 Kimberly M. Davis
Kimberly M. Davis- W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, United States
Yersinia pseudotuberculosis replicates within mammalian tissues to form clustered bacterial replication centers, called microcolonies. A subset of bacterial cells within microcolonies interact directly with host immune cells, and other subsets of bacteria only interact with other bacteria. This establishes a system where subsets of Yersinia have distinct gene expression profiles, which are driven by their unique microenvironments and cellular interactions. When this leads to alterations in virulence gene expression, small subsets of bacteria can play a critical role in supporting the replication of the bacterial population, and can drive the overall disease outcome. Based on the pathology of infections with each of the three Yersinia species that are pathogenic to humans, it is likely that this specialization of bacterial subsets occurs during all Yersiniae infections. This review will describe the pathology that occurs during infection with each of the three human pathogenic Yersinia, in terms of the structure of bacterial replication centers and the specific immune cell subsets that bacteria interact with, and will also describe the outcome these interactions have or may have on bacterial gene expression.
Introduction
The genus Yersinia contains three species that are pathogenic to humans: Yersinia pestis, Yersinia pseudotuberculosis, and Yersinia enterocolitica. Y. pseudotuberculosis and Y. enterocolitica are intestinal pathogens that typically cause self-limiting gastroenteritis and mesenteric lymphadenitis (Hubbert et al., 1971; Paff et al., 1976). Y. pestis causes bubonic, septicemic, and pneumonic plague, and has acquired a distinct inoculation route over the course of its divergence from an ancestral Y. pseudotuberculosis, despite relatively few genetic changes (Achtman et al., 1999). Y. pestis is inoculated intradermally into a mammalian host through injection during a flea bite, and Y. pestis can also be directly inhaled to cause primary pneumonic plague. Despite distinctions in inoculation route, all three Yersinia species have a lymphotropism, meaning they quickly traffic to lymph nodes (LNs) or lymphoid tissues and preferentially colonize these tissues. All Yersinia can also spread systemically by accessing the bloodstream and colonizing deep tissue sites, such as the spleen and liver.
Several well-characterized virulence factors play an important role in the disease progression of Yersinia and the ability of the bacteria to survive and proliferate within multiple host tissues. The human pathogenic Yersinia all contain a virulence plasmid termed either pCD1 or pYV, which contains genes encoding a central virulence factor for Yersinia: the type-III secretion system (T3SS) and its associated effector proteins, Yops (Gemski et al., 1980; Portnoy et al., 1981). The Yersinia T3SS is a needle-like structure that injects Yops directly into the host cytosol. Yops collectively function to modulate host signaling pathways to either inhibit or promote inflammation, to inhibit phagocytosis and motility of host cells, and can also inhibit the production and release of antimicrobial substances, such as reactive oxygen species (ROS) (Schesser et al., 1998; Viboud and Bliska, 2005; Songsungthong et al., 2010, Zhang and Bliska, 2010.)
Despite many genetic similarities, there are several well-defined genetic differences between Yersinia species. Y. pestis has acquired two additional virulence plasmids and 32 additional chromosomal genes since its divergence from Y. pseudotuberculosis, but also lost the functionality of as many as 13% of Y. pseudotuberculosis gene products (Achtman et al., 1999; Parkhill et al., 2001; Deng et al., 2002; Chain et al., 2004). Y. pestis pseudogenes include adhesins, and the lack of these genes may impact the ability of the bacterium to adhere to host cells and inject the T3SS (Simonet et al., 1996; Chain et al., 2004). Multiple adhesins: invasin, Ail, YadA, plasminogen activator protease (Pla, expressed from one of the Y. pestis-specific virulence plasmids), and pH 6 antigen, have been shown to play a critical role in adherence to host cells and T3SS injection (Yang and Isberg, 1993; Marra and Isberg, 1997; Felek and Krukonis, 2009; Durand et al., 2010; Felek et al., 2010; Maldonado-Arocho et al., 2013). Y. enterocolitica and Y. pseudotuberculosis utilize invasin, Ail and YadA (Yang and Isberg, 1993; Marra and Isberg, 1997; Hudson and Bouton, 2006; Durand et al., 2010; Maldonado-Arocho et al., 2013; Paczosa et al., 2014; Mühlenkamp et al., 2015). Y. pestis has acquired inactivating mutations in both yadA and invasin, and instead uses Ail, Pla, and pH 6 antigen to promote host cell interactions (Bartra et al., 2008; Felek and Krukonis, 2009; Felek et al., 2010). Ail also plays a major role in promoting serum resistance and preventing complement deposition in all three Yersinia, which allows bacteria to survive within the bloodstream and resist neutrophil-mediated killing (Miller and Falkow, 1988; Bliska and Falkow, 1992; Pierson and Falkow, 1993; Kolodziejek et al., 2007; Bartra et al., 2008).
Yersinia replicate extracellularly within host tissue sites to form clonal bacterial clusters (Simonet et al., 1990; Logsdon and Mecsas, 2006; Oellerich et al., 2007; Crimmins et al., 2012; Davis et al., 2015). The sites of Y. pseudotuberculosis replication have been termed lesions, microcolonies, and recently, pyogranulomas, but each term refers to the same structure, described in Figure 1 (Davis et al., 2015; Peterson et al., 2017; Zhang et al., 2018). This review will first describe disease progression in mammalian hosts, to introduce the different tissues Yersinia replicates within, and then discuss similarities and differences in the inflammatory lesions that form during Yersinia infection, with a focus on how interactions with distinct immune cell subsets may alter the gene expression profile of individual Yersinia cells within a replicating population.
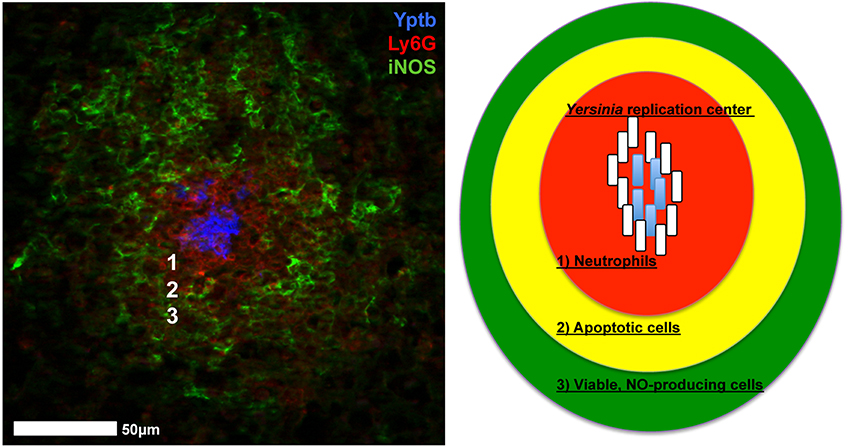
Figure 1. Yersinia replicate to form inflammatory lesions that contain several different types of immune cells. Left: Adapted from Davis et al. (2015). Neutrophils (Ly6G, red) and iNOS+ cells (iNOS, green) are recruited to replicating centers of Y. pseudotuberculosis (Yptb, blue). Right: Diagram of inflammatory lesions. (1) Neutrophils (red) form an inner layer surrounded by (2) Apoptotic cells (yellow; monocytes, some neutrophils). (3) Viable, NO producing cells (green; macrophages, dendritic cells, some neutrophils) form an outer layer of phagocytes. Peripheral bacterial cells (white rods) respond to host cell contact and diffusible antimicrobials, and allow interior bacteria (blue rods) to replicate. White numbers in the micrograph correspond with the numbered regions in the diagram.
Yersinia Human Disease Progression
Enteropathogenic Yersinia Disease Progression
Y. pseudotuberculosis in an enteric pathogen that infects the terminal ileum of the small intestine and also colonizes associated lymphoid tissues, such as Peyer's patches and mesenteric lymph nodes (LNs) (Hubbert et al., 1971; Paff et al., 1976). In healthy individuals, robust neutrophil recruitment is typically sufficient to limit bacterial growth, and can result in clearance. However, in immunocompromised individuals, individuals with hemochromatosis (iron-overload disorder), or in a mouse model of infection, bacteria access the bloodstream and spread to deep tissue sites, such as the spleen and liver (Capron et al., 1981; Abbott et al., 1986; Barnes et al., 2006; Miller et al., 2016). Bacteria replicate within deep tissues to high numbers, which can cause a lethal infection in the mouse model. Replication within intestinal tissues also leads to spread to the cecum, where bacteria associate with cecal lymphoid follicles and can establish a long term, persistent infection in the mouse (Fahlgren et al., 2014; Avican et al., 2015).
Like Y. pseudotuberculosis, Y. enterocolitica is an intestinal pathogen that initially colonizes the intestinal lumen and associated lymphoid tissues, such as Peyer's patches and mesenteric LNs. Y. enterocolitica can also spread to deep tissues following replication at intestinal sites (Heesemann et al., 1993; Viboud and Bliska, 2005; Oellerich et al., 2007). The disease progression for enteric Yersinia appears to be very similar, however, Y. enterocolitica is a relatively common cause of gastroenteritis, whereas patients rarely present with Y. pseudotuberculosis infection. This may be because Y. enterocolitica commonly colonizes many different species of livestock and wild game animals, which could lead to increased exposure rates within human populations, or because Y. enterocolitica may elicit a heightened inflammatory response resulting in more severe disease (Dube et al., 2001; Chlebicz and Slizewska, 2018; Syczylo et al., 2018).
Y. pestis Disease Progression
Y. pestis is inoculated into the dermis of mammalian hosts after replication within the midgut of the flea. Within the flea, bacteria replicate within dense aggregates that partially block the proventricular valve of the flea, thus promoting dissemination into the mammalian host during a flea bloodmeal (Hinnebusch et al., 2017). Y. pestis travels from the injection site rapidly into local draining LNs (dLNs), where colonization is established within the mammalian host. In mouse models of infection, bacteria have been shown to traffic to dLNs intracellularly within dendritic cells and monocytes, and also extracellularly within the lymphatic system (Shannon et al., 2013, 2015; St. John et al., 2014; Gonzalez et al., 2015). The exact route depends on the inoculation (intradermal or subcutaneous) and also the virulence of the bacterial strain. Following initial trafficking, bacteria appear to occupy an extracellular niche. Bacteria initially replicate within dLNs with limited inflammation, however as bacteria continue to replicate, high levels of inflammatory immune cell recruitment and tissue damage occurs. This forms the characteristic inflamed LNs, or buboes, that define bubonic plague (Butler, 1994). Tissue damage within LNs promotes bacterial bloodstream access, which initiates septicemic plague, and leads to colonization of deep tissues such as the spleen and liver (Perry and Fetherston, 1997). Septicemic plague can also occur immediately following flea-borne transmission, if the flea bite occurs deep within dermal tissues (Sebbane et al., 2006a). High levels of bacteria within the bloodstream can promote the development of a secondary pneumonic plague. Following bacterial replication in the lung, infected individuals can expel aerosols containing Y. pestis, which disseminate and cause primary pneumonic plague in new hosts. Septicemic plague also contributes to dissemination when a flea takes a bloodmeal from an infected host, which then restarts the Y. pestis life cycle in the flea vector (Perry and Fetherston, 1997; Hinnebusch et al., 2017).
Host Response to Yersinia, and Yersinia Responses to the Host
Yersinia encounter many different tissue sites during infection of mammalian hosts, which forces bacteria to adapt to multiple different environments within a single host organism. Recruited inflammatory cells attempt to kill Yersinia through phagocytosis or by releasing antimicrobial substances to eliminate extracellular bacteria. Yersinia, in turn, can inhibit phagocytosis and detoxify many of these antimicrobials. The genes required to battle the host response have been largely defined by transcriptional data and mutational analyses, that indicate a given gene or pathway contributes to virulence at the population level. Interestingly, distinct subsets of the bacterial population respond differently, depending on their interactions with, and proximity to, different host cell subsets (Marteyn et al., 2010; Burton et al., 2014; Davis et al., 2015). These studies have described host-driven heterogeneity during Salmonella enterica serovar Typhimurium and Shigella flexineri infections, in addition to Yersinia, which may indicate this is a widespread phenomenon within pathogenic bacterial populations. Although much is still unknown concerning Yersinia heterogeneity within different tissue sites, we will describe what has been shown thus far, and which pathways may also be utilized in other tissues based on similarities in host cell interactions.
Lymph Nodes
It has been hypothesized that the ability of Y. pestis to disseminate from dermal tissues and replicate within draining lymph nodes (dLNs) is due to the presence of Pla protease, which was acquired on an additional virulence plasmid after Y. pestis diverged from Y. pseudotuberculosis (Welkos et al., 1997; Lathem et al., 2007). However, Y. pseudotuberculosis can also effectively replicate within the dermis and traffic to dLNs following intradermal inoculation (Guinet et al., 2008, 2015). The major pathological difference during infection with the two species was the ability of the immune response to contain bacterial replication. Y. pseudotuberculosis elicits robust neutrophil recruitment that surrounds and contains bacteria, whereas Y. pestis exists as individual bacteria and reaches heightened bacterial loads (Guinet et al., 2008). Y. pestis has acquired the ability to produce LPS with poor stimulatory activity and also produces an anti-phagocytic capsule, both of which could limit neutrophil interactions (Montminy et al., 2006; Dudte et al., 2017). There were also many examples of Y. pestis clustered together within the same infected tissues, but it was unclear if clustering promotes bacterial growth or clearance by the host. In the absence of clustering, Y. pseudotuberculosis replication within mesenteric LNs causes a lethal infection in the mouse, suggesting clustering promotes host survival (Peterson et al., 2017). Y. pseudotuberculosis was contained by a layer of apoptotic monocytes (Figure 1), which may also contain Y. pseudotuberculosis within dLNs. These studies suggest that the formation of microcolony, or pyogranuloma, structure contains infection and protects the host, however this structure also allows bacteria to cooperate and promote their own growth (Davis et al., 2015).
Y. pestis replication within dLNs is characterized by high bacterial loads and high levels of tissue destruction. Replication of Y. pestis within dLNs initially induces very little host cytokine expression, which is actively inhibited by components of the virulence plasmid including the T3SS (Comer et al., 2010). At later timepoints, neutrophils and additional phagocytes are recruited to infected LNs, and IL-17 is produced (Comer et al., 2010). In response, Y. pestis increases expression of reactive nitrogen species (RNS) detoxifying genes and iron scavenging genes, indicating Y. pestis is being exposed to the antimicrobial diffusible gas, nitric oxide (NO), and has limited access to iron during replication within the dLN (Sebbane et al., 2006b). NO was produced by recruited neutrophils, and likely other phagocytes, within the rat bubo, and the detoxifying gene, hmp, played an important role in promoting Y. pestis virulence (Sebbane et al., 2006b). It is likely that bacteria in the closest proximity to NO-producing phagocytes may specifically respond to this stress, and detoxify NO to protect other members of the population (Davis et al., 2015), although this hasn't yet been investigated. It will also be interesting to determine if iron limitation is uniformly experienced across LNs, or if this is specific to a subset of cells. Interestingly there was little evidence for a bacterial response to reactive oxygen species (ROS) in dLNs, and only one superoxide dismutase increased in transcript levels. A mutant strain lacking this superoxide dismutase (sodA) was fully virulent, which indicates Y. pestis effectively inhibits this response within LNs (Sebbane et al., 2006b).
Deep Tissues
Multiple immune cell types are recruited to deep tissues during infection, and Yersinia effectively responds to create a hospitable environment for bacterial replication. Neutrophils directly interact with clustered centers of extracellular Y. pseudotuberculosis, and additional phagocytes are maintained at a distance relative to the bacterial centers (Figure 1). The layer of NO-producing cells likely contains a mixed population of monocytes, macrophages, and dendritic cells, and is adjacent to an apoptotic cell layer likely composed of monocytes (Davis et al., 2015; Peterson et al., 2017; Zhang et al., 2018). In the spleen, only the bacteria at the periphery of replicating centers respond to NO, and detoxify NO to prevent further diffusion into the microcolony, thus protecting interior bacteria (Davis et al., 2015). NO was detoxified by Hmp, which was expressed in response to the Nos2 gene product, inducible nitric oxide synthase, indicating hmp expression was a specific response to NO. This example of cooperative behavior likely also exists within mesenteric LNs, based on the recruitment of NO-producing cells, and the similarities in these microcolony or pyogranuloma structures (Zhang et al., 2018). Peripheral Y. pseudotuberculosis cells also respond to neutrophil contact in the spleen by expressing heightened levels of the T3SS (Pettersson et al., 1996; Davis et al., 2015). It remains unclear if heightened expression signifies a T3SS translocation event, or if this is due to prolonged host cell contact. Heightened T3SS expression could occur in multiple tissue sites and response to many host cell types, as it is clear that Yersinia can inject T3SS effector proteins into many different immune cell types during infection (Durand et al., 2010).
Y. pestis and Y. enterocolitica actively inhibit immune cell recruitment to the spleen by limiting CCR2 signaling and promoting Gal-1 expression, respectively (Kerschen et al., 2004; Ye et al., 2009, 2011; Davicino et al., 2017). In the liver, inflammatory monocytes and dendritic cells are recruited to sites of Y. pestis infection, and YopM promotes cell death of neutrophils and macrophages leading to the formation of an apoptotic layer of host cells around replicating bacterial centers (Ye et al., 2014). This mirrors the layer of apoptotic cells observed within Y. pseudotuberculosis-infected mesenteric LNs (Figure 1) (Peterson et al., 2017). iNOS+ cells are also recruited to Y. pestis and Y. enterocolitica lesions, suggesting that subsets of peripheral cells may also express hmp to protect other individuals within the population. This also suggests that there may be quite a few similarities in the host response to Yersinia within deep tissues, unlike the pathological distinctions that are observed within infected LNs. This also indicates that all Yersinia may respond to similar stresses within deep tissues, and supports the idea that the cooperative behavior described for Y. pseudotuberculosis may also occur within other Yersinia infections, potentially even by expressing the same stress response pathways in distinct subsets of bacteria.
Neutrophils are a major producer of reactive oxygen species (ROS), but production can be inhibited by the T3SS, so it remains unclear if ROS restricts Yersinia growth in tissues. Mutant Y. enterocolitica lacking superoxide dismutase (sodA) were attenuated within the spleen and liver, but this could be due to sensitivity to endogeneous ROS generated during bacterial aerobic respiration, or host-derived ROS (Roggenkamp et al., 1997). Transcriptional responses to ROS have not been observed with Y. pestis or Y. pseudotuberculosis, and a Y. pestis sodA mutant was not attenuated for growth in buboes or systemic infection (Sebbane et al., 2006b; Davis et al., 2015). However, a ROS-sensitive Y. pseudotuberculosis dusB-fis mutant was attenuated in immunocompetent mice, and rescued in the absence of ROS production, suggesting ROS was produced during infection (Green et al., 2016). Fis is a nucleoid-associated protein that likely has pleiotropic effects on the bacterial response to ROS, which may explain why the dusB-fis mutant uncovered an impact of ROS on Y. pseudotuberculosis growth. WT Y. pseudotuberculosis growth was not impacted by the presence or absence of host-derived ROS, indicating the WT strain can effectively defend itself from ROS without significant transcriptional changes (Davis et al., 2015; Green et al., 2016).
Intestinal Tissue and Peyer's Patches
Following intestinal colonization, enteropathogenic Yersinia traverse M cells to form micrcolonies within Peyer's patches, which are specialized lymphoid tissues associated with the intestinal epithelium. The immune response to Y. pseudotuberculosis and Y. enterocolitica in Peyer's patches is characterized by neutrophil influx and a mixed TH17/TH1 response, in contrast to the TH17-specific response to Y. pestis within dLNs (Comer et al., 2010; Davicino et al., 2017; Nuss et al., 2017). Y. pseudotuberculosis responds by expressing genes to prevent phagocytosis and counteract iron deprivation and RNS, similar to Y. pestis within dLNs (Sebbane et al., 2006b; Nuss et al., 2017). Y. enterocolitica also actively inhibits neutrophil recruitment by a YopH-dependent mechanism that reduces CXCR2 surface expression on neutrophils (Dave et al., 2016). Microcolonies within the lamina propria contain CD4+ T cells and inflammatory macrophage and/or dendritic cell populations, in addition to CD8+ T cells. Interaction with CD8+ tissue-resident memory T cells within the lamina propria can effectively contain Y. pseudotuberculosis replication within the terminal ileum (Bergsbaken and Bevan, 2015).
Intestinal infection with sublethal doses of Y. pseudotuberculosis can establish a persistent, asymptomatic infection with sustained fecal shedding of bacteria, which may promote dissemination to additional hosts (Fahlgren et al., 2014; Avican et al., 2015). Persistence led to a dramatic shift in bacterial gene expression, which resembled growth at room temperature in bacteriological media, and was characterized by heightened expression of flagella and invasin, and downregulation of the T3SS (Avican et al., 2015; Heine et al., 2018). RovA was involved in these changes, and is known to positively regulate invasin and pH6 antigen expression. RovA expression is bistable in bacteriological media between 30 and 34°C, indicating bacteria utilize a bet-hedging approach to prepare a subset of the population to invade host cells, potentially as bacteria are moving through the host environment and the temperature is increasing (Quade et al., 2012; Nuss et al., 2016). RovA expression was also detected within a subset of the bacterial population replicating within the cecum, suggesting that RovA expression could be another example of cooperative behavior (Nuss et al., 2016).
Lungs
Very little inflammation is observed early during primary pneumonic Y. pestis infection, similar to early timepoints within LNs during bubonic plague (Lathem et al., 2005; Guinet et al., 2008; Comer et al., 2010). However, by 48 h post-infection, inflammatory cells are localized around centers of replicating bacteria and bacteria are responding to the host environment (Lathem et al., 2005). Within the lung, Y. pestis upregulates expression of the T3SS, iron acquisition genes, and RNS detoxifying genes, including hmp (Lathem et al., 2005). In contrast, expression of pH 6 antigen, Pla, and ROS detoxifying genes were downregulated, indicating bacteria were not responding to ROS, and these adhesins was not required to replicate within the lungs (Lathem et al., 2005). Similar results were seen in a Y. pseudotuberculosis lung infection model, where Ail and YadA contribute to replication and dissemination to deep tissues, but pH 6 antigen was dispensible (Paczosa et al., 2014). It will be interesting to determine if heightened gene expression occurs within all the individual Yersinia cells replicating within the lung, or if distinct subsets of cells respond in different ways. Based on experiments with Y. pseudotuberculosis, individual Y. pestis cells may be expressing heightened levels of the T3SS or heightened levels of hmp depending on their spatial location relative to immune cell subsets.
Concluding Remarks
As Yersinia replicate within tissues, they form microcolonies where some individual cells interact directly with host immune cells, and other cells only interact with other bacteria. Yersinia species elicit similar inflammatory responses, characterized by recruitment of neutrophils, monocytes, macrophages, and dendritic cells, and all Yersinia appear to react similarly by upregulating expression of anti-phagocytic factors, RNS detoxifying genes, and iron scavenging genes. Several key questions remain: Are microcolonies protective for the host, or do they promote bacterial replication? Can we alter the host response to better contain bacterial growth? Limiting the host response impacts the containment of Yersinia within microcolonies and increases virulence, suggesting microcolonies protect the host (Peterson et al., 2017). However, disruption of microcolonies by attenuating Yersinia can promote clearance of bacteria, suggesting there is also a protective effect for bacteria (Davis et al., 2015). More future studies will be needed to determine if the host immune response can be manipulated to better contain and clear infection.
We are only beginning to understand how host cell interactions are driving differences in bacterial gene expression, and how small subsets of the bacterial population may be producing virulence genes that are critical for promoting disease. Better understanding and characterization of the heterogeneity in gene expression across bacterial populations, specifically in virulence gene expression, will have important implications for identifying potential drug targets for novel therapeutics. We believe the heterogeneity within Yersinia populations is likely also present within populations of other bacterial pathogens, and may be a widespread phenomenon that needs to be taken into account when designing antimicrobial therapeutics.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author of this manuscript declares no conflict of interest. This work was supported by the NIAID Career Transition award, 1K22AI123465-01.
References
Abbott, M., Galloway, A., and Cunningham, J. (1986). Haemochromatosis presenting with a double Yersinia infection. J. Infect. 13, 143–145. doi: 10.1016/S0163-4453(86)92869-0
Achtman, M., Zurth, K., Morelli, G., Torrea, G., Guiyoule, A., and Carniel, E. (1999). Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U.S.A. 96, 14043–14048. doi: 10.1073/pnas.96.24.14043
Avican, K., Fahlgren, A., Huss, M., Heroven, A., Beckstette, M., Dersch, P., et al. (2015). Reprogramming of Yersinia from virulent to persistent mode revealed by complex in vivo RNA-seq analysis. PLoS Pathog. 11:e1004600. doi: 10.1371/journal.ppat.1004600
Barnes, P. D., Bergman, M. A., Mecsas, J., and Isberg, R. R. (2006). Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J. Exp. Med. 203, 1591–1601. doi: 10.1084/jem.20060905
Bartra, S. S., Styer, K. L., O'Bryant, D. M., Nilles, M. L., Hinnebusch, B. J., Aballey, A., et al. (2008). Resistance of Yersinia pestis to complement-dependent killing is mediated by the ail outer membrane protein. Infect. Immun. 76, 612–622. doi: 10.1128/IAI.01125-07
Bergsbaken, T., and Bevan, M. (2015). Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat. Immunol. 16, 406–414. doi: 10.1038/ni.3108
Bliska, J. B., and Falkow, S. (1992). Bacterial resistance to complement killing mediated by the ail protein of Yersinia enterocolitica. Proc. Natl. Acad. Sci. U.S.A. 89, 3561–3565. doi: 10.1073/pnas.89.8.3561
Burton, N., Schürmann, N., Casse, O., Steeb, A. K., Claudi, B., Zankl, J., et al. (2014). Disparate impact of oxidative host defenses determines the fate of salmonella during systemic infection of mice. Cell Host Microbe 15, 72–83. doi: 10.1016/j.chom.2013.12.006
Butler, T. (1994). Yersinia infections: centennial of the discovery of the plague bacillus. Clin. Infect. Dis. 19, 655–661. doi: 10.1093/clinids/19.4.655
Capron, J., Delamarre, J., Delcenserie, R., Gineston, J., Dupas, J., and Lorriaux, A. (1981). Liver absess complicating Yersinia pseudotuberculosis ileitis. Gastroenterology 81, 150–152.
Chain, P. S., Carniel, E., Larimer, F. W., Lamerdin, J., Stoutland, P. O., Regala, W. M., et al. (2004). Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U.S.A. 101, 13826–13831. doi: 10.1073/pnas.0404012101
Chlebicz, A., and Slizewska, K. (2018). Campylobacteriosis, salmonellosis, yersiniosis, and listeriosis as zoonotic foodborne diseases: a review. Int. J. Environ. Res. Public Health 15:E863. doi: 10.3390/ijerph15050863
Comer, J. E., Sturdevant, D. E., Carmody, A. B., Virtaneva, K., Gardner, D., Long, D., et al. (2010). Transcriptomic and innate immune responses to Yersinia pestis in the lymph node during bubonic plague. Infect. Immun. 78, 5086–5098. doi: 10.1128/IAI.00256-10
Crimmins, G. T., Mohammadi, S., Green, E. R., Bergman, M. A., Isberg, R. R., and Mecsas, J. (2012). Identification of MrtAB, an ABC transporter specifically required for Yersinia pseudotuberculosis to colonize the mesenteric lymph nodes. PLoS Pathog. 8:e1002828. doi: 10.1371/journal.ppat.1002828
Dave, M. N., Silva, J. E., Eliçabe, R. J., Jeréz, M. B., Filippa, V. P., Gorlino, C. V., et al. (2016). Yersinia enterocolitica YopH-deficient strain activates neutrophil recruitment to Peyer's patches and promotes clearance of the virulent strain. Infect. Immun. 84, 3172–3181. doi: 10.1128/IAI.00568-16
Davicino, R. C., Méndez-Huergo, S. P., Eliçabe, R. J., Stupirski, J. C., Autenrieth, I., De Genaro, M. S., et al. (2017). Galectin-1-driven tolerogenic programs aggravate Yersinia enterocolitica infection by repressing antibacterial immunity. J. Immunol. 199, 1382–1392. doi: 10.4049/jimmunol.1700579
Davis, K. M., Mohammadi, S., and Isberg, R. R. (2015). Community behavior and spatial regulation within a bacterial microcolony in deep tissue sites serves to protect against host attack. Cell Host Microbe 17, 21–31. doi: 10.1016/j.chom.2014.11.008
Deng, W., Burland, V., Plunkett, G., Boutin, A., Mayhew, G. F., Liss, P., et al. (2002). Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184, 4601–4611. doi: 10.1128/JB.184.16.4601-4611.2002
Dube, P. H., Revell, P. A., Chaplin, D. D., Lorenz, R. G., and Miller, V. L. (2001). A role for IL-1 alpha in inducing pathologic inflammation during bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 98, 10880–10885. doi: 10.1073/pnas.191214498
Dudte, S. C., Hinnebusch, B. J., and Shannon, J. G. (2017). Characterization of Yersinia pestis interactions with human neutrophils in vitro. Front. Cell. Infect. Microbiol. 7:358. doi: 10.3389/fcimb.2017.00358
Durand, E. A., Maldonado-Arocho, F. J., Castillo, C., Walsh, R. L., and Mecsas, J. (2010). The presence of professional phagocytes dictates the number of host cells targeted for Yop translocation during infection. Cell. Microbiol. 12, 1064–1082. doi: 10.1111/j.1462-5822.2010.01451.x
Fahlgren, A., Avican, K., Westermark, L., Nordfeith, R., and Fällman, M. (2014). Colonization of cecum is important for development of persistent infection by Yersinia pseudotuberculosis. Infect. Immun. 82, 3471–3482. doi: 10.1128/IAI.01793-14
Felek, S., and Krukonis, E. (2009). The Yersinia pestis ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77, 825–836. doi: 10.1128/IAI.00913-08
Felek, S., Tsang, T. M., and Krukonis, E. S. (2010). Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 78, 4134–4150. doi: 10.1128/IAI.00167-10
Gemski, P., Lazere, J. R., and Casey, T. (1980). Plasmid associated with pathogenicity and calcium dependency of Yersinia enterocolitica. Infect. Immun. 27, 682–685.
Gonzalez, R. J., Lane, M. C., Wagner, N. J., and Weening, E. H, Miller, V. L. (2015). Dissemination of a highly virulent pathogen: tracking the early events that define infection. PLoS Pathog. 11:e1004587. doi: 10.1371/journal.ppat.1004587
Green, E. R., Clark, S., Crimmins, G. T., Mack, M., Kumamoto, C. A., and Mecsas, J. (2016). Fis is essential for Yersinia pseudotuberculosis virulence and protects against reactive oxygen species produced by phagocytic cells during infection. PLoS Pathog. 12:e1005898. doi: 10.1371/journal.ppat.1005898
Guinet, F., Avé, P., Filali, S., Huon, C., Savin, C., Huerre, M., et al. (2015). Dissociation of tissue destruction and bacterial expansion during bubonic plague. PLoS Pathog. 11:e1005222. doi: 10.1371/journal.ppat.1005222
Guinet, F., Avé, P., Jones, L., Huerre, M., and Carniel, E. (2008). Defective innate cell response and lymph node infiltration specify Yersinia pestis infection. PLoS ONE 3:e1688. doi: 10.1371/journal.pone.0001688
Heesemann, J., Gaede, K., and Autenrieth, I. (1993). Experimental Yersinia enterocolitica infection in rodents: a model for human yersiniosis. APMIS 101, 417–429. doi: 10.1111/j.1699-0463.1993.tb00130.x
Heine, W., Beckstette, M., Heroven, A. K., Thiemann, S., Heise, U., Nuss, A. M., et al. (2018). Loss of CNFY toxin-induced inflammation drives Yersinia pseudotuberculosis into persistency. PLoS Pathog. 14:e1006858. doi: 10.1371/journal.ppat.1006858
Hinnebusch, B. J., Jarrett, C. O., and Bland, D. M. (2017). “Fleaing” the plague: adaptations of Yersinia pestis to its insect vector that lead to transmission. Annu. Rev. Microbiol. 71, 215–232. doi: 10.1146/annurev-micro-090816-093521
Hubbert, W., Petenyi, C., Glasgow, L., Uyeda, C., and Creighton, S. (1971). Yersinia pseudotuberculosis infection in the United States. Septicemia, appendicitis, and mesenteric lymphadenitis. Am. J. Trop. Med. Hyg. 20, 679–684. doi: 10.4269/ajtmh.1971.20.679
Hudson, K. J., and Bouton, A. H. (2006). Yersinia pseudotuberculosis adhesins regulate tissue-specific colonization and immune cell localization in a mouse model of systemic infection. Infect. Immun. 74, 6487–6490. doi: 10.1128/IAI.00718-06
Kerschen, E. J., Cohen, D. A., Kaplan, A. M., and Straley, S. C. (2004). The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect. Immun. 72, 4589–4602. doi: 10.1128/IAI.72.8.4589-4602.2004
Kolodziejek, A. M., Sinclair, D. J., Seo, K. S., Schnider, D. R., Deobald, C. F., Rohde, H. N., et al. (2007). Phenotypic characterization of OmpX, an ail homologue of Yersinia pestis KIM. Microbiology 153, 2941–2951. doi: 10.1099/mic.0.2006/005694-0
Lathem, W. W., Crosby, S. D., Miller, V. L., and Goldman, W. E. (2005). Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 102, 17786–17791. doi: 10.1073/pnas.0506840102
Lathem, W. W., Price, P. A., Miller, V. L., and Goldman, W. E. (2007). A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315, 509–513. doi: 10.1126/science.1137195
Logsdon, L. K., and Mecsas, J. (2006). The proinflammatory response induced by wild-type Yersinia pseudotuberculosis infection inhibits survival of yop mutants in the gastrointestinal tract and Peyer's patches. Infect. Immun. 74, 1516–1527. doi: 10.1128/IAI.74.3.1516-1527.2006
Maldonado-Arocho, F. J., Green, C., Fisher, M. L., Paczosa, M. K., and Mecsas, J. (2013). Adhesins and host serum factors drive Yop translocation by Yersinia into professional phagocytes during animal infection. PLoS Pathog. 9:e1003415. doi: 10.1371/journal.ppat.1003415
Marra, A., and Isberg, R. (1997). Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer's patch intestinal epithelium. Infect. Immun. 65, 3412–3421.
Marteyn, B., West, N. P., Browning, D. F., Cole, J. A., Shaw, J. G., Palm, F., et al. (2010). Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465, 355–358. doi: 10.1038/nature08970
Miller, H. K., Schwiesow, L., Au-Yeung, W., and Auerbuch, V. (2016). Hereditary hemochromatosis predisposes mice to Yersinia pseudotuberculosis infection even in the absence of the type III secretion system. Front. Cell. Infect. Microbiol. 6:69. doi: 10.3389/fcimb.2016.00069
Miller, V. L., and Falkow, S. (1988). Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 56, 1242–1248.
Montminy, S. W., Khan, N., McGrath, S., Walkowicz, M. J., Sharp, F., Conlon, J. E., et al. (2006). Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 7, 1066–1073. doi: 10.1038/ni1386
Mühlenkamp, M., Oberhettinger, P., Leo, J. c., Linke, D., and Schütz, M. S. (2015). Yersinia adhesin A (YadA)–beauty & beast. Int. J. Med. Microbiol. 305, 252–258. doi: 10.1016/j.ijmm.2014.12.008
Nuss, A. M., Beckstette, M., Pimenova, M., Schmühl, C., Opitz, W., Pisano, F., et al. (2017). Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 114, E791–E800. doi: 10.1073/pnas.1613405114
Nuss, A. M., Schuster, F., Roselius, L., Klein, J., Bücker, R., Herbst, K., et al. (2016). A precise temperature-responsive bistable switch controlling Yersinia virulence. PLoS Pathog. 12:e1006091. doi: 10.1371/journal.ppat.1006091
Oellerich, M. F., Jacobi, C. A., Freund, S., Niedung, K., Bach, A., Heesemann, J., et al. (2007). Yersinia enterocolitica infection of mice reveals clonal invasion and abscess formation. Infect. Immun. 75, 3802–3811. doi: 10.1128/IAI.00419-07
Paczosa, M. K., Fisher, M. L., Maldonado-Arocho, F. J., and Mecsas, J. (2014). Yersinia pseudotuberculosis uses Ail and YadA to circumvent neutrophils by directing Yop translocation during lung infection. Cell Microbiol. 16, 247–268. doi: 10.1111/cmi.12219
Paff, J. R., Triplett, D. A., and Saari, T. N. (1976). Clinical and laboratory aspects of Yersinia pseudotuberculosis infections, with a report of two cases. Am. J. Clin. Pathol. 66, 101–110. doi: 10.1093/ajcp/66.1.101
Parkhill, J., Wren, B. W., Thomson, N. R., Titball, R. W., Holden, M. T., Prentice, M. B., et al. (2001). Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523–527. doi: 10.1038/35097083
Perry, R. D., and Fetherston, J. D. (1997). Yersinia pestis–etiologic agent of plague. Clin. Microbiol. Rev. 10, 35–66.
Peterson, L., Philip, N., DeLaney, A., Wynosky-Dolfi, M., Asklof, K., Gray, F., et al. (2017). RIPK1-dependent apoptosis bypasses pathogen blockage of innate signaling to promote immune defense. J. Exp. Med. 214, 3171–3182. doi: 10.1084/jem.20170347
Pettersson, J., Nordfeith, R., Dubinina, E., Bergman, T., Gustafsson, M., Magnusson, K. E., et al. (1996). Modulation of virulence factor expression by pathogen target cell contact. Science 273, 1231–1233. doi: 10.1126/science.273.5279.1231
Pierson, D. E., and Falkow, S. (1993). The ail gene of Yersinia enterocolitica has a role in the ability of the organism to survive serum killing. Infect. Immun. 61, 1846–1852.
Portnoy, D. A., Moseley, S. L., and Falkow, S. (1981). Characterization of plasmids and plasmid-associated determinants of Yersinia enterocolitica pathogenesis. Infect. Immun. 31, 775–782.
Quade, N., Mendonca, C., Herbst, K., Heroven, A. K., Ritter, C., Heinz, D. W., et al. (2012). Structural basis for intrinsic thermosensing by the master virulence regulator RovA of Yersinia. J. Biol. Chem. 287, 35796–35803. doi: 10.1074/jbc.M112.379156
Roggenkamp, A., Bittner, T., Leitritz, L., Sing, A., and Heesemann, J. (1997). Contribution of the Mn-cofactored superoxide dismutase (SodA) to the virulence of Yersinia entercolitica serotype O8. Infect. Immun. 65, 4705–4710.
Schesser, K., Splik, A. K., Dukuzumuremyi, J. M., Neurath, M. F., Pettersson, S., and Wolf-Watz, H. (1998). The yopJ locus is required for Yersinia-mediated inhibition of NF-kappaB activation and cytokine expression: YopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol. Microbiol. 28, 1067–1079. doi: 10.1046/j.1365-2958.1998.00851.x
Sebbane, F., Jarrett, C. O., Gardner, D., Long, D., and Hinnebusch, B. J. (2006a). Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. U.S.A. 103, 5526–5530. doi: 10.1073/pnas.0509544103
Sebbane, F., Lemaître, N., Sturdevant, D. E., Rebeil, R., Virtaneva, K., Porcella, S. F., et al. (2006b). Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. U.S.A. 103, 11766–11771. doi: 10.1073/pnas.0601182103
Shannon, J. G., Bosio, C. F., and Hinnebusch, B. J. (2015). Dermal neutrophil, macrophage and dendritic cell responses to Yersinia pestis transmitted by fleas. PLoS Pathog. 11:e1004734. doi: 10.1371/journal.ppat.1004734
Shannon, J. G., Hasenkrug, A. M., Dorward, D. W., Nair, V., Carmody, A. B., and Hinnebusch, B. J. (2013). Yersinia pestis subverts the dermal neutrophil response in a mouse model of bubonic plague. MBio 4, e00170–e00113. doi: 10.1128/mBio.00170-13
Simonet, M., Richard, S., and Berche, P. (1990). Electron microscopic evidence for in vivo extracellular localization of Yersinia pseudotuberculosis harboring the pYV plasmid. Infect. Immun. 58, 841–845.
Simonet, M., Riot, B., Fortineau, N., and Berche, P. (1996). Invasin production by Yersinia pestis is abolished by insertion of an IS200-like element within the inv gene. Infect. Immun. 64, 375–379.
Songsungthong, W., Higgins, M. C., Rolán, H. G., Murphy, J. L., and Mecsas, J. (2010). ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell. Microbiol. 12, 988–1001. doi: 10.1111/j.1462-5822.2010.01448.x
St. John, A. L., Ang, W. X. G., Huang, M. N., Kunder, C. A., Chan, E. W., Gunn, M. D., et al. (2014). S1P-dependent trafficking of intracellular Yersinia pestis through lymph nodes establishes bubos and systemic infection. Immunity 41, 440–450. doi: 10.1016/j.immuni.2014.07.013
Syczyło, K., Platt-Samoraj, A., Bancerz-Kisiel, A., Szczerba-Turek, A., Pajdak-Czaus, J., Łabuć, S., et al. (2018). The prevalence of Yersinia enterocolitica in game animals in Poland. PLoS ONE 13:e0195136. doi: 10.1371/journal.pone.0195136
Viboud, G. I., and Bliska, J. B. (2005). Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89. doi: 10.1146/annurev.micro.59.030804.121320
Welkos, S., Friedlander, A., and Davis, K. (1997). Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain CO92. Microb. Pathog. 23, 211–223. doi: 10.1006/mpat.1997.0154
Yang, Y., and Isberg, R. (1993). Cellular internalization in the absence of invasin expression is promoted by the Yersinia pseudotuberculosis yadA product. Infect. Immun. 61, 3907–3913.
Ye, Z., Gorman, A. A., Uittenbogaard, A. M., Myers-Morales, T., Kaplan, A. M., Cohen, D. A., et al. (2014). Caspase-3 mediates the pathogenic effect of Yersinia pestis YopM in liver of C57BL/6 mice and contributes to YopM's function in spleen. PLoS ONE 9:e110956. doi: 10.1371/journal.pone.0110956
Ye, Z., Kerschen, E. J., Cohen, D. A., Kaplan, A. M., van Rooijen, N., and Straley, S. C. (2009). Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect. Immun. 77, 3791–3806. doi: 10.1128/IAI.00284-09
Ye, Z., Uittenbogaard, A. M., Cohen, D. A., Kaplan, A. M., Ambati, J., and Straley, S. C. (2011). Distinct CCR2(+) Gr1(+) cells control growth of the Yersinia pestis ΔyopM mutant in liver and spleen during systemic plague. Infect. Immun. 79, 674–687. doi: 10.1128/IAI.00808-10
Zhang, Y., and Bliska, J. (2010). YopJ-promoted cytotoxicity and systemic colonization are associated with high levels of murine interleukin-18, gamma interferon, and neutrophils in a live vaccine model of Yersinia pseudotuberculosis infection. Infect. Immun. 78, 2329–2341. doi: 10.1128/IAI.00094-10
Zhang, Y., Khairallah, C., Sheridan, B. S., van der Velden, A. W. M., and Bliska, J. B. (2018). CCR2+ inflammatory monocytes are recruited to Yersinia pseudotuberculosis pyogranulomas and dictate adaptive responses at the expense of innate immunity during oral infection. Infect. Immun. 86, e00782–e00717. doi: 10.1128/IAI.00782-17
Keywords: Yersinia infections, inflammation, phagocytes, gene expression, heterogeneity
Citation: Davis KM (2018) All Yersinia Are Not Created Equal: Phenotypic Adaptation to Distinct Niches Within Mammalian Tissues. Front. Cell. Infect. Microbiol. 8:261. doi: 10.3389/fcimb.2018.00261
Received: 07 May 2018; Accepted: 13 July 2018;
Published: 03 August 2018.
Edited by:
Matthew S. Francis, Umeå University, SwedenReviewed by:
Igor Brodsky, University of Pennsylvania, United StatesPetra Dersch, Helmholtz Center for Infection Research, Germany
Matthew B. Lawrenz, University of Louisville, United States
Copyright © 2018 Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly M. Davis, a2RhdmkxNDBAamh1LmVkdQ==