 Bing Lu1
Bing Lu1 Changying Lin1
Changying Lin1 Haican Liu2Xin Zhang1Yi Tian1Ying Huang1Hanqiu Yan1
Haican Liu2Xin Zhang1Yi Tian1Ying Huang1Hanqiu Yan1 Mei Qu1Lei Jia1
Mei Qu1Lei Jia1 Quanyi Wang1*
Quanyi Wang1*- 1Institute for Infectious Disease and Endemic Disease Control, Beijing Center for Disease Prevention and Control, Beijing Research Center for Preventive Medicine, Beijing Key Laboratory of Diagnostic and Traceability Technologies for Food Poisoning, Beijing, China
- 2State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Background: Klebsiella pneumoniae is an opportunistic pathogen associated with community-acquired and nosocomial infections. Since 2010, K. pneumoniae testing has been included into an existing diarrhea-syndrome surveillance system for estimating the prevalence of K. pneumoniae in diarrhea-syndrome patients, assessing antibiotic susceptibility, and investigating molecular characteristics of K. pneumoniae.
Methods: Klebsiella pneumoniae strains were isolated from stool specimens from diarrhea-syndrome outpatients in Beijing, China. Isolates were tested for antibiotic susceptibility, and phylogenetic relationships were explored though whole genome sequence analysis. Multi-locus sequence type (MLST) alleles were extracted from the whole genome sequence (WGS) data. A maximum likelihood tree was generated by MEGAX. Genomes were annotated by Prokka; core genes were produced by Roary; a maximum likelihood phylogenetic tree was generated using FastTree.
Results: Forty-four K. pnuemoniae strains were isolated from 2010 to July 2019; of these 37 were K. pneumoniae and seven were K. variicola. Antibiotic susceptibility testing showed that all 44 strains were sensitive to gentamicin, imipenem, amikacin, meropenem, kanamycin; 97.73% were sensitive to cefoxitin andlavo-ofloxacin; the highest antibiotic resistance rate was 79.55%, which was to ampicillin. We found three extended-spectrum beta-lactamase (ESBL) producing strains; we identified high-virulence ST types, including ST307 and ST65; and we found that ST23 has been the epidemic clone since 2010. MLST and core genome sequence analysis showed two distinct clusters of 44 K. pnuemoniae; 40 alleles were identified in core genome sequence analysis, while 36 alleles were identified in MLST typing.
Conclusions: There is an urgent need for epidemiological and molecular studies to understand the dynamics of antibiotic resistance and virulence gene transmission to guide strategies for K. pneumoniae surveillance. WGS analysis provided high discrimination power and reliable and robust data useful for molecular epidemiology.
Background
Klebsiella pneumoniae is ubiquitous in the environment. K. pneumoniae is a Gram-negative opportunistic pathogen associated with community-acquired and nosocomial infections (Moradigaravand et al., 2017). Clinically, K. pneumoniae causes pneumoniae, abscesses, bacteremia, urinary tract infections (Podschun and Ullmann, 1998; Wyres and Holt, 2016), and occasionally, diarrhea (Moradigaravand et al., 2017). Nosocomial infections caused by K. pneumoniae impose an increasing risk of community infection.
Since 2010, K. pneumoniae testing has been included in an existing enteric pathogen surveillance system focused on diarrhea-syndrome outpatients of all ages in 245 sentinel hospitals of the 16 districts of Beijing (Lu et al., 2017). The aim of the system is to monitor the prevalence of K. pneumoniae in diarrhea-syndrome outpatients, assess antimicrobial resistance, and explore molecular characteristics of community-acquired K. pneumoniae infection strains.
Methods
Identification of Bacterial Strains
From 2010 to July 2019, stool specimens collected from diarrhea-syndrome outpatients in sentinel hospitals were analyzed using a reverse transcription polymerase chain reaction (RT-PCR) for diarrhea-generating viruses (e.g., rotavirus, norovirus, and calicivirus) (Deng et al., 2012; Gao et al., 2012; Ying et al., 2017) and cultured for isolation of diarrhea-generating bacteria. Any isolated bacteria strains were further tested to identify the pathogens (e.g., Salmonella, Shigella, Escherichia coli, Vibrio parahemolyticus, or K. pneumoniae) usingVitek2 Compact Instrument (bioMérieux; Marcy, France).
Isolated K. pneumoniae strains were tested for antibiotic susceptibility, deoxyribonucleic acid (DNA) extraction, whole-genome sequencing (WGS) analysis, and determination of their molecular characteristics.
Antimicrobial Resistance Testing
Antimicrobial resistance testing for K. pneumoniae strains was assessed using the minimal inhibitory concentration (MIC) method. MICs were interpreted in accordance with the Clinical and Laboratory Standards Institute (CLSI) document, M100-S29:2019. Twenty-seven antimicrobials obtained from Shanghai Xingbai Co. (AST Panel for Aerobic Gram Negative bacilli) were used for antimicrobial resistance testing: ampicillin, ampicillin-sulbactam, amoxicillin with clavulanate potassium, cephazoline, cefepime, cefotaxime, cefoxitin, ceftazidime, aztreonam, imipenem, meropenem, gentamicin, amikacin, kanamycin, azithromycin, tetracycline, minocycline, doxycycline, nalidixic acid, ciprofloxacin, lavofloxacin, gemifloxacin, trimethoprim-sulphamethoxazole, sulfisoxazole, chloramphenicol, cefotaxime with clavulanate, and ceftazidime with clavulanate. Escherichia coli ATCC 25922 was used as a quality-control strain. MIC levels at 2 μg/mL or above for cefotaxime indicated a possible extended-spectrum beta-lactamase (ESBLs)-producing strain, requiring further confirmation. MIC for ceftazidime combined with clavulanatede creasing at least three two-fold concentrations compared with the MIC value for ceftazidime alone (e.g., ceftazidime MIC = 8 μg/mL; ceftazidime-clavulanate MIC = 1 μg/mL) confirmed an ESBL-producing strain.
DNA Extraction and WGS
DNA was extracted by QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Quantification of extracted genomic DNA (gDNA) was determined on a NanoDrop spectrophotometer, with verification by agarose gel electrophoresis and fluorometric analysis (Qubit2.0).
Multiplexed paired-end libraries (2 × 300 bp) were prepared for DNA sequencing using the NEBNext®Ultra™ DNA Library Prep Kit for Illumina (NEB, USA). Sequences were determined on an Illumina PE150 platform with 100 × coverage at Beijing Novogene technology Co., Ltd.
Raw sequencing data were checked for quality, trimmed, and assembled de novo into contiguous segments using CLC Genomics Workbench version 10.1.1 (CLC, Bio-QIAGEN, Aarhus, Denmark) and SPAdes version 3.13 (Bankevich et al., 2012).
The WGS data were matched in the NCBI BLAST database to identify three distinct species of K. pneumoniae: K. pneumoniae (KpI), K. quasipneumoniae (KpII), and K. variicola (KpIII) (Holt et al., 2015).
Plasmid, Antimicrobial Resistant Genes and Multi-Locus Sequence Type (MLST) Analysis
The genomic analysis was based on the Center for Genomic Epidemiology web server (https://cge.cbs.dtu.dk/services/cge/), in which web-based multi-locus sequence type (MLST) 2.0 (Larsen et al., 2012), ResFinder 3.2 (Zankari et al., 2012), and PlasmidFinder 2.1 (Carattoli et al., 2014) were used for cluster sequencing types, investigating antimicrobial resistant genes, and defining content of plasmid replicon types, respectively.
MLST analyses were performed using seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpob, and tonB) to characterize diversity and epidemiology of K. pneumoniae isolates (Diancourt et al., 2005). WGS data were used to generate MLST assignments for each isolate; unknown STs were sent to the Klebsiella pneumoniae MLST database at the Pasteur Institute (https://bigsdb.pasteur.fr/klebsiella/klebsiella.html). Genotyping analysis was based on MLST sequences; maximum likelihood trees were generated by MEGA-X (Kumar et al., 2018).
Annotation and Core Genome Analysis
Genomes were annotated by Prokka, a tool for rapid prokaryotic genome annotation (Seemann, 2014). Phylogenetic analyses were produced by Roary, a tool that rapidly builds large-scale pan genomes and identifies core genes (shared by all strains) and accessory genes (Page et al., 2015). A maximum likelihood phylogenetic tree was generated by FastTree version 2.1.10 (Price et al., 2010) to assess relatedness among genomes in the isolated bacteria and to approximate the species tree.
Results
Surveillance led to isolation of 1 to 11 K. pneumoniae strains each year, identifying 44 K. pneumoniae strains from 25,411 stool specimens in 10 years, the detection rate was 0.17% (44/25,411).
Antimicrobial Resistance
All 44 K. pneumoniae strains were sensitive to gentamicin, imipenem, amikacin, meropenem, kanamycin; 97.7% were sensitive to cefoxitin andlavo-ofloxacin; 95.5% were sensitive to nalidixic acid, azithromycin, and ciprofloxacin; 79.6% of isolated K. pnuemoniae strains manifested resistance to ampicillin, and 13.6% of isolated staring showed resistance to sulfisoxazole, trimethoprim, and sulphame-thoxazole (Table 1). Three K. pneumoniae strains were confirmed as ESBL-producing strains (Table 2).
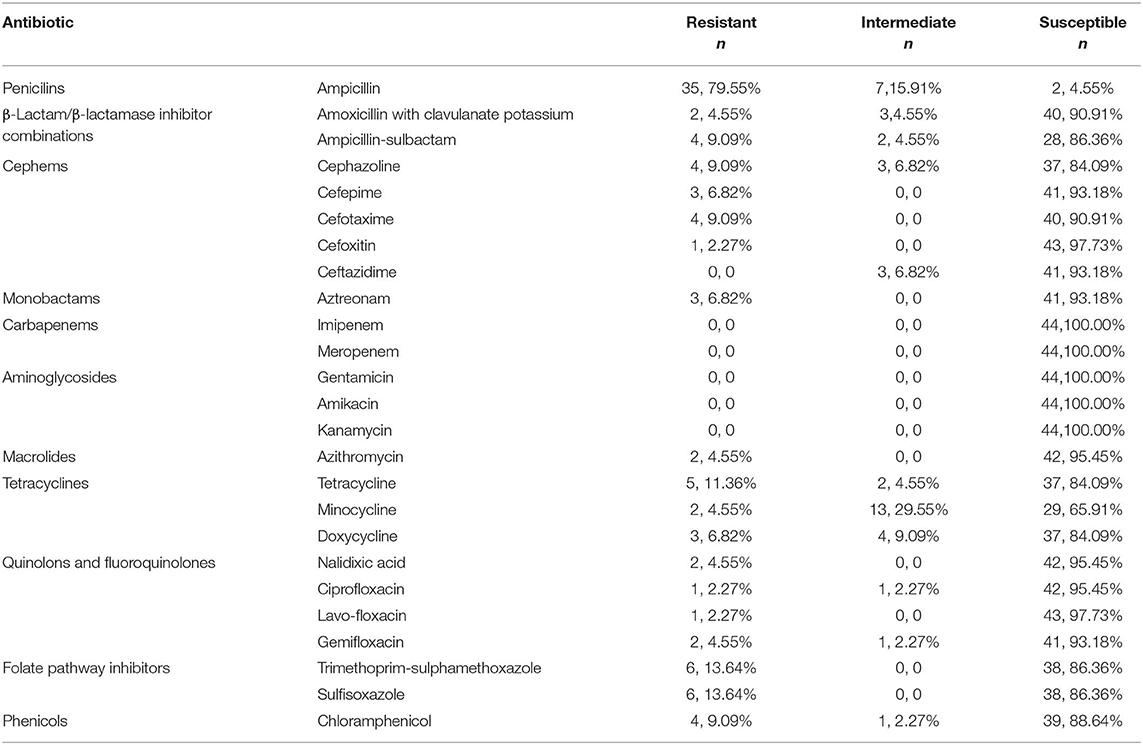
Table 1. Antibiotic susceptibility results for 44 K. pneumoniae strains.
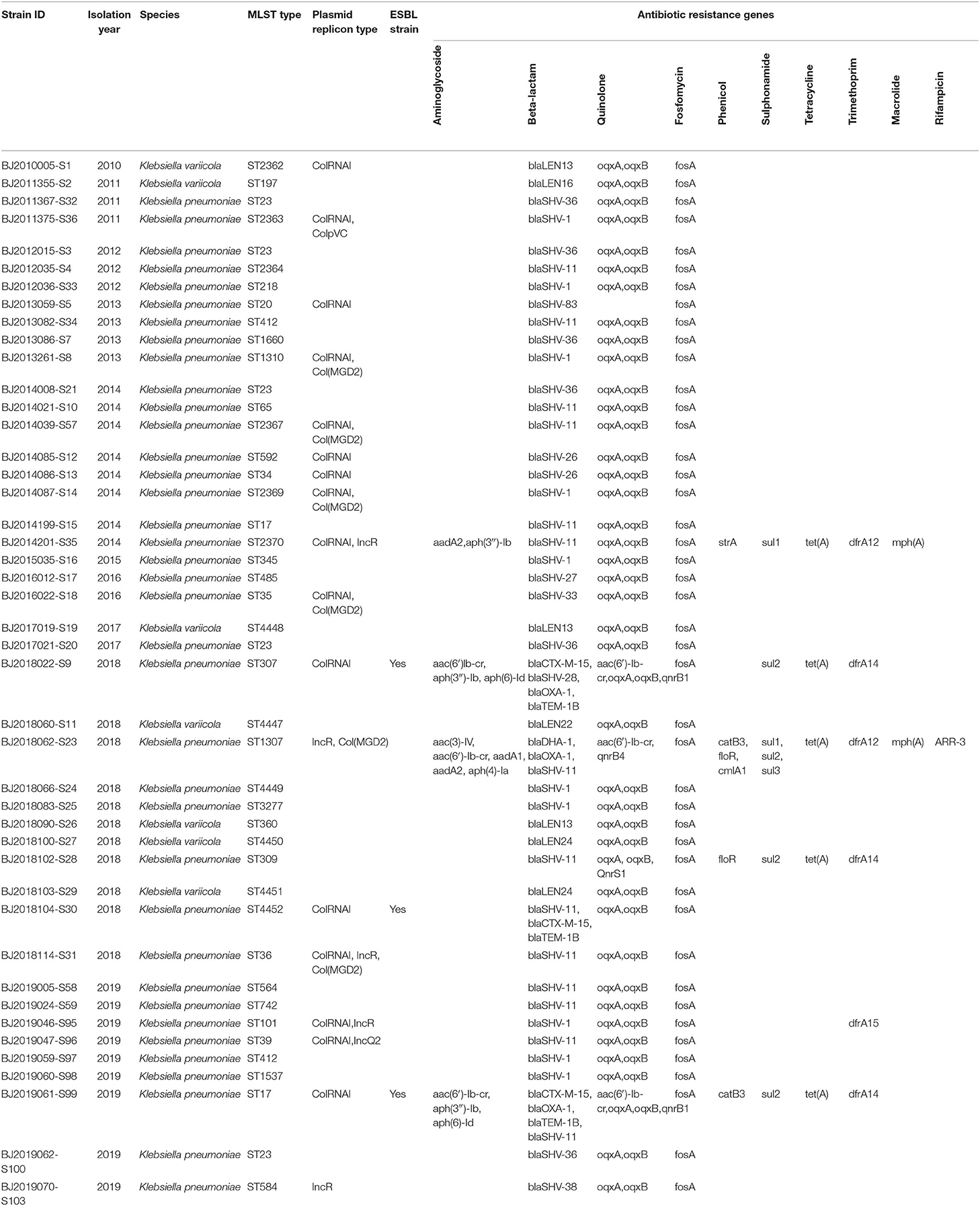
Table 2. Summary of species, and genotypic characteristics for 44 K. pneumoniae strains.
WGS NCBI Blast Results
Forty-four K. pneumoniae strains were disambiguated into two species: 37 K. pneumoniae (KpI) strains and 7 K. variicola (KpIII) strains. Surveillance did not identify any K. quasipneumoniae (KpII) strains (Table 2).
MLST Results and MEGA Analysis
MLST of the 44 strains revealed 36 different sequence types (STs), including ST23, which has been detected five times in Beijing in the most recent 10 years, and ST4447, ST4448, ST4449, ST4450, ST4451, and ST4452, which were seen for the first time in the global database. The maximum likelihood tree identified 36 MLST alleles, and 44 strains were disambiguated into two clonal groups: Cluster M1 (containing 7 strains, K. variicola strains) and Cluster M2 (containing 37 strains, K. pneumoniae strains) (Figure 1).
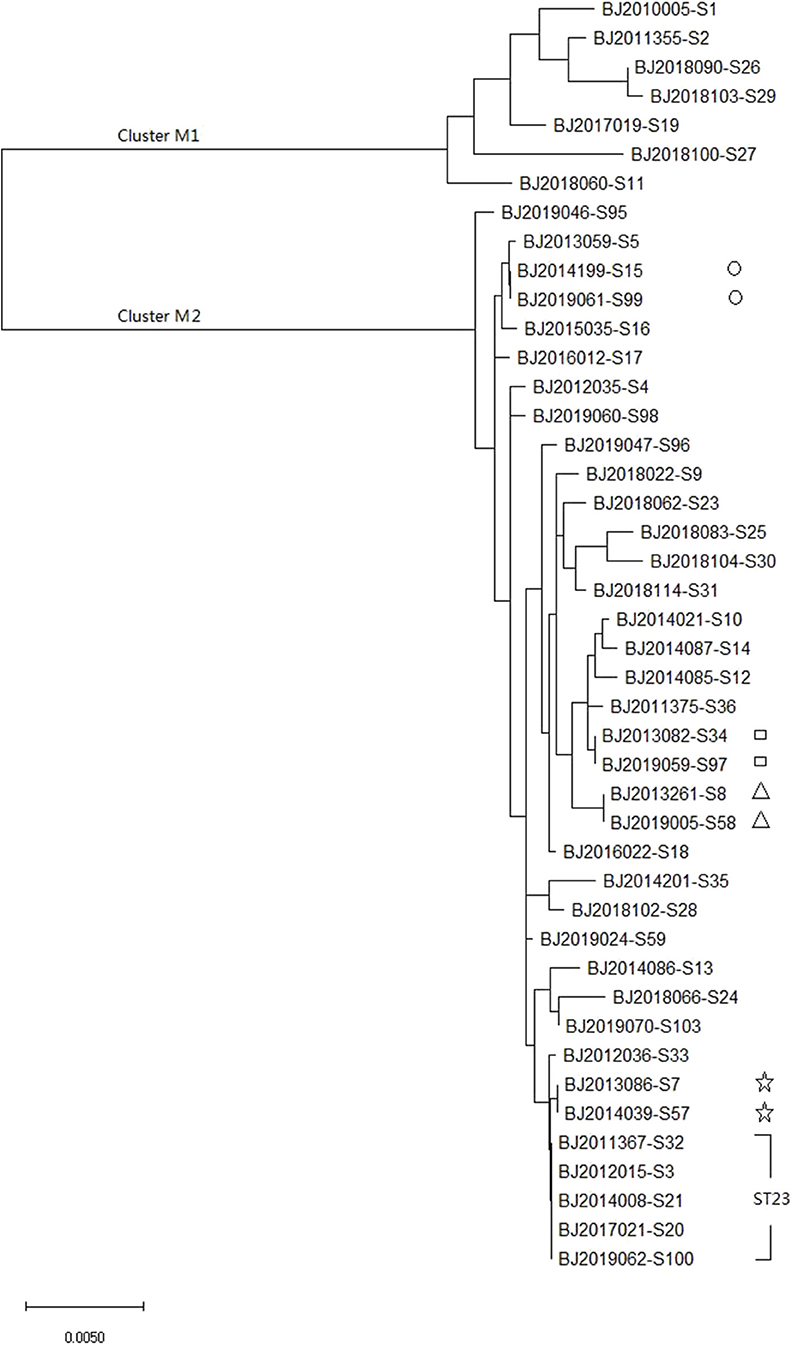
Figure 1. Molecular phylogenetic analysis by maximum likelihood method based on MLST sequences of 44 K. pneumoniea strains.
Plasmid and Drug Resistance Genes Identification
ColRNAI, Col(MGD2), ColpVC, IncR, and IncQ2 plasmid replicons were identified, encompassing 40.9, 13.6, 2.3, 11.4, and 2.3% of the 44 strains, respectively. Among the resistance genes to Beta-lactams, Quinolone, Fosfomycin, Phenicol, Sulphonamide, Tetracycline, Trimethoprim, Macrolide, and Rifampicin, resistance genesto Beta-lactam, Quinolone, and Fosfomycin were predominant. Among the resistance genes identified from 3 ESBL-producing strains, blaCTX-M-15 and blaTEM-1B were unique resistance genes to Beta-lactams (Table 2).
Core Genome Analysis
The whole genome sequence of the 44 strains identified 3,428 core genes. In maximum-likelihood phylogenies trees, these core genome sequences showed 40 allele differences that grouped into two distinct clusters: cluster C1 (containing7 K. varricola strains), and cluster C2 (containing 37 K. pneumoniae strains) (Figure 2).
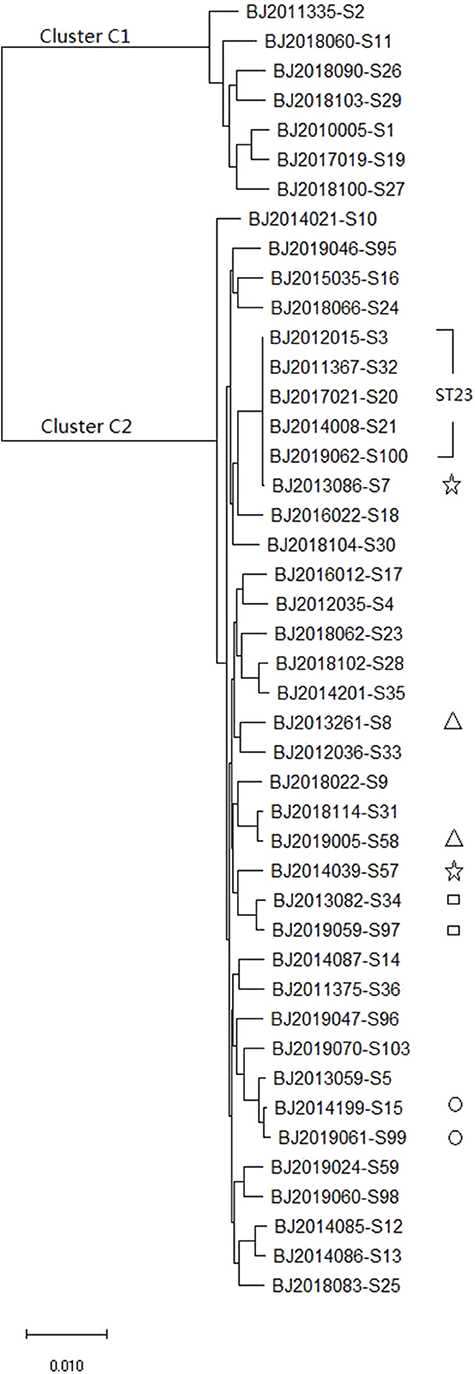
Figure 2. The maximum likelihood phylogenetic tree based on the core genome sequences of 44 K. pneumoniae strains.
Discussion
K. pneumoniae has been reported to be a leading cause of hospital associated infections and a common cause of community-acquired infections in many countries (Pendleton et al., 2013; Moradigaravand et al., 2017; Musicha et al., 2017). Beijing outpatient-based diarrhea-syndromes surveillance detected K. pneumoniae every year since 2010 demonstrating the existence of community-acquired infection caused by K. pneumoniae. Detection of five ST23 strains from 2010 to 2019 further demonstrated that the ST23 strain has persisted in Beijing throughout these years. Our results should alert public health officials since ST23 of K. pneumoniae has well-known virulence and is able to cause severe disease in otherwise healthy individuals (Turton et al., 2007; Brisse et al., 2009; Holt et al., 2015). It typically carries all four acquired siderophore systems as well as rmpA (Brisse et al., 2009). K. pneumoniae ST23 is the most predominant sequence type causing invasive community-acquired infections in Asia (Chung et al., 2012). Surveillance also detected an ST65 strain, which carries colibactin and rmpA (Brisse et al., 2009), and which is associated with lethal infections in humans and marine mammals (Liao et al., 2014).
The three community-acquired K. pneumoniae ESBL-producing strains (ST307, ST4452, and ST17) that were identified in the most recent 2-years period provide a significant signal of drug resistance in the population. All three ESBLs producing strains harbor blaCTX-M-15 and blaTEM-1B antibiotic resistance genes. CTX-M-15 belonged to the CTX-M-1 group, and is widespread in east Asia (Bonnet, 2004). The blaCTX-M was first reported in 1990 in a cefotaxime resistant E. coli strain isolated from the fecal flora of a laboratory dog (Bauernfeind et al., 1990). Since then, the CTX-M enzymes have formed a rapidly growing family of ESBLs distributed over wide geographic areas and among a wide range of clinical bacteria, particularly among members of the Enterobacteriaceae family (Bonnet, 2004). Outbreaks have been described in several countries (Yan et al., 2000; Baraniak et al., 2002). Since 1999, CTX-M has been reported to have become the most frequent ESBL in the Enterobacteriaceae in China (Chanawong et al., 2002; Xiong et al., 2002; Wang et al., 2003). Notably, the K. pneumoniae ST307 ESBL-producing strain has a novel lineage with potential to become an epidemic or “high-risk” clone. It has been recognized as a candidate for becoming one of the most clinically-relevant clones since its worldwide emergence during recent years (Villa et al., 2017). The ST307 lineage displays an association with CTX-M-15- and Carbapenemase (KPC)-producing encoding plasmids (Villa et al., 2017). The K. pneumoniae ST307 detected in our study did not harbor the blaKPC gene, but KPC producing factor could be acquired through horizontal plasmids transfer. The ability of this clone lineage to acquire novel genetic features may contribute to its increased persistence in the environment and highlights its potential public health threat of dramatically disseminated multiple drug resistance among bacteria.
MLST and core genomes sequences consistently differentiated 44 K. pneumoniae into 7 K. varricola strains and 37 K. pneumoniae strains. However, the core genome sequences increase discriminatory power for bacterial pathogen subtyping. For example, BJ2013086-S7 strain is very close to BJ2014039-S57 in an MLST molecular phylogenetic tree (see Figure 1), however, BJ2013086-S7 was separated from BJ2014039-S57in the phylogenetic tree generated by the core genome, and was closer to ST23 strain. Similar distinction was made for BJ2013261-S8 and BJ2019005-S58 stains, BJ2019059-S97 and BJ2013082-S34, and BJ2019061-S99 and BJ2014199-S15. Since WGS consists of sequencing chromosome information, both inherited from ancestors and their mutations, in theory, this powerful tool can deduce the chains of potential cross transmission of K. pneumoniae infection (Croucher and Didelot, 2015) and facilitate study of the population structure and pathogen evolution (Bialek-Davenet et al., 2014; Struve et al., 2015; Zhou et al., 2017). However, its discriminatory power relies on reliable, and robust, and long-term WGS data from different geographic areas. It will be valuable to establish a K. pneumoniae identification network for information sharing.
This study suffers two main limitations. First, the sentinel surveillance could have under-estimated the prevalence of K. pneumoniae in diarrhea-syndrome outpatients since K. pneumoniae in most of the circumstance is not the predominant causative-pathogen. Second, lack of comparation with molecular characteristics of hospital-acquired K. pneumoniae infection strains encourages more effort should be made to provide complete molecular spectrum in future studies.
Conclusions
Outpatient-based diarrhea-syndrome surveillance in Beijing China identified 3 ESBLs-producing strains in 2018 and 2019 that had not been detected previously. We identified high virulence ST types, such as ST307 and ST65, and we showed that ST23 has been the epidemic clone since 2010. There is an urgent need for epidemiological and molecular studies to understand the dynamics of antibiotic resistance and virulence gene transmission to guide strategies for K. pneumoniae surveillance. WGS analysis provides high discrimination power, and reliable and robust data for molecular epidemiology.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The study was approved by the Ethics Committee of the Beijing Center for Disease Prevention and Control.
Author Contributions
BL participated in data analysis and drafted the manuscript. CL and HL managed the bio information analysis. XZ and YH carried out the molecular genetic studies. HY participated in sample isolation. LJ and YT managed the strains and data collection. MQ and QW participated in the design of the study. All authors read and approved the final manuscript.
Funding
This work was supported by National Science and Technology Support Projects for the Thirteenth Five-Year Plan for Infectious Diseases of China (2018ZX10714002-003-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the team of curators of the Institute Pasteur MLST and whole genome MLST databases for curating the data and making them publicly available at http://bigsdb.pasteur.fr/. We thank the epidemiological investigation teams of all 16 districts in Beijing, China.
Abbreviations
K. pneumoniae, Klebsiella pneumonia; K. quasipneumoniae, Klebsiella quasipneumoniae; K. variicola, Klebsiella variicola; MLST, multi-locus sequence typing; RT-PCR, reverse transcription polymerase chain reaction; DNA, deoxyribonucleic acid; WGS, whole genome sequence; CLSI, Clinical and Laboratory Standards Institute; ESBL, extended-spectrum beta-lactamase; MIC, minimal inhibitory concentration; STs, sequence types; KPC, Carbapenemase.
References
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baraniak, A., Fiett, J., Sulikowska, A., Hryniewicz, W., and Gniadkowski, M. (2002). Countrywide spread of CTX-M-3 extended-spectrum beta-lactamase-producing microorganisms of the family Enterobacteriaceae in Poland. Antimicrob. Agents Chemother. 46, 151–159. doi: 10.1128/aac.46.1.151-159.2002
Bauernfeind, A., Grimm, H., and Schweighart, S. (1990). A new plasmidic cefotaximase in a clinical isolate of Escherichia coli. Infection 18, 294–298. doi: 10.1007/bf01647010
Bialek-Davenet, S., Criscuolo, A., Ailloud, F., Passet, V., Jones, L., Delannoy-Vieillard, A. S., et al. (2014). Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 20, 1812–1820. doi: 10.3201/eid2011.140206
Bonnet, R. (2004). Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob. Agents Chemother. 48, 1–14. doi: 10.1128/aac.48.1.1-14.2004
Brisse, S., Fevre, C., Passet, V., Issenhuth-Jeanjean, S., Tournebize, R., Diancourt, L., et al. (2009). Virulent clones of Klebsiella pneumoniae: identification and evolutionary scenario based on genomic and phenotypic characterization. PLoS ONE 4:e4982. doi: 10.1371/journal.pone.0004982
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chanawong, A., M'Zali, F. H., Heritage, J., Xiong, J. H., and Hawkey, P. M. (2002). Three cefotaximases, CTX-M-9, CTX-M-13, and CTX-M-14, among Enterobacteriaceae in the people's Republic of China. Antimicrob. Agents Chemother. 46, 630–637. doi: 10.1128/aac.46.3.630-637.2002
Chung, D. R., Park, M. H., Kim, S. H., Ko, K. S., Kang, C. I., Peck, K. R., et al. (2012). Prevalence and molecular characterization of serotype K1 Klebsiella pneumoniae strains from various clinical specimen sources in 11 Asian countries. J. Infect. 64, 622–625. doi: 10.1016/j.jinf.2012.02.007
Croucher, N. J., and Didelot, X. (2015). The application of genomics to tracing bacterial pathogen transmission. Curr. Opin. Microbiol. 23, 62–67. doi: 10.1016/j.mib.2014.11.004
Deng, Y., Gao, Z., Yan, H., Li, X., LI, W., Li, W., et al. (2012). Investigation of human calicivirus infections for diarrhea patients visiting enteric clinics in Beijing. Int. J. Virol. 19, 193–197. doi: 10.3760/cma.j.issn.1673-4092.2012.05.001
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A., and Brisse, S. (2005). Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J. Clin. Microbiol. 43, 4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005
Gao, Z., Li, X., Yan, H., Jia, L., LI, G., Wu, X., et al. (2012). Surveillance of group A rotavirus diarrhea in children under 5 years old in Beijing, 2011. Int. J. Virol. 19, 153–156. doi: 10.3760/cma.j.issn.1673-4092.2012.04.003
Holt, K. E., Wertheim, H., Zadoks, R. N., Baker, S., Whitehouse, C. A., Dance, D., et al. (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. U.S.A. 112, E3574–3581. doi: 10.1073/pnas.1501049112
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Liao, C. H., Huang, Y. T., Chang, C. Y., Hsu, H. S., and Hsueh, P. R. (2014). Capsular serotypes and multilocus sequence types of bacteremic Klebsiella pneumoniae isolates associated with different types of infections. Eur. J. Clin. Microbiol. Infect. Dis. 33, 365–369. doi: 10.1007/s10096-013-1964-z
Lu, B., Zhou, H., Zhang, X., Qu, M., Huang, Y., and Wang, Q. (2017). Molecular characterization of Klebsiella pneumoniae isolates from stool specimens of outpatients in sentinel hospitals Beijing, China, 2010-2015. Gut. Pathog. 9:39. doi: 10.1186/s13099-017-0188-7
Moradigaravand, D., Martin, V., Peacock, S. J., and Parkhill, J. (2017). Evolution and epidemiology of multidrug-resistant klebsiella pneumoniae in the United Kingdom and Ireland. MBio. 8:e01976–16. doi: 10.1128/mBio.01976-16
Musicha, P., Cornick, J. E., Bar-Zeev, N., French, N., Masesa, C., Denis, B., et al. (2017). Trends in antimicrobial resistance in bloodstream infection isolates at a large urban hospital in Malawi (1998–2016): a surveillance study. Lancet. Infect. Dis. 17, 1042–1052. doi: 10.1016/S1473-3099(17)30394-8
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pendleton, J. N., Gorman, S. P., and Gilmore, B. F. (2013). Clinical relevance of the ESKAPE pathogens. Expert. Rev. Anti. Infect. Ther. 11, 297–308. doi: 10.1586/eri.13.12
Podschun, R., and Ullmann, U. (1998). Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603. doi: 10.1128/CMR.11.4.589
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Struve, C., Roe, C. C., Stegger, M., Stahlhut, S. G., Hansen, D. S., Engelthaler, D. M., et al. (2015). Mapping the evolution of hypervirulent Klebsiella pneumoniae. MBio. 6:e00630. doi: 10.1128/mBio.00630-15
Turton, J. F., Englender, H., Gabriel, S. N., Turton, S. E., Kaufmann, M. E., and Pitt, T. L. (2007). Genetically similar isolates of Klebsiella pneumoniae serotype K1 causing liver abscesses in three continents. J. Med. Microbiol. 56(Pt 5), 593–597. doi: 10.1099/jmm.0.46964-0
Villa, L., Feudi, C., Fortini, D., Brisse, S., Passet, V., Bonura, C., et al. (2017). Diversity, virulence, and antimicrobial resistance of the KPC-producing Klebsiella pneumoniae ST307 clone. Microb. Genom. 3:e000110. doi: 10.1099/mgen.0.000110
Wang, H., Kelkar, S., Wu, W., Chen, M., and Quinn, J. P. (2003). Clinical isolates of Enterobacteriaceae producing extended-spectrum beta-lactamases: prevalence of CTX-M-3 at a hospital in China. Antimicrob. Agents Chemother. 47, 790–793. doi: 10.1128/aac.47.2.790-793.2003
Wyres, K. L., and Holt, K. E. (2016). Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 24, 944–956. doi: 10.1016/j.tim.2016.09.007
Xiong, Z., Zhu, D., Wang, F., Zhang, Y., Okamoto, R., and Inoue, M. (2002). Investigation of extended-spectrum beta-lactamase in Klebsiellae pneumoniae and Escherichia coli from China. Diagn. Microbiol. Infect. Dis. 44, 195–200. doi: 10.1016/s0732-8893(02)00441-8
Yan, J. J., Ko, W. C., Tsai, S. H., Wu, H. M., Jin, Y. T., and Wu, J. J. (2000). Dissemination of CTX-M-3 and CMY-2 beta-lactamases among clinical isolates of Escherichia coli in southern Taiwan. J. Clin. Microbiol. 38, 4320–4325. doi: 10.1128/JCM.38.12.4320-4325.2000
Ying, Z., Liang, Q. I., Junrong, L., Hao, S., and Baolin X. U. (2017). Etiological study of viral diarrhea among children under 5 years of age in Dongcheng district of Beijing. Int. J. Virol. 24, 119–122. doi: 10.3760/cma.j.issn.1673-4092.2017.02.012
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Klebsiella pneumoniae, multi-locus sequencing type (MLST), whole genome sequencing (WGS), molecular typing, ESBL
Citation: Lu B, Lin C, Liu H, Zhang X, Tian Y, Huang Y, Yan H, Qu M, Jia L and Wang Q (2020) Molecular Characteristics of Klebsiella pneumoniae Isolates From Outpatients in Sentinel Hospitals, Beijing, China, 2010–2019. Front. Cell. Infect. Microbiol. 10:85. doi: 10.3389/fcimb.2020.00085
Received: 21 November 2019; Accepted: 19 February 2020;
Published: 28 February 2020.
Edited by:
Ghassan M. Matar, American University of Beirut, LebanonReviewed by:
Graciela Castro Escarpulli, National Polytechnic Institute, MexicoRima Abdallah Moghnieh, Makassed General Hospital, Lebanon
Copyright © 2020 Lu, Lin, Liu, Zhang, Tian, Huang, Yan, Qu, Jia and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanyi Wang, YmpjZGN4bUAxMjYuY29t