 Asa W. Nichols
Asa W. Nichols Charles W. Machan
Charles W. Machan- Department of Chemistry, University of Virginia, Charlottesville, VA, United States
The generation of fuels and value-added chemicals from carbon dioxide (CO2) using electrocatalysis is a promising approach to the eventual large-scale utilization of intermittent renewable energy sources. To mediate kinetically and thermodynamically challenging transformations of CO2, early reports of molecular catalysts focused primarily on precious metal centers. However, through careful ligand design, earth-abundant first-row transition metals have also demonstrated activity and selectivity for electrocatalytic CO2 reduction. A particularly effective and promising approach for enhancement of reaction rates and efficiencies of molecular electrocatalysts for CO2 reduction is the modulation of the secondary coordination sphere of the active site. In practice, this has been achieved through the mimicry of enzyme structures: incorporating pendent Brønsted acid/base sites, charged residues, sterically hindered environments, and bimetallic active sites have all proved to be valid strategies for iterative optimization. Herein, the development of secondary-sphere strategies to facilitate rapid and selective CO2 reduction is reviewed with an in-depth examination of the classic [Fe(tetraphenylporphyrin)]+, [Ni(cyclam)]2+, Mn(bpy)(CO)3X, and Re(bpy)(CO)3X (X = solvent or halide) systems, including relevant highlights from other recently developed ligand platforms.
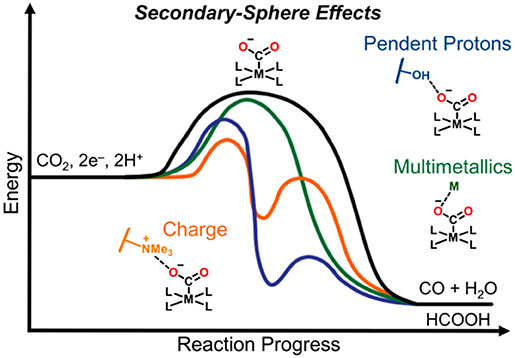
Graphical Abstract. Secondary-sphere strategies known to improve catalytic selectivity and activity include pendent proton donors/shuttles in the form of Brønsted sites, charged moieties, sterically demanding functional groups, bimetallic active species, and stoichiometric participation of alkali and alkaline earth metal cations.
Introduction
The development of scalable and cost-effective processes to store electrical energy in chemical bonds using CO2 as a primary feedstock remains a significant challenge for energy research (Centi and Perathoner, 2009; Senftle and Carter, 2017). Of particular interest are homogeneous catalysts for CO2 reduction, as their well-defined structures give chemists an opportunity to effectively characterize intermediates relevant to the operating mechanism and further optimize active catalyst families through iterative ligand design (Benson et al., 2009; Rountree et al., 2014). While the influence of different ligand types in the primary coordination sphere can be observed in the catalytic activity and selectivity of transition metals across the d-block towards CO2 reduction, more subtle effects—including rate enhancement and lowered overpotentials—can be obtained through modulation of the secondary coordination sphere within specific classes of metal complexes (Jiang et al., 2019). This has been particularly successful in the design of highly active and selective catalysts for CO2 reduction, directing a paradigmatic shift in the general understanding of “design principles” (Costentin et al., 2012a, 2014a,b; Sampson et al., 2014; Azcarate et al., 2016; Ngo et al., 2017).
While this review focuses on molecular catalysts which were specifically examined for electrochemical CO2 reduction, secondary-sphere effects have been successfully harnessed in related catalytic processes, including thermal CO2 hydrogenation (Himeda et al., 2004, 2005, 2007; Hull et al., 2012; Wang et al., 2012, 2013, 2014; Manaka et al., 2014; Suna et al., 2014; Cammarota et al., 2017), hydrogen evolution (Curtis et al., 2003; Henry et al., 2005, 2006; Wilson et al., 2006; Fraze et al., 2007; Jacobsen et al., 2007a,b; DuBois and DuBois, 2009; Gloaguen and Rauchfuss, 2009; Helm et al., 2011; Reback et al., 2013; Ginovska-Pangovska et al., 2014), hydrogen oxidation (Curtis et al., 2003; Henry et al., 2005, 2006; Wilson et al., 2006; Fraze et al., 2007; Jacobsen et al., 2007a,b; Dutta et al., 2013, 2014; Ginovska-Pangovska et al., 2014), formate oxidation (Galan et al., 2011, 2013; Seu et al., 2012), and oxygen reduction (Collman, 1977; Collman et al., 2004; Lewis and Tolman, 2004; Mirica et al., 2004; Fukuzumi, 2013; Ray et al., 2014; Fukuzumi et al., 2015; Nam, 2015; Sahu and Goldberg, 2016; Elwell et al., 2017; Hong et al., 2017; Sinha et al., 2019) reactions. In this review, we focus on how the mechanism of CO2 reduction relates to the type of secondary-sphere effects employed in molecular systems. Strategies discussed here which have been shown to increase catalytic activity and selectivity include pendent proton donors/shuttles in the form of Brønsted sites, charged moieties, sterically demanding functional groups, bimetallic active species, and stoichiometric participation of alkali and alkaline earth metal cations (Graphical Abstract).
To contextualize the motivation and principles, examples of secondary-sphere effects in enzymes which catalyze the interconversion of CO2 with either CO or formic acid are discussed. This overview is followed by a careful examination of secondary-sphere effects in several abiotic molecular electrocatalyst examples, beginning with the [Fe(tetraphenylporphyrin)]+ [Fe(TPP)]+ systems pioneered by Savéant, Robert, and Constentin, including a discussion of the effects of pendent proton source placement and the distance dependence of through-space effects induced by charged residues (Costentin et al., 2014a,b; Manbeck et al., 2015; Mohamed et al., 2015; Azcarate et al., 2016; Zahran et al., 2016; Margarit et al., 2018; Nichols et al., 2018b; Sinha and Warren, 2018). Next, M(bpy)(CO)3X catalysts (M = Mn or Re; X = solvent molecule or halide) (Wong et al., 1998; Bourrez et al., 2011; Smieja et al., 2013; Chabolla et al., 2014, 2017; Franco et al., 2014; Machan et al., 2014a, 2015, 2016; Riplinger et al., 2014; Agarwal et al., 2015; Manbeck et al., 2015; Riplinger and Carter, 2015; Machan and Kubiak, 2016; Ngo et al., 2017; Sahu et al., 2017; Sinha et al., 2019) in which steric parameters, pendent Lewis acid effects, and charged residues have been shown to be effective will be discussed. Finally, [Ni(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane), which contains pendent proton donors on the coordinating N atoms of the macrocycle, is discussed (Beley et al., 1984; Barefield et al., 1986; Balazs and Anson, 1993; Kelly et al., 1999; Froehlich and Kubiak, 2012; Song et al., 2014; Nichols and Chang, 2018). These systems are among the most highly studied in the field and representative of the progress that has been made in understanding how secondary-sphere coordination effects enhance molecular electrocatalysis. Additional discussions on emerging systems for CO2 reduction which utilize secondary-sphere effects are included to summarize some of the current work in the field.
Enzymes for the Interconversion of CO2 and CO or Formic Acid
Cu,Mo-Containing Carbon Monoxide Dehydrogenase (Cu,Mo-CODH)
The structure and function of Cu,Mo-CODH enzymes have been previously reviewed in great detail (Appel et al., 2013; Kroneck and Torres, 2014). Of this class of enzyme, the Cu,Mo-CODH in O. carboxydovorans has been the most extensively studied (Figure 1A) (Dobbek et al., 2001a, 2002; Hille, 2013). The active site of this enzyme contains Mo and Cu ions, which are bridged by a μ2-sulfido ligand. Mo is coordinated in a distorted square pyramidal fashion by an enedithiolate moiety (from the pyran ring found in the pyranopterin cofactor), a μ2-sulfido ligand, and an oxo/hydroxo ligand in the equatorial plane. The apical ligand is an oxo, with a glutamine residue within hydrogen-bonding distance and a glutamate residue in a trans position. A unique structural feature of Cu,Mo-CODH in comparison to other Mo-containing hydrogenases is its covalent linkage through a cysteine residue to the Cu ion, connecting the heterobimetallic active site to the large subunit of the CODH enzyme.
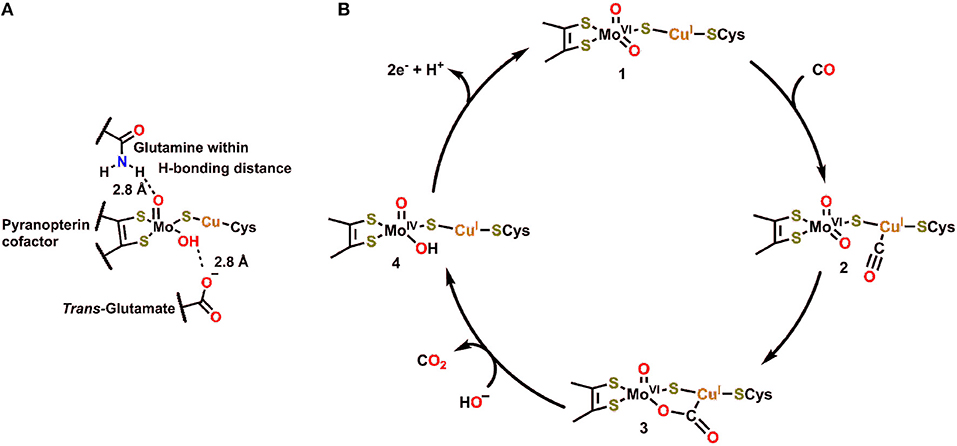
Figure 1. (A) Active site of Cu,Mo-CODH. (B) Proposed catalytic cycle for oxidation of CO by Cu,Mo-CODH (Dobbek et al., 2002; Kroneck and Torres, 2014).
In the catalytic cycle for transformation of CO to CO2 by Cu,Mo-CODH, CO is proposed to initially bind to the Cu(I) ion 2, followed by nucleophilic attack by the equatorial oxo ligand to generate the bridging μ2-η2-CO2 adduct 3 (Figure 1B) (Dobbek et al., 2002). This five membered metallacycle results from a redox-neutral reaction which maintains the Mo(VI) state (Figure 1B). Following rearrangement and net oxidation of CO, CO2 release occurs with hydroxide binding to produce the Mo(IV) species 4. Subsequent 2e− oxidation returns the Mo active site to the initial Mo(VI) oxidation state. Notably, the glutamine residue, which is in contact with the equatorial oxo ligand (Figure 1B), can act as a Brønsted base (proton acceptor) to regenerate the more nucleophilic oxo ligand when transitioning from 4 to 1. If the reverse of this catalytic cycle is imagined, it is clear that a proton-donating residue would be beneficial for CO2 reduction and C–O bond cleavage to produce CO.
Monofunctional Ni,Fe-Containing CODHs
The active site of Ni,Fe-containing CODHs, known as cluster C, is proposed to cycle between three separate redox states during catalytic CO oxidation (Cred1, Cint, and Cred2; Figure 2) (Jeoung and Dobbek, 2007). The behavior and activity of cluster C from Carboxydothermus hydrogenoformans have been experimentally interrogated in these three different redox states utilizing chemical-reducing agents (Jeoung and Dobbek, 2007). The cofactor contains an Fe4Ni cluster bridged by sulfide ligands with a single Fe and Ni atom in the active site, which also contains histidine and lysine residues in close proximity to interact with active site-bound substrate molecules. In Cred1, the active site contains Fe2+ and Ni2+ ions with an Fe-bound hydroxo ligand that is within hydrogen-bonding distance of the proximal lysine residue. Upon exposure to CO, the Fe-bound hydroxide is deprotonated, and the resultant oxo species can form a new C–O bond to generate the μ2-η2-CO2 adduct Cred2-CO2, where CO2 is bound through C by Ni and O by Fe (Figure 2) (Jeoung and Dobbek, 2007). This μ2-η2-CO2 binding mode is stabilized by hydrogen-bonding interactions with the pendent histidine and lysine residues (Jeoung and Dobbek, 2007). A 2e− oxidation with concomitant binding of an aquo ligand reduces the Ni2+ to Ni0 in Cred1, triggering release of CO2. Oxidation of the reduced cofactor by 2e− regenerates the Cred1 catalytic resting state.
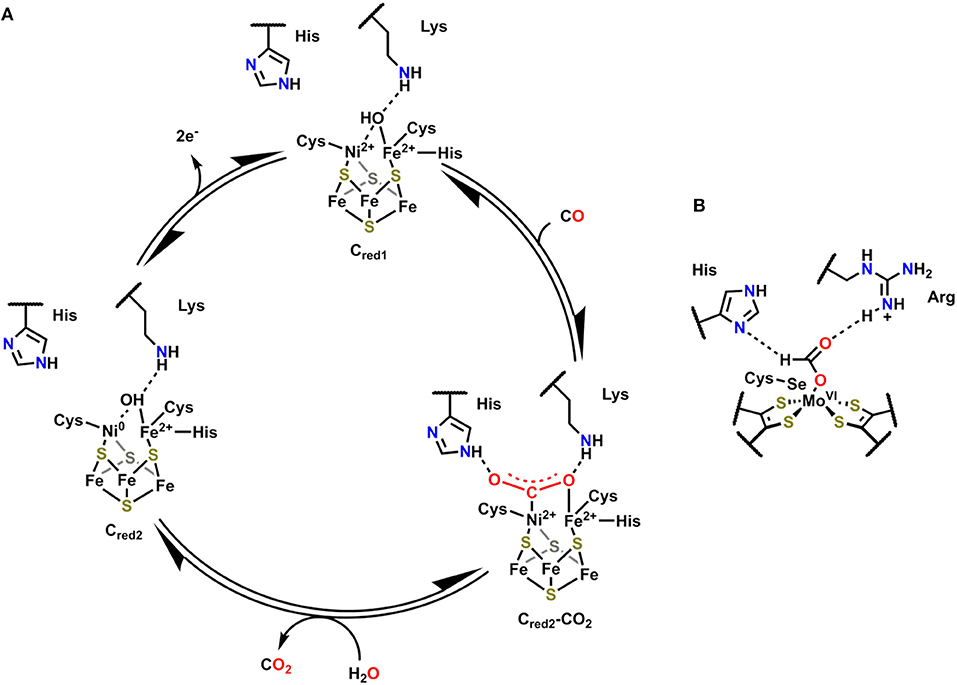
Figure 2. (A) Proposed mechanism for CO oxidation by Ni,Fe CODH (Jeoung and Dobbek, 2007). (B) Active site of FADH in Escherichia coli with bound formate substrate (Boyington et al., 1997).
Formic Acid Dehydrogenase
Formic acid dehydrogenase (FADH) enzymes catalyze the reversible 2e−/1H+ interconversion of CO2 and formate, another reaction of interest to CO2 utilization (Sawers, 1994). The active site of the FADH enzyme in Escherichia coli contains a single Mo(VI) atom coordinated to four S atoms from two cis-dithiolene moieties originating from the bis(molybdopterin guanine dinucleotide) cofactor, a hydroxyl ligand, and a SeCysteine (Boyington et al., 1997). The active site also contains histidine and arginine residues in a position to interact with activated Mo-bound intermediates (Figure 2B). The catalytic oxidation of formate begins by displacement of the hydroxyl ligand with an equivalent of formate, which binds as an η1-OCHO ligand; the η1-O binding mode is stabilized through hydrogen-bonding interactions between the arginine and histidine residues and the unbound H and O atoms of formate (Figure 2B). Subsequent oxidation and transfer of 2e− from formate to the Mo center occurs with the release of CO2 and proton transfer to the SeCysteine residue. The Mo(IV) center can then be returned to the resting state through the loss of 2e− as the histidine deprotonates the Mo-bound SeCysteine. Abiotic structural motifs similar to these three examples are discussed in the subsequent sections where either a) bimetallic active sites or b) secondary-sphere moieties are used to mediate electrochemical CO2 reduction.
Benchmarking Molecular Electrocatalysts for CO2 Reduction
Electrochemical techniques can facilitate the determination of kinetic and thermodynamic parameters for both Faradaic and catalytic reactions. Cyclic voltammetry (CV), a nondestructive potentiostatic technique, is particularly informative. Indeed, the breadth and importance of rigorous CV analysis have been reviewed in great detail (Savéant, 2008, 2018; Costentin et al., 2012b; Costentin and Savéant, 2014, 2018a; Rountree et al., 2014). Experimental determination of fundamental reaction parameters is essential for comparing the effects of pendent functional groups on the activity of electrocatalysts.
The effective catalytic overpotential (η) is the difference between the standard potential of CO2 reduction () and the potential at half catalytic current height (Ecat/2) as described in Equation (1) and describes a thermodynamic quantity: the electrochemical energy beyond the standard potential which is required to drive a reaction of interest at an appreciable rate. We note that some prefer to define the overpotential term as ECO2−E1/2. We distinguish between these through the use of “effective catalytic overpotential” to describe the overpotential calculated utilizing Ecat/2 (Appel and Helm, 2014).
Another reaction parameter commonly measured through CV is the maximal turnover frequency (TOFmax), which can also be described as the observed catalytic rate constant (kobs) with units of s−1 (Costentin and Savéant, 2014). In a Nernstian electrocatalytic reaction, TOF is related to overpotential by the catalytic Tafel equation, Equation (2). TOF is the turnover frequency at the applied potential, F is Faraday's constant, R is the ideal gas constant, T is the temperature, E1/2 is the catalyst standard reduction potential, and ηapp is the difference between and the applied potential. It is worth noting, however, that the limitations of the molecular catalytic reactions, including diffusion and side phenomena, can cause ‘peaks’ in catalytic CV waves and truncate the region where the Tafel relationship results in increased activity as the applied potential becomes more negative.
To benchmark the kinetic and thermodynamic parameters of different electrocatalysts, Equation (2) can be used to generate catalytic Tafel plots (nota bene, these explicitly include the limitations imposed on molecular Tafel behavior mentioned above and enable comparison across different experimental conditions). Figure 3 shows an example of a catalytic Tafel plot for a generic molecular catalyst: “better” catalysts are located at the top left of catalytic Tafel plots, where overpotential is low and TOFmax is large and “worse” catalysts are located at the bottom right of a catalytic Tafel plot where the trends in overpotential and TOFmax are reversed. Figure 4 shows a Tafel plot comparing reported catalysts.
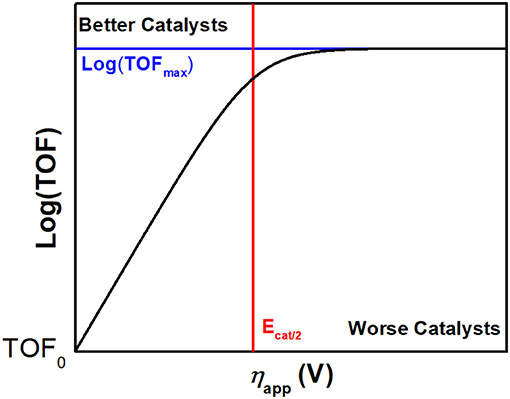
Figure 3. Example of a catalytic Tafel plot of a generic molecular species with regions where better and worse catalysts are located explicitly labeled, along with the position of important benchmarking parameters.
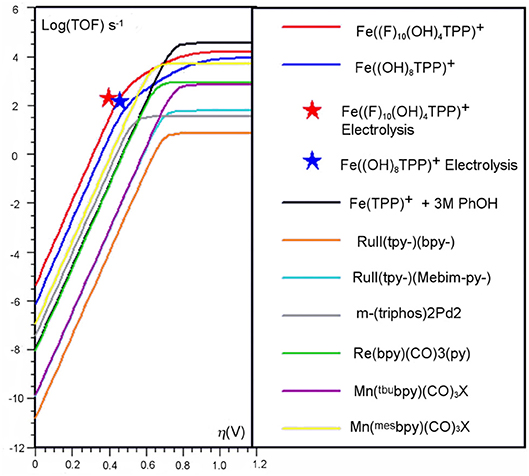
Figure 4. Catalytic Tafel plot showing enhancements from the inclusion of a secondary coordination sphere in the [Fe(TPP)]+ system (Costentin et al., 2014b).
Finally, simple plots of log(TOFmax) vs. Ecat/2 can be used to compare catalysts in the same family to look for secondary-sphere effects (Pegis et al., 2017; Costentin and Savéant, 2018b; Nichols et al., 2018b). If there is no secondary-sphere effect, then a linear scaling relationship based on electrochemical driving force should occur; however, if a secondary-sphere effect is present, a deviation from the linearity achieved by comparing inductive substitution effects can be observed. One should utilize caution using this method for comparing catalysts, however, as changes to the reaction mechanism that alter cosubstrate identity, concentration dependence, solvent, and cosolvent effects can greatly impact the catalytic activity of a series of complexes through scaling relationships that are unrelated to the secondary-sphere interactions of interest. Likewise, the effects of Nernstian changes in the experimentally observed potential based on changes in the reaction equilibrium, Keq, can obscure details if not properly accounted for (Costentin and Savéant, 2018b).
Secondary-Sphere Effects in [Fe(TPP)]+
For [Fe(TPP)]+ complexes, increased activity for the electrocatalytic reduction of CO2 to CO is observed with the addition of both Lewis and Brønsted acids (Bhugun et al., 1994, 1996; Gennaro et al., 1996). As a result, [Fe(TPP)]+ complexes bearing secondary-sphere functionalities exploiting classical Brønsted acid/base push–pull reactions have been the subject of extensive studies (Costentin et al., 2012a, 2014a,b; Sinha and Warren, 2018). In particular, iron 5, 10, 15, 20-tetrakis-(2,6-dihydroxyphenyl)-porphyrin ([Fe((OH)8TPP)]+) was identified as a promising candidate for electrochemical CO2 reduction because it positioned proton donors oriented toward the active site in close enough proximity to interact with bound substrate. Experimentally, the pendent –OH moieties were observed to cause a large catalytic current enhancement and a decrease in overpotential (Costentin et al., 2012a). Control experiments using [Fe((OMe)8TPP)]+, where the –OH groups were converted to –OMe ether moieties, also showed an increased TOFmax relative to [Fe(TPP)]+; however, the overpotential was much larger than both [Fe(TPP)]+ and [Fe((OH)8TPP)]+. These differences relate to the mechanism of CO2 reduction by [Fe(TPP)]+ and how key steps are affected by the presence of the pendent –OH moieties (Bonin et al., 2017).
The proposed mechanism for the reduction of CO2 by [Fe(TPP)]+ requires the electrochemical generation of [Fe(0)TPP]2− at the electrode surface, followed by CO2 binding and activation to generate [Fe(I)(TPP)(η1-)]2−. The sacrificial proton donor, AH, stabilizes the binding of the radical anion through hydrogen bonding. Concerted protonation and electron transfer from the metal center cause C–O bond cleavage, leading to the formation of [Fe(II)(TPP)(CO)]0. Release of CO is facilitated by a comproportionation reaction with a second equivalent of [Fe(0)TPP]2− to generate two equivalents of [Fe(I)TPP]− and one equivalent of CO. We note that a second mechanism has been proposed for Fe(TPP), wherein upon CO2 binding, the metal center is oxidized by 2e− from Fe(0) to Fe(II) in the η1-CO2 adduct before interaction with the proton donor, rather than generating an Fe(I) species (Fukuzumi et al., 2018).
The introduction of a pendent proton source minimally alters the mechanism for CO2 reduction but causes catalytic rate increases by favoring several steps of the reaction. Upon the generation of [Fe(0)((OH)8TPP)]2−, CO2 also binds in η1 fashion and is activated to the radical anion, but stabilization occurs through hydrogen-bonding interactions with the pendent proton donors (Costentin et al., 2012a). Experimentally, this is observed as a prewave to the catalytic feature in CV experiments, which can be more easily examined through additional modulation of electron density at the metal center with the related partially fluorinated derivative [Fe((F)10(OH)8TPP)]+ (Costentin et al., 2014a,b). Subsequent intramolecular protonation balanced by the exogenous proton source can occur, generating η1-•CO2H at an Fe(I) center (Costentin et al., 2014a). The cleavage of the C–OH bond is induced upon further reduction and a concerted intramolecular protonation reaction (again balanced by proton transfer from the sacrificial donor) to regenerate the resting state, Fe(I), of the electrocatalytic cycle and CO product. The enhanced catalytic activity at more negative potentials of the ether-containing control complex may also be explained through an enhancement of this mechanism (the ether groups function as a Brønsted base to orient and enhance the proton activity of the exogeneous proton donor), but this has a relatively lower enhancement effect on the CO2 reduction reaction. A comparison of the catalytic activity increases from these effects can be seen through the catalytic Tafel analysis shown in Figure 4.
Another area of study for secondary-sphere effects focuses on mono-functionalized porphyrins, which also orient functional groups toward the metal active site. Both the positioning of these functional groups relative to the active site and the pKa of the pendent proton source are of importance to the electrocatalytic activity for CO2 reduction. In a study by Chang and co-workers the positional effects of pendent amide groups were investigated. It was found that orientation toward the active site and positioning above the active site were both important for catalytic enhancement, as these stabilized catalytic intermediates through hydrogen bonding and facilitated efficient proton transfer (Nichols et al., 2018b). This was quantified experimentally through equilibrium binding constants for CO2 determined by rapid-scan CV techniques. When a pendent amide group was attached to the meso-phenyl of the porphyrin in the ortho position, [(Fe(o-2-amide-TPP)]+, the highest catalytic activity was observed. In comparison with the other reported derivatives, a “Goldilocks” relationship was observed, where positioning the amide group either closer or farther did not result in comparable catalytic current enhancement (Figure 5).
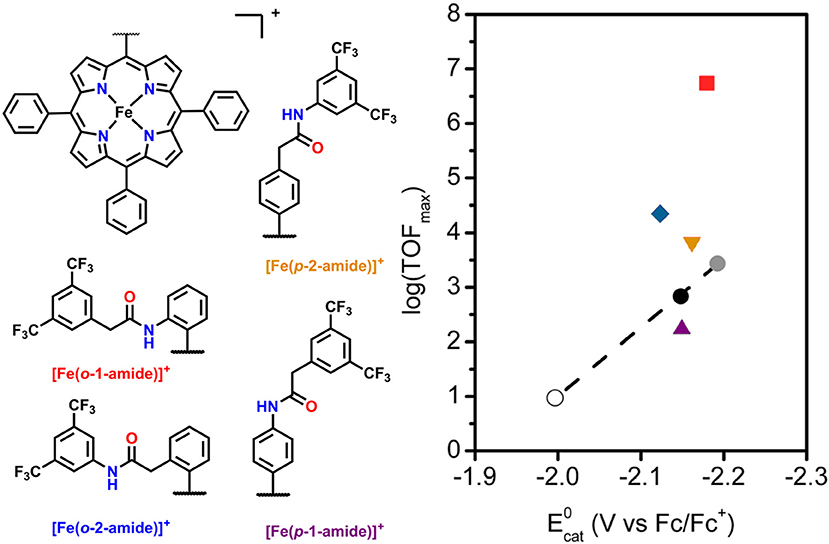
Figure 5. Plot of log(TOFmax) vs. for mono-functionalized porphyrins used to study positional dependence of catalytic enhancement for this system. Circles are [Fe(TPP)]+ derivatives which utilize electronic substitution of the phenyl rings for modulation of the redox potential of the FeI/0 couple. The upward purple triangle is [Fe(p-1-amide-TPP)]+, the downward orange triangle is [Fe(p-2-amide-TPP)]+, the blue diamond is [Fe(o-1–amide-TPP)]+, and the red square is [Fe(o-2-amide-TPP)]+. Reprinted with permission from Nichols et al. (2018b)—published by the Royal Society of Chemistry.
Nocera and co-workers found the pKa of the pendent proton source was important for the stabilization of CO2 binding in the active site of related “hangman-type” porphyrin architectures (Figure 6) (Margarit et al., 2018). Pendent phenol- and guanidinium-based hangman functional groups were predicted by DFT to cause a 2.1–6.6 kcal/mol stabilization of bound CO2 within the hangman pocket. Conversely, a sulfonic acid derivative was found to function as a proton donor under the experimental conditions in the absence of applied potential, which was attributed to an estimated pKa of ~3 under experimental conditions. The resultant anionic sulfonate derivative showed diminished catalytic activity relative to the porphyrins functionalized with phenol and guanidinium. The anionic charge of the conjugate base, combined with the overall steric bulk of the sulfonate, was proposed to prevent CO2 binding within the hangman cleft and contribute minimally to the stabilization of the CO2 adduct (Margarit et al., 2018). The pendent phenol-based “hangman-type” architecture was predicted to have the greatest stabilization of CO2 binding by DFT calculations and was observed to have the fastest catalytic rate constant by CV methods.
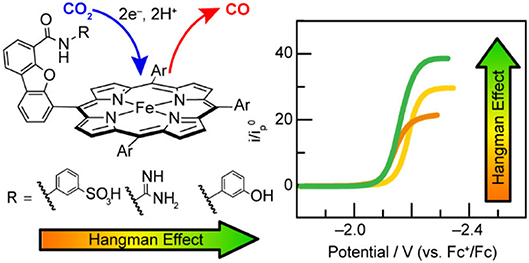
Figure 6. Figure showing effect of pKa of the hanging group on CO2 reduction activity in hangman [Fe(TPP)]+. Reprinted with permission from Margarit et al. (2018). Copyright 2018 American Chemical Society.
Others have examined the relationship between electrocatalytic activity and the H-bonding ability of pendent residues on triazole-based picket-fence-type [Fe(TPP)]+ derivatives with pendent amide functional groups (Sen et al., 2019). In this report, a correlation between log(TOFmax) and the pKa of pendent proton donors was established for a picket fence amide with an encapsulated water molecule in comparison to previously reported [Fe(TPP)]+ derivatives. The pendent groups are proposed to contribute to the stabilization of the Fe-bound CO2 molecule through hydrogen-bonding interactions and facilitate proton transfer to mediate the rate-determining C–O bond cleavage step.
The studies discussed above have shown that pendent protons can be beneficial for [Fe(TPP)]+-based electrocatalysts, but also that careful consideration of steric constraints and the pKa of the chosen pendent proton donor under experimental conditions is necessary. Installation of pendent proton groups in porphyrin ligands can have a detrimental effect if their pKa and spatial orientation are not chosen carefully (e.g., sulfonic acid, vide supra). A recent study has also shown that the choice of solvent can largely alter the catalytic response of [Fe(TPP)]+ with an asymmetrically substituted pendent proton moiety (Sinha and Warren, 2018). Using a porphyrin containing a single pendent hydroxy functionality in the 2-position of a single meso-phenyl ring, it was demonstrated that the electrocatalytic activity of this system can be greatly hindered by utilizing solvents with strong hydrogen-bonding properties through a comparison of activity in MeCN, DMF, and DMSO. Strong hydrogen bond acceptor solvents like DMF and DMSO lead to a decrease in activity of the catalyst, while in MeCN, the activity approached that reported for [Fe((ortho-TMA)TPP)]5+, which is the fastest molecular electrocatalyst for CO2 reduction reported to date. The authors postulated that the interference of competitive hydrogen bonding between solvent molecules leads to slowed electrochemical kinetic parameters (Sinha and Warren, 2018). This suggests that more rigorous interrogations of functional group cooperativity (specifically the number and type of functional groups) and the interactions of functional groups with solvent and proton donors will offer additional insight into these mechanisms. Indeed, Costentin and Savéant described the origin of catalytic enhancement by pendent proton relays (Costentin and Savéant, 2018b). Boosting of electrocatalytic activity by pendent proton donors occurs when the forward rate constant of proton transfer from the pendent group to the active site is competitive with that of exogeneous proton donor directly to the active site (under the assumption that reprotonation of the pendent donor is extremely rapid). Should these forward rate constants not be well-matched, inefficiency in the proton relay mechanism will limit the ultimate catalytic current enhancement.
Further studies on this class of catalysts have modulated the potential of the FeI/0 reduction where the catalytic response occurs by examining electron-withdrawing perfluorophenyl-substituted tetraphenylporphyrins ([Fe(F5TPP)]+, [Fe(F10TPP)]+, and [Fe(F20TPP)]+, where F5 corresponds to a single perfluorinated phenyl ring, etc.) (Azcarate et al., 2016). This is a purely electronic effect, perturbing the continuum of electronic distribution in the ligand–metal manifold. The inclusion of proximal ionic charges has also been explored with trimethylanilinium- ([Fe(-ortho-TMA-TPP)]5+ and [Fe(-para-TMA-TPP)]5+) and sulfonato-functionalized porphyrins ([Fe(-para-sulfonato-TPP)]3−) to understand the role of through-space electrostatic interactions (Azcarate et al., 2016). The perfluorinated derivatives demonstrated a relationship between the catalytic response and the FeI/0 potential: as the potential of that redox couple becomes more positive, log(TOFmax) decreases in a linear fashion, corresponding to a relative decrease in added electron density at the metal center upon electrochemical reduction (Azcarate et al., 2016; Costentin et al., 2018). However, for [Fe(TPP)]+ substituted with a charged functional group (para–, para–, and ortho–), log(TOFmax) increases linearly as the redox potential of the FeI/0 couple shifts to more positive potentials. [Fe-ortho-TMA-TPP]5+ has the most positive FeI/0 redox potential of any [Fe(TPP)]+-based electrocatalyst reported to date, as well as largest log(TOFmax) value. To understand this, one can once again imagine the CO2 binding mode wherein a single electron generates radical anion, which is stabilized by the charged groups close to the active site. It is compelling that such dramatic enhancements should be observed, but this could suggest that the reaction pathway might also be significantly altered from other [Fe(TPP)]+ derivatives, as the effect is an inverse scaling relationship to that predicted for purely inductive reasons (Azcarate et al., 2016).
The data points from charge-based functionalization are relatively limited; the proposal of design principles for new systems for molecular CO2 reduction systems requires additional information on these effects. For instance, the synthesis of further positional isomers of charged systems could give more evidence that a scaling relationship exists based on the distance between the charged moiety and the active site. Ultimately, it is clear that the inclusion of charge should seek to explore deliberate manipulation of the known mechanism to achieve the greatest enhancement effect.
Re(bpy)(CO)3X
Re(bpy)(CO)3X (where bpy is a 2,2′-bipyridine, often additionally functionalized in a symmetric fashion, and X is a halide anion or solvent molecule) is active for the electrocatalytic reduction of CO2 to CO in near quantitative fashion (Hawecker et al., 1984; Grice and Kubiak, 2013). Under Faradaic conditions, this complex is proposed to undergo reduction according to Figure 7. Initial reduction is localized at the bpy ligand, followed by loss of Cl− from [Re(bpy)(CO)3Cl]− via an overall EC mechanism to generate the neutral five-coordinate species [Re(bpy)(CO)3]0. At this stage, two separate mechanisms for reduction can occur: 1) a single reduction assigned to the ligand framework can occur, generating the catalytically active monoanionic species [Re(bpy)(CO)3]−. Commonly, the [Re(bpy)(CO)3Cl]− species is stable long enough on the CV timescale that [Re(bpy)(CO)3]− forms instead at the second reduction with Cl− loss. 2) Following initial single-electron reduction and Cl− loss, a Re–Re bond between two equivalents of [Re(bpy)(CO)3]0 can form to generate Re(bpy)(CO)3]2. Dimer formation requires two sequential reductions to cleave the Re–Re bond and form the [Re(bpy)(CO)3]− active species. The formation of the metal–metal bond is slow under most conditions because of the persistence of [Re(bpy)(CO)3Cl]− on the CV timescale and is most often outcompeted by the unimolecular pathway to [Re(bpy)(CO)3]− (Grills et al., 2014).
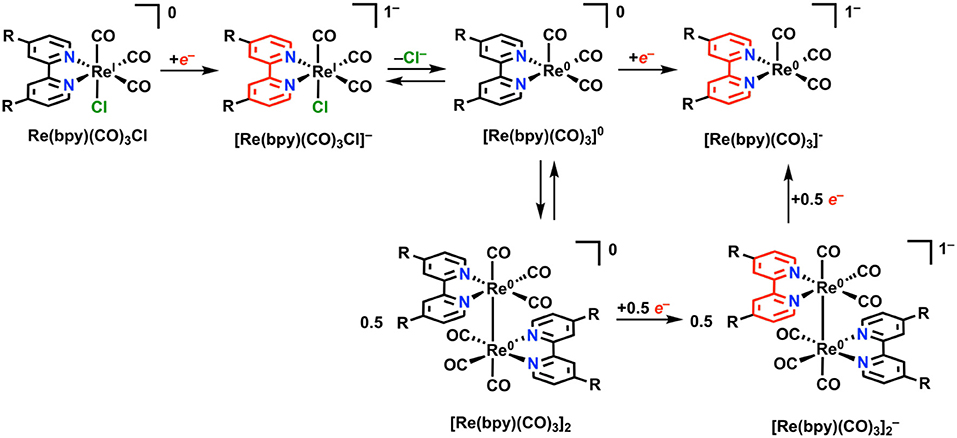
Figure 7. Faradaic reduction of Re(bpy)(CO)3X. Adapted with permission from Machan et al. (2014b). Copyright 2014 American Chemical Society.
There are also two possible mechanisms for CO2 reduction under electrocatalytic conditions. First, a relatively slower bimolecular process involving two equivalents of [Re(bpy)(CO)3]0 can occur, resulting in a net reductive disproportionation of two equivalents of CO2 into one each of CO and (Sullivan et al., 1985). Second, a unimolecular catalytic process with [Re(bpy)(CO)3]− as the resting state, where the two-electron reduction of CO2 occurs at a single Re center (Sullivan et al., 1985; Keith et al., 2013). As is observed with the [Fe(TPP)]+-based electrocatalysts above, there are observable enhancements in TOFmax and catalyst stability for CO2 reduction upon the addition of a sacrificial proton donor (Wong et al., 1998).
In the proposed mechanism for electrocatalytic CO2 reduction, a monoanionic five-coordinate species [Re(bpy)(CO)3]− is invoked as the active species which binds CO2 (Figure 8) (Keith et al., 2013). This has been validated experimentally through direct synthesis of [Re(bpy)(CO)3]− using chemical-reducing agents, with subsequent spectrochemical studies demonstrating a kinetic preference for CO2 over H+ (Smieja et al., 2012; Sampson et al., 2013). Re(bpy)(CO)3(η1-CO2H) is initially formed upon the binding and activation of CO2 by [Re(bpy)(CO)3]− with a proton donor present. Further reduction generates an anionic species, [Re(bpy)(CO)3(η1-CO2H)]−, at which point C–O bond cleavage is facilitated by an exogenous proton source to generate water and the neutral species, [Re(bpy)(CO)4]0. CO release from the 19e− complex [Re(bpy)(CO)4]0 is facile, and additional reduction regenerates the resting [Re(bpy)(CO)3]− state. In the Re(bpy)(CO)3X catalyst family, most work has focused on modulating the steric and electronic properties of the bipyridine ligand in attempts to modulate the reducing power and activity of Re. Generally, the use of electron-rich bpy ligands like 4,4′-di-tert-butylbpy enhances the activity of the catalyst by creating a more nucleophilic Re center upon reduction, albeit at larger overpotentials (Clark et al., 2018).
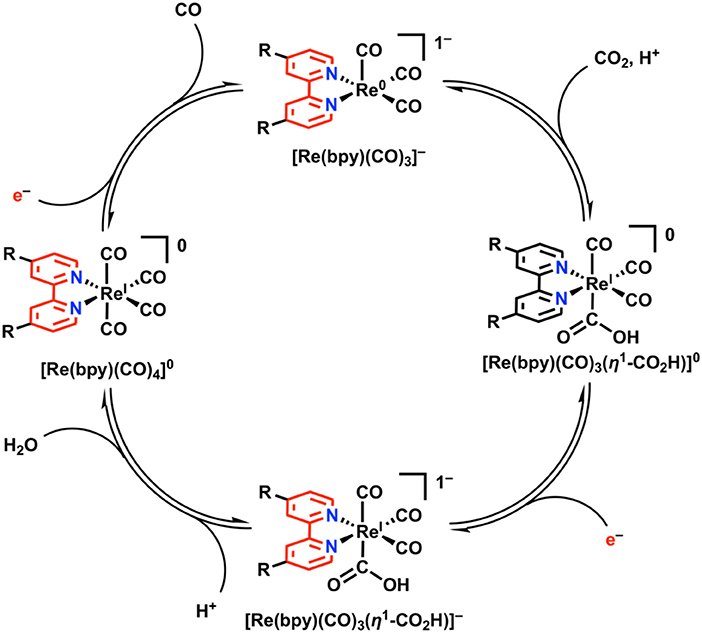
Figure 8. 2e−/2H+-dependent catalytic cycle for Re(bpy)(CO)3X in the reduction of CO2 to CO. Adapted with permission from Machan et al. (2014b). Copyright 2014 American Chemical Society.
Kubiak and co-workers have demonstrated, however, that steric considerations can supersede electronic effects in this class of catalysts. Re(bpy)(CO)3X-type complexes were synthesized with 3,3′- and 5,5′-dimethyl bpy (Chabolla et al., 2014). The methyl groups in those ring positions have identical Hammett parameters, so for purely electronic reasons, their respective electrocatalytic activities with a [Re(CO)3]+ core would be expected to be similar. When compared to the unsubstituted parent compound, however, the 5,5′-dimethyl bpy complex shows increased catalytic current at slightly more negative potentials, while the 3,3′-dimethyl bpy shows decreased catalytic current at nearly the same potential. This is rationalized through the steric hindrance from the methyl groups at the 3,3′-positions, which is observed in the non-planarity of the bpy ligand in the crystal structure of the precatalyst. Previous experimental and computational studies have postulated that a key mechanistic component is the participation of π* orbitals in the bpy ligand in the redox mechanism, suggesting that an inability to achieve a higher degree of planarity upon reduction diminishes catalytic activity for 3,3′-dimethyl bpy-based Re catalysts (Smieja et al., 2012; Benson et al., 2013; Keith et al., 2013; Chabolla et al., 2014). Since the distribution of added electron density between the bpy ligand and the Re metal center has been identified as important to the selectivity for CO2 over H+ as the electrophile of choice; it is noteworthy that an steric perturbation of the HOMO of the active species should have this effect.
The effect of secondary-sphere modifications on the Re(bpy)CO3X system was explored through the incorporation of –OH groups at the 4,4′- and 6,6′-positions of the bpy, with the goal of using them both as a pendent proton source and to contribute electron density to the bpy ligand (Manbeck et al., 2015). In this case, the authors found atypical behavior during electrocatalysis: the O–H bonds of the bipyridine ligand are cleaved by stepwise electrochemical reduction, which they propose leads to dearomatization of the doubly reduced bpy ligand. By isolating the deprotonated species in a chemical fashion, the authors were able to give both UV-vis and IR spectroscopic evidence—which aligned well with infrared spectroelectrochemistry (IR-SEC), UV-vis spectroelectrochemistry (UV-vis-SEC), and DFT calculations—supporting this hypothesis. Interestingly, only the 4,4′-dihydroxybpy complex is active as an electrocatalyst for CO2 reduction, while the 6,6′-dihydroxybpy complex completes only ~1 TON for CO, despite having nearly identical electrochemical properties under Faradaic conditions by CV. This result suggests slow CO release and decomposition of intermediates at applied potentials limit activity for the 6,6′-dihydroxybpy derivative.
Further work with monomeric Re(bpy)(CO)3X electrocatalysts showed promising results with the incorporation of peptide linkages of varying lengths containing proton relays and hydrogen-bonding groups on the bpy backbone (Chabolla et al., 2017). Through electrochemical experiments, 2D NMR spectroscopy, and molecular dynamics modeling, the study showed that an optimal chain length of five peptides allows for the peptide backbone to adopt conformations which allow for intramolecular interactions on the NMR timescale. Importantly, this study showed the Re(bpy)(CO)3X system to be stable under peptide synthesis conditions, allowing for insertion of the complex into peptide linkages at any desired point (Chabolla et al., 2017).
The asymmetric incorporation of a thiourea tether into Re(bpy)(CO)3X systems has been shown to be a successful strategy for enhancing the CO2 reduction activity (Haviv et al., 2018). Interestingly, the sulfur atom of the thiourea tether was shown to bind CO2 prior to reduction of the Re catalyst, which is expected to lower the reorganization energy penalty expected for the reduction of the linear CO2 molecule. This interaction was shown to work synergistically with the reduced Re state, as the thiourea moiety also enhanced the catalytic response by acting as a pendent proton donor capable of beneficial hydrogen-bonding interactions and facilitating C–O bond cleavage. Added Brønsted acids inhibited catalytic activity, likely the result of competitive interactions with the pendent thiourea tether. In a separate study on the asymmetric incorporation of phenolic pendent proton sources to Re(bpy)(CO)3X, catalysis was ‘turned on’ at lower overpotentials, specifically the first reduction potential by CV in the two complexes studied (Rotundo et al., 2019). These complexes did, however, suffer from low Faradaic efficiencies for CO when no external proton source was present. Each of these catalysts also had issues with electrode absorption phenomena, a deleterious reaction with inhibitory consequences for nominally molecular electrocatalysts.
A Re(bpy)-based CO2 reduction catalyst which features an imidazolium group as a charged residue in the secondary sphere reported by Nippe and co-workers was observed to cause changes in redox properties and mechanism compared to unfunctionalized Re(bpy)(CO)3X (Sung et al., 2017). It was proposed that the C2-H carbon of the imidazolium moiety was important for the catalytic enhancement through an alteration of mechanism: theoretical methods suggested that hydrogen bond-like or electrostatic C2-H—X (X = Cl−, , or H2O) interactions change the ground state energies of intermediates relevant to the catalytic cycle. These assignments were supported experimentally through testing of a control complex where the imidazolium C2-H was replaced by C2-CH3, and the non-linear dependence of the catalytic activity of each complex on [H2O], which is anomalous to the archetypal electrocatalytic response of Re(bpy)(CO)3X. It was postulated that a reduction-first mechanism for CO2 reduction was occurring, where reduction of the Re(I)(η1-CO2H) adduct preceded protonation and C–O bond cleavage, as is classically seen in the unfunctionalized complex (Figure 8).
The effects of charge on the electrocatalytic activity of Re(bpy)(CO)3X were studied in a series of polymeric frameworks using a series of charged monomers (Sahu et al., 2017). Three norbornenyl-based polymers containing either positively charged quaternary ammonium, neutral phenyl, or negatively charged trifluoroborate moieties were generated through ROMP and covalently end-labeled with a Re(bpy)(CO)3X-based terminating reagent. Electrochemical studies in acetonitrile indicated that the polymers containing quaternary ammonium salts exhibited catalytic behavior at a significantly more positive potential (~300 mV) than the neutral polymer, which behaved consistently with unfunctionalized Re(bpy)(CO)3X. The incorporation of negatively charged groups caused a shift to more reducing potentials, and catalytic activity was not observed in the solvent window. The incorporation of known catalysts onto a polymeric framework with the ability to tune reduction potential is a possible precursor to highly ordered structures such as thin films, abiotic metalloproteins, porous catalytic membranes, and cationic nanoparticles for use in devices.
Mn(bpy)(CO)3X
Unpublished results referenced by Johnson et al. in 1996 stated that Mn(bpy)(CO)3X, unlike its third-row congener Re, was inactive for electrocatalytic CO2 reduction under aprotic conditions (Johnson et al., 1996). A more recent examination by Deronzier and co-workers in 2011 repeated this result, but also showed that the addition of a weak proton donor facilitated a significant and selective electrocatalytic response for CO2 reduction to CO (Bourrez et al., 2011). One important mechanistic detail about the Mn(bpy)(CO)3X system is that upon reduction by a single electron and subsequent loss of X, a Mn–Mn dimer [Mn(bpy)(CO)3]2 can rapidly form with rates approaching the diffusion limit (Figure 9) (Grills et al., 2014). The formation of this dimer has two detrimental effects: (1) it increases the electrochemical driving force required to generate the catalytically active monoanionic five-coordinate [Mn(bpy)(CO)3]− species required for CO2 reduction (the Mn–Mn bond requires more reducing potentials to cleave), and (2) it reduces the activity of the complex toward CO2 reduction as a non-catalytic competing pathway (Grice and Kubiak, 2013; Smieja et al., 2013).
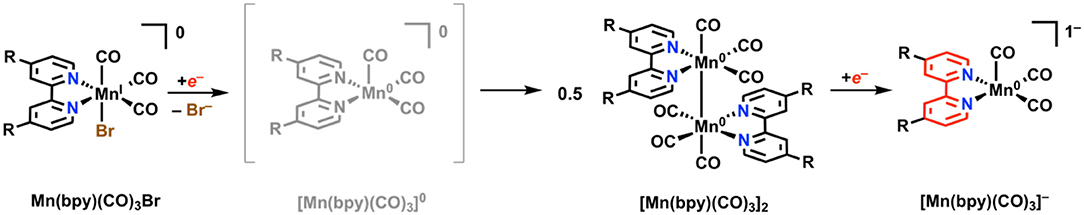
Figure 9. Faradaic reduction mechanism of Mn(bpy)(CO)3X. Adapted with permission from Machan et al. (2014b). Copyright 2014 American Chemical Society.
In attempts to combat these detrimental effects from dimerization, a bulky bpy analog was designed (6,6′-dimesitylbpy) (Sampson et al., 2014; Sampson and Kubiak, 2016). This ligand framework proved to be effective in eliminating the dimerization reaction. Rather than two irreversible 1e− waves on the reductive sweep as seen in Mn(bpy)(CO)3X, a single reversible 2e− wave was observed (Sampson et al., 2014). This leads to the formation of monomeric, anionic [Mn(mesbpy)(CO)3]− at 300 mV more positive potentials than in the case of the original Mn(bpy)(CO)3X complexes (Sampson et al., 2014). In the presence of CO2 and a proton source, the 2e− reversible feature becomes irreversible and shifts toward positive potentials, which is indicative of CO2 binding (Sampson et al., 2014). This mechanistic difference was confirmed through control experiments, IR-SEC (Ashley and Pons, 1988; Zavarine and Kubiak, 2001; Best et al., 2008; Kaim and Fiedler, 2009; Machan et al., 2014b), and the direct synthesis of the active species. A significant catalytic response does not occur in the presence of Brønsted acids until potentials are similar to those that are catalytic for the unfunctionalized parent complex (Sampson et al., 2014). Rapid C–O bond cleavage in the hydroxycarbonyl intermediate [Mn(mesbpy)(CO)3(η1-CO2H)] does not occur until “overreduction” to generate [Mn(mesbpy)(CO)3(η1-CO2H)]− at potentials 400 mV more negative than the 2e− reversible feature (Figure 10) (Riplinger et al., 2014). To take advantage of the initial CO2 binding event by [Mn(mesbpy)(CO)3]− at more positive potentials, a subsequent report used Lewis acid additives (Sampson and Kubiak, 2016). This strategy proved successful, as the addition of Mg2+ ions as cosubstrate to solution aided in C–O bond cleavage at the potential where CO2 binding occurs, greatly reducing the overpotential required for the generation of CO from CO2 (Sampson and Kubiak, 2016).
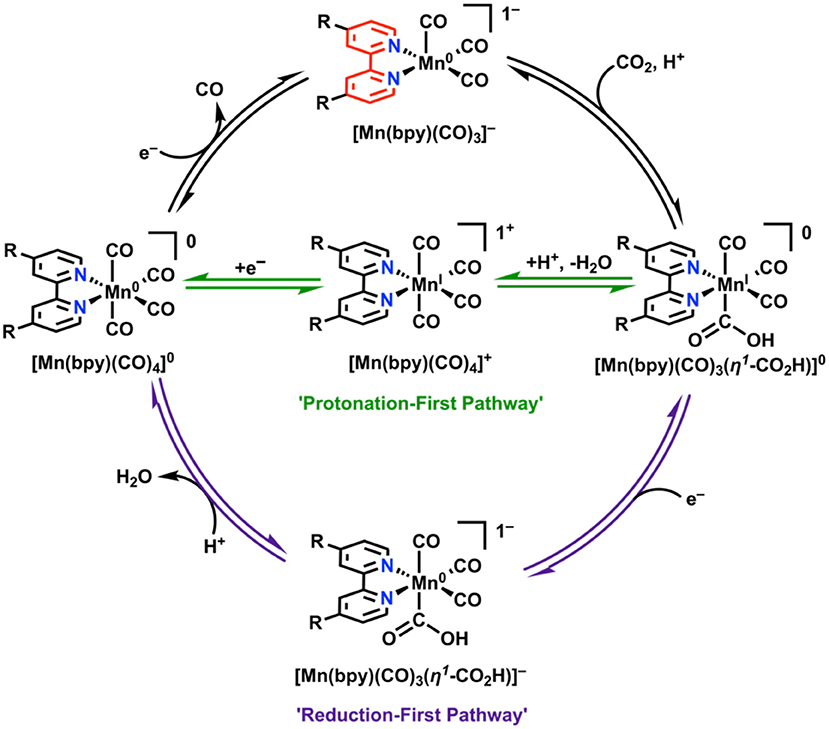
Figure 10. “Protonation-first” and “reduction-first” pathways for CO2 reduction by Mn(bpy)(CO)3X (Riplinger et al., 2014).
Initial attempts to incorporate pendent proton relays into the Mn(bpy)(CO)3X framework involved asymmetric attachment of phenol moieties onto the bpy ligand (Franco et al., 2014; Agarwal et al., 2015). This led to enhanced activity in comparison to the parent Mn(bpy)(CO)3X complex, including activity in the absence of a proton source; however, the competitive dimerization reaction was still apparent in each case (Franco et al., 2014; Agarwal et al., 2015). Converting the pendent –OH groups to ethers with methoxy groups showed insignificant or no catalytic activity in control studies for these derivatives.
To extend these studies to symmetrically functionalized systems, Rochford and co-workers synthesized a methoxy ether-containing analog of the bulky bpy ligand, 6,6′-(2,6-dimethoxyphenyl)bpy [(MeO)2Ph]2bpy (Ngo et al., 2017). A combined experimental and theoretical analysis showed that through the introduction of sufficiently acidic exogeneous proton sources, the slower “protonation-first” pathway seen in the original bulky bpy study could be “turned on” at lower overpotentials in comparison to the “reduction-first” pathway at more negative potentials (Ngo et al., 2017). In the protonation-first pathway, a bound hydroxycarbonyl is formed, and further protonation by a sufficiently strong acid can lead to the facilitation of C–O bond cleavage prior to reduction and release of CO at more positive potentials than the reduction-first pathway (Figure 10). In the reduction-first pathway, a bound hydroxycarbonyl is again formed; however, without a sufficiently strong acid present, reduction of the hydroxycarbonyl must occur at more negative potentials prior to protonation and C–O bond cleavage.
The acid dependence of the “protonation-first” and “reduction-first” pathways for Mn([(MeO)2Ph]2bpy)(CO)3X was rationalized through a hydrogen-bonding interaction between the Mn-bound η1-CO2H hydroxycarbonyl species and the pendent Lewis base groups located on the ligand (Figure 11). This hydrogen bond donor–acceptor interaction was also proposed to facilitate the subsequent transfer of a second equivalent of a sufficiently strong Brønsted acid additive to protonate and facilitate C–O bond cleavage, ultimately producing H2O and Mn-bound CO (Ngo et al., 2017). In the presence of a proton source of insufficient acidity, the reduction of the bound η1-CO2H hydroxycarbonyl species is instead required before protonation and C–O bond cleavage can occur, as is observed with the “overreduction” of the Mn catalyst with the “bulky” bpy ligand (Ngo et al., 2017). Importantly, no metal–metal dimerization reaction was observed for this ligand.
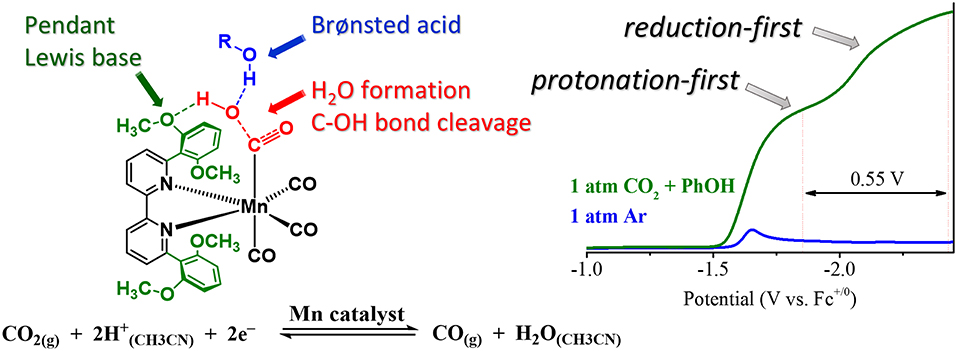
Figure 11. Figure showing proposed transition state which occurs to turn on a low-overpotential, secondary-sphere-promoted mechanism for CO2 reduction with Mn((OMe)2bpy)(CO)3X. Adapted with permission from Ngo et al. (2017). Copyright American Chemical Society 2017.
Recently, a charged imidazolium functionality was also introduced asymmetrically into the Mn(bpy)(CO)3X system (Sung et al., 2019). A series of derivatives examining hydrogen-bonding effects showed activity for CO2 reduction at potentials of only −1.4 V vs. Fc+/Fc in the presence of water. This was postulated to originate from a synergistic effect between the pendent imidazolium functionality and a network of water molecules in the solvation shell that facilitated CO2 reduction. The combined use of charge and hydrogen-bonding donors shows promise for lowering the catalytic overpotential, but further work is needed to enhance Faradaic efficiencies to match other catalyst platforms in this family.
Ni(cyclam)]2+
The electrocatalytic activity of a [Ni(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) derivative was first reported in 1980 by Fisher and Eisenberg (1980). It has been the subject of continuous study because of its tolerance for a wide range of acid strengths and solvent systems without a loss of catalytic activity (Beley et al., 1984; Barefield et al., 1986; Balazs and Anson, 1993; Kelly et al., 1999; Schneider et al., 2012). The activity of Ni(cyclam)]2+ for electrocatalytic reduction of CO2 to CO in the presence of water as a Brønsted acid source was attributed specifically to the Trans I isomer (one of six possible isomers based on the orientation of the H atoms on the four metal-coordinated secondary amines in the macrocycle; all four H atoms are cofacial). This isomer is the most favorable for high CO2 reduction activity due to the hydrogen bond donor–acceptor interactions between the ligand NH groups and a Ni-bound CO2 molecule (Froehlich and Kubiak, 2012). Furthermore, DFT calculations have indicated that the trans I isomer has a more stable CO2 adduct than the trans III isomer (two H atoms are cofacial) by approximately 3 kcal/mol (Song et al., 2014).
Further evidence for the importance of the amine protons and these hydrogen-bonding interactions is found through a comparison with the N-alkylated derivatives [Ni(dimethylcyclam)]2+ and [Ni(tetramethylcyclam)]2+, where greatly diminished activity for electrocatalytic CO2 reduction is observed (Froehlich and Kubiak, 2012). The absence of hydrogen bond-induced stabilization effects and increased steric parameters make CO2 binding less facile in the N-alkylated derivatives (Figure 12). Pendent proton donor effects have been shown with other functional groups as well: the introduction of a carboxylic acid on the carbon backbone of Ni(cyclam)]2+ improves its activity by making it stable and selective for CO2 reduction down to pH 2 at similar overpotentials to other reported water-soluble CO2 reduction catalysts (Neri et al., 2016).
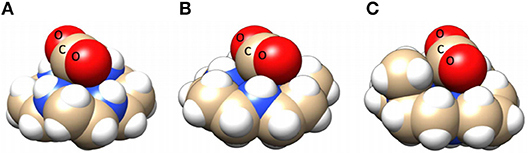
Figure 12. Space-filling models of trans I [Ni(cyclam)(CO2)]+ (A), trans III [Ni(cyclam)(CO2)]+ (B), and [Ni(DMC)(CO2)]+ (C). Structures indicate that enhanced CO2 reduction in the case of trans I [Ni(cyclam)(CO2)]+ may be due to increased stabilization of the CO2 adduct through increased hydrogen bond donors oriented toward the hydrogen bond accepting O atoms on the CO2 adduct. Used with permission from Song et al. (2014). Copyright American Chemical Society 2014.
The use of an exogenous urea additive with Ni(cyclam)]2+ was shown to improve catalytic activity for CO2 reduction for similar reasons (Nichols and Chang, 2018). This study demonstrated that the urea additive acted as a cocatalyst for the system, and it was proposed that its unique structure allows for it to promote CO2 reduction through the formation of multipoint hydrogen bonds with the bound CO2 adduct [Ni(cyclam)(η1-CO2)]+ (Nichols and Chang, 2018). This conclusion was supported through the introduction of multiple cationic and neutral additives with similar pKas. In each case, the cocatalytic response observed with urea was not present (Nichols and Chang, 2018). Although the urea additive was not tethered to cyclam, it is conspicuous that it should have a cocatalytic role with both Ni(cyclam)]2+- and Re(bpy)-based catalysts for CO2 reduction, vide supra.
Multimetallic Systems
Multimetallic systems are important examples of secondary-sphere interactions, because they are common to the enzymes which catalyze CO2 reduction chemistry (Dobbek et al., 2001b, 2002). In many cases, these multimetallic systems work cooperatively to both store excess charge and to activate the molecule of interest using “push–pull” donor–acceptor effects. Homobimetallic cofacial [Fe(TPP)]+ systems connected by a phenylene bridge were synthesized to generate a bimetallic species from the well-known [Fe(TPP)]+ system discussed above (Mohamed et al., 2015). By tuning the Fe–Fe distance through synthetic modification, CO2 binding could be induced at the Fe3+/2+ wave rather than the Fe1+/0 as observed in most [Fe(TPP)]+ systems (Mohamed et al., 2015; Bonin et al., 2017). This was rationalized through the expected Fe–Fe distance of 3.2–4.0 Å in the ortho-bridged system, which would be suitable for binding the linear CO2 molecule. In contrast, the meta-bridged system was expected to have a significantly shorter separation and showed diminished activity similar to monomeric [Fe(TPP)]+ (Mohamed et al., 2015). The ortho-bridged system has significant catalytic activity (TOFmax = 4300 s−1) and high Faradaic efficiency for CO (95%) at an overpotential of ~0.7 V in the presence of 10% H2O in DMF, which is a significant improvement over monomeric [Fe(TPP)]+. The overpotential for this class of homobimetallic catalyst could be further tuned using electron-withdrawing and -donating substituents on the phenyl rings of each [Fe(TPP)]+ unit (Figure 13) (Zahran et al., 2016). When the overpotential was synthetically tuned to ~0.4 V with electron-withdrawing groups, a ~3-fold decrease in activity in comparison to the parent phenyl-functionalized dimer was observed (Zahran et al., 2016). Chang and co-workers have recently demonstrated that these beneficial multimetallic effects extend beyond bimetallic systems using a porous organic cage containing six [Fe(TPP)]+ units, which was active for CO2 reduction in aqueous solutions (Smith et al., 2018).
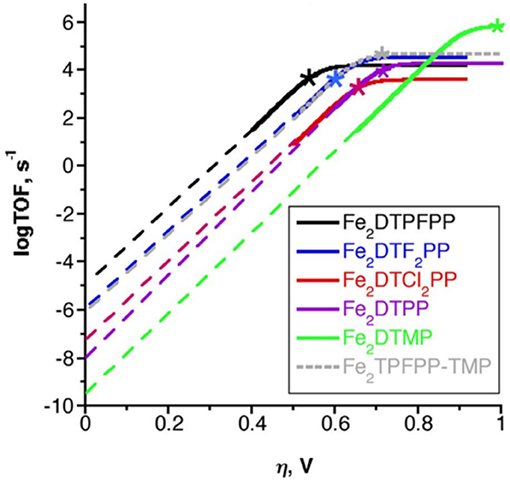
Figure 13. Catalytic Tafel plot showing overpotential tuning in cofacial Fe(TTP) through the introduction of electron with drawing substituents on the phenyl groups of the porphyrin system. Adapted from Zahran et al. (2016).
A multimetallic Fe carbonyl cluster system with an interstitial main group element [Fe4X(CO)12−nLn]− (X = N or C, L = another ligand, n = 1 or O) has been developed by the Berben group for the reduction of CO2 to formate under both aqueous and non-aqueous conditions (Rail and Berben, 2011; Taheri et al., 2015; Taheri and Berben, 2016; Loewen et al., 2017). This cluster is proposed to generate formate via an intermediate bridging hydride. The bridging hydride on the cluster motif has the ideal hydricity and pKa to selectively generate formate in the presence of CO2, preventing the competitive formation of H2 (Loewen et al., 2017). Pendent proton groups in this system alter the selectivity of this catalyst family from nearly quantitative generation of formate to the generation of H2, which highlights the importance of controlling the kinetics of substrate delivery in determining selectivity for competitive CO2 and H+ reduction reactions (Loewen et al., 2016).
The development of bimetallic Re complexes based on Re(bpy)(CO)3X and its derivatives has been of recent interest for both photocatalytic (Bruckmeier et al., 2012) and electrocatalytic processes due to the concentration-dependent formation of binuclear intermediates as part of the catalytic cycle (Machan et al., 2014a, 2016; Wilting et al., 2017; Yang et al., 2018). In electrocatalytic systems, the first bimolecular Re system studied utilized acetoamidomethyl modified bpy to generate a supramolecular catalyst system in situ. This system operated at more positive potentials (~250 mV) in MeCN than the 4,4′-dimethylbpy-based control complex as the result of a hydrogen-bonded dimer active state, albeit with a lower TOF and FE than the unimolecular process (Machan et al., 2014a). To further probe this hydrogen bond-based dimer system, a subsequent report focused on a heterobimetallic Re–Mn construct using a 1:1 mixture of acetoamidomethyl-modified Re(bpy)(CO)3X with acetoamidomethyl-modified Mn(bpy)(CO)3X (Machan and Kubiak, 2016). Results from this study indicated a cooperative heterobimetallic pathway was operative, and it was proposed that the Mn center was activating CO2 followed by protonation to generate a Mn-bound hydroxycarbonyl species within the heterobimetallic dimer. Enhanced reduction-first pathway kinetics were initiated in this case by electron transfer from the reduced Re species present in the dimer.
Further modifications to this supramolecular system replaced the acetoamidomethyl unit with an amide-linked PhOH-containing tyrosine functional group (Machan et al., 2016). This modification led to an increased TOFmax in comparison to the initial complex and near quantitative Faradaic efficiency; mechanistic studies where the PhOH unit was substituted for a phenyl ring showed the pendent –OH functionality was essential for improving catalytic activity in the bimetallic mechanism (Machan et al., 2016). The “soft” non-covalent linkages used in this strategy are reminiscent of biological active sites: the catalytic system can adopt a variety of conformations on the potential energy surface facilitated by weaker interactions instead of more rigid systems reliant on distance or conformational tuning through purely synthetic means. The success of this approach is dependent on how well the weak interactions overcome the added diffusional component of the bimolecular reaction mechanism.
Work to generate rigid homobimetallic Re(bpy)(CO)3X systems where the metal centers are in close proximity has been achieved using an anthracene linker by Jurss and co-workers (Yang et al., 2018). This complex can be isolated as cis or trans isomers through chromatography, which alters the relative positioning of the Re centers (Yang et al., 2018). The cis isomer, in which the Re centers are in close proximity to one another, outperformed both a monometallic anthracene control complex and the trans isomer of the homobimetallic species in catalytic studies. Another covalently linked homobimetallic system with an imidazole–pyridine bridge was examined by Siewert and co-workers (Wilting et al., 2017). The observed catalytic activity of the homobimetallic species outperformed the mononuclear control complex, which was inactive for CO2 reduction (Sinha et al., 2017). The introduction of the phenol linker between the two imidazole–pyridine arms to generate a proton relay in close proximity to the active site of the two metal centers was also examined (Wilting et al., 2017). The pendent proton source enhanced the activity of the bimetallic Re complex, with Faradaic efficiencies for CO of ~60% (Wilting et al., 2017).
Emerging Systems
Several relatively new systems have also been reported where pendent functional group interactions are essential to the overall mechanism (Figure 14). A recent report on a series of cobalt(cyclopentadienyl)(P2N2) complexes by Artero and co-workers showed that pendent tertiary amines enabled selective formate generation from CO2 (Roy et al., 2017). DFT calculations suggest that a reaction-defining transition state occurs where one of the pendent amines forms a hydrogen-bonding interaction with water to align it with CO2 as simultaneous hydride transfer occurs from the Co center, lowering the overall transition state energy and enhancing activity (Roy et al., 2017). Work in our own group has identified a Schiff base-type ligand based on bpy as a promising new direction: the Fe(III) derivative is active for the reduction of CO2 to formate with PhOH as a proton source. Mechanistic investigations suggest the Fe-bound oxygen atoms act as a site for protonation upon initial reduction of the complex, generating a pendent proton source for the reaction at applied potential (Nichols et al., 2018a). A macrocyclic, aminopolypyridyl Co complex from Marinescu and co-workers generated CO in a near quantitative fashion in the presence of MeOH as a proton source (Chapovetsky et al., 2016, 2018). Upon alkylation of the pendent amine functionalities, a two-fold decrease in activity occurred, suggesting that the pendent protons on the amines linking the pyridyl groups play an important role as hydrogen bond donors during CO2 reduction (Chapovetsky et al., 2016, 2018). The activity and selectivity of all these platforms are promising for future studies on optimizing secondary-sphere effects.
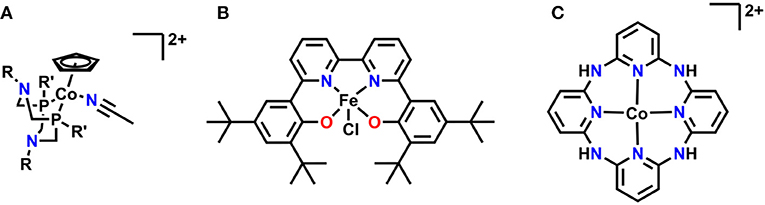
Figure 14. (A) Co(Cp)(P2N2) utilizing pendent tertiary amines for reduction of CO2 to formate. (B) Fe(tbudhbpy)CI is protonated at the Fe-coordinating O atoms upon reduction and reduces CO2 to formate. (C) Macrocyclic–aminopyridyl complex which reduces CO2 to CO.
Conclusions and Outlook
Molecular electrocatalysts for CO2 reduction are of continuing interest for their possible utility in storing renewable energy in chemical bonds. Through careful mechanistic observations and design principles inspired by nature, researchers have managed to improve many of the known catalyst systems for CO2 reduction. This iterative optimization of catalysts has demonstrated that the incorporation of pendent Brønsted acids/bases, charged groups, sterically bulky groups, Lewis basic sites, and the use of multimetallic sites with careful solvent choice can lead to improved catalytic activity and even new mechanisms through secondary-sphere effects reminiscent of biological systems.
With these successes in mind, it is useful to acknowledge that there is still much work to do: CODHs can reversibly interconvert CO2/CO in aqueous systems with a TOFmax of 0.5 s−1 (CO2 to CO) and 31,000 s−1 (CO to CO2) with minimal overpotentials (Maynard and Lindahl, 2001). However, the use of secondary-sphere effects in abiotic molecular electrocatalysts has already been shown to be important for enhancing selectivity and activity. Further development in this area may lead to the development of catalysts capable of reducing products beyond CO and HCO2H, which are a burgeoning area for molecular electrocatalysts. The study of immobilized molecular Cu(TPP) systems has shown to be a successful strategy in generating higher ordered products like methane and ethylene in an electrocatalytic fashion (Weng et al., 2016), which is interesting, since crystalline copper electrodes are known to do this chemistry as heterogenous electrode materials (Kuhl et al., 2012). Bringing molecular design principles to materials seems to be another viable strategy for using secondary-sphere effects in electrocatalysis: Gong et al. (2017) immobilized porphyrin cages on Cu electrodes to tune activity and selectivity for carbon–carbon coupling products from CO2 reduction through supramolecular effects. Recent reports have also described photocatalysts which convert CO2 to methane from molecular catalysts related to those described here, which is promising for developing eventual electrocatalytic behavior (Rao et al., 2017; Shirley et al., 2019).
Running CO2 reduction reactions reversibly with abiotic systems could close the “loop” on the energy cycle, enabling the development of new fuel cell technologies beyond H2 (Matsumoto et al., 2011, 2013; Kadirov et al., 2018). New catalyst systems which approach these biological efficiencies are unlikely to be rapidly found through purely synthetic routes due to the inherent depth and synthetic difficulty of the parameter space. Rather, the most efficient approach to future catalyst development should utilize a multidisciplinary approach which combines statistical, computational, and experimental methods to assist in the search for new CO2 reduction catalysts unique from the current catalytic systems by improving predictive power.
Author Contributions
The manuscript was written through contributions of all the authors. All authors have given approval to the final version of the manuscript.
Funding
Funding was obtained from the University of Virginia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the University of Virginia for the generous funding.
References
Agarwal, J., Shaw, T. W., Schaefer, H. F., and Bocarsly, A. B. (2015). Design of a catalytic active site for electrochemical CO2 reduction with Mn(I)-tricarbonyl species. Inorg. Chem. 54, 5285–5294. doi: 10.1021/acs.inorgchem.5b00233
Appel, A. M., Bercaw, J. E., Bocarsly, A. B., Dobbek, H., DuBois, D. L., Dupuis, M., et al. (2013). Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658. doi: 10.1021/cr300463y
Appel, A. M., and Helm, M. L. (2014). Determining the overpotential for a molecular electrocatalyst. ACS Catal. 4, 630–633. doi: 10.1021/cs401013v
Ashley, K., and Pons, S. (1988). Infrared spectroelectrochemistry. Chem. Rev. 88, 673–695. doi: 10.1021/cr00086a006
Azcarate, I., Costentin, C., Robert, M., and Saveant, J.-M. (2016). Through-space charge interaction substituent effects in molecular catalysis leading to the design of the most efficient catalyst of CO2-to-CO electrochemical conversion. J. Am. Chem. Soc. 138, 16639–16644. doi: 10.1021/jacs.6b07014
Balazs, G. B., and Anson, F. C. (1993). Effects of CO on the electrocatalytic activity of Ni (cyclam)2+ toward the reduction of CO2. J. Electroanal. Chem. 361, 149–157. doi: 10.1016/0022-0728(93)87049-2
Barefield, E. K., Freeman, G. M., and Van Derveer, D. G. (1986). Electrochemical and structural studies of nickel (II) complexes of N-alkylated cyclam ligands: x-ray structures of trans-[Ni (C14H32N4)(OH2) 2] Cl2. 2H2O and [Ni (C14H32N4)](O3SCF3)2. Inorg. Chem. 25, 552–558. doi: 10.1021/ic00224a033
Beley, M., Collin, J.-P., Ruppert, R., and Sauvage, J.-P. (1984). Nickel (II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1315–1316. doi: 10.1039/c39840001315
Benson, E. E., Kubiak, C. P., Sathrum, A. J., and Smieja, J. M. (2009). Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev 38, 89–99. doi: 10.1039/B804323J
Benson, E. E., Sampson, M. D., Grice, K. A., Smieja, J. M., Froehlich, J. D., Friebel, D., et al. (2013). The electronic states of rhenium bipyridyl electrocatalysts for CO2 reduction as revealed by x-ray absorption spectroscopy and computational quantum chemistry. Angew. Chem. Int. 52, 4841–4844. doi: 10.1002/anie.201209911
Best, S., Crumbliss, A., Dunsch, L., Haga, M.-A., Hartl, F., Kubiak, C., et al. (2008). Spectroelectrochemistry. Cambridge, UK: Royal Society of Chemistry.
Bhugun, I., Lexa, D., and Saveant, J.-M. (1994). Ultraefficient selective homogeneous catalysis of the electrochemical reduction of carbon dioxide by an iron (0) porphyrin associated with a weak Broensted acid cocatalyst. J. Am. Chem. Soc. 116, 5015–5016. doi: 10.1021/ja00090a068
Bhugun, I., Lexa, D., and Savéant, J.-M. (1996). Catalysis of the electrochemical reduction of carbon dioxide by iron (0) porphyrins: synergystic effect of weak Brönsted acids. J. Am. Chem. Soc. 118, 1769–1776. doi: 10.1021/ja9534462
Bonin, J., Maurin, A., and Robert, M. (2017). Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances. Coord Chem. Rev. 334, 184–198. doi: 10.1016/j.ccr.2016.09.005
Bourrez, M., Molton, F., Chardon-Noblat, S., and Deronzier, A. (2011). [Mn (bipyridyl)(CO) 3Br]: an abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. 123, 10077–10080. doi: 10.1002/ange.201103616
Boyington, J. C., Gladyshev, V. N., Khangulov, S. V., Stadtman, T. C., and Sun, P. D. (1997). Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275, 1305–1308. doi: 10.1126/science.275.5304.1305
Bruckmeier, C., Lehenmeier, M. W., Reithmeier, R., Rieger, B., Herranz, J., and Kavakli, C. (2012). Binuclear rhenium (I) complexes for the photocatalytic reduction of CO2. Dalton Trans. 41, 5026–5037. doi: 10.1039/c2dt30273j
Cammarota, R. C., Vollmer, M. V., Xie, J., Ye, J., Linehan, J. C., Burgess, S. A., et al. (2017). A bimetallic nickel–gallium complex catalyzes CO2 hydrogenation via the intermediacy of an anionic d10 nickel hydride. J. Am. Chem. Soc. 139, 14244–14250. doi: 10.1021/jacs.7b07911
Centi, G., and Perathoner, S. (2009). Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal Today 148, 191–205. doi: 10.1016/j.cattod.2009.07.075
Chabolla, S. A., Dellamary, E. A., Machan, C. W., Tezcan, F. A., and Kubiak, C. P. (2014). Combined steric and electronic effects of positional substitution on dimethyl-bipyridine rhenium (I) tricarbonyl electrocatalysts for the reduction of CO2. Inorganica Chim. Acta 422, 109–113. doi: 10.1016/j.ica.2014.07.007
Chabolla, S. A., Machan, C. W., Yin, J., Dellamary, E. A., Sahu, S., Gianneschi, N. C., et al. (2017). Bio-inspired CO2 reduction by a rhenium tricarbonyl bipyridine-based catalyst appended to amino acids and peptidic platforms: incorporating proton relays and hydrogen-bonding functional groups. Faraday Discuss. 198, 279–300. doi: 10.1039/C7FD00003K
Chapovetsky, A., Do, T. H., Haiges, R., Takase, M. K., and Marinescu, S. C. (2016). Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles. J. Am. Chem. Soc. 138, 5765–5768. doi: 10.1021/jacs.6b01980
Chapovetsky, A., Welborn, M., Luna, J. M., Haiges, R., Miller, T. F., and Marinescu, S. C. (2018). Pendant hydrogen-bond donors in cobalt catalysts independently enhance CO2 reduction. ACS Cent. Sci. 4, 397–404. doi: 10.1021/acscentsci.7b00607
Clark, M. L., Cheung, P. L., Lessio, M., Carter, E. A., and Kubiak, C. P. (2018). Kinetic and mechanistic effects of bipyridine (bpy) substituent, labile ligand, and Brønsted acid on electrocatalytic CO2 reduction by Re(bpy) complexes. ACS Catal. 8, 2021–2029. doi: 10.1021/acscatal.7b03971
Collman, J. P. (1977). Synthetic models for the oxygen-binding hemoproteins. Acc. Chem. Res. 10, 265–272. doi: 10.1021/ar50115a006
Collman, J. P., Boulatov, R., Sunderland, C. J., and Fu, L. (2004). Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem. Rev. 104, 561–588. doi: 10.1021/cr0206059
Costentin, C., Drouet, S., Robert, M., and Savéant, J.-M. (2012a). A local proton source enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 338:90. doi: 10.1126/science.1224581
Costentin, C., Drouet, S., Robert, M., and Savéant, J.-M. (2012b). Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 134, 11235–11242. doi: 10.1021/ja303560c
Costentin, C., Passard, G., Robert, M., and Savéant, J.-M. (2014a). Pendant acid–base groups in molecular catalysts: H-bond promoters or proton relays? Mechanisms of the conversion of CO2 to CO by electrogenerated iron(0)porphyrins bearing prepositioned phenol functionalities. J. Am. Chem. Soc. 136, 11821–11829. doi: 10.1021/ja506193v
Costentin, C., Passard, G., Robert, M., and Savéant, J.-M. (2014b). Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion. Proc. Natl. Acad. Sci. U.S.A. 111, 14990–14994. doi: 10.1073/pnas.1416697111
Costentin, C., and Savéant, J.-M. (2018a). Homogeneous catalysis of electrochemical reactions: the steady-state and nonsteady-state statuses of intermediates. ACS Catal. 8, 5286–5297. doi: 10.1021/acscatal.8b01195
Costentin, C., and Savéant, J.-M. (2018b). Homogeneous molecular catalysis of electrochemical reactions: manipulating intrinsic and operational factors for catalyst improvement. J. Am. Chem. Soc. 140, 16669–16675. doi: 10.1021/jacs.8b09154
Costentin, C., Savéant, J.-M., and Tard, C. (2018). Ligand “noninnocence” in coordination complexes vs. kinetic, mechanistic, and selectivity issues in electrochemical catalysis. Proc. Natl. Acad. Sci. U.S.A. 115, 9104–9109. doi: 10.1073/pnas.1810255115
Costentin, C., and Savéant, J. M. (2014). Multielectron, multistep molecular catalysis of electrochemical reactions: benchmarking of homogeneous catalysts. ChemElectroChem 1, 1226–1236. doi: 10.1002/celc.201300263
Curtis, C. J., Miedaner, A., Ciancanelli, R., Ellis, W. W., Noll, B. C., Rakowski DuBois, M., et al. (2003). [Ni (Et2PCH2NMeCH2PEt2) 2] 2+ as a functional model for hydrogenases. Inorg. Chem. 42, 216–227. doi: 10.1021/ic020610v
Dobbek, H., Gremer, L., Kiefersauer, R., Huber, R., and Meyer, O. (2002). Catalysis at a dinuclear [CuSMo (O) OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 15971–15976. doi: 10.1073/pnas.212640899
Dobbek, H., Gremer, L., Meyer, O., and Huber, R. (2001a). Handbook of Metalloproteins. Chichester: John Wiley & Sons Ltd.
Dobbek, H., Svetlitchnyi, V., Gremer, L., Huber, R., and Meyer, O. (2001b). Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science 293, 1281–1285. doi: 10.1126/science.1061500
DuBois, M. R., and DuBois, D. L. (2009). The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem. Soc. Rev. 38, 62–72. doi: 10.1039/B801197B
Dutta, A., Lense, S., Hou, J., Engelhard, M. H., Roberts, J. A., and Shaw, W. J. (2013). Minimal proton channel enables H2 oxidation and production with a water-soluble nickel-based catalyst. J. Am. Chem. Soc. 135, 18490–18496. doi: 10.1021/ja407826d
Dutta, A., Roberts, J. A., and Shaw, W. J. (2014). Arginine-containing ligands enhance H2 oxidation catalyst performance. Angew. Chem. Int. 53, 6487–6491. doi: 10.1002/anie.201402304
Elwell, C. E., Gagnon, N. L., Neisen, B. D., Dhar, D., Spaeth, A. D., Yee, G. M., et al. (2017). Copper–oxygen complexes revisited: structures, spectroscopy, and reactivity. Chem. Rev. 117, 2059–2107. doi: 10.1021/acs.chemrev.6b00636
Fisher, B. J., and Eisenberg, R. (1980). Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 102, 7361–7363. doi: 10.1021/ja00544a035
Franco, F., Cometto, C., Vallana, F. F., Sordello, F., Priola, E., Minero, C., et al. (2014). A local proton source in a [Mn(bpy-R)(CO)3 Br]-type redox catalyst enables CO2 reduction even in the absence of Brønsted acids. Chem. Commun. 50, 14670–14673. doi: 10.1039/C4CC05563B
Fraze, K., Wilson, A. D., Appel, A. M., Rakowski DuBois, M., and DuBois, D. L. (2007). Thermodynamic properties of the Ni–H bond in complexes of the type [HNi (P2RN2R')2](BF4) and evaluation of factors that control catalytic activity for hydrogen oxidation/production. Organometallics 26, 3918–3924. doi: 10.1021/om070143v
Froehlich, J. D., and Kubiak, C. P. (2012). Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg. Chem. 51, 3932–3934. doi: 10.1021/ic3001619
Fukuzumi, S. (2013). Electron-transfer properties of high-valent metal-oxo complexes. Coord Chem Rev 257, 1564–1575. doi: 10.1016/j.ccr.2012.07.021
Fukuzumi, S., Lee, Y.-M., Ahn, H. S., and Nam, W. (2018). Mechanisms of catalytic reduction of CO2with heme and nonheme metal complexes. Chem. Sci 9, 6017–6034. doi: 10.1039/C8SC02220H
Fukuzumi, S., Ohkubo, K., Lee, Y. M., and Nam, W. (2015). Lewis acid coupled electron transfer of metal–oxygen intermediates. Chem. Eur. J. 21, 17548–17559. doi: 10.1002/chem.201502693
Galan, B. R., Reback, M. L., Jain, A., Appel, A. M., and Shaw, W. J. (2013). Electrocatalytic oxidation of formate with nickel diphosphine dipeptide complexes: effect of ligands modified with amino acids. Eur. J. Inorg. Chem. 2013, 5366–5371. doi: 10.1002/ejic.201300751
Galan, B. R., Schöffel, J., Linehan, J. C., Seu, C., Appel, A. M., and Roberts, J. A., et al. (2011). Electrocatalytic oxidation of formate by [Ni (PR2NR′ 2) 2 (CH3CN)] 2+ complexes. J. Am. Chem. Soc. 133, 12767–12779. doi: 10.1021/ja204489e
Gennaro, A., Isse, A. A., Severin, M.-G., Vianello, E., Bhugun, I., and Saveant, J.-M. (1996). Mechanism of the electrochemical reduction of carbon dioxide at inert electrodes in media of low proton availability. J. Chem. Soc. Faraday Trans. 92, 3963–3968. doi: 10.1039/FT9969203963
Ginovska-Pangovska, B., Dutta, A., Reback, M. L., Linehan, J. C., and Shaw, W. J. (2014). Beyond the active site: the impact of the outer coordination sphere on electrocatalysts for hydrogen production and oxidation. Acc. Chem. Res. 47, 2621–2630. doi: 10.1021/ar5001742
Gloaguen, F., and Rauchfuss, T. B. (2009). Small molecule mimics of hydrogenases: hydrides and redox. Chem. Soc. Rev. 38, 100–108. doi: 10.1039/B801796B
Gong, M., Cao, Z., Liu, W., Nichols, E. M., Smith, P. T., Derrick, J. S., et al. (2017). Supramolecular porphyrin cages assembled at molecular–materials interfaces for electrocatalytic CO reduction. ACS Cent. Sci. 3, 1032–1040. doi: 10.1021/acscentsci.7b00316
Grice, K., and Kubiak, C. (2013). Chapter five—recent studies of rhenium and manganese bipyridine carbonyl catalysts for the electrochemical reduction of CO2. Adv. Inorg. Chem. 66, 163–188. doi: 10.1016/B978-0-12-420221-4.00005-6
Grills, D. C., Farrington, J. A., Layne, B. H., Lymar, S. V., Mello, B. A., Preses, J. M., et al. (2014). Mechanism of the formation of a Mn-based CO2 reduction catalyst revealed by pulse radiolysis with time-resolved infrared detection. J. Am. Chem. Soc. 136, 5563–5566. doi: 10.1021/ja501051s
Haviv, E., Azaiza-Dabbah, D., Carmieli, R., Avram, L., Martin, J. M. L., and Neumann, R. (2018). A thiourea tether in the second coordination sphere as a binding site for CO2 and a proton donor promotes the electrochemical reduction of CO2 to CO catalyzed by a rhenium bipyridine-type complex. J. Am. Chem. Soc. 140, 12451–12456. doi: 10.1021/jacs.8b05658
Hawecker, J., Lehn, J.-M., and Ziessel, R. (1984). Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Am. Chem. Soc. Chem. Commun. 6, 328–330. doi: 10.1039/C39840000328
Helm, M. L., Stewart, M. P., Bullock, R. M., DuBois, M. R., and DuBois, D. L. (2011). A synthetic nickel electrocatalyst with a turnover frequency above 100,000s−1 for H2 production. Science 333, 863. doi: 10.1126/science.1205864
Henry, R. M., Shoemaker, R. K., DuBois, D. L., and DuBois, M. R. (2006). Pendant bases as proton relays in iron hydride and dihydrogen complexes. J. Am. Chem. Soc. 128, 3002–3010. doi: 10.1021/ja057242p
Henry, R. M., Shoemaker, R. K., Newell, R. H., Jacobsen, G. M., DuBois, D. L., and Rakowski DuBois, M. (2005). Stereochemical control of iron (II) complexes containing a diphosphine ligand with a pendant nitrogen base. Organometallics 24, 2481–2491. doi: 10.1021/om050071c
Hille, R. (2013). The molybdenum oxotransferases and related enzymes. Dalton Trans. 42, 3029–3042. doi: 10.1039/c2dt32376a
Himeda, Y., Onozawa-Komatsuzaki, N., Sugihara, H., Arakawa, H., and Kasuga, K. (2004). Half-sandwich complexes with 4,7-dihydroxy-1,10-phenanthroline: water-soluble, highly efficient catalysts for hydrogenation of bicarbonate attributable to the generation of an oxyanion on the catalyst ligand. Organometallics 23, 1480–1483. doi: 10.1021/om030382s
Himeda, Y., Onozawa-Komatsuzaki, N., Sugihara, H., and Kasuga, K. (2005). Recyclable catalyst for conversion of carbon dioxide into formate attributable to an oxyanion on the catalyst ligand. J. Am. Chem. Soc. 127, 13118–13119. doi: 10.1021/ja054236k
Himeda, Y., Onozawa-Komatsuzaki, N., Sugihara, H., and Kasuga, K. (2007). Simultaneous tuning of activity and water solubility of complex catalysts by acid–base equilibrium of ligands for conversion of carbon dioxide. Organometallics 26, 702–712. doi: 10.1021/om060899e
Hong, S., Lee, Y.-M., Ray, K., and Nam, W. (2017). Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coord Chem Rev 334, 25–42. doi: 10.1016/j.ccr.2016.07.006
Hull, J. F., Himeda, Y., Wang, W. H., Hashiguchi, B., Periana, R., Szalda, D. J., et al. (2012). Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 4, 383–388. doi: 10.1038/nchem.1295
Jacobsen, G. M., Shoemaker, R., Rakowski DuBois, M., and DuBois, D. L. (2007a). Syntheses and reactions of iron (II) complexes containing diphosphine ligands with pendant nitrogen bases. Organometallics 26, 4964–4971. doi: 10.1021/om700510b
Jacobsen, G. M., Shoemaker, R. K., McNevin, M. J., Rakowski DuBois, M., and DuBois, D. L. (2007b). Syntheses and structural characterizations of iron (II) complexes containing cyclic diphosphine ligands with positioned pendant nitrogen bases. Organometallics 26, 5003–5009. doi: 10.1021/om700601h
Jeoung, J.-H., and Dobbek, H. (2007). Carbon Dioxide Activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 318, 1461–1464. doi: 10.1126/science.1148481
Jiang, C., Nichols, A. W., and Machan, C. W. (2019). A look at periodic trends in d-block molecular electrocatalysts for CO2 reduction. Dalton Trans. doi: 10.1039/C9DT00491B. [Epub ahead of print].
Johnson, F. P., George, M. W., Hartl, F., and Turner, J. J. (1996). Electrocatalytic reduction of CO2 using the complexes [Re(bpy)(CO)3L]n (n = +1, L = P (OEt)3, CH3CN; n = 0, L = Cl−, Otf−; bpy = 2,2′-bipyridine; Otf− = CF3SO3) as catalyst precursors: infrared spectroelectrochemical investigation. Organometallics 15, 3374–3387. doi: 10.1021/om960044+
Kadirov, M. K., Karasik, A. A., Nizameev, I. R., Strelnik, I. D., Kholin, K. V., Kadirov, D. M., et al. (2018). Organometallic polymer electrolyte membrane fuel cell bis-ligand nickel(Ii) complex of 1,5-di-P-tolyl-3,7-dipyridine-1,5,3,7-diazadiphosphacyclo-octane catalyst. Energy Technol. 6, 1088–1095. doi: 10.1002/ente.201700711
Kaim, W., and Fiedler, J. (2009). Spectroelectrochemistry: the best of two worlds. Chem. Soc. Rev. 38, 3373–3382. doi: 10.1039/b504286k
Keith, J. A., Grice, K. A., Kubiak, C. P., and Carter, E. A. (2013). Elucidation of the selectivity of proton-dependent electrocatalytic CO2 reduction by fac-Re(bpy)(CO)3Cl. J. Am. Chem. Soc. 135, 15823–15829. doi: 10.1021/ja406456g
Kelly, C. A., Blinn, E. L., Camaioni, N., D'Angelantonio, M., and Mulazzani, Q. G. (1999). Mechanism of CO2 and H+ reduction by ni (cyclam)+ in aqueous solution. A pulse and continuous radiolysis study. Inorg. Chem. 38, 1579–1584. doi: 10.1021/ic980902p
Kroneck, P. M., and Torres, M. E. S. (2014). The Metal-driven Biogeochemistry of Gaseous Compounds in the Environment. Dordrecht: Springer. doi: 10.1007/978-94-017-9269-1
Kuhl, K. P., Cave, E. R., Abram, D. N., and Jaramillo, T. F. (2012). New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059. doi: 10.1039/c2ee21234j
Lewis, E. A., and Tolman, W. B. (2004). Reactivity of dioxygen–copper systems. Chem. Rev. 104, 1047–1076. doi: 10.1021/cr020633r
Loewen, N. D., Neelakantan, T. V., and Berben, L. A. (2017). Renewable formate from C–H bond formation with CO2: using iron carbonyl clusters as electrocatalysts. Acc. Chem. Res. 50, 2362–2370. doi: 10.1021/acs.accounts.7b00302
Loewen, N. D., Thompson, E. J., Kagan, M., Banales, C. L., Myers, T. W., Fettinger, J. C., et al. (2016). A pendant proton shuttle on [Fe4N(CO)12]– alters product selectivity in formate vs. H2 production via the hydride [H–Fe4N(CO)12]−. Chem. Sci. 7, 2728–2735. doi: 10.1039/C5SC03169A
Machan, C. W., Chabolla, S. A., and Kubiak, C. P. (2015). Reductive disproportionation of carbon dioxide by an alkyl-functionalized pyridine monoimine Re(I) fac-tricarbonyl electrocatalyst. Organometallics 34, 4678–4683. doi: 10.1021/acs.organomet.5b00406
Machan, C. W., Chabolla, S. A., Yin, J., Gilson, M. K., Tezcan, F. A., and Kubiak, C. P. (2014a). Supramolecular assembly promotes the electrocatalytic reduction of carbon dioxide by Re(I) bipyridine catalysts at a lower overpotential. J. Am. Chem. Soc. 136, 14598–14607. doi: 10.1021/ja5085282
Machan, C. W., and Kubiak, C. P. (2016). Interrogating heterobimetallic co-catalytic responses for the electrocatalytic reduction of CO2 using supramolecular assembly. Dalton Trans. 45, 15942–15950. doi: 10.1039/C6DT01956K
Machan, C. W., Sampson, M. D., Chabolla, S. A., Dang, T., and Kubiak, C. P. (2014b). Developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 33, 4550–4559. doi: 10.1021/om500044a
Machan, C. W., Yin, J., Chabolla, S. A., Gilson, M. K., and Kubiak, C. P. (2016). Improving the efficiency and activity of electrocatalysts for the reduction of CO2 through supramolecular assembly with amino acid-modified ligands. J. Am. Chem. Soc. 138, 8184–8193. doi: 10.1021/jacs.6b03774
Manaka, Y., Wang, W.-H., Suna, Y., Kambayashi, H., Muckerman, J. T., Fujita, E., et al. (2014). Efficient H2 generation from formic acid using azole complexes in water. Catal. Sci. Technol. 4, 34–37. doi: 10.1039/C3CY00830D
Manbeck, G. F., Muckerman, J. T., Szalda, D. J., Himeda, Y., and Fujita, E. (2015). Push or pull? Proton responsive ligand effects in rhenium tricarbonyl CO2 reduction catalysts. J. Phys. Chem. 119, 7457–7466. doi: 10.1021/jp511131x
Margarit, C. G., Schnedermann, C., Asimow, N. G., and Nocera, D. G. (2018). Carbon dioxide reduction by iron hangman porphyrins. Organometallics 38, 1219–1223. doi: 10.1021/acs.organomet.8b00334
Matsumoto, T., Kim, K., Nakai, H., Hibino, T., and Ogo, S. (2013). Organometallic catalysts for use in a fuel cell. ChemCatChem 5, 1368–1373. doi: 10.1002/cctc.201200595
Matsumoto, T., Kim, K., and Ogo, S. (2011). Molecular catalysis in a fuel cell. Angew. Chem. Int. 123, 11398–11401. doi: 10.1002/ange.201104498
Maynard, E. L., and Lindahl, P. A. (2001). Catalytic coupling of the active sites in acetyl-CoA synthase, a bifunctional CO-channeling enzyme. Biochemistry 40, 13262–13267. doi: 10.1021/bi015604+
Mirica, L. M., Ottenwaelder, X., and Stack, T. D. P. (2004). Structure and spectroscopy of copper–dioxygen complexes. Chem. Rev. 104, 1013–1046. doi: 10.1021/cr020632z
Mohamed, E. A., Zahran, Z. N., and Naruta, Y. (2015). Efficient electrocatalytic CO2 reduction with a molecular cofacial iron porphyrin dimer. Chem. Commun. 51, 16900–16903. doi: 10.1039/C5CC04273A
Nam, W. (2015). Synthetic mononuclear nonheme iron–oxygen intermediates. Acc. Chem. Res. 48, 2415–2423. doi: 10.1021/acs.accounts.5b00218
Neri, G., Aldous, I. M., Walsh, J. J., Hardwick, L. J., and Cowan, A. J. (2016). A highly active nickel electrocatalyst shows excellent selectivity for CO2 reduction in acidic media. Chem. Sci 7, 1521–1526. doi: 10.1039/C5SC03225C
Ngo, K. T., McKinnon, M., Mahanti, B., Narayanan, R., Grills, D. C., Ertem, M. Z., et al. (2017). Turning on the protonation-first pathway for electrocatalytic CO2 reduction by manganese bipyridyl tricarbonyl complexes. J. Am. Chem. Soc. 139, 2604–2618. doi: 10.1021/jacs.6b08776
Nichols, A. W., Chatterjee, S., Sabat, M., and Machan, C. W. (2018a). Electrocatalytic reduction of CO2 to formate by an iron Schiff base complex. Inorg. Chem. 57, 2111–2121. doi: 10.1021/acs.inorgchem.7b02955
Nichols, E. M., and Chang, C. J. (2018). Urea-based multipoint hydrogen-bond donor additive promotes electrochemical CO2 reduction catalyzed by nickel cyclam. Organometallics 38, 1213–1218. doi: 10.1021/acs.organomet.8b00308
Nichols, E. M., Derrick, J. S., Nistanaki, S. K., Smith, P. T., and Chang, C. J. (2018b). Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins. Chem. Sci. 9, 2952–2960. doi: 10.1039/C7SC04682K
Pegis, M. L., Wise, C. F., Koronkiewicz, B., and Mayer, J. M. (2017). Identifying and breaking scaling relations in molecular catalysis of electrochemical reactions. J. Am. Chem. Soc. 139, 11000–11003. doi: 10.1021/jacs.7b05642
Rail, M. D., and Berben, L. A. (2011). Directing the reactivity of [HFe4N(CO)12]− toward H+ or CO2 reduction by understanding the electrocatalytic mechanism. J. Am. Chem. Soc. 133, 18577–18579. doi: 10.1021/ja208312t
Rao, H., Schmidt, L. C., Bonin, J., and Robert, M. (2017). Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 548:74. doi: 10.1038/nature23016
Ray, K., Pfaff, F. F., Wang, B., and Nam, W. (2014). Status of reactive non-heme metal–oxygen intermediates in chemical and enzymatic reactions. J. Am. Chem. Soc. 136, 13942–13958. doi: 10.1021/ja507807v
Reback, M. L., Ginovska-Pangovska, B., Ho, M. H., Jain, A., Squier, T. C., Raugei, S., et al. (2013). The role of a dipeptide outer-coordination sphere on H2-production catalysts: influence on catalytic rates and electron transfer. Chem. Eur. J. 19, 1928–1941. doi: 10.1002/chem.201202849
Riplinger, C., and Carter, E. A. (2015). Influence of weak Brønsted acids on electrocatalytic CO2 reduction by manganese and rhenium bipyridine catalysts. ACS Catal. 5, 900–908. doi: 10.1021/cs501687n
Riplinger, C., Sampson, M. D., Ritzmann, A. M., Kubiak, C. P., and Carter, E. A. (2014). Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide. J. Am. Chem. Soc. 136, 16285–16298. doi: 10.1021/ja508192y
Rotundo, L., Garino, C., Priola, E., Sassone, D., Rao, H., Ma, B., et al. (2019). Electrochemical and photochemical reduction of CO2 catalyzed by Re(I) complexes carrying local proton sources. Organometallics 38, 1351–1360. doi: 10.1021/acs.organomet.8b00588
Rountree, E. S., McCarthy, B. D., Eisenhart, T. T., and Dempsey, J. L. (2014). Evaluation of homogeneous electrocatalysts by cyclic voltammetry. Inorg. Chem. 53, 9983–10002. doi: 10.1021/ic500658x
Roy, S., Sharma, B., Pécaut, J., Simon, P., Fontecave, M., Tran, P. D., et al. (2017). Molecular cobalt complexes with pendant amines for selective electrocatalytic reduction of carbon dioxide to formic acid. J. Am. Chem. Soc. 139, 3685–3696. doi: 10.1021/jacs.6b11474
Sahu, S., Cheung, P. L., Machan, C. W., Chabolla, S. A., Kubiak, C. P., and Gianneschi, N. C. (2017). Charged macromolecular rhenium bipyridine catalysts with tunable CO2 reduction potentials. Chem. Eur. J. 23, 8619–8622. doi: 10.1002/chem.201701901
Sahu, S., and Goldberg, D. P. (2016). Activation of dioxygen by iron and manganese complexes: a heme and nonheme perspective. J. Am. Chem. Soc. 138, 11410–11428. doi: 10.1021/jacs.6b05251
Sampson, M. D., Froehlich, J. D., Smieja, J. M., Benson, E. E., Sharp, I. D., and Kubiak, C. P. (2013). Direct observation of the reduction of carbon dioxide by rhenium bipyridine catalysts. Energy Environ. Sci. 6, 3748–3755. doi: 10.1039/c3ee42186d
Sampson, M. D., and Kubiak, C. P. (2016). Manganese electrocatalysts with bulky bipyridine ligands: utilizing lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 138, 1386–1393. doi: 10.1021/jacs.5b12215
Sampson, M. D., Nguyen, A. D., Grice, K. A., Moore, C. E., Rheingold, A. L., and Kubiak, C. P. (2014). Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: eliminating dimerization and altering catalysis. J. Am. Chem. Soc. 136, 5460–5471. doi: 10.1021/ja501252f
Savéant, J.-M. (2008). Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 108, 2348–2378. doi: 10.1021/cr068079z
Savéant, J.-M. (2018). Molecular catalysis of electrochemical reactions. Cyclic voltammetry of systems approaching reversibility. ACS Catal. 8, 7608–7611. doi: 10.1021/acscatal.8b02007
Sawers, G. (1994). The hydrogenases and formate dehydrogenases of Escherichia coli. Antonie Van Leeuwenhoek 66, 57–88. doi: 10.1007/BF00871633
Schneider, J., Jia, H., Kobiro, K., Cabelli, D. E., Muckerman, J. T., and Fujita, E. (2012). Nickel (II) macrocycles: highly efficient electrocatalysts for the selective reduction of CO2 to CO. Energy Environ. Sci. 5, 9502–9510. doi: 10.1039/c2ee22528j
Sen, P., Mondal, B., Saha, D., Rana, A., and Dey, A. (2019). Role of 2nd sphere H-bonding residues in tuning the kinetics of CO2 reduction to CO by iron porphyrin complexes. Dalton Trans. 48, 5965–5977. doi: 10.1039/C8DT03850C
Senftle, T. P., and Carter, E. A. (2017). The holy grail: chemistry enabling an economically viable CO2 capture, utilization, and storage strategy. Acc. Chem. Res. 50, 472–475. doi: 10.1021/acs.accounts.6b00479
Seu, C. S., Appel, A. M., Doud, M. D., DuBois, D. L., and Kubiak, C. P. (2012). Formate oxidation via β-deprotonation in [Ni(PR2NR′2)2(CH3CN)]2+ complexes. Energy Environ. Sci. 5, 6480–6490. doi: 10.1039/c2ee03341k
Shirley, H., Su, X., Sanjanwala, H., Talukdar, K., Jurss, J. W., and Delcamp, J. H. (2019). Durable solar-powered systems with Ni-catalysts for conversion of CO2 or CO to CH4. J. Am. Chem. Soc. 141, 6617–6622. doi: 10.1021/jacs.9b00937
Sinha, S., Berdichevsky, E. K., and Warren, J. J. (2017). Electrocatalytic CO2 reduction using rhenium(I) complexes with modified 2-(2′-pyridyl)imidazole ligands. Inorganica Chim. Acta 460, 63–68. doi: 10.1016/j.ica.2016.09.019
Sinha, S., Ghosh, M., and Warren, J. J. (2019). Changing the selectivity of O2 reduction catalysis with one ligand heteroatom. ACS Catal. 9, 2685–2691. doi: 10.1021/acscatal.8b04757
Sinha, S., and Warren, J. J. (2018). Unexpected solvent effect in electrocatalytic CO2 to CO conversion revealed using asymmetric metalloporphyrins. Inorg. Chem. 57, 12650–12656. doi: 10.1021/acs.inorgchem.8b01814
Smieja, J. M., Benson, E. E., Kumar, B., Grice, K. A., Seu, C. S., Miller, A. J. M., et al. (2012). Kinetic and structural studies, origins of selectivity, and interfacial charge transfer in the artificial photosynthesis of CO. Proc. Natl. Acad. Sci. U.S.A. 109, 15646–15650. doi: 10.1073/pnas.1119863109
Smieja, J. M., Sampson, M. D., Grice, K. A., Benson, E. E., Froehlich, J. D., and Kubiak, C. P. (2013). Manganese as a substitute for rhenium in CO2 reduction catalysts: the importance of acids. Inorg. Chem. 52, 2484–2491. doi: 10.1021/ic302391u
Smith, P. T., Benke, B. P., Cao, Z., Kim, Y., Nichols, E. M., Kim, K., et al. (2018). Iron porphyrins embedded into a supramolecular porous organic cage for electrochemical CO2 reduction in water. Angew. Chem. Int. 57, 9684–9688. doi: 10.1002/anie.201803873
Song, J., Klein, E. L., Neese, F., and Ye, S. (2014). The mechanism of homogeneous CO2 reduction by Ni(cyclam): product selectivity, concerted proton–electron transfer and C–O bond cleavage. Inorg. Chem. 53, 7500–7507. doi: 10.1021/ic500829p
Sullivan, B. P., Bolinger, C. M., Conrad, D., Vining, W. J., and Meyer, T. J. (1985). One-and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine). J. Am. Chem. Soc. Chem. Commun. 20, 1414–1416. doi: 10.1039/C39850001414
Suna, Y., Ertem, M. Z., Wang, W.-H., Kambayashi, H., Manaka, Y., Muckerman, J. T., et al. (2014). Positional effects of hydroxy groups on catalytic activity of proton-responsive half-sandwich Cp* Iridium (III) complexes. Organometallics 33, 6519–6530. doi: 10.1021/om500832d
Sung, S., Kumar, D., Gil-Sepulcre, M., and Nippe, M. (2017). Electrocatalytic CO2 reduction by imidazolium-functionalized molecular catalysts. J. Am. Chem. Soc. 139, 13993–13996. doi: 10.1021/jacs.7b07709
Sung, S., Li, X., Wolf, L. M., Meeder, J. R., Bhuvanesh, N. S., Grice, K. A., et al. (2019). Synergistic effects of imidazolium-functionalization on fac-Mn(CO)3 bipyridine catalyst platforms for electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 141, 6569–6582. doi: 10.1021/jacs.8b13657
Taheri, A., and Berben, L. A. (2016). Tailoring electrocatalysts for selective CO2 or H+ reduction: iron carbonyl clusters as a case study. Inorg. Chem. 55, 378–385. doi: 10.1021/acs.inorgchem.5b02293
Taheri, A., Thompson, E. J., Fettinger, J. C., and Berben, L. A. (2015). An iron electrocatalyst for selective reduction of CO2 to formate in water: including thermochemical insights. ACS Catal. 5, 7140–7151. doi: 10.1021/acscatal.5b01708
Wang, W.-H., Muckerman, J. T., Fujita, E., and Himeda, Y. (2013). Mechanistic insight through factors controlling effective hydrogenation of CO2 catalyzed by bioinspired proton-responsive iridium (III) complexes. ACS Catal. 3, 856–860. doi: 10.1021/cs400172j
Wang, W. H., Hull, J., Muckerman, J. T., Fujita, E., and Himeda, Y. (2012). Second-coordination-sphere and electronic effects enhance iridium(III)-catalyzed homogeneous hydrogenation of carbon dioxide in water near ambient temperature and pressure. Energy Environ. Sci. 5, 7923–7926. doi: 10.1039/C2EE21888G
Wang, W. H., Xu, S., Manaka, Y., Suna, Y., Kambayashi, H., Muckerman, J. T., et al. (2014). Formic acid dehydrogenation with bioinspired iridium complexes: a kinetic isotope effect study and mechanistic insight. ChemSusChem 7, 1976–1983. doi: 10.1002/cssc.201301414
Weng, Z., Jiang, J., Wu, Y., Wu, Z., Guo, X., Materna, K. L., et al. (2016). Electrochemical CO2 reduction to hydrocarbons on a heterogeneous molecular Cu catalyst in aqueous solution. J. Am. Chem. Soc. 138, 8076–8079. doi: 10.1021/jacs.6b04746
Wilson, A. D., Newell, R. H., McNevin, M. J., Muckerman, J. T., Rakowski DuBois, M., and DuBois, D. L. (2006). Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J. Am. Chem. Soc. 128, 358–366. doi: 10.1021/ja056442y
Wilting, A., Stolper, T., Mata, R. A., and Siewert, I. (2017). Dinuclear rhenium complex with a proton responsive ligand as a redox catalyst for the electrochemical CO2 reduction. Inorg. Chem. 56, 4176–4185. doi: 10.1021/acs.inorgchem.7b00178
Wong, K.-Y., Chung, W.-H., and Lau, C.-P. (1998). The effect of weak Brönsted acids on the electrocatalytic reduction of carbon dioxide by a rhenium tricarbonyl bipyridyl complex. J. Electroanal. Chem. 453, 161–170. doi: 10.1016/S0022-0728(98)00116-8
Yang, W., Sinha Roy, S., Pitts, W. C., Nelson, R. L., Fronczek, F. R., and Jurss, J. W. (2018). Electrocatalytic CO2 reduction with cis and trans conformers of a rigid dinuclear rhenium complex: comparing the monometallic and cooperative bimetallic pathways. Inorg. Chem. 57, 9564–9575. doi: 10.1021/acs.inorgchem.8b01775
Zahran, Z. N., Mohamed, E. A., and Naruta, Y. (2016). Bio-inspired cofacial Fe porphyrin dimers for efficient electrocatalytic CO2to CO conversion: overpotential tuning by substituents at the porphyrin rings. Sci. Rep. 6:24533. doi: 10.1038/srep24533
Keywords: inorganic, electrocatalysis, CO2, secondary-sphere, molecular
Citation: Nichols AW and Machan CW (2019) Secondary-Sphere Effects in Molecular Electrocatalytic CO2 Reduction. Front. Chem. 7:397. doi: 10.3389/fchem.2019.00397
Received: 30 January 2019; Accepted: 17 May 2019;
Published: 13 June 2019.
Edited by:
Soumyajit Roy, Indian Institute of Science Education and Research Kolkata, IndiaCopyright © 2019 Nichols and Machan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles W. Machan, bWFjaGFuJiN4MDAwNDA7dmlyZ2luaWEuZWR1
orcid.org/0000-0002-5182-1138