr.2006.10.020
20. Taniguchi M, Wilson C, Hunter CA, Pehowich DJ, Clanachan AS, Lopaschuk GD. Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. Am J Physiol Heart Circ Physiol. (2001) 280:H1762–9. doi: 10.1152/ajpheart.2001.280.4.H1762
21. Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. (2012) 94:359–69. doi: 10.1093/cvr/cvs129
22. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
23. Carley AN, Taglieri DM, Bi J, Solaro RJ, Lewandowski ED. Metabolic efficiency promotes protection from pressure overload in hearts expressing slow skeletal troponin I. Circ Heart Fail. (2015) 8:119–27. doi: 10.1161/CIRCHEARTFAILURE.114.001496
24. Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, et al. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J. Am. Coll. Cardiol. (1994) 23:1617–24. doi: 10.1016/0735-1097(94)90665-3
25. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
26. Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. (2006) 17(Suppl. 1):S169–S77. doi: 10.1111/j.1540-8167.2006.00401.x
27. Chandler MP, Stanley WC, Morita H, Suzuki G, Roth BA, Blackburn B, et al. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ Res. (2002) 91:278–80. doi: 10.1161/01.RES.0000031151.21145.59
28. Fragasso G, Perseghin G, De Cobelli F, Esposito A, Palloshi A, Lattuada G, et al. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J. (2006) 27:942–8. doi: 10.1093/eurheartj/ehi816
29. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. (2000) 86:580–8. doi: 10.1161/01.RES.86.5.580
30. Meredith LR, Janani S, Yuanteng Li, Dale JH, Howard OF, Heinrich T. A PKM2 signature in the failing heart. Biochem Biophys Res Commun. (2015) 495:430–6. doi: 10.1016/j.bbrc.2015.02.122
31. Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. (1986) 261:3068–74.
32. Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. (2003) 93:105–15. doi: 10.1034/j.1600-0773.2003.930301.x
33. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JA, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. (2016) 90:2063–76. doi: 10.1007/s00204-016-1759-y
34. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Cancer Rev. (2007) 7:332–44. doi: 10.1038/nrc2106
35. Zarko B, Dominik S, Alena S, Omid A, Sabine S, Julia H, et al. Radiation–induced signaling results in mitochondrial impairment in mouse heart at 4 weeks after exposure to X-rays. PLoS ONE (2011) 6:e27811. doi: 10.1371/journal.pone.0027811
36. Marie-Laure Crouch, Gary K, Rudolph S, Nolan GE, Jason H, Bielas DJM, et al. Cyclophosphamide leads to persistent deficits in physical performance and in vivo mitochondria function in a mouse model of chemotherapy late effects. PLoS ONE (2017) 12:e0181086. doi: 10.1371/journal.pone.0181086
37. Ana Filipa R-M, Emilia S, Maria de Lourdes B, Vera Marisa C. The role of the metabolism of anticancer drugs in their induced-cardiotoxicity. Curr Drug Metabol. (2016) 17:75–90. doi: 10.2174/1389200216666151103114926
38. Zoltán V, Varga PF, Lucas L, Pál P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. (2015) 309:H1453–H67. doi: 10.1152/ajpheart.00554.2015
39. Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. (2013) 3:38. doi: 10.3389/fonc.2013.00038
40. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
41. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
42. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi: 10.1038/nrc1478
43. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. (2011) 43:869–74. doi: 10.1038/ng.890
44. Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene (2013) 32:1638–50. doi: 10.1038/onc.2012.198
45. Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate (2008) 68:1223–31. doi: 10.1002/pros.20788
46. Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. (2008) 109:394–402. doi: 10.1016/j.ygyno.2008.01.038
47. Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer (2010) 102:1746–52. doi: 10.1038/sj.bjc.6605701
48. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. (2010) 2:31ra4. doi: 10.1126/scitranslmed.3000677
49. Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. (2012) 8:839–47. doi: 10.1038/nchembio.1060
50. Anastasiou D, Cantley LC. Breathless cancer cells get fat on glutamine. Cell Res. (2012) 22:443–6. doi: 10.1038/cr.2012.5
51. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481:380–4. doi: 10.1038/nature10602
52. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481:385–8. doi: 10.1038/nature10642
Keywords: cardiotoxicity, cardiac metabolism, heart failure, altered metabolism, Warburg effect, cardio-oncology
Citation: Saleme B and Sutendra G (2018) A Similar Metabolic Profile Between the Failing Myocardium and Tumor Could Provide Alternative Therapeutic Targets in Chemotherapy-Induced Cardiotoxicity. Front. Cardiovasc. Med. 5:61. doi: 10.3389/fcvm.2018.00061
Received: 03 April 2018; Accepted: 15 May 2018;
Published: 11 June 2018.
Edited by:
Thomas Pulinilkunnil, Dalhousie University, CanadaReviewed by:
Scott A. Grandy, Dalhousie University, CanadaBeshay Zordoky, College of Pharmacy, University of Minnesota, United States
Copyright © 2018 Saleme and Sutendra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gopinath Sutendra, sutendra@ualberta.ca