 Masaya Yamaguchi1*
Masaya Yamaguchi1* Yujiro Hirose1
Yujiro Hirose1 Moe Takemura1,2
Moe Takemura1,2 Masayuki Ono1,3
Masayuki Ono1,3 Tomoko Sumitomo1
Tomoko Sumitomo1 Masanobu Nakata1
Masanobu Nakata1 Yutaka Terao4
Yutaka Terao4 Shigetada Kawabata1*
Shigetada Kawabata1*- 1Department of Oral and Molecular Microbiology, Osaka University Graduate School of Dentistry, Osaka, Japan
- 2Department of Oral and Maxillofacial Surgery II, Osaka University Graduate School of Dentistry, Osaka, Japan
- 3Department of Fixed Prosthodontics, Osaka University Graduate School of Dentistry, Osaka, Japan
- 4Division of Microbiology and Infectious Diseases, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan
Streptococcus pneumoniae is a Gram-positive bacterium belonging to the oral streptococcus species, mitis group. This pathogen is a leading cause of community-acquired pneumonia, which often evades host immunity and causes systemic diseases, such as sepsis and meningitis. Previously, we reported that PfbA is a β-helical cell surface protein contributing to pneumococcal adhesion to and invasion of human epithelial cells in addition to its survival in blood. In the present study, we investigated the role of PfbA in pneumococcal pathogenesis. Phylogenetic analysis indicated that the pfbA gene is highly conserved in S. pneumoniae and Streptococcus pseudopneumoniae within the mitis group. Our in vitro assays showed that PfbA inhibits neutrophil phagocytosis, leading to pneumococcal survival. We found that PfbA activates NF-κB through TLR2, but not TLR4. In addition, TLR2/4 inhibitor peptide treatment of neutrophils enhanced the survival of the S. pneumoniae ΔpfbA strain as compared to a control peptide treatment, whereas the treatment did not affect survival of a wild-type strain. In a mouse pneumonia model, the host mortality and level of TNF-α in bronchoalveolar lavage fluid were comparable between wild-type and ΔpfbA-infected mice, while deletion of pfbA decreased the bacterial burden in bronchoalveolar lavage fluid. In a mouse sepsis model, the ΔpfbA strain demonstrated significantly increased host mortality and TNF-α levels in plasma, but showed reduced bacterial burden in lung and liver. These results indicate that PfbA may contribute to the success of S. pneumoniae species by inhibiting host cell phagocytosis, excess inflammation, and mortality by interacting with TLR2.
Introduction
Streptococcus pneumoniae is a Gram-positive bacterium belonging to the mitis group that colonizes the human nasopharynx in ~20% of children without causing clinical symptoms (Kawamura et al., 1995; Bogaert et al., 2004; Richards et al., 2014). On the other hand, S. pneumoniae is also a leading cause of bacterial pneumonia, meningitis, and sepsis worldwide. The pathogen is estimated to be responsible for the deaths of ~1,190,000 people annually from lower respiratory infection (GBD 2015 LRI Collaborators, 2017). Following the introduction of pneumococcal conjugate vaccines, S. pneumoniae is still responsible for two thirds of all cases of meningitis (McIntyre et al., 2012). In addition, antibiotic selective pressure causes resistant pneumococcal clones to emerge and expand all over the world and the World Health Organization listed S. pneumoniae as one of antibiotic-resistant “priority pathogens” (WHO, 2017). Centers for Disease Control and Prevention data from active bacterial core surveillance for 2009 to 2013 indicated that pneumococcal conjugate vaccines work as a useful tool against antibiotic resistance (Kim et al., 2016). However, these vaccines also generate selective pressure, and non-vaccine serotypes of S. pneumoniae are increasing worldwide (Flasche et al., 2011; Golubchik et al., 2012).
During the process of invasive infection, S. pneumoniae needs to evade host immunity and replicate in the host after colonization. In these steps, pneumococcal cell surface proteins work as adhesins and/or anti-phagocytic factors. There are two types of motifs for pneumococcal cell surface localization, a cell wall anchoring motif, LPXTG (Lofling et al., 2011), and choline-binding repeats interacting with pneumococcal phosphorylcholine (Hakenbeck et al., 2009). Choline-binding proteins (CBPs) localize on the pneumococcal cell wall via the phosphorylcholine moiety of teichoic acids, while LPXTG-anchored proteins are covalently attached to the cell wall. Several LPXTG-anchored proteins and CBPs contribute to the adhesion to host epithelial cells through the interaction with host factors (Hakenbeck et al., 2009; Mitchell and Mitchell, 2010; Lofling et al., 2011; Weiser et al., 2018). Some pneumococcal cell surface proteins also contribute to bacterial survival by limiting complement deposition or inhibiting phagocytosis (Dave et al., 2004; Ren et al., 2004; Hakenbeck et al., 2009; Gutiérrez-Fernández et al., 2016; Yamaguchi et al., 2019). On the other hand, the host recognizes S. pneumoniae and regulates immune responses using pattern recognition receptors, including the Toll-like receptors (TLRs), nucleotide oligomerization domain-like receptors, and retinoic acid-inducible gene-I-like receptors (Koppe et al., 2012). In addition, extracellular bacteria are recognized by TLR2 and TLR4 located on the host cell surface. TLR2 recognizes pneumococcal cell wall components and lipoproteins, while TLR4 senses a pore-forming toxin, pneumolysin (Koppe et al., 2012; Tomlinson et al., 2014). Generally, both TLR2 and TLR4 agonists induce neutrophil activation and inhibit the apoptosis (Sabroe et al., 2005). However, in mouse influenza A virus and S. pneumoniae co-infection model, a TLR2 agonist decreased inflammation and reduced bacterial shedding and transmission (Richard et al., 2014). TLRs play important, but redundant, roles in host defense and regulating inflammatory responses against pneumococcal infection. Appropriate immune responses contribute to pneumococcal clearance, while excessive inflammation can lead to serious tissue damage.
We previously reported that plasmin- and fibronectin-binding protein A (PfbA) plays a role in fibronectin-dependent adhesion to and invasion of epithelial cells, and that an S. pneumoniae PfbA-deficient mutant strain exhibited decreased survival in human blood (Yamaguchi et al., 2008; Yamaguchi, 2018). PfbA is an LPXTG-anchored protein that features a right-handed parallel β-helix with a groove or cleft, formed by three parallel β-sheets and connecting loops (Suits and Boraston, 2013; Beulin et al., 2014). Since the distribution and structural arrangement of the groove residues in the β-helix make it favorable for binding to carbohydrates, PfbA binds to D-galactose, D-mannose, D-glucosamine, D-galactosamine, N-acetylneuraminic acid, D-sucrose, and D-raffinose (Beulin et al., 2017). PfbA also binds to human erythrocytes by interacting with N-acetylneuraminic acids on the cells (Radhakrishnan et al., 2018).
In this study, we investigated the role of PfbA in pneumococcal pathogenesis. Phylogenetic analysis indicated that pfbA genes in S. pneumoniae and Streptococcus pseudopneumoniae were highly conserved and formed an independent cluster from a cluster formed by most mitis group species (Streptococcus mitis, Streptococcus oralis, Streptococcus infantis, Streptococcus gordonii, and Streptococcus anginosus). Our in vitro analysis revealed that PfbA works as an anti-phagocytic factor and that the protein causes NF-κB activation via TLR2. In addition, Toll-interleukin 1 receptor adaptor protein (TIRAP) inhibition increased the survival rate of the pfbA mutant strain after incubation with neutrophils, while the wild-type (WT) strain was not affected. Mouse infection assays suggested that PfbA contributes to pneumococcal survival in at least some organs. However, in a mouse sepsis model, pfbA mutant strain-infected mice showed significantly higher mortality and TNF-α levels in blood. Our findings indicate that PfbA is pneumococcal conserved anti-phagocytic factor and suppresses host excess inflammation.
Materials and Methods
Bacterial Strains and Construction of Mutant Strain
Streptococcus pneumoniae strains were cultured in Todd-Hewitt broth (BD Biosciences, San Jose, CA, USA) supplemented with 0.2% yeast extract THY medium (BD Biosciences) at 37°C. For selection and maintenance of mutants, spectinomycin (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) was added to the medium at 120 μg/mL. The Escherichia coli strain XL10-Gold (Agilent, Santa Clara, CA, USA) was used as a host for derivatives of plasmid pQE-30. All E. coli strains were cultured in Luria-Bertani broth supplemented with 100 μg/mL carbenicillin (Nacalai Tesque, Kyoto, Japan) at 37°C with agitation.
S. pneumoniae TIGR4 isogenic pfbA mutant strain was generated as previously described with minor modifications (Bricker and Camilli, 1999; Yamaguchi et al., 2008; Mori et al., 2012). Briefly, the upstream region of pfbA, an aad9 cassette, the downstream region of pfbA, and pGEM-T Easy vector (Promega, Madison, WI, USA) were amplified by PrimeSTAR® MAX DNA Polymerase (TaKaRa Bio, Shiga, Japan) using the specific primers listed in Supplementary Table 1. The DNA fragments were assembled using a GeneArt® Seamless Cloning and Assembly Kit (Thermo Fisher Scientific, Waltham, MA, USA). The constructed plasmid was then transformed into E. coli XL-10 Gold, and the inserted DNA region was amplified by PCR. The products were used to construct mutant strains by double-crossover recombination with the synthesized competence-stimulating peptide-2. Mutation was confirmed by PCR amplification of genomic DNA isolated from the mutant strain.
Cell Culture
Human promyelocytic leukemia cells (HL-60, RCB0041) were purchased from RIKEN Cell Bank (Ibaraki, Japan). HL-60 cells were maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS, and were incubated at 37°C in 5% CO2. HL-60 cells were differentiated into neutrophil-like cells for 5 days in culture media containing 1.2% DMSO (Collins et al., 1979; Wen et al., 2016). Cell differentiation was confirmed by nitro blue tetrazolium reduction assay (Collins et al., 1979).
Human TLR2/NF-κB/SEAP stably transfected HEK293 cells and human TLR4/MD-2/CD14/NF-κB/SEAP stably transfected HEK293 cells (Novus Biologicals, Centennial, CO, USA, currently sold by InvivoGen, San Diego, CA, USA) were maintained in DMEM with 4.5 g/L glucose, 10% FBS, 4 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin, 100 μg/mL streptomycin, 10 μg/mL blasticidin, and 500 μg/mL G418 and DMEM with 4.5 g/L glucose, 10% FBS, 4 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin, 100 μg/mL streptomycin, 10 μg/mL blasticidin, 2 μg/mL puromycin, 200 μg/mL zeocin, and 500 μg/mL G418, respectively. A secreted alkaline phosphatase reporter assay was performed according to the manufacturer's instructions (Novus Biologicals).
Phylogenetic Analysis
Phylogenetic analysis was performed as described previously (Yamaguchi et al., 2016, 2017, 2019), with minor modifications. Briefly, homologs and orthologs of the pfbA gene were searched using tBLASTn and DELTA-BLAST (Gertz et al., 2006; Boratyn et al., 2012). Sequences from tBLASTn and DELTA-BLAST results with e-values <1 × 10−80 and >40% query coverage were selected for phylogenetic tree analysis. Sequences from incomplete coding sequences were excluded from the DELTA-BLAST results. The sequences were aligned using Phylogears2 (Venditti et al., 2006; Tanabe, 2008) and MAFFT v.7.221 using an L-INS-i strategy (Katoh and Standley, 2013), and ambiguously aligned regions were removed using Jalview or trimAl (Talavera and Castresana, 2007; Capella-Gutiérrez et al., 2009; Waterhouse et al., 2009). The best-fitting codon evolutionary models for phylogenetic analyses were determined using Kakusan4 (Tanabe, 2011). Bayesian Markov chain Monte Carlo analyses were performed with MrBayes v.3.2.6 (Ronquist et al., 2012), with sampling until the standard deviation of split frequencies was 0.01 or 8 × 106 generations. To validate phylogenetic inferences, maximum likelihood phylogenetic analyses were performed with RAxML v.8.1.20 (Stamatakis, 2014). Phylogenetic trees were generated using FigTree v.1.4.4 (Rambaut, 2014) based on the calculated data. The pfbA genes of Gram-positive Granulicatella strains and Streptococcus merionis were used to root as outgroups.
Human Neutrophil and Monocyte Preparation
Human blood was obtained via venipuncture from healthy donors after obtaining informed consent. The protocol was approved by the institutional review boards of Osaka University Graduate School of Dentistry (H26-E43). Human neutrophils and monocytes were prepared using Polymorphprep (Alere Technologies AS, Oslo, Norway), according to the manufacturer's instructions. Human blood was carefully layered on the Polymorphprep solution in centrifugation tubes, which were then centrifuged at 450 × g for 30 min in a swing-out rotor at 20°C. Monocyte and neutrophil fractions were transferred into tubes containing ACK buffer (0.15 M NH4Cl, 0.01 M KHCO3, 0.1 mM EDTA), then centrifuged, washed in phosphate-buffered saline (PBS), and resuspended in RPMI 1640 medium.
Neutrophil Bactericidal Assays
The pneumococcal cells grown to the mid-log phase were resuspended in PBS. TIGR4 strains (3–11 × 103 CFUs/well) with or without rPfbA (0, 10, or 100 nM) were combined with human neutrophils or neutrophil like-differentiated HL-60 cells (2 × 105 cells/well), and R6 strains (1.4–2.0 × 102 CFUs/well) were combined with human neutrophils (1 × 105 cells/well). The mixture was incubated at 37°C in 5% CO2 for 1, 2, and 3 h. Viable cell counts were determined by plating diluted samples onto TS blood agar. The growth index was calculated as the number of CFUs at the specified time point/number of CFUs in the initial inoculum. Bacterial phagocytosis was blocked by addition of cytochalasin D (20 μM), and pneumococcal killing was blocked by protease inhibitor cocktail set V (Merck, Darmstat, Germany; 500 μM AEBSF, 150 nM Aprotinin, 1 μM E-64, and 1 μM leupeptin hemisulfate, EDTA-free) at 1 h before incubation. To determine whether TLR2 and TLR4 signaling affect pneumococcal survival, 100 μM TIRAP (TLR2 and TLR4) inhibitor peptide or control peptide (Novus Biologicals) were added to neutrophils at 1 h before incubation.
Time-Lapse Microscopic Analysis
For time-lapse observations, isolated neutrophils were resuspended in RPMI 1640 at 1 × 106 cells/mL. Next, 10 μL of S. pneumoniae R6 wild type or ΔpfbA strains (1 × 106 CFUs) was added to 2 mL of the cells, and the mixture was incubated and observed at 37°C. Time-lapse images were captured using an Axio Observer Z1 microscope system (Carl Zeiss, Oberkochen, Germany).
Flow Cytometric Analysis of Phagocytes
Recombinant PfbA (rPfbA) or BSA was coated onto 0.5 μm-diameter fluorescent beads (FluoroSphere, Thermo Fisher Scientific), according to the manufacturer's instructions. rPfbA was purified as previously described (Yamaguchi et al., 2008). Isolated neutrophils or monocytes were then resuspended in RPMI 1640 at 1.0 × 107 cells/mL, after which 900 μL of RPMI 1640 containing 1 μL of rPfbA-, BSA-, or non-coated fluorescent beads was added to 100 μL of cells, and then the mixtures were rotated at 37°C for 1 h. The cells were washed twice and fixed with 2% glutaraldehyde-RPMI 1640 at 37°C for 1 h, then washed again three times and analyzed with a CyFlow flow cytometer (Sysmex, Hyogo, Japan) using FlowJo software ver. 8.3.2 (BD Biosciences, Franklin Lakes, NJ, USA).
TLR2/4 SEAPorter Assay
HEK cells expressing TLR2 or TLR4 were stimulated with S. pneumoniae and/or rPfbA for 16 h, according to the manufacturer's instructions (Novus Biologicals). To avoid the effect of bacterial replication on this assay, S. pneumoniae were pasteurized by incubation at 56°C for 30 min. To perform the assay under the same condition, rPfbA was also incubated at 56°C for 30 min. Lipopolysaccharides from E. coli O111:B4 (Sigma-Aldrich Japan Inc., Tokyo, Japan) for the TLR-4 cell line and Pam3CSK4 and Zymozan (Novus Biologicals) for the TLR-2 cell line were used as positive controls under the same conditions. Secreted alkaline phosphatase (SEAP) was analyzed using the SEAPorter Assay (Novus Biologicals) according to the manufacturer's instructions. Quantitative data (ng/mL) were obtained using a standard curve for the SEAP protein.
RNA Extraction and microRNA (miRNA) Array
We performed miRNA array analysis using neutrophil like-differentiated HL-60 cells incubated with S. pneumoniae strains and/or 100 nM rPfbA for 1 h. We compared rPfbA-treated and non-treated cells, wild type and ΔpfbA-infected cells, and ΔpfbA with and without rPfbA-infected cells. In each cell sample, six replicates were pooled and total RNA including microRNA was isolated from the pooled cells by miRNeasy Mini Kit (Qiagen, Hilden, Germany). Approximately 1,000 ng RNA was used for microarray analysis using Affymetrix GeneChip miRNA 4.0 arrays (Affymetrix, Santa Clara, CA, USA) through Filgen Inc. (Nagoya, Japan). Briefly, the quality of total RNA was assessed using a Bioanalyzer 2100 (Agilent). Hybridization was performed using a FlashTag Biotin HSR RNA Labeling Kit, GeneChip Hybridization Oven 645, and GeneChip Fluidics Station 450. The arrays were scanned by Affymetrix GeneChip Scanner 3000 7G. The GeneChip miRNA 4.0 arrays contain 30,424 total mature miRNA probe sets including 2,578 mature human miRNAs, 2,025 pre-miRNA human probes, and 1,196 Human snoRNA and scaRNA probe sets.
Mouse Infection Assays
Mouse infection assays were performed as previously described (Okerblom et al., 2017; Yamaguchi et al., 2017, 2019; Hirose et al., 2018). For the lung infection model, CD-1 mice (Slc:ICR, 8 weeks, female) were infected intratracheally with 4.3–6.7 × 106 CFUs of S. pneumoniae. For intratracheal infection, the vocal cords were visualized using an operating otoscope (Welch Allyn, NY, USA), and 40 μL of bacteria was placed onto the trachea using a plastic gel loading pipette tip. Mouse survival was monitored twice daily for 14 days. At 24 h after intratracheal infection, bronchoalveolar lavage fluid (BALF) was collected following perfusion with PBS.
For the sepsis model, CD-1 mice (Slc:ICR, 8 weeks, female) were infected intravenously with 3.3–6.5 × 105 CFUs of S. pneumoniae via the tail vein. Mouse survival was monitored twice daily for 14 days. At 24 and 48 h after infection, blood aliquots were collected from mice following induction of general euthanasia. Brain, lung, and liver samples were collected following perfusion with PBS. Brain and lung whole tissues as well as the anterior segment of the liver were resected. Bacterial counts in the blood as well as organ homogenates were determined by separately plating serial dilutions, with organ counts corrected for differences in organ weight. Detection limits were 50 CFUs/organ and 50 CFUs/mL in blood.
The concentrations of TNF-α in BALF and plasma were determined using a Duoset® ELISA Kit (R&D Systems, Minneapolis, MN, USA). Mice plasma was obtained by centrifuging the heparinized blood. All mouse experiments were conducted in accordance with animal protocols approved by the Animal Care and Use Committees at Osaka University Graduate School of Dentistry (28-002-0).
Statistical Analysis
Statistical analysis of in vitro and in vivo experiments was performed using a non-parametric analysis, Mann-Whitney U-test, or Kruskal-Wallis test with Dunn's multiple comparisons test. Mouse survival curves were compared using a log-rank test. p < 0.05 was considered to indicate a significant difference. The tests were carried out with Graph Pad Prism version 6.0h or 8.1.2 (GraphPad Software, Inc., San Diego, CA, USA).
Results
The pfbA Gene Is Highly Conserved in S. pneumoniae Among Mitis Group Streptococcus
We searched pfbA-homologs using tBLASTn and DELTA-BLAST and performed phylogenetic analysis (Figure 1 and Supplementary Figure 1). The tBLASTn search of the NCBI Nucleotide collection database showed that pfbA-homologs were present only in S. pneumoniae, S. pseudopneumoniae, S. gordonii, and Streptococcus merionis. To expand the targeted information, we performed DELTA-BLAST using the NCBI Non-redundant proteins sequences database, and utilized the obtained sequences for phylogenetic analysis after excluding incomplete coding sequences. The pfbA gene homologs were identified in mitis group Streptococcus (S. pneumoniae, S. pseudopneumoniae, S. mitis, S. oralis, S. infantis, S. gordonii, and S. anginosus), Sphingomonas paucimobilis, Haemophilus haemolyticus, Granulicatella species, and S. merionis. In these bacteria, S. paucimobilis and H. haemolyticus would have obtained the pfbA gene by occasional horizontal gene transfer from S. pneumoniae, given that the gene was only detected in one strain in each species and showed 100% identity with the pneumococcal variant. Genus Granulicatella bacteria were previously identified as a nutritionally variant Streptococcus (Christensen and Facklam, 2001), and Granulicatella species and S. merionis each contain a gene according to the query, with coverage and identity >50%. S. merionis strain NCTC13788 (also known as WUE3771, DSM 19192, and CCUG 54871), isolated from the oropharynges of Mongolian jirds (Meriones unguiculatus), contained 16S rRNA that belongs in a cluster distinct from the mitis group (Tappe et al., 2009). The pfbA genes in S. pneumoniae and S. pseudopneumoniae are highly conserved and formed an independent cluster from a cluster of mostly other mitis-group strains, whereas this gene showed genetic diversity in other mitis-group bacteria. Interestingly, the pfbA gene of some S. mitis strains belongs to a cluster between pneumococcal clusters. These results indicated that during the evolutionary process, some S. mitis strains lost and re-gained the gene via horizontal gene transfer from S. pneumoniae.

Figure 1. Bayesian phylogenetic analysis of the pfbA gene. The codon-based Bayesian phylogenetic relationship was calculated using the MrBayes program. Strains with identical sequences are listed on the same branch. S. pneumoniae and S. pseudopneumoniae pfbA genes are shaded in cyan. Other mitis group bacterial pfbA genes are shaded in magenta. S. mitis pfbA genes among S. pneumoniae are shaded in yellow. The posterior probabilities are shown near the nodes. The scale bar indicates nucleotide substitutions per site.
PfbA Contributes to Evasion of Neutrophil Killing
To investigate whether PfbA contributes to evasion of neutrophil killing, we determined pneumococcal survival rates after incubation with human neutrophils. After 3 h incubation, the TIGR4 ΔpfbA strain showed a significantly decreased bacterial survival rate. In addition, to clarify whether the observed effects were attributable to PfbA, we also performed the assay using rPfbA. In the presence of 100 nM rPfbA, TIGR4 ΔpfbA strain demonstrated a recovered survival rate nearly equal to that of the wild-type strain (Figure 2A). In pneumococcal survival assays with neutrophil-like differentiated HL-60 cells, TIGR4 strains showed similar results (Figure 2B). We also performed the assay using the non-encapsulated strain R6 and human neutrophils. The R6 ΔpfbA strain showed significantly decreased survival rates as compared to the wild-type strain after incubation for 1, 2, and 3 h (Figure 2C). As the R6 strain showed this phenotype at earlier time points than the TIGR4 strain, we performed pneumococcal survival assays using R6 strains with inhibitors (Figure 2D). Neutrophil phagocytic killing of S. pneumoniae requires the serine proteases (Standish and Weiser, 2009). Thus, in the present study, we used a protein inhibitor cocktail as a positive control of a neutrophil killing inhibitor. While the R6 ΔpfbA strain showed significantly decreased survival rates at 1 h after incubation with human fresh neutrophils in the absence of inhibitors, treatment with an actin polymerization inhibitor, cytochalasin D, reduced the differences among the wild-type and ΔpfbA strains as well as the protein inhibitor cocktail.
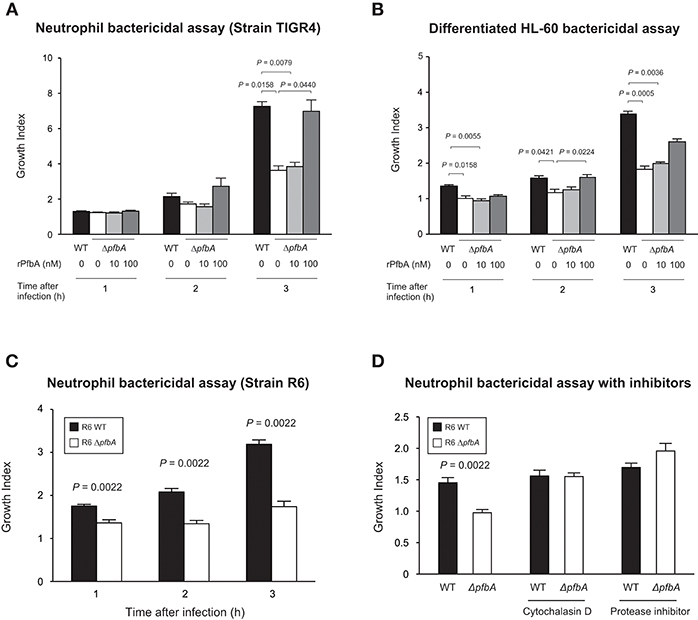
Figure 2. PfbA contributes to pneumococcal survival after incubation with neutrophils. (A) Growth of TIGR4 strains incubated with human fresh neutrophils. (B) Growth of TIGR4 strains incubated with neutrophil-like differentiated HL-60 cells. Bacterial cells were incubated with human neutrophils or differentiated HL-60 cells in the presence or absence of rPfbA for 1, 2, and 3 h at 37°C in a 5% CO2 atmosphere. Next, the mixture was serially diluted and plated on TS blood agar. Following incubation, the number of CFUs was determined. Growth index was calculated by dividing CFUs after incubation by CFUs of the original inoculum. (C) Growth of R6 strains incubated with human fresh neutrophils. S. pneumoniae strains were added to human neutrophils without serum and gently mixed for 1, 2, or 3 h at 37°C. Next, the mixtures were serially diluted and plated on TS blood agar. After incubation, the number of CFUs was determined. (D) Growth of R6 strains incubated with human fresh neutrophils in the presence of inhibitors. S. pneumoniae strains were added to human neutrophils with or without cytochalasin D, or protease inhibitor cocktail in the absence of serum, then gently mixed for 1 h at 37°C. The percent bacterial survival was calculated based on viable counts relative to the wild-type strain. These data are presented as the mean values of six samples, with S.E. values represented by vertical lines. Differences between several groups were analyzed using a Kruskal-Wallis test followed by Dunn's multiple comparisons test (A,B). The Mann-Whitney's U-test was used to compare differences between two independent groups (C,D). Three experiments were performed, with data from a representative experiment shown.
We confirmed the anti-phagocytic activity of PfbA using flow cytometry and PfbA-coated fluorescent beads (Figure 3A). The fluorescence intensity of neutrophils and monocytes incubated with PfbA-coated beads was substantially lower as compared with cells incubated with non- or BSA-coated beads. These results indicated that neutrophils and monocytes phagocytosed the non- and BSA-coated fluorescent beads, whereas the PfbA-coated fluorescent beads escaped phagocytosis by neutrophils and monocytes.
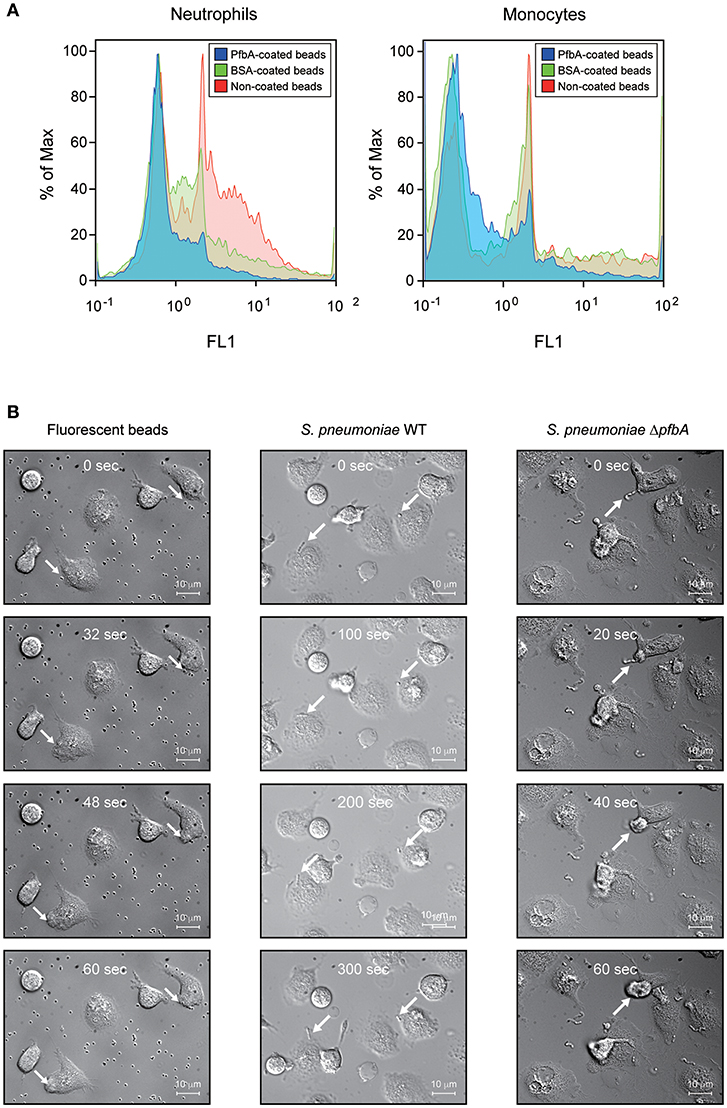
Figure 3. PfbA suppresses host cell phagocytosis. (A) Uptake of fluorescent PfbA-coated beads by neutrophils and monocytes. Human neutrophils and monocytes were separately incubated with PfbA-, BSA-, or non-coated fluorescent beads for 1 h at 37°C. Phagocytic activities were analyzed using flow cytometry. Data are presented as histograms. The value shown for the maximum percentage was determined by normalizing against the peak height (100%). Experiments were performed in triplicate, with data from a representative experiment shown. (B) Time-lapse analysis of the interaction between S. pneumoniae and neutrophils. S. pneumoniae wild-type and ΔpfbA strains were incubated with neutrophils. The elapsed times from contact with neutrophils are shown in the upper part of the figures. Arrows indicate when S. pneumoniae cells or beads contacted neutrophils, or engulfed by a neutrophil phagosome.
We performed real-time observations for time-lapse analysis of the interaction between S. pneumoniae and neutrophils (Figure 3B). S. pneumoniae strain R6 wild-type and ΔpfbA strains were separately incubated with fresh human neutrophils in RPMI 1640 medium. After coming into contact with neutrophils, the ΔpfbA strain was phagocytosed within 1 min, whereas the wild-type strain was not phagocytosed after more than 5 min. Time-lapse analysis also showed the ΔpfbA strain engulfed by neutrophil phagosomes.
In summary, these results showed that pfbA deficiency in both S. pneumoniae TIGR4 and R6 strains decreased the survival rate after incubation with human neutrophils and differentiated HL-60 cells. Furthermore, rPfbA-addition recovered the survival rate of the ΔpfbA strain, with this recovery also observed following treatment with cytochalasin D and a protein-inhibitor cocktail. Additionally, PfbA-coated beads escaped phagocytosis, and time-lapse analysis showed that the ΔpfbA strain was more easily phagocytosed than the WT strain. These findings suggested that PfbA can directly inhibit phagocytosis.
PfbA Works as a TLR2 Ligand and May Inhibit Phagocytosis Through TLR2
Some lectins of pathogens work as ligand for TLR2 and TLR4 (Ricci-Azevedo et al., 2017). We previously reported that PfbA can interact with glycolipid and glycoprotein fractions of red blood cells, several monosaccharides, D-sucrose, and D-raffinose (Beulin et al., 2017; Radhakrishnan et al., 2018). Hence, to determine whether PfbA works as a TLR ligand, we performed a SEAP assay using HEK-293 cells stably transfected with either TLR2 or TLR4, NF-κB, and SEAP (Figure 4A). Pam3CSK4 and Zymozan were used as positive controls for the TLR2 ligand, while LPS was used for TLR4. The SEAP assay indicated that pasteurized S. pneumoniae TIGR4 wild-type cells activated NF-κB via TLR2, whereas ΔpfbA cells did not stimulate cells expressing either TLR2 or TLR4. Pasteurized rPfbA also activated NF-κB dose-dependently through TLR2, but not TLR4. In addition, in the presence of pasteurized rPfbA, ΔpfbA cells activated the cells expressing TLR2. To confirm the effect of pasteurization, we performed this assay using intact and pasteurized bacteria or recombinant proteins (Supplementary Figure 2). Use of the same number of live bacteria generated less amounts of SEAP as compared to that of a smaller number of bacteria, possibly due to cell death caused by the introduction of abundant live bacteria. To minimize this effect, we performed the assay using a smaller number of bacteria and observed that both pasteurized proteins and bacteria exhibited significantly decreased activity relative to intact variants. These results indicated that pasteurization at least partially denatured PfbA and decreased its recognition by TLR2, and that PfbA is responsible for pneumococcal NF-κB activation through TLR2 recognition of the structure.
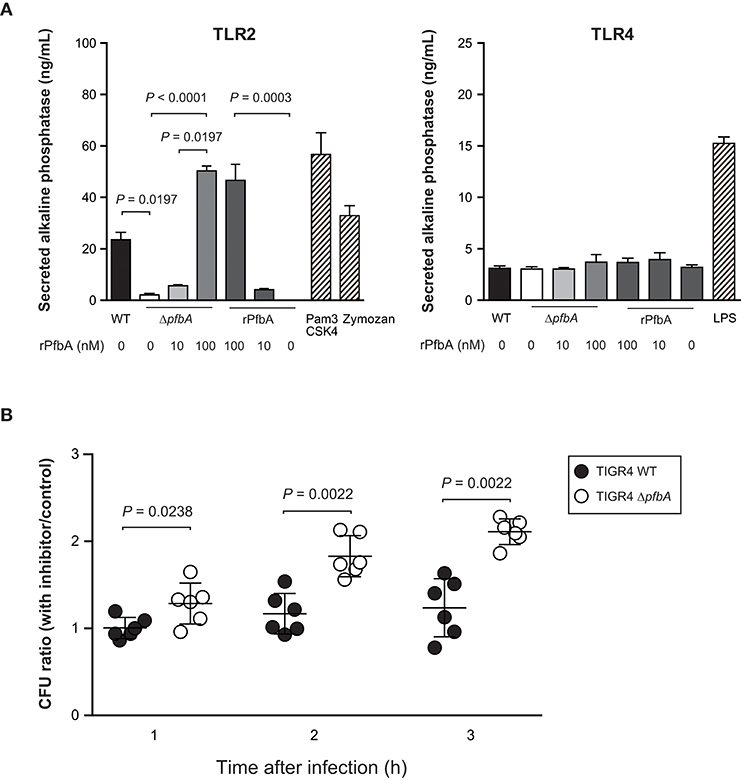
Figure 4. PfbA activates NF-κB via TLR2 and TLR2/4 inhibitor enhances ΔpfbA strain survival. (A) Secreted alkaline phosphatase (SEAP) porter assay using TLR2/NF-κB/SEAPorter or TLR4/MD-2/CD14/NF-κB SEAPorter HEK293 cell lines. The cells were plated in 24-well plates at 5 × 105 cells/well. After 24 h, cells were stimulated with various amount of rPfbA, pasteurized S. pneumoniae (~5 × 106 CFU), 1 μg/mL Pam3CSK4, 10 μg/mL Zymozan, or 25 ng/mL LPS for 24 h. SEAP was analyzed using the SEAPorter Assay Kit. Data are presented as the mean of six wells. SE values are represented by vertical lines. Differences in pneumococcal infection group and rPfbA addition group were analyzed using a Kruskal-Wallis test followed by Dunn's multiple comparisons test, respectively. (B) TLR2/4 inhibitor peptide enhances survival of the TIGR4 ΔpfbA strain incubated with human neutrophils. S. pneumoniae TIGR4 wild type strain or ΔpfbA strain bacteria were incubated with human neutrophils in the presence of TLR2/4 inhibitor peptide or control peptide. After 1, 2, and 3 h, the mixture was serially diluted and plated on TS blood agar. Following incubation, the number of CFUs was determined. The CFU ratio was calculated by dividing CFUs in the presence of inhibitor peptide by CFUs in the presence of control peptide. Data are presented as the mean of six wells. S.E. values are represented by vertical lines. Differences between groups were analyzed using Mann-Whitney's U-test.
Next, to determine whether TLR signaling suppresses survival of pneumococci incubated with neutrophils, we performed a neutrophil survival assay using a TIRAP inhibitor peptide (Figure 4B). Data are presented as the ratio calculated by dividing CFUs in the presence of inhibitor peptide by CFUs in the presence of control peptide. TIRAP is an adaptor protein involved in MyD88-dependent TLR2 and TLR4 signaling pathways. Since the TIRAP inhibitor peptide blocks the interaction between TIRAP and TLRs, the peptide works as a TLR2 and TLR4 inhibitor. The inhibitor peptide treatment increased survival rates of the ΔpfbA strain, but did not affect wild-type survival rates. These results indicated that PfbA contributes to the evasion of neutrophil phagocytosis, and TIRAP inhibitor treatment did not change survival rates of pneumococci incubated with neutrophils. On the other hand, the S. pneumoniae ΔpfbA strain was more easily killed by neutrophils as compared to the wild-type strain, and this phenotype was abolished by the TIRAP inhibitor.
PfbA Deficiency Reduces Pneumococcal Burden in Balf but Does Not Alter Host Survival Rate in a Mouse Pneumonia Model
To investigate the role of PfbA in pneumococcal pathogenesis, we infected mice with S. pneumoniae strains intratracheally and compared bacterial CFUs and TNF-α levels in BALF from mice at 24 h after infection. There were no differences observed in survival time between mice infected with wild type and ΔpfbA strains (Figure 5A). However, recovered CFUs of wild-type bacteria were significantly greater than those of ΔpfbA strains in mouse BALF. In addition, the level of TNF-α in BALF was almost the same in wild type and ΔpfbA infection (Figure 5B).
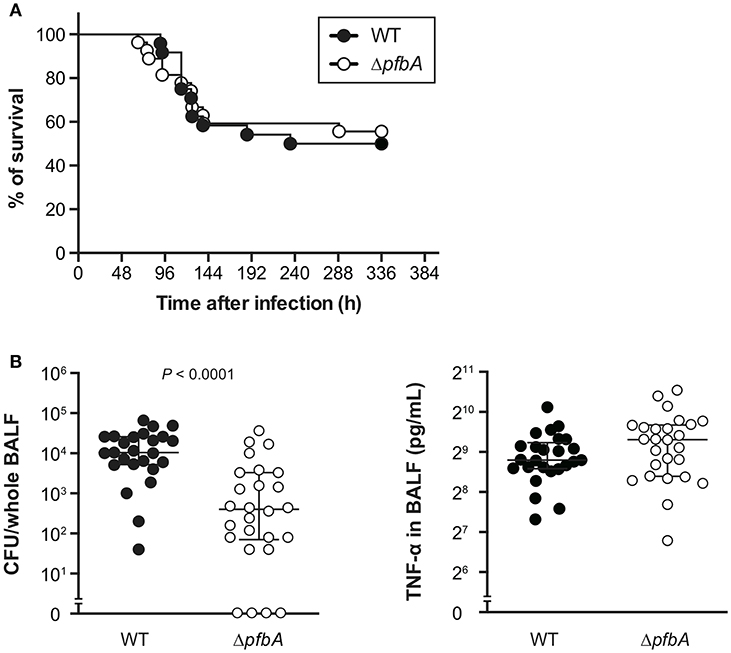
Figure 5. In a mouse pneumonia model, deficiency of pfbA decreases pneumococcal burden in the lung but does not affect host mortality. (A) CD-1 mice were infected intratracheally with the S. pneumoniae TIGR4 wild-type or ΔpfbA strain (3–18 × 106 CFUs). Mice survival was recorded for 14 days. The differences between groups were analyzed using a log-rank test. (B) Bacterial CFUs and TNF-α in BALF collected from CD-1 mice after intratracheal infection with S. pneumoniae. CD-1 mice were infected intratracheally with the S. pneumoniae TIGR4 wild type or ΔpfbA strain (4–7 × 106 CFUs). BALF was collected at 24 h after pneumococcal infection, and bacterial CFUs and TNF-α levels in the BALF were determined. The median and interquartile range (IQR) values are represented by vertical lines. Statistical differences between groups were analyzed using Mann-Whitney's U-test. The data obtained from three independent experiments were pooled.
PfbA Deficiency Increases Pneumococcal Pathogenicity in a Mouse Sepsis Model
We also investigated the role of PfbA in mice following intravenous infection as a model of sepsis. In the infection model, the ΔpfbA strain showed significantly higher levels of virulence as compared to the wild-type strain (Figure 6A). Furthermore, we compared the TNF-α levels in plasma and examined the bacterial burden in blood, brain, lung, and liver samples obtained at 24 and 48 h after intravenous infection (Figures 6B,C and Supplementary Figure 3). At 24 h after infection, TNF-α ELISA findings showed a significantly greater level in the plasma of pfbA mutant strain-infected mice as compared to the wild-type strain-infected mice. The numbers of CFUs of both the wild-type and pfbA mutant strains in the blood and brain samples were comparable. On the other hand, in the lung and liver samples, the pfbA mutant strain-infected mice showed slightly but significantly reduced numbers of CFUs as compared with the wild-type strain-infected mice. At 48 h after infection, there were no significant differences in TNF-α level and bacterial burden in each organ between the wild-type- and pfbA mutant strain-infected mice (Supplementary Figure 3). Bacteria were not detected in the blood of two of the wild-type strain-infected mice and five of the pfbA mutant strain-infected mice. Meanwhile, three of the wild-type strain-infected mice yielded more than 106 CFUs/mL, while seven of the pfbA mutant strain-infected mice did (Supplementary Table 2). The pfbA mutant strain infection caused a polarized bacterial burden in the hosts at 48 h after infection as compared with this not being observed following wild-type strain infection. PfbA deficiency did not increase bacterial burden in mouse organs, but increases pneumococcal pathogenicity in intravenous infection.
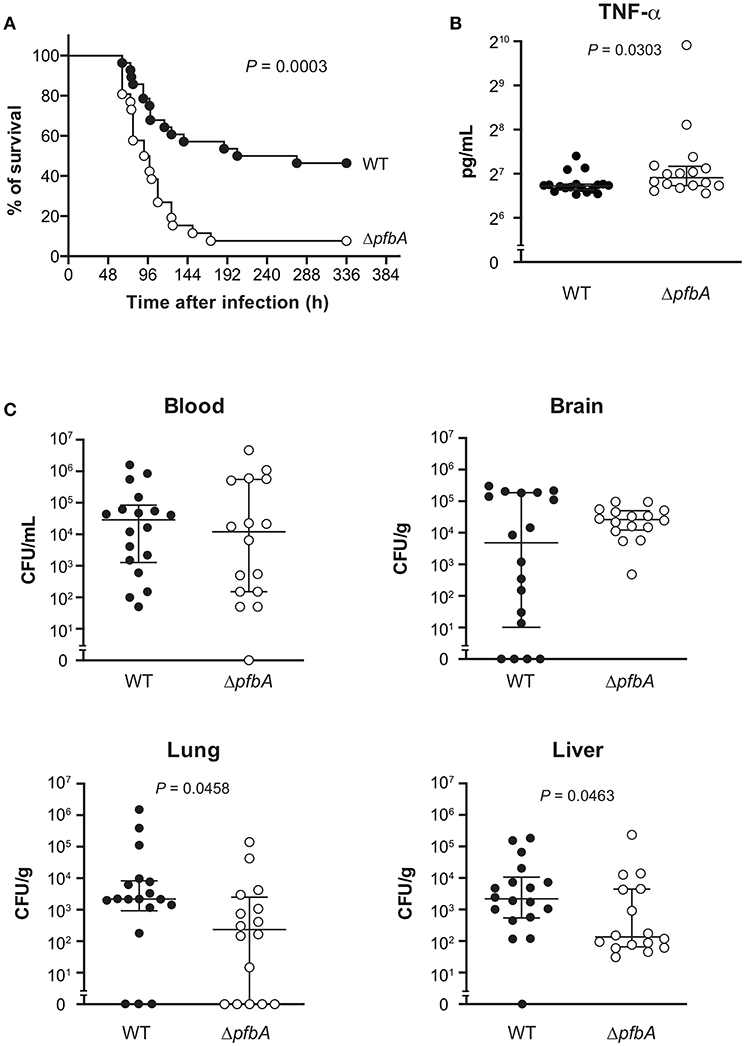
Figure 6. In a mouse sepsis model, the deficiency of pfbA increases the virulence and TNF-α level in blood but decreases the bacterial burden in the lung and liver. CD-1 mice were infected intravenously with the S. pneumoniae TIGR4 wild type or ΔpfbA strain (3–6 × 106 CFUs). (A) Mouse survival was monitored for 14 days. Statistical differences between groups were analyzed using a log-rank test. (B) CD-1 mice were infected intravenously with the S. pneumoniae TIGR4 wild type or ΔpfbA strain (6–9 × 106 CFUs). Plasma samples were collected from these mice at 24 h after infection. Values are presented as the mean of 16 or 18 samples. Vertical lines represent the median ± IQR. Statistical differences between groups were analyzed using Mann-Whitney's U-test. (C) The bacterial burden in the blood, brain, lung, and liver were assessed after 24 h of infection. The median and IQR values are represented by vertical lines. All mice were perfused with PBS after blood collection, organ samples were collected. Statistical differences between groups were analyzed using Mann-Whitney's U-test. The mouse survival data were obtained from three independent experiments, and the TNF-α level and bacterial burden values obtained from two independent experiments were pooled.
Discussion
In the present study, we identified pfbA as a highly conserved gene in S. pneumoniae that contributes to evasion of neutrophil phagocytosis. We determined that PfbA can activate NF-κB through TLR2. TIRAP inhibition increased the survival rate of ΔpfbA strain incubated with neutrophils, while this inhibition did not affect a wild-type strain survival. In a mouse model of lung infection, the bacterial burden of the ΔpfbA strain was significantly reduced as compared with that of the wild-type strain, but the TNF-α level was comparable between the strains. Overall, there was no significant difference in the survival rates of mice infected with the wild-type S. pneumoniae strain and those infected with the ΔpfbA strain. Furthermore, in a mouse model of blood infection, the ΔpfbA strain showed a significantly higher TNF-α level than the wild-type strain. These results suggest that PfbA may suppress the host innate immune response by acting as an anti-phagocytic factor interacting with TLR2.
Prior studies have shown that S. pneumoniae under selective pressure can adapt to the environment by importing genes from other related streptococci, such as those in the mitis group (Bek-Thomsen et al., 2012; Kilian et al., 2014; Jensen et al., 2015; Skov Sorensen et al., 2016). Although S. mitis and S. oralis are oral commensal bacteria, these species contain various pneumococcal virulence factor homologs. Some mitis group strains harbor several CBPs including autolysins and pneumolysin, and cell wall anchoring sialidases (Kilian et al., 2008; Hakenbeck et al., 2009; Johnston et al., 2010). In the present study, we found that the pfbA gene forms an independent cluster and is highly conserved in S. pneumoniae and S. pseudopneumoniae, whereas the gene shows genetic diversity in other mitis group bacteria. Interestingly, species with clear evolutionary separation from the mitis group, Granulicatella species and S. merionis, contained a pfbA ortholog. This result indicates that during the evolutionary process, pfbA was conserved in S. pneumoniae, while other mitis species lost the gene, maintained the gene with genetic diversity, or lost and re-gained the gene by horizontal gene transfer from S. pneumoniae.
Although lipoproteins are major TLR2 ligands as well as peptidoglycans in S. pneumoniae (Tomlinson et al., 2014), we found that rPfbA can activate NF-κB solely in HEK293 cells expressing TLR2, but not those expressing TLR4. As E. coli does not have the capacity to glycosylate proteins (Clausen et al., 2015) and rPfbA cannot be glycosylated, rPfbA-mediated TLR2 activation would be independent of pneumococcal glycosylation. Plant and pathogen lectins can induce NF-κB activation through binding to TLR2 N-glycans, while a classical ligand such as Pam3CSK4 can induce glycan-independent NF-κB activation (Ricci-Azevedo et al., 2017). TLR2 has four N-glycans whose structures still remain unknown, and the N-glycans are critical for the lectins to induce TLR2-mediated activation (Ricci-Azevedo et al., 2017). PfbA binds to various carbohydrates via the groove residues in the β-helix (Beulin et al., 2017; Radhakrishnan et al., 2018). There is a possibility that PfbA induces TLR2 signaling by binding to TLR2 N-glycans.
Although PfbA was capable of activating NF-κB through TLR2, treatment with a TIRAP inhibitor peptide increased the survival rate of the ΔpfbA strain but did not affect the survival rate of the wild type strain in the presence of human neutrophils. This discrepancy might be explained by PfbA-mediated suppression of phagocytosis via the induction of miRNA expression in a TLR2-dependent fashion. Human macrophages challenged with S. pneumoniae induce a negative feedback loop, preventing excessive inflammation via miR-146a and potentially other miRNAs on the TLR2-MyD88 axis (Griss et al., 2016). On the other hand, pneumococcal endopeptidase O enhances macrophage phagocytosis in a TLR2- and miR-155-dependent manner (Yao et al., 2017). Furthermore, miR-9 is induced by TLR agonists and functions in feedback control of the NF-κB-dependent responses in human monocytes and neutrophils (Bazzoni et al., 2009). These studies indicate that host phagocytes are regulated by a complex combination of pattern recognition receptor signaling and miRNA induction. On the other hand, an miRNA array (Supplementary Figure 4) showed that the levels of the involved miRNAs were not changed over 2-fold in the presence or absence of PfbA. One possible hypothesis is that PfbA induces different miRNA responses from classical TLR ligands via glycan-dependent recognition. Although PfbA can downregulate miR-1281 in differentiated HL-60 cells, the role of miR-1281 in phagocytes remains unclear. Further comprehensive studies are required to investigate the role of miRNAs in host innate immunity.
Our results indicated that PfbA contributes to the evasion of opsonin-independent phagocytosis. In S. pneumoniae, several proteins work as anti-opsonophagocytic factors. For example, Pneumococcal surface protein A, Choline-binding protein A, and three surface exoglycosidases, NanA, BgaA, and StrH, inhibit complement-dependent opsonization (Dave et al., 2004; Ren et al., 2004; Dalia et al., 2010). In addition, S. pneumoniae expresses an IgA1 protease capable of cleaving human immunoglobulin A1 (Kilian et al., 1988). On the other hand, there are not so many reports concerning pneumococcal factors against opsonin-independent phagocytosis. Hyams et al. reported that the pneumococcal capsule can inhibit non-opsonic phagocytosis in addition to opsonophagocytosis (Hyams et al., 2010). Moreover, pneumococcal fragments from autolyzed bacteria inhibit phagocytosis of intact pneumococcal cells in human blood (Martner et al., 2009). Furthermore, Choline-binding protein J inhibits neutrophil bactericidal activity (Yamaguchi et al., 2019). However, the detailed mechanisms associated with these activities remain unknown. Recently, receptors involved in non-opsonic phagocytosis were identified. G-protein-coupled formyl peptide receptors directly mediate neutrophil phagocytosis (Wen et al., 2019), and carcinoembryonic antigen-related cell-adhesion molecule 3, a family member expressed on neutrophils, is the receptor responsible for the rapid opsonin-independent phagocytosis and one of the fastest evolving human genes (Adrian et al., 2019). Our study did not exclude the possibility that PfbA is involved in the interaction between these receptors and pneumococcal cells; therefore, further studies are required in this area.
Unexpectedly, our mouse pneumonia and sepsis models indicated that pfbA deficiency reduces pneumococcal survival in the host, but does not decrease or increases host mortality. We previously reported that PfbA works as an adhesin and invasin of host epithelial cells (Yamaguchi et al., 2008). The reduction of bacterial burden in host organs can be explained by the synergy of adhesive and anti-phagocytic abilities. On the other hand, the S. pneumoniae ΔpfbA strain showed equivalent or greater induction of TNF-α as compared with the wild-type strain. Generally, a deficiency of TLR ligands would suppress inflammatory responses. However, a deficiency of PfbA would cause more efficient bacterial uptake by phagocytes and promote inflammatory responses. In addition, there is a possibility that the negative feedback loop induced by PfbA is lost and causes excess inflammation. High mortality does not mean bacterial success, as host death leads to the limitation of bacterial reproduction. PfbA may be beneficial for pneumococcal species by increasing the bacterial reproductive number through suppression of host cell phagocytosis and host mortality. PfbA showed high specificity for and conservation in S. pneumoniae species. The assumed negative feedback loop may not be as significant in non-pathogenic mitis group Streptococcus, given that these commensal bacteria basically do not cause severe inflammation.
In single toxin-induced infectious diseases such as diphtheria and tetanus, highly safe and protective vaccines are established. On the other hand, in multiple factor-induced diseases such as those caused by S. pneumoniae and S. pyogenes, there are either no approved vaccines or existing vaccines still need optimization. Our study indicates that PfbA is a pneumococcal specific cell surface protein, which contributes to evasion from phagocytosis. Additionally, we found that PfbA contributed to pneumococcal evasion from non-opsonic phagocytosis probably through the interaction with TLR2. Therefore, PfbA would not be suitable as a vaccine antigen, since the protein suppresses pneumococcal virulence in a mouse sepsis model. Further investigation of the intricate balance between host immunity and pathogenesis is required to establish the basis for drug and vaccine design.
Data Availability
The datasets generated for this study can be found in the GSE128341.
Ethics Statement
All mouse experiments were conducted in accordance with animal protocols approved by the Animal Care and Use Committees at Osaka University Graduate School of Dentistry (28-002-0). Human blood was obtained via venipuncture from healthy donors after obtaining informed consent. The protocol was approved by the institutional review boards of Osaka University Graduate School of Dentistry (H26-E43).
Author Contributions
MY and SK designed the study. MY performed bioinformatics analyses. MY, YH, MT, and MO performed the experiments. MY, TS, MN, YT, and SK contributed to the setup of the experiments. MY wrote the manuscript. YH, MT, MO, TS, MN, YT, and SK contributed to the writing of the manuscript.
Funding
This study was supported by the Japanese Society for the Promotion of Science (JSPS), KAKENHI [grant numbers 26861546, 15H05012, 16H05847, 16H06279 (PAGS), 16K15787, 17H05103, 17K11666, and 18K19643], the SECOM Science and Technology Foundation, Takeda Science Foundation, GSK Japan Research Grant, Asahi Glass Foundation, Kurata Memorial Hitachi Science and Technology Foundation, Kobayashi International Scholarship Foundation, and the Naito Foundation. Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00301/full#supplementary-material
References
Adrian, J., Bonsignore, P., Hammer, S., Frickey, T., and Hauck, C. R. (2019). Adaptation to host-specific bacterial pathogens drives rapid evolution of a human innate immune receptor. Curr. Biol. 29, 616–630 e615. doi: 10.1016/j.cub.2019.01.058
Bazzoni, F., Rossato, M., Fabbri, M., Gaudiosi, D., Mirolo, M., Mori, L., et al. (2009). Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. U.S.A. 106, 5282–5287. doi: 10.1073/pnas.0810909106
Bek-Thomsen, M., Poulsen, K., and Kilian, M. (2012). Occurrence and evolution of the paralogous zinc metalloproteases IgA1 protease, ZmpB, ZmpC, and ZmpD in Streptococcus pneumoniae and related commensal species. MBio 3:e00303–12. doi: 10.1128/mBio.00303-12
Beulin, D. S., Yamaguchi, M., Kawabata, S., and Ponnuraj, K. (2014). Crystal structure of PfbA, a surface adhesin of Streptococcus pneumoniae, provides hints into its interaction with fibronectin. Int. J. Biol. Macromol. 64, 168–173. doi: 10.1016/j.ijbiomac.2013.11.035
Beulin, D. S. J., Radhakrishnan, D., Suresh, S. C., Sadasivan, C., Yamaguchi, M., Kawabata, S., et al. (2017). Streptococcus pneumoniae surface protein PfbA is a versatile multidomain and multiligand-binding adhesin employing different binding mechanisms. FEBS J. 284, 3404–3421. doi: 10.1111/febs.14200
Bogaert, D., van Belkum, A., Sluijter, M., Luijendijk, A., de Groot, R., Rümke, H. C., et al. (2004). Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363, 1871–1872. doi: 10.1016/S0140-6736(04)16357-5
Boratyn, G. M., Schäffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct 7:12. doi: 10.1186/1745-6150-7-12
Bricker, A. L., and Camilli, A. (1999). Transformation of a type 4 encapsulated strain of Streptococcus pneumoniae. FEMS Microbiol. Lett. 172, 131–135. doi: 10.1111/j.1574-6968.1999.tb13460.x
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Christensen, J. J., and Facklam, R. R. (2001). Granulicatella and Abiotrophia species from human clinical specimens. J. Clin. Microbiol. 39, 3520–3523. doi: 10.1128/JCM.39.10.3520-3523.2001
Clausen, H., Wandall, H. H., Steentoft, C., Stanley, P., and Schnaar, R. L. (2015). “Glycosylation engineering,” in Essentials of Glycobiology, 3rd Edn, eds A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, et al. (New York, NY: Cold Spring Harbor Laboratory Press, 713–728.
Collins, S. J., Ruscetti, F. W., Gallagher, R. E., and Gallo, R. C. (1979). Normal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxide. J. Exp. Med. 149, 969–974. doi: 10.1084/jem.149.4.969
Dalia, A. B., Standish, A. J., and Weiser, J. N. (2010). Three surface exoglycosidases from Streptococcus pneumoniae, NanA, BgaA, and StrH, promote resistance to opsonophagocytic killing by human neutrophils. Infect. Immun. 78, 2108–2116. doi: 10.1128/IAI.01125-09
Dave, S., Carmicle, S., Hammerschmidt, S., Pangburn, M. K., and McDaniel, L. S. (2004). Dual roles of PspC, a surface protein of Streptococcus pneumoniae, in binding human secretory IgA and factor H. J. Immunol. 173, 471–477. doi: 10.4049/jimmunol.173.1.471
Flasche, S., Van Hoek, A. J., Sheasby, E., Waight, P., Andrews, N., Sheppard, C., et al. (2011). Effect of pneumococcal conjugate vaccination on serotype-specific carriage and invasive disease in England: a cross-sectional study. PLoS Med. 8:e1001017. doi: 10.1371/journal.pmed.1001017
GBD 2015 LRI Collaborators (2017). Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 17, 1133–1161. doi: 10.1016/S1473-3099(17)30396-1
Gertz, E. M., Yu, Y. K., Agarwala, R., Schäffer, A. A., and Altschul, S. F. (2006). Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4:41. doi: 10.1186/1741-7007-4-41
Golubchik, T., Brueggemann, A. B., Street, T., Gertz, R. E. Jr., Spencer, C. C., Ho, T., et al. (2012). Pneumococcal genome sequencing tracks a vaccine escape variant formed through a multi-fragment recombination event. Nat. Genet. 44, 352–355. doi: 10.1038/ng.1072
Griss, K., Bertrams, W., Sittka-Stark, A., Seidel, K., Stielow, C., Hippenstiel, S., et al. (2016). MicroRNAs constitute a negative feedback loop in Streptococcus pneumoniae-induced macrophage activation. J. Infect. Dis. 214, 288–299. doi: 10.1093/infdis/jiw109
Gutiérrez-Fernández, J., Saleh, M., Alcorlo, M., Gómez-Mejia, A., Pantoja-Uceda, D., Trevino, M. A., et al. (2016). Modular architecture and unique teichoic acid recognition features of Choline-binding protein L (CbpL) contributing to pneumococcal pathogenesis. Sci. Rep. 6:38094. doi: 10.1038/srep38094
Hakenbeck, R., Madhour, A., Denapaite, D., and Bruckner, R. (2009). Versatility of choline metabolism and choline-binding proteins in Streptococcus pneumoniae and commensal streptococci. FEMS Microbiol. Rev. 33, 572–586. doi: 10.1111/j.1574-6976.2009.00172.x
Hirose, Y., Yamaguchi, M., Goto, K., Sumitomo, T., Nakata, M., and Kawabata, S. (2018). Competence-induced protein Ccs4 facilitates pneumococcal invasion into brain tissue and virulence in meningitis. Virulence 9, 1576–1587. doi: 10.1080/21505594.2018.1526530
Hyams, C., Camberlein, E., Cohen, J. M., Bax, K., and Brown, J. S. (2010). The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 78, 704–715. doi: 10.1128/IAI.00881-09
Jensen, A., Valdorsson, O., Frimodt-Moller, N., Hollingshead, S., and Kilian, M. (2015). Commensal streptococci serve as a reservoir for β-lactam resistance genes in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 59, 3529–3540. doi: 10.1128/AAC.00429-15
Johnston, C., Hinds, J., Smith, A., van der Linden, M., Van Eldere, J., and Mitchell, T. J. (2010). Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J. Clin. Microbiol. 48, 2762–2769. doi: 10.1128/JCM.01746-09
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kawamura, Y., Hou, X. G., Sultana, F., Miura, H., and Ezaki, T. (1995). Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int. J. Syst. Evol. Micrbiol. 45, 406–408. doi: 10.1099/00207713-45-2-406
Kilian, M., Mestecky, J., and Russell, M. W. (1988). Defense mechanisms involving Fc-dependent functions of immunoglobulin A and their subversion by bacterial immunoglobulin A proteases. Microbiol. Rev. 52, 296–303.
Kilian, M., Poulsen, K., Blomqvist, T., Havarstein, L. S., Bek-Thomsen, M., Tettelin, H., et al. (2008). Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS ONE 3:e2683. doi: 10.1371/journal.pone.0002683
Kilian, M., Riley, D. R., Jensen, A., Bruggemann, H., and Tettelin, H. (2014). Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. MBio 5:e01490–14. doi: 10.1128/mBio.01490-14
Kim, L., McGee, L., Tomczyk, S., and Beall, B. (2016). Biological and epidemiological features of antibiotic-resistant Streptococcus pneumoniae in pre- and post-conjugate vaccine eras: a United States perspective. Clin. Microbiol. Rev. 29, 525–552. doi: 10.1128/CMR.00058-15
Koppe, U., Suttorp, N., and Opitz, B. (2012). Recognition of Streptococcus pneumoniae by the innate immune system. Cell. Microbiol. 14, 460–466. doi: 10.1111/j.1462-5822.2011.01746.x
Lofling, J., Vimberg, V., Battig, P., and Henriques-Normark, B. (2011). Cellular interactions by LPxTG-anchored pneumococcal adhesins and their streptococcal homologues. Cell. Microbiol. 13, 186–197. doi: 10.1111/j.1462-5822.2010.01560.x
Martner, A., Skovbjerg, S., Paton, J. C., and Wold, A. E. (2009). Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect. Immun. 77, 3826–3837. doi: 10.1128/IAI.00290-09
McIntyre, P. B., O'Brien, K. L., Greenwood, B., and van de Beek, D. (2012). Effect of vaccines on bacterial meningitis worldwide. Lancet 380, 1703–1711. doi: 10.1016/S0140-6736(12)61187-8
Mitchell, A. M., and Mitchell, T. J. (2010). Streptococcus pneumoniae: virulence factors and variation. Clin. Microbiol. Infect. 16, 411–418. doi: 10.1111/j.1469-0691.2010.03183.x
Mori, Y., Yamaguchi, M., Terao, Y., Hamada, S., Ooshima, T., and Kawabata, S. (2012). α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 287, 10472–10481. doi: 10.1074/jbc.M111.280321
Okerblom, J. J., Schwarz, F., Olson, J., Fletes, W., Ali, S. R., Martin, P. T., et al. (2017). Loss of CMAH during human evolution primed the monocyte-macrophage lineage toward a more inflammatory and phagocytic state. J. Immunol. 198, 2366–2373. doi: 10.4049/jimmunol.1601471
Radhakrishnan, D., Yamaguchi, M., Kawabata, S., and Ponnuraj, K. (2018). Streptococcus pneumoniae surface adhesin PfbA and its interaction with erythrocytes and hemoglobin. Int. J. Biol. Macromol. 120(Pt A), 135–143. doi: 10.1016/j.ijbiomac.2018.08.080
Rambaut, A. (2014). FigTree Ver.1.4.2. Available online at: https://github.com/rambaut/figtree/releases (accessed May 17, 2019).
Ren, B., McCrory, M. A., Pass, C., Bullard, D. C., Ballantyne, C. M., Xu, Y., et al. (2004). The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 173, 7506–7512. doi: 10.4049/jimmunol.173.12.7506
Ricci-Azevedo, R., Roque-Barreira, M. C., and Gay, N. J. (2017). Targeting and recognition of Toll-like receptors by plant and pathogen lectins. Front. Immunol. 8:1820. doi: 10.3389/fimmu.2017.01820
Richard, A. L., Siegel, S. J., Erikson, J., and Weiser, J. N. (2014). TLR2 signaling decreases transmission of Streptococcus pneumoniae by limiting bacterial shedding in an infant mouse Influenza A co-infection model. PLoS Pathog. 10:e1004339. doi: 10.1371/journal.ppat.1004339
Richards, V. P., Palmer, S. R., Pavinski Bitar, P. D., Qin, X., Weinstock, G. M., Highlander, S. K., et al. (2014). Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol. Evol. 6, 741–753. doi: 10.1093/gbe/evu048
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sabroe, I., Dower, S. K., and Whyte, M. K. (2005). The role of Toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clin Infect Dis 41(Suppl. 7), S421–S426. doi: 10.1086/431992
Skov Sorensen, U. B., Yao, K., Yang, Y., Tettelin, H., and Kilian, M. (2016). Capsular polysaccharide expression in commensal Streptococcus species: genetic and antigenic similarities to Streptococcus pneumoniae. MBio 7:e01844–16. doi: 10.1128/mBio.01844-16
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Standish, A. J., and Weiser, J. N. (2009). Human neutrophils kill Streptococcus pneumoniae via serine proteases. J. Immunol. 183, 2602–2609. doi: 10.4049/jimmunol.0900688
Suits, M. D., and Boraston, A. B. (2013). Structure of the Streptococcus pneumoniae surface protein and adhesin PfbA. PLoS ONE 8:e67190. doi: 10.1371/journal.pone.0067190
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tanabe, A. S. (2008). Phylogears2 Ver. 2.0. Available online at: http://www.fifthdimension.jp/ (accessed May 10, 2019).
Tanabe, A. S. (2011). Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol. Ecol. Resour. 11, 914–921. doi: 10.1111/j.1755-0998.2011.03021.x
Tappe, D., Pukall, R., Schumann, P., Gronow, S., Spiliotis, M., Claus, H., et al. (2009). Streptococcus merionis sp. nov., isolated from Mongolian jirds (Meriones unguiculatus). Int. J. Syst. Evol. Microbiol. 59(Pt. 4), 766–770. doi: 10.1099/ijs.0.65823-0
Tomlinson, G., Chimalapati, S., Pollard, T., Lapp, T., Cohen, J., Camberlein, E., et al. (2014). TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J. Immunol. 193, 3736–3745. doi: 10.4049/jimmunol.1401413
Venditti, C., Meade, A., and Pagel, M. (2006). Detecting the node-density artifact in phylogeny reconstruction. Syst. Biol. 55, 637–643. doi: 10.1080/10635150600865567
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Weiser, J. N., Ferreira, D. M., and Paton, J. C. (2018). Streptococcus pneumoniae: transmission, colonization and invasion. Nat. Rev. Microbiol. 16, 355–367. doi: 10.1038/s41579-018-0001-8
Wen, X., Jin, T., and Xu, X. (2016). Imaging G protein-coupled receptor-mediated chemotaxis and its signaling events in neutrophil-like HL60 cells. J. Vis. Exp. 115:e54511. doi: 10.3791/54511
Wen, X., Xu, X., Sun, W., Chen, K., Pan, M., Wang, J. M., et al. (2019). G-protein-coupled formyl peptide receptors play a dual role in neutrophil chemotaxis and bacterial phagocytosis. Mol. Biol. Cell 30, 346–356. doi: 10.1091/mbc.E18-06-0358
WHO (2017). WHO Priority Pathogens List for RandD of New Antibiotics. Available online at: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/
Yamaguchi, M. (2018). Synergistic findings from microbiological and evolutional analyses of virulence factors among pathogenic streptococcal species. J. Oral Biosci. 60, 36–40. doi: 10.1016/j.job.2018.02.004
Yamaguchi, M., Goto, K., Hirose, Y., Yamaguchi, Y., Sumitomo, T., Nakata, M., et al. (2019). Identification of evolutionarily conserved virulence factor by selective pressure analysis of Streptococcus pneumoniae. Commun. Biol. 2:96. doi: 10.1038/s42003-019-0340-7
Yamaguchi, M., Hirose, Y., Nakata, M., Uchiyama, S., Yamaguchi, Y., Goto, K., et al. (2016). Evolutionary inactivation of a sialidase in group B Streptococcus. Sci. Rep. 6:28852. doi: 10.1038/srep28852
Yamaguchi, M., Nakata, M., Sumioka, R., Hirose, Y., Wada, S., Akeda, Y., et al. (2017). Zinc metalloproteinase ZmpC suppresses experimental pneumococcal meningitis by inhibiting bacterial invasion of central nervous systems. Virulence 8, 1516–1524. doi: 10.1080/21505594.2017.1328333
Yamaguchi, M., Terao, Y., Mori, Y., Hamada, S., and Kawabata, S. (2008). PfbA, a novel plasmin- and fibronectin-binding protein of Streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J. Biol. Chem. 283, 36272–36279. doi: 10.1074/jbc.M807087200
Keywords: Streptococcus pneumoniae (pneumococcus), neutrophils (PMNs), phagocytosis, PfbA, TLR (Toll like receptors)
Citation: Yamaguchi M, Hirose Y, Takemura M, Ono M, Sumitomo T, Nakata M, Terao Y and Kawabata S (2019) Streptococcus pneumoniae Evades Host Cell Phagocytosis and Limits Host Mortality Through Its Cell Wall Anchoring Protein PfbA. Front. Cell. Infect. Microbiol. 9:301. doi: 10.3389/fcimb.2019.00301
Received: 10 May 2019; Accepted: 05 August 2019;
Published: 20 August 2019.
Edited by:
Yasuko Rikihisa, The Ohio State University, United StatesReviewed by:
Izabela Sitkiewicz, National Medicines Institute, PolandKondwani C. Jambo, Liverpool School of Tropical Medicine, United Kingdom
Copyright © 2019 Yamaguchi, Hirose, Takemura, Ono, Sumitomo, Nakata, Terao and Kawabata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaya Yamaguchi, yamaguchi@dent.osaka-u.ac.jp; Shigetada Kawabata, kawabata@dent.osaka-u.ac.jp