 Carlos Eduardo M. Salvador
Carlos Eduardo M. Salvador Carlos Kleber Z. Andrade
Carlos Kleber Z. Andrade
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 30 July 2019
Sec. Organic Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00531
This article is part of the Research Topic Isocyanide-based Multicomponent Reactions View all 10 articles
A continuous flow approach for the synthesis of α-acyloxy ketone derivatives from the corresponding arylglyoxals, isocyanides, and carboxylic acids is described. The target products were obtained in excellent yields in short residence times and with high purities via the first transcription of the microwave-to-flow paradigm to the isocyanide-based Passerini reaction. Furthermore, this methodology allowed a 10-fold scale-up using the same experimental conditions initially established.
Over the last decades, α-acyloxy ketones and their derivatives have become outstanding building blocks in organic synthesis due to their presence in several natural products and active pharmaceutical ingredients (APIs) (Prasad et al., 2018). Moreover, they participate in a wide range of synthetic transformations as starting materials or reactants, which result in important compounds such as cyclobutanes, aminoalcohols, hydroxy ketones, and heterocycles (furans, pyrroles, oxazoles, thiophenes, among others) (Shindo et al., 2007; Prasad et al., 2018).
Several reagents and reaction conditions to synthesize α-acyloxy ketones have been described in the literature (Prasad et al., 2018). However, most of them involve metal catalyzed oxidative coupling or electrophilic halogenations followed by nucleophilic displacement, which suffer from low atom economy and employ high amounts of oxidizing agents (Du et al., 2015; Hunter et al., 2016). Alternatively, functionalized α-acyloxy ketones may be easily accessed in a more sustainable way through the Passerini reaction (Neo et al., 2005, 2011).
The Passerini reaction is an efficient and versatile synthetic methodology and belongs to the class of isocyanide-based multicomponent reactions (IMCRs) (Dömling et al., 2012; Kazemizadeh and Ramazani, 2012). This one-pot reaction allows the obtention of α-acyloxy ketones bearing an α-carboxamide moiety from arylglyoxals, aldehydes and carboxylic acids (Scheme 1). In contrast to other processes described in the literature, this reaction not only offers an excellent atom economy without using oxidizing agents but also allows the obtention of functionalized products that can be explored in further transformations. However, the reported reaction times are longer (usually 2 to 3 days) and the Passerini products are obtained in moderate yields and in small scales (Neo et al., 2005, 2011). Compounds 4 are versatile intermediates and can be further converted to β-keto amides by Zn reduction in the presence of NH4Cl.
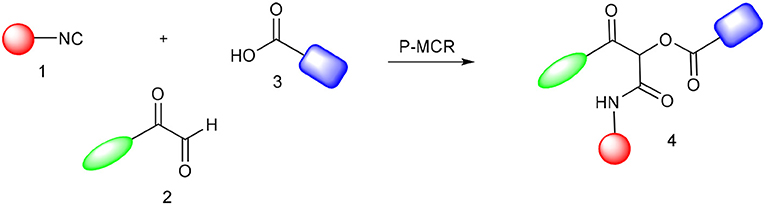
Scheme 1. Synthesis of α-acyloxy ketones (4) through Passerini reaction between isocyanides (1), arylglyoxals (2), and carboxylic acids (3).
The continuous flow approach has been a relevant strategy in organic synthesis to access a plethora of compounds in a faster and scaled-up manner (Gutmann et al., 2015; Ley et al., 2015; Plutschack et al., 2017), including natural products and APIs (De Souza et al., 2018). Application of this strategy to IMCRs has already been described (Sharma et al., 2013; Heublein et al., 2014; Salvador et al., 2015; Vasconcelos et al., 2017) but its employment in Passerini reactions is still scarce (Sharma et al., 2013).
Herein, we describe a continuous flow approach for the synthesis of α-acyloxy ketone derivatives from the corresponding arylglyoxals, isocyanides and carboxylic acids. The first transcription of the microwave-to-flow paradigm (Glasnov and Kappe, 2011) toward the isocyanide-based Passerini MCR was used to access the target products in excellent yields, short residence times and with high purities through simple evaporation of the solvent at the end (Andrade and Dar, 2016). Furthermore, a 10-fold scale-up was accomplished using the same experimental conditions initially established.
To evaluate the most appropriate reaction conditions for the synthesis of α-acyloxy ketones, an initial search for the best solvent was carried out in a batch microwave (MW) preliminary optimization using solvents typically applied in Passerini reactions (Wu et al., 2018) (Table 1) and considering our previous experience in MW-mediated MCR reactions (Barreto et al., 2011a,b, 2014; Salvador et al., 2015; Barreto and Andrade, 2018, 2019). Formation of the Passerini product in toluene at 110°C was not observed probably be due to the low microwave dielectric heating property of this solvent (Gabriel et al., 1998; Kappe et al., 2012) (entry 1). In CHCl3 at 60°C, low conversions were observed (entry 2). However, using MeCN at this same temperature, a good conversion of the desired product was achieved (entry 3). In all cases, the applied experimental conditions were the same using equimolar amounts of the starting materials (0.5 mmol), 2 mL of solvent and reaction time of 30 min. Thus, MeCN was chosen as the best solvent for this reaction. Then, we decided to move forward and raise the temperature near the boiling point of MeCN (80°C). To our delight, at this temperature full conversion to product 4a was achieved (entry 4). By decreasing the reaction time (entries 5–7), we found out that only 5 min were required for a full conversion of phenylglyoxal 2a to product 4a, which was isolated essentially pure in 94% yield (entry 7).
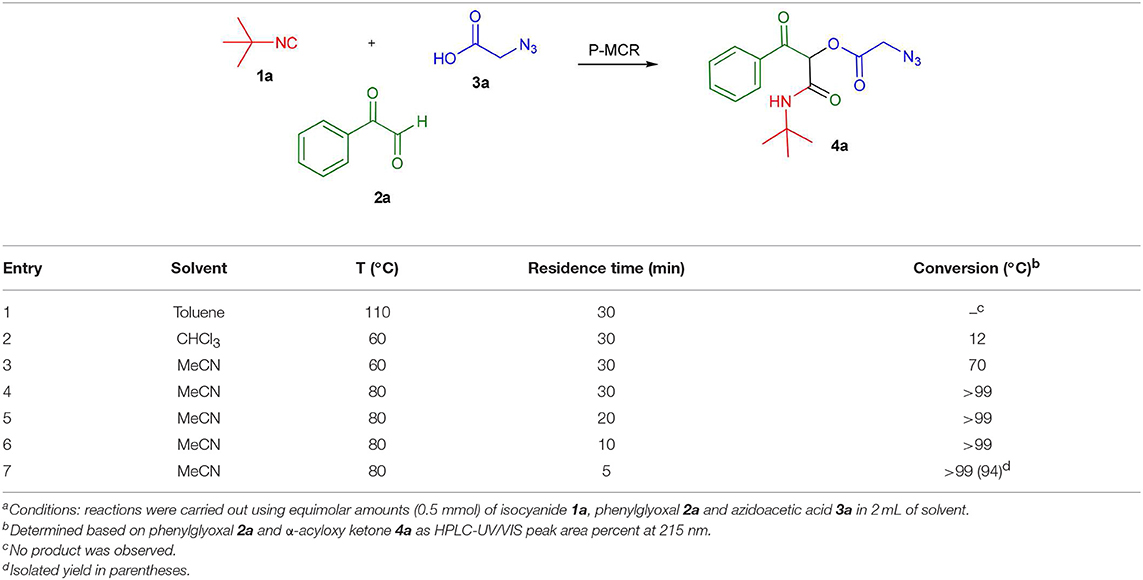
Table 1. Batch optimization of substituted α-acyloxy ketones synthesis using microwave heatinga.
As already known, organic reactions performed in short times and well-established temperatures at microwave reactors are prone to be applied to a continuous flow setup device due to the effective residence time control and high thermal transfer achieved in this method (Damn et al., 2010; Kappe et al., 2013). With this in mind, we decided to transpose the microwave batch procedure just established for the synthesis of α-acyloxy ketones 4a (Table 1, entry 7) into a continuous flow process (Scheme 2). Thus, a 0.5 M solution of tert-butyl isocyanide 1a in MeCN (1 mL) in an injection loop (SL1) was mixed via a syringe pump in a flow rate of 0.5 mL min−1 (feed A in MeCN) with a 0.5 M solution of phenylglyoxal 2a and azidoacetic acid 3a (in 1 mL), which was pumped simultaneously from the second injection loop (SL2) at the same flow rate (feed B in MeCN). The combined solution possesses the same molar concentration (0.25 M) used in the microwave batch optimization and was pumped through a 5 mL perfluoroalkoxy (PFA) reactor coil (heated at 80°C in an oil bath) with outside (OD) and inner (ID) diameters of 1.6 and 0.8 mm, respectively. Additionally, a 3 bar back pressure regulator (BPR) was used to guarantee greater precision in the follow-up of the residence time. The design of the reactor capacity (5 mL) and the resulting flow rate (1 mL min−1) furnished a total residence time unit (RTU) of 5 min (same reaction time used in the microwave optimization process). Satisfyingly, a similar and reproducible yield of compound 4a was obtained with the continuous flow system as compared to the microwave-assisted synthesis. For comparison, the same reaction was carried out at room temperature and at reflux condition. The first condition gave the product in only 68% yield after 7 h (~10% of non-reacted glyoxal). The second one gave the product in 94% yield but took 4 h to get to completion. Similar results were obtained for arylglyoxal 4c bearing a strong electron donating group (p-OMe). These results show a significant improvement in the reaction outcome using the flow conditions developed in this study.
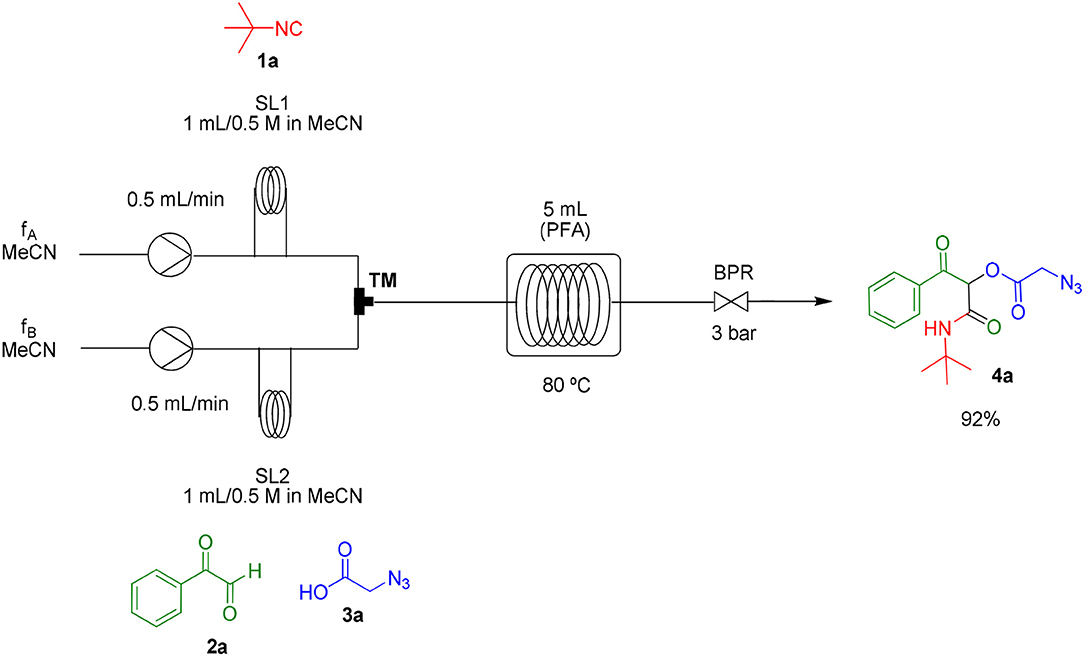
Scheme 2. Continuous flow synthesis of α-acyloxy ketone 4a. Conditions: Feed A (1.0 mL of a 0.5 M solution of 1a in MeCN) and feed B (1.0 mL of a 0.5 M solution of 2a and 3a in MeCN) pumped through a 5 mL PFA reactor at 80°C and 3 bar back-pressure regulator.
Inspired by these promising results, we decided to broaden the scope of this methodology and use this flow approach to synthesize various α-acyloxy ketones 4 using isocyanides 1a and 1b, arylglyoxals 2a-c, and carboxylic acids 3a-g. Numerous α-acyloxy ketones were obtained in excellent yields without the need for any re-optimization of the flowing experimental conditions (Table 2). All compounds from Table 2 were isolated with high purity by a simple evaporation of the solvent (see NMR spectra in the Supplementary Material).
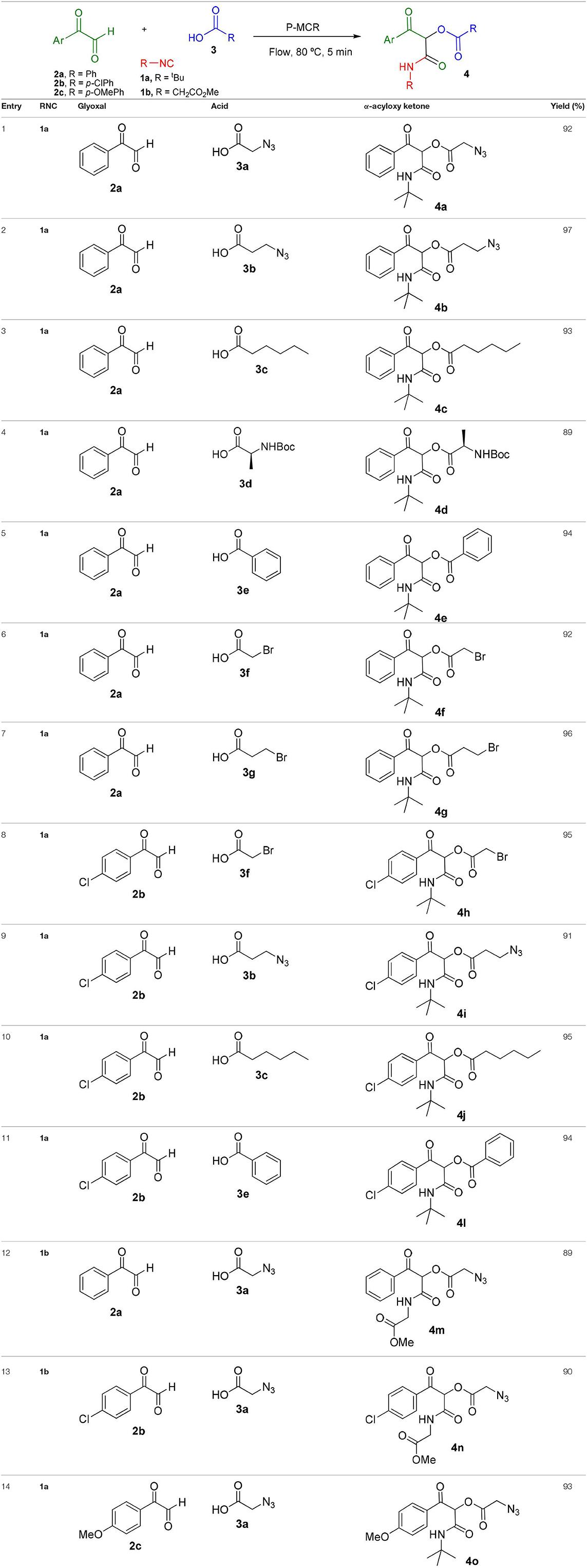
Table 2. Scope for the Passerini-MCR flow synthesis of substituted α-acyloxy ketones.
Most organic reactions performed in traditional glassware usually work well at small-scale. Nevertheless, when an increase in scale is required, for instance for a chain development of distinct processes in pharmaceutical and fine chemicals, a great challenge arises in most cases due to inherent problems of scaling up processes such as heat generated and transferred, potential hazards of chemical compounds and environmental issues (Geyer et al., 2006). In contrast, the scale-up reaction performed in flow technology work very well due to the use of versatile approaches, which have similarities with the conditions used in small scale reactions (May et al., 2012, 2016). According to the features of flow processes, the scale-up synthesis of compound 4b was used as a proof of concept and was achieved by increasing the length of the injection loop (2 mL) for better solubility of the starting materials and the length of the reactor coil (10 mL) to keep the same RTU used before (5 min). With these small adjustments, it was possible to achieve a reproducible 10-fold increase in the scale for the synthesis of compound 4b in a productivity of 0.312 g/min (Scheme 3).
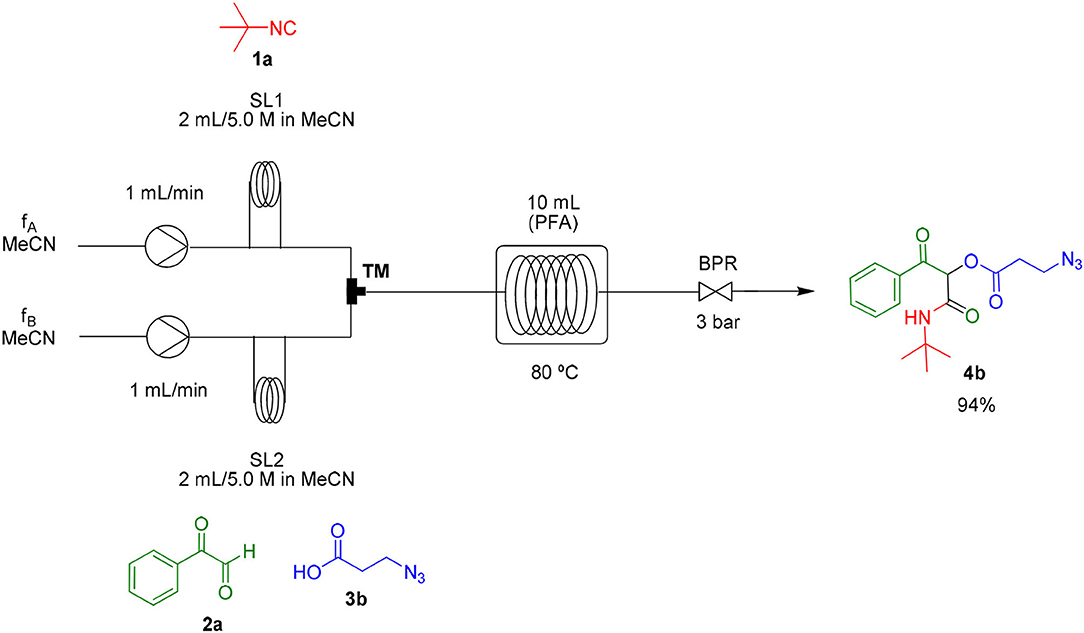
Scheme 3. Continuous flow scale-up for the synthesis of α-acyloxy ketone 4b.
In summary, we have applied the first transcription of the microwave-to-flow paradigm for the isocyanide-based Passerini reaction to the synthesis of substituted α-acyloxy ketones via a continuous flow process. The target products can be further functionalized and were easily obtained in low residence times and excellent yields without the need for purification, which constitutes a significant improvement over the existing methods for preparing these compounds. Furthermore, the success of this methodology was corroborated by a 10-fold increase in the scale under similar reaction conditions leading to a productivity of 0.312 g/min.
1H-NMR spectra were recorded on a Varian Mercury Plus 300 MHz instrument. 13C-NMR spectra were recorded on this same instrument at 75 MHz. Chemical shifts (δ) are expressed in ppm downfield from TMS as internal standard. The letters s, brs, t, dd and m are used to indicate singlet, broad singlet, triplet, double of doublets, and multiplet, respectively. HRMS experiments were performed on a TOF LC/MS instrument equipped with an ESI ion source (positive ionization mode) from Bruker. Analytical HPLC (Shimadzu LC20) analysis was carried out on a C18 reversed-phase (RP) analytical column (150 × 4.6 mm, particle size 5 μm) at 37°C using a mobile phase A (water/acetonitrile 90:10 (v/v) + 0.1% TFA) and B (MeCN + 0.1% TFA) at a flow rate of 1.5 mL/min. The following gradient was applied: linear increase from 30 to 100% B in 17 min. All microwave-mediated reactions were carried out in a CEM, Co. Discover microwave reactor in borosilicate closed glass reaction vessels (10 mL). For continuous flow procedures, KDS 210 Legacy dual syringe pump was used. Static backpressure, modular mixers, sample manual injectors, fittings, and tubings are commercially available from Idex Health & Science, Inc. All solvents and chemicals were obtained from standard commercial suppliers and were used without any further purification.
2-azidoacetic acid (Yu et al., 2011), 3-azidopropionic acid (Yang et al., 2013), 2-oxo-2-phenylacetaldehyde, 2-(4-chlorophenyl)-2-oxoacetaldehyde and 2-(4-methoxyphenyl)-2-oxoacetaldehyde (Wang et al., 2013) were prepared and characterized according to literature procedures.
A sealed 10 mL borosilicate glass reaction vessel, equipped with a magnetic stir bar and containing a mixture of equimolar amounts (0.5 mmol) of isocyanide 1a, phenylglyoxal 2a, and azidoacetic acid 3a in 2 mL of MeCN was irradiated in a CEM Co. Discover reactor for 5 min at 80°C. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure and the desired product (4a) was isolated in essentially pure form (entry 7).
The PFA reactor coil (0.8 mm inner diameter; 5.0 mL) was completely filled with solvent (MeCN) and then heated in an oil bath at 80°C. Isocyanide 1a (0.5 mmol) was diluted in 1.0 mL of MeCN (0.5 M) and stored in a 1 mL sample loop (SL1). Phenylglyoxal 2a and azidoacetic acid 3a (0.5 mmol each) were diluted in 1.0 mL of MeCN (0.5 M) and stored in a 1 mL sample loop (SL2). The sample loops were connected via a 6-way manual valve and were at the same time pumped into a T-shaped mixer (TM). After mixing, the resulting solution was fed (1 mL.min−1 stream) into the PFA reactor coil, thus remaining for 5 min of residence time under heating (80°C). A static back pressure regulator (3 bar) was used to allow the best fluid flow condition (Kappe et al., 2013). The desired products (4a−4o) were obtained after solvent evaporation under reduced pressure.
Same as above using 5.0 M solutions of compounds 1a, 2a, and 3b, a 10.0 mL PFA reactor coil and 2 mL sample loops. Compound 4b was obtained in 94% yield (1.56 g) with a productivity of 0.312 g/min (see Scheme 3).
Very viscous yellow oil (0.146 g, 92% yield); HRMS (ESI): m/z: calcd for C15H18N4O4Na [M + Na+]: 341.1220, found: 341.1226. 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.17–8.10 (m, 2H), 7.67–7.58 (m, 1H), 7.54–7.46 (m, 2H), 6.30 (s, 1H), 6.14 (brs, 1H), 4.08 (s, 2H), 1.33 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.3, 166.3, 161.7, 134.3, 134.1, 129.7, 128.7, 77.5, 52.2, 50.1, 28.4.
Very viscous yellow oil (0.161 g, 97% yield); HRMS (ESI): m/z: calcd for C16H20N4O4Na [M + Na+]: 355.1377, found: 355.1387. 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.15–8.10 (m, 2H), 7.64–7.58 (m, 1H), 7.53–7.47 (m, 2H), 6.26 (s, 1H), 6.22 (brs, 1H), 3.61 (t, J = 6.2 Hz, 2H), 2.75 (t, J = 6.2 Hz, 2H), 1.34 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.8, 168.8, 162.3, 134.3, 134.1, 129.6, 128.6, 76.5, 52.0, 46.4, 33.6, 28.4.
Very viscous colorless oil (0.155 g, 93% yield); HRMS (ESI): m/z: calcd for C19H27NO4Na [M + Na+]: 356.1832, found: 356.1829; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.18–8.08 (m, 2H), 7.65–7.55 (m, 1H), 7.54–7.43 (m, 2H), 6.21 (s, 1H), 6.21–6.17 (brs, 1H), 2.54–2.44 (m, 2H), 1.74–1.59 (m, 2H), 1.36–1.31 (m, 13H), 0.95–0.85 (m, 3H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 192.5, 171.6, 162.8, 134.5, 134.0, 129.6, 128.5, 76.1, 51.9, 33.6, 31.1, 28.5, 24.3, 22.2, 13.8.
Very viscous brown oil (0.181 g, 89% yield); HRMS (ESI): m/z: calcd for C21H30N2O6Na [M + Na+]: 429.1996, found: 429.1998; Mixture of diastereomers: 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.11–8.09 (m, 4H), 7.65–7.57 (m, 2H), 7.53–74.6 (m, 4H), 6.48 (brs, 1H), 6.35 (brs, 1H), 6.31 (s, 1H), 6.23 (s, 1H), 4.54–4.37 (m, 2H), 1.48–1.43 (m, 24H), 1.35 (d, J = 2.0 Hz, 18H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.7, 191.5, 171.8, 170.9, 162.6, 162.6, 134.6, 134.4, 134.1, 134.0, 129.7, 129.7, 128.6, 128.6, 80.3, 76.4, 52.0, 49.1, 28.5, 28.5, 28.3, 28.5, 17.8, 17.7.
Very viscous yellow oil (0.170 g, 94% yield); HRMS (ESI): m/z: calcd for C20H21NO4Na [M + Na+]: 362.1363, found: 362.1365; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.24–8.04 (m, 2H), 8.15–8.01 (m, 2H), 7.67–7.57 (m, 2H), 7.55–7.46 (m, 4H), 6.43 (s, 1H), 6.28 (brs, 1H), 1.39 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 192.4, 164.6, 162.9, 134.6, 134.1, 133.9, 133.4, 129.7, 129.7, 128.7, 128.6, 76.8, 52.0, 28.6.
Very viscous yellow oil (0.164 g, 92% yield); HRMS (ESI): m/z: calcd for C15H18BrNO4Na [M + Na+]: 380.0296, found: 380.0296; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.13–8.10 (m, 2H), 7.66–7.59 (m, 2H), 7.55–7.47 (m, 2H), 6.27 (s, 1H), 6.26 (brs, 1H), 4.09–3.86 (m, 2H), 1.35 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.9, 165.0, 162.0, 134.3, 134.2, 129.8, 128.6, 77.1, 52.1, 28.5, 24.8.
Very viscous colorless oil (0.178 g, 96% yield); HRMS (ESI): m/z: calcd for C16H20BrNO4Na [M + Na+]: 392.0468, found: 392.0473; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.15–8.10 (m, 2H), 7.66–7.57 (m, 1H), 7.54–7.45 (m, 2H), 6.27 (s, 1H), 6.25 (brs, 1H), 3.61 (t, J = 6.6 Hz, 2H), 3.14 (t, J = 6.6 Hz, 2H), 1.34 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.8, 168.5, 162.3, 134.4, 134.2, 129.7, 128.6, 76.5, 52.1, 37.2, 28.5, 25.1.
Very viscous colorless oil (0.186 g, 95% yield); HRMS (ESI): m/z: calcd for C15H17BrClNO4Na [M + Na+]: 411.9922, found: 411.9918; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.12–8.06 (m, 8H), 7.51–7.44 (m, 8H), 6.24 (brs, 1H), 6.21 (s, 1H), 3.99 (m, 2H), 1.35 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 190.0, 164.9, 161.8, 141.0, 132.6, 131.2, 129.3, 129.0, 128.8, 76.9, 52.2, 28.5, 24.7.
Very viscous yellow oil (0.175 g, 91% yield); HRMS (ESI): m/z: calcd for C16H19N4O4Na [M + Na+]: 389.0987, found: 389.1002; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.12–8.06 (m, 2H), 7.51–7.44 (m, 2H), 6.25 (brs, 1H), 6.20 (s, 1H), 3.64 (t, J = 6.2 Hz, 2H), 2.78 (t, J = 6.2 Hz, 2H), 1.34 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 190.7, 168.8, 162.1, 140.8, 132.7, 131.6, 129.0, 76.4, 52.1, 46.5, 33.6, 28.5.
Very viscous yellow oil (0.175 g, 95% yield); HRMS (ESI): m/z: calcd for C19H26ClNO4Na [M + Na+]: 390.1443, found: 390.1446; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.11–8.06 (m, 2H), 7.49–7.44 (m, 2H), 6.19 (brs, 1H), 6.14 (s, 1H), 2.54–2.45 (m, 2H), 1.73–1.59 (m, 2H), 1.36–1.31 (m, 13H), 0.95–0.85 (m, 3H).13C-NMR (75 MHz, CDCl3) δ (ppm) 191.3, 171.5, 162.6, 140.5, 132.9, 131.1, 128.8, 75.9, 51.9, 33.6, 31.0, 28.4, 24.3, 22.2, 13.8.
Very viscous brown oil (0.176 g, 94% yield); HRMS (ESI): m/z: calcd for C20H20ClNO4Na [M + Na+]: 396.0973, found: 396.0966; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.18–8.11 (m, 2H), 8.09–8.04 (m, 2H), 7.68–7.60 (m, 1H), 7.55–7.46 (m, 4H), 6.36 (s, 1H), 6.29 (bs, 1H), 1.39 (s, 9H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 191.2, 164.5, 162.7, 140.7, 134.0, 132.9, 131.1, 129.8, 129.0, 128.7, 76.7, 52.1, 28.5.
Very viscous colorless oil (0.151 g, 89% yield); HRMS (ESI): m/z: calcd for C14H14N4O6 [M + H+]: 341.1220, found: 341.1226. 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.14–8.10 (m, 2H), 7.66–7.61 (m, 1H), 7.54–7.47 (m, 2H), 6.47 (s, 1H), 6.47 (brs, 1H), 4.16–4.08 (dd, J = 18.0, 6.0 Hz, 1H), 4.12 (s, 2H), 3.99–3.91 (dd, J = 18.0, 6.0 Hz, 1H), 3.73 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 190.3, 169.4, 166.5, 163.2, 134.5, 133.8, 129.8, 128.7, 76.7, 52.6, 50.0, 41.1.
Very viscous yellow oil (0.168 g, 90% yield); HRMS (ESI): m/z: calcd for C14H13N4O6Cl [M + H+]: 369.0596, found: 369.0604. 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.09–8.05 (m, 2H), 7.50–7.46 (m, 1H), 7.54–7.47 (m, 2H), 7.13 (brs, 1H), 6.42 (s, 1H), 4.16–4,08 (dd, J = 18.0, 6.0 Hz, 1H), 4.14 (s, 2H), 4.02–3.96 (dd, J = 18.0, 6.0 Hz, 1H), 3.95 (s, 3H). 13C-NMR (75 MHz, CDCl3) δ (ppm) 189.1, 169.4, 166.4, 163.0, 141.3, 132.1, 131.2, 129.1, 76.7, 52.7, 50.0, 41.1.
Very viscous brown oil (0.162 g, 93% yield); HRMS (ESI): m/z: calcd for C16H20N4O5 [M + Na+]: 371.1326, found: 371.1332. 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.15–8.12 (m, 2H), 6.98–6.96 (m, 2H), 6.24 (brs, 1H), 6.12 (s, 1H), 4.10 (s, 2H), 3.90 (s, 2H), 1.33 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 189.3, 166.3, 164.6, 162.1, 132.3, 126.9, 113.9, 77.5, 55.5, 52.2, 50.1, 28.5.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
CS was responsible for designing and performing the experiments and helped writing the publication. CA directed the project and wrote the publication.
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grant n° 423491/2016-4, and Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), grant n° 0193.000900/2015, for research funds. Universidade de Brasília for open access publication fees.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Universidade de Brasília, FAPDF, and CNPq for financial help. We also thank Prof. Laila Darvene from Universidade de Brasília for mass spectra.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00531/full#supplementary-material
Andrade, C. K. Z., and Dar, A. R. (2016). Applying green processes and techniques to simplify reaction work-ups. Tetrahedron 72, 7375–7391. doi: 10.1016/j.tet.2016.09.055
Barreto, A. F. S., and Andrade, C. K. Z. (2018). Microwave-mediated synthesis of a cyclic heptapeptoid through consecutive Ugi reactions. Tetrahedron 74, 6861–6865. doi: 10.1016/j.tet.2018.10.018
Barreto, A. F. S., and Andrade, C. K. Z. (2019). Synthesis of (macro)heterocycles by consecutive/repetitive isocyanide-based multicomponent reactions. Beilstein J. Org. Chem. 15, 906–930. doi: 10.3762/bjoc.15.88
Barreto, A. F. S., Vercillo, O. E., and Andrade, C. K. Z. (2011a). Microwave-assisted Passerini reactions under solvent-free conditions. J. Braz. Chem. Soc. 22, 462–467. doi: 10.1590/S0103-50532011000300008
Barreto, A. F. S., Vercillo, O. E., Birkett, M. A., Caulfield, J. C., Wessjohann, L. A., and Andrade, C. K. Z. (2011b). Fast and efficient microwave-assisted synthesis of functionalized peptoids via Ugi reactions. Org. Biomol. Chem. 9, 5024–5027. doi: 10.1039/C1OB05471F
Barreto, A. F. S., Vercillo, O. E., Wessjohann, L. A., and Andrade, C. K. Z. (2014). Consecutive isocyanide-based multicomponent reactions: synthesis of cyclic pentadepsipeptoids. Beilstein J. Org. Chem. 10, 1017–1022. doi: 10.3762/bjoc.10.101
Damn, M., Glasnov, T. N., and Kappe, C. O. (2010). Translating high-temperature microwave chemistry to scalable continuous flow process. Org. Process Res. Dev. 14, 215–224. doi: 10.1021/op900297e
De Souza, J. M., Galaverna, R., de Souza, A. A. N., Brocksom, T. J., Pastre, J. C., de Souza, R. O. M. A., et al. (2018). Impact of continuous flow chemistry in the synthesis of natural products and active pharmaceutical ingredients. Ann. Braz. Acad. Sci. 90, 1131–1134. doi: 10.1590/0001-3765201820170778
Dömling, A., Wang, W., and Wang, K. (2012). Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3185. doi: 10.1021/cr100233r
Du, J., Zhang, X., Suna, X., and Wang, L. (2015). Copper-catalyzed direct α-ketoesterification of propiophenones with acetophenones via C(sp3)–H oxidative cross-coupling. Chem. Commun. 51, 4372–4375. doi: 10.1039/C4CC09524C
Gabriel, C., Gabriel, S., Grant, E. H., Halstead, B. S. J., and Mingos, D. M. P. (1998). Dielectric parameters relevant to microwave dielectric heating. Chem. Soc. Rev. 27, 213–223.
Geyer, K., Codée, J. D., and Seeberger, P. H. (2006). Microreactors as tools for synthetic chemists-the chemists' round-bottomed flask of the 21st century? Chem. Eur. J. 12, 8434–8442. doi: 10.1002/chem.200600596
Glasnov, T. N., and Kappe, C. O. (2011). The microwave-to-flow paradigm: translating high-temperature batch microwave chemistry to scalable continuous-flow processes. Chem. Eur. J. 17, 11956–11968. doi: 10.1002/chem.201102065
Gutmann, B., Cantillo, D., and Kappe, C. O. (2015). Continuous-flow technology - a tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 54, 6688–6728. doi: 10.1002/anie.201409318
Heublein, N., Moore, J. S., Smith, C. D., and Jensen, K. F. (2014). Investigation of Petasis and Ugi reactions in series in an automated microreactor system. RSC Adv. 4, 63627–63631. doi: 10.1039/c4ra13626h
Hunter, A. C., Chinthapally, K., and Sharma, I. (2016). Rh2(esp)2: an efficient catalyst for O–H insertion reactions of carboxylic acids into acceptor/acceptor diazo compounds. Eur. J. Org. Chem. 2016, 2260–2263. doi: 10.1002/ejoc.201600064
Kappe, C. O., Pieber, B., and Dallinger, D. (2013). Microwave effects in organic synthesis: myth or reality? Angew. Chem. Int. Ed. 52, 1088–1094. doi: 10.1002/anie.201204103
Kappe, C. O., Stadler, A., and Dallinger, D. (2012). Microwaves in Organic and Medicinal Chemistry, 2nd Edn. Weinheim: Wiley-VCH.
Kazemizadeh, A. R., and Ramazani, A. (2012). Synthetic applications of Passerini reaction. Curr. Org. Chem. 16, 418–450. doi: 10.2174/138527212799499868
Ley, S. V., Fitzpatrick, D. E., Ingham, R. J., and Myers, R. M. (2015). Organic synthesis: march of the machines. Angew. Chem. Int. Ed. 54, 3449–3464. doi: 10.1002/anie.201410744
May, S. A., Johnson, M. D., Braden, T. M., Calvin, J. R., Haeberle, B. D., Jines, A. R., et al. (2012). Rapid development and scale-up of a 1H-4-substituted imidazole intermediate enabled by chemistry in continuous plug flow reactors. Org. Process Res. Dev. 14, 215–224. doi: 10.1021/op900297e
May, S. A., Johnson, M. D., Buser, Y. J., Campbell, A. N., Frank, S. A., Haeberle, B. D., et al. (2016). Development and manufacturing GMP scale-up of a continuous Ir-catalyzed homogeneous reductive amination reaction. Org. Process Res. Dev. 20, 1870–1898. doi: 10.1021/acs.oprd.6b00148
Neo, A. G., Carillo, R. M., Delgado, J., Marcaccini, S., and Marcos, C. F. (2011). A multicomponent approach to the synthesis of 1,3-dicarbonylic compounds. Mol. Divers. 15, 529–539. doi: 10.1007/s11030-010-9277-y
Neo, A. G., Delgado, J., Polo, C., Marcaccini, S., and Marcos, C. F. (2005). A new synthesis of β-keto amides by reduction of Passerini adducts. Tetrahedron Lett. 46, 23–26. doi: 10.1016/j.tetlet.2004.11.041
Plutschack, M. B., Pieber, B., Gilmore, K., and Seeberger, P. H. (2017). The hitchhiker's guide to flow chemistry. Chem. Rev. 117, 11796–11893. doi: 10.1021/acs.chemrev.7b00183
Prasad, P. K., Reddi, R. N., and Arumugam, S. (2018). Recent methods for the synthesis of α-acyloxy ketones. Org. Biomol. Chem. 16, 9334–9348. doi: 10.1039/c8ob02881h
Salvador, C. E. M., Pieber, B., Neu, P. M., Torvisco, A., Andrade, C. K. Z., and Kappe, C. O. (2015). A sequential Ugi multicomponent/Cu-catalyzed azide–alkyne cycloaddition approach for the continuous flow generation of cyclic peptoids. J. Org. Chem. 80, 4590–4602. doi: 10.1021/acs.joc.5b00445
Sharma, S., Maurya, R. A., Min, K.-I., Jeong, G.-Y., and Kim, D.-P. (2013). Odorless isocyanide chemistry: an integrated microfluidic system for a multistep reaction sequence. Angew. Chem. Int. Ed. 52, 7564–7568. doi: 10.1002/anie.201303213
Shindo, M., Yoshimura, Y., Hayashi, M., Soejima, H., Yoshikawa, T., Matsumoto, K., et al. (2007). Synthesis of multisubstituted furans, pyrroles, and thiophenes via ynolates Org. Lett. 9, 1963–1966. doi: 10.1021/ol0705200
Vasconcelos, S. N. S., Fornari, E., Caracelli, I., and Stefani, H. A. (2017). Synthesis of α-amino-1,3-dicarbonyl compounds via Ugi flow chemistry reaction: access to functionalized 1,2,3-triazoles. Mol. Divers. 21, 893–902. doi: 10.1007/s11030-017-9764-5
Wang, P., Tao, W.-J., Sun, X.-L., Liao, S., and Tang, Y. (2013). A highly efficient and enantioselective intramolecular Cannizzaro reaction under TOX/Cu(II) catalysis. J. Am. Chem. Soc. 135, 16849–16852. doi: 10.1021/ja409859x
Wu, D., Li, J., and Wang, W. (2018). Selective formation of monoacylated diols through a mild Passerini reaction. Eur. J. Org. Chem. 2018, 3022–3030. doi: 10.1002/ejoc.201800499
Yang, C., Mi, J., Feng, Y., Ngo, L., Gao, T., Yan, L., et al. (2013). Labeling lysine acetyltransferase substrates with engineered enzymes and functionalized cofactor surrogates. J. Am. Chem. Soc. 135, 7791–7794. doi: 10.1021/ja311636b
Keywords: continuous flow, α-acyloxy ketones, Passerini reaction, microwaves, multicomponent reactions
Citation: Salvador CEM and Andrade CKZ (2019) A Mild, Fast, and Scalable Synthesis of Substituted α-Acyloxy Ketones via Multicomponent Reaction Using a Continuous Flow Approach. Front. Chem. 7:531. doi: 10.3389/fchem.2019.00531
Received: 25 April 2019; Accepted: 11 July 2019;
Published: 30 July 2019.
Edited by:
Alexander Dömling, University of Groningen, NetherlandsReviewed by:
Valentine Nenajdenko, Lomonosov Moscow State University, RussiaCopyright © 2019 Salvador and Andrade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Kleber Z. Andrade, Y2tsZWJlckB1bmIuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.