 Christoph Suster
Christoph Suster Ian R. Baxendale2
Ian R. Baxendale2 Christian Stanetty
Christian Stanetty- 1Faculty of Technical Chemistry, Institute of Applied Synthetic Chemistry, TU Wien, Vienna, Austria
- 2Department of Chemistry, University of Durham, Durham, United Kingdom
Bacterial lipopolysaccharides (LPS) are important bio-medical structures, playing a major role in the interaction with human immune systems. Their core regions, containing multiple units of L-glycero-D-manno heptoses (L,D-heptose), are highly conserved structurally (with O3 and O7 glycosidic bonds), making them an epitope of high interest for the potential development of new antibiotics and vaccines. Research in this field has always been restricted by the limited availability of the parent L,D-heptose as well as its biochemical epimeric precursor D-glycero-D-manno heptose (D,D-heptose). This problem of availability has recently been solved by us, through a rapid and efficient practical synthesis of L,D-manno-heptose peracetate demonstrated at scale. Herein we report an optimized, technically simple and versatile synthetic strategy for the differentiation of both the L-glycero and D-glycero-D-manno heptose scaffolds. Our approach is based on an orthoester methodology for the differentiation of all three positions of the sugar core using a O6, O7-tetraisopropyl disiloxyl (TIPDS) protecting group for the exocyclic positions. Furthermore, the regioselective opening toward 7-OH acceptors (6O-FTIPDS ethers) differentiates the exocyclic diol which has been demonstrated with a broader set of substrates and for both manno-heptoses for the first time.
Introduction
Microorganisms are able to generate a variety of sugars which, to our current knowledge are absent in vertebrate organisms. Among these, L-glycero-D-manno-heptose (L,D-heptose) has been identified as a major constituent of the lipopolysaccharide (LPS) of Gram-negative bacteria, an important mediator for numerous interactions with the native and adaptive immune system of the host (Holst, 2007; Kosma, 2008). The inner core region is based on L,D-heptoses and 3-deoxy-D-manno-2-octulosonic acid (Kdo) and is exhibited in a highly conserved manner in many enterobacterial strains (exemplified in Figure 1; Holst, 2007). In LPS-substructures O3 and O7 glycosylations are the most important with O4 phosphorylation being a widespread motif. However, O6 and O2 modifications (e.g., deoxy) have also been targeted for specific biological purposes in the past (Kosma, 2008; Tikad and Vincent, 2013).
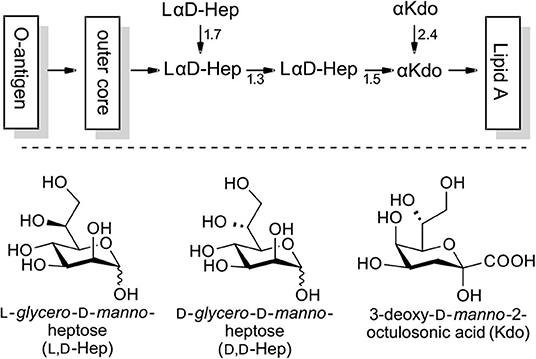
Figure 1. Exemplary structure of LPS with the L,D-heptose containing inner core region (phosphorylation not shown).
In Nature, L,D-heptose substructures are produced from the 6-epimeric structure D-glycero-D-manno-heptose. Thus, structures containing both D-manno-heptoses have been targeted and have for example been studied in terms of binding to the cross-reactive antibacterial monoclonal antibody WN1 222-5 (Di Padova et al., 1993; Gomery et al., 2012), for their interactions with C-type lectins (Jaipuri et al., 2008; Wang et al., 2008; Šulák et al., 2011; Marchetti et al., 2012) and in respect to their roles as potential diagnostic tools for bacterial infections (Anish et al., 2013).
Historically, access to the parent L,D- and D,D-heptoses has required 6-8 step synthesis from commercially available shorter chain sugars, thus creating a substantial entry-burden to this increasingly important research area. In the light of increasing antibiotic resistance, the awareness of the importance of heptoside containing fragments has grown rapidly and as such synthetic efforts in the field have evolved from niche-existence into more mainstream focus, reflected by recent reports across a wider application base. However, these new approaches generally stuck with the principle strategy of long synthetic routes, establishing complex protecting group designs alongside the chain elongation steps toward the heptose backbone. These synthetic efforts, although chemically elegant, do not constitute a general solution to the lack of easy access to common heptose motifs being applicable for multiple purposes (Brimacombe and Kabir, 1986; Segerstedt et al., 2004; Ohara et al., 2010; Anish et al., 2013; Inuki et al., 2017).
We strongly believed that what was required was a fundamental alteration in mind-set and revolutionary change of approach to create a simplified entry to this important field. Ideally, the desired heptoses should be available directly off the shelf as are standard hexoses constituting mammalian glycans, for which efficient and partly automated approaches are already state of the art today. This would allow for an entirely different level of sophistication in terms of questions posed and streamlining the methods applied in answering them.
Toward our aspirations in this area we recently reported the first short and fully scalable synthetic approach to L,D-heptose peracetate based on indium mediated acyloxyallylation of L-lyxose and demonstrated it on a >100 mmol (45 g) scale (Stanetty and Baxendale, 2015). We are currently developing methodology for the analogous preparation of D,D-heptose peracetate (Aronow et al., 2019), with the intention of paving the way to make both D-manno-heptoses commercially available as their crystalline, bench-stable pyranose peracetate derivatives. Availability, be it commercial or at least through synthetically straightforward chemistry, is however only the first piece of a much larger puzzle (Scheme 1, bottom-left).
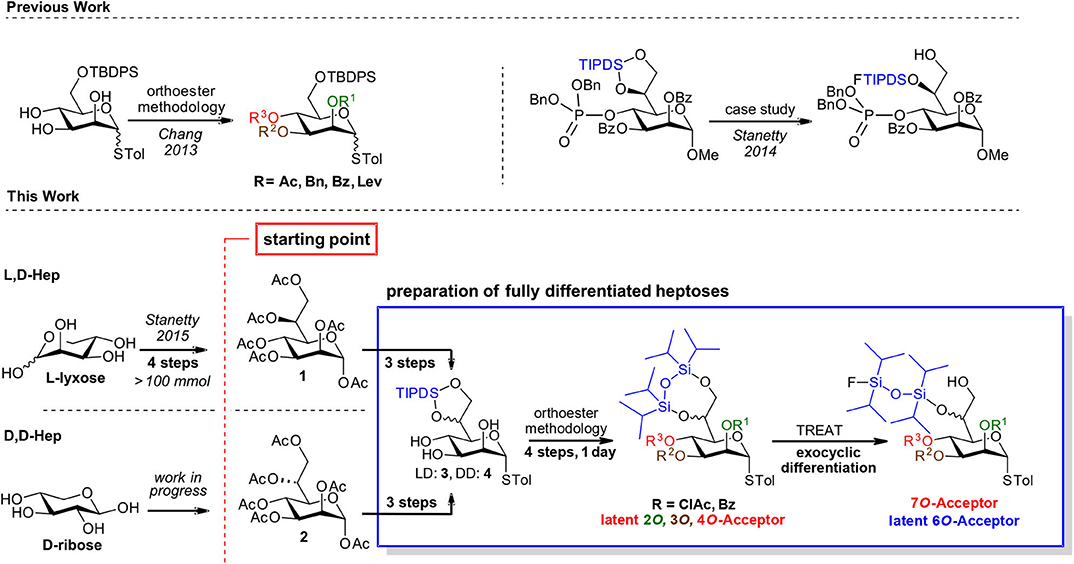
Scheme 1. General strategy based on bulk manno-heptose starting pools 3 and 4 and a common differentiation methodology for both D,D and L,D-manno heptosides.
Thus, the focus of this publication is to underline the suitability of the peracetates 1 and 2 as starting points for different types of decorated heptose structures required for molecular probes or as donor/acceptors for oligoheptoside assembly. Despite tremendous advances in glycosylation techniques, it is still common to resort to a systematic trial and error search to come up with optimized conditions to install in high yield a specific glycosidic bond in a stereoselective fashion. Rapid change of decoration pattern can be a decisive element toward ultimate success in high yielding oligosaccharide formation.
We therefore believe that common starting materials for the late-stage decoration of the D,D- as well as the L,D-scaffolds are required and to this need we suggest the tetraisopropyl disiloxyl (TIPDS) protected manno-heptoses (3 and 4) as suitable examples of such compounds. We suggest diversification based on orthoester methodology and a recently introduced regioselective TIPDS-cleavage.
In cis-trans-triols, the cis-diol can be selectively addressed to form orthoesters which upon acidic hydrolysis yield the axial esters in high selectivity (King and Allbutt, 1970). In the presence of the intermediate orthoester several protecting groups can be installed under basic conditions and after orthoester cleavage another group can be installed at the equatorial hydroxyl group. Such an approach was recently demonstrated comprehensively on mannosides (Chang et al., 2013), and has also been used in target oriented syntheses of heptosides (Stanetty et al., 2014; Walter et al., 2017). The regioselective TIPDS cleavage on the other hand was originally introduced by the Ziegler group (Ziegler and Eckhardt, 1992; Ziegler et al., 1994) on standard hexoses and was successfully transferred to the exocyclic diol of the manno-heptose scaffold within a case study with an exceptionally bulky O4 group in place (see Scheme 1, top-right) and demonstrated there at preparative scale (Stanetty et al., 2014).
Within this current work, we set out to demonstrate the general value of unifying these two methods, exemplifying the orthoester methodology by permutating the labile chloroacetate group over the heptose core, and showcasing that the regioselective partial cleavage of the TIPDS at the exocyclic diol is applicable with other smaller 4O-substitutents as well as for both the L,D- and D,D-heptose scaffold (see Scheme 1, bottom-right).
Results and Discussion
Preparation of Starting Materials for Diversification
Starting from the peracetylated heptose species 1 and 2 standard reaction conditions were used to introduce the STol group at the anomeric center (5 and 6). Further deacetylation using Zemplén conditions gave the pentaols 7 and 8. At this stage the L,D-material 7 was obtained in near quantitative yield after recrystallization, while for the recrystallization of D,D-material only moderate recoveries (64%) were accomplished (see Scheme 2).
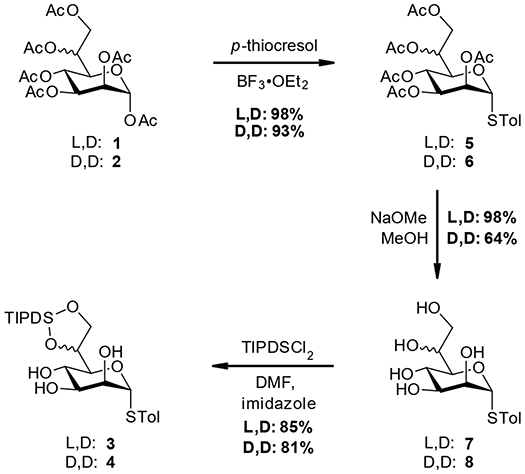
Scheme 2. Preparation of triol-intermediates 3 and 4.
Next, the TIPDS group was selectively introduced using a combination of TIPDSCl2 and imidazole at −78 °C, following conditions originally developed by Corey and Venkateswarlu (1972). Alternative conditions as described by Ziegler et al. (1994) employing pyridine at 0 °C are operationally simpler but gave lower yields for our compounds (ca. 60%). In addition to the thioglycosides 3 and 4, we also included the TIPDS protected methyl heptoside 9, which had also been used in the original case study to evaluate potential effects arising from the anomeric position (Stanetty et al., 2014; see Scheme 3 for structure 9).
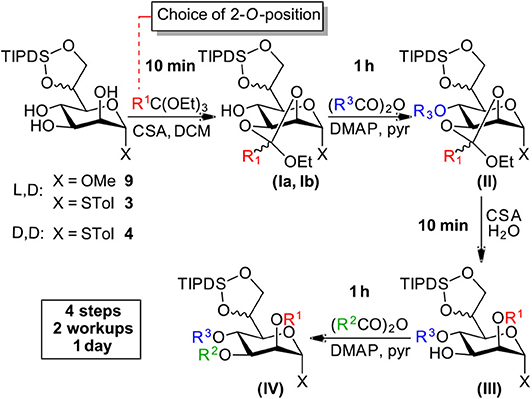
Scheme 3. Efficient and versatile orthoester methodology.
Core Differentiation by an Orthoester Methodology
Starting from compounds 3, 4, and 9 we set out to develop a robust protocol for the rapid differentiation of the manno-configured triol based on an orthoester methodology and to exemplify this chemistry by the installation of the labile chloroacetate ester on all endocyclic ring-positions. Herein, we present a reliable and well-understood protocol specifically optimized for this class of the bacterial manno-heptoses (Scheme 3).
All four reaction steps of the protocol can be performed in dry DCM and intermediate aqueous workup is only required after the protection at O4 (structure II) and O3 (structure IV). The overall process can be accomplished within a working day with subsequent purification by standard column chromatography. However, steps 2 and 4 can also be safely left running overnight and intermediate purification is possible after step 2 and/or 3 if desired.
Step 1—Orthoester Formation
The formation of the orthoester intermediates is highly selective and is completed within minutes. Subsequently the sulfonic acid catalyst reacts readily with the orthoester reagents to form the sulfonic acid esters1. Generally, the two diastereomers (Ia and Ib) are observed as separate spots on TLC (e.g., LP/EtOAc 4:1, Rf 0.55 and 0.60). We found the addition of a small amount of Et3N prior to evaporation of the solvent is advisable to prevent any undesired orthoester cleavage. Co-evaporation from toluene facilitates removal of residual ethanol, present from the orthoester introduction, which if left, would consume acylating agent in the subsequent step2.
Step 2—Acylation at O4
At the O4 position acetates, benzoates and chloroacetates were introduced via the corresponding anhydrides using pyridine [Et3N is not compatible with (ClAc)2O!] and DMAP as a catalyst, which proved necessary for the sterically restricted position close to the TIPDS-group. Interestingly, while 2O/3O-orthoacetates and orthobenzoates with unprotected 4-OH group (I) were instantaneously hydrolysed in the presence of water and acid, upon 4O-protection the orthoester moieties (II) was stable to washing with 1 N HCl during extractive workup and column chromatography (Stanetty et al., 2014).
Step 3—Orthoester Hydrolysis
Under homogenous conditions in the presence of catalytic amounts of CSA or TsOH and with only a few equivalents of water the orthoester moiety was cleaved within minutes to provide 2O-ester (III).
Step 4—Acylation at O3
The final acylation follows the same principles as in step 2 and can generally follow step 3 directly, but evaporation and re-dissolution in DCM prevents hydrolysis of the acylating agent which gives a cleaner reaction.
We have applied this 4-step protocol at 1 mmol scale to starting materials 3, 4, and 9 to prepare a set of compounds (see Table 1) with overall yields of 60–81% reflecting at least 88% yield per step. The process can equally be applied to the methyl glycoside (entries 1 and 2) and thioglycosides and allows the installation for example the labile chloroacetyl group on position O3 and O4 but proved troublesome at the O2-position. We included a 4O-chloroacetate (entry 7) as an example with D,D-configuration for comparison in particular in the later TIPDS-cleavage.
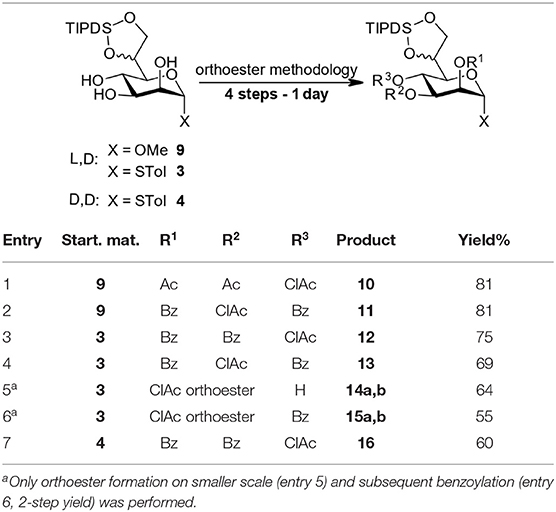
Table 1. Decoration of L,D-Hep (1 mmol scale).
Stability of the Ortho Chloroacetate
The aim of introducing the labile chloroacetate ester at the O2-position could not be directly accomplished as the intermediate ortho chloroacetate exhibited surprising stability and withstood the standard conditions both as 4O-Bz (15) as well as the 4-OH (14) intermediate. Under harsher, previously reported conditions (80% AcOH or CF3COOH) (Oscarson and Tedebark, 1996; Ueki et al., 2010) multiple unidentified products were observed with both orthoesters 14a,b and 15a,b. The problem was identified as arising from the presence of the TIPDS group (see Supplementary Material).
Nevertheless, the high acid stability of the ortho chloroacetate moieties allowed chromatographic separation of both diastereomeric pairs 14a,b and 15a,b and full characterization thereof. We suggest, that the ortho chloroacetate can be considered as a selective long-term protecting group for the 2O,3O-diol of the manno-scaffold. It tolerates transformations under slightly acidic conditions which cannot be performed with the usual orthoesters which we also demonstrated by including 15 in our TIPDS cleavage study (vide infra). To also be able to include the originally targeted 3O,4O-dibenzoyl-2O-chloroacetyl species 16 (see Table 2 for structure) in the survey we have synthesized it by a lengthier synthetic route (see Supplementary Material).
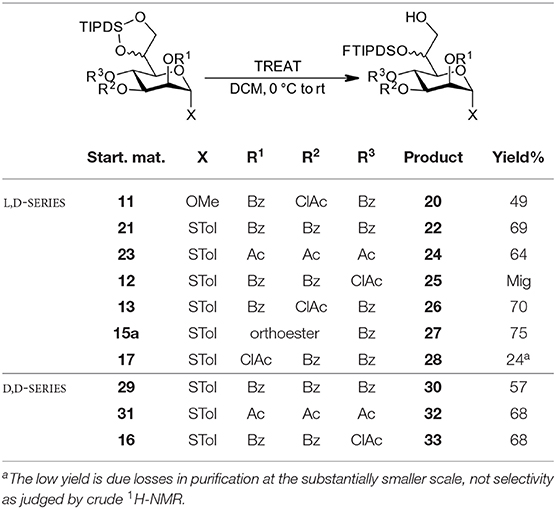
Table 2. Regioselective cleavage of the TIPDS group.
Regioselective TIPDS-Cleavage to Provide 7-OH Acceptors
With the compounds obtained via the orthoester protocol and the additional per-benzoylated and per-acetylated model compounds (21, 23, 29, 31 in Table 2), we set out to systematically investigate the regioselective TIPDS cleavage toward 7-OH acceptors using triethylamine trihydrofluoride (TREAT) as a mild fluoride source. We commenced the investigation with the L,D-configured methyl and STol glycosides (see Table 2, top entries) under the conditions from the case study. The targeted compound could generally be obtained as the major product in yields between 50–75% independent of the ester group at O4 (confirming that the bulky dibenzyl phosphate group of the case study was not a prerequisite for this attractive transformation, see Scheme 1). Successful TIPDS cleavage at the O7 position can be confirmed in the NMR by a new COSY signal between 7-OH and the H7 protons. Further, a diagnostic shift in the 13C-NMR by 5 ppm highfield for C7 is observed. Experimentally, the reactions were performed in dry DCM at 0 °C in a teflon vessel to prevent uncontrolled quenching of the fluoride reagent on glass. There is an inherent over-reaction toward complete desilylation (there was no indication of opening toward 6-OH acceptors) which requires TLC monitoring of the three main components (starting material, target compound, and fully deprotected by-product) for optimal results. Sufficient stirring combined with slow addition of the TREAT prevents overreaction. When prolonged stirring at 0 °C led to sluggish conversion more TREAT was added in portions until completion as determined by TLC.
When studying this reaction for the D,D-heptoses, a simple modification of conditions was applied. Slow warming of the reaction to room temperature following addition of the TREAT led to complete conversion in a short time frame; comparable yields to other protocol were also obtained. In Table 2 we have summarized our findings.
Independent of the protocol, most transformations performed as expected and with good selectivity toward initial attack at the “primary” side of the disiloxane ring system. However, the reaction toward L,D-configured compound 25, where the labile chloroacetate group is attached to the 4O-position, partial migration during workup, and/or purification to the 7O-position was observed. Interestingly, in the reaction with the D,D-analog 16 this migration was not observed and a good isolated yield was obtained. We assume that the stereochemistry at 6O-position with the bulky FTIPDSO-group leads to a conformational change in the side-chain and thus prevents proximity between the 7-OH and the particularly reactive chloroacetate at O4 which tentatively favors migration in the L,D-case. Also, the other two examples with D,D-configuration, perbenzoate 29, and peracetate 31 gave comparable selectivities and isolated yields to their L,D-counterparts, confirming the independence of the size of the O4-substitutent also with this configuration. When the ortho chloroacetate 15a was submitted to the reaction conditions, we were pleased to note that the acid stability was sufficient to cleanly prepare the corresponding 7-OH acceptor 27.
Despite the somewhat lower yields compared to the original example in our case study, the simultaneous O6/O7-protection together with the differentiation between those two positions in one step is an attractive feature of the overall approach. Noteworthy, performance in this interesting transformation was comparable between the two epimeric families, which supports this methodology for future use in both manno-heptose families alike.
Conclusion
We are convinced that the field of preparative manno-heptose chemistry would benefit greatly from a shift of paradigm toward common parent starting materials (e.g., peracetates) and differentiation thereof. In this light, we present short and scalable protocols to both (L,D, D,D) crystalline STol-heptosides and promote the utilization of the TIPDS-group as a protection for their exocyclic diols. This is due to its attractive partial regioselective cleavage which was proven to be a generally applicable solution for the differentiation between O6/O7. Additionally, we report an efficient 4-steps/1-purification protocol based on orthoester methodology to achieve the prior differentiation of the sugar core triol. The unforeseen stability of the ortho chloroacetates in this context was exploited by showing its applicability as stable protecting group during the TIPDS cleavage. We hope that the presented approach will pave the way towards a more unified starting point into biologically relevant structures based on manno-heptoses.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
CSu and CSt have been preparing and analyzing the materials. MM and IB have contributed with advice. All authors contributed to the article and approved the submitted version.
Funding
The Austrian Science Fund (FWF, Grant No. J 3449-N28) is gratefully acknowledged for financial support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marc Hofbauer and Isabella Sickha for technical support and the Austrian Science Fund (FWF, Grant No. J 3449-N28) was gratefully acknowledged for financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00625/full#supplementary-material
Footnotes
1. ^Due to this side reaction the choice of sulfonic acid catalyst was made based on the chromatographic properties of the resultant ester in respect to the final target compound which is to be columned.
2. ^Formation of 2O-esters is also partly observed on TLC, which should not be mistaken for an incomplete orthoester formation. After addition of TEA such decomposition on TLC is not observed any more.
References
Anish, C., Guo, X., Wahlbrink, A., and Seeberger, P. H. (2013). Plague detection by anti-carbohydrate antibodies. Angew. Chem. Int. Ed. 52, 9524–9528. doi: 10.1002/anie.201301633
Aronow, J., Stanetty, C., Baxendale, I. R., and Mihovilovic, M. D. (2019). Methyl glycosides via Fischer glycosylation: translation from batch microwave to continuous flow processing. Monatshefte für Chemie Chem. Monthly 150, 11–19. doi: 10.1007/s00706-018-2306-8
Brimacombe, J. S., and Kabir, A. K. M. (1986). Convenient syntheses of L-glycero-D-manno-heptose and D-glycero-D-manno-heptose. Carbohydr. Res. 152, 329–334. doi: 10.1016/S0008-6215(00)90316-1
Chang, S.-M., Tu, Z., Jan, H.-M., Pan, J.-F., and Lin, C.-H. (2013). Rapid synthesis of oligomannosides with orthogonally protected monosaccharides. Chem. Commun. 49, 4265–4267. doi: 10.1039/C2CC37099A
Corey, E. J., and Venkateswarlu, A. (1972). Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J. Am. Chem. Soc. 94, 6190–6191. doi: 10.1021/ja00772a043
Di Padova, F. E., Brade, H., Barclay, G. R., Poxton, I. R., Liehl, E., Schuetze, E., et al. (1993). A broadly cross-protective monoclonal antibody binding to Escherichia coli and Salmonella lipopolysaccharides. Infect. Immun. 61, 3863–3872. doi: 10.1128/IAI.61.9.3863-3872.1993
Gomery, K., Muller-Loennies, S., Brooks, C. L., Brade, L., Kosma, P., Di, P. F., et al. (2012). Antibody WN1 222-5 mimics Toll-like receptor 4 binding in the recognition of LPS. Proc. Natl. Acad. Sci. U.S.A. 109, 20877–20882. doi: 10.1073/pnas.1209253109
Holst, O. (2007). The structures of core regions from enterobacterial lipopolysaccharides – an update. FEMS Microbiol. Lett. 271, 3–11. doi: 10.1111/j.1574-6968.2007.00708.x
Inuki, S., Aiba, T., Kawakami, S., Akiyama, T., Inoue, J.-I., and Fujimoto, Y. (2017). Chemical synthesis of D-glycero-D-manno-heptose 1,7-bisphosphate and evaluation of its ability to modulate NF-κB activation. Org. Lett. 19, 3079–3082. doi: 10.1021/acs.orglett.7b01158
Jaipuri, F. A., Collet, B. Y. M., and Pohl, N. L. (2008). Synthesis and quantitative evaluation of glycero-D-manno-heptose binding to concanavalin A by fluorous-tag assistance. Angew. Chem. Int. Ed. 47, 1707–1710. doi: 10.1002/anie.200704262
King, J. F., and Allbutt, A. D. (1970). Remarkable stereoselectivity in the hydrolysis of dioxolenium ions and orthoesters fused to anchored six-membered rings. Can. J. Chem. 48, 1754–1769. doi: 10.1139/v70-288
Kosma, P. (2008). Occurrence, synthesis and biosynthesis of bacterial heptoses. Curr. Org. Chem. 12, 1021–1039. doi: 10.2174/138527208785161169
Marchetti, R., Malinovska, L., Lameignére, E., Adamova, L., De Castro, C., Cioci, G., et al. (2012). Burkholderia cenocepacia lectin A binding to heptoses from the bacterial lipopolysaccharide. Glycobiology 22, 1387–1398. doi: 10.1093/glycob/cws105
Ohara, T., Adibekian, A., Esposito, D., Stallforth, P., and Seeberger, P. H. (2010). Towards the synthesis of a Yersinia pestis cell wall polysaccharide: enantioselective synthesis of an L-glycero-D-manno-heptose building block. Chem. Commun. 46, 4106–4108. doi: 10.1039/c000784f
Oscarson, S., and Tedebark, U. (1996). Synthesis and acidic opening of chlorinated carbohydrate orthoacetates. J. Carbohydr. Chem. 15, 507–512. doi: 10.1080/07328309608005670
Segerstedt, E., Mannerstedt, K., Johansson, M., and Oscarson, S. (2004). Synthesis of the branched trisaccharide L-Glycero-α-D-manno-heptopyranosyl-(1 → 3)- [β-D-glucopyranosyl-(1 → 4)]-L-glycero-α-D-manno-heptopyranose, protected to allow flexible access to Neisseria and Haemophilus LPS inner core structures. J. Carbohydr. Chem. 23, 443–452. doi: 10.1081/CAR-200044580
Stanetty, C., and Baxendale, I. R. (2015). Large-scale synthesis of crystalline 1,2,3,4,6,7-hexa-O-acetyl-L-glycero-α-D-manno-heptopyranose. Eur. J. Org. Chem. 2015, 2718–2726. doi: 10.1002/ejoc.201500024
Stanetty, C., Walter, M., and Kosma, P. (2014). Convergent synthesis of 4-O-phosphorylated L-glycero-D-manno-heptosyl lipopolysaccharide core oligosaccharides based on regioselective cleavage of a 6,7-O-tetraisopropyldisiloxane-1,3-diyl protecting group. J. Org. Chem. 79, 582–598. doi: 10.1021/jo402312x
Šulák, O., Cioci, G., Lameignère, E., Balloy, V., Round, A., Gutsche, I., et al. (2011). Burkholderia cenocepacia BC2L-C is a super lectin with dual specificity and proinflammatory activity. PLoS Pathog. 7:e1002238. doi: 10.1371/journal.ppat.1002238
Tikad, A., and Vincent, S. P. (2013). Constrained 3,6-anhydro-heptosides: synthesis by a DAST-induced debenzylative reaction, and reactivity profile. Eur. J. Org. Chem. 2013, 7593–7603. doi: 10.1002/ejoc.201301071
Ueki, A., Takano, Y., Kobayashi, A., Nakahara, Y., Hojo, H., and Nakahara, Y. (2010). Solid-phase synthesis of glycopeptide carrying a tetra-N-acetyllactosamine-containing core 2 decasaccharide. Tetrahedron 66, 1742–1759. doi: 10.1016/j.tet.2009.12.031
Walter, M., Kohout, C., Blaukopf, M., and Kosma, P. (2017). Synthesis of 3-O- and 4-O-(2-aminoethylphosphono) derivatives of methyl L-glycero-α-D-manno-heptopyranoside. Monatshefte fur Chemie 148, 111–119. doi: 10.1007/s00706-016-1868-6
Wang, H., Head, J., Kosma, P., Brade, H., Mueller-Loennies, S., Sheikh, S., et al. (2008). Recognition of heptoses and the inner core of bacterial lipopolysaccharides by surfactant protein D. Biochemistry 47, 710–720. doi: 10.1021/bi7020553
Ziegler, T., and Eckhardt, E. (1992). A novel convergent strategy for the construction of oligosaccharides using TIPS-protected glycosides. Synthesis of fragments related to glycolipids of Mycobacterium smegmatis. Tetrahedron Lett. 33, 6615–6618. doi: 10.1016/S0040-4039(00)60999-1
Keywords: heptose, higher carbon sugars, orthoester derivatives, lipopolysaccharides (LPS), carbohydrate chemistry, synthetic methodology
Citation: Suster C, Baxendale IR, Mihovilovic MD and Stanetty C (2020) Straight Forward and Versatile Differentiation of the L-glycero and D-glycero-D-manno Heptose Scaffold. Front. Chem. 8:625. doi: 10.3389/fchem.2020.00625
Received: 01 May 2020; Accepted: 15 June 2020;
Published: 31 July 2020.
Edited by:
Nuno Manuel Xavier, Universidade de Lisboa, PortugalReviewed by:
Charles Gauthier, Institut National de la Recherche Scientifique (INRS), CanadaMarkus Blaukopf, University of Natural Resources and Life Sciences Vienna, Austria
Copyright © 2020 Suster, Baxendale, Mihovilovic and Stanetty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Stanetty, Y2hyaXN0aWFuLnN0YW5ldHR5QHR1d2llbi5hYy5hdA==