 Zili Li
Zili Li Nuchareenat Wiratpruk
Nuchareenat Wiratpruk Peter J. Barnard
Peter J. Barnard- Department of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC, Australia
A modular stepwise synthetic method has been developed for the preperation of tetra-imidazolium macrocycles. Initially a series of three bis(imidazolylmethyl)benzene precursors were alkylated with 1,2-dibromoethane to produce the corresponding bis-bromoethylimidazolium bromide salts. In the second step the bis-bromoethylimidazolium bromide salts were reacted with selected bis(imidazolylmethyl)benzene molecules to produce a series of two symmetrical and three asymmetrical tetra-imidazolium macrocycles. These tetra-imidazolium salts act receptors for anions and 1H-NMR titration studies were used to determine the association constants between two of the macrocycles and the halide anions chloride, bromide and iodide. The tetra-imidazolium salts are precursors for N-heterocyclic carbene (NHC) ligands and the corresponding silver(I), gold(I), and palladium(II) NHC complexes have been prepared. Varied structures were obtained, which depend on the chosen macrocyclic ligand and metal ion and in the case of the coinage metals Ag(I) and Au(I), mono, di, and hexanuclear complexes were formed.
Introduction
Imidazolium linked macrocycles have attracted significant recent attention because of their capacity to act as anion receptors and to function as pro-ligands for the synthesis of N-heterocyclic carbene metal complexes. Due to the great importance of negatively charged anionic species in biology, the preparation of receptor molecules designed to recognize and sense anions is an area of great research interest (Beer and Gale, 2001; Gale, 2003; Martínez-Máñez and Sancenón, 2003). Imidazolium groups are now well-recognized for their favorable features for the generation of anion receptors, which result both from electrostatic and hydrogen bonding interactions (Alcalde et al., 1999, 2007; Chellappan et al., 2005; Wong et al., 2005; Yoon et al., 2006; Xu et al., 2010). For example, a series of tetra-imidazolium linked macrocyclic compounds e.g., I (Figure 1) were shown to bind to biologically relevant anions such as chloride and hydrogen sulfate (Wong et al., 2005; Serpell et al., 2011). Additionally, compounds of this type have been used as selective luminescent sensors for nucleic acids and nucleotide derivatives such as DNA, RNA, ATP, and GTP and to sense and image RNA in the living cells, as a result of strong C-H+···A− hydrogen bonding interations (Neelakandan and Ramaiah, 2008; Ahmed et al., 2011, 2012; Shirinfar et al., 2013).
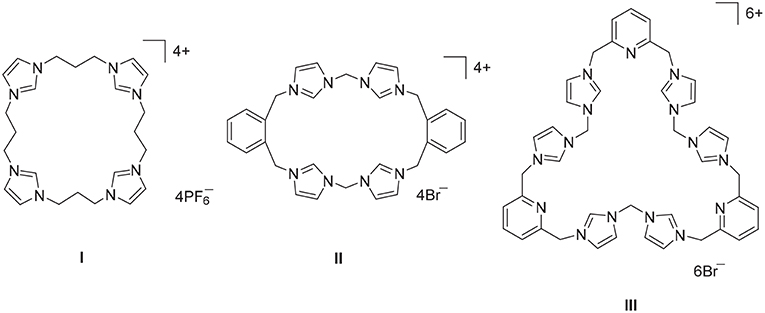
Figure 1. Polyimidazolium linked macrocyclic compound prepared by stepwise macrocyclization (I) and direct macrocyclization (II) and (III) methodologies.
A range of imidazolium linked macrocyclic compounds have been previously reported and both direct macrocyclization (Bass et al., 2010; Altmann et al., 2015; Toure et al., 2016) and stepwise macrocyclization (Mesquida et al., 2013) processes have been previously described for the synthesis of compounds of this type. In the direct macrocyclization approach, equal quantities of a bis-imidazole precursor, and a dihaloalkane are combined under high dilution conditions to produce the desired macrocyclic compound. For example, the imidazolium linked macrocycles II and III were prepared using the direct macrocyclization approach (Hahn et al., 2008; Schulte to Brinke et al., 2013). This approach can be limited due to unintended reactivity between the precursor compounds, which can produce complex reaction mixtures and poor yields of the desired product. Alternatively, stepwise macrocyclization can be used for the synthesis of polyimidazolium linked macrocyclic compounds (Schulte to Brinke and Hahn, 2015) and the synthesis of the tetra-imidazolium macrocycle I was achieved via sequential alkylation of bis-imidazolium precursors (Wong et al., 2005).
Polyimidazolium linked macrocycles have also been utilized as precursors for N-heterocyclic carbene metal complexes and previously the Au(III) complex IV (Figure 2) was prepared the reaction of KAuCl4 with a macrocyclic tetra-imidazolium salt II (Mageed et al., 2017). Additionally, dinuclear Au(II) and mixed valence Au(I)/Au(III) complexes of have been prepared by the oxidation of dinuclear Au(I) complexes of macrocyclic NHC ligands (Mageed et al., 2018). In the past decade, a number of Ag(I) complexes of macrocyclic NHC ligands that display wide range of structures have been reported (Mckie et al., 2007; Hahn et al., 2008; Schulte to Brinke et al., 2013; Altmann et al., 2016; Fei et al., 2017; Lu et al., 2017, 2018). For example, a sandwich like tetranuclear Ag(I) complex featuring two tetra-NHC ligands was prepared V (Figure 2) and this complex was utilized as a precursor for the preparation of Au(I), Ni(II), Pd(II), and Pt(II) complexes (Altmann et al., 2016). In addition, macrocyclic tetra-NHC ligands have been shown to act as tetradentate ligand for square-planer metals such as Pd(II), Ni(II), Cu(I), and Pt(II) (Fei et al., 2017) and the interesting square-planar Pt(II) complex VI (Figure 2) was prepared by metal-template reaction from the tetrakis(trimethylphosphane)platinum(II) triflate and 2-azidophenylisocyanide (Hahn et al., 2005). The biological properties of metal complexes of NHC-based cyclophane and macrocyclic ligands have also been of significant recent interest and Ag(I) and Au(I) complexes have been shown to possess potent antimicrobial and anticancer activities, respectively (Aweda et al., 2013; Shah et al., 2013; Nomiya et al., 2018; Pöthig et al., 2018). Youngs et al. have been particularly active in this field (Hindi et al., 2009; Johnson et al., 2017) and have described the potent antimicrobrial properties of a Ag(I)-NHC complexes of cyclophane ligands (Melaiye et al., 2005). Additionally, a series of Au(I) complexes of related cyclophane-based NHC ligand systems were shown to be selectively toxic to cancer cells as a result of an antimitochondrial mechanism (Barnard et al., 2004, 2006). Meyer and more recently Kühn have also extended the application of macrocyclic tetra-NHC ligands to the synthesis of iron complexes, that provide fascinating models of reactive intermediates that are generated in the catalytic cycles of a range of heme and non-heme iron enzymes (Meyer et al., 2013; Anneser et al., 2015).
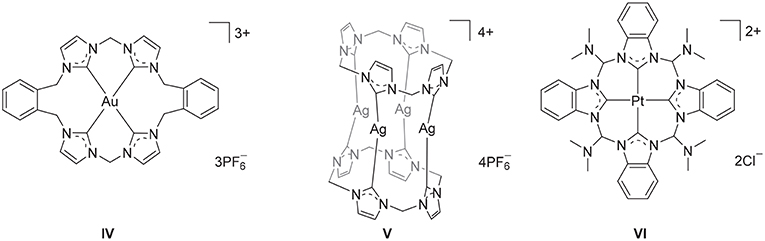
Figure 2. Selected metal complexes derived from macrocyclic imidazolium pro-ligands.
As imidazolium linked macrocycles offer the potential for the generations of novel sensors for biologically significant anions and as precursors for NHC metal complexes of metals that are well-known for their biological properties, we became interested in developing new strategies for the synthesis of compounds of this type. In the present paper, we report a novel modular stepwise synthetic approach for the synthesis of tetra-imidazolium macrocycles. This approach involves the initial synthesis of bis-bromoethylimidazolium bromide precursors, which can then be utilized for the formation of either symmetrical or asymmetrical tetra-imidazolium macrocycles. These tetra-imidazolium salts bind anions in solution and association constants between two of the macrocycles and the halide anions chloride, bromide, and iodide were determined for two macrocycles. A range of silver(I), gold(I), and palladium(II) NHC complexes have been prepared from these pro-ligands.
Results and Discussion
Synthesis of Tetra-imidazolium Macrocycles
The ortho-phenylene linked tetra-imidazolium macrocycle 9·Br4 was prepared via a stepwise macrocyclization procedure. Initially, 1,2-bis(imidazolylmethyl)benzene 1(Baker et al., 2001) was alkylated with an excess of 1,2-dibromoethane (30 equivalents) to obtain the bis-bromoethylimidazolium bromide salt 5·Br2 in a moderate yield (Scheme 1). The 1H-NMR spectrum of 5·Br2 shows the imidazolium C2-H proton resonates as a singlet signal at 9.50 ppm, while the ethylene group protons resonate as two upfield shifted triplet signals at 4.00 and 4.68 ppm. The macrocycle 9·Br4, was prepared by heating an equimolar mixture of 5·Br2 and 1 in a solvent system consisting of a 1:4 mixture of DMF and acetonitrile under high dilution conditions (Scheme 1) and was isolated in a moderate yield of 29.7%. Compound 9·Br4 gave a simple 1H-NMR spectrum consistent with its high symmetry (point group D2h) with downfield shifted signals for the imidazolium protons which resonated at 9.45 (C2-H) and 7.98 and 7.88 ppm (C4-H/C5-H). The protons of the ethylene linker groups resonated as a singlet signal at 4.81 ppm. A similar method was adopted for the synthesis of the meta-phenylene linked macrocycle 10·Br4 from a mixture of 2 and 6·Br2 (Scheme 1).
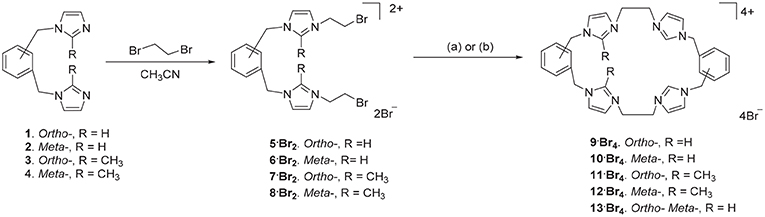
Scheme 1. Synthesis of tetra-imidazolium macrocyclic compounds 9·Br4-13·Br4. (a) DMF/CH3CN 1:4, 110°C. (b) Bu4N·Br (6.0 eq.), DMF/CH3CN 1:4, 110°C.
With the successful synthesis of the symmetrical macrocycles 9·Br4 and 10·Br4, we were interested in further exploring the versatility of our stepwise synthetic methodology with the synthesis of asymmetric tetra-imidazolium salts. In initial studies, reaction of either 3 with 5·Br2 or 1 with 7·Br2 (Scheme 1) under high dilution condition did not successfully produce the desired asymmetrical tetra-imidazolium salts. Additionally, attempts to use tetra-n-butylammonium bromide (Bu4N·Br) as a templation reagent in the reaction of 3 with 5·Br4 also did not give the desired product. By contrast, the reaction of compound 1 with 7·Br2 under high dilution condition in presence of Bu4N·Br gave the desired product 11·Br4 in a low yield. The 1H-NMR spectrum of 11·Br4 showed the imidazolium C2-H proton as singlet signal at 9.76 ppm and the methyl group on the imidazolium C2 carbon resonated as a singlet signal at 2.64 ppm. In addition, consistent with the lower symmetry structure (point group C2h) the benzylic proton resonates as two singlet signals at 5.47 and 5.52 ppm. The asymmetrical pro-ligands 12·Br4 and 13·Br4 were prepared in a similar manner from the reaction of either 2 and 8·Br2 or 2 and 5·Br2, respectively, in presence of Bu4N·Br. Again, the 1H-NMR spectrum for 13·Br4 is consistent with the lower symmetry structure, with the C2-H protons resonating as two singlet signals at 8.99 and 9.13 ppm.
Synthesis of Ag(I), Au(I), and Pd(II) Complexes
The Ag(I) complex 14·(PF6)6 was prepared by the reaction of pro-ligand 9·Br4 with Ag2O in DMF with the exclusion of light (Scheme 2) and product was isolated as a white crystalline solid in a yield of 21%. The 1H-NMR spectrum of 14·(PF6)6 showed no imidazolium C2-H proton signal, indicating that the C2 carbon is deprotonated and coordinated to the metal center as a carbene. The ortho-substituted phenyl-linker group protons resonate as two set of doublets (5.68 and 6.94 ppm) and triplet (7.17 and 7.52 ppm) signals, consistent with a more complex magnetic environment for the Ag(I) complex when compared to the pro-ligand. The 13C-NMR spectrum for 14·(PF6)6 revealed a downfield shifted signal at 182 ppm, for which 107Ag-13C (d, 1J = 182.32 Hz) and 109Ag-13C (d, 1J = 209.99 Hz) couplings were observed, which is also consistent with coordination of the C2 carbon to Ag(I). The high-resolution mass spectrum for 14·(PF6)6 produced a series of peaks consistent with a hexanuclear structure with the general formula [Ag6L3]6+ (where L is the macrocyclic tetra-carbene ligand). For example, a peak was observed at m/z = 372.0334, which corresponds to the formula [C96H96N24Ag6]6+ (calculated = 372.0419).
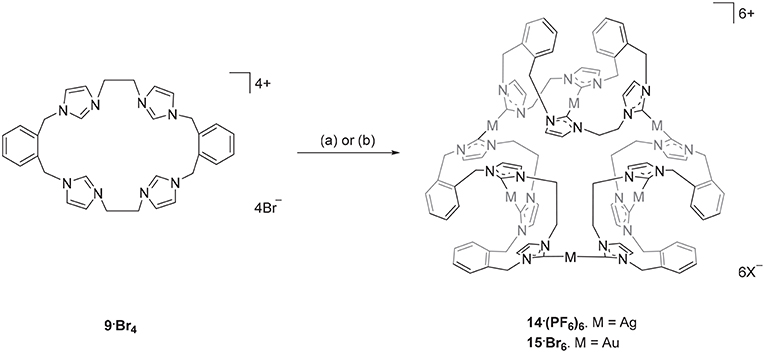
Scheme 2. Synthesis of the Ag(I) and Au(I) complexes derived from pro-ligand 9·Br4. (a) Ag2O (4.0 eq.), DMF, 50°C, 3 d. (b) (THT)AuCl (2.2 eq.), NaOAc, DMF, 110°C, 1 h.
Using a similar approach, the Ag(I) complex 17·(PF6)3 was prepared from the pro-ligand 10·Br4, however for this compound the 1H-NMR spectrum showed a signal at 8.37 ppm, consistent with the C2-H proton being present in the complex. However, the 13C-NMR spectrum of 17·(PF6)3 showed a downfield shifted signal at 180.4 ppm that displays 107Ag-13C (d, 1J = 183.58 Hz) and 109Ag-13C (d, 1J = 211.24 Hz) couplings, indicating coordination of the imidazole C2 carbon to Ag(I). These NMR results are consistent with a mononuclear complex, where two of the imidazole units are coordinated to the metal, while the other two remain uncoordinated imidazolium units (Scheme 3). To further investigate this result, a different synthetic method was undertaken where the pro-ligand 10·(PF6)4 was reacted with AgNO3 in present of NH4OH, however the same mononuclear Ag(I) complex was obtained (Scheme 3).
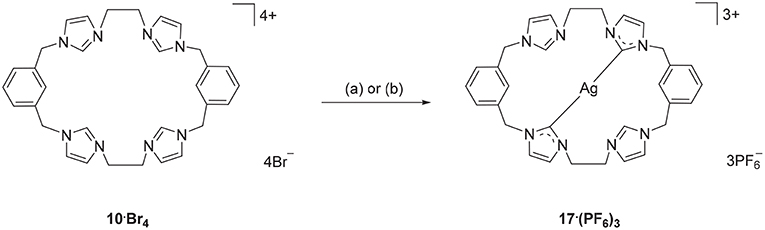
Scheme 3. Synthesis of Ag(I) complex 17·(PF6)3 from the pro-ligand 10·Br4. (a) Ag2O (4.0 eq.), DMF, 70°C, 3 d. (b) AgNO3 (4.0 eq.), NH4OH (eq.), CH3CN, RT, overnight.
The hexanuclear Au(I) complex 15·Br6 was prepared by the reaction of 9·Br4 with (THT)AuCl in present of the mild base sodium acetate (Scheme 2) and the complex was obtained as an off-white solid in 54.2% yield. The same approach was used in an attempt to prepare the Au(I) complex of pro-ligand 10·Br4, however no complex could be isolated from the reaction mixture. In the next set of reactions, the pro-ligand 11·Br4 (with both normal and C2-blocked imidazolium groups) was reacted with (THT)AuCl in presence of sodium acetate. It was anticipated that this ligand might produce a complex displaying both “normal” and “abnormal” NHC coordination modes, however the dinuclear Au(I) complex 18·(PF6)2 was obtained, which displayed only the “normal” NHC coordination mode (Scheme 4). Using the same method, a dinuclear Au(I) complex derived from 19·(PF6)4 was also prepared.
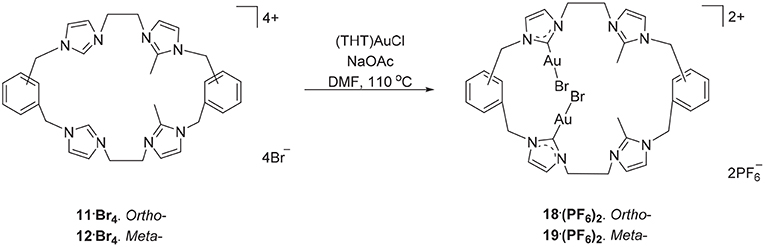
Scheme 4. Synthesis of dinuclear Au(I) complex derived from 11·Br4 to 12·Br4.
Due to their potential to act as tetradentate ligands with metals that adopt square-planar coordination geometries, the Pd(II) complex of pro-ligand 9·Br4 was prepared. A range of approached have been previously employed for the preparation of Pd(II)-NHC complexes, including in situ deprotonation and metallation (Baker et al., 2001; Fei et al., 2017) and transmetallation via an intermediate Ag(I) complex (Schulte to Brinke et al., 2013; Andrew et al., 2016). In this work the former in situ deprotonation and metallation approach was initially investigated by reacting 9·(PF6)4 with Pd(OAc)2 in DMSO, however the desired Pd(II) complex could not be isolated. As described previously, a hexanuclear Ag(I) complex could be prepared from the pro-ligand 9·(PF6)4 and in a second attempt to synthesize the Pd(II) complex, pro-ligand 9·Br4 was first reacted with Ag2O to form the Ag(I) complex in situ followed by addition of K2PdCl4 (Scheme 5). 1H-NMR analysis of Pd(II) complex 16·(PF6)2 showed a relatively simple spectrum with the C4/5 protons of the NHC groups resonating as two sets of doublets at 7.48 and 7.83 ppm. The benzylic protons resonate as two sets of doublet signals at 5.20 and 6.44 ppm (AX pattern), which is consistent with a rigid molecular structure in solution. Furthermore, the 13C-NMR spectrum showed a downfield shifted signal at 167.41 ppm, which corresponds to the NHC carbene carbon coordinated to the Pd(II) metal center.
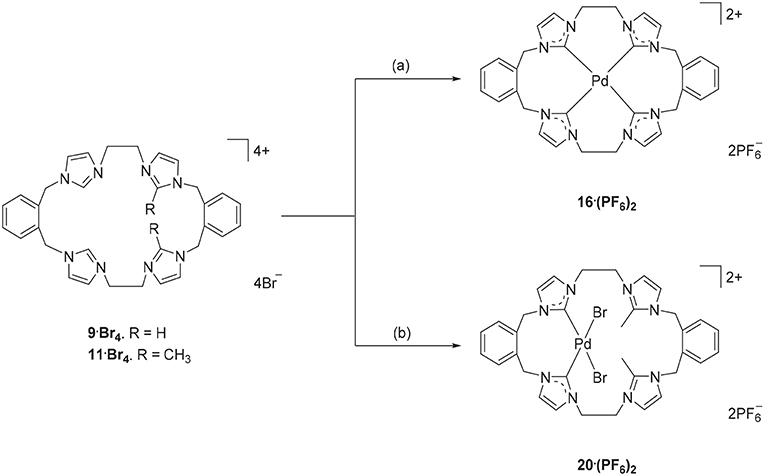
Scheme 5. Synthesis of Pd(II) complex 16·(PF6)2 and 20·(PF6)2. (a) 9·Br4, Ag2O (4.0 eq.), K2PdCl4, DMF, 85°C. (b) 11·Br4, K2PdCl4, DMSO, 85°C, overnight.
The Ag(I) transmetallation approach did not successfully produced a Pd(II) complex of pro-ligand 11·Br4. In this attempt, Ag2CO3 was used instead of Ag2O to avoid the undesirable oxidative cleavage of the C2-blocking methyl group which has been observed previously (Chianese et al., 2004). In a second synthetic attempt, the pro-ligand 11·Br4 was reacted with K2PdCl4 in presence of NaOAc. The 1H-NMR of the crude product showed no peak for the C2-H proton for the “unblocked” imidazole units indicating that these groups are coordinated to the metal center. The C4/5 protons of the C2 “blocked” imidazolium group at 7.34 and 7.66 ppm suggesting that C2 blocked imidazolium unit did not react with the metal. Furthermore, the benzylic proton signals at 5.24 and 5.26 ppm and the ethylene linker signal at 4.61–4.70 ppm also indicated an asymmetrical structure of the title complex. The crystal structure in Figure 5 also suggested the Pd(II) complex 20·(PF6)2 was successfully synthesized. Unfortunately, it was unable to separate this complex as pure product.
Structural Studies
Compounds 5·Br2, 9·Br4, 10·Br3PF6, 11·Br4, 15·Br2(PF6)4, and 20·(PF6)2 were characterized by X-ray crystallography. A representation of the precursor compound 5·Br2 is shown in Figure S1 while representations of the tetra-imidazolium macrocycles 9·Br4, 10·Br3PF6, and 11·Br4 are shown in Figure 3. In all cases these imidazolium salts display hydrogen bonding interactions between various hydrogens on the cationic imidazolium units and the bromide counter anions. For example, the shortest C2···Br distances for 5·Br2, 9·Br4, 10·Br3PF6, and 11·Br4, respectively, are: 3.4496(3); 3.42219(5) Å; 3.4822(4) Å; and 3.6552 (5) Å, respectively, which fall within a typical range of C-H···Br distance caused by hydrogen bonding (Yuan et al., 2002).
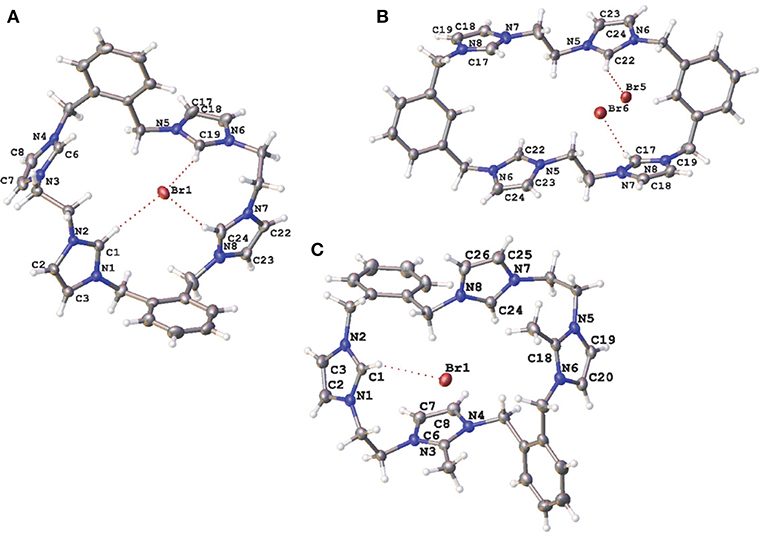
Figure 3. Representations of the X-ray crystal structures of (A) 9·Br4, (B) 10·Br3PF6, and (C) 11·Br4. Only counter ions involved in hydrogen bonding are included for clarity. Atomic displacement ellipsoids are shown at the 50% probability level.
Two representations of the X-ray crystal structure for the Au(I) complex 15·Br2(PF6)4 are shown in Figure 4. This remarkable structure reveals that the Au(I) complex adopts a cyclic hexanuclear supramolecular assembly with the overall formula [Au6L3]6+. In the cation, each of the Au(I) centers adopt linear two-coordinate geometries, and they can be divided into two distinct groups of three atoms each. For the first group, each Au(I) atoms (Au1, Au4, and Au5) is bound to two NHC units on opposite sides of each of the three ligand molecules. While each of the Au(I) atoms in the second group (Au2, Au3, and Au6) are coordinated to NHC donors from adjacent ligand molecules, resulting in the formation of a metallo-macrocycle. In each case, the ligand molecules are bowl shaped to accommodate the two Au(I) coordination modes. Interestingly, the metallo-macrocycle is arrayed around an encapsulated central bromide counterion. This bromide ion displays interactions with Au(I) atoms Au1, Au4, and Au5 with the distances being 3.19488(5), 3.24731(5), and 3.23064(4) Å, respectively. Previously, a hexanuclear Ag(I) complex was reported for a tetra-carbene ligand linked by aliphatic butyl chains (Fei et al., 2017).
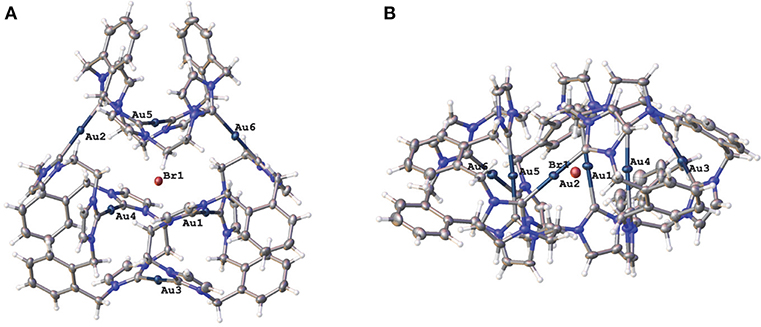
Figure 4. (A) Two representations of the X-ray crystal structures of 15·Br2(PF6)4 (A) top view and (B) edge view. Hydrogen atoms are excluded and only the encapsulated bromide counterion included for clarity. Atomic displacement ellipsoids are shown at the 50% probability level.
A representation of the X-ray crystal structure for the Pd(II) 20·(PF6)2 is shown in Figure 5. The molecular structure shows that the ligand is coordinated to the metal center through the “normal” NHC groups, with the C2 “blocked” groups present as cationic imidazolium units. The Pd(II) center is four-coordinate, with the two-remaining sited being occupied by bromide ions.
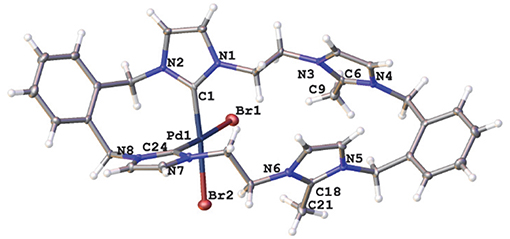
Figure 5. Representations of the X-ray crystal structures of the Pd(II) complex 20·(PF6)2. Atomic displacement ellipsoids are shown at the 50% probability level.
Anion Binding Studies
Due to the tetracationic charge of the imidazolium linked macrocycles prepared in this work, the propensity of 9·(PF6)4 and 10·(PF6)4 to bind to the halide anions F− Cl−, Br−, and I− (as their tetra-n-butylammonium halide salts) was evaluated using 1H-NMR titration experiments. In initial studies, it was found that the addition of Bu4N·F to 10·(PF6)4 in d6-DMSO caused an immediate color change to pale yellow. The 1H-NMR analysis showed that the color change occurred concurrently with a significant broadening and downfield shift of the imidazolium C2-H signal. In addition, the appearance of new unidentified 1H-NMR signals were observed which were consistent with decomposition of the macrocyclic receptor. As such this anion was not studied further.
Addition of increasing equivalents of Bu4N·Cl (0.25–14.0 eq.) to a solution of 9·(PF6)4 in d6-DMSO caused a significant downfield shift of the resonance corresponding to the imidazolium C2-H signal from 8.96 to 9.64 ppm. In addition, the benzylic proton signal was also shifted downfield from 5.34 to 5.76 ppm. Unfortunately, this study was hampered by the gradual precipitation of the imidazolium salt at higher Cl− concentrations. In a similar manner, addition of increasing equivalents of Bu4N·Br and Bu4N·I to 9·(PF6)4 caused downfield shifts in the imidazolium C2-H signal, although to a lesser extent than that seen for Bu4N·Cl, however for these anions no precipitation of the macrocycle was seen. Figure 6 shows the 1H-NMR titration between 9·(PF6)4 and Bu4N·Br, while Figure 7 shows the change in the imidazolium C2-H chemical shift for 9·(PF6)4 in response to increasing equivalents of the added halide anion (solid black lines). Similar results were seen for the macrocyclic receptor 10·(PF6)4 and the plot of experimental titration data is shown in Figure S2. Job plot analysis was then used to evaluate the stoichiometry of the interactions between the macrocyclic receptors and the anions Cl−, Br−, and I−. The results of these studies Figures S3, S4 showed that maxima occurred at close to χ = 0.5 indicating 1:1 stoichiometry for both receptor 94+ and 104+ with these anions.
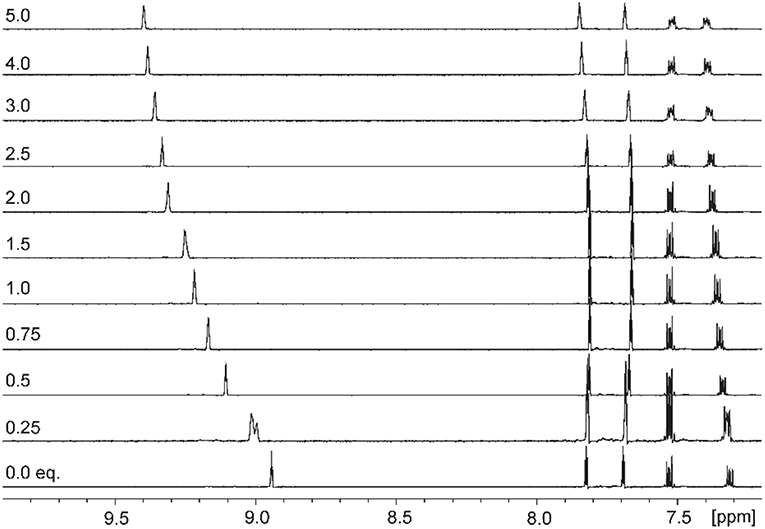
Figure 6. 1H-NMR titration of 9·(PF6)4 in d6-DMSO in the presence of increasing concentrations of Bu4N·Br.
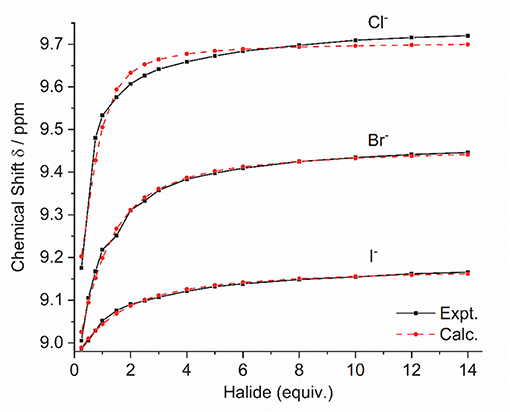
Figure 7. Experimental titration data (black line, squares) and fitted binding isotherms (red dashed line, circles) for the addition of the Bu4N+ halide anion to a solution of 9·(PF6)4 in d6-DMSO.
Binding (association) constants Ka (M−1) for the tetra-imidazolium macrocycles 9·(PF6)4 and 10·(PF6)4 were determined by analysis of the 1H-NMR titration data using the computer program HypNMR 2018 (Frassineti et al., 1995, 2003). The fitted binding isotherms are shown in Figure 6 and Figure S2 (red dashed lines) and the calculated binding constants are given in Table 1. The values obtained for the compounds studied in this work are similar in magnitude to those reported previously for related compounds. For example, Beer et al. reported association constants of 420(23), 241(3), and 120(1) M−1 for a tetra-imidazolium macrocycle in d6-DMSO solution with the anions Cl−, Br− and I−, respectively (Serpell et al., 2011).
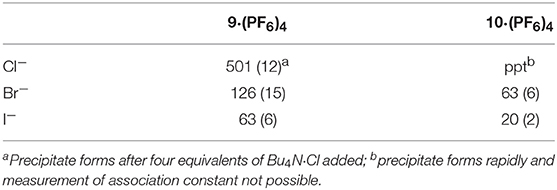
Table 1. Association constants of the tetra-imidazolium macrocycles 9·(PF6)4 and 10·(PF6)4 with F−, Cl−, Br−, and I−.
Conclusion
In conclusion, a novel stepwise synthetic strategy is reported that allows for the synthesis of both symmetrical and asymmetrical tetra-imidazolium linked macrocyclic compounds. The synthetic strategy is modular as initially a range of bis-bromoethylimidazolium bromide precursors were synthesized, which when combined with chosen bis(imidazolylmethyl)benzene molecules produced a range of tetra-imidazolium linked macrocycles. These tetra-imidazolium salts are of significant interest as they bind anions in solution and offer the potential for the development of sensors for biologically relevant anions. Using 1H-NMR titration studies, the association constants between 9·(PF6)4 and 10·(PF6)4 and the halide anions Cl−, Br−, and I− were determined and these ranged between 501 and 20 M−1. Imidazolium salts are precursors for N-heterocyclic carbene (NHC) ligands and given that Ag(I) and Au(I) complexes of NHC ligands have been shown to display antimicrobial (Aweda et al., 2013; Shah et al., 2013) and anticancer(Barnard et al., 2004) properties, respectively, the Ag(I), Au(I), and Pd(II) NHC complexes derived from these tetra-imidazolium linked macrocycles were prepared. The Ag(I) complexes 146+ and the analogous Au(I) complex 156+ adopted intriguing hexanuclear structures with the general formula [M6L3]6+. Currently we are exploring the development of silver complexes of NHC ligands as potential antimicrobial agents and supramolecular assemblies such as complex 146+ are a particular focus for the slow release of silver ions and the results of these studies will be reported in due course.
Experimental
General Details
All solvents and chemicals were purchased from Sigma-Aldrich, Chem Supply, Alfa Aesar, and were used as received unless otherwise stated. Where necessary, solvents were further purified using an Innovative Technology Pure Solv solvent purification system. All experiments were performed under an atmosphere of N2 unless otherwise stated. 1H-NMR and 13C-NMR spectra were recorded on Bruker Advance 500 (500.023 MHz for 1H and 125.74 MHz for 13C) and spectra were referenced to solvent resonances. Where required, COZY, HSQC, HMBC, and NOESY 2-dimensional experiments were used to assist assignments. Mass spectra were obtained using an Agilent 6530 Q-TOF LC/MS mass spectrometer fitted with an Agilent electrospray ion (ESI) source.
X-ray Crystallography
Single crystals suitable for X-ray diffraction studies were grown as follows: 5·Br2 and 9·Br4 diffusion of diethyl ether into an methanol solution of the title compound; 10·Br3PF6 diffusion of diethyl ether into an acetonitrile solution of the title compound containing two drops of a solution of tetra-n-butylammonium bromide in acetonitrile; 11·Br4 slow evaporation of a methanol solution of the title compound; 15·Br2(PF6)4 diffusion of ethyl acetate into an acetonitrile solution of the title compound; 20·(PF6)2 was grown by slow diffusion of diethyl ether into an acetonitrile solution of the titled compound. Crystallographic data for all structures determined are given in Table S1. For all samples, crystals were removed from the crystallization vial and immediately coated with paratone oil on a glass slide. A suitable crystal was mounted in Paratone oil on a glass fiber and cooled rapidly to 173 K in a stream of cold N2 using an Oxford low temperature device. Diffraction data were measured using an Rigaku Oxford Diffraction SuperNova X-ray Diffraction System mounted with Mo-Kα λ = 0.71073 Å and Cu-Kα λ = 1.54184. Data were reduced and corrected for absorption using the CrysAlis Pro program. The SHELXL2013-2 program was used to solve the structures with Direct Methods, with refinement by the Full-Matrix Least-Squares refinement techniques on F2. The non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed geometrically and refined using the riding model. Coordinates and anisotropic thermal parameters of all non-hydrogen atoms were refined. All calculations were carried out using the program Olex2. Further XRD details are provided in the Supporting Information. CCDC 1885400-1885405 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
1H-NMR Titration Studies
A solution of the title compound (10 mg) in d6-DMSO (600 μL) and a 1.5 M solution of Bu4N·X (X = Cl, Br and I) in d6-DMSO were prepared, respectively. To the solution of title compound, increasing equivalents (0.25–14.0 eq.) of 1.5 M Bu4N·X solution was added and the resultant solution was thoroughly mixed. The 1H-NMR spectrum was recorded ~2 min after each addition at 302 K.
Jobs Plot Analysis
A solution of the title compound (10 mg/mL) in d6-DMSO and a 0.050 M solution of Bu4N·X (X = Cl, Br and I) in d6-DMSO were prepared, respectively. A varied fraction of title compound solution and Bu4N·X solution was added and diluted with d6-DMSO to 600 μL to maintain the total concentration of substance at 10 mM. The resultant solution was thoroughly mixed and 1H-NMR spectrum was recorded at 302 K.
Synthesis
4. Sodium hydride (1.14 g, 47.35 mmol) was added to a solution of 2-methylimidazole (3.11 g, 37.88 mmol) in DMF (50 mL) cooled to 0°C and the resultant mixture was stirred at RT for 1 h and α,α′-dibromo-m-xylene (5.00 g, 18.94 mmol) was then added. Stirring was continued at RT for 12 h and the mixture was then diluted with water (100 mL). The mixture was then extracted with CH2Cl2 (5 × 10 mL) and the combined organic extracts were washed with water (5 × 50 mL) and then brine (20 mL). The organic layer was dried with MgSO4 and the solvent was evaporated in vacuo yielding a yellow oil. Yield: 2.18 g, 43.2%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.33 (t, 3JH−H = 7.5 Hz, 1H, ArH), 7.08 (d, 3JH−H = 1.0 Hz, 2H, Himi), 7.02 (d, 3JH−H = 8.0 Hz, 2H, ArH), 6.96 (s, 1H, ArH), 6.76 (d, 3JH−H = 1.5 Hz, 2H, Himi), 5.12 (s, 4H, CH2), 2.19 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 144.36 (Cq), 138.55 (Cq), 129.64 (CAr), 126.96 (Cimi), 126.59 (CAr), 126.07 (CAr), 120.70 (Cimi), 48.94 (CH2), 13.18 (CH3). HRESI-MS+ (CH3CN): C14H15 m/z = 267.1543, calcd = 267.1604.
5·Br2. To a solution of 1,2-dibromoethane (21.69 mL, 251.7 mmol) in CH3CN (50 mL) stirred at 110°C, was added dropwise a solution of 1 (2.0 g, 8.39 mmol) in CH3CN (100 mL) over a period of 3 h. The resultant mixture was stirred at the same temperature for 12 h and then filtered whilst still hot. The filtrate was then evaporated in vacuo and the resulting solid was recrystallized from hot ethanol (20 mL) yielding a white crystalline solid. Yield: 2.19 g, 41.2%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 9.50 (s, 2H, Himi), 7.98 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.88 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.49 (dd, 3JH−H = 5.8 Hz, 4JH−H = 3.5 Hz, 2H, ArH), 7.30 (dd, 3JH−H = 5.5 Hz, 4JH−H = 3.5 Hz, 2H, ArH), 5.75 (s, 4H, CH2), 4.69 (t, 3JH−H = 6.0 Hz, 4H, CH2), 4.01 (t, 3JH−H = 6.0 Hz, 4H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 137.56 (Cimi), 133.38 (Cq), 130.10 (CAr), 129.83 (CAr), 123.42 (Cimi), 123.38 (Cimi), 50.76 (CH2), 49.61 (CH2), 32.90 (CH2). HRESI-MS+ (CH3OH): C18H22N4 m/z = 227.0094, calcd = 227.0090, C18H22N4 m/z = 532.9367, calcd = 532.9369.
6·Br2. This compound was prepared using the same method as described for 5·Br2 from 1,2-dibromoethane (10.80 mL, 125.90 mmol) and 2 (1.00 g, 4.20 mmol). The crude product was purified by a trituration with diethyl ether (3 × 10 mL) yielding a light brown oil. Yield: 0.62 g, 24.0%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 9.63 (s, 2H, Himi), 7.95 – 7.97 (m, 4H, Himi), 7.65 (s, 1H, ArH), 7.44 – 7.50 (m, 3H, ArH), 5.56 (s, 4H, CH2), 4.69 (t, 3JH−H = 5.9 Hz, 4H, CH2), 4.01 (t, 3JH−H = 5.9 Hz, 4H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 137.31 (Cimi), 136.02 (Cq), 130.19 (CAr), 129.08 (CAr), 128.90 (CAr), 123.36 (Cimi), 123.16 (Cimi), 52.05 (CH2), 50.76 (CH2), 32.90 (CH2). HRESI-MS+ (CH3OH): C18H22N4 m/z = 227.0102, calcd = 227.0090.
7·Br2. This compound was using the same method as described for 5·Br2 from 1,2-dibromoethane (4.85 mL, 56.31 mmol) and 3 (0.50 g, 1.88 mmol). The crude product was recrystallized from ethanol yielding a white solid. Yield: 0.62 g, 51.6%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.90 (d, 3JH−H = 2.1 Hz, 2H, Himi), 7.68 (d, 3JH−H = 2.1 Hz, 2H, Himi), 7.40 (dd, 3JH−H = 5.5 Hz, 4JH−H = 3.4 Hz, 2H, ArH), 6.82 (dd, 3JH−H = 5.7 Hz, 4JH−H = 3.5 Hz, 2H, ArH), 5.67 (s, 4H, CH2), 4.68 (t, 3JH−H = 5.9 Hz, 4H, CH2), 4.00 (t, 3JH−H = 5.9 Hz, 4H, CH2), 2.69 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 146.15 (Cq), 132.62 (Cq), 129.28 (CAr), 126.95 (CAr), 122.65 (Cimi), 122.43 (Cimi), 49.29 (CH2), 48.85 (CH2), 31.96 (CH2), 10.70 (CH3). HRESI-MS+ (CH3OH): C20H26N4 m/z = 241.0306, calcd = 241.0246.
8·Br2. This compound was prepared using the same method as described for 5·Br2 from 1,2-dibromoethane (8.15 mL, 94.61 mmol) and 4 (0.84 g, 3.15 mmol). The crude product was recrystallized from ethanol yielding a white solid. Yield: 0.76 g, 37.5%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.81 (s, 2H, Himi), 7.79 (s, 2H, Himi), 7.46 (t, 3JH−H = 7.8 Hz, 1H, ArH), 7.33 (s, 1H, ArH), 7.27 (d, 3JH−H = 7.7 Hz, 2H, ArH), 5.47 (s, 4H, CH2), 4.62 (t, 3JH−H = 6.0 Hz, 4H, CH2), 3.93 (t, 3JH−H = 5.9 Hz, 4H, CH2), 2.67 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 145.47 (Cq), 135.76 (Cq), 130.32 (CAr), 128.15 (CAr), 127.59 (CAr), 122.40 (Cimi), 122.35 (Cimi), 50.88 (CH2), 49.17 (CH2), 31.78 (CH2), 10.37 (CH3). HRESI-MS+ (CH3OH): C20H26N4 m/z = 241.0294, calcd = 241.0246, HRESI-MS+ (CH3OH): C20H26N4 m/z = 560.9702, calcd = 560.9682.
9·Br4. Solutions of 1 (0.31 g, 1.30 mmol) in CH3CN (100 mL) and 5·Br2 (0.80 g, 1.30 mmol) in DMF (100 mL) were added simultaneously dropwise to 150 mL of CH3CN heated at 110°C over a period of 3 h. The mixture was then stirred at the same temperature for a further 5 d during which time a precipitate formed. The precipitate was collected and washed with CH3CN (3 × 5 mL) and recrystallized from a mixture of methanol and isopropanol yielding a white crystalline solid. Yield: 0.33 g, 29.7%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 9.45 (s, 4H, Himi), 7.90 (t, 3JH−H = 1.5 Hz, 4H, Himi), 7.77 (t, 3JH−H = 1.5 Hz, 4H, Himi), 7.52 (dd, 3JH−H = 5.8 Hz, 4JH−H = 3.0 Hz, 4H, ArH), 7.40 (dd, 3JH−H = 5.5 Hz, 4JH−H = 3.5 Hz, 4H, ArH), 5.68 (s, 8H, CH2), 4.81 (s, 8H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 136.75 (Cimi), 133.13 (Cq), 131.04 (CAr), 130.38 (CAr), 123.66 (Cimi), 123.51(Cimi), 49.95 (CH2). HRESI-MS+ (CH3CN): C32H36 m/z = 133.0655, calcd = 133.0760, C32H36N8P3 m/z = 967.2020, calcd = 967.1967.
10·Br4. This compound was prepared using the same method as described for 9·Br4. from 2 (0.24 g, 1.01 mmol) and 6·Br2 (0.62 g, 1.01 mmol). Yield: 0.16 g, 18.0%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 9.53 (s, 4H, Himi) 7.83 (t, 3JH−H = 1.7 Hz, 4H, Himi), 7.80 (t, 3JH−H = 1.7 Hz, 4H, Himi), 7.51 (s, 2H, ArH), 7.36 (t, 3JH−H = 7.7 Hz, 4H, ArH), 7.26–7.28 (m, 4H, ArH), 5.45 (s, 8H, CH2), 4.84 (s, 8H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 137.50 (Cimi), 135.80 (Cq), 130.70 (CAr), 128.92 (CAr), 128.87 (CAr), 128.39 (Cimi), 52.21 (CH2), 48.83 (CH2). HRESI-MS+ (CH3CN): C32H36 m/z = 133.0686, C32H36 calcd = 133.0760, C32H36N8P2 m/z = 411.1099, C32H36N8P2 calcd = 411.1163, C32H36N8P3 m/z = 967.1990, C32H36N8P3 calcd = 967.1967.
11·Br4. Solutions of 1 (0.19 g, 0.80 mmol) in CH3CN (50 mL) and 7·Br2 (0.50 g, 0.80 mmol) in DMF (50 mL) were added simultaneously dropwise to a solution of Bu4N·Br (1.55 g, 4.80 mmol) in CH3CN (150 mL) heated at 110°C over a period of 3 h. The mixture was stirred at the same temperature for a further 5 d during which time a precipitate formed. The precipitate was collected and washed with CH3CN (3 × 5 mL) and then recrystallized from a mixture of methanol and diethyl ether yielding a white crystalline solid. Yield: 0.10g, 7.5%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 9.76 (s, 2H, Himi), 8.00 (s, 2H, Himi), 7.65 (s, 2H, Himi), 7.49–7.53 (m, 10H, ArH, Himi), 7.23 (dd, 3JH−H = 5.4 Hz, 4JH−H = 3.6 Hz, 4H, ArH), 5.52 (s, 4H, CH2), 5.47 (s, 4H, CH2), 4.81 – 4.83 (m, 4H, CH2), 4.70 – 4.72 (m, 4H, CH2), 2.64 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 145.98 (Cq), 137.44 (Cimi), 132.92 (Cq), 132.34 (Cq), 131.15 (CAr), 130.47 (CAr), 129.97 (CAr), 129.56 (CAr), 123.98 (Cimi), 123.42 (Cimi), 122.83 (Cimi), 122.67 (Cimi), 49.48 (CH2), 49.38 (CH2), 49.19 (CH2), 47.90 (CH2), 10.02 (CH3). HRESI-MS+ (CH3CN): C32H36N8P2 m/z = 425.1335, calcd = 425.1324, C34H40N8P3 m/z = 995.2323, calcd 995.2296.
12·Br4. This compound was prepared using the same method described for 11·Br4 from 2 (0.11g, 0.47 mmol) and 8·Br2 (0.30 g, 0.47 mmol). The crude product was recrystallized from methanol yielding a white crystalline solid. Yield: 0.068 g, 16.5%. 1H-NMR (500.023 MHz, d6-DMSO): δ = 9.53 (s, 2H, Himi), 7.80-7.84 (m, 4H, Himi), 7.62 (d, 3JH−H = 2.2 Hz, 2H, Himi), 7.48 (d, 3JH−H = 2.2 Hz, 2H, Himi), 7.46 (s, 1H, ArH), 7.40 (t, 3JH−H = 7.7 Hz, 1H, ArH), 7.34 (s, 1H, ArH), 7.24 – 7.30 (m, 3H, ArH), 7.12 (dd, 3JH−H = 7.7 Hz, 4JH−H = 1.3 Hz, 2H, ArH), 5.42 (s, 4H, CH2), 5.38 (s, 4H, CH2), 4.74 (s, 8H, CH2), 2.63 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 145.85 (Cq), 137.61 (Cimi), 135.54 (Cq), 135.39 (Cq), 128.47 (CAr), 128.04 (CAr), 123.50 (Cimi), 123.41 (Cimi), 122.30 (Cimi), 122.23 (Cimi), 52.19 (CH2), 50.92 (CH2), 48.33 (CH2), 47.73 (CH2), 10.49 (CH3). HRESI-MS+ (CH3CN): C32H36N8 m/z = 360.0861, calcd = 360.0865.
13·(PF6)4 This compound was prepared using the same method described for 11·Br4 from 2 and 5·Br2 (0.52 g, 0.85 mmol) and Bu4N·Br (1.37 g, 4.25 mmol). The crude product was then dried in vacuo and re-dissolved in water (5 mL) and then filtered through a plug of celite. To this solution, a solution of KPF6 saturated in aqueous (3 mL) was added to obtain a white precipitate. The precipitate was washed with isopropanol (5 mL) and recrystallized by vapor diffusion of CH3CN/diethyl ether to obtain a white crystalline. Yield: 0.15 g, 16.0%. 1H-NMR (500.023 MHz, d6-DMSO): δ = 9.13 (s, 2H, Himi), 8.99 (s, 2H, Himi), 7.84 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.72 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.71 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.65 (t, 3JH−H = 1.8 Hz, 2H, Himi), 7.39 – 7.43 (m, 3H, ArH), 7.11 (s, 1H, ArH), 7.02 (dd, 3JH−H = 7.7 Hz, 4JH−H = 1.1 Hz, 2H, ArH), 6.92 – 6.96 (m, 2H, ArH), 5.37 (s, 4H, CH2), 5.36 (s, 4H, CH2), 4.72–4.80 (m, 8H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 137.40 (Cimi), 137.28 (Cimi), 135.56 (Cq), 132.55 (Cq), 130.16 (CAr), 130.04 (CAr), 129.30 (CAr), 127.75 (CAr), 127.35(CAr), 123.99 (Cimi), 123.74 (Cimi), 123.72 (Cimi), 123.57 (Cimi), 52.14 (CH2), 49.50 (CH2), 49.20 (CH2). HRESI-MS+ (CH3CN): C32H36 m/z = 133.0750, calcd = 133.0760, C32H36N8P2 m/z = 411.1158, calcd = 411.1168, C32H36N8P3 m/z = 967.1985, calcd = 967.1983.
14·(PF6)6. A slurry of 9·Br4 (0.10 g, 0.12 mmol) and Ag2O (0.11 g, 0.47 mmol) in DMF (10 mL) was stirred at 50°C for 3 d with the exclusion of light. Diethyl ether (~50 mL) was then added to the mixture and a gray precipitate formed which was collected and dissolved in hot water (5 mL). The solution was clarified by filtration through syringe filter and a saturated solution of KPF6 (2 mL) was added to obtain an off-white precipitate. The solid collected and washed with hot isopropanol (2 mL) and then recrystallized from a mixture of CH3CN and diethyl ether to obtain a white crystalline solid. Yield: 0.026 g, 21.4%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.70 (s, 6H, Himi), 7.68 (s, 6H, Himi), 7.52 (d, 3JH−H = 7.5 Hz, 6H, ArH), 7.17 (tapp, J = 7.5 Hz, 6H, ArH,), 7.09 (s, 6H, Himi), 6.94 (tapp, J = 7.5 Hz, 6H, ArH), 6.76 (s, 6H, Himi), 5.75 (d, 2JH−H = 15.0 Hz, 12H, CH2), 5.68 (d, 3JH−H = 7.5 Hz, 6H, ArH), 5.60 (d, 2JH−H = 17.0 Hz, 6H, CH2), 4.98 (d, 2JH−H = 14.5 Hz, 6H, CH2), 4.65 (t, 3JH−H = 12.5 Hz, 6H, CH2), 4.52 (t, 3JH−H = 12.5 Hz, 6H, CH2), 4.21 (d, 3JH−H = 12.0 Hz, 6H, CH2), 2.48 (s, 6H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 186.31 (d, 1J = 182.32 Hz, 107Ag-Ccarbene), 186.31 (d, 1J = 209.99 Hz, 109Ag-Ccarbene), 178.75 (d, 1J = 182.32 Hz, 107Ag-Ccarbene), 178.75 (d, 1J = 209.99 Hz, 109Ag-Ccarbene), 135.80 (Cq), 131.60 (Cq), 130.88 (CAr), 129.41 (CAr), 128.14 (CAr), 124.63 (CAr), 123.90 (Cimi), 123.78(Cimi), 121.75 (Cimi), 55.37 (CH2), 52.58 (CH2), 51.66 (CH2), 51.24 (CH2), 50.50 (CH2). HRESI-MS+ (CH3CN): C96H96N24 m/z = 372.0334, calcd = 372.0419, C96H96N24Ag6P3 m/z = 889.0490, calcd = 889.0485.
15·Br6. A slurry of 9·Br4 (0.10 g, 0.12 mmol) and (THT)AuCl (0.083 g, 0.26 mmol) in DMF (10 mL) was stirred at 110°C for 0.5 h. To this mixture NaOAc (0.050 g, 0.59 mmol) was added and the solution was stirred at the same temperature for 1 h. The resultant mixture was cooled to RT and then diethyl ether (50 mL) was added to obtain a white precipitate. The precipitate was collected and recrystallized from hot methanol (~5 mL). Yield: 0.048 g, 54.2%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.83 (s, 6H, Himi), 7.72 (s, 6H, Himi), 7.57 (d, 3JH−H = 7.5 Hz, 6H, ArH), 7.21 (tapp, J = 7.5 Hz, 6H, ArH) 7.11 (s, 6H, Himi), 6.93 (tapp, J = 7.5 Hz, 6H, ArH) 6.84 (s, 6H, Himi), 6.07 (d, 2JH−H = 16.5 Hz, 6H, CH2), 5.74 – 5.78 (m, 12H, ArH, CH2), 5.66 (d, 3JH−H = 16.5 Hz, 6H, CH2), 5.06 (d, 3JH−H = 14.0 Hz, 12H, CH2), 4.49 (t, 3JH−H = 12.8 Hz, 6H, CH2), 4.37 (d, 3JH−H = 12.5 Hz, 6H, CH2), 2.33 (d, 3JH−H = 12.5 Hz, 6H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 186.31 (Ccarbene), 181.48 (Ccarbene), 135.84 (Cq), 131.37 (Cq), 131.02 (CAr), 129.62 (CAr), 127.99 (CAr), 124.59 (CAr), 123.95 (Cimi), 123.64 (Cimi), 121.86 (Cimi), 51.89 (CH2), 51.27 (CH2), 50.22 (CH2). HRESI-MS+ (CH3CN): C96H96N24 m/z = 461.1022, calcd = 461.1035.
16·(PF6)2. A slurry of 9·Br4 (0.050 g, 0.059 mmol) and Ag2O (0.030 g, 0.13 mmol) in DMF (5 mL) was stirred at 50°C for 12 h with the exclusion of light. To this mixture K2PdCl4 (0.019 g, 0.059 mmol) was added and stirring was continued for a further stirred for 12 h at 80°C. The reaction mixture was clarified by centrifugation and diethyl ether (30 mL) was added to the supernatant yielding a gray precipitate. The precipitate was collected and re-dissolved in hot water (5 mL) and the solution filtered through a syringe filter. To the filtrate, a saturated solution of KPF6 (2 mL) was added to obtain an off-white precipitate. The precipitate was dried in vacuo and then recrystallized from a mixture of CH3CN and diethyl ether to obtain a white crystalline solid. Yield: 0.0052 g, 9.6%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.87 – 7.92 (m, 4H, ArH), 7.83 (d, 3JH−H = 2.0 Hz, 4H, Himi), 7.48 (d, 3JH−H = 2.0 Hz, 4H, Himi), 7.42 – 7.47 (m, 4H, ArH), 6.44 (d, 3JH−H = 15.0 Hz, 4H, CH2), 5.20 (d, 3JH−H = 14.7 Hz, 4H, CH2), 4.98 – 5.06 (m, 4H, CH2), 4.54 – 4.61 (m, 4H, CH2). 13C-NMR (125.74 MHz, d6-DMSO): δ = 167.41 (Ccarbene), 135.56 (Cq), 131.73 (CAr), 129.83 (CAr), 124.96 (Cimi), 122.68 (Cimi), 50.79 (CH2), 47.39 (CH2). HRESI-MS+ (CH3CN): C32H32N8Pd2+ m/z = 317.0889, calcd = 317.0887.
17·(PF6)3. This compound was prepared using the same method as described for 14·(PF6)6 from 10·Br4 (0.050 g, 0.059 mmol) and Ag2O (0.057 g, 0.24 mmol). Yield: 0.0050 g, 6.2%. 1H-NMR (500.02 MHz, CD3CN): δ = 8.37 (t, J = 1.5 Hz, 2H, Himi), 7.45 – 7.53 (m, 12H, ArH, Himi), 7.21 (t, 3JH−H = 1.8 Hz, 2H, Himi), 6.98 (s, 2H, ArH), 6.95 (t, 3JH−H = 1.9 Hz, 2H, Himi) 5.31 (s, 4H, CH2), 5.08 (s, 4H, CH2), 4.60 (d, 3JH−H = 4.3 Hz, 4H, CH2), 4.55 (d, 3JH−H = 4.7 Hz, 4H, CH2). 13C-NMR (125.74 MHz, CD3CN): δ = 180.39 (d, 1J = 183.58 Hz, 107Ag-Ccarbene), 180.39 (d, 1J = 211.24 Hz, 109Ag-Ccarbene), 138.62 (Cq), 136.79 (Cq), 136.18 (Cimi), 130.86 (CAr), 129.82 (Cimi), 129.48 (Cimi), 126.74 (CAr), 125.47 (CAr), 125.42 (CAr), 124.47 (Cimi), 124.12 (Cimi), 122.39 (CAr), 122.35 (CAr), 55.33 (CH2), 53.45 (CH2), 52.73 (CH2), 50.96 (CH2). HRESI-MS+ (CH3CN): C32H34N8Ag3+ m/z = 213.0643, calcd = 213.0646, C32H34N8AgP m/z = 391.2845, calcd = 391.0794, C32H34N8AgP2 m/z = 927.1228, calcd = 927.1235.
18·(PF6)2. This compound was prepared using the same method as described for 15·Br6 from 11·Br4 (0.050 g, 0.057 mmol), (THT)AuCl (0.040 g, 0.13 mmol), and NaOAc (0.019 g, 0.23 mmol). For exchange of the bromide anion to hexafluorophosphate the crude product was dissolved in water (3 mL) and the solution filtered through celite. To this solution, a saturated solution of KPF6 (2 mL) was added to obtain a white precipitate which was then recrystallized from a mixture of CH3CN and diethyl ether yielding a white crystalline solid. Yield: 0.020 g, 29.9%. 1H-NMR (500.02 MHz, CD3CN): δ = 7.55 – 7.57 (m, 1H, ArH), 7.46 – 7.48 (m, 1H, ArH), 7.44 (d, 3JH−H = 2.0 Hz, 1H, Himi), 7.41−7.42 (m, 1H, ArH), 7.38 (s, 1H, Himi), 7.23 (d, 3JH−H = 2.1 Hz, 1H, Himi), 7.21-7.22 (m, 1H, ArH), 7.15 (d, 3JH−H = 1.9 Hz, 1H, Himi), 5.53 (s, 2H, CH2), 5.49 (s, 2H, CH2), 4.66 – 4.67 (m, 2H, CH2), 4.57 – 4.59 (m, 2H, CH2), 2.54 (s, 3H, CH3). 13C-NMR (125.74 MHz, CD3CN): δ = 145.20 (Ccarbene), 133.61 (Cq), 130.51 (CAr), 130.43 (CAr), 130.22 (CAr), 129.75 (CAr), 122.74 (Cimi), 122.50 (Cimi), 122.24 (Cimi), 122.05 (Cimi), 51.84 (CH2), 50.05 (CH2), 49.69 (CH2), 48.48 (CH2), 10.36 (CH3). HRESI-MS+ (CH3CN): C34H38N8Au2 m/z = 556.0369, calcd = 556.0443.
19·(PF6)2. This compound was prepared using the same method as that described for 15·Br6 from 11·Br4 (0.030 g, 0.034 mmol), (THT)AuCl (0.024 g, 0.075 mmol), and NaOAc (0.11 g, 0.14 mmol). For exchange of the bromide anion to hexafluorophosphate the crude product was dissolved in water (3 mL) and the solution filtered through celite. To this solution, a saturated solution of KPF6 (2 mL) was added to obtain a white precipitate which was then recrystallized from a mixture of CH3CN and diethyl ether yielding a white crystalline solid. Yield: 0.0070 g, 1.5%. 1H-NMR (500.02 MHz, d6-DMSO): δ = 7.71 (d, 3JH−H = 1.9 Hz, 2H, Himi), 7.62 (d, 3JH−H = 1.9 Hz, 2H, Himi), 7.56 (d, 3JH−H = 1.9 Hz, 2H, Himi), 7.39−7.43 (m, 3H, ArH, Himi), 7.28 – 7.38 (m, 3H, ArH), 7.14 – 7.19 (m, 1 H, ArH), 7.07 – 7.14 (m, 1H, ArH), 6.56 – 6.69 (m, 2H, ArH), 5.31 (s, 4H, CH2), 5.06 (s, 4H, CH2), 4.67 (s, 4H, CH2), 4.66 (s, 4H, CH2), 2.40 (s, 6H, CH3). 13C-NMR (125.74 MHz, d6-DMSO): δ = 172.52 (Ccarbene), 144.61 (Cq), 136.79 (Cq), 134.11 (Cq), 130.12 (CAr), 127.96 (CAr), 125.29 (CAr), 123.45 (Cimi), 122.28 (Cimi), 121.98 (Cimi), 121.91 (Cimi), 52.82 (CH2), 50.44 (CH2), 49.84 (CH2), 48.11 (CH2), 9.19 (CH3). HRESI-MS+ (CH3CN): C34H38N8Au2 m/z = 556.0422, calcd = 556.0443.
20·(PF6)2. To a solution of 11·Br4 (0.050 g, 0.057 mmol) and K2PdCl4 (0.019 g, 0.057 mmol) in DMSO (5 mL) at 85°C, NaOAc (0.019 g, 0.23 mmol) was added and the reaction mixture was stirred same temperature for 12 h. The resultant mixture was then diluted with acetone (5 mL) followed by adding diethyl ether (20 mL) to form a precipitate. The crude precipitate was collected and then re-dissolved in water and filtered through a plug of Celite followed by addition of a saturated solution of KPF6 (3 mL). The resultant precipitate was collected and washed with isopropanol (5 mL) and diethyl ether (2 × 5 mL) and then recrystallized from a mixture of CH3CN and diethyl ether yielding the product as a pale yellow solid. NMR analysis showed this material to be impure and the 1H-NMR spectrum is reported for the impure material. 1H-NMR (500.02 MHz, CD3CN): δ = 7.66 (s, 2H, Himi), 7.55 – 7.57 (m, 4H, ArH), 7.35−7.40 (m, 2H, ArH), 7.34 (s, 2H, Himi), 7.22 – 7.24 (m, 2H, Himi), 7.03 (s, 2H, Himi), 7.02 (s, 2H, Himi), 5.26 (s, 4H, CH2), 5.24 (s, 4H, CH2), 4.61 – 4.70 (m, 8H, CH2), 2.57 (s, 6H, CH3). A crystal suitable for single crystal X-ray diffraction analysis of 20·(PF6)2 was grown from diffusion of diethyl ether into an acetonitrile solution of 20·(PF6)2.
Author Contributions
PB conceived of the presented idea. ZL synthesized compounds and recorded NMR spectra. NW recorded and analyzed high resolution mass spectra. PB and ZL collected X-ray diffraction data. All authors discussed the results and contributed to the final manuscript.
Funding
This research was financially supported by The Australia Research Council, DP150102741 and ZL acknowledges receipt of a La Trobe University postgraduate award.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00270/full#supplementary-material
Supporting Information. Synthetic details for compounds 1-3, Crystallographic data for compounds 5·Br2, 9·Br4, 10·Br3PF6, 11·Br4, 15·Br2(PF6)4, and 20·(PF6)2 and further X-ray crystallographic details and anion binding Job plots for compounds 9·(PF6)4 and 10·(PF6)4 and 1H and 13C NMR spectra for tetra-imidazolium salts and metal complexes.
References
Ahmed, N., Shirinfar, B., Geronimo, I., and Kim, K. S. (2011). Fluorescent imidazolium-based cyclophane for detection of guanosine-5′-triphosphate and I– in aqueous solution of physiological pH. Org. Lett. 13, 5476–5479. doi: 10.1021/ol202183t
Ahmed, N., Shirinfar, B., Youn, I. S., Bist, A., Suresh, V., and Kim, K. S. (2012). A highly selective fluorescent chemosensor for guanosine-5′-triphosphatevia excimer formation in aqueous solution of physiological pH. Chem. Commun. 48, 2662–2664. doi: 10.1039/c2cc17145g
Alcalde, E., Mesquida, N., Perez-Garcia, L., Alvarez-Rua, C., Garcia-Granda, S., and Garcia-Rodriguez, E. (1999). Hydrogen bonded driven anion binding by dicationic [14]imidazoliophanes. Chem. Commun. 0, 295–296. doi: 10.1039/a808503j
Alcalde, E., Mesquida, N., Vilaseca, M., Alvarez-Rúa, C., and García-Granda, S. (2007). Imidazolium-based dicationic cyclophanes. solid-state aggregates with unconventional (C–H)+···Cl– hydrogen bonding revealed by X-ray diffraction. Supramol. Chem. 19, 501–509. doi: 10.1080/10610270601132624
Altmann, P. J., Jandl, C., and Pöthig, A. (2015). Introducing a pyrazole/imidazole based hybrid cyclophane: a hydrogen bond sensor and binucleating ligand precursor. Dalton Trans. 44, 11278–11281. doi: 10.1039/C5DT01775K
Altmann, P. J., Weiss, D. T., Jandl, C., and Kühn, F. E. (2016). Exploring coordination modes: late transition metal complexes with a methylene-bridged macrocyclic tetra-NHC ligand. Chem. Asian J. 11, 1597–1605. doi: 10.1002/asia.201600198
Andrew, R. E., Storey, C. M., and Chaplin, A. B. (2016). Well-defined coinage metal transfer agents for the synthesis of NHC-based nickel, rhodium and palladium macrocycles. Dalton Trans. 45, 8937–8944. doi: 10.1039/C6DT01263A
Anneser, M. R., Haslinger, S., Pöthig, A., Cokoja, M., Basset, J.-M., and Kühn, F. E. (2015). Synthesis and characterization of an iron complex bearing a cyclic tetra-N-heterocyclic carbene ligand: an artifical heme analogue? Inorg. Chem. 54, 3797–3804. doi: 10.1021/ic503043h
Aweda, T. A., Ikotun, O., Mastren, T., Cannon, C. L., Wright, B., Youngs, W. J., et al. (2013). The use of 111Ag as a tool for studying biological distribution of silver-based antimicrobials. Med. Chem. Commun. 4, 1015–1017. doi: 10.1039/c3md00082f
Baker, M. V., Skelton, B. W., White, A. H., and Williams, C. C. (2001). Palladium carbene complexes derived from imidazolium-linked ortho-cyclophanes. J. Chem. Soc. Dalton Trans. 111–120. doi: 10.1039/b007293l
Barnard, P. J., Baker, M. V., Berners-Price, S. J., and Day, D. A. (2004). Mitochondrial permeability transition induced by dinuclear gold(I)–carbene complexes: potential new antimitochondrial antitumour agents. J. Inorg. Biochem. 98, 1642–1647. doi: 10.1016/j.jinorgbio.2004.05.011
Barnard, P. J., Wedlock, L. E., Baker, M. V., Berners-Price, S. J., Joyce, D. A., Skelton, B. W., et al. (2006). Luminescence studies of the intracellular distribution of a dinuclear Gold(I) N-heterocyclic carbene complex. Angew. Chem. Int. Ed. 45, 5966–5970. doi: 10.1002/anie.200601526
Bass, H. M., Cramer, S. A., Price, J. L., and Jenkins, D. M. (2010). 18-atom-ringed macrocyclic tetra-imidazoliums for preparation of monomeric tetra-carbene complexes. Organometallics 29, 3235–3238. doi: 10.1021/om100625g
Beer, P. D., and Gale, P. A. (2001). Anion recognition and sensing: the state of the art and future perspectives. Angew. Chem. int. Ed. 40, 486–516. doi: 10.1002/1521-3773(20010202)40:3<486::AID-ANIE486>3.0.CO;2-P
Chellappan, K., Singh, N. J., Hwang, I. C., Lee, J. W., and Kim, K. S. (2005). A Calix[4]imidazolium[2]pyridine as an anion receptor. Angew. Chem. Int. Ed. 44, 2899–2903. doi: 10.1002/anie.200500119
Chianese, A. R., Zeglis, B. M., and Crabtree, R. H. (2004). Unexpected oxidative C–C cleavage in the metallation of 2-substituted imidazolium salts to give N-heterocyclic carbene complexes. Chem. Commun. 2176–2177. doi: 10.1039/B409672J
Fei, F., Lu, T., Chen, X., and Xue, Z. (2017). Synthesis and structural characterization of metal complexes with macrocyclic tetracarbene ligands. N. J. Chem. 41, 13442–13453. doi: 10.1039/C7NJ02485A
Frassineti, C., Alderighi, L., Gans, P., Sabatini, A., Vacca, A., and Ghelli, S. (2003). Determination of protonation constants of some fluorinated polyamines by means of 13C NMR data processed by the new computer program HypNMR2000. Protonation sequence in polyamines. Anal. Boianal. Chem. 376, 1041–1052. doi: 10.1007/s00216-003-2020-0
Frassineti, C., Ghelli, S., Gans, P., Sabatini, A., Moruzzi, M. S., and Vacca, A. (1995). Nuclear magnetic resonance as a tool for determining protonation constants of natural polyprotic bases in solution. Anal. Biochem. 231, 374–382. doi: 10.1006/abio.1995.9984
Gale, P. A. (2003). Anion and ion-pair receptor chemistry: highlights from 2000 and 2001. Coord. Chem. Rev. 240, 191–221. doi: 10.1016/S0010-8545(02)00258-8
Hahn, F. E., Langenhahn, V., Lügger, T., Pape, T., and Le Van, D. (2005). Template synthesis of a coordinated tetracarbene ligand with crown ether topology. Angew. Chem. Int. Ed. 44, 3759–3763. doi: 10.1002/anie.200462690
Hahn, F. E., Radloff, C., Pape, T., and Hepp, A. (2008). Synthesis of silver(I) and gold(I) complexes with cyclic tetra- and hexacarbene ligands. Chem. Eur. J. 14, 10900–10904. doi: 10.1002/chem.200801877
Hindi, K. M., Panzner, M. J., Tessier, C. A., Cannon, C. L., and Youngs, W. J. (2009). The medicinal applications of imidazolium carbene–metal complexes. Chem. Rev. 109, 3859–3884. doi: 10.1021/cr800500u
Johnson, N. A., Southerland, M. R., and Youngs, W. J. (2017). Recent developments in the medicinal applications of silver-NHC complexes and imidazolium salts. Molecules 22:1263. doi: 10.3390/molecules22081263
Lu, T., Yang, C., Steren, C. A., Fei, F., Chen, X., and Xue, Z. (2018). Synthesis and characterization of Ag(I) and Au(I) complexes with macrocyclic hybrid amine N-heterocyclic carbene ligands. N. J. Chem. 42, 4700–4713. doi: 10.1039/C7NJ04020B
Lu, T., Yang, C. F., Zhang, L. Y., Fei, F., Chen, X. T., and Xue, Z. L. (2017). Metal complexes with a hexadentate macrocyclic diamine-tetracarbene ligand. Inorg. Chem. 56, 11917–11928. doi: 10.1021/acs.inorgchem.7b01896
Mageed, A. H., Skelton, B. W., and Baker, M. V. (2017). Stable AuIII complexes with four N-heterocyclic carbene groups can be prepared in high yield directly from KAuCl4. Dalton Trans. 46, 7844–7856. doi: 10.1039/C7DT01272A
Mageed, A. H., Skelton, B. W., Sobolev, A. N., and Baker, M. V. (2018). Formation of dinuclear AuII and AuI/AuIII mixed-valence complexes is directed by structural constraints imposed by cyclophane-NHC ligands. Eur. J. Inorg. Chem. 2018, 109–120. doi: 10.1002/ejic.201701272
Martínez-Máñez, R., and Sancenón, F. (2003). Fluorogenic and chromogenic chemosensors and reagents for anions. Chem. Rev. 103, 4419–4476. doi: 10.1021/cr010421e
Mckie, R., Murphy, J. A., Park, S. R., Spicer, M. D., and Zhou, S. Z. (2007). Homoleptic crown N-heterocyclic carbene complexes. Angew. Chem. Int. Ed. 46, 6525–6528. doi: 10.1002/anie.200702138
Melaiye, A., Sun, Z., Hindi, K., Milsted, A., Ely, D., Reneker, D. H., et al. (2005). Silver(I)–imidazole cyclophane gem-diol complexes encapsulated by electrospun tecophilic nanofibers: formation of nanosilver particles and antimicrobial activity. J. Am. Chem. Soc. 127, 2285–2291. doi: 10.1021/ja040226s
Mesquida, N., Dinarès, I., Ibáñez, A., and Alcalde, E. (2013). [14]Heterophane prototypes containing azolium and/or azole anion-binding motifs. Org. Biomol. Chem. 11, 6385–6396. doi: 10.1039/c3ob41214h
Meyer, S., Klawitter, I., Demeshko, S., Bill, E., and Meyer, F. (2013). A tetracarbene–oxoiron(IV) complex. Angew. Chem. Int. Ed. 52, 901–905. doi: 10.1002/anie.201208044
Neelakandan, P. P., and Ramaiah, D. (2008). DNA-assisted long-lived excimer formation in a cyclophane. Angew. Chem. Int. Ed. 47, 8407–8411. doi: 10.1002/anie.200803162
Nomiya, K., Morozumi, S., Yanagawa, Y., Hasegawa, M., Kurose, K., Taguchi, K., et al. (2018). Syntheses, structures, and antimicrobial activities of gold(I)– and copper(I)–N-heterocyclic carbene (NHC) complexes derived from basket-shaped dinuclear Ag(I)–NHC complex. Inorg. Chem. 57, 11322–11332. doi: 10.1021/acs.inorgchem.8b00011
Pöthig, A., Ahmed, S., Winther-Larsen, H. C., Guan, S., Altmann, P. J., Kudermann, J., et al. (2018). Antimicrobial activity and cytotoxicity of Ag(I) and Au(I) pillarplexes. Front. Chem. 6:584. doi: 10.3389/fchem.2018.00584
Schulte to Brinke, C., and Hahn, F. (2015). Synthesis of a flexible macrocyclic tetraimidazolium salt–precursor for a tetracarbene ligand with metal dependent coordination modes. Dalton Trans. 44, 14315–14322. doi: 10.1039/C5DT02115D
Schulte to Brinke, C., Pape, T., and Hahn, F. E. (2013). Synthesis of polynuclear Ag(I) and Au(I) complexes from macrocyclic tetraimidazolium salts. Dalton Trans. 42, 7330–7337. doi: 10.1039/c2dt32905k
Serpell, C. J., Cookson, J., Thompson, A. L., and Beer, P. D. (2011). A dual-functional tetrakis-imidazolium macrocycle for supramolecular assembly. Chem. Sci. 2, 494–500. doi: 10.1039/C0SC00511H
Shah, P. N., Lin, L. Y., Smolen, J. A., Tagaev, J. A., Gunsten, S. P., Han, D. S., et al. (2013). Synthesis, characterization, and in vivo efficacy of shell cross-linked nanoparticle formulations carrying silver antimicrobials as aerosolized therapeutics. ACS Nano. 7, 4977–4987. doi: 10.1021/nn400322f
Shirinfar, B., Ahmed, N., Park, Y. S., Cho, G. S., Youn, I. S., Han, J. K., et al. (2013). Selective fluorescent detection of RNA in living cells by using imidazolium-based cyclophane. J. Am. Chem. Soc. 135, 90–93. doi: 10.1021/ja3112274
Toure, M., Charles, L., Chendo, C., Viel, S., Chuzel, O., and Parrain, J.-L. (2016). Straightforward and controlled shape access to efficient macrocyclic imidazolylboronium anion receptors. Chem. Eur. J. 22, 8937–8942. doi: 10.1002/chem.201601174
Wong, W. W., Vickers, M. S., Cowley, A. R., Paul, R. L., and Beer, P. D. (2005). Tetrakis-imidazolium macrocyclic receptors for anion binding. Org. Biomol. Chem. 3, 4201–4208. doi: 10.1039/b510068b
Xu, Z., Kim, S. K., and Yoon, J. (2010). Revisit to imidazolium receptors for the recognition of anions: highlighted research during 2006–2009. Chem. Soc. Rev. 39, 1457–1466. doi: 10.1039/b918937h
Yoon, J., Kim, S. K., Singh, N. J., and Kim, K. S. (2006). Imidazolium receptors for the recognition of anions. Chem. Soc. Rev. 35, 355–360. doi: 10.1039/b513733k
Keywords: N-heterocycle carbene, macrocycle, anion receptor, N-heterocarbene-gold(I) complexes, tetra-imidazolium
Citation: Li Z, Wiratpruk N and Barnard PJ (2019) Stepwise Synthesis of Tetra-imidazolium Macrocycles and Their N-Heterocyclic Carbene Metal Complexes. Front. Chem. 7:270. doi: 10.3389/fchem.2019.00270
Received: 17 December 2018; Accepted: 02 April 2019;
Published: 24 April 2019.
Edited by:
Angela Casini, Cardiff University, United KingdomReviewed by:
Alexander Pöthig, Technische Universität München, GermanyGreta Bergamaschi, Istituto di Chimica del Riconoscimento Molecolare (ICRM), Italy
Copyright © 2019 Li, Wiratpruk and Barnard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Barnard, cC5iYXJuYXJkQGxhdHJvYmUuZWR1LmF1