 Shinya Furukawa
Shinya Furukawa Genki Nishimura3
Genki Nishimura3 Tomoaki Takayama
Tomoaki Takayama- 1Institute for Catalysis, Hokkaido University, Sapporo, Japan
- 2Elements Strategy Initiative for Catalysts and Batteries, Kyoto University, Kyoto, Japan
- 3Department of Chemistry, School of Science, Tokyo Institute of Technology, Tokyo, Japan
Ammonia-borane is one of the most promising candidates for hydrogen carriers. A series of Ni- and Co-based bimetallic catalysts supported on SiO2 (Ni–M/SiO2 and Co–M/SiO2; M = Ga, Ge, Sn, Zn) was prepared and tested as catalysts for hydrogen production from ammonia-borane (AB) in water or methanol. Ni–Zn/SiO2 and Co–Ge/SiO2 exhibited catalytic activities much higher than those of monometallic Ni/SiO2 and Co/SiO2, respectively. Ni–Zn/SiO2 showed a high catalytic activity when water was used as a solvent, where the reaction was completed within 6 min at room temperature with a specific reaction rate of 4.3 ml min−1 mmol-cat−1 mM-AB−1. To the best of our knowledge, this is the highest value among those reported using 3d metal-based catalysts. Co–Ge/SiO2 afforded a five-fold higher reaction rate than that of the corresponding monometallic Co/SiO2. XRD, TEM, and HAADF-STEM-EDS analyses revealed that Ni0.75Zn0.25 and Co0.8Ge0.2 solid-solution alloys were formed with high phase purities. An XPS study showed that Co atoms in Co0.8Ge0.2 were electron-enriched due to electron transfer from Ge to Co, which may be the origin of the improved catalytic activity.
Introduction
Hydrogen has been considered as one of the best energy carrier alternatives to fossil fuels because of its high energy density, clean combustion product (only water), and environmental friendliness (Schlapbach and Züttel, 2011). Hydrogen is currently produced by steam reforming of methane contained in natural gas (Heinzel et al., 2002), which is not renewable and sustainable. Therefore, alternative methodologies such as photocatalytic (Moniz et al., 2015; Chen et al., 2017; Wang et al., 2017) and photoelectrochemical (Zhang et al., 2014; Zheng and Zhang, 2016; Han et al., 2017) water splitting have recently attracted increasing attention. On the other hand, storage, transport, and release of hydrogen are known as technological barriers to practical application in view of cost and safety (Züttel, 2003). Ammonia–borane (H3N·BH3, hereafter AB) is one of the more promising candidates as a hydrogen carrier or source (Landge et al., 2018) owing to its high hydrogen content (19.6 wt%), high thermal stability, and low toxicity (Marder, 2007). AB can release three equimolar amounts of hydrogen at ambient temperatures using an appropriate catalyst through solvolysis with protic solvents such as water and methanol. Using noble metals such as Pt, Rh, Ir enables the achievement of rapid hydrogen release typically within several minutes (Chandra and Xu, 2007; Xu and Chandra, 2007). Recently, development of non-noble 3d transition metal-based catalysts that are active for hydrogen production from AB has been increasingly focused (Xu and Chandra, 2006; Yan et al., 2008, 2009; Kalidindi et al., 2009; Metin et al., 2010; Patel et al., 2010; Ozay et al., 2011; Peng et al., 2015). To develop highly efficient catalytic systems using base metal elements, the catalytic activity of these metals should be greatly enhanced by appropriate catalyst design such as the modification of morphology, the addition of some cocatalysts, or the formation of alloy phases (Furukawa and Komatsu, 2017).
In this study, a series of Ni- and Co-based bimetallic catalysts was prepared (Ni–M/SiO2 and Co–M/SiO2; M = Ga, Ge, Sn, and Zn) and tested as catalysts for hydrogen production from AB in water or methanol as a solvent. The observed catalytic performances were discussed in view of the reaction mechanism. Herein, we report a novel and highly efficient catalytic system for hydrogen production from AB using non-noble metal catalysts.
Materials and Methods
Catalyst Preparation
Monometallic Ni and Co catalysts were prepared by pore-filling impregnation using silica as a support. Aqueous solutions of Ni(NO3)2·6H2O (Wako, 99%) or Co(NO3)2·3H2O (Sigma Aldrich, 99%) were added to dried silica gel (CARiACT G-6, Fuji Silysia, SBET = 470 m2 g−1) so that the solutions filled the silica pores. The mixtures were sealed overnight at room temperature and dried over a hot plate, followed by reduction under flowing H2 at 600°C for 1 h. Silica-supported Ni- and Co-based catalysts (Ni–M/SiO2 and Co–M/SiO2; M = Ga, Ge, Sn, and Zn) were prepared by pore-filling co-impregnation. Mixed aqueous solutions of Ni(NO3)2·6H2O or Co(NO3)2·3H2O and a second metal salt, Ga(NO3)3·nH2O (Wako, 99.9%), (NH4)2GeF6 (Sigma Aldrich, 99.99%), SnCl2 (Kanto, 97%), and Zn(NO3)2·6H2O (Kanto, 99%) were used in a manner similar to that of the monometallic catalyst. The metal loading of Ni or Co, the atomic ratio of Ni/M or Co/M, and the reduction temperature during the catalyst preparation were set to 3 wt%, 3.0, and 600°C except for Ni–Ge and Co–Ge, which had a reduction temperature of 800°C, and Co–Sn with an atomic ratio of 1.0.
Characterizations
The crystal structures of the catalysts were determined by powder X-ray diffraction (XRD) with a Rigaku RINT2400 diffractometer using a Cu Kα X-ray source. Difference XRD patterns were obtained by subtracting the pattern for the SiO2 support from those of the supported catalysts. Transmission electron microscopy (TEM) was conducted using a JEOL JEM-2010F microscope at an accelerating voltage of 200 kV. To prepare the TEM specimen, all samples were sonicated in tetrachloromethane and then dispersed on a Cu grid supported by an ultrathin carbon film. X-ray photoelectron spectra (XPS) of the bimetallic compounds were measured with an ULVAC PHI 5000 VersaProbe spectrometer. The catalyst was pressed into a pellet and placed into a quartz reactor, where it was reduced under flowing hydrogen (60 ml min−1) at 450°C for 0.5 h prior to the measurement. The sample was put into a transfer vessel in a grove box (O2 concentration: < 1 ppm) and then introduced into the spectrometer without exposure to air. Spectra were obtained with an Al Kα X-ray source, using C 1s as a reference for binding energy (284.8 eV). The reduction behavior of the catalyst was examined by temperature-programmed reduction (TPR). Under flowing H2 (5%)/Ar, the temperature of the sample bed was raised from room temperature to 900°C at a heating rate of 10°C·min−1 and the consumption of hydrogen was continuously measured by a thermal conductivity detector (TCD).
Catalytic Reaction
A catalyst (100 mg) was placed into a 50 mL three-necked round-bottom flask equipped with a silicone rubber septum and a gas burette and pretreated under an H2 stream (60 mL·min−1) at 450°C for 0.5 h using a mantle heater. After the pretreatment, dry Ar (20 mL·min−1) was passed into the flask to replace the residual H2, and the flask was cooled to room temperature. A reaction mixture containing a solvent (deionized water or dehydrated methanol, Kanto 99.8%, 10 mL) and AB (Sigma-Aldrich, 97%, 2.0 mmol) was added into the flask through the septum at 25°C. The volume of the evolved H2 was measured using the gas burette. The total volume of H2 is expected to be 147 ml (6.0 mmol) at 25°C for the complete conversion of AB (2.0 mmol).
Results and Discussion
The prepared Ni- and Co-based catalysts were tested for H2 production from AB using methanol as a solvent (Figure 1). The monometallic Ni catalyst showed moderate catalytic activity with a short induction period. The Ni–Sn catalyst showed a very low catalytic activity with a long induction period. On the other hand, the Ni–Zn, Ni–Ga, and Ni–Ge catalysts exhibited high catalytic activities without an induction period. Particularly for the Ni–Zn catalyst, the reaction was completed within ca. 10 min. Co-based catalysts generally gave catalytic activities lower than those of Ni-based catalysts, which reflects the intrinsic difference of catalytic activity between Ni and Co for this reaction. Moreover, the catalytic activities were quite different depending on the second metal (Co–Ge > Co–Ga >> Co >> Co–Sn). Induction periods were observed for some catalysts (Ni, Ni–Sn, Co, Co–Ga, and Co–Zn), indicating that, for these catalysts, some real active species (probably, zero-valent Ni or Co species) were formed or increased during the catalytic reaction. A remarkable enhancement in the reaction rate was achieved by addition of Ge to Co, suggesting the contribution of a specific effect of Ge on the catalysis. The discovered active catalysts, Ni–Zn and Co–Ge, were also tested in hydrogen production from AB in water as a solvent (Figure 2). For each catalyst, H2 evolution in water was much faster than that in methanol as reported for Ni- and Co-based catalysts in the literature. Note that the reaction was completed within 6 min when the Ni–Zn catalyst was used. This is one of the best performances in AB hydrolysis reported to this day. The obtained catalytic performance was listed in Table 1 with those of reported catalysts. Because the reaction conditions were different depending on each study, we quantitatively compared them based on the specific H2 formation rates (rcat[AB]) calculated by dividing H2 formation rates (rH2) by the amount of catalyst (mmol) and concentration of AB (mM). Since the number of active sites, such as metal atoms on the surface, was not clearly mentioned, the total number of metal atoms was used for this calculation. The difference in AB concentration was compensated for by assuming the first-order dependence of the formation rate on AB concentration. The Ni–Zn catalyst exhibited an rcat[AB] value higher than that of Ni2P, which was reported to be the most active base-metal catalyst for AB hydrolysis (Peng et al., 2015). Therefore, the Ni–Zn catalyst is the most active 3d metal-based catalyst for H2 production by hydrolysis of AB. We also examined the reusability of the catalysts. Figure 3 shows the result of the reuse test for the Co–Ge/SiO2 catalyst in H2 production in methanol. At the first reuse at 110 min, we added 1.33 mol of AB (2/3 equivalent to the standard condition). The total volume H2 evolved decreased from 130.5 to 87 ml, showing complete conversion of AB at reuse. The reaction rate also became approximately two-thirds of the original (2.03–1.75), indicating that rH2 strongly depends on AB concentration. Although the reaction rate was slightly decreased at the second and third reuse, the Co–Ge/SiO2 catalyst could be reused without any regeneration procedure. Thus, it was found that the Co–Ge/SiO2 was a recyclable heterogeneous catalyst for H2 production from AB.
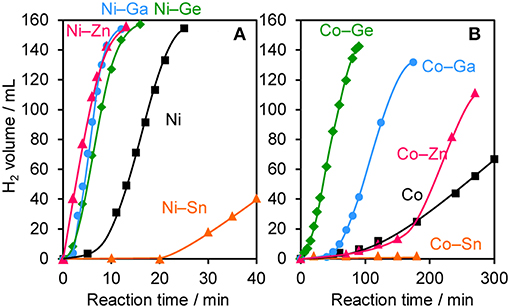
Figure 1. Time course of H2 evolution during catalytic methanolysis of AB using (A) Ni- and (B) Co-based catalysts.
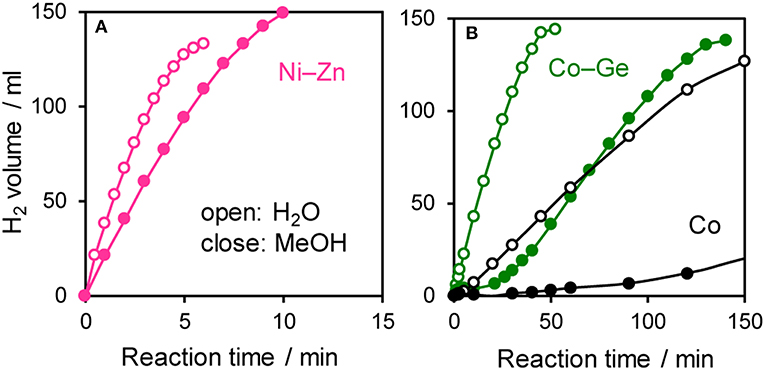
Figure 2. Time course of H2 evolution during catalytic hydrolysis (H2O) and methanolysis (MeOH) of AB using (A) Ni–Zn, (B) Co–Ge, and Co catalyst.
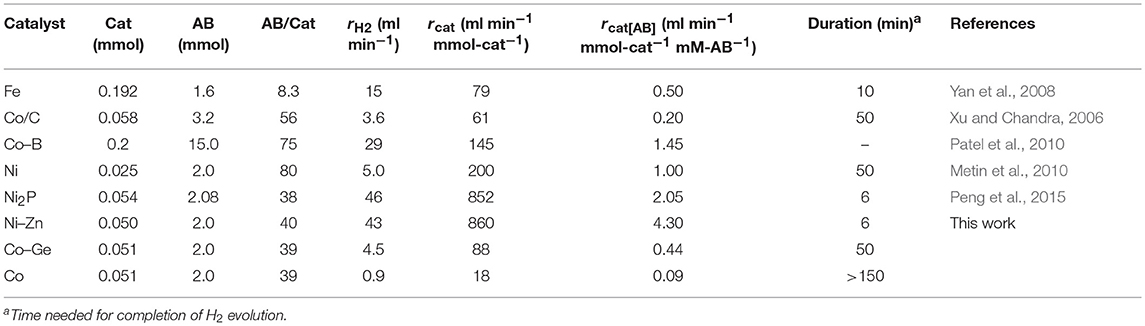
Table 1. Summary of reaction condition and rate of AB hydrolysis at 25°C using various catalysts.
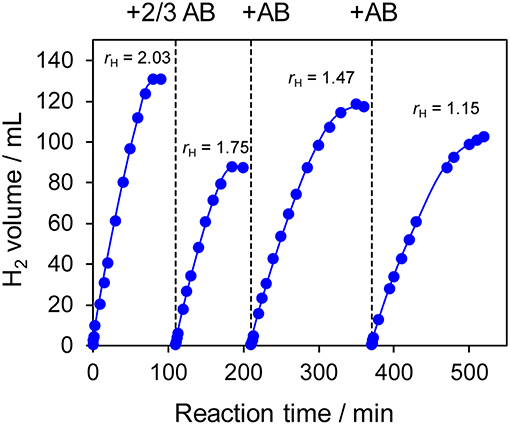
Figure 3. Reuse tests for H2 production in methanol using Co–Ge/SiO2 catalyst. For the first reuse at 110 min, the amount of AB was reduced to 2/3 (1.33 mmol).
Then, the discovered active catalysts were characterized to clarify their structures. Figure 4 shows XRD patterns of the Ni–Zn/SiO2 and Co–Ge/SiO2 catalysts. For Ni–Zn/SiO2, a solid-solution alloy between Ni and Zn with a 3: 1 ratio, namely Ni0.75Zn0.25 phase (Vassilev, 1992), was observed as single phase. A similar alloy phase (Co0.8Ge0.2) (Ishida and Nishizawa, 1991) was also observed as a main species for Co–Ge/SiO2. One-to-one intermetallic phase of CoGe was also detected as a minor species. Thus, XRD analysis confirmed the formation of alloy phases with high purities. The crystallite sizes of Ni–Zn and Co–Ge were estimated using Scherrer equation as <3 and 9 nm, respectively. The larger crystallite size of Co–Ge may stem from the higher reduction temperature (800°C) during the catalyst preparation. Figure 5 shows TEM and STEM images of Co–Ge/SiO2, size distribution of nanoparticles, and the elemental map of Co and Ge acquired using EDS. Particle size ranged from 2 to 20 nm with a mean diameter of 8.5 nm (Figures 5a,b), which is consistent with the crystallite size estimated by the Scherrer equation (9 nm). Figure 5c displays the high-resolution TEM image of a single Co–Ge nanoparticle. Lattice fringes with 2.06 spacing were clearly observed, which agrees finely with the interplanar distance of the (111) plane of Co0.8Ge0.2 solid-solution alloy with an fcc structure (2.07) (Ishida and Nishizawa, 1991). The elemental maps of Co and Ge that were acquired using the EDS analysis revealed that the Co and Ge atoms comprising the nanoparticles were homogeneously dispersed (Figures 5d–f). These results strongly suggest that the Co0.8Ge0.2 alloy nanoparticles were formed with high phase purities. Thus, the results obtained from the STEM-EDS analysis were consistent with that of XRD.
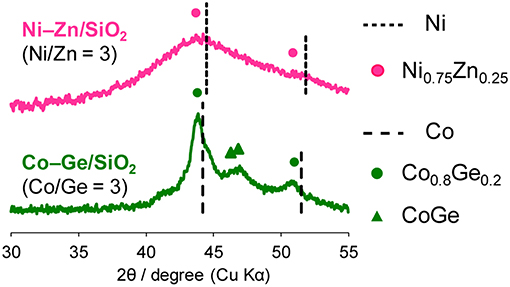
Figure 4. XRD patterns of Ni–Zn/SiO2 (Ni/Zn = 3) and Co–Ge/SiO2 (Co/Ge = 3).
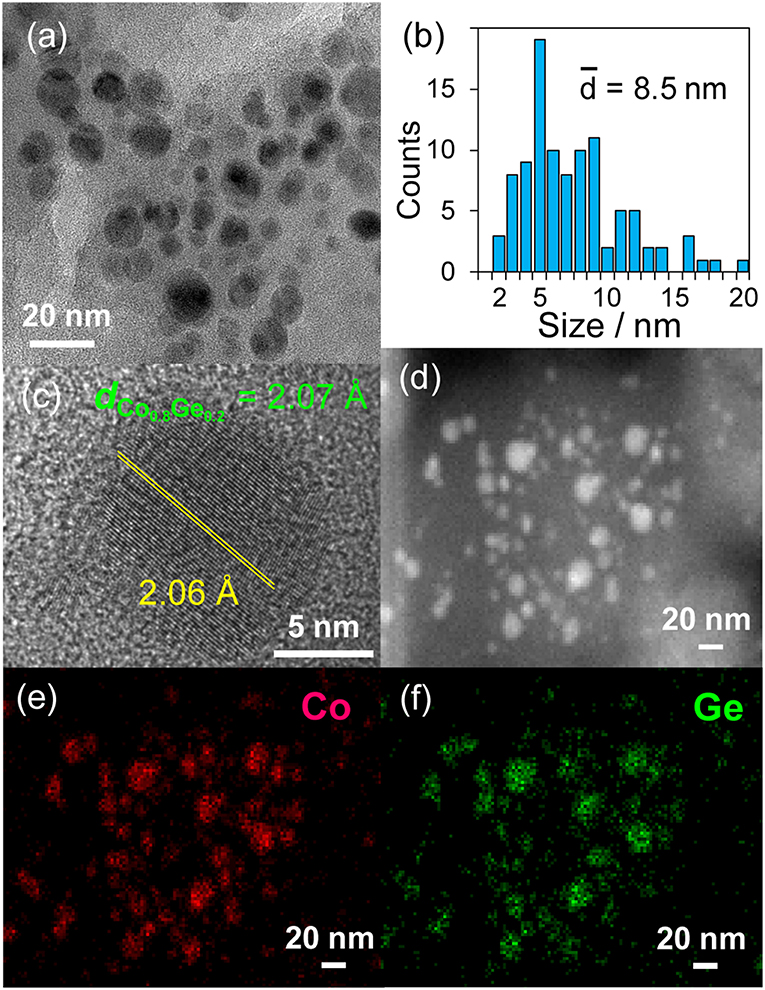
Figure 5. (a) TEM image and the (b) size distribution of Co–Ge/SiO2 catalyst. (c) High resolution TEM image of a single nanoparticle of Co–Ge. (d) HAAF-STEM image of Co–Ge/SiO2 catalyst and elemental maps of (e) Co, and (f) Ge for the nanoparticles acquired using EDS.
We then studied the reason why the catalytic activity was significantly enhanced by the formation of the alloy phase. Because the Co–Ge system exhibited a remarkable increase in the reaction rate (five times higher) compared to pure Co, we focused on the difference between the Co and Co–Ge systems.
Figure 6 shows the TPR profiles of the Co/SiO2 and Co–Ge/SiO2 catalysts. To evaluate the reduction temperatures of the component metals, as-impregnated catalysts were used for the TPR experiments. For both catalysts, intense peaks were observed at 150 and 200°C, which may be attributed to the reduction of O2 or NO2 derived from the decomposition of the Co(NO3)2 (van Steen et al., 1996). For Co/SiO2, two reduction peaks were observed at 260 and 760°C, which are assigned to the reduction of Co2+ species that weakly and strongly interact with the silica surface, respectively (van Steen et al., 1996). On the contrary, for Co–Ge/SiO2, three different reduction peaks were observed at 370, 550, and 740°C, which could be assigned to the reduction of Co2+, Co3+, and Ge4+ species. We previously reported a TPR profile of Ge/SiO2, with reduction peaks appearing at temperatures higher than 650°C (Komatsu et al., 1997). Therefore, the reduction peak at 740°C can be attributed to the reduction of Ge4+. The other reduction peaks assignable for Co species appeared at much higher temperatures than that for Co/SiO2, suggesting that the strong interaction between Co and Ge inhibits the reduction of Co2+. It can be said that Co species in Co/SiO2 were not completely reduced because the reduction temperature for the preparation of Co/SiO2 (600°C) is not sufficient to completely reduce all the Co species. This may be one of the reasons why the Co/SiO2 catalysts showed low catalytic activity. On the contrary, for Co–Ge/SiO2, the reduction temperature for the preparation (800°C) is enough to reduce all the Co2+ and Ge4+ species.
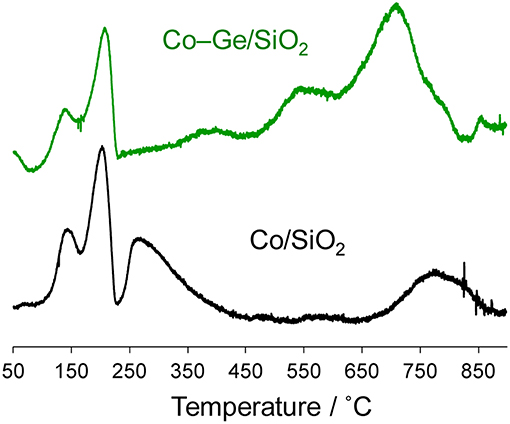
Figure 6. TPR profiles for as-impregnated Co(NO3)2/SiO2 and Co(NO3)2-(NH4)2GeF6/SiO2 catalysts.
We also investigated the electronic states of the Co/SiO2 and Co–Ge/SiO2 catalysts by XPS (Figure 7). For Co/SiO2, the fraction of Co2+ or Co3+ species (Sexton et al., 1986) was much higher than that of Co(0), suggesting that a large part of the catalyst surface is oxidized. By contrast, for Co–Ge/SiO2, Co(0) (Sexton et al., 1986) was detected as a main species. These results are consistent with the expectation derived from TPR mentioned above. It is also likely that Co atoms on the Co–Ge/SiO2 catalyst are more resistant to aerobic oxidation than those on Co/SiO2. Jagirdar et al. reported that Co2+ or Ni2+ ions could be reduced by the evolved H2 during hydrolysis of AB to form Ni or Co nanoparticles (Kalidindi et al., 2008). Therefore, it is likely that similar in situ reduction of Co2+ could occur to completely reduce the catalyst surface in our system. This may be the reason why the induction period was observed in H2 production from AB over Co/SiO2. The binding energies of Co2+ or Co3+ for Co/SiO2 and Co–Ge/SiO2 were almost same, suggesting that Ge species do not affect the electronic state of Co cations. However, a different trend was observed for Co(0) species: Co–Ge/SiO2 exhibited a lower binding energy than Co/SiO2, indicating that Co atoms in Co–Ge are electron-enriched compared with pure Co. This is probably because electron transfer from Ge to Co-occurs due to alloy formation. Similar electron transfer has also been reported for the relevant systems such as Pt–Ge alloys (Komatsu et al., 1997). Thus, XPS analysis revealed that the formation of Co–Ge alloy drastically changed the electronic state of Co. On the basis of these results, we considered that the difference in the electron density of Co(0) species, namely, electron-enrich Co by Ge via alloying, is the key factor for the remarkable enhancement in the catalytic activity.
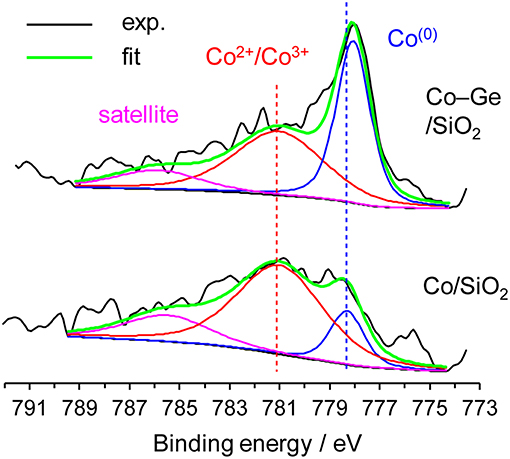
Figure 7. Co 2p3/2 XPS of Co/SiO2 and Co–Ge/SiO2 catalysts.
The reaction mechanism of AB hydrolysis has been reported as follows: (1) the formation of an activated complex between AB and the metal surface, (2) B–N bond scission assisted by H2O attack, and (3) hydrolysis of the resulting BH3 moiety to form H2 and (Xu and Chandra, 2006; Mahyari and Shaabani, 2014). It is also known that H atoms bound to the B and N atoms are slightly electropositive and electronegative, respectively, due to the different electronegativities of B and N. Therefore, it is likely that the alloy surface with polar active sites (Coδ−–Geδ+) facilitates the formation of the active complex (1), thus enhancing the following steps and the overall reaction rate. A similar reaction mechanism has also been reported in the system of Ruδ−–Niδ+ bimetallic catalysts for AB hydrolysis (Mori et al., 2016).
Conclusion
In this study, we prepared a series of Ni- and Co-based bimetallic catalysts (Ni–M/SiO2 and Co–M/SiO2; M = Ga, Ge, Sn, and Zn) and tested them in H2 production from AB in water or methanol. Catalytic activity for hydrogen production is enhanced by the addition of second metals except Sn. Particularly, the addition of Ge to Co enables great enhancement in the catalytic activity, namely a five-fold higher rH2 than the monometallic Co/SiO2 catalyst. The active species is electron-enriched Co atoms constituting the Co0.8Ge0.2 solid solution alloy phase. The Ni–Zn/SiO2 catalyst exhibited an rcat[AB] value higher than those ever reported for hydrolysis of AB to the best of our knowledge. The Ni0.75Zn0.25 solid solution alloy phase acts as the active species.
Author Contributions
SF designed the research and experiments. GN performed all the experiments. TT managed the HAADF-STEM-EDS analysis. SF and TK prepared the manuscript.
Funding
This work was supported by JSPS KAKENHI grant numbers 26820350 and 17H0496508.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We deeply appreciate the Center for Advanced Materials Analysis of the Tokyo Institute of Technology for the aid of TEM and STEM observation.
References
Chandra, M., and Xu, Q. (2007). Room temperature hydrogen generation from aqueous ammonia-borane using noble metal nano-clusters as highly active catalysts. J. Power Sources 168, 135–142. doi: 10.1016/j.jpowsour.2007.03.015
Chen, S. S., Takata, T., and Domen, K. (2017). Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2:17050. doi: 10.1038/Natrevmats.2017.50
Furukawa, S., and Komatsu, T. (2017). Intermetallic compounds: promising inorganic materials for well-structured and electronically modified reaction environments for efficient catalysis. ACS Catalysis 7, 735–765. doi: 10.1021/acscatal.6b02603
Han, J., Zheng, X., Zhang, L., Fu, H., and Chen, J. (2017). Removal of SO2 on a nanoporous photoelectrode with simultaneous H2 production. Environ. Sci. Nano 4, 834–842. doi: 10.1039/C6EN00638H
Heinzel, A., Vogel, B., and Hübner, P. (2002). Reforming of natural gas—hydrogen generation for small scale stationary fuel cell systems. J. Power Sources 105, 202–207. doi: 10.1016/S0378-7753(01)00940-5
Ishida, K., and Nishizawa, T. (1991). The Co-Ge (cobalt-germanium) system. J. Phase Equilibria 12, 77–83. doi: 10.1007/BF02663679
Kalidindi, S. B., Indirani, M., and Jagirdar, B. R. (2008). First row transition metal ion-assisted ammonia-borane hydrolysis for hydrogen generation. Inorgan. Chem. 47, 7424–7429. doi: 10.1021/ic800805r
Kalidindi, S. B., Vernekar, A. A., and Jagirdar, B. R. (2009). Co-Co2B, Ni-Ni3B and Co-Ni-B nanocomposites catalyzed ammonia-borane methanolysis for hydrogen generation. Phys. Chem. Chem. Phys. 11, 770–775. doi: 10.1039/b814216e
Komatsu, T., Hyodo, S., and Yashima, T. (1997). Catalytic properties of Pt-Ge intermetallic compounds in the hydrogenation of 1,3-butadiene. J. Phys. Chem. B 101, 5565–5572. doi: 10.1021/Jp971117l
Landge, V. G., Pitchaimani, J., Midya, S. P., Subaramanian, M., Madhu, V., and Balaraman, E. (2018). Phosphine-free cobalt pincer complex catalyzed Z-selective semi-hydrogenation of unbiased alkynes. Catalysis Sci. Technol. 8, 428–433. doi: 10.1039/c7cy01994g
Mahyari, M., and Shaabani, A. (2014). Nickel nanoparticles immobilized on three-dimensional nitrogen-doped graphene as a superb catalyst for the generation of hydrogen from the hydrolysis of ammonia borane. J. Mater. Chem. A 2, 16652–16659. doi: 10.1039/c4ta03940h
Marder, T. B. (2007). Will we soon be fueling our automobiles with ammonia–borane? Angew. Chem. Int. Ed. 46, 8116–8118. doi: 10.1002/anie.200703150
Metin, O., Mazumder, V., Ozkar, S., and Sun, S. S. (2010). Monodisperse nickel nanoparticles; and their catalysis in hydrolytic dehydrogenation of ammonia borane. J. Am. Chem. Soc. 132, 1468–1469. doi: 10.1021/ja909243z
Moniz, S. J. A., Shevlin, S. A., Martin, D. J., Guo, Z. X., and Tang, J. W. (2015). Visible-light driven heterojunction photocatalysts for water splitting—a critical review. Energy Environ. Sci. 8, 731–759. doi: 10.1039/c4ee03271c
Mori, K., Miyawaki, K., and Yamashita, H. (2016). Ru and Ru-Ni nanoparticles on TiO2 support as extremely active catalysts for hydrogen production from ammonia-borane. ACS Catalysis 6, 3128–3135. doi: 10.1021/acscatal.6b00715
Ozay, O., Inger, E., Aktas, N., and Sahiner, N. (2011). Hydrogen production from ammonia borane via hydrogel template synthesized Cu, Ni, Co composites. Int. J. Hydrogen Energy 36, 8209–8216. doi: 10.1016/j.ijhydene.2011.04.140
Patel, N., Fernandes, R., Guella, G., and Miotello, A. (2010). Nanoparticle-assembled Co-B thin film for the hydrolysis of ammonia borane: a highly active catalyst for hydrogen production. Appl. Catalysis B Environ. 95, 137–143. doi: 10.1016/j.apcatb.2009.12.020
Peng, C. Y., Kang, L., Cao, S., Chen, Y., Lin, Z. S., and Fu, W. F. (2015). Nanostructured Ni2P as a robust catalyst for the hydrolytic dehydrogenation of ammonia-borane. Angew. Chem. Int. Ed. 54, 15725–15729. doi: 10.1002/anie.201508113
Schlapbach, L., and Züttel, A. (2011). “Hydrogen-storage materials for mobile applications,” in Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, ed V. Dusastre (London: World Scientific), 265–270.
Sexton, B., Hughes, A., and Turney, T. (1986). An XPS and TPR study of the reduction of promoted cobalt-kieselguhr Fischer-Tropsch catalysts. J. Catalysis 97, 390–406. doi: 10.1016/0021-9517(86)90011-4
van Steen, E., Sewell, G. S., Makhothe, R. A., Micklethwaite, C., Manstein, H., de Lange, M., et al. (1996). TPR study on the preparation of impregnated Co/SiO2 catalysts. J. Catalysis 162, 220–229. doi: 10.1006/jcat.1996.0279
Vassilev, G. P. (1992). Thermodynamic evaluation of the Ni-Zn system. J. Alloys Compounds 190, 107–112. doi: 10.1016/0925-8388(92)90184-B
Wang, Q., Hisatomi, T., Suzuk, Y., Pan, Z. H., Seo, J., Katayama, M., et al. (2017). Particulate photocatalyst sheets based on carbon conductor layer for efficient Z-scheme pure-water splitting at ambient pressure. J. Am. Chem. Soc. 139, 1675–1683. doi: 10.1021/jacs.6b12164
Xu, Q., and Chandra, M. (2006). Catalytic activities of non-noble metals for hydrogen generation from aqueous ammonia-borane at room temperature. J. Power Sources 163, 364–370. doi: 10.1016/j.jpowsour.2006.09.043
Xu, Q., and Chandra, M. (2007). A portable hydrogen generation system: catalytic hydrolysis of ammonia–borane. J. Alloys Compounds 446, 729–732. doi: 10.1016/j.jallcom.2007.01.040
Yan, J. M., Zhang, X. B., Han, S., Shioyama, H., and Xu, Q. (2008). Iron-nanoparticle-catalyzed hydrolytic dehydrogenation of ammonia borane for chemical hydrogen storage. Angew. Chem. Int. Ed. 47, 2287–2289. doi: 10.1002/anie.200704943
Yan, J. M., Zhang, X. B., Han, S., Shioyama, H., and Xu, Q. (2009). Synthesis of longtime water/air-stable ni nanoparticles and their high catalytic activity for hydrolysis of ammonia-borane for hydrogen generation. Inorgan. Chem. 48, 7389–7393. doi: 10.1021/ic900921m
Zhang, L., Reisner, E., and Baumberg, J. J. (2014). Al-doped ZnO inverse opal networks as efficient electron collectors in BiVO4 photoanodes for solar water oxidation. Energy Environ. Sci. 7, 1402–1408. doi: 10.1039/C3EE44031A
Zheng, X. Z., and Zhang, L. W. (2016). Photonic nanostructures for solar energy conversion. Energy Environ. Sci. 9, 2511–2532. doi: 10.1039/c6ee01182a
Keywords: hydrogen production, ammonia borane, hydrolysis, alloy, catalyst
Citation: Furukawa S, Nishimura G, Takayama T and Komatsu T (2019) Highly Active Ni- and Co-Based Bimetallic Catalysts for Hydrogen Production From Ammonia-Borane. Front. Chem. 7:138. doi: 10.3389/fchem.2019.00138
Received: 27 November 2018; Accepted: 25 February 2019;
Published: 20 March 2019.
Edited by:
Steve Suib, University of Connecticut, United StatesReviewed by:
Balaraman Ekambaram, National Chemical Laboratory (CSIR), IndiaLiwu Zhang, Fudan University, China
Guohong Wang, Hubei Normal University, China
Copyright © 2019 Furukawa, Nishimura, Takayama and Komatsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shinya Furukawa, ZnVydWthd2FAY2F0Lmhva3VkYWkuYWMuanA=
Takayuki Komatsu, a29tYXRzdS50LmFkQG0udGl0ZWNoLmFjLmpw