Abstract
Pyranose dehydrogenases (PDHs; EC 1.1.99.29; AA3_2) demonstrate ability to oxidize diverse carbohydrates. Previous studies of these enzymes have also uncovered substrate-dependent regioselectivity, along with potential to introduce more than one carbonyl into carbohydrate substrates. Enzymatic oxidation of carbohydrates facilitates their further derivatization or polymerization into bio-based chemicals and materials with higher value; accordingly, PDHs that show activity on xylooligosaccharides could offer a viable approach to extract higher value from hemicelluloses that are typically fragmented during biomass processing. In this study, AbPDH1 from Agaricus bisporus and AmPDH1 from Leucoagaricus meleagris were tested using linear xylooligosaccharides, along with xylooligosaccharides substituted with either arabinofuranosyl or 4-O-(methyl)glucopyranosyluronic acid residues with degree of polymerization of two to five. Reaction products were characterized by HPAEC-PAD to follow substrate depletion, UPLC-MS-ELSD to quantify the multiple oxidation products, and ESI-MSn to reveal oxidized positions. A versatile method based on product reduction using sodium borodeuteride, and applicable to carbohydrate oxidoreductases in general, was established to facilitate the identification and quantification of oxidized products. AbPDH1 activity toward the tested xylooligosaccharides was generally higher than that measured for AmPDH1. In both cases, activity values decreased with increasing length of the xylooligosaccharide and when using acidic rather than neutral substrates; however, AbPDH1 fully oxidized all linear xylooligosaccharides, and 60–100% of all substituted xylooligosaccharides, after 24 h under the tested reaction conditions. Oxidation of linear xylooligosaccharides mostly led to double oxidized products, whereas single oxidized products dominated in reactions containing substituted xylooligosaccharides. Notably, oxidation of specific secondary hydroxyls vs. the reducing end C-1 depended on both the enzyme and the substrate. For all substrates, however, oxidation by both AbPDH1 and AmPDH1 was clearly restricted to the reducing and non-reducing xylopyranosyl residues, where increasing the length of the xylooligosaccharide did not lead to detectable oxidation of internal xylopyranosyl substituents. This detailed analysis of AbPDH1 and AmPDH1 action on diverse xylooligosaccharides reveals an opportunity to synthesize bifunctional molecules directly from hemicellulose fragments, and to enrich for specific products through appropriate PDH selection.
Introduction
Hemicelluloses comprise a diverse group of polysaccharides present in plant cell walls, and are thought to strengthen cell wall structures through interactions with both cellulose and lignin (Ebringerová, 2006; Scheller and Ulvskov, 2010). Xylans are the dominant hemicellulose in cell walls of both grasses and deciduous trees. In deciduous trees, the main xylan is glucuronoxylan, where the β-(1 → 4)-linked D-xylopyranose (Xylp) backbone can be substituted with 4-O-methylated glucopyranosyluronic acid (MeGlcpA); the Xylp backbone can also be acetylated (Ebringerová, 2006; Scheller and Ulvskov, 2010). Arabinoxylans (AXs) are the main xylans in grasses, where the Xylp backbone can be substituted with L-arabinofuranose (Araf). In addition to Araf, arabinoglucuronoxylans found in cell walls of both grasses and coniferous trees, contain glucuronic acid and MeGlcpA substitutions (Scheller and Ulvskov, 2010).
So far, biocatalysts that transform hemicelluloses have mainly been studied and developed to deconstruct corresponding polysaccharides to sugars that can be fermented to fuels and chemicals (Chundawat et al., 2011). These enzymes include glycoside hydrolases (GHs), carbohydrate esterases (CEs), and auxiliary activities (AAs) that are classified by the Carbohydrate-active enzyme (CAZy) database (www.cazy.org) into multiple GH, CE, and AA families (Lombard et al., 2014). Alternatively, enzymatic functionalization of hemicelluloses, including oligosaccharides liberated during lignocellulose processing, opens new possibilities to make high value bioproducts that retain the carbon and energy stored in starting carbohydrate structures.
Carbohydrate oxidoreductases that act on hemicelluloses without cleaving them are attractive targets for further functionalization (Parikka et al., 2012). Among these are AA3 and AA7 FAD-containing oxidoreductases. Briefly, AA7 activity is restricted to the anomeric carbon of carbohydrate substrates (Huang et al., 2005; Vuong et al., 2013); the resulting lactone undergoes spontaneous ring-opening to the carboxylic acid that can be used in polymerization reactions (MacCormick et al., 2018). On the other hand, AA3 oxidoreductases are reported to oxidize the anomeric carbon and secondary hydroxyls to carboxylic acid or ketone functional groups, respectively (Giffhorn et al., 2000; Levasseur et al., 2013; Sützl et al., 2018). Based on phylogenetic analyses, AA3 sequences have been divided into four subfamilies, including activities such as cellobiose dehydrogenase, alcohol oxidase and pyranose oxidase in subfamilies 1, 3, and 4, respectively. Subfamily 2 is the most complex one of these, comprising enzymes with aryl-alcohol oxidase, aryl-alcohol dehydrogenase, glucose oxidase, glucose dehydrogenases, or pyranose dehydrogenase (PDH) activities (Levasseur et al., 2013; Sützl et al., 2018). From these, AA3_2 PDHs have been shown to oxidize several monosaccharides and some oligosaccharides through single or double oxidation and substrate dependent regioselectivity (Peterbauer and Volc, 2010; Rafighi et al., 2018), which increases their attractiveness on oligosaccharide modification.
PDHs have been found in a narrow group of litter-decomposing fungi within the fungal division Basidiomycota (Peterbauer and Volc, 2010). These extracellular enzymes contain two domains, an FAD-binding and substrate-binding domain (Tan et al., 2013). The active site contains two conserved histidines. His512 of AmPDH1 from Leucoagaricus meleagris acts as the main base in the first reductive half-reaction and His556 together with Gln392 and Tyr510 take part in substrate interactions that likely influence the observed substrate specific regioselectivity (Graf et al., 2013, 2015; Tan et al., 2013). After the first half-reaction, the reduced flavin is oxidized by an electron acceptor which acts as the second substrate in the reaction. Along with AmPDH1, AbPDH1 from Agaricus bisporus are the two most studied PDHs. AbPDH1 and AmPDH1 oxidize D-xylose to 2,3-diketo-D-xylose (Volc et al., 2000; Sedmera et al., 2006) and display slightly higher relative activity toward D-xylose compared to D-glucose (Volc et al., 2000; Sedmera et al., 2006). In addition to D-xylose, AmPDH1 was shown to oxidize xylobiose (X2) (Sygmund et al., 2008). PDH activity on oligosaccharides with degree of polymerization (DP) greater than two has been tested using cello- and maltooligosaccharides. Those studies confirm that AbPDH1 and AmPDH1 are both active toward cellotetraose and maltotriose (Volc et al., 1997; Peterbauer and Volc, 2010), and that AmPDH1 is also active toward maltooligosaccharides up to maltoheptaose (Tasca et al., 2007; Peterbauer and Volc, 2010; Rafighi et al., 2018). Among the characterized PDHs, AmPDH1 showed highest catalytic efficiency (kcat/Km) toward D-xylose (Sygmund et al., 2012), whereas AbPDH1 showed highest catalytic efficiency toward cellobiose, maltose and lactose (Gonaus et al., 2016).
PDH action toward xylooligosaccharides common to hardwood and agricultural resources has not been investigated. Herein, AbPDH1 and AmPDH1 were directly compared in terms of activity toward linear and substituted xylooligosaccharides, and ability to oxidize corresponding substrates at more than one position to create a new class of telechelic building blocks. The analysis of corresponding oligosaccharide products is complicated by the multiple hydroxyl groups that could potentially be oxidized. Moreover, the oxidized products are inherently unstable in water because they can further transform into various end products. For example, oxidation of the anomeric carbon leads to a lactone which spontaneously hydrolyzes to the carboxylic acid in water (Vuong et al., 2013). Furthermore, secondary hydroxyls that are oxidized to ketones and primary hydroxyls that are oxidized to aldehydes, exist primarily as hydrates (geminal diols) in water (Volc et al., 2002; Andberg et al., 2017). In this study, a new analytical method was developed to simplify and clarify the analysis of oxidized xylooligosaccharides utilizing deuterium to label the oxidized position. By following the deuterated residues by HILIC-MS-ELSD and ESI-MSn, the oxidation positions were identified and the proportion of each reaction product was quantified. Both AbPDH1 and AmPDH1 were shown to oxidize X2, xylotriose (X3) and xylotetraose (X4) with and without Araf substitution; however, activity toward acidic glucuronoxylooligosaccharides was 10-times lower than with neutral substrates.
Materials and Methods
Materials
Below mentioned growth medium chemicals, yeast extract, yeast nitrogen base, and peptone were purchased from Lab M Ltd. (UK). Salts and vitamins were obtained from Sigma-Aldrich or Merck (Germany). Neutral substrates X2, X3, X4, 32-α-L-arabinofuranosyl-xylobiose (A3X), 23-α-L-arabinofuranosyl-xylotriose (A2XX), 33-α-L-arabinofuranosyl-xylotetraose (XA3XX) were purchased from Megazyme (UK). 23-(4-O-methyl-α-D-glucuronyl)-xylotriose (U4m2XX) and 23-(4-O-methyl-α-D-glucuronyl)-xylotetraose (XU4m2XX) substrates were prepared as in Koutaniemi et al. (2012) by Dr. T. Vuong, University of Toronto and Dr. S. Koutaniemi, University of Helsinki and kindly provided to the study. The commercial laccase from Trametes versicolor (Sigma-Aldrich, Germany) was used in oxidation reactions (described below) to recycle 1,4-benzoquinone (BQ; Sigma-Aldrich, Germany) electron acceptor.
PDH Production and Purification
Agaricus bisporus and Agaricus meleagris pyranose dehydrogenases (AbPDH1 and AmPDH1; pyranose:acceptor oxidoreductase, EC 1.1.99.29, CAZy family AA3_2) were expressed in Pichia pastoris strain KM71H. Codon optimized genes encoding AbPDH1 and AmPDH1 amino acid sequences (AAW92124 and AAW82997, respectively) were obtained as subcloned in pPICZB plasmids with C-terminal 6 x His tag (GenScript, New Jersey, USA). PDHs were produced in eight 2 l shake-flasks each containing 250 ml of medium. Precultures were grown over night in buffered glycerol-complex medium [BMGY; 100 mM potassium phosphate buffer, pH 6.0, 2% (w/v) peptone, 1% (w/v) yeast extract, 1.34% (w/v) yeast nitrogen base, 4 × 10−5% (w/v) biotin, 1% (v/v) glycerol] at 30°C and 220 rpm. Cells were then transferred to methanol-complex medium (BMMY) containing 0.5% (v/v) methanol instead of glycerol. Methanol was added to 0.5% (v/v) every 24 h and induction was continued 4 d at 25°C and 220 rpm. After the induction, culture supernatants were recovered and filtered, and the secreted recombinant proteins were purified by affinity chromatography; AbPDH1 was further purified by anion exchange and size exclusion (Figure S2). Final protein concentrations were measured using the Bradford method (Bio-Rad Laboratories, US); purified proteins were then aliquoted and stored in −80°C.
Initial Activity Measurements
To select the optimal conditions for the xylooligosaccharide oxidation, the activity of AbPDH1 and AmPDH1 was screened at 30°C with 25 mM substrate (D-xylose or D-glucose) and 5 mM BQ in 50 mM sodium acetate, 50 mM ammonium acetate (at pH values 3.0–5.5), and in 50 mM sodium phosphate (at pH values 6.0–7.0) buffers. Reduction of BQ (εabs290nm = 2.24 mM−1 cm−1) in 250 μl reaction was followed at 290 nm; all reactions were performed in triplicate.
Enzyme loading for PDH conversion of xylooligosaccharides was based on activity units determined in 50 mM ammonium acetate buffer (pH 5.5). PDH activity was measured using 25 mM D-xylose and 5 mM BQ similarly as mentioned above. Laccase activity was measured using 5 mM hydroquinone (HQ, Sigma Aldrich, Germany), and oxidation of HQ (εabs249 = 17.25 mM−1 cm−1) in 250 μl reaction was followed at 249 nm.
Initial AbPDH1 and AmPDH1 activity toward xylooligosaccharides was measured using 10 mM neutral or 2 mM of acidic substrates, and 0.2 mM ferrocenium hexafluorophosphate (Fc+, Sigma Aldrich, Germany) as the electron acceptor. Reactions (100 μl) were performed at 30°C in 50 mM sodium phosphate buffer (pH 7.5), and the reduction of the Fc+ ion (εabs250nm = 10.6 mM−1 cm−1) was measured for up to 1 h at 250 nm. All the activity measurements were followed using an Eon plate reader (BioTek, USA).
Enzymatic Conversion of Xylooligosaccharides
Reactions (30–400 μl reaction volume in 1.5 ml Eppendorf tubes) were performed at 30°C for up to 48 h with shaking (500 rpm) in 10 mM ammonium-acetate buffer (pH 5.5) containing 1 mM BQ as the electron acceptor, and 5 mM of neutral xylooligosaccharides or 4 mM of acidic xylooligosaccharides (Table 1). Enzyme loadings for reactions containing neutral xylooligosaccharides were 50 mU of AbPDH or AmPDH, and 50 mU of T. versicolor laccase. Enzyme loadings for reactions containing acidic xylooligosaccharides were 800 mU of AbPDH1 or AmPDH1, and 800 mU of T. versicolor laccase. Time course sampling (25 or 10 μl) for the HPAEC-PAD analyses was done at 1, 4, and 8 h for the neutral substrates and 24 h for acidic substrates. Total reaction time was 24 or 48 h. Oxygen availability was not controlled during the reactions. Reactions were stopped by filtrating the samples through 10 kDa cut off Vivaspin 500 spin columns (Sartorius, Germany) and then kept frozen at −80°C before analysis by High-Performance Anion-Exchange Chromatography Coupled with Pulsed Electrochemical Detection (HPAEC-PAD) and mass spectrometry as described below.
Table 1
| Substrate | Substrate (mM) | PDH (U/ml) | Reactiona time (h) |
|---|---|---|---|
Xylobiose (X2) 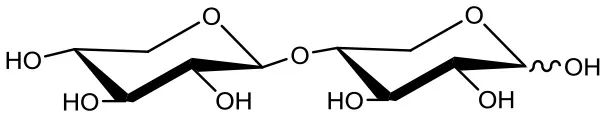 | 5 | 0.2 | Up to 24 |
Xylotriose (X3)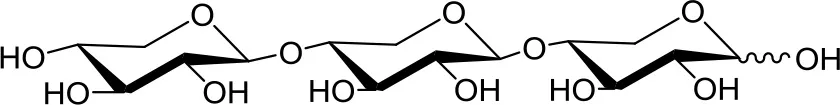 | 5 | 0.2 | Up to 24 |
Xylotetraose (X4) 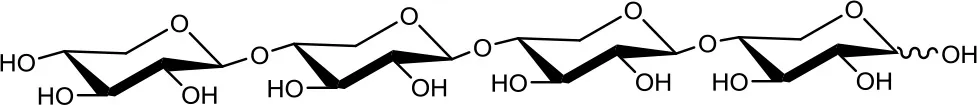 | 5 | 0.2 | Up to 24 |
32-α-L-arabinofuranosyl-xylobiose (A3X) 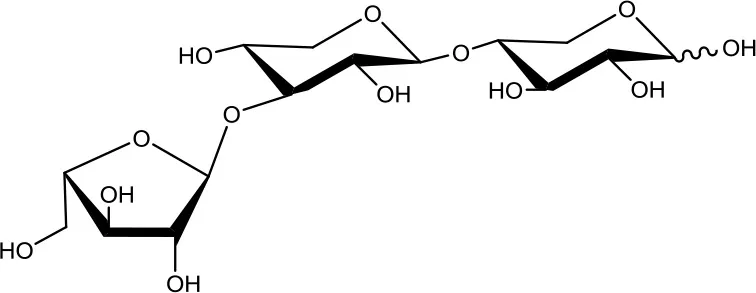 | 5 | 0.2 | Up to 24 |
23-α-L-arabinofuranosyl-xylotriose (A2XX) 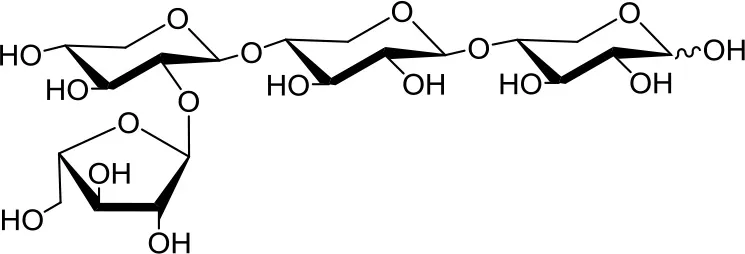 | 5 | 0.2 | Up to 24 |
33-α-L-arabinofuranosyl-xylotetraose (XA3XX) 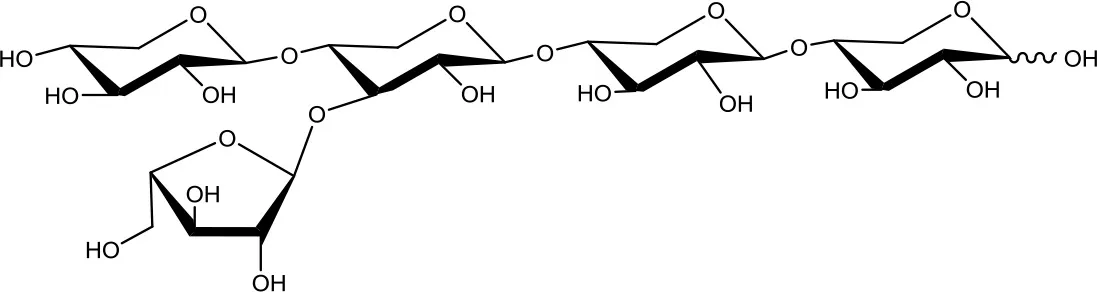 | 5 | 0.2 | Up to 24 |
23-(4-O-methyl-α-D-glucuronyl)-xylotriose (U4m2XX) 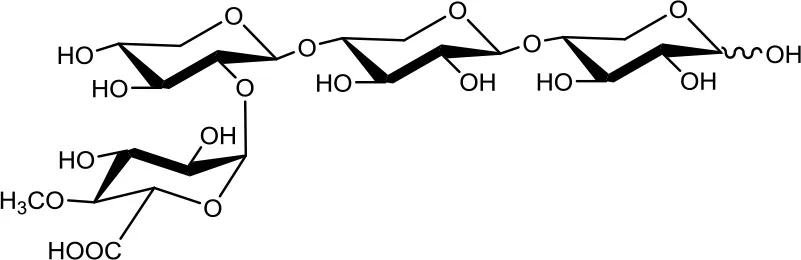 | 4 4 | 0.2 2 | Up to 48 Up to 24 |
23-(4-O-methyl-α-D-glucuronyl)-xylotetraose (XU4m2XX) 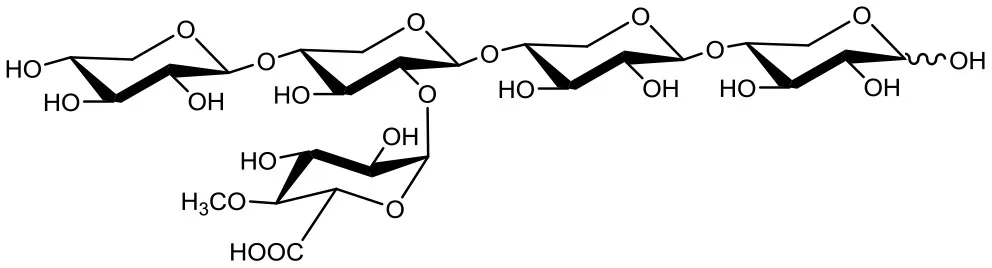 | 4 4 | 0.2 2 | Up to 48 Up to 24 |
Structures of xylooligosaccharides and oxidation reaction details.
All the reactions contained 0.2 U/ml of laccase and 1 mM BQ as electron acceptor.
Quantification of Substrate Depletion by HPAEC-PAD
Products of reactions described above (section Enzymatic Conversion of Xylooligosaccharides) were diluted in ultrapure water to 50–100 ppm concentrations depending on the sensitivity of substrate detection by HPAEC-PAD. Substrate depletion over the course of the reaction was followed using a DionexTM-5000+ system equipped with a DionexTM CarboPacTM PA1 IC column and corresponding precolumn (Thermo Scientific, USA). The samples were eluted at 1 ml/min with eluent A (0.1 M NaOH) in a linear gradient toward an increasing proportion of eluent B (1 M NaOAc in 0.1 M NaOH). The gradient reached 21.7% B at 20 min after injection, 38% B at 25 min and 100% B at 27 min after which it was kept at 100% for 7 min. Data were analyzed using the Thermo Scientific Dionex Chromeleon 7 Chromatography Data System (version 7.2 SR4, Thermo Fisher Scientific). Corresponding xylooligosaccharides with three known concentrations were used to create a standard curve for each run. Integrated peak areas of the enzyme treated xylooligosaccharides were compared against the corresponding control reactions without the PDH enzyme.
Mass Spectrometric Analysis of Oxidized Products by Direct Infusion
A 5 μl sample from 24 h reactions described above (section Enzymatic Conversion of Xylooligosaccharides) was mixed with 5 μl of 10 mg/ml NH4Cl and 500 μl 50% acetonitrile (ACN) in water. Each sample solution was then introduced into Quadruple Time-of-flight (Q-Tof) mass spectrometry with an ESI source (SYNAPT G2-Si, Waters, MA, USA) at a flow rate of 5 μl/min. The capillary was set to 3 kV; source temperature to 80°C and desolvation temperature to 150°C. The cone gas was 100 l/h and desolvation gas was 600 l/h with nebulizer set at 6.5 bar. The analysis was done in negative mode and the ions were collected in m/z range of 50–800. The products of acidic xylooligosaccharides oxidized by 2 U/ml PDH for 24 h were analyzed directly by ESI-Q-Tof-MS without the addition of NH4Cl.
Identification of Oxidized Positions Through Fragmentation Analysis of Products After NaBD4 Reduction
The multiple potential oxidations and reactivity of oxidized products in water complicate the interpretation of MS spectra. Therefore, after confirming the production of oxidized xylooligosaccharides by direct infusion mass spectrometry, reaction products were reduced using NaBD4 to facilitate their identification. Specifically, NaBD4 was added to the 24 h reactions described above (section Enzymatic Conversion of Xylooligosaccharides) at 3 mol equivalent NaBD4 per mole of substrate in the initial reaction; the resulting solution was then stirred overnight to reduce the carbonyls back to hydroxyls.
After reduction, samples were desalted using Porous Graphitic Carbon (PGC) columns (Hyper carb PGC 50 mg, Thermo Fisher, MA, USA). Briefly, a column was pre-washed with 0.1% trifluoroacetic acid (TFA) 80% ACN and water before loading a sample. The impurities were washed away with Milli Q water and the reaction products were eluted with 50% ACN. The purified samples were then lyophilized, dissolved in 200 μl MilliQ-water, and diluted to 10 μg/ml using 50% ACN, after which ammonium chloride (10 mg/ml) was added to a final concentration of 40 μg /ml. The negative ion MS and MSn spectra were obtained by direction infusion of the solutions to the Finnigan LXQ Ion Trap (IT) mass spectrometer equipped with ESI source (Thermo Fisher, MA, USA) at a flow rate of 5 μl/min. The parameters were automatically tuned by the instrument based on X3. The collision energy for fragmentation was optimized based on each substrate.
X2 and its oxidized products after reduction were not successfully purified by the PGC column. Instead, they were analyzed by Acquity Ultra High Performance Liquid Chromatography (UPLC) coupled to an ESI-Q-Tof mass spectrometer (Waters, MA. USA) as described in Figure S12.
Quantification of Oxidized Products by UPLC-ESI-Q-Tof-MS and UPLC Evaporative Light Scattering Detection (ELSD)
The reaction products after NaBD4 reduction and PGC purification were quantified using an Acquity UPLC coupled with ELSD (Waters, MA, USA). A 1.7 μm, 2.1*150 mm Acquity UPLC BEH Amide column (Waters, MA, USA) was used to separate the reaction products. The mobile phases were (A) ACN with 0.1% ammonium hydroxide and (B) 20% ACN with 0.1% ammonium hydroxide. The elution gradient was as follows: from 96% ACN to 50% ACN in 10 min, isocratic (50% ACN) for 2 min, back to 96% ACN in 0.01 min, and 18 min re-equilibrium in initial conditions. The flow was at 250 μl/min with column temperature at 35°C. The ELSD drift tube temperature was set to 40°C and the gain to 200. The nebulizer was set to cooling and pressure to 40 psi. Reaction products were diluted with pure ACN to ~3,000 ng of reaction products per 7 μl of injected sample. External standards were made by reducing and purifying the pure substrates using the PGC column as described in section Identification of Oxidized Positions Through Fragmentation Analysis of Products After NaBD4Reduction. Standard curves were made by injecting 200–3,750 ng of each reduced substrate for each injection. The Acquity UPLC coupled with ESI-Q-Tof-MS was used to identify the peaks in ELSD chromatogram. The parameters for ESI-Q-Tof were set to the same as mentioned in section Mass Spectrometric Analysis of Oxidized Products by Direct Infusion, except that the desolvation temperature was set to 400°C. Quantitative analysis of the reaction products using the UPLC-ELSD and mass spectra collected in section Identification of Oxidized Positions Through Fragmentation Analysis of Products After NaBD4Reduction was then achieved as described below (section Quantitative Interpretation of Mass Spectra).
Quantitative Interpretation of Mass Spectra
Following NaBD4 reduction of oxidized xylooligosaccharides, the same m/z value can be obtained for isotopic non-oxidized xylooligosaccharides and specific oxidized products. For example, the monoisotopic mass of X3 has an m/z of 452, and the naturally present isotopic chloride, carbon, hydrogen and oxygen generate m/z values of 453 and 454. At the same time, single oxidation of a secondary hydroxyl in X3 followed by NaBD4 reduction will also generate a peak at m/z 453, and double oxidation of secondary hydroxyls in X3 followed by NaBD4 reduction will generate a peak at m/z 454 (Figure S1). Therefore, the following system of equations (Equations 1–4) was established to calculate the percent of single oxidized and double oxidized products, taking into account the abundance of naturally present isotopic substrate:
where a, b, and c are the abundance of m/z values corresponding to non-oxidized, single oxidized, and double oxidized oligosaccharides in reactions following enzymatic oxidation (e.g., m/z 452, 453, and 454, respectively, for X3); and r1 and r2 are the abundances of corresponding m/z values naturally present the isotopic substrate.
Therefore:
where:
When an oligosaccharide is oxidized at the reducing end C-1, the reaction product containing the carboxyl acid forms an anionic, deprotonated species. For example, X3 oxidized at the C-1 position will have an m/z of 429. Herein, the theoretical isotopic distribution of C-1 oxidized oligosaccharides was generated by Masslynx V4.1. When additional oxidations take place at the secondary hydroxyls, the deuterium label would increase the corresponding m/z value by a corresponding number of Daltons. Therefore, Equations (1)–(4) can be extended by Equations (5)–(8), to calculate the extent of oxidized products with at least one oxidation at the reducing end C-1:
where a′, b′, and c′ are the abundance of m/z values corresponding to C-1 oxidized, C-1 oxidized + single oxidation at a secondary hydroxyl, and C-1 oxidized + double oxidation at secondary hydroxyls (e.g., m/z 429, 430, and 431, respectively, for X3); and r1′ and r2′ are the relative abundance of theoretically generated isotopic oligosaccharide with one oxidation at reducing end C-1.
In this case, the double oxidation consists of one reducing end C-1 oxidation and one secondary hydroxyl oxidation, and a the triple oxidation consists of one reducing end C-1 oxidation and two secondary hydroxyl oxidation.
Therefore:
where:
See Figure S1 for example calculations.
Results
Initial Rates of PDH Action on Xylooligosaccharides
The yield of recombinantly produced and purified AbPDH1 and AmPDH1 was 1.65 and 11.5 mg/l, respectively, where over 90% purity was reached for both enzymes (Figure S2). The activity and pH optima of each PDH was comparable to previous studies when using glucose as the substrate (Table S1). Activity on xylose and xylooligosaccharides was then measured using Fc+ ion as the electron acceptor, given the particular sensitivity of the corresponding assay (Sygmund et al., 2008). Both AbPDH1 and AmPDH1 were active on 10 mM X-X4 and A3X; furthermore, AbPDH1 was distinguished by its additional activity on A2XX and XA3XX under the conditions used (Figure 1).
Figure 1
Activity of both enzymes on xylose (43–45 U/mg; Figure 1) were similar to those previously reported for the native AmPDH1 (39 U/mg; Sygmund et al., 2008). Overall, AbPDH1 had higher activities toward X3, X4, and A3X compared to corresponding activity of AmPDH1. Notably, the activity of AbPDH1 toward A3X was comparable to unsubstituted X2, and AbPDH1 activity toward A2XX and XA3XX was ~40 and 55% of the corresponding unsubstituted X3 and X4 substrates. By contrast, the activity of AmPDH1 on A3X was only 38% of its activity on X2 and initial activity values could not be obtained for AmPDH1 oxidation of A2XX or XA3XX under these conditions. Similarly, initial activities were not obtained for AbPDH1 and AmPDH1 toward U4m2XX or XU4m2XX; however, oxidation of all tested xylooligosaccharides by AbPDH1 and AmPDH1 was observed after reaction optimization and prolonged incubation time, as described below.
Measuring Xylooligosaccharide Conversion by AbPDH1 and AmPDH1
To maximize xylooligosaccharide oxidation by AmPDH1 and AbPDH1, the Fc+ electron acceptor used to measure initial rates was replaced with BQ that can be regenerated using laccase (Baminger et al., 2001). In an effort to optimize PDH and laccase activities, glucose and xylose oxidation by AmPDH1 and AbPDH1 was tested at pH values ranging from pH 3.0 to 7.0 (Figure S3). Based on these analyses, subsequent reactions were performed at pH 5.5. Notably, the addition of laccase clearly increased substrate conversions by both enzymes (Figure S4), where up to 85% depletion of xylose (50 mM starting concentration) was reached in 24 h using 5 mM BQ.
Both AbPDH1 and AmPDH1 fully depleted X2, X3, and A3X after 24 h; AbPDH1 also fully depleted X4 after 24 h (Figure 2). With the exception of X2, substrate depletion was more rapid in reactions containing AbPDH1 than AmPDH1 (Figure 2). Differences in substrate conversion were most apparent after 8 h, where X3 and X4 conversion by AbPDH1 was 1.2 and 3.9 times higher than by AmPDH1, respectively (Figures 2B,C).
Figure 2
When testing AbPDH1 and AmPDH1 activity over 24 h on A3X, A2XX, and XA3XX, minor α-arabinofuranosidase activity was detected in the commercial laccase. Specifically, after 24 h in reactions containing laccase alone, 7.5, 11.5, and 20% of Araf was hydrolyzed from A3X, A2XX, and XA3XX, respectively (Figures S5–S7). This reduction of substrate by hydrolysis was taken into account by comparing the substrate depletion in PDH reactions to corresponding control reactions containing laccase alone. Whereas, both AbPDH1 and AmPDH1 were able to fully deplete A3X by 8 h (Figure 2D), A2XX conversion by AbPDH1 was 1.6 times higher than AmPDH1 after 24 h (Figure 2E), and both AbPDH1 and AmPDH1 transformed ~60% of XA3XX after 24 h (Figure 2F).
When using the same enzyme loading (0.2 U/ml) as for neutral xylooligosaccharides, only AbPDH1 showed clear conversion of both U4m2XX and XU4m2XX (Figure S8). Detectable conversion of U4m2XX and XU4m2XX by AmPDH1 was only observed after increasing the enzyme loading 10 times (to 2 U/ml). When comparing AbPDH1 and AmPDH1 after 24 h at the higher enzyme loading, AbPDH1 depleted nearly 100% of both U4m2XX and XU4m2XX whereas substrate depletion by AmPDH1 was ~30 and 20% for U4m2XX and XU4m2XX, respectively (Figure S8).
Direct Infusion ESI-Q-Tof Mass Spectrometry Confirms the Formation of Oxidized Xylooligosaccharides
Direct infusion ESI-Q-Tof analyses confirmed that all the tested substrates were oxidized by both AbPDH1 and AmPDH1. The C-1 oxidation products were negatively charged and detected in their anionic form, while neutral oligosaccharides were detected as chlorine adducts. X2, X3, X4, A3X, A2XX, XA3XX gave peaks at mass-to-charge ratios (m/z) 317, 449, 581, 449, 581, and 713, respectively (Figures S9, S10). Consistent with the HPAEC-PAD analyses summarized above, low levels of products with a loss of pentose (132 Da) were detected in reactions containing Araf substituted xylooligosaccharides, resulting from α-arabinofuranosidase side activity in the commercial laccase preparation. U4m2XX and XU4m2XX were detected deprotonated [M-H]− with peaks at m/z 603 and 735, respectively (Figure S11).
Both AbPDH1 and AmPDH1 completely consumed X2 and X3 after 24 h treatment. By contrast, varying amounts of residual substrate could be detected for X4 and all substituted xylooligosaccharides. AbPDH1 and AmPDH1 treatment of X2, A3X, and XA3XX resulted in similar MS spectra and thus end products; however, clear differences in product profiles were seen for the other tested substrates. For example, highest peaks following X3 oxidation by AbPDH1 and AmPDH1 were at m/z 463 and 427, respectively; and highest peaks following X4 oxidation were at m/z 595/597 and 561, respectively (Figure S9). Considering substituted xylooligosaccharides, a peak at m/z 597 represented the highest product peak from A2XX following 24 h treatment with AbPDH1, whereas the most abundant peak after 24 h AmPDH1 treatment was the non-oxidized A2XX (m/z 581; Figure S10). Consistent with the greater oxidation of neutral xylooligosaccharides by AbPDH1 compared to AmPDH1, the ESI-Q-Tof analyses confirmed significant oxidation of U4m2XX and XU4m2XX by the higher dose of AbPDH1, resulting in peaks at m/z 619 and 751, respectively. By contrast, AmPDH1 oxidized U4m2XX and XU4m2XX to a small percentage (Figure S11). Given the higher oxidation of neutral substrates by both enzymes, the corresponding products were analyzed in greater detail.
Qualitative Comparison of Oxidized Xylooligosaccharides Generated by AbPDH1 and AmPDH1
Due to the multiple potential oxidations and reactivity of the oxidized products in water, MS spectra interpretation is challenging. Taking the AbPDH1 oxidized X3 as an example (Figure S1C), the main peak m/z 463 is expectedly representing a chlorine adduct of a double oxidized product with two carbonyl groups of which the other one has reacted with water to a hydrate (compound A in Table S2). Peak m/z 481 is presumably the chorine adduct of the same double oxidized product in which both carbonyls are in the hydrate form. The second most abundant peak m/z 427 is most probably the double oxidized product carrying carboxylic acid group at the reducing end C-1 and one carbonyl group (compound B in Table S2) whereas peak m/z 445 can represent the same product in a hydrate form. Mass to charge ratio of 445 can also represent a chlorine adduct of the original double oxidized product with two carbonyl groups. Thus, one compound that is oxidized at two secondary hydroxyls is shown as three peaks in the mass spectrum (compound A in Table S2).
To facilitate the interpretation of MS spectra, the oxidized products were treated with NaBD4, and analyzed by ESI-MS after purification. Thus, a PDH oxidation at secondary hydroxyl resulting in a ketone is equivalent to one Dalton difference in mass after NaBD4 reduction compared to substrate (Figure 3A). Instead, the oxidation at the reducing end C-1 forms a carboxyl group, which is resistant to NaBD4 reduction (Figure 3B). Compared to the chloride adduct of NaBD4 reduced substrate, the oxidation at reducing end C-1 will result in a deprotonated m/z peak that is 23 Da less. Therefore, by counting the Dalton difference between the reduced oxidation products and the reduced original substrates, the number of oxidations occurring in one molecule can be determined.
Figure 3
After NaBD4 reduction, the predominant peak in mass spectra of oxidized X2 by both PDHs was m/z 322, which is 2 Da more than the control X2 indicating double oxidation at two secondary hydroxyls (Figure S12). The 2 Da increase in mass was also seen for oxidized X3. Oxidation of X4 by both PDHs resulted in single and double oxidized products after 24 h reaction. Notably, Araf substitution inhibited multiple oxidation of corresponding substrates; A3X and A2XX were mostly single oxidized at a secondary hydroxyl while the XA3XX was still largely non-oxidized by both PDHs after 24 h reaction (Figure S13).
In addition to oxidizing secondary hydroxyls, both AbPDH1 and AmPDH1 oxidized the reducing end C-1 of all substrates. Moreover, mass spectra of X2, X3, and X4 products revealed that reducing end C-1 oxidation and secondary hydroxyl oxidations could co-exist in one molecule, resulting in final products with a mass that is 22 Da (reducing end C-1 oxidation + one secondary hydroxyl oxidation) or 21 Da (reducing end C-1 oxidation + two secondary hydroxyls oxidation) less than the control substrate (Figure S12).
Direct infusion mass spectrometry after NaBD4 reduction thus confirmed the ability of both AbPDH1 and AmPDH1 to oxidize xylooligosaccharides at multiple positions. Quantification of the reaction products was subsequently important to uncover the specificity of each PDH.
Separate Quantification of Xylooligosaccharides Oxidized at Secondary Hydroxyl Positions and the Reducing End C-1
A two-stage MS-based approach was developed in this study to quantify single and multiply oxidized oligosaccharides, and as well as oxidations at secondary hydroxyls vs. the reducing end C-1 (section Quantitative Interpretation of Mass Spectra; see Figure S1 for example calculation).
Product profiles were first calculated based on mass spectra, where different ionization intensities are obtained for neutral and acidic products, preventing the direct quantitative comparison of residual substrate products with ketone groups, and acidic products with reducing end C-1 oxidation. As a result, the extent of oxidized secondary hydroxyls and the reducing end C-1 were calculated separately (Figure 4). Overall, products with two ketones (i.e., oxidation of two secondary hydroxyls) were dominant for linear xylooligosaccharides. The only exception was AmPDH1 oxidation of X4, which was only partially oxidized and mainly to a single oxidized product (Figure 4A). For all linear xylooligosaccharides treated for 24 h with AbPDH1, products oxidized at the reducing end C-1 were mostly also oxidized at a secondary hydroxyl group (i.e., were double oxidized products, Figure 4B). This same pattern was observed for AmPDH1 oxidation of X3; however, a triple oxidized product (C-1 oxidation + two ketones) represented more than 50% of acidic products generated through AmPDH1 oxidization of X2. Whereas, oxidation of linear xylooligosaccharides mostly led to double oxidized products, single oxidized products dominated in reactions containing Araf substituted xylooligosaccharides (Figures 4A,B).
Figure 4
Total Comparative Quantification of Oxidized Xylooligosaccharides Generated by AbPDH1 and AmPDH1
Using hydrophilic interaction chromatography (HILIC), acidic and neutral reaction products were successfully separated and identified by MS (Figures S14, S15). The acidic products (C-1 oxidation) eluted earlier than the neutral ones and these two classes of products could be quantified after HILIC separation with on-line ELSD using external standards. The ELSD quantification was successfully done for X3, X4, A3X, A2XX, and XA3XX products. Due to co-elution with salts, X2 products could not be quantified. Six reaction species could be quantified: non-oxidized substrate, one oxidation at a secondary hydroxyl, two oxidations at secondary hydroxyls, one oxidation at the reducing end C-1, one oxidation at reducing end C-1 with one oxidation at secondary hydroxyl, and one oxidation at reducing end C-1 with two oxidations at secondary hydroxyls (Figure 5).
Figure 5
AbPDH1 and AmPDH1 primarily oxidized secondary hydroxyls, and to a lesser extent the anomeric C-1. The main, double oxidized derivative with two ketone groups represented 81 and 56% of X3 products after 24 h treatment by AbPDH1 and AmPDH1, respectively (Figure 5). AbPDH1 also mainly oxidized X4 at secondary hydroxyls with 38% being single ketone and 48% being double ketone products, whereas the most abundant reaction product after AmPDH1 treatment was C-1 oxidized X4 (34%). Dominating A3X products (73–78%) carried single ketone group whereas A2XX was primary oxidized by AbPDH1 to one (50%) and two (21%) ketone derivatives. After AmPDH1 treatment, 48% of A2XX remained non-oxidized and 39% of products carried single ketone group. The composition of reaction products from XA3XX treatments by both PDHs was rather similar, with ~50% of substrates being non-oxidized after 24 h reaction; ~25% converted to products with one ketone group, and 22% were oxidized only at the reducing end C-1 (Figure 5).
MS Fragmentation to Determine the Oxidized Residues
The oxidized products after reduction were analyzed using Q-Tof or ion trap by following the deuterium label in the fragment ions. For example, the MS/MS spectrum of reduced X2 shows the glycosidic linkage cleavage generating Y1 ion containing the reduced Xylp residues at m/z 152 (Figure 6A). The molecular masses of Y1 ions carrying 1 Da more than the control sample (Figures 6B,C), indicate that one oxidation had taken place at the reducing Xylp residue by both AbPDH1 and AmPDH1. The mass of precursor ions increased by 2 Da for oxidized samples, thus the other oxidation was at the non-reducing end Xylp residue. The C-1 oxidized X2 would have m/z 297 and generate the Y1 ion at m/z 165 (data not shown). The m/z 298 ion produced the Y1 ion also at m/z 165 (Figure 6D), suggesting one secondary hydroxyl oxidation took place at the non-reducing Xylp moiety together with the oxidation at reducing end C-1. The Y1 ion shown in Figure 6E increased by 1 Da compared to the compound shown in Figure 6D, indicating that AmPDH1 can even generate triple oxidized X2 with a ketone group at the non-reducing Xylp residue, a ketone at the reducing Xylp residue, and a reducing end carboxylic acid.
Figure 6
Following the same procedure as described above for X2, the oxidized positions for other linear xylooligosaccharides were determined (Figure 5; see example MSn spectra in Figures S16, S17). Oxidation by both AbPDH1 and AmPDH1 was clearly restricted to the reducing and non-reducing Xylp residues. Consistent with quantification using ELSD (section Separate Quantification of Xylooligosaccharides Oxidized at Secondary Hydroxyl Positions and the Reducing End C-1), the MS fragmentation confirmed that linear xylooligosaccharides were mostly multi-oxidized by both PDHs, with the exception of X4 oxidation by AmPDH1 which was dominated by single oxidation. The MS fragmentation further revealed that the oxidation of X4 by AmPDH1 was localized to the reducing end secondary hydroxyls or reducing end C-1. Surprising, whereas single oxidation of X4 by both AbPDH1 and AmPDH1 was restricted to the reducing Xylp, single oxidation of X3 by both PDHs was mainly at the non-reducing end Xylp secondary hydroxyls (Figure 5). Double oxidation by PDHs included products oxidized at (1) secondary hydroxyls of both the reducing and non-reducing Xylp, or (2) the reducing C-1 position and a secondary hydroxyl at the non-reducing end. Increasing the length of the xylooligosaccharide to X3 and X4 did not lead to detectable oxidation of internal Xylp substituents. Interestingly, in addition to the regioselectivity of single and double oxidations, a product of m/z 299 was detected in the AmPDH1 oxidized X2 after reduction, representing products that were triple oxidized. In this case, it is conceivable that the reducing Xylp is oxidized at both the reducing end C-1, and a secondary hydroxyl. This was the only detected triple oxidized product.
The Araf substituted substrates were mainly single oxidized or non-oxidized after 24 h treatment. A3X containing α-(1 → 3)-linked Araf substitution at the non-reducing end Xylp residue, was single oxidized by both PDHs at the reducing end Xylp resulting in a ketone group or C-1 oxidized carboxylic acid. A2XX with α-(1 → 2)-linked Araf substitution was similarly oxidized at the secondary hydroxyl or C-1 of the reducing end Xylp by both PDHs. Interestingly, in some A2XX products, a second ketone group was detected at the non-reducing end Xylp, which carries the Araf substituent. Moreover, AmPDH1 showed unique ability to form products with a single ketone group at the non-reducing end Xylp of A2XX. No clear oxidation of Araf was detected in any of the samples.
Discussion
AbPDH1 was first isolated over 20 years ago (Volc et al., 1997), and was followed nearly 10 years later by the isolation of AmPDH1 (Sedmera et al., 2006). Since then, AbPDH1 and AmPDH1 have been biochemically characterized using a broad collection of monosaccharides, heteroglycosides, and selected oligosaccharides, revealing their potential for single and double oxidation of many plant-derived carbohydrates, and motivating their application in biofuel cells and organic syntheses (Giffhorn et al., 2000; Peterbauer and Volc, 2010). Herein, AbPDH1 and AmPDH1 were directly compared in terms of potential to oxidize diverse xylooligosaccharides, both linear and branched, acid and neutral. In particular, we investigated the impact of xylooligosaccharide length and substitution on the extent and position of oxidation by each enzyme. In this way, we evaluated the potential of AbPDH1 and AmPDH1 to serve as catalysts in the synthesis of telechelic building blocks from xylan fragments common to wood and agricultural residues.
Consistent with reported AmPDH1 activity toward X2 (Sygmund et al., 2008) and glucose-containing oligosaccharides (Volc et al., 1997; Tasca et al., 2007; Peterbauer and Volc, 2010; Rafighi et al., 2018) both AbPDH1 and AmPDH1 produced herein oxidized X2 as well as X3 and X4. As observed for PDH activity toward cellooligosaccharides and maltooligosaccharides (Kujawa et al., 2007; Sygmund et al., 2008; Peterbauer and Volc, 2010; Graf et al., 2017), the specific activity (U/mg) of both AbPDH1 and AmPDH1, as well as the rate of the substrate depletion detected with HPAEC-PAD, decreased with increasing degree of polymerization, where the impact of substrate length on activity was greater for AmPDH1 than for AbPDH1. Notably, AmPDH1 depleted X2 faster than AbPDH1, which is in line with the previous studies, where AmPDH1 shows higher catalytic turnover toward disaccharides, cellobiose, maltose and lactose than AmPDH1 (Sygmund et al., 2008; Gonaus et al., 2016). However, the reverse pattern was observed for longer linear oligosaccharides; AbPDH1 activity toward substituted xylooligosaccharides was also substantially higher than that measured for AmPDH1. In both cases, however, activities toward A3X were higher than X3, and activities toward neutral substrates were higher than for the acidic xylooligosaccharides U4m2XX and XU4m2XX. The structural basis for these differences in AbPDH1 and AmPDH1 activity toward oligosaccharides awaits a solved structure for AbPDH1 and of enzyme-substrate complexes for this enzyme family. Still, as found in Gonaus et al. (2016), a homology model of AbPDH1 based on the solved structure of AmPDH1 revealed two amino acid deletions in the loop 1 region, differences in the loop 3 region, and substitution of Phe508 in AmPDH1 to the smaller Val505 in AbPDH1. These differences vary the amino acid content in loop regions at the entrance of the substrate binding pocket, and could impact accessibility of the active site (Gonaus et al., 2016).
When considering the synthesis of telechelic molecules from xylooligosaccharides, it is especially important to evaluate the extent and positions of oxidation in each reaction product. To facilitate this analysis, we established an alternative method for the identification and quantification of oxidized carbohydrates, which utilizes NaBD4 reduction and is solely based on MS and UPLC systems. This method enables fast quantification of formed carbonyl groups as the results are readily interpreted by counting the Dalton difference. In addition, the analysis can be accomplished with reaction products in microgram levels, allowing it to be an attractive technique for characterizing carbohydrate oxidoreductases where enzyme or substrate quantities are limiting. For example, even though specific activity values could not be obtained using the acidic xylooligosaccharides U4m2XX and XU4m2XX, the near-complete oxidation of both substrates by AbPDH1 after 24 h was confirmed using this method. Given the higher activity of both AbPDH1 and AmPDH1 on neutral xylooligosaccharides, corresponding product profiles were quantified in more detail. Consistent with activity values, AbPDH1 reached a higher level of double oxidized end products from X4 and A2XX compared to AmPDH1. An exception was for X2, where treatment with AmPDH1 led even to a triple-oxidized product. The ESI-MSn analyses confirmed that oxidations were targeted to the reducing and non-reducing ends of all tested xylooligosaccharide substrates. Interestingly, yet unexplained, selectivity was noticed by both enzymes. For example, a single keto group was formed at the non-reducing Xylp residue of X3, whereas in X4 a single keto group was found solely at the reducing end Xylp. Also, AmPDH1 produced more C-1 oxidized acidic products from X3 and X4 than AbPDH1. As previously reported for activity toward cellobiose, maltose and lactose (Volc et al., 2004; Sygmund et al., 2012; Gonaus et al., 2016), AbPDH1 and AmPDH1 oxidized the anomeric carbon together with secondary hydroxyls of xylooligosaccharides.
Although cross-ring fragmentation was observed in some oxidation products (e.g., with C-1 oxidation), we were not able to identify which secondary hydroxyl was targeted by each enzyme due to proton transfer during the fragmentation (Domon and Costello, 1988). Nevertheless, as reported for glucose-containing oligosaccharides (Sedmera et al., 2006; Peterbauer and Volc, 2010; Rafighi et al., 2018), the context of the secondary hydroxyls at C-2 and C-3 positions clearly impacted AbPDH1 and AmPDH1 activity toward xylooligosaccharides. Most notably, the alpha-(1 → 3)-linked Araf in A3X and XA3XX shifted AbPDH1 and AmPDH1 activity toward the reducing end of both substrates. By contrast, both the reducing and non-reducing ends of A2XX were oxidized.
In summary, with the newly developed method to identify and quantify oxidized carbohydrates, we successfully determined the degree of oxidation and analyzed the regioselectivity of AbPDH1 and AmPDH1 toward xylooligosaccharides present in wood and agricultural fiber. The widened carbohydrate profile of AbPDH1 and AmPDH1 provides further support for their proposed biological function in evading plant defense mechanisms through reducing plant-derived quinones, or promoting lignin depolymerization through reducing lignin-derived radicals generated by lignin-active peroxidases and laccases (Peterbauer and Volc, 2010; Sützl et al., 2018; Herzog et al., 2019). Confirming herein that oxidative activity is restricted to the ends of xylooligosaccharides, and that both linear and substituted xylooligosaccharides are accepted, also opens new applications of PDHs that transform underused xylan streams into telechelic molecules primed for polymerization.
Statements
Data availability statement
The datasets generated for this study are available on request to the corresponding author.
Author contributions
JK, HZ, AK, MT, and EM contributed conception and design of the study. JK, HZ, and AK performed the experiments. JK, HZ, S-LC, and AK analyzed the data. MT and EM contributed reagents, materials, and analysis tools. JK, HZ, MT, and EM wrote the paper. All authors read and approved the submitted version.
Funding
This work was conducted with the funding from Academy of Finland for COCOA (project codes 308996 and 308997).
Acknowledgments
Dr. Thu Vuong and Dr. Sanna Koutaniemi are acknowledged for kindly providing acidic substrates for this study. Dr. Anne Usvalampi was acknowledged for the eminent technical support and expertise in HPAEC-PAD analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00011/full#supplementary-material
References
1
AndbergM.MollerupF.ParikkaK.KoutaniemiS.BoerH.JuvonenM.et al. (2017). A novel Colletotrichum graminicola raffinose oxidase in the AA5 family. Appl. Environ. Microbiol.83, 1–17. 10.1128/AEM.01383-17
2
BamingerU.LudwigR.GalhaupC.LeitnerC.KulbeK. D.HaltrichD. (2001). Continuous enzymatic regeneration of redox mediators used in biotransformation reactions employing flavoproteins. J. Mol. Catal.11, 541–550. 10.1016/S1381-1177(00)00034-5
3
ChundawatS.BeckhamG. T.HimmelM. E.DaleB. E. (2011). Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol.2, 121–145. 10.1146/annurev-chembioeng-061010-114205
4
DomonB.CostelloC. E. (1988). A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J.5, 397–409. 10.1007/BF01049915
5
EbringerováA. (2006). Structural diversity and application potential of hemicelluloses. Macromol. Symp.232, 1–12. 10.1002/masy.200551401
6
GiffhornF.KöpperS.HuwigA.FreimundS. (2000). Rare sugars and sugar-based synthons by chemo-enzymatic synthesis. Enzyme Microb. Technol.27, 734–742. 10.1016/S0141-0229(00)00293-3
7
GonausC.KittlR.SygmundC.HaltrichD.PeterbauerC. (2016). Transcription analysis of pyranose dehydrogenase from the basidiomycete Agaricus bisporus and characterization of the recombinantly expressed enzyme. Protein Expr. Purif.119, 36–44. 10.1016/j.pep.2015.11.003
8
GrafM. M.BrenU.HaltrichD.OostenbrinkC. (2013). Molecular dynamics simulations give insight into d-glucose dioxidation at C2 and C3 by Agaricus meleagris pyranose dehydrogenase. J. Comput. Aided Mol. Des.27, 295–304. 10.1007/s10822-013-9645-7
9
GrafM. M.WeberS.KracherD.KittlR.SygmundC.LudwigR.et al. (2017). Characterization of three pyranose dehydrogenase isoforms from the litter-decomposing basidiomycete Leucoagaricus meleagris (syn. Agaricus meleagris). Appl. Microbiol. Biotechnol.101, 2879–2891. 10.1007/s00253-016-8051-1
10
GrafM. M. H.SucharitakulJ.BrenU.ChuD. B.KoellenspergerG.HannS.et al. (2015). Reaction of pyranose dehydrogenase from Agaricus meleagris with its carbohydrate substrates. FEBS J.282, 4218–4241. 10.1111/febs.13417
11
HerzogP. L.SützlL.EisenhutB.MareschD.HaltrichD.ObingerC.et al. (2019). Versatile oxidase and dehydrogenase activities of bacterial pyranose 2-oxidase facilitate redox cycling with manganese peroxidase in vitro. Appl. Environ. Microbiol.85, 1–15. 10.1128/AEM.00390-19
12
HuangC. H.LaiW. L.LeeM. H.ChenC. J.VasellaA.TsaiY. C.et al. (2005). Crystal structure of glucooligosaccharide oxidase from Acremonium strictum: a novel flavinylation of 6-S-cysteinyl, 8α-N1-histidyl FAD. J. Biol. Chem.280, 38831–38838. 10.1074/jbc.M506078200
13
KoutaniemiS.GuillonF.TranquetO.BouchetB.TuomainenP.VirkkiL.et al. (2012). Substituent-specific antibody against glucuronoxylan reveals close association of glucuronic acid and acetyl substituents and distinct labeling patterns in tree species. Planta236, 739–751. 10.1007/s00425-012-1653-7
14
KujawaM.VolcJ.HaladaP.SedmeraP.DivneC.SygmundC.et al. (2007). Properties of pyranose dehydrogenase purified from the litter-degrading fungus Agaricus xanthoderma. FEBS J.274, 879–894. 10.1111/j.1742-4658.2007.05634.x
15
LevasseurA.DrulaE.LombardV.CoutinhoP. M.HenrissatB. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels6:41. 10.1186/1754-6834-6-41
16
LombardV.Golaconda RamuluH.DrulaE.CoutinhoP. M.HenrissatB. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res.42, D490–D495. 10.1093/nar/gkt1178
17
MacCormickB.VuongT. V.MasterE. R. (2018). Chemo-enzymatic Synthesis of Clickable Xylo-oligosaccharide Monomers from Hardwood 4-O-Methylglucuronoxylan. Biomacromolecules19, 521–530. 10.1021/acs.biomac.7b01642
18
ParikkaK.LeppänenA. S.XuC.PitkänenL.EronenP.ÖsterbergM.et al. (2012). Functional and anionic cellulose-interacting polymers by selective chemo-enzymatic carboxylation of galactose-containing polysaccharides. Biomacromolecules13, 2418–2428. 10.1021/bm300679a
19
PeterbauerC. K.VolcJ. (2010). Pyranose dehydrogenases: biochemical features and perspectives of technological applications. Appl. Microbiol. Biotechnol.85, 837–848. 10.1007/s00253-009-2226-y
20
RafighiP.BollellaP.PankratovaG.PeterbauerC. K.ConghaileP. Ó.LeechD.et al. (2018). Substrate preference pattern of Agaricus meleagris pyranose dehydrogenase evaluated through bioelectrochemical flow injection amperometry. ChemElectroChem6, 801–809. 10.1002/celc.201801194
21
SchellerH. V.UlvskovP. (2010). Hemicelluloses. Annu. Rev. Plant Biol.61, 263–289. 10.1146/annurev-arplant-042809-112315
22
SedmeraP.HaladaP.KubátováE.HaltrichD.PrikrylováV.VolcJ. (2006). New biotransformations of some reducing sugars to the corresponding (di)dehydro(glycosyl) aldoses or aldonic acids using fungal pyranose dehydrogenase. J. Mol. Catal.41, 32–42. 10.1016/j.molcatb.2006.04.004
23
SützlL.LaurentC. V. F. P.AbreraA. T.SchützG.LudwigR.HaltrichD. (2018). Multiplicity of enzymatic functions in the CAZy AA3 family. Appl. Microbiol. Biotechnol.102, 2477–2492. 10.1007/s00253-018-8784-0
24
SygmundC.GutmannA.KrondorferI.KujawaM.GliederA.PscheidtB.et al. (2012). Simple and efficient expression of Agaricus meleagris pyranose dehydrogenase in Pichia pastoris. Appl. Microbiol. Biotechnol.94, 695–704. 10.1007/s00253-011-3667-7
25
SygmundC.KittlR.VolcJ.HaladaP.KubátováE.HaltrichD.et al. (2008). Characterization of pyranose dehydrogenase from Agaricus meleagris and its application in the C-2 specific conversion of D-galactose. J. Biotechnol.133, 334–342. 10.1016/j.jbiotec.2007.10.013
26
TanT. C.SpadiutO.WongnateT.SucharitakulJ.KrondorferI.SygmundC.et al. (2013). The 1.6 Å crystal structure of pyranose dehydrogenase from Agaricus meleagris rationalizes substrate specificity and reveals a flavin intermediate. PLoS ONE8:e53567. 10.1371/journal.pone.0053567
27
TascaF.TimurS.LudwigR.HaltrichD.VolcJ.AntiochiaR.et al. (2007). Amperometric biosensors for detection of sugars based on the electrical wiring of different pyranose oxidases and pyranose dehydrogenases with osmium redox polymer on graphite electrodes. Electroanalysis19, 294–302. 10.1002/elan.200603740
28
VarkiA.CummingsR. D.AebiM.PackerN. H.SeebergerP. H.EskoJ. D.et al. (2015). Symbol nomenclature for graphical representations of glycans. Glycobiology25, 1323–1324. 10.1093/glycob/cwv091
29
VolcJ.KubátováE.WoodD. A.DanielG. (1997). Pyranose 2-dehydrogenase, a novel sugar oxidoreductase from the basidiomycete fungus Agaricus bisporus. Arch. Microbiol.167, 119–125. 10.1007/s002030050424
30
VolcJ.SedmeraP.HaladaP.DanielG.PrikrylováV.HaltrichD. (2002). C-3 oxidation of non-reducing sugars by a fungal pyranose dehydrogenase: spectral characterization. J. Mol. Catal.17, 91–100. 10.1016/S1381-1177(02)00014-0
31
VolcJ.SedmeraP.HaladaP.PrikrylováV.HaltrichD. (2000). Double oxidation of D-xylose to D-glycero-pentos-2,3-diulose (2,3-diketo-D-xylose) by pyranose dehydrogenase from the mushroom Agaricus bisporus. Carbohydr. Res.329, 219–225. 10.1016/S0008-6215(00)00167-1
32
VolcJ.SedmeraP.KujawaM.HaladaP.KubátováE.HaltrichD. (2004). Conversion of lactose to β-D-galactopyranosyl-(1 → 4)-D-arabino-hexos-2-ulose-(2-dehydrolactose) and lactobiono-1,5-lactone by fungal pyranose dehydrogenase. J. Mol. Catal.30, 177–184. 10.1016/j.molcatb.2004.05.006
33
VuongT. V.VesterinenA. H.FoumaniM.JuvonenM.SeppäläJ.TenkanenM.et al. (2013). Xylo- and cello-oligosaccharide oxidation by gluco-oligosaccharide oxidase from Sarocladium strictum and variants with reduced substrate inhibition. Biotechnol. Biofuels6:148. 10.1186/1754-6834-6-148
Summary
Keywords
carbohydrate oxidoreductases, pyranose dehydrogenase, xylooligosaccharide, degree of oxidation, telechelic molecules
Citation
Karppi J, Zhao H, Chong S-L, Koistinen AE, Tenkanen M and Master E (2020) Quantitative Comparison of Pyranose Dehydrogenase Action on Diverse Xylooligosaccharides. Front. Chem. 8:11. doi: 10.3389/fchem.2020.00011
Received
07 October 2019
Accepted
07 January 2020
Published
28 January 2020
Volume
8 - 2020
Edited by
Caroline Remond, Université de Reims Champagne-Ardenne, France
Reviewed by
Dietmar Haltrich, University of Natural Resources and Life Sciences Vienna, Austria; Paul-Henri Ducrot, INRA UMR1318 Institut Jean Pierre Bourgin, France
Updates
Copyright
© 2020 Karppi, Zhao, Chong, Koistinen, Tenkanen and Master.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma Master emma.master@utoronto.ca
This article was submitted to Chemical and Process Engineering, a section of the journal Frontiers in Chemistry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.