 Maria Ochs
Maria Ochs Bernhard Mayer
Bernhard Mayer Thomas J. J. Müller
Thomas J. J. Müller- 1Institut für Organische Chemie and Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Düsseldorf, Germany
- 2Ernst-Berl Institut für Technische and Makromolekulare Chemie, Technische Universität Darmstadt, Darmstadt, Germany
Exciplex or excited complex emission is an excited state process, arising from considerable charge transfer of an excited energy donor to an acceptor, which can be identified by the occurrence of a redshifted emission band that is absent in the individual constituents. Particularly interesting are exciplexes that are formed by intramolecular excited state interaction, which are inherently concentration independent. Based upon our previous experience in the Ugi-4CR syntheses of donor-acceptor conjugates capable of photo-induced intramolecular electron transfer (PIET), that is, generation of light-induced charge separation, we now disclose the diversity-oriented approach on unimolecular exciplex emitters and their reference systems by Ugi-4CR. The photophysics is studied by absorption and emission spectroscopy and accompanied by density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations.
Introduction
Molecular luminescence (Lakowicz, 2006; Valeur and Berberan-Santos, 2012) is a widespread phenomenon of functional organic materials (Müller and Bunz, 2007) and finds broad application in many fields of science and technology, ranging from fundamental science (luminescence spectroscopy) (Wolfbeis, 1993; Valeur and Brochon, 2012) over biophysical analytics (Chen et al., 1998; Nilsson et al., 2002; Wagenknecht, 2008; Demchenko et al., 2009; Kim and Park, 2009; Cairo et al., 2010), diagnostics (Kobayashi et al., 2010; Carter et al., 2014), and dye lasers (Thiel, 2000; Shankarling and Jarag, 2010) to sensors (Loving et al., 2010; Klymchenko, 2017; Zhang et al., 2017) and organic light-emitting diodes (OLED) (Müllen and Scherf, 2006; Park et al., 2011; Thejo Kalayani and Dhoble, 2012; Li, 2015). Besides fluorescence and phosphorescence as radiative deactivation of electronically excited singlet or triplet states, also radiative states arising from inter- or intramolecular electronic interaction, eventually by energy or electron transfer, are particularly interesting. With this respect, exciplexes (excited complexes) and excimers (excited dimers) are emissive charge-transfer complexes, which are formed by excitation of one of the constituting chromophores that in turn collides with a second chromophore in its electronic ground state (Balzani et al., 2003; Balzani, 2008). In contrast to charge-separated charge transfer states, exciplexes and excimers can deactivate by broad structureless emission (Lakowicz, 2006; Valeur and Berberan-Santos, 2012), however, redshifted with respect to the constituting individual chromophores (Figure 1) (Balzani et al., 2003; Balzani, 2008). While excimers constitute from identical chromophores in exciplexes, the two colliding chromophores differ in their structure and nature. In either case, stability results from Coulomb and favorable orbital interactions at distances between 3 and 4 Å. Since exciplex emission represents a pathway of quenching the excited molecule's fluorescence, this phenomenon might be employed for sensing proximity of two interacting chromophores by a radiative process. This excited state phenomenon is particularly interesting—absorption spectra are simple superpositions of their constituting chromophores—as the emission will be dependent on the solvent polarity in terms of energy of the exciplex state as well as in terms of the intensity (Kavarnos, 1993).
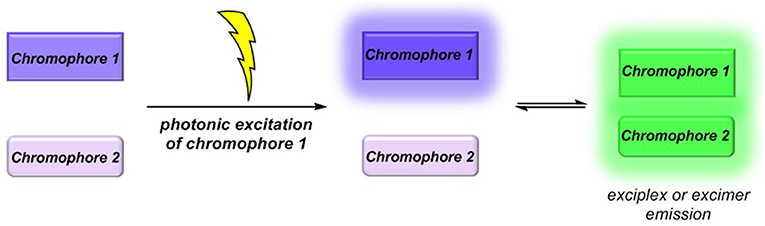
Figure 1. Exciplex or excimer emission as a radiative quenching pathway by intermolecular chromophore-chromophore interaction in the excited state.
If two chromophores are placed into a unimolecular setting, that is, by covalent ligation, the unimolecular exciplex emission is inherently independent of concentration and diffusion control, in contrast to exciplex emission arising from two particles. The positive solvatochromicity of the exciplex emission band, that is, a redshift of the longest wavelength emission band with increasing solvent polarity, as well as its decreasing effect on the fluorescence quantum yield and fluorescence lifetimes has impressively been shown for ether-tethered dyads consisting of pyrene and 2-benzylidenemalononitrile as constituting chromophores (Piuzzi, 1993; Zhang et al., 1997; Piuzzi et al., 1999). Likewise, alkyl-tethered pyrene/anthracene-N,N-dimethylaniline dyads were studied in detail by time-resolved laser spectroscopy (Mataga and Miyasaka, 1999). In hexane, the formation of the compact exciplex, where the hydrocarbon and the N,N-dimethylaniline moieties are sandwiched, occurs in the nanosecond time regime, while in acetonitrile, the loose exciplex forms within picoseconds after excitation, obviously due to a facilitated charge-transfer proceeding without conformational changes. Time-resolved transient absorption spectroscopy moreover supports that the compact exciplex bands are strongly broadened in contrast to the acetonitrile spectra, which clearly support the well-resolved signature of superimposing radical ion spectra, that is, a strong ion pair character. Intermediate solvent polarities account for signal broadening, that is, the population of more ordered compacted conformations.
As part of our program to methodologically develop concise syntheses of functional chromophores by multicomponent reactions (MCR) (Levi and Müller, 2016a), we are particularly interested in MCR syntheses of fluorophores (Levi and Müller, 2016b; Riva et al., 2016; Merkt and Müller, 2018; Müller, 2018) and donor-acceptor systems that interact in the excited state via photo-induced electron transfer (PIET) (Kavarnos, 1993; Lemmetyinen et al., 2011; Wenger, 2011; Ricks et al., 2012; Vauthey, 2012), as shown by non-radiative fluorescence quenching (Bucci and Müller, 2006; Bay et al., 2013, 2014; Bay and Müller, 2014). This can be considered as chromophore-chromophore interactions at short distances, that is, between Dexter and Förster radii.
Conceptually, we reasoned that employing a scaffold-forming MCR approach (Levi and Müller, 2016a) to exciplex-forming bichromophores could establish a concise general synthetic approach to these types of unimolecular exciplex emitters (Figure 2). In addition, if emission of non-interacting and exciplex conformers occurs simultaneously from this Boltzmann distribution, dual emission may arise, furnishing emission color mixing. Here, we report the Ugi-4CR synthesis of several representative unimolecular exciplex emitters consisting of an N,N-dimethyl aniline moiety as a donor and anthracene, naphthalene, or pyrene as acceptor chromophores, as well as reference chromophores. Furthermore, the absorption and emission spectroscopic characteristics are reported and discussed in the light of DFT and TDDFT calculations of the donor-acceptor systems.
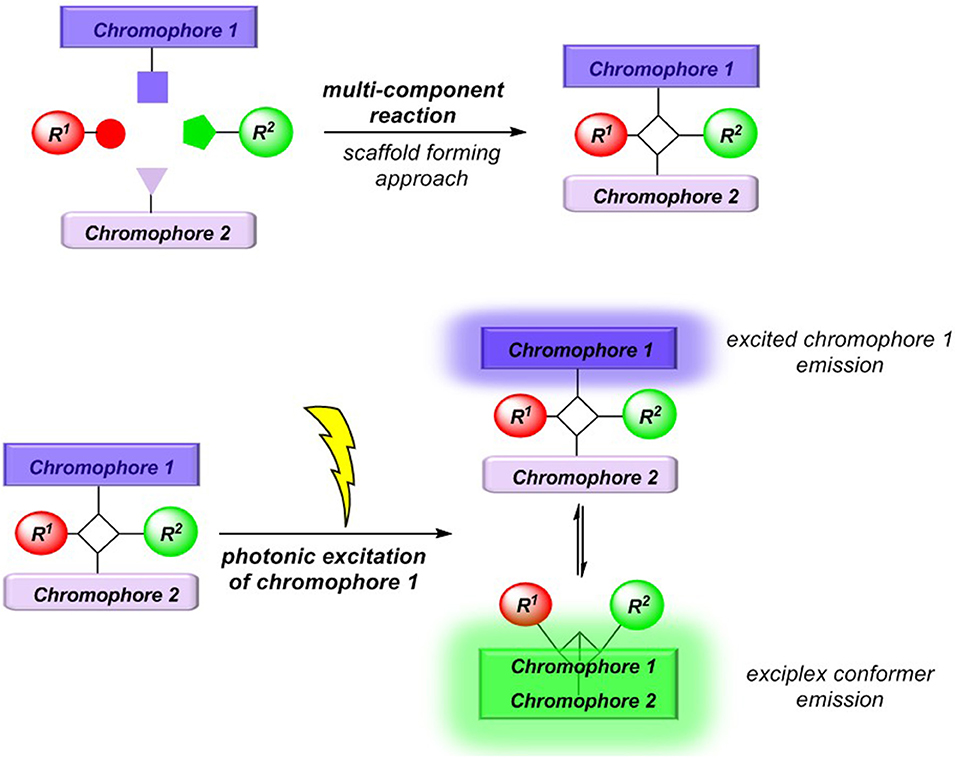
Figure 2. MCR synthesis (scaffold-forming approach) of exciplex forming bichromophores.
Results and Discussion
Synthesis
The Ugi four-component reaction (4CR) (Dömling and Ugi, 2000; Dömling, 2006; Biggs-Houck et al., 2010) generates the chemically robust α-aminoacyl amide scaffold with a high level of diversity and has widely and successfully been applied in biological and medicinal chemistry (Dömling et al., 2012). Encouraged by Ugi 4CR syntheses of a phenothiazine-anthraquinone-based PIET system and related dyads (Bay et al., 2013, 2014; Bay and Müller, 2014), characterized by cyclic voltammetry, steady-state UV/vis, and fluorescence spectroscopy, as well as femtosecond transient absorption spectroscopy for identification of the desired charge separated state after light excitation, we decided to transpose the Ugi 4CR to a novel synthesis of N,N-dimethylaniline-anthracene dyads, which are known to form exciplex emitters (Hui and Ware, 1976).
Methanol as a solvent is often most ideal for Ugi 4CR, however, portions of dichloromethane were found to beneficially increase the solubility and to ensure a homogeneous solution. In a model reaction employing 9-aminomethylanthracene hydrochloride (1a) (Stack et al., 2002), commercially available 4-(N,N-dimethylamino)benzaldehyde (2a), acetic acid (3), and tert-butylisocyanide (4), we set out to identify optimal conditions for the preparation of the targeted potential exciplex system 5 by variation of the base, reaction time, and conditions (see Table S1).
The optimal conditions at the equistoichiometric ratio (Table S1, entry 6) were therefore employed for the synthesis of other bichromophores and their reference systems 5 in this study, which were obtained in moderate to good yield (Scheme 1). For the donor-acceptor bichromophores, 4-N,N-dimethylamino phenyl derivatives were used as donor moieties, whereas the acceptor unit was altered from anthracene over pyrene to naphthalene. For solubility issues of pyrene derivatives in methanol, the amount of dichloromethane had to be increased.
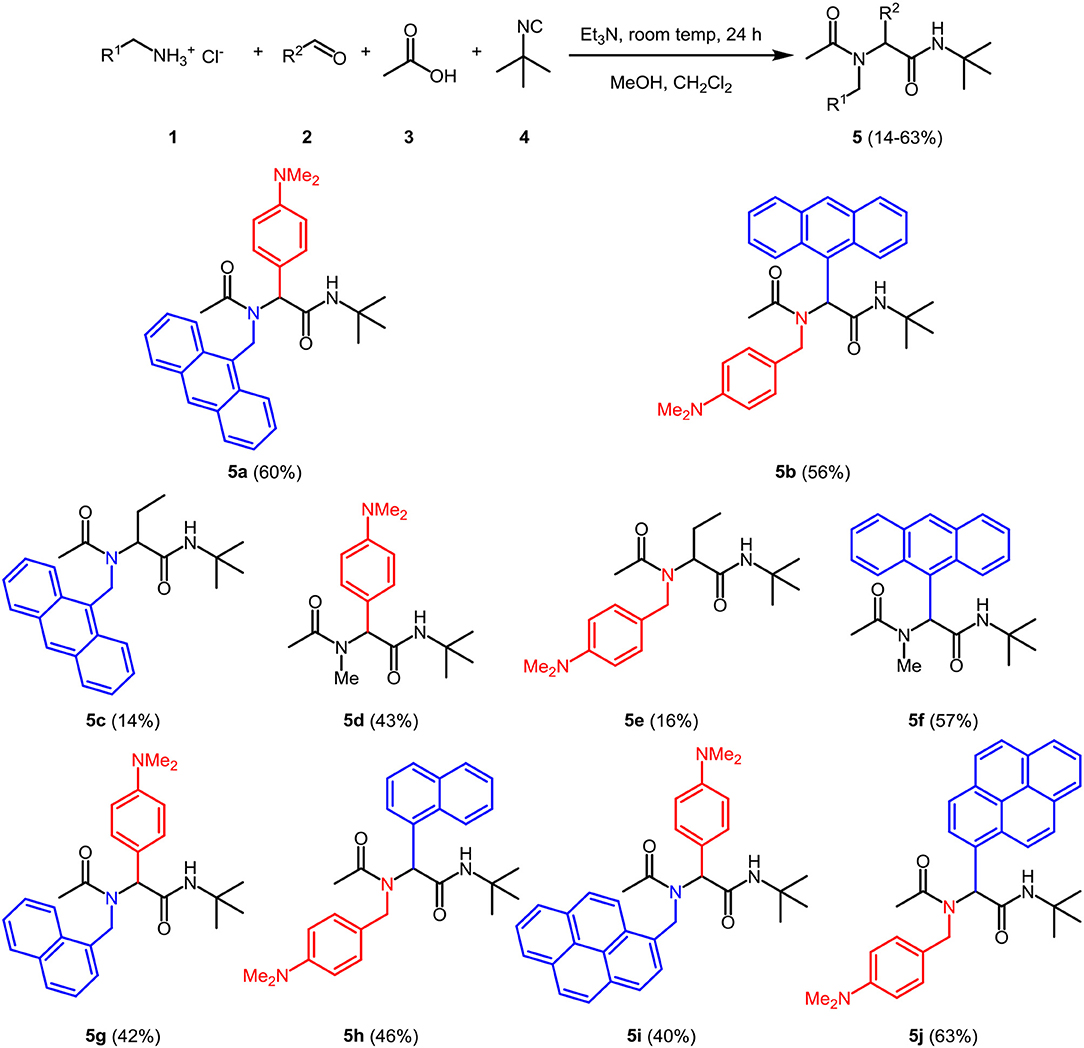
Scheme 1. Ugi 4CR synthesis of donor-acceptor dyads and reference compounds 5.
The appearance of single-signal sets in the 1H and 13C NMR spectra of 5 unambiguously supports the structural assignment of the bichromophores, and rotamer mixtures due to restricted amide bond rotation can be excluded. The depicted structural assignments were made by identifying the lowest energy conformations by force field calculation scans of the corresponding structures 5. Distinct resonances in the aromatic region of the spectra account for 9-anthracenyl, 3-pyrenyl, and 1-naphthyl and the p-N,N-dimethylamino phenyl substitutions, respectively. Mass spectrometry and combustion analysis are additionally in agreement with the structures of bichromophores 5a,b,j-h and the corresponding reference compounds 5c-f.
Photophysical Properties
The absorption and emission spectra of all donor-acceptor dyads and their reference compounds were measured in dichloromethane at room temperature (Table 1). By comparison of the UV/Vis spectra of N,N-dimethylaniline-anthracene dyads 5a and 5b with N,N-dimethylaniline (5d and 5e) and anthracene (5c and 5f) reference compounds, it can be clearly seen that in the electronic ground state, which is reflected by absorption spectroscopy, no interaction of donor and acceptor subchromophores can be detected by the absence of charge transfer bands. In addition, the spectra of the dyads 5a and 5b show an additive behavior with respect to the spectra of the reference compounds 5c and 5d and 5e and 5f, respectively (Table 1). However, in the excited state upon excitation at λexc = 391 nm, that is, the longest wavelength absorption band of the anthracene, in the emission spectra besides the anthracene characteristic vibrationally resolved fluorescence for both dyads, 5a and 5b, a broad unstructured and redshifted emission band at λmax,em = 561 (dyad 5a) and 568 nm (dyad 5b) is detected (Figure 3). This redshifted band is most characteristic for the N,N-dimethylaniline-anthracene exciplex. With respect to the isomer correlation of the dyads 5a and 5b and the essentially identical energy of this transition, it can be concluded that the placement of the aniline and the anthracene can be introduced in either case via the aldehyde or the primary amine.
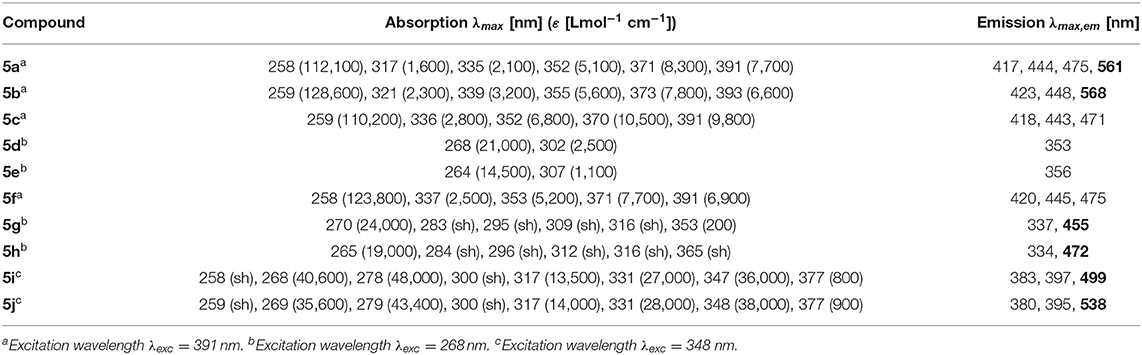
Table 1. Selected absorption and emission spectra of exciplex bichromophores (exciplex emission bands are highlighted in bold face) and reference compounds 5 (recorded in CH2Cl2 (chromasolv), T = 298 K).
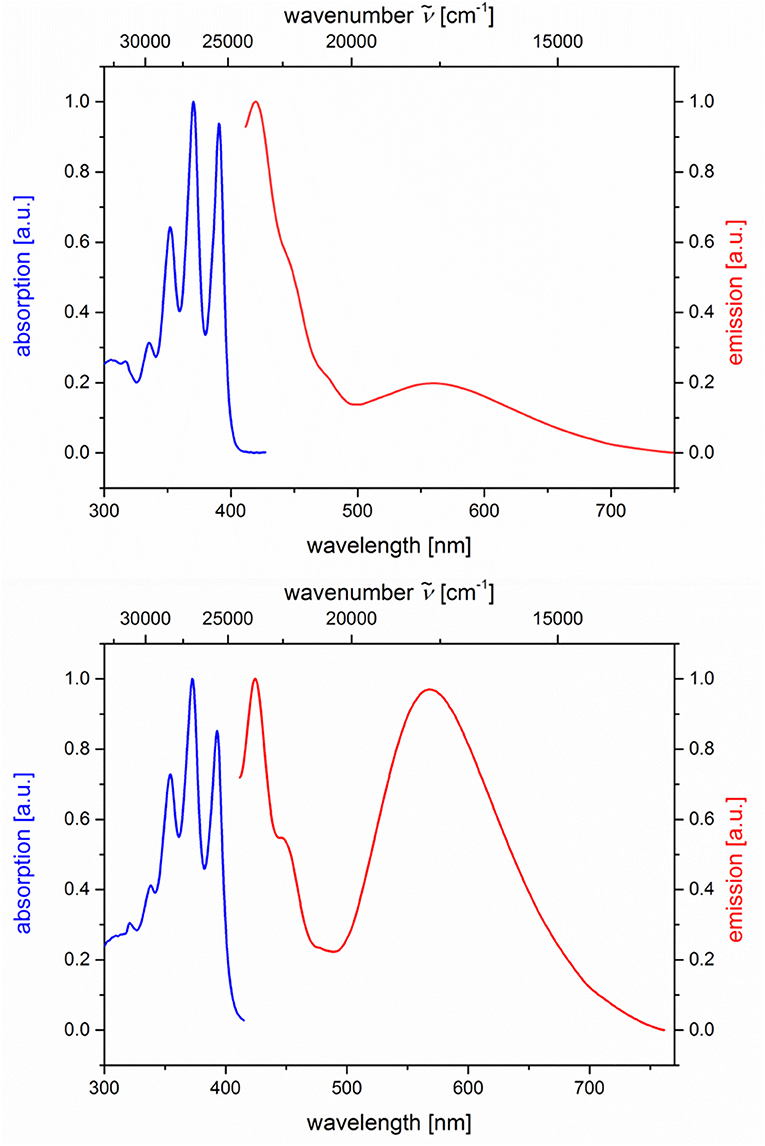
Figure 3. Normalized absorption (blue lines) and emission (red lines) spectra of compounds 5a (top) and 5b (bottom) (recorded in CH2Cl2 (chromasolv), T = 298 K, λexc = 391 nm).
From the comparison of the emission spectra of dyads 5a and 5b and the anthracene reference compounds 5c and 5f at identical concentration and identical excitation wavelength [c(5) = 10−5 M, T = 298 K, λexc = 391 nm], it can be clearly seen that for both dyads, a radiative deactivation pathway of the anthracene luminescence by structureless broad exciplex emission bands exists (Figure 4). From the relative emission intensities of the exciplex bands and the residual anthracene emission, it can be seen that the exciplex band of dyad 5b is more intense than that of dyad 5a. Upon excitation of the dyads 5a and 5b at the main absorption bands of the N,N-dimethylaniline references 5d and 5e (λexc = 268 nm), the comparison of the emission spectra reveals that for the dyads, the anthracene emission arises from complete energy transfer from the excited N,N-dimethylaniline moieties on expense of the characteristic aniline emission, which can be identified in the emission spectra of the reference compounds 5d and 5e (Figure 5). Furthermore, the emission spectrum of dyad 5a when excited at 268 nm reveals at this excitation wavelength that both the anthracene emission bands and the broad exciplex emission band are can be unambiguously detected (see Figure S24). The energy excitation of the exciplex emission arising from reductive electron transfer (from N,N-dimethylaniline to excited anthracene) clearly is apparent.
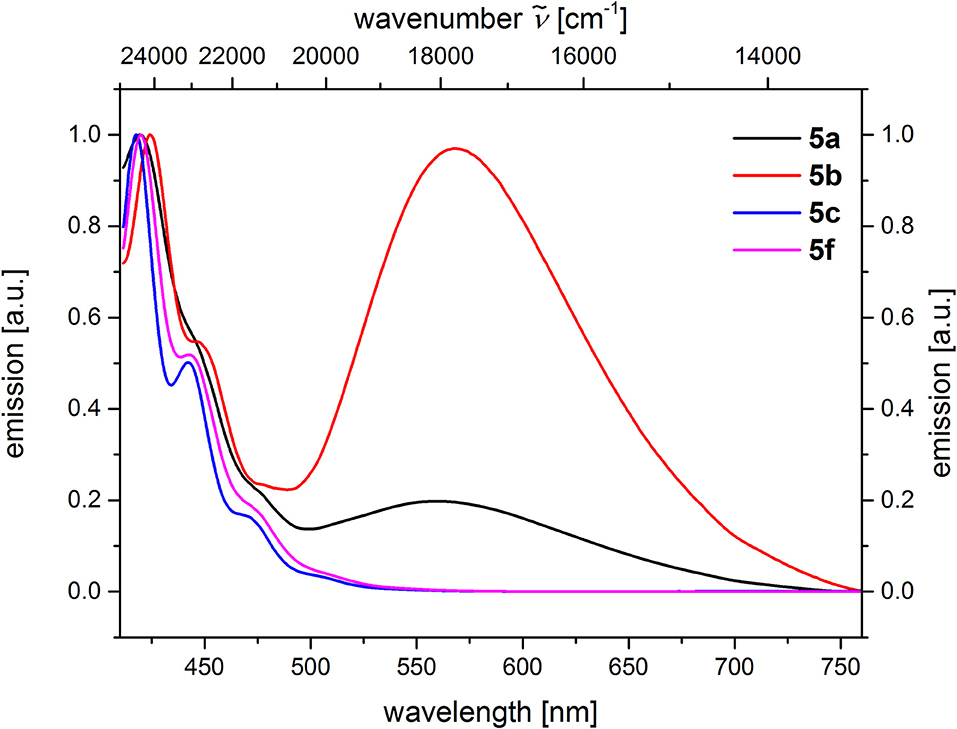
Figure 4. Comparison of emission spectra of compounds 5ac, f (recorded in CH2Cl2 (chromasolv), c(5) = 10−5 M, T = 298 K, λexc = 391 nm).
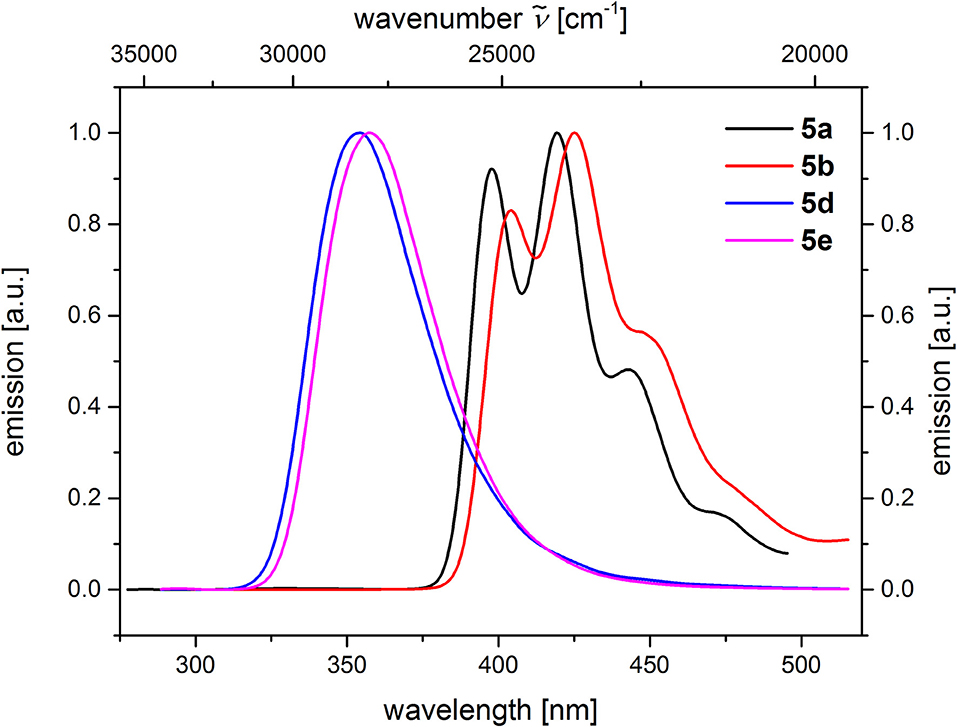
Figure 5. Comparison of emission spectra of compounds 5a and 5b (c(5) = 10−5 M, λexc = 268 nm), as well as compounds 5d and 5e (c(5) = 10−4 M, λexc = 258 nm) (recorded in CH2Cl2 (chromasolv), T = 298 K).
The dyads 5g and 5h consist of naphthalene as an acceptor and N,N-dimethylaniline as a donor (Table 2; Figure 6). Upon excitation at λexc = 268 nm, the emission spectra reveal weak naphthalene emission bands that are quenched in favor of exciplex emission at 455 and 472 nm, respectively. These differences already indicate that the overlap of naphthalene and N,N-dimethylaniline in the sandwich conformer might differ in both isomers.
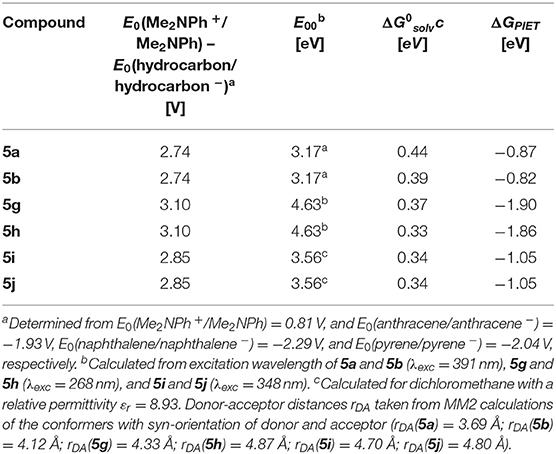
Table 2. Rehm-Weller estimation of the PIET's Gibbs free energies ΔGPIET calculated from the energy differences of the redox potentials E0(Me2NPh+/Me2NPh) – E0(hydrocarbon/hydrocarbon −), the excitation wavelengths E00, and the solvent correctional term for syn-conformers of selected dyads 5.
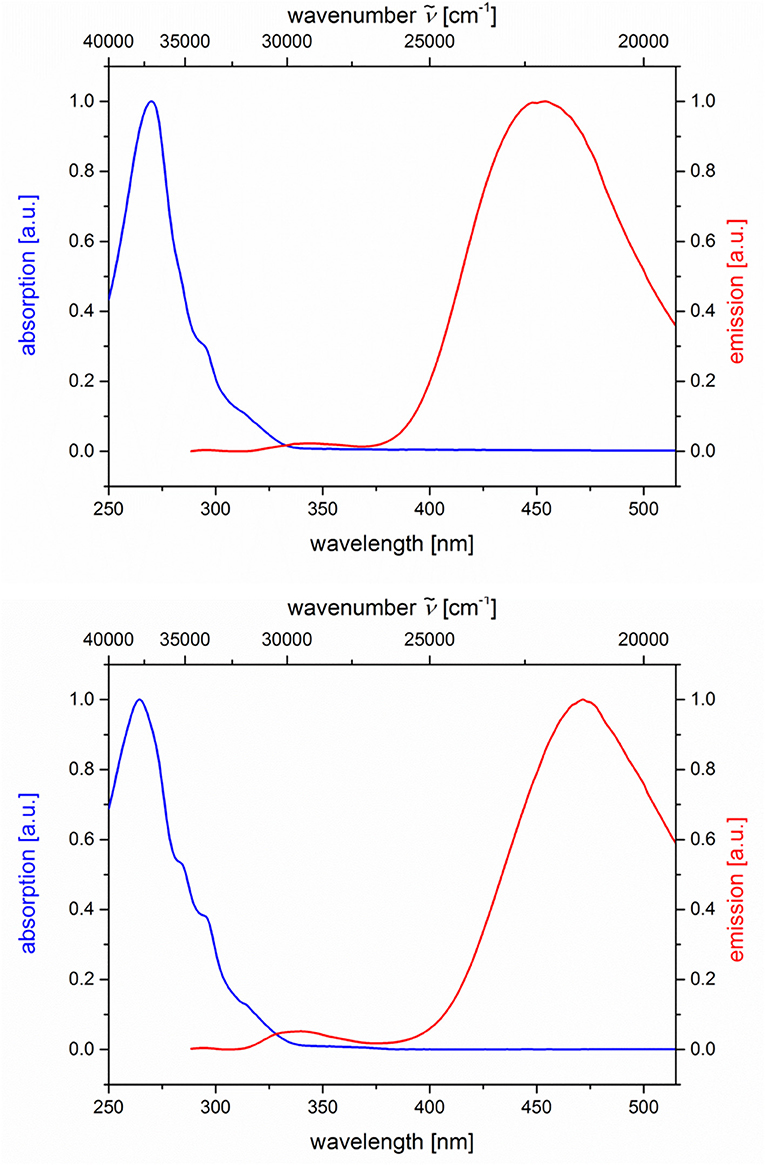
Figure 6. Normalized absorption (blue lines) and emission (red lines) spectra of compounds 5g (top) and 5h (bottom) (recorded in CH2Cl2 (chromasolv), T = 298 K, λexc = 268 nm).
Likewise and even more pronounced is the difference in exciplex emission of the pyrene-based dyads 5i and 5j, which, upon excitation at λexc = 348 nm, show well vibrationally resolved pyrene emission bands for isomer 5i and a broad weaker exciplex band at 499 nm, whereas for isomer 5j displays a broadened weaker pyrene emission and a more intense, broad exciplex band at 538 nm (Table 2; Figure 7).
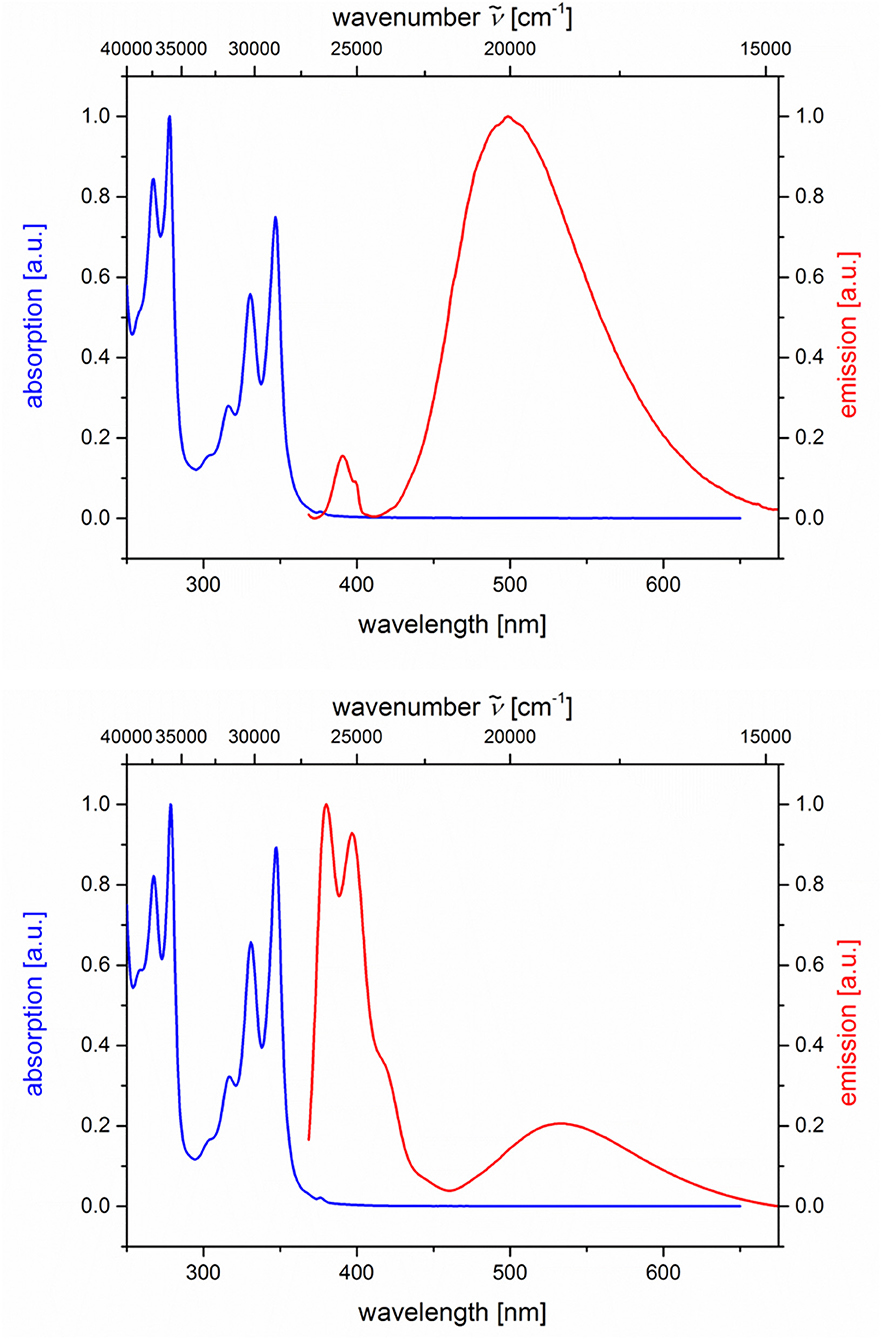
Figure 7. Normalized absorption (blue lines) and emission (red lines) spectra of compounds 5i (top) and 5j (bottom) (recorded in CH2Cl2 (chromasolv), T = 298 K, λexc = 348 nm).
For all three combinations of N,N-dimethylaniline-hydrocarbon dyads, exciplex formation occurs by photoexcitation of the hydrocarbon moieties and subsequent interaction with the ground state of the N,N-dimethylaniline moiety (Ceroni and Balzani, 2012). The exciplex formation depends on spatial proximity and as a partial electron transfer by the redox potentials of donor and acceptor moieties. The Rehm-Weller approximation (Rehm and Weller, 1970) allows an estimation of the PIET's Gibbs free enthalpy ΔGPIET (Equation 1) by
where E0(D+/D) is the donor's normal oxidation potential, E0(A/A−) is the acceptor's normal reduction potential (both are often determined from cyclic voltammetry and are given in V), E00 is the excitation energy of absorption band in eV (Nishizawa et al., 1998), and (in eV) is a correctional term (Equation 2) describing the Coulomb stabilization of two point charges at a distance rDA in a solvent according to
with εr (solvent's relative permittivity), ε0 (vacuum's permittivity, 8.8542 · 10−12 C V−1 m−1), e (elementary charge, 1.6022 · 10−19 C), and rDA (donor-acceptor distance of the corresponding centroids of the molecule parts).
For N,N-dimethylaniline-hydrocarbon dyad conformers with a syn-orientation, the Rehm-Weller approximation with literature redox potentials of the constituting donor and acceptor moieties (Kavarnos and Turro, 1986) and MM2-calculated donor-acceptor distances rDA reveals that ΔGPIET is exergonic in all cases (Table 2).
The peculiar emission behavior of the dyads 5a, b, g-j can already be seen upon eyesight under a handheld UV lamp as shown for dyad 5a in solvents of different polarity and in the solid state (Figure 8). A pronounced positive emission solvatochromism, that is, a red shift of the emission with increasing polarity, immediately becomes apparent. The solid state emission band is very broad and unstructured, and the maximum is detected at 474 nm (λexc = 391 nm) (see Figure S32) and luminesces intensively blue with an external quantum yield Φf = 0.320, as determined by an integrating sphere. The broad structureless emission accounts for π-π-stacking of anthracene moieties, as shown for single crystals of 1-acetyl-3-phenyl-5-(9-anthryl)-2-pyrazoline (Feng et al., 2014). As a consequence, the dyad 5a preferentially adopts the anti-conformation in the solid state, where the exciplex emission is absent.

Figure 8. Dyad 5a in the solid state and in cyclohexane, toluene, 1,4-dioxane, and dichloromethane solutions (from left to right) under a handheld UV lamp (λexc = 365 nm).
The change of dipole moment from ground to excited state μE − μG can be calculated using the Lippert-Mataga equation (Equation 3),
where a and e are the absorption and emission maxima (in cm−1), ε0 is the vacuum permittivity constant (8.8542 · 10−12AsV−1m−1), c is the speed of light (2.9979 · 1010 cms−1), h is the Planck's constant (6.6256 · 10−34Js−1), and the orientation polarizability Δf (Equation (4),
The solvent orientation polarizability Δf of four representative solvents (1,4-dioxane, dichloromethane, acetone, and acetonitrile) gives a very good fit (r2 = 0.9594) using the Lippert-Mataga plot (Mataga et al., 1956; Lippert, 1957) (see Figure S42), the linear correlation of the Stokes shifts Δ with the relative permittivity εr and the optical refractive index n of the corresponding solvent (for details, see Supplementary Material). Since syn- and anti-conformers might be involved in the electronic ground and excited states, we decided to calculate the Onsager radii a for both syn- and anti-conformations from the optimized ground state structures of dyad 5a by DFT calculations (Frisch et al., 2016) (vide infra). Assuming a syn-conformation for dyad 5a with an Onsager radius of 3.42 Å (3.42 · 10−10 m), a change of dipole moment upon excitation from the ground state to the emissive exciplex state Δμ of 6.02 D (2.008 · 10−29 Cm) can be calculated. Likewise, for an anti-conformation for dyad 5a with an Onsager radius of 7.92 Å (7.92 · 10−10 m), Δμ can be calculated to 21.19 D (7.067 · 10−29 Cm).
Calculated Electronic Structure
For further elucidation of the electronic structure, the geometries of three representative dyads 5a, 5g, and 5i in their syn- and anti-conformations were optimized by DFT calculations using Gaussian 16 (Frisch et al., 2016) with the B3LYP functional (Lee et al., 1988; Becke, 1993; Kim and Jordan, 1994; Stephens et al., 1994) and the Pople 6-311++G** basis set (Krishnan et al., 1980), applying vacuum calculations as well as the Polarizable Continuum Model (PCM) with dichloromethane as a solvent (Scalmani and Frisch, 2010) (for details on the DFT calculations, see Supplementary Material). The optimized geometries were verified by frequency analyses of the local minima. Comparison of the calculated ground state energies of the syn- and anti-conformers reveals that for the anthracene dyad 5a and for the pyrene dyad 5i, the syn-conformers are stabilized while for the naphthalene dyad 5g, the anti-conformer is slightly favored (Figure 9). Equilibrium constants account for a contribution of both conformers in the absorption and, thus, emission behavior of the dyads 5a, 5g, and 5i. The Onsager radii a for the syn- and anti-ground state conformations of dyad 5a were estimated from the calculated geometry optimization, which led to the Lippert-Mataga plot to the calculation of the change of dipole moment Δμ between the electronic ground state and the vibrationally relaxed exciplex state (vide supra).
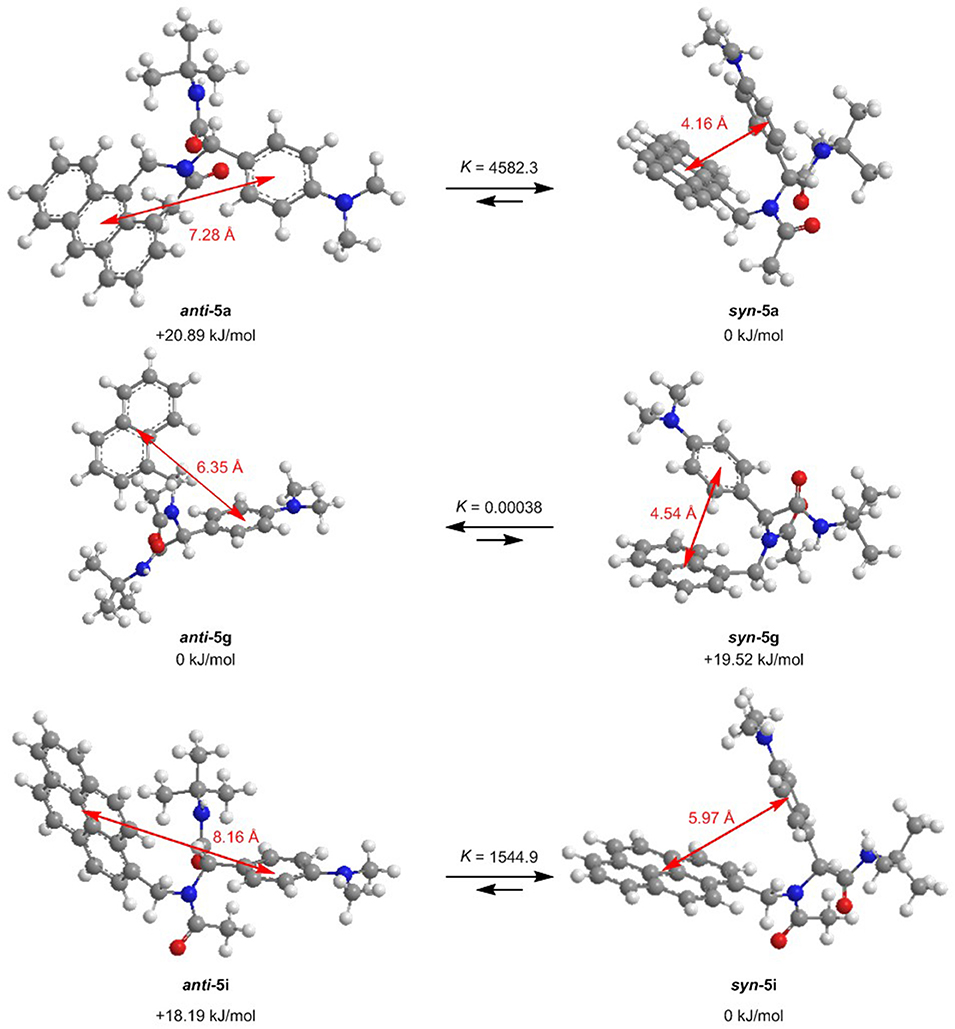
Figure 9. Ground state anti-syn-conformation equilibria (ΔG, K) and donor-acceptor centroid distances according to DFT calculations on the minimum energy conformers of dyads 5a, 5g, and 5i (B3LYP/6-311++G** at T = 298 K).
Based on the optimized ground state structures of the anti- and syn-conformers, the electronic absorptions of the three dyads 5a, 5g, and 5i were calculated on the level of TDDFT theory employing the gradient-corrected exchange and correlation Perdew-Burke-Ernzerhof functionals PBE1PBE/6-31G**) (Krishnan et al., 1980; Perdew et al., 1996, 1997; Adamo and Barone, 1999; Ernzerhof and Scuseria, 1999), with dichloromethane (IEFPCM) (Scalmani and Frisch, 2010) as a solvent (for details on the TDDFT calculations, see Table S4). The TDDFT calculation of structure syn-5a gives a lowest energy transition at 431 nm for the S1 Franck-Condon absorption, which is represented to 99% as a HOMO to LUMO transition with a significant oscillatory strength of 0.0167 due to coefficient density on both donor and acceptor moieties. This transition represents considerable charge transfer character from the N,N-dimethylaniline substituent to the anthracene unit. The experimental spectrum displays an intense structureless band with a maximum at 391 nm, which is in agreement with an excitation of the anthracene part. The calculated S2 Franck-Condon absorption, which is represented to 98% as a HOMO-1 to LUMO transition with an oscillatory strength of 0.0850, accounts for a dominant anthracene excitation and appears at 388 nm. This transition correlates with the anthracene-type transition at 371 nm in the experimental spectrum. Likewise, the TDDFT calculation of structure anti-5a gives a lowest energy transition at 415 nm for the S1 Franck-Condon absorption, which is represented to 99% as a HOMO to LUMO transition with a significant oscillatory strength of 0.003, due to coefficient density on both donor and acceptor moieties. This transition represents considerable charge transfer character from the N,N-dimethylaniline substituent to the anthracene unit. The experimental spectrum displays an intense structureless band with a maximum at 391 nm, which is in agreement with an excitation of the anthracene part.
For a general discussion of a model system, the absorption and exciplex emission of dyads syn-5a and anti-5a were visualized by plotting in a Jablonski term scheme (Figure 10). Starting for structure syn-5a from the ground state S0 according to TDDFT calculations Franck-Condon absorption to the vibrationally excited state proceeds at 380 nm (experiment: 391 nm) as represented by a HOMO-1-LUMO transition, which simultaneously comprises local excitation in the anthracene moiety and coefficient density transfer from N,N-dimethylaniline to anthracene (Figure 10A). The vibrationally relaxed S1 state lies 0.751 eV lower in energy. This geometry corresponds to that of the vibrationally excited state . Therefore, the excitation from the vibrationally excited ground state to the relaxed first excited state S1 translates into the process of exciplex fluorescence, which proceeds at 571 nm (experiment: 561 nm). The geometry of the vibrationally excited ground state lies 0.347 eV above the vibrationally relaxed ground state S0 and is slightly tighter. This exciplex emission retransmits coefficient density from the anthracene part to the N,N-dimethylaniline unit and locally in the anthracene moiety.
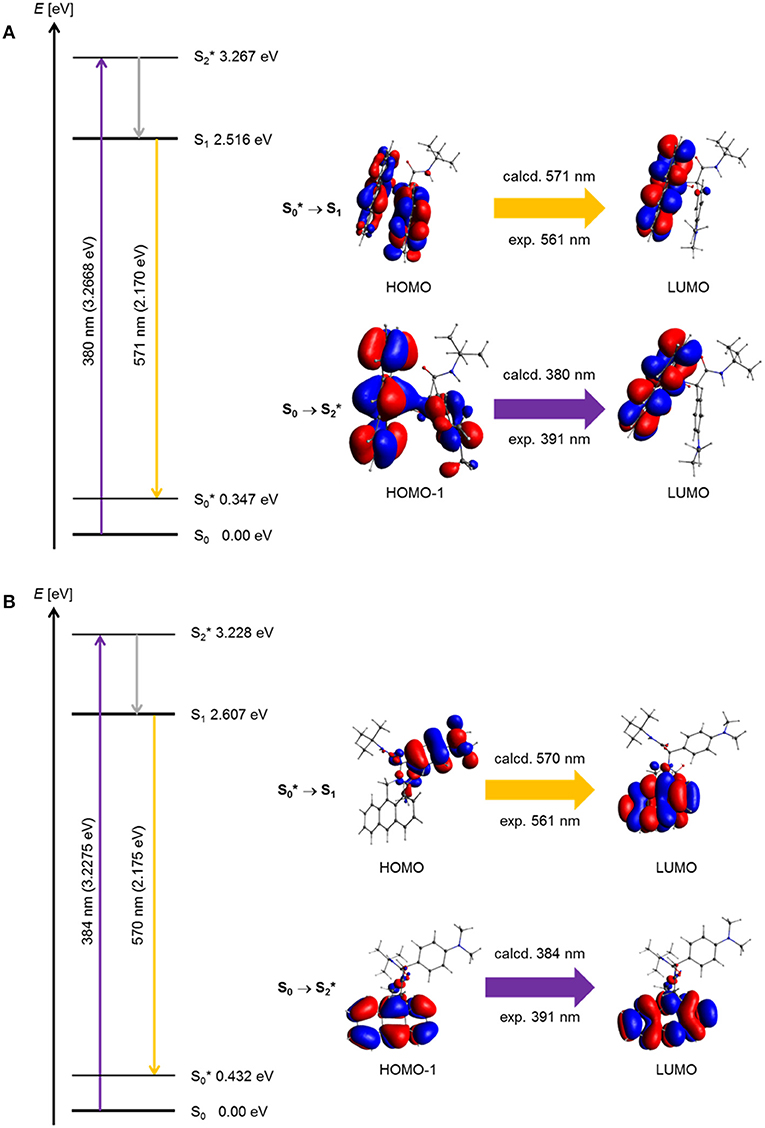
Figure 10. Jablonski diagrams of dyads (A) syn-5a and (B) syn-5b with assignment of the FMO-transitions in the experimental and calculated longest wavelength absorption bands and the exciplex emission bands [E(S0) = 0 eV; PBE1PBE 6-31G** IEFPCM CH2Cl2, isosurface value at 0.03 a.u.].
Geometrically, the coefficient density in structure anti-5a is clearly separated in HOMO and LUMO (Figure 10B). However, a similar Franck-Condon absorption energy to the vibrationally excited state proceeds at 384 nm (experiment: 391 nm), and an almost identical exciplex emission from the vibrationally relaxed S1 state to the vibrationally excited state is calculated at 570 nm (experiment: 561 nm). As can be seen from the calculated oscillatory strength (vide supra), the dominant absorption of the anti-5a conformer is a local excitation of the anthracene moiety, whereas the exciplex emission is completely charge transfer in its character. Expectedly, the oscillatory strength for this process is considerably smaller. Therefore, the experimentally observed exciplex emission preferentially might occur rather from the syn-conformer.
Indeed, the simultaneous occurrence of exciplex and hydrocarbon emission in all dyads 5 represents an interesting design principle of novel types of dually emissive bichromophores, arising from selective local excitation followed by acceptor emission as well as redshifted exciplex emission.
Conclusion
The Ugi 4CR is an excellent synthetic tool for accessing libraries of donor-acceptor dyads that are covalently ligated, yet, electronically decoupled in the ground state, but electronically coupled in the excited state by virtue of the appearance of exciplex emission. The placement of donor and acceptor moieties has been probed in this study for N,N-dimethylaniline as a donor moiety and anthracene, naphthalene, and pyrene as hydrocarbon acceptors with favorable redox potentials for enabling exergonic photo-induced intramolecular electron transfer (PIET) upon local hydrocarbon excitation and estimated by Rehm-Weller approximation. The polar nature of the excited state responsible for exciplex emission is unambiguously assigned by emission solvatochromicity measurement and finally by Lippert-Mataga analysis of the Stokes shifts. The change of dipole moment upon excitation from the ground state to the emissive exciplex state can be calculated to Δμ = 6.02 D for the syn-conformer of the N,N-dimethylaniline-anthracene dyad and to Δμ = 21.19 D for the corresponding anti-conformer. For the same dyad, TDDFT calculations with the gradient-corrected exchange and correlation Perdew-Burke-Ernzerhof functionals PBE1PBE were carried out, allowing a qualitative assignment of the experimental absorption and emission bands and, thus, rationalizing the particular relevance of the exciplex' electronic structure. In conclusion, the synthetic scaffold concept employing the isonitrile-based Ugi 4CR enables the design and photophysical proof of concept of novel types of unimolecular exciplex emitters. In principle, even unimolecular multichromophore arrays can be quickly assembled according to the same principle. Synthetic and photophysical studies of dually emissive unimolecular multichromophores are currently underway.
Experimental
General Considerations
All reactions were performed in oven-dried Schlenk vessels under a nitrogen atmosphere. The employed solvents were dried with a solvent drying system MB-SPS 800 by M. Braun Inertgas-Systeme GmbH and used as obtained. Commercial grade reagents were used as supplied without further purification and were purchased from abcr GmbH & Co. KG, Acros Organics, Alfa Aesar GmbH & Co. KG, Sigma-Aldrich Chemie GmbH. The purification of Ugi-compounds 5 was performed by column chromatography on silica gel 60 M (0.04–0.063 mm) from Macherey-Nagel GmbH & Co. KG using flash technique under pressure of 2 bar. The crude mixtures were adsorbed on Celite® 545 from Carl Roth GmbH Co. KG before chromatographic purification. The reaction progress was monitored qualitatively using TLC Silica gel 60 F254 aluminum sheets obtained from Merck KGaA, Darmstadt. The spots were detected with UV light at 254 nm and using an iodine chamber. 1H, 13C, and 135-DEPT 13C NMR spectra were recorded on Bruker Avance DRX 300 and Bruker Avance DRX 500. CDCl3 and DMSO-d6 were used as deuterated solvents. The resonances of the solvents were locked as internal standards (CDCl3: 1H δ 7.27, 13C δ 77.23; DMSO-d6: 1H δ 2.50, 13C δ 39.5). The muliplicities of the signals were abbreviated as follows: s: singlet; d: doublet; dd: doublet of doublets; t: triplet; m: multiplet. The type of carbon nucleus was determined on the basis of 135-DEPT 13C NMR spectra. For the description of the 13C NMR spectra primary carbon nuclei are abbreviated with CH3, secondary carbon nuclei with CH2, tertiary carbon nuclei with CH, and quaternary carbon nuclei with Cquat.. MALDI mass spectra were measured on a Bruker Ultraflex spectrometer, ESI mass spectra were measured on an Ion-Trap-API-mass spectrometer of Finnigan LCQ Deca (Thermo Quest). IR spectra were obtained on a Shimadzu IRAffinity-1, which works with the attenuated total reflection (ATR) method. The intensity of signals is abbreviated as follows: s (strong), m (medium), w (weak). UV/Vis spectra were recorded on a Lambda 19 spectrometer (Perkin Elmer) in dichloromethane at T = 293 K. Emission spectra were recorded on a Hitachi F-7000 instrument in dichloromethane and concentrations of 10−6 mol L−1 at T = 293 K. Data analysis was done with the software FL solutions 4.0 by Hitachi. The melting points (uncorrected) were measured on a Büchi Melting Point B-540 apparatus. Combustion analyses were carried out on a vario Micro Cube by Elementaranalysentechnik in the Microanalytical Laboratory of the Institut für Pharmazeutische und Medizinische Chemie der Heinrich-Heine-Universität Düsseldorf.
General Procedure (GP) for the Ugi-4CR Synthesis of Exciplex Bichromophores and Reference Compounds 5
In a 25-mL Schlenk tube with a magnetic stir bar were placed methylamine hydrochloride 1 (0.50 mmol), triethylamine (0.07 mL, 0.50 mmol), and methanol, and the mixture was stirred for 30 min (for experimental details, see Table 3). Then, aldehyde 2 (0.50 mmol) dissolved in dichloromethane was added dropwise, and the reaction mixture was stirred for 1 h. Finally, acetic acid (3) (0.50 mmol) and tert-butyl isocyanide (4) (0.06 mL, 0.50 mmol) were added, and reaction mixture was stirred at room temperature for 24 h. After removal of the solvents, the crude products were purified by flash chromatography on silica gel (n-hexane/ethyl acetate) and crystallized from various solvents or solvent mixtures to give analytically pure Ugi-4CR products as colorless to yellow solids.
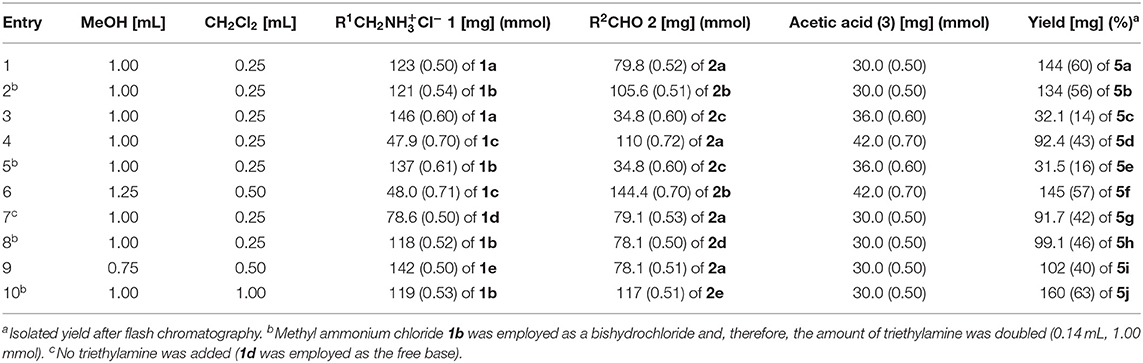
Table 3. Experimental details of the Ugi-4CR synthesis of exciplex bichromophores and reference compounds 5.
2-(N-(9-Anthrylmethyl)acetamido)-N-(tert-butyl)-2-(4-(dimethylamino)phenyl) acetamide (5a)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 3:2) and crystallization from hexane/ethanol (50:3), compound 5a (144 mg, 60%) was obtained as a light yellow solid, Mp 186–187°C, Rf (n-hexane/ethyl acetate 1:1) = 0.40.
1H NMR (300 MHz, CDCl3): δ = 0.83 (s, 9 H), 1.98 (s, 3 H), 2.90 (s, 6 H), 4.69 (d, J = 14.7 Hz, 1 H), 4.69 (s, 1 H), 5.38 (d, J = 14.7 Hz, 1 H), 6.26 (d, J = 14.1 Hz, 1 H), 6.51–6.69 (m, 2 H), 6.74–6.92 (m, 2 H), 7.387.63 (m, 4 H), 8.03 (d, J = 7.5 Hz, 2 H), 8.22 (d, J = 7.2 Hz, 2 H), 8.48 (s, 1 H). 13C NMR (75 MHz, CDCl3): δ = 24.1 (CH3), 28.3 (CH3), 40.6 (CH3), 41.8 (CH2), 51.3 (Cquat), 62.9 (CH), 112.4 (CH), 122.0 (Cquat), 124.0 (CH), 125.5 (CH), 127.5 (CH), 128.0 (Cquat), 128.6 (CH), 129.0 (CH), 129.6 (CH), 131.6 (Cquat), 131.7 (Cquat), 150.0 (Cquat), 168.3 (Cquat), 173.5 (Cquat). MALDI-TOF-MS (m/z): 480.1 ([M]+). IR: [cm−1] = 3,298 (w); 3,055 (w); 2,957 (w); 2,910 (w); 2,863 (w); 2,806 (w); 1,678 (s); 1,614 (s); 1,549 (m); 1,526 (s); 1,481 (w); 1,445 (m); 1,418 (w); 1,391 (w); 1,358 (m); 1,339 (w); 1,325 (w); 1,306 (w); 1,285 (w); 1,256 (w); 1,227 (m); 1,209 (w); 1,193 (w); 1,188 (w); 1,167 (w); 1,167 (w); 1,140 (w); 1,063 (w); 1,040 (w); 1,024 (w); 988 (w); 949 (w); 881 (m); 841 (w); 827 (w); 812 (w); 791 (s); 735 (s); 694 (w); 660 (w); 642 (w); 625 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 352 (5,100), 417 (8,300), 391 (7,700). Fluorescence (CH2Cl2): λmax,em [nm] = 417, 444, 475, 561; Stokes shift Δ = 1,600 cm−1. Anal. calcd. for C31H35N3O2 (481.3): C 77.31, H 7.32, N 8.72; Found: C 77.03, H 7.31, N 8.57.
2-(9-Anthryl)-N-(tert-butyl)-2-(N-(4-(dimethylamino)phenyl) acetamide) (5b)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 3:2) and crystallization from hexane/ethyl acetate (55:5), compound 5b (134 mg, 56%) was obtained as a yellow solid, Mp 192°C, Rf (n-hexane/ethyl acetate 1:1) = 0.50.
1H NMR (300 MHz, CDCl3): δ = 1.211.23 (m, 0.5 H), 1.23 (s, 9 H), 2.02 (s, 0.5 H), 2.16 (s, 3 H), 2.63 (s, 6 H), 4.10 (q, J = 7.2 Hz, 0.3 H) 4.464.71 (m, 2 H), 5.06 (s, 1 H), 6.02 (d, J = 8.7 Hz, 2 H), 6.14 (d, J = 8.6 Hz, 2 H), 7.407.59 (m, 4 H), 7.63 (s, 1 H), 7.92 (d, J = 7.6 Hz, 2 H), 8.36 (s, 1 H), 8.37 (d, J = 8.5 Hz, 2 H). 13C NMR (75 MHz, CDCl3): δ = 14.35 (CH3), 21.18 (CH3), 22.94 (CH3), 28.71 (CH3), 40.74 (CH3), 50.14 (CH2), 51.94 (Cquat), 57.62 (CH), 60.53 (CH2), 112.27 (CH), 124.96 (CH), 125.14 (CH), 125.73 (Cquat), 126.25 (Cquat), 126.56 (CH), 127.28 (CH), 129.58 (CH), 130.57 (CH), 131.63 (Cquat), 132.29 (Cquat), 149.24 (Cquat), 171.05 (Cquat), 171.28 (Cquat), 173.01 (Cquat). MALDI-TOF-MS (m/z): 480.1 ([M]+), 450.9 ([M – 2 CH3]+), 269.9 ([M – C11H16N2O]+). IR: [cm−1] = 3,422 (w) 2,979 (w); 2,882 (w); 2,849 (w); 2,799 (w); 1,736 (s); 1,680 (s); 1,620 (s); 1,514 (s); 1,449 (m); 1,406 (m); 1,391 (m); 1,364 (m); 1,256 (m); 1,341 (m); 1,327 (w); 1,314 (w); 1,287 (w); 1,275 (w); 1,258 (w); 1,223 (m); 1,192 (m); 1,169 (w); 1,070 (w); 1,036 (w); 1,022 (w); 997 (w); 968 (w); 949 (w); 939 (w); 908 (w); 853 (w); 812 (w); 785 (m); 741 (s). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 355 (5,600), 373 (7,800), 393 (6,600). Fluorescence (CH2Cl2): λmax, em [nm] = 423, 448, 568. Stokes shift Δ = 1,800 cm−1. Anal. calcd. for C31H35N3O2 1/6 CH3CO2C2H5 (481.3 + 14.7): C 76.59, H 7.35, N 8.51; Found: C 76.24, H 7.21, N 8.54.
2-(N-(9-Anthrylmethyl)acetamido)-N-tert-butylbutanamide (5c)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 2:1) and crystallization from hexane/ethyl acetate (6.5:0.5), compound 5c (32.1 mg, 14%) was obtained as a yellow crystalline solid, Mp 150°C, Rf (n-hexane/ethyl acetate 2:1) = 0.20.
1H NMR (300 MHz, CDCl3): δ = 0.52 (s, 9 H), 1.09 (t, J = 7.5 Hz, 3 H), 1.35 (m, 1H), 2.04 (s, 3 H), 2.132.34 (m, 1H), 3.48 (t, J = 7.2 Hz, 1 H), 4.38 (s, 1 H), 4.91 (d, J = 15.0 Hz, 1 H), 6.59 (d, J = 15.0 Hz, 1 H), 7.407.65 (m, 4 H), 8.03 (d, J = 8.4 Hz, 2 H), 8.33 (d, J = 8.8 Hz, 2 H), 8.48 (s, 1 H). 13C NMR (75 MHz, CDCl3): δ = 12.3 (CH3), 22.8 (CH3), 24.1 (CH2), 27.8 (CH3), 42.0 (CH2), 50.6 (Cquat), 61.3 (CH), 123.6 (CH), 125.7 (CH), 127.6 (Cquat), 128.0 (CH), 129.2 (CH), 130.0 (CH), 131.5 (Cquat), 131.7 (Cquat), 169.4 (Cquat), 171.8 (Cquat). MALDI-TOF-MS (m/z): 391.0 ([M]+), 347.0 ([M – OC2H3]+). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 352 (6,800), 370 (10,500), 391 (9,800). Fluorescence (CH2Cl2): λmax,em [nm] = 418, 443, 471. Stokes shift Δ = 1,700 cm−1. Anal. calcd. for C25H30N2O2 (390.2): C 76.89, H 7.74, N 7.17; Found: C 76.88, H 7.78, N 6.99.
N-tert-butyl-2-(4-(dimethylamino)phenyl)-2-(N-methylacetamido)acetamide (5d)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 1:1) and crystallization from hexane/ethyl acetate (25:2.5), compound 5d (92.4 mg, 43%) was obtained as a light brown solid, Mp 155°C, Rf (n-hexane/ethyl acetate 1:1) = 0.10.
1H NMR (300 MHz, CDCl3): δ = 1.30 (s, 9 H), 2.09 (s, 3 H), 2.80 (s, 3 H), 2.93 (s, 6 H), 5.52 (s, 1H), 6.07 (s, 1 H), 6.66 (d, J = 8.7 Hz, 2 H), 7.15 (d, J = 8.7 Hz, 2 H). 13C NMR (75 MHz, CDCl3): δ = 22.36 (CH3), 28.84 (CH3), 33.16 (CH3), 40.52 (CH3), 51.67 (Cquat), 60.13 (CH), 112.43 (CH), 122.96 (Cquat), 130.44 (CH), 150.36 (Cquat), 169.84 (Cquat), 171.56 (Cquat). MALDI-TOF-MS (m/z): 304.3 ([M]+). IR: [cm−1] = 3,287 (w); 2.965 (w); 2.918 (w); 2,895 (w); 2,806 (w); 1,680 (s); 1,612 (s); 1,551 (m); 1,522 (s); 1,503 (w); 1,474 (w); 1,443 (w); 1,431 (w); 1,404 (m); 1,387 (w); 1,375 (w); 1,350 (m); 1,281 (w); 1,254 (w); 1,227 (m); 1,204 (w); 1,182 (w); 1,169 (w); 1,144 (w); 1,126 (w); 1,065 (w); 1,022 (m); 959 (w); 945 (m); 829 (w); 812 (w); 797 (m); 745 (w); 694 (w); 640 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 268 (21,000). Fluorescence (CH2Cl2): λmax,em [nm] = 353. Stokes shift Δ = 9,000 cm−1. Anal. calcd. for C17H27N3O2 (305.2): C 66.85, H 8.91, N 13.76; Found: C 66.58, H 8.86, N 13.46.
N-tert-butyl-2-(N-(4-(dimethylamino)benzyl)acetamido)butanamide (5e)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 2:1) and crystallization from hexane/ethyl acetate (5:0.5), compound 5e (31.5 mg, 16%) was obtained as a dark yellow solid, Mp 106°C, Rf (n-hexane/ethyl acetate 1:1) = 0.30.
1H NMR (300 MHz, CDCl3): δ = 0.80 (t, J = 7.3 Hz, 3 H), 1.25 (s, 9 H), 1.731.98 (m, 2 H), 2.06 (s, 3 H), 2.88 (s, 6 H), 4.48 (s, 2 H), 4.584.67 (m, 1 H), 6.34 (s, 1 H), 6.64 (d, J = 8.7 Hz, 2 H), 7.00 (d, J = 8.6 Hz, 2 H). 13C NMR (75 MHz, CDCl3): δ = 11.1 (CH3), 21.9 (CH2), 22.6 (CH3), 28.8 (CH3), 40.6 (CH3), 48.9 (CH2), 51.2 (Cquat), 60.5 (CH), 112.9 (CH), 125.3 (Cquat), 127.2 (CH), 150.0 (Cquat), 170.1 (Cquat), 173.1 (Cquat). MALDI-TOF-MS (m/z): 332.0 ([M]+). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 264 (14,500), 321 (1,300). Fluorescence (CH2Cl2): λmax,em [nm] = 356. Stokes shift Δ = 9,800 cm−1. Anal. calcd. for C19H31N3O2 (333.2): C 68.43, H 9.37, N 12.60; Found: C 68.18, H 9.36, N 12.35.
2-(9-Anthryl)-N-(tert-butyl)-2-(N-methylacetamido)acetamide (5f)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 2:1) and crystallization from hexane/ethyl acetate (30:3), compound 5f (145 mg, 57%) was obtained as a colorless solid, Mp 260–262°C, Rf (n-hexane/ethyl acetate 2:1) = 0.10.
1H NMR (300 MHz, CDCl3): δ = 1.21 (s, 9 H), 2.22 (s, 3 H), 2.85 (s, 3 H), 4.95 (s, 1 H), 7.397.58 (m, 4 H), 7.49 (s, 1 H), 7.988.05 (m, 2 H), 8.28 (d, J = 9.0 Hz, 2 H), 8.52 (s, 1 H). 13C NMR (75 MHz, CDCl3): δ = 22.4 (CH3), 28.7 (CH3), 33.3 (CH3), 51.86 (Cquat), 56.6 (CH), 125.0 (CH), 125.3 (CH), 126.2 (Cquat), 127.3 (CH), 129.6 (CH), 130.4 (CH), 131.7 (Cquat), 132.2 (Cquat), 170.6 (Cquat), 171.3 (Cquat). MALDI-TOF-MS (m/z): 363.2 ([M]+), 290.2 ([M – NHC(CH3)3]+). IR: [cm−1] = 3,327 (w); 3.046 (w); 2,967 (w); 2,930 (w); 2,899 (w); 2,870 (w); 1,684 (s); 1,632 (s); 1,533 (m); 1,472 (w); 1,452 (m); 1,375 (w); 1,358 (m); 1,335 (w); 1,323 (m); 1,281 (w); 1,256 (w); 1,227 (w); 1,188 (w); 1,163 (w); 1,130 (w); 1,107 (w); 1,055 (w); 1,020 (w); 1,001 (w); 991 (w); 961 (w); 941 (w); 926 (w); 897 (m); 874 (w); 851 (m); 820 (w); 787 (w); 733 (s); 638 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 353 (5,200), 371 (7,700), 391 (6,900). Fluorescence (CH2Cl2): λmax,em [nm] = 420, 445, 475. Stokes shift Δ = 1,800 cm−1. Anal. calcd. for C23H26N2O2 (362.2): C 76.21, H 7.23, N 7.73; Found: C 75.93, H 7.18, N 7.72.
N-(tert-butyl)-2-(4-(dimethylamino)phenyl)-2-(N-(1-naphthylmethyl)acetamide) acetamide (5g)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 1:1) and crystallization from hexane/ethyl acetate (30:2) compound 5g (91.7 mg, 42%) was obtained as a colorless solid, Mp 174°C, Rf (n-hexane/ethyl acetate 1:1) = 0.50.
1H NMR (300 MHz, CDCl3): δ = 1.34 (s, 9 H), 2.01 (s, 3 H), 2.77 (s, 6 H), 4.875.32 (m, 2 H), 5.53 (s, 1 H), 5.90 (s, 1 H), 6.42 (d, J = 8.5 Hz, 2 H), 7.19 (d, J = 8.5 Hz, 2 H), 7.267.34 (m, 2 H), 7.377.50 (m, 2 H), 7.557.67 (m, 1 H), 7.737.86 (m, 2 H). 13C NMR (75 MHz, CDCl3): δ = 22.3 (CH3), 28.8 (CH3), 40.4 (CH3), 47.9 (CH2), 51.7 (Cquat), 62.9 (CH), 112.3 (CH), 122.1 (Cquat), 122.3 (CH), 123.4 (CH), 125.5 (CH), 125.7 (CH), 126.1 (CH), 127.2 (CH), 128.9 (CH), 130.3 (Cquat), 130.9 (CH), 133.0 (Cquat), 133.5 (Cquat), 150.4 (Cquat), 169.9 (Cquat), 172.9 (Cquat). MALDI-TOF-MS (m/z): 430.1 ([M]+). IR: [cm−1] = 3,314 (w); 3,049 (w); 2,968 (w); 2,912 (w); 2,886 (w); 2,808 (w); 1,676 (m); 1,632 (s); 1,616 (m); 1,601 (w); 1,541 (m); 1,522 (m); 1,507 (w); 1,477 (w); 1,447 (w); 1,423 (m); 1,391 (w); 1,358 (m); 1,319 (w); 1,261 (w); 1,250 (w); 1,227 (m); 1,206 (w); 1,190 (m); 1,165 (w); 1,126 (w); 1,063 (w); 1,047 (w); 1,032 (w); 978 (w); 939 (w); 827 (w); 802 (m); 789 (s); 772 (s); 750 (w); 735 (w); 687 (w); 640 (w); 625 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 270 (24,000). Fluorescence (CH2Cl2): λmax,em [nm] = 337, 455. Stokes shift Δ = 7,400 cm−1. Anal. calcd. for C27H33N3O2 (431.3): C 75.14, H 7.71, N 9.74; Found: C 75.15, H 7.58, N 9.67.
N-tert-butyl-2-(N-(4-(dimethylamino)benzyl)acetamido)-2-(1-naphthyl)acetamide (5h)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 1:1) and crystallization from hexane/ethyl acetate (33:2.5), compound 5h (99.1 mg, 46%) was obtained as a light brown solid, Mp 179°C, Rf (n-hexane/ethyl acetate 1:1) = 0.50.
1H NMR (300 MHz, CDCl3): δ = 1.57 (s, 9 H), 2.31 (s, 3 H), 2.93 (s, 6 H), 4.714.90 (m, 2 H), 5.76 (s, 1 H), 6.39 (d, J = 8.7 Hz, 2 H), 6.57 (d, J = 8.7 Hz, 2 H), 7.548.03 (m, 7 H), 8.31 (d, J = 8.4 Hz, 1 H). 13C NMR (75 MHz, CDCl3): δ = 23.0 (CH3), 28.8 (CH3), 40.8 (CH3), 49.9 (CH2), 51.9 (Cquat), 58.0 (CH), 112.4 (CH), 123.9 (CH), 125.2 (CH), 125.6 (Cquat), 126.2 (CH), 127.1 (CH), 127.2 (CH), 127.9 (CH), 128.9 (CH), 129.7 (CH), 132.0 (Cquat), 132.8 (Cquat), 133.8 (Cquat), 149.3 (Cquat), 170.2 (Cquat), 173.0 (Cquat). MALDI-TOF-MS (m/z): 430.1 ([M]+). IR: [cm−1] = 3,647 (w); 3,283 (w); 3,233 (w); 3,069 (w); 2,972 (w); 2,909 (w); 2,801 (w); 2,309 (w); 1,684 (s); 1,632 (m); 1,618 (s); 1,597 (w); 1,558 (m); 1,456 (w); 1,412 (m); 1,364 (m); 1,350 (w); 1,337 (m); 1,314 (w); 1,273 (w); 1,260 (w); 1,225 (m); 1,192 (w); 1,180 (w); 1,142 (w); 1,053 (w); 1,024 (w); 976 (w); 941 (w); 910 (w); 795 (s); 777 (s); 754 (w); 735 (w); 667 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 265 (19,000). Fluorescence (CH2Cl2): λmax,em [nm] = 334, 472. Stokes shift Δ = 7,800 cm−1. Anal. calcd. for C27H33N3O2 (431.3): C 75. 14, H 7.71, N 9.74; Found: C 74.88, H 7.67, N 9.46.
N-tert-butyl-2-(4-(dimethylamino)phenyl)-2-(N-(1-pyrenylmethyl)acetamido) acetamide (5i)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 1:1) and crystallization from hexane/ethyl acetate (45:3.5), compound 5i (102 mg, 40%) was obtained as a light brown solid, Mp 217–219°C, Rf (n-hexane/ethyl acetate 1:1) = 0.20.
1H NMR (300 MHz, CDCl3): δ = 1.24 (t, J = 7.1 Hz, 0.6 H), 1.36 (s, 9 H), 2.02 (s, 0.6 H), 2.10 (s, 3 H), 2.51 (s, 6 H), 4.10 (q, J = 7.1 Hz, 0.4 H), 5.245.52 (m, 2 H), 5.57 (s, 1 H), 6.04 (s, 1 H), 6.25 (d, J = 8.6 Hz, 2 H), 7.17 (d, J = 8.5 Hz, 2 H), 7.748.17 (m, 9 H). 13C NMR (75 MHz, CDCl3): δ = 14.3 (CH3), 21.2 (CH3), 22.6 (CH3), 28.9 (CH3), 40.0 (CH3), 48.0 (CH2), 51.7 (Cquat), 60.5 (CH2), 62.6 (CH), 112.1 (CH), 121.8 (Cquat), 122.0 (CH), 123.8 (CH), 124.6 (Cquat), 124.8 (CH), 125.1 (CH), 125.3 (CH), 126.0 (CH), 126.9 (CH), 127.2 (CH), 127.5 (Cquat), 130.2 (CH), 130.7 (Cquat), 130.9 (Cquat), 131.3 (CH), 131.4 (Cquat), 150.1 (Cquat), 170.07 (Cquat), 172.9 (Cquat), 173.0 (Cquat). MALDI-TOF-MS (m/z): 504.2 ([M]+). IR: [cm−1] = 3,289 (w); 3,040 (w); 2,961 (w); 2,893 (w); 2,805 (w); 1,739 (w); 1,670 (m); 1,612 (s); 1,587 (w); 1,547 (m); 1,522 (s); 1,433 (m); 1,414 (m); 1,391 (w); 1,360 (m); 1,339 (w); 1,304 (w); 1,267 (w); 1,254 (w); 1,225 (m); 1,204 (m); 1,167 (m); 1,132 (w); 1,063 (w); 1,032 (w); 980 (w); 934 (w); 843 (s); 812 (m); 797 (m); 710 (s); 640 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 317 (13,000), 331 (27,000), 347 (36,000). Fluorescence (CH2Cl2): λmax,em [nm] = 383, 397, 499. Stokes shift Δ = 2,700 cm−1. Anal. calcd. for C33H35N3O2 1/5 CH3CO2C2H5 (505.3 + 17.6): C 77.58, H 7.05, N 8.02; Found: C 77.11, H 7.00, N 8.02.
N-(tert-butyl)-2-(N-(4-(dimethylamino)benzyl)acetamido)-2-(1-pyrenyl)acetamide (5j)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 1:1) and crystallization from hexane/ethyl acetate (62:4.5), compound 5j (160 mg, 63%) was obtained as a light brown solid, Mp 202–205°C, Rf (n-hexane/ethyl acetate 1:1) = 0.30.
1H NMR (300 MHz, CDCl3): δ = 1.201.25 (m, 1.2 H), 1.37 (s, 9 H), 2.00 (s, 1.2 H), 2.20 (s, 6 H), 2.21 (s, 3 H), 4.08 (q, J = 7.1 Hz, 0.8 H), 4.454.76 (m, 2 H), 5.70 (s, 1 H), 5.70 (d, J = 8.5 Hz, 2 H), 6.21 (d, J = 8.6 Hz, 2 H), 7.25 (s, 1 H), 7.888.27 (m, 9 H). 13C NMR (75 MHz, CDCl3): δ = 14.28 (CH3), 21.0 (CH3), 22.9 (CH3), 28.9 (CH3), 40.2 (CH3), 50.2 (CH2), 52.0 (Cquat), 58.3 (CH), 60.5 (CH2), 111.8 (CH), 123.3 (CH), 124.5 (Cquat), 124.7 (Cquat), 124.8 (CH), 125.0 (Cquat), 125.6 (CH), 126.1 (CH) 126.7 (CH), 127.2 (CH), 127.3 (CH), 127.9 (CH), 128.7 (CH), 129.1 (Cquat), 130.8 (Cquat), 130.9 (Cquat), 131.2 (Cquat), 131.8 (Cquat), 148.5 (Cquat), 170.1 (Cquat), 170.4 (Cquat), 172.8 (Cquat). MALDI-TOF-MS (m/z): 504.2 ([M]+). IR: [cm−1] = 3,304 (w); 3,040 (w); 2,965 (w); 2,897 (w); 2,884 (w); 2,874 (w); 1,733 (w); 1,684 (m); 1,674 (w); 1,616 (s); 1,549 (m); 1,524 (m); 1,506 (w); 1,468 (w); 1,435 (m); 1423 (m), 1393 (w),1366 (w), 1341 (w), 1314 (w), 1277 (w), 1253 (w), 1225 (m), 1175 (w), 1126 (w), 1,053 (w); 1,034 (w); 980 (w); 972 (w); 943 (w); 910 (w); 845 (s); 812 (w); 799 (m); 758 (w); 710 (w); 681 (w). UV/Vis (CH2Cl2): λmax (ε [Lmol−1 cm−1]) [nm] = 317 (14,000), 331 (28,000), 348 (38,000). Fluorescence (CH2Cl2): λmax,em [nm] = 380, 395, 538. Stokes shift Δ = 2,400 cm−1. Anal. calcd. for C33H35N3O2 1/2 CH3CO2C2H5 (505.3 + 44.1): C 76.47, H 7.15, N 7.64; Found: C 76.51, H 7.15, N 8.10.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
The project was conceptualized by TM for the MSc thesis of MO, who developed the synthetic approach and conducted the photophysical studies and their evaluation. BM performed all DFT and TDDFT calculations and assigned the experimental absorption and emission bands transitions. Based upon the master thesis of MO, the manuscript was written and corrected by TM and BM.
Funding
Fonds der Chemischen Industrie (ad personam funding of TM) and Deutsche Forschungsgemeinschaft (Mu 1088/9-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We cordially thank Fonds der Chemischen Industrie and Deutsche Forschungsgemeinschaft (Mu 1088/9-1) for the financial support. Computational support and infrastructure was provided by the Centre for Information and Media Technology (ZIM) at the University of Düsseldorf (Germany). We also thank MSc Lukas Biesen for measuring the solid state emission and the fluorescence quantum yield of the N,N-dimethylaniline-anthracene dyad 5a.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00717/full#supplementary-material
Supplementary Information contains the optimization of the synthesis of compound 5a, 1H and 13C NMR spectra of compounds 5, the absorption and emission spectra of compounds 5, the Lippert-Mataga analysis of dyad 5a and the data and evaluation of the DFT and TDDFT calculations on the syn- and anti-structures 5a, 5g, and 5i.
References
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. doi: 10.1063/1.478522
Balzani, V. (2008). Electron Transfer in Chemistry. Weinheim: Wiley-VCH Verlag GmbH. doi: 10.1002/9783527618248
Balzani, V., Credi, A., and Venturi, M. (2003). Molecular Devices and Machines - A Journey into the Nano World, Weinheim: Wiley-VCH. doi: 10.1002/3527601600
Bay, S., Makhloufi, G., Janiak, C., and Müller, T. J. J. (2014). Concise modular synthesis of donor-anthraquinone dyads by Ugi four-component reaction. Beilstein J. Org. Chem. 10, 1006–1016. doi: 10.3762/bjoc.10.100
Bay, S., and Müller, T. J. J. (2014). Phenothiazine-aromatic hydrocarbon acceptor dyads as photo-induced electron transfer systems by Ugi four-component reaction. Z. Naturforsch. 69b, 541–553. doi: 10.5560/ZNB.2014-4060
Bay, S., Villnow, T., Ryseck, G., Rai-Constapel, V., Gilch, P., and Müller, T. J. J. (2013). The Ugi 4CR route to photo-inducible electron transfer systems. ChemPlusChem 78, 137–141. doi: 10.1002/cplu.201200279
Becke, A. D. (1993). A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377. doi: 10.1063/1.464304
Biggs-Houck, J. E., Younai, A., and Shaw, J. T. (2010). Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr. Opin. Chem. Biol. 14, 371–382. doi: 10.1016/j.cbpa.2010.03.003
Bucci, N., and Müller, T. J. J. (2006). First Syntheses and electronic properties of (Oligo)Phenothiazine C60 dyads. Tetrahedron Lett. 47, 8323–8327. doi: 10.1016/j.tetlet.2006.09.076
Cairo, C. W., Key, J. A., and Sadek, C. M. (2010). Fluorescent small-molecule probes of biochemistry at the plasma membrane. Curr. Opin. Chem. Biol. 14, 57–63. doi: 10.1016/j.cbpa.2009.09.032
Carter, K. P., Young, A. M., and Palmer, A. E. (2014). Fluorescent sensors for measuring metal ions in living systems. Chem. Rev. 114, 4564–4601. doi: 10.1021/cr400546e
Ceroni, P., and Balzani, V. (2012). “chpt. 2: Photoinduced energy and electron transfer processes,” in The Exploration of Supramolecular Systems and Nanostructures by Photochemical Techniques, ed P.Ceroni (New York, NY; Heidelberg: Springer), 21–38. doi: 10.1007/978-94-007-2042-8_2
Chen, C., Wagner, H., and Still, W. C. (1998). Fluorescent, sequence-selective peptide detection by synthetic small molecules. Science 279, 851–853. doi: 10.1126/science.279.5352.851
Demchenko, A. P., Mély, Y., Duportail, G., and Klymchenko, A. S. (2009). Monitoring biophysical properties of lipid membranes by environment-sensitive fluorescent probes. Biophys. J. 96, 3461–3470. doi: 10.1016/j.bpj.2009.02.012
Dömling, A. (2006). Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 106, 17–89. doi: 10.1021/cr0505728
Dömling, A., and Ugi, I. (2000). Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 39, 3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Dömling, A., Wang, W., and Wang, K. (2012). Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3135. doi: 10.1021/cr100233r
Ernzerhof, M., and Scuseria, G. E. (1999). Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 110, 5029–5036. doi: 10.1063/1.478401
Feng, Q., Wang, M., Xu, C., Khan, A., Wu, X., Lu, J., et al. (2014). Investigation of molecular arrangements and solid-state fluorescence properties of solvates and cocrystals of 1-acetyl-3-phenyl-5-(9-anthryl)-2-pyrazoline. Cryst. Eng. Commun. 16, 5820–5826. doi: 10.1039/C3CE42210K
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). GAUSSIAN 16 (Revision A.03). Wallingford CT: Gaussian Inc.
Hui, M. H., and Ware, W. R. (1976). Exciplex photophysics. V. The kinetics of fluorescence quenching of anthracene by N,N-dimethylaniline in cyclohexane. J. Am. Chem. Soc. 98, 4718–4727. doi: 10.1021/ja00432a005
Kavarnos, G. J. (1993). Fundamentals of Photoinduced Electron Transfer. Weinheim; New York, NY: VCH.
Kavarnos, G. J., and Turro, N. J. (1986). Photosensitization by reversible electron transfer: theories, experimental evidence, and examples. Chem. Rev. 86, 401–449. doi: 10.1021/cr00072a005
Kim, E., and Park, S. B. (2009). Chemistry as a prism: a review of light-emitting materials having tunable emission wavelengths. Chem. Asian J. 4, 1646–1658. doi: 10.1002/asia.200900102
Kim, K., and Jordan, K. D. (1994). Comparison of density functional and MP2 calculations on the water monomer and dimer. J. Phys. Chem. 98, 10089–10094. doi: 10.1021/j100091a024
Klymchenko, A. S. (2017). Solvatochromic and fluorogenic dyes as environment-sensitive probes: design and biological applications. Acc. Chem. Res. 50, 366–375. doi: 10.1021/acs.accounts.6b00517
Kobayashi, H., Ogawa, M., Alford, R., Choyke, P. L., and Urano, Y. (2010). New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 110, 2620–2640. doi: 10.1021/cr900263j
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654. doi: 10.1063/1.438955
Lakowicz, J. R. (2006). Principles of Fluorescence Spectroscopy, 3rd Edn. New York, NY: Springer. doi: 10.1007/978-0-387-46312-4
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 37, 785–789. doi: 10.1103/PhysRevB.37.785
Lemmetyinen, H., Tkachenko, N. V., Efimov, A., and Niemi, M. (2011). Photoinduced intra- and intermolecular electron transfer in solutions and in solid organized molecular assemblies. Phys. Chem. Chem. Phys. 13, 397–412. doi: 10.1039/C0CP01106A
Levi, L., and Müller, T. J. J. (2016a). Multicomponent syntheses of functional chromophores. Chem. Soc. Rev. 45, 2825–2846. doi: 10.1039/C5CS00805K
Levi, L., and Müller, T. J. J. (2016b). Multicomponent syntheses of fluorophores initiated by metal catalysis. Eur. J. Org. Chem. 2907–2918. doi: 10.1002/ejoc.201600409
Lippert, E. Z. (1957). Spektroskopische Bestimmung des Dipolmomentes aromatischer Verbindungen im ersten angeregten Singulettzustand. Z. Elektrochem. 61, 962–975. doi: 10.1002/bbpc.19570610819
Loving, G. S., Sainlos, M., and Imperiali, B. (2010). Monitoring protein interactions and dynamics with solvatochromic fluorophores. Trends Biotechnol. 28, 73–83. doi: 10.1016/j.tibtech.2009.11.002
Mataga, N., Kaifu, Y., and Koizumi, M. (1956). Solvent effects upon fluorescence spectra and the dipolemoments of excited molecules. Bull. Chem. Soc. Jpn. 29, 465–470. doi: 10.1246/bcsj.29.465
Mataga, N., and Miyasaka, H. (1999). “Electron Transfer and Exciplex Chemistry,” in Advances in Chemical Physics: Electron Transfer from Isolated Molecules to Biomolecules Part 2, Vol. 107, eds I. Prigogine, and S. A. Rice (New York, NY; Chichester; Weinheim; Brisbane, QLD; Singapore; Toronto, ON: John Wiley and Sons, Inc.), 431–496. doi: 10.1002/9780470141663.ch8
Merkt, F. K., and Müller, T. J. J. (2018). Solid state and aggregation induced emissive chromophores by multi-component syntheses. Isr. J. Chem. 58, 889–900. doi: 10.1002/ijch.201800058
K.. Müllen, and U.. Scherf (eds.) (2006). Organic Light-Emitting Diodes - Synthesis, Properties, and Applications. Weinheim: Wiley-VCH. doi: 10.1002/3527607986
Müller, T. J. J. (2018). Multi-component synthesis of fluorophores via catalytic generation of alkynoyl intermediates. Drug Discov. Today Technol. 29, 19–26. doi: 10.1016/j.ddtec.2018.06.003
T. J. J.. Müller, and U. H. F.. Bunz (eds.) (2007). Functional Organic Materials. Syntheses, Strategies, and Applications. Weinheim: Wiley-VCH. doi: 10.1002/9783527610266
Nilsson, D., Kugler, T., Svensson, P.-O., and Berggren, M. (2002). An all-organic sensor–transistor based on a novel electrochemical transducer concept printed electrochemical sensors on paper. Sens. Actuators B 86, 193–197. doi: 10.1016/S0925-4005(02)00170-3
Nishizawa, S., Kaneda, H., Uchida, T., and Teramae, N. (1998). Anion sensing by a donor–spacer–acceptor system: an intra-molecular exciplex emission enhanced by hydrogen bond-mediated complexation. J. Chem. Soc. Perkin Trans. 2, 2325–2328. doi: 10.1039/A805075I
Park, J.-S., Chae, H., Chung, H. K., and Lee, S. I. (2011). Thin film encapsulation for flexible AM-OLED: a review. Semicond. Sci. Technol. 26, 034001–034008. doi: 10.1088/0268-1242/26/3/034001
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Errata: Generalized gradient approximation made simple. Phys. Rev. Lett. 78:1396. doi: 10.1103/PhysRevLett.78.1396
Perdew, P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Piuzzi, F. (1993). Hole-burning spectra of the isomeric forms of jet-cooled anthracene complexes with aniline derivatives. Chem. Phys. Lett. 209, 484–492. doi: 10.1016/0009-2614(93)80122-6
Piuzzi, F., Uridat, D., Dimicoli, I., Mons, M., Tramer, A., LeBarbu, K., et al. (1999). Photoinduced electron transfer in jet cooled molecular complexes. Acta Phys. Pol. A, 95, 121. doi: 10.12693/APhysPolA.95.121
Rehm, D., and Weller, A. (1970). Kinetics of fluorescence quenching by electron and H-atom transfer. Isr. J. Chem. 8, 259–271. doi: 10.1002/ijch.197000029
Ricks, A. B., Brown, K. E., Wenninger, M., Karlen, S. D., Berlin, Y. A., Co, D. T., et al. (2012). Exponential distance dependence of photoinitiated stepwise electron transfer in donor–bridge–acceptor molecules: implications for wirelike behavior. J. Am. Chem. Soc. 134, 4581–4588. doi: 10.1021/ja205913q
Riva, R. L, Moni, L., and Müller, T. J. J. (2016). Multicomponent strategies for the diversity-oriented synthesis of blue-emissive heterocyclic chromophores. Targets Heterocyclic Syst. 20, 85–112. doi: 10.17374/targets.2017.20.85
Scalmani, G., and Frisch, M. J. (2010). Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 132:114110. doi: 10.1063/1.3359469
Shankarling, G. S., and Jarag, K. J. (2010). Laser dyes. Resonance 15, 804–818. doi: 10.1007/s12045-010-0090-
Stack, D. E., Hill, A. L., Diffendaffer, C. B., and Burns, N. M. (2002). Synthesis of a new fluorescent probe specific for catechols. Org. Lett. 4, 4487–4490. doi: 10.1021/ol027000j
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi: 10.1021/j100096a001
Thejo Kalayani, N., and Dhoble, S. J. (2012). Organic light emitting diodes: energy saving lighting technology - A review. Renew. Sust. Energ. Rev. 16, 2696–2723. doi: 10.1016/j.rser.2012.02.021
Thiel, E. (2000). “Laser Dyes,” in Ullmann's Encyclopedia of Industrial Chemistry, ed B. Elvers (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 315–335. doi: 10.1002/14356007.a15_15
Valeur, B., and Berberan-Santos, M. N. (2012). Molecular Fluorescence. Principles and Applications, 2nd Edn. Weinheim: WileyVCH Verlag GmbH. doi: 10.1002/9783527650002
B.. Valeur, and J.-C.. Brochon (eds.) (2012). New Trends in Fluorescence Spectroscopy: Applications to Chemical and Life Sciences. Berlin; Heidelberg: Springer Science and Business Media.
Vauthey, E. (2012). Photoinduced symmetry-breaking charge separation. ChemPhysChem 13, 2001–2011. doi: 10.1002/cphc.201200106
Wagenknecht, H.-A. (2008). Fluorescent DNA base modifications and substitutes: multiple fluorophore labeling and the DETEQ concept. Ann. N. Y. Acad. Sci. 1130, 122–130. doi: 10.1196/annals.1430.001
Wenger, O. S. (2011). Photoinduced electron and energy transfer in phenylene oligomers. Chem. Soc. Rev. 40, 3538–3550. doi: 10.1039/C1CS15044H
O. S.. Wolfbeis(ed.). (1993). Fluorescence Spectroscopy: New Methods and Applications. Berlin; Heidelberg: Springer. doi: 10.1007/978-3-642-77372-3
Zhang, B. W., Cao, Y., Bai, J. W., and Chen, J. R. (1997). Photoinduced intramolecular electron transfer and exciplex formation in anthracene and pyrene binary compounds. J. Photochem. Photobiol. A 106, 169–175. doi: 10.1016/S1010-6030(97)00055-5
Keywords: absorption, bichromophores, DFT, emission, energy transfer (ET) dyes, exciplexes, isonitrile, multicomponent reaction
Citation: Ochs M, Mayer B and Müller TJJ (2019) Unimolecular Exciplexes by Ugi Four-Component Reaction. Front. Chem. 7:717. doi: 10.3389/fchem.2019.00717
Received: 12 June 2019; Accepted: 10 October 2019;
Published: 01 November 2019.
Edited by:
Alexander Dömling, University of Groningen, NetherlandsReviewed by:
Valentine Nenajdenko, Lomonosov Moscow State University, RussiaLuca Banfi, University of Genoa, Italy
Copyright © 2019 Ochs, Mayer and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. J. Müller, VGhvbWFzSkouTXVlbGxlckBoaHUuZGU=