 Xinhao Zhang1,2
Xinhao Zhang1,2 Ruifa Jin1,2*
Ruifa Jin1,2*- 1Inner Mongolia Key Laboratory of Photoelectric Functional Materials, Chifeng University, Chifeng, China
- 2College of Chemistry and Chemical Engineering, Chifeng University, Chifeng, China
A series of D–A novel star-shaped molecules with 2,4,6-triphenyl-1,3,5-triazine (TPTA) as core, diketopyrrolo[3,4-c]pyrrole (DPP) derivatives as arms, and triphenylamine (TPA) derivatives as end groups have been systematically investigated for organic solar cells (OSCs) applications. The electronic, optical, and charge transport properties were studied using density functional theory (DFT) and time-dependent DFT (TD-DFT) approaches. The parameters such as energetic driving force ΔEL−L, adiabatic ionization potential AIP, and adiabatic electron affinity AEA were also calculated at the same level. The calculated results show that the introduction of different groups to the side of DPP backbones in the star-shaped molecules can tune the frontier molecular orbitals (FMOs) energy of the designed molecules. The designed molecules can provide match well with those of typical acceptors PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) and PC71BM ([6,6]-phenyl-C71-butyric acid methyl ester). Additionally, the absorption wavelengths of the designed molecules show bathochromic shifts compared with that of the original molecule, respectively. The introduction of different groups can extend the absorption spectrum toward longer wavelengths, which is beneficial to harvest more sunlight. The calculated reorganization energies suggest that the designed molecules are expected to be the promising candidates for ambipolar charge transport materials except molecule with benzo[c]isothiazole group can be used as hole and electron transport material. Moreover, the different substituent groups do not significantly affect the stability of the designed molecules.
Introduction
Nowadays, organic π-conjugated small molecules (SMs) used as the donors in organic solar cells (OSCs) have drawn intense attention because of their outstanding advantages, such as excellent reproducibility, easy purification, well-defined chemical and optoelectronic properties (Coughlin et al., 2014; Yao et al., 2016; Bin et al., 2017). Owing to the tremendous efforts on improving the performance of OSCs based on SMs, their power conversion efficiency (PCE) has surpassed over 10% recently (Zhou et al., 2012; Kan et al., 2015). However, it is worth noting that their overall performance still falls behind that of their polymer counterparts (Ni et al., 2013; Lin and Zhan, 2016). Accordingly, to address this issue, it is a big challenge to design and synthesize high-performance and desirable donor novel SMs (Chaudhry et al., 2018; Irfan et al., 2018; Wazzan et al., 2018). In general, the high-efficiency SMs donor materials should possess suitable frontier molecular orbital (FMOs) (including the highest occupied molecular orbital, HOMO, and lowest unoccupied molecular orbital, LUMO) energy levels, high charge carrier mobility, broad absorption region, and miscibility with fullerene derivatives. In this regard, the HOMO level of the designed donor materials should been reduced to increase the open circuit voltage (Voc), because the HOMO of donor and the LUMO of acceptor are closely relate to the Voc. With the aim to harvest more sunlight, the energy gaps of the designed donor materials should been decreased, which results in an increase in the short circuit current density (Jsc) (Loser et al., 2017; Maglione et al., 2017; Zhang et al., 2017). Moreover, a key factor that impact on the efficient exciton splitting and charge dissociation is the downhill energetic driving force (ΔEL−L), which is the energy differences between the LUMOs of the donor and acceptor. The ΔEL−L value should be about 0.3 eV to ensure efficient charge transfer, exciton splitting, and charge dissociation (Scharber et al., 2006). Therefore, an ideal donor material should have narrowing the HOMO-LUMO gap (Eg) and suitable FMOs energy levels with PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) and PC71BM ([6,6]-phenyl-C71 butyric acid methyl ester), which are widely employed as acceptors in OSCs (He et al., 2007; Lenes et al., 2008). Among the various approaches to design organic π-conjugated SMs materials with the long range absorption, one of the successful approaches is to incorporate the electron-donating (D) and electron-accepting (A) moieties in π-conjugated SMs (Qu and Tian, 2012; Guo et al., 2017; Wang et al., 2017). The FMOs energy levels, absorption and emission properties as well as intermolecular charge transfer of these materials can be tuned effectively by altering the chemistries of the donor and acceptor units. At the same time, adjusting the donor and acceptor units can also affect their self-assembly in the solid state. Among the various D–A type SMs donors for OSCs, diketopyrrolo[3,4-c]pyrrole (DPP)-based molecules are promising building blocks owing to their excellent coplanarity, broader absorption region, and thermal stability (Chen et al., 2013; Lin et al., 2013; Zhang et al., 2014). Furthermore, the introduction of the planar heteroarenes into the strong electron-withdrawing DPP-based molecules backbones can lead to lower the band gap because of increasing effective conjugation length (Dutta et al., 2012; Patra et al., 2013). In addition, star-shaped SMs materials with π-conjugated arms can harvest sunlight effectively because of their extended dimensionality. Meanwhile, their steric hindrances can prevent the formation of an ordered, long-range, and coplanar π-π stacking, which are beneficial for their charge transport property (Irfan et al., 2017). Therefore, the star-shaped D–A type DPP-based molecules may possess narrower band gap, broader absorption region, strong light absorption, and high charge carrier mobility (Sharma et al., 2014; Shiau et al., 2015).
Considering these merits and characteristics mentioned above, in this contribution, we report the design of a series of novel star-shaped DPP-based molecules with electron-accepting 2,4,6-triphenyl-1,3,5-triazine (TPTA) as core, electron accepting DPP derivatives as arms, and electron-donating triphenylamine derivatives (TPA) as end groups for OSCs applications (as shown in Scheme 1). With the aim to investigate the relationships between structure and properties of the designed molecules, the different planar heteroarenes have been introduced into the side of DPP molecules backbones in the star-shaped molecules. The HOMO energy (EHOMO), LUMO energy (ELUMO), HOMO–LUMO gap (Eg), energetic driving force ΔEL−L, and absorption spectra of the designed molecules were systematically investigated by applying density functional theory (DFT) and time-dependent DFT (TD-DFT) methodology. The charge transfer properties (reorganization energy, λ) were also simulated.
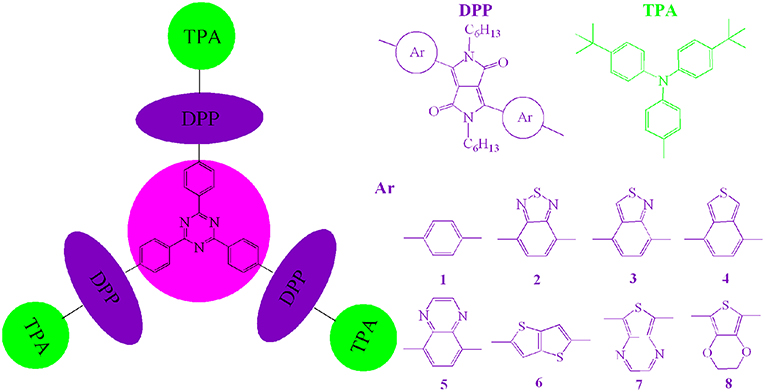
Scheme 1. Molecule models of 1–8 investigated in this work.
Computational Details
Using the Gaussian 09 W software package (Frisch et al., 2009), all the geometry optimizations and frequency for the designed molecules in the gas phase were performed with the DFT method. No imaginary frequency was used to ensure the nature of the stationary point for the optimized molecules. On the basis of the optimized structures, the absorption spectra of the designed molecules were predicted using the TD-DFT method. The 6-31G (d,p) basis set was employed for all calculations in this work. For the FMOs energy levels of the designed molecules, because it is difficult to describe the virtual orbitals theoretically (Wu et al., 2013). The LUMO energy levels can be calculated with the equation, ELUMO = EHOMO + Eex, where Eex represents the first vertical excited energy (Zhang and Musgrave, 2007; Ku et al., 2011; Zhang et al., 2012). A crucial step in the theoretical investigations is to select an appropriate exchange correlation functional. With the aim to select an appropriate approach, we chose various functionals such as B3LYP (Lee et al., 1988), PBE0 (Adamo and Barone, 1999), LC-wPBE (Tawada et al., 2004), M062X (Zhao and Truhlar, 2008), and CAM-B3LYP (Yanai et al., 2004) to optimize the geometries of the parent molecule 1. Based on the optimized geometries, the absorptions were predicted using the TD-DFT method. The longest wavelengths of absorption (λabs) as well as the experimental data are shown in Figure 1. As showing Figure 1, the calculated λabs value obtained at PBE0 (543 nm) level provided better agreement with the experimental value (523 nm) (Shiau et al., 2015) than those obtained with other levels of theory, with the deviation being 20 nm. Although B3LYP appeared adapted to 1,3,5-triazine and DPP derivatives in literature (Feng et al., 2014; Vala et al., 2014; Jin, 2015; Jin and Irfan, 2015; Jin and Xiao, 2015; Fujii et al., 2016), the λabs value obtained at the B3LYP/6-31G (d,p) level is worsen accordance with the experimental data (the deviation is 48 nm) than that for at the PBE0/6-31G (d,p) level (the deviation is 20 nm). Additionally, we also calculated the FMOs energy levels of molecule 1 using both at PBE0 and B3LYP methods. The calculated EHOMO and ELUMO values (−5.04 and −2.76 eV) at the PBE0/6-31G (d,p) level are more close to the electrochemical measurements data (−5.47 and −3.41 eV) (Shiau et al., 2015) than those obtained at the B3LYP/6-31G (d,p) level (−4.82 and −2.67 eV), respectively. Furthermore, in order to make further investigation of the validity of the selected approach, both PBE0 and B3LYP methods were also employed to optimize the structure of PCBM and PC71BM. The calculated EHOMO and ELUMO of PCBM and PC71BM along with available experimental data are listed in Table S1. Inspection of Table S1 reveals clearly that the EHOMO and ELUMO at the PBE0/6-31G (d,p) level of PCBM are −5.98 and −3.99 eV, and the corresponding values of PC71BM are −5.92 and −3.82 eV, respectively. These are well reproduce the experimental values of PCBM (−6.00 and −3.80 eV) (Jeon et al., 2016) and PC71BM (−6.00 and −3.95 eV) (Chandrasekharam et al., 2014), respectively. However, at B3LYP/6-31G (d,p) level, the calculated EHOMO and ELUMO of PCBM are −5.67 and −3.75 eV, while the corresponding values of PC71BM are −5.61 and −3.60 eV, respectively. It was noticed that B3LYP overestimate the EHOMO and ELUMO of PCBM and PC71BM. The B3LYP overestimate the EHOMO and ELUMO compared with experimental value, as reported in the literature (Blouin et al., 2008; Xiao et al., 2010; Abbotto et al., 2012). Therefore, PBE0 functional is reasonable to investigate the current system. In order to obtain insight into the method to describe and the influence of functionals on the optical properties, the absorption spectrum of the designed molecules were also simulated at B3LYP/6-31G (d,p) levels.
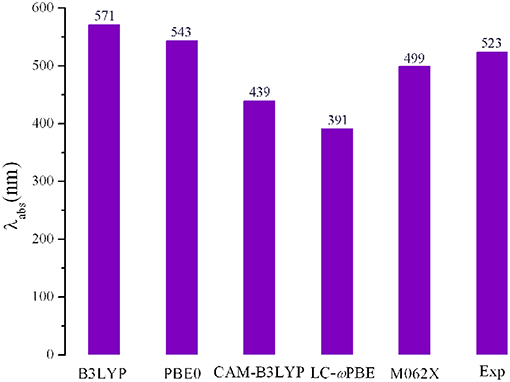
Figure 1. Calculated absorption wavelengths (λabs) of 1 using various functionals, together with the experimental result.
It is well-known that the reorganization energy (λ) play the dominant role in the effective charge transfer according to the Marcus theory (Marcus, 1964, 1993). A good charge transfer materials should possess lower λ values, which led to higher charge transfer rate. We only pay attention to the internal reorganization energy in this work owing to the low dielectric constant of medium in OSCs materials (Marcus, 1964). The electron (λe) and hole (λh) reorganization energy can be expressed as follows (Köse et al., 2007; Sancho-García, 2007):
Here, and are the energies of the cationic (anionic) states with the optimized neutral and cationic (anionic) geometry, respectively. and represent the energy of the neutral states with the optimized geometry of the cationic (anionic) and neutral structures, respectively. The λe and λh of the designed molecules were predicted at the PBE0/6-31G(d,p) level.
It is noteworthy that the stability is the most important criteria to evaluate the nature of devices for OSCs. Generally, the absolute hardness (η) was applied to explore the stability of the materials. From a viewpoint of conceptual density functional theory, the η values of the designed molecules were calculated with the following equation (Cheung and Troisi, 2010):
Here, μ and N correspond to the chemical potential and total electron number, respectively. The adiabatic ionization potential (AIP) is the energy difference between the cation radical specie and its neutral specie, while the adiabatic electron affinity (AEA) represents the energy difference between the neutral molecule and its anion radical molecule.
Results and Discussion
Frontier Molecular Orbitals and Band Gaps
In order to characterize the optical and electronic properties, we investigated the distributions of the FMOs for the designed molecules. The distribution of HOMOs and LUMOs are plotted in Figure 2. Based on Mulliken population analysis, molecular orbital contribution (%) from core TPTA, arms DPP, and end groups TPA to the FMOs of 1–8 are given in Table 1. The corresponding contributions (%) from TPTA, DPP, and TPA groups to the HOMOs-1 and LUMOs+1 of 1–8 are given in Table S2. As visualized in Figure 2, the distribution of HOMOs and LUMOs are spread over the conjugated backbone and show π orbital features. The HOMOs are mainly localized on the arm groups DPP and end groups TPA with only minor contributions from the core fragments TPTA. The sum contributions of DPP and TPA fragments are larger than 96.1%, while the corresponding values of TPTA fragments are within 3.9% for HOMOs. On the contrary, the LUMOs are mainly distributed on the DPP and TPTA moieties with minor contributions from TPA fragments. The sum contributions of DPP and TPTA fragments for LUMOs are larger than 94.4%, while the corresponding values of TPA fragments are within 5.6%. Obviously, the contributions of both DPP and TPTA fragments for LUMOs are larger than those of for HOMOs, respectively. The contributions of TPA fragments to LUMOs are decreased compared with those of to HOMOs, respectively. Similar phenomena are found for the HOMOs-1 and LUMOs+1 of 1–8. The changes in contributions suggest that the electronic density flow from the end groups TPA to the arms groups DPP and cores groups TPTA for HOMOs → LUMOs excitations. This indicates that the end groups TPA serve as donors, whereas, the arm groups DPP and core groups TPTA serve as acceptors, respectively.
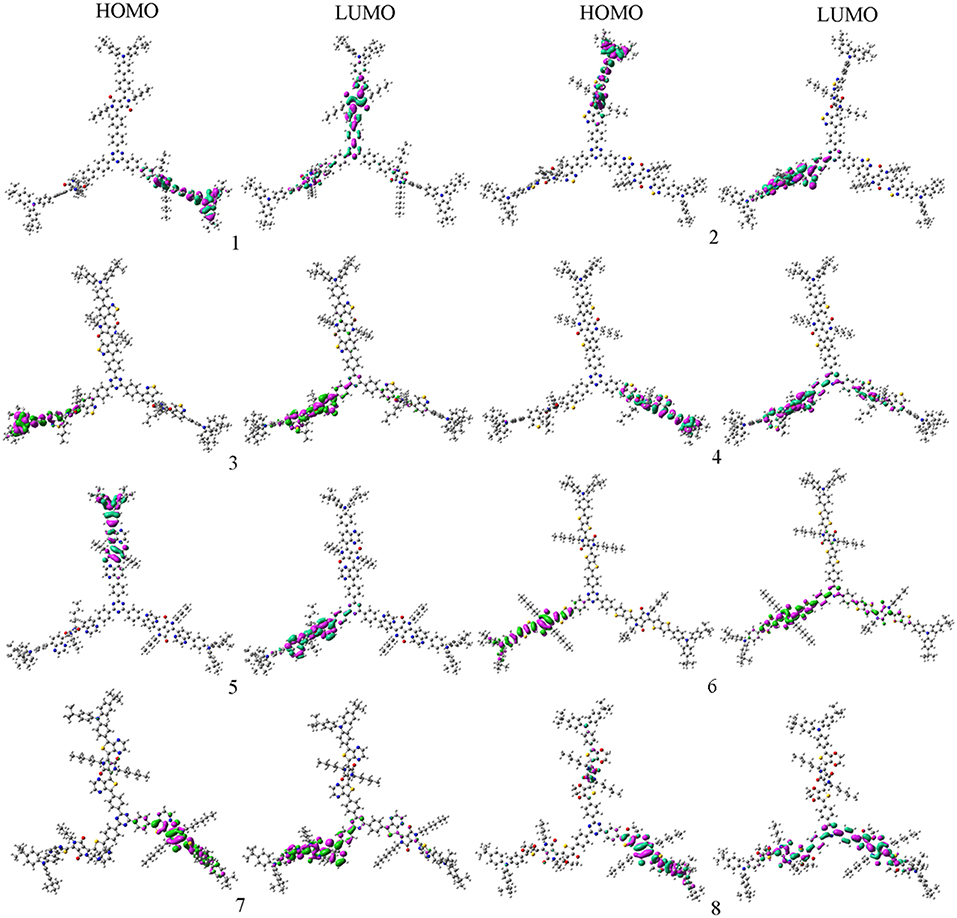
Figure 2. The FMOs of the designed molecules at the PBE0/6-31G(d,p) level.
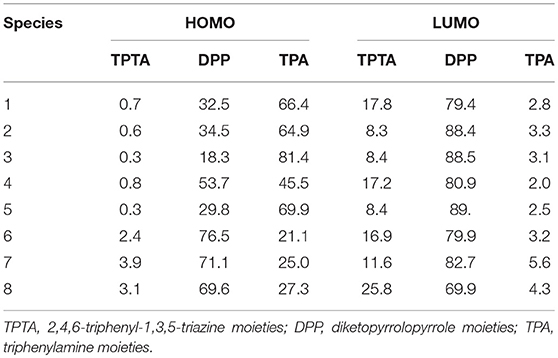
Table 1. Molecular orbital contribution (%) from core TPTA, arms DPP, and end groups TPA to the FMOs of 1–8 at the PBE0/6-31G(d,p).
It is worth noting that the EHOMO, ELUMO, Eg, and ΔEL−L are strongly related to the optical and electronic properties. The calculated values of EHOMO, ELUMO, Eg, and ΔEL−L of the designed molecules are given in Table 2 and depicted in Figure 3. As shown in Figure 3, it is clear that the EHOMO values of 3–8 increase, while the corresponding value of 2 decreases compared with that of 1. The EHOMO values is in the order of 8 > 7 > 6 > 4 ≈ 5 > 3 > 1 > 2. On the other hand, the ELUMO values of 2–7 decrease, while the corresponding value of 8 increases compared with that of 1. The sequence of ELUMO values is 8 > 1 > 4 > 5 > 6 > 3 > 2 > 7. Therefore, the Eg values of 2–8 decrease compared with that of 1. The Eg values are in the order of 1 < 4 < 5 < 8 < 3 < 6 < 2 < 7. The analysis indicates that the decrease of Eg is mainly attributable to the increased EHOMO and declined ELUMO. The reducing the Eg of the designed molecules should leads to bathochromic shifts of the maximum absorption compared with that of 1. Consequently, the introduction of different groups to the side of DPP molecules backbones in the star-shaped molecules can tune the FMOs energy and Eg values of the original molecule. It provides a powerful strategy for design high-performance and desirable donor novel SMs. Furthermore, in order to ensure efficient charge transfer, the ΔEL−L values must exceed the binding energy (0.2 ~ 1.0 eV) (Hill et al., 2000; Knupfer, 2003). From Table 2, it is noteworthy that the ΔEL−L values of the designed molecules are all beyond the binding energy with regard to PCBM and PC71BM as acceptors. It is clear that the sequence of the values of ΔEL−L with regard to PCBM and PC71BM are all 8 > 1 > 4 > 5 > 6 > 3 > 7 > 2. In addition, the differences between the EHOMO of 1–8 and the ELUMO of PCBM and PC71BM are larger than 0.73 and 0.56 eV, respectively. Thus, it is quite clear that the designed molecules can provide match well with PCBM and PC71BM as acceptors.
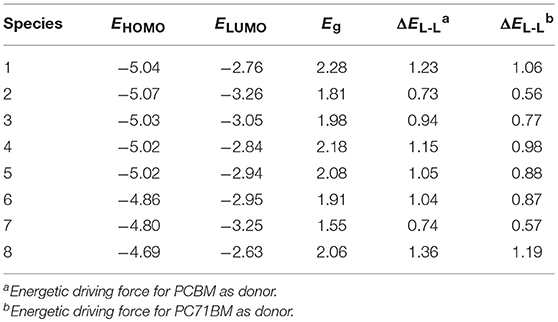
Table 2. Calculated EHOMO, ELUMO, Eg, and ΔEL−L (all in eV) for investigated molecules at the PBE0/6-31G(d,p).
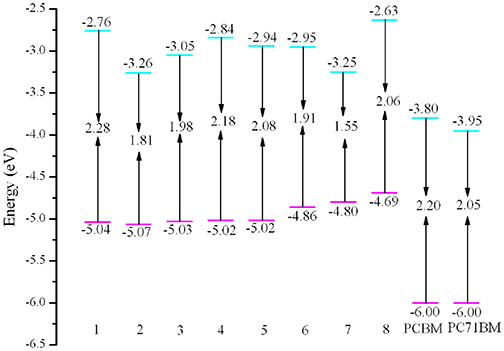
Figure 3. Evaluation of calculated FMO energies for the designed molecules as well as FMO energies for PCBM and PC70BM at the PBE0/6-31G(d,p) level.
Absorption Spectra
The absorption wavelengths λabs (in nm), the oscillator strength f, and main assignments (coefficient), and the absorption region R of 1–8 at the PBE0/6-31G(d,p) level are listed in Table 3. R denotes for the difference of the longest and shortest wavelength values with oscillator strength larger than 0.01 considering the first 15 excited states (see Table S3). The simulated absorption spectra of 1–8 are shown in Figure 4, which were plotted by using the GaussSum 1.0 program (O'Boyle and Vos, 2003). As expected, the results displayed in Table 3 reveals that the λabs of 2–8 exhibit bathochromic shifts compared with that of 1. The bathochromic shifts values of 2–8 are 143, 83, 26.1, 53.8, 104.6, 256.2, and 59.0 nm (3834, 2439, 844, 1658, 2971, 5897, and 1803 cm−1), respectively. Moreover, the λabs values are in the order of 7 > 2 > 6 > 3 > 8 > 5 > 4 > 1, which is in excellent agreement with the corresponding reverse sequence of their Eg values. It reveals that the introduction of different groups to the side of DPP molecules backbones leads to bathochromic shifts of the maximum absorption for the original molecule. The order of the bathochromic shifts values compared with that of 1 is thieno[3,4-b]pyrazine (7) > benzo[c][1,2,5]thiadiazole (2) > thieno[3,2-b]thiophene (6) > benzo[c]isothiazole (3) > 2,3-dihydrothieno[3,4-b][1,4]dioxine (8) > quinoxaline (5) > benzo[c]thiophene (4). Additionally, one can find that 6–8 have larger oscillator strengths, while the corresponding values of 2, 3, and 5 possess slightly < that of 1. The oscillator strength value of 4 is almost equal to that of 1, indicating that the designed molecules shown large absorption intensity. At the same time, the designed molecules have large absorption region R (82.3–225.7 nm). The R values of 2–8 are larger than that of parent compound 1. It suggests that the introduction of different groups to the side of DPP molecules backbones lead to the increase of R values compared with parent molecule 1. The order of R values compared with that of 1 is thieno[3,4-b]pyrazine (7) > benzo[c]isothiazole (3) > 2,3-dihydrothieno[3,4-b][1,4]dioxine (8) > benzo[c][1,2,5]thiadiazole (2) > thieno[3,2-b]thiophene (6) > quinoxaline (5) > benzo[c]thiophene (4). It is noticeable that a good overlap between the absorption spectrum of the designed molecules and the solar emission spectrum, which can improve the light-absorption efficiency. It clearly shows that the introduction of different groups can extend the absorption spectrum toward longer wavelengths, which is beneficial to harvest more sunlight. These results imply that the designed compounds have strong absorption and are expected to be the promising candidates for donor materials in OSCs applications.
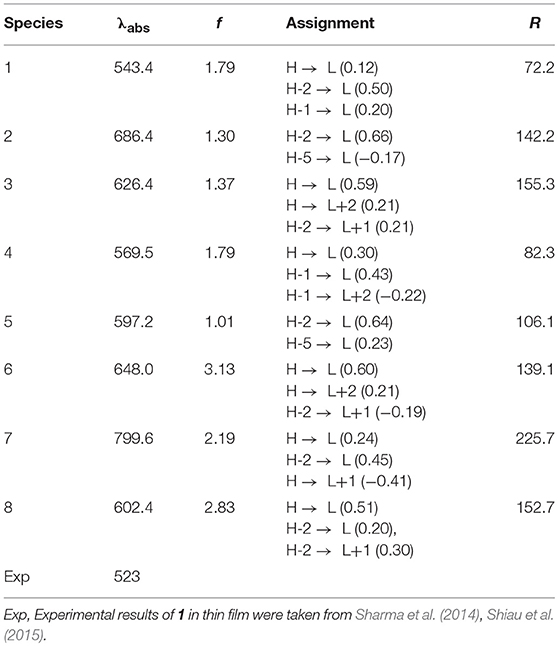
Table 3. The electronic transition, absorption wavelengths λabs (in nm), the oscillator strength f, main assignments (coefficient), and the absorption region R of 1–8 at the TD-PBE0/6-31G(d,p)//PBE0/6-31G(d,p) level, along with available experimental data.
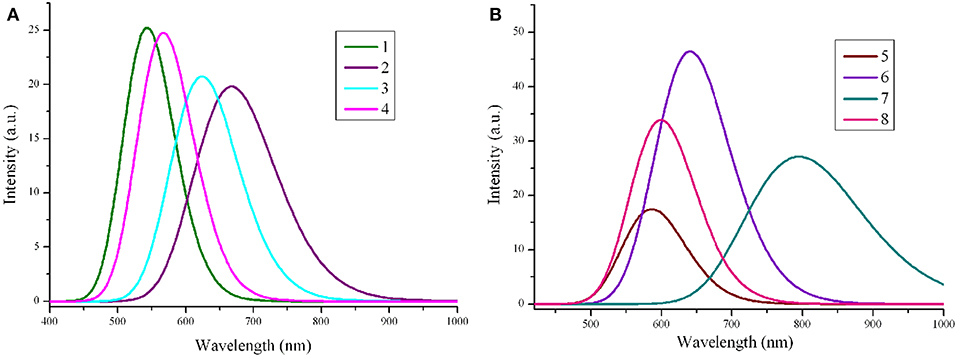
Figure 4. The calculated absorption spectra of the investigated molecules (value of full width at half maximum is 3,000 cm−1). (A) Molecules 1–4; (B) Molecules 5–8.
The calculated λabs, f, and main assignments (coefficient) of 1–8 at the B3LYP/6-31G(d,p) level are listed in Table S4. Comparing the results shown in Table 3 with Table S4, one can find that the calculated λabs values of 1–8 at the B3LYP/6-31G(d,p) are larger than those obtained at the PBE0/6-31G(d,p), respectively. The differences between λabs at the B3LYP/6-31G(d,p) and PBE0/6-31G(d,p) levels are about 30 ~ 50 nm. It should be mentioned that although the EHOMO and ELUMO are overestimated with both the PBE0 and B3LYP functionals, the predicted λabs values using PBE0 are found to be closer to the experimental findings. The trend for λabs at B3LYP/6-31G(d,p) is similar to using PBE0/6-31G(d,p) method. Obviously, B3LYP functional underestimate the Eg value, resulting in the large λabs compared with experimental value. Considering the FMOs energy levels and the predicted absorption spectra mentioned above, the PBE0/6-31G(d,p) approach is the best choice to well reproduce the experimental results. Therefore, the λ, η, AIP, and AEA of the designed molecules were computed at PBE0/6-31G(d,p).
Adiabatic Ionization Potential and Electron Affinity
It is well known that AIP and AEA are two major parameters that determine the charge transfer behavior for materials. The carrier polarity of materials can be adjusted by the AIP and AEA values (Chen and Chao, 2005; Liu et al., 2010). The lower AIP and higher AEA revealed that material would be better hole and electron transporter, respectively (Li et al., 2012). The calculated AIP and AEA of 1–8 are collected in Table 4. Obviously, the results displayed in Table 4 show that the increasing sequence of AIP values is 8 < 7 < 6 < 4 < 5 < 1 < 3 < 2. On the other hand, the decreasing order of AEA values is 7 > 2 > 3 > 6 > 5 > 4 > 1 > 8. It indicates that the introduction of benzo[c]thiophene (4), quinoxaline (5), thieno[3,2-b]thiophene (6), and thieno[3,4-b]pyrazine (7) groups can decrease/increase AIP/AEA values compared with that of 1. However, the benzo[c][1,2,5]thiadiazole (2) and benzo[c]isothiazole (3) groups can increase both AIP and AEA values, whereas 2,3-dihydrothieno[3,4-b][1,4]dioxine (8) group can decrease both AIP and AEA values compared with that of 1. It is noticeable that the introduction of different aromatic heterocyclic group to the side of DPP molecules backbones can affect the AIP and AEA of the designed molecules.
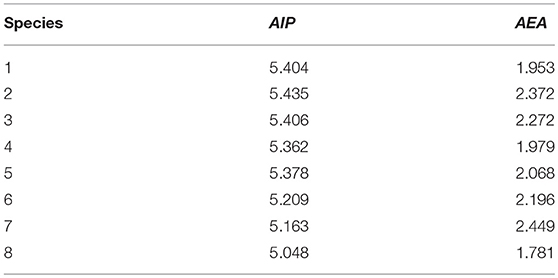
Table 4. Calculated molecular AIP and AEA (both in eV) of 1–8 at the PBE0/6-31G(d,p) level.
Reorganization Energies and Stability Properties
The calculated λe, λh, and η of 1–8 are listed in Table 5. It is worth noting that the lower the reorganization energy values can be beneficial to the higher charge transfer rate (Marcus, 1964, 1993). Usually, tris(8-hydroxyquinolinato)aluminum(III) (Alq3, λe = 0.276 eV) and N,N′-diphenyl-N,N′-bis(3- methlphenyl)-(1,1′-biphenyl)-4,4′-diamine (TPD, λh = 0.290 eV) are taken as typical electron and hole transport materials, respectively (Gruhn et al., 2002; Lin et al., 2005). It is clear from Table 5 that the λh values of 1–8 (0.046–0.129 eV) are smaller than that of TPD. It indicates that the hole transfer rates of 1–8 are higher than that of TPD. On the other hand, the λe values of 1–8 (0.091–0.209 eV) are smaller than that of Alq3. It implies that the electron transfer rates of 1–8 might be higher than that of Alq3. The λh values of 1–5 are slightly smaller than those of 6–8, suggesting that the hole transfer rates of 1–5 should be higher than those of 6–8, respectively. It indicates that the introduction of benzene (1), benzo[c][1,2,5]thiadiazole (2), benzo[c]isothiazole (3), benzo[c]thiophene (4), and quinoxaline (5) groups may lead to higher charge transfer rates than that of thieno[3,2-b]thiophene (6), thieno[3,4-b]pyrazine (7), and 2,3-dihydrothieno[3,4-b][1,4]dioxine (8) groups, respectively. The λe values is in the order of 3 > 5 > 4 > 1 > 8 > 7 > 2 > 6. It suggests that molecules with benzo[c][1,2,5]thiadiazole (2), thieno[3,2-b]thiophene (6), thieno[3,4-b]pyrazine (7), and 2,3-dihydrothieno[3,4-b][1,4]dioxine (8) possess higher electron transfer rates, while molecules with benzo[c]isothiazole (3), benzo[c]thiophene (4), and quinoxaline (5) groups have lower electron transfer rates compared with that of 1, respectively. Additionally, the λh values of 1–5 are smaller than those of their λe values, suggesting that the carrier mobility of the hole is larger than that of the electron. However, the λe values of 6 and 7 are smaller than those of their λh values, implying that the carrier mobility of the electron is larger than that of the hole. Moreover, the differences between λe and λh values of the designed molecules are in the region of 0.00 ~ 0.087 eV except the corresponding value of 3 is 0.163 eV, respectively. It indicates that they exhibit better equilibrium feature for hole and electron transport. Therefore, 1, 2, and 4–8 are expected to be the promising candidates for ambipolar charge transports materials, whereas 3 can be used as hole and electron transport material.
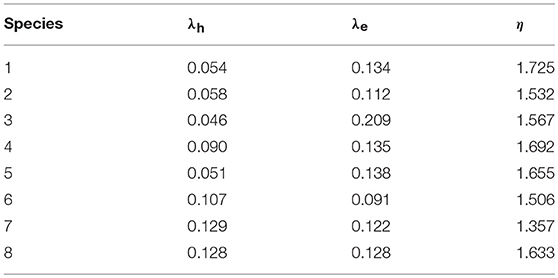
Table 5. Calculated molecular λe, λh, and η (all in eV) of 1–8 at the PBE0/6-31G(d,p) level.
The absolute hardness η of 1–8 were calculated and shown in Table 5. The η values is in the order of 1 > 4 > 5 > 8 > 3 > 2 > 6 > 7. Inspection of Table 5 reveals clearly that the η values of 2–8 are smaller slightly than the value of 1, which may be owing to the steric hindrances of the heterocyclic groups introduced to the side of DPP backbones in star-shaped DPP-based molecules. It implies that the introduction of different heterocyclic groups do not significantly affect the stability of the designed molecules.
Conclusion
In this contribution, a series of novel star-shaped molecules have been systematically investigated. Their electronic, optical, and charge transport properties studied using DFT and TD-DFT approaches. The calculated results show that the introduction of different groups to the side of DPP backbones in the star-shaped molecules can tune the FMOs energy and Eg values of the original molecule. The designed molecules can provide match well with PCBM and PC71BM as acceptors. Additionally, the λabs of 2–8 show bathochromic shifts compared with that of the original molecule 1, respectively. The introduction of different groups can extend the absorption spectrum toward longer wavelengths, which is beneficial to harvest more sunlight. Our results suggest that the designed molecules are expected to be the promising candidates for ambipolar charge transport materials except molecule with benzo[c]isothiazole group (3) can be used as hole and electron transport material. Moreover, the different substituent groups do not significantly affect the stability of the designed molecules.
Author Contributions
RJ conceived and designed the research and headed, wrote, and revised the manuscript. XZ contributed to the performance and analysis of the frontier molecular orbitals, absorption spectra the reorganization energies. Both authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NSFC (No. 21563002) and the Research Program of Sciences at Universities of Inner Mongolia Autonomous Region (No. NJZY19223).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00122/full#supplementary-material
References
Abbotto, A., Seri, M., Dangate, M. S., De Angelis, F., Manfredi, N., Mosconi, E., et al. (2012). A vinylene-linked benzo[1,2-b:4,5-b′]dithiophene-2,1,3-benzothiadiazole low-bandgap polymer. J. Polym. Sci. Pol. Chem. 50, 2829–2840. doi: 10.1002/pola.26046
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. HJ. Chem. Phys. 110, 6158–6170. doi: 10.1063/1.478522
Bin, H., Yang, Y., Zhang, Z. G., Ye, L., Ghasemi, M., Chen, S., et al. (2017). 9.73% efficiency nonfullerene all organic small molecule solar cells with absorption-complementary donor and acceptor. J. Am. Chem. Soc. 139, 5085–5094. doi: 10.1021/jacs.6b12826
Blouin, N., Michaud, A., Gendron, D., Wakim, S., Blair, E., Neagu-Plesu, R., et al. (2008). Toward a rational design of poly(2,7-carbazole) derivatives for solar cells. J. Am. Chem. Soc. 130, 732–742. doi: 10.1021/ja0771989
Chandrasekharam, M., Anil Reddy, M., Ganesh, K., Sharma, G. D., Singh, S. P. J., and Rao, L. (2014). Synthesis and photovoltaic properties of D-A-D type small molecules containing diketopyrrolopyrrole (DPP) acceptor central unit with different donor terminal units. Org. Electron. 15, 2116–2125. doi: 10.1016/j.orgel.2014.05.033
Chaudhry, A. R., Muhammad, S., Irfan, A., Al-Sehemi, A. G., Haq, B. U., and Hussain, S. (2018). Structural, electronic, and nonlinear optical properties of novel derivatives of 9,12-diiodo-1,2-dicarba-closo-dodecaborane: density functional theory approach. Z. Naturforsch. A 73, 1037–1045. doi: 10.1515/zna-2018-0123
Chen, H. Y., and Chao, I. (2005). Effect of perfluorination on the charge-transport properties of organic semiconductors: density functional theory study of perfluorinated pentacene and sexithiophene. Chem. Phys. Lett. 401, 539–545. doi: 10.1016/j.cplett.2004.11.125
Chen, Y., Wan, X., and Long, G. (2013). High performance photovoltaic applications using solution-processed small molecules. Acc. Chem. Res. 46, 2645–2655. doi: 10.1021/ar400088c
Cheung, D. L., and Troisi, A. (2010). Theoretical study of the organic photovoltaic electron acceptor PCBM: morphology, electronic structure, and charge localization. J. Phys. Chem. C 114, 20479–20488. doi: 10.1021/jp1049167
Coughlin, J. E., Henson, Z. B., Welch, G. C., and Bazan, G. C. (2014). Design and synthesis of molecular donors for solution-processed high-efficiency organic solar cells. Acc. Chem. Res. 47, 257–270. doi: 10.1021/ar400136b
Dutta, P., Yang, W., Eom, S. H., Lee, W. H., Kang, I. N., and Lee, S. H. (2012). Development of naphtho[1,2-b:5,6-b′]dithiophene based novel small molecules for efficient bulk-heterojunction organic solar cells. Chem. Commun. 48 573–575. doi: 10.1039/C1CC15465F
Feng, H. F., Fu, W. F., Li, L., Yu, Q. C., Lu, H., Wan, J. H., et al. (2014). Triphenylamine modified bis-diketopyrrolopyrrole molecular donor materials with extended conjugation for bulk heterojunction solar cells. Org. Electron. 15, 2575–2586. doi: 10.1016/j.orgel.2014.07.020
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford, CT: Gaussian, Inc.
Fujii, M., Yasuda, T., and Yamashita, K. (2016). Photon-absorbing charge-bridging states in organic bulk heterojunctions consisting of diketopyrrolopyrrole derivatives and PCBM. Phys. Chem. Chem. Phys. 18, 9514–9523. doi: 10.1039/C5CP06183K
Gruhn, N. E., da Silva Filho, D. A., Bill, T. G., Malagoli, M., Coropceanu, V., Kahn, A., et al. (2002). The vibrational reorganization energy in pentacene: molecular influences on charge transport. J. Am. Chem. Soc. 124, 7918–7919. doi: 10.1021/ja0175892
Guo, Y. Q., Wang, Y., Song, L. C., Liu, F., Wan, X., Zhang, H., et al. (2017). Small molecules with asymmetric 4-alkyl-8-alkoxybenzo[1,2-b:4,5-b′]dithiophene as the central unit for high-performance solar cells with high fill factors. Chem. Mater. 29, 3694–3703. doi: 10.1021/acs.chemmater.7b00642
He, C., He, Q. G., Yang, X. D., Wu, G. L., Yang, C. H., Bai, F. L., et al. (2007). Synthesis and photovoltaic properties of a solution-processable organic molecule containing triphenylamine and DCM moieties. J. Phys. Chem. C 111, 8661–8666. doi: 10.1021/jp070714x
Hill, I. G., Kahn, A., Soos, Z. G., and Pascal, R. A Jr. (2000). Charge-separation energy in films of π-conjugated organic molecules. Chem. Phys. Lett. 327, 181–188. doi: 10.1016/S0009-2614(00)00882-4
Irfan, A., Assiri, M., and Al-Sehemi, A. G. (2018). Exploring the optoelectronic and charge transfer performance of diaza[5]helicenes at molecular and bulk level. Org. Electron. 57, 211–220. doi: 10.1016/j.orgel.2018.03.022
Irfan, A., Muhammad, S., Chaudhry, A. R., Al-Sehemi, A. G., and Jin, R. (2017). Tuning of optoelectronic and charge transport properties in star shaped anthracenothiophene-pyrimidine derivatives as multifunctional materials. Optik 149, 321–331. doi: 10.1016/j.ijleo.2017.09.065
Jeon, I., Delacou, C., Nakagawa, T., and Yutaka Matsuo, Y. (2016). Enhancement of open-circuit voltage by using the 58-p silylmethyl fullerenes in small-molecule organic solar cells. Chem. Asian J. 11, 1268–272. doi: 10.1002/asia.201501400
Jin, R. (2015). Theoretical study of the optical and charge transport properties of star-shaped molecules with 1,3,5-triazine-core derivatives as organic light-emitting and organic solar cells materials. C. R. Chimie. 18, 954–959. doi: 10.1016/j.crci.2015.05.021
Jin, R., and Irfan, A. (2015). Theoretical study on photophysical properties of multifunctional star-shaped molecules with 1,8-naphthalimide core for organic light-emitting diode and organic solar cell application. Theor. Chem. Acc. 134:89. doi: 10.1007/s00214-015-1693-8
Jin, R., and Xiao, W. (2015). Rational design of organoboron heteroarene derivatives as luminescent and charge transport materials for organic light-emitting diodes. New J. Chem. 39, 8188–8194. doi: 10.1039/C5NJ01499A
Kan, B., Li, M., Zhang, Q., Liu, F., Wan, X., Wang, Y., et al. (2015). A series of simple oligomer-like small molecules based on oligothiophenes for solution-processed solar cells with high efficiency. J. Am. Chem. Soc. 137, 3886–3893. doi: 10.1021/jacs.5b00305
Knupfer, M. (2003). Exciton binding energies in organic semiconductors. Appl. Phys. A 77, 623–626. doi: 10.1007/s00339-003-2182-9
Köse, M. E., Mitchell, W. J., Kopidakis, N., Chang, C. H., Shaheen, S. E., Kim, K., et al. (2007). Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 129, 14257–14270. doi: 10.1021/ja073455y
Ku, J., Lansac, Y., and Jang, Y. H. (2011). Time-dependent density functional theory study on benzothiadiazole-based low-band-gap fused-ring copolymers for organic solar cell applications, J. Phys. Chem. C 115, 21508–21516. doi: 10.1021/jp2062207
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the colle–salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi: 10.1103/PhysRevB.37.785
Lenes, M., Wetzelaer, G. -J. A. H., Kooistra, F. B., Veenstra, S. C., Hummelen, J. C., and Blom, P. W. M. (2008). Fullerene bisadducts for enhanced open-circuit voltages and efficiencies in polymer solar cells. Adv. Mater. 20, 2116–2119. doi: 10.1002/adma.200702438
Li, G., Zhu, R., and Yang, Y. (2012). Polymer solar cells. Nat. Photonics. 6, 153–161. doi: 10.1038/nphoton.2012.11
Lin, B. C., Cheng, C. P., You, Z. Q., and Hsu, C. P. (2005). Charge transport properties of tris(8-hydroxyquinolinato)aluminum(III): why it is an electron transporter. J. Am. Chem. Soc. 127, 66–67. doi: 10.1021/ja045087t
Lin, Y., and Zhan, X. (2016). Oligomer molecules for efficient organic photovoltaics. Acc. Chem. Res. 49, 175–183. doi: 10.1021/acs.accounts.5b00363
Lin, Y. Z., Ma, L. C., Li, Y. F., Liu, Y. Q., Zhu, D. B., and Zhan, X. W. (2013). A solution-processable small molecule based on benzodithiophene and diketopyrrolopyrrole for high-performance organic solar cells. Adv. Energy. Mater. 3, 1166–1170. doi: 10.1002/aenm.201300181
Liu, C. C., Mao, S. W., and Kuo, M. Y. (2010). Cyanated pentaceno[2,3-c]chalcogenophenes for potential application in air-stable ambipolar organic thin-film transistors. J. Phys. Chem. C 114, 22316–22321. doi: 10.1021/jp1099464
Loser, S., Lou, S. J., Savoie, B. M., Bruns, C. J., Timalsina, A., Leonardi, M. J., et al. (2017). Systematic evaluation of structure–property relationships in heteroacene–diketopyrrolopyrrole molecular donors for organic solar cell. J. Mater. Chem. A 5, 9217–9232. doi: 10.1039/C7TA02037F
Maglione, C., Carella, A., Centore, R., Chávez, P., Lévêque, P., Fall, S., et al. (2017). Novel low bandgap phenothiazine functionalized DPP derivatives prepared by direct heteroarylation: application in bulk heterojunction organic solar cells. Dyes. Pigments. 141, 169–178. doi: 10.1016/j.dyepig.2017.02.012
Marcus, R. A. (1964). Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 15, 155–196.
Marcus, R. A. (1993). Electron transfer reactions in chemistry. theory and experiment. Rev. Mod. Phys. 65, 599–610.
Ni, D., Zhao, B., Shi, T., Ma, S., Tu, G., and Wu, H. (2013). Monodisperse low-bandgap macromolecule-based 5,5′-bibenzo[c][1,2,5]thiadiazole swivel cruciform for organic solar cells. ACS. Macro. Lett. 2, 621–624. doi: 10.1021/mz4002436
Patra, D., Huang, T. Y., Chiang, C. C., Maturana, R. O., Pao, C. W., Ho, K. C., et al. (2013). 2-Alkyl-5-thienyl-substituted benzo[1,2-b:4,5-b]dithiophene-based donor molecules for solution-processed organic solar cells. ACS Appl. Mater. Interfaces 5, 9494–9500. doi: 10.1021/am4021928
Qu, S., and Tian, H. (2012). Diketopyrrolopyrrole (dpp)-based materials for organic photovoltaics. Chem. Commun. 48, 3039–3051. doi: 10.1039/c2cc17886a
Sancho-García, J. C. (2007). Assessment of density-functional models for organic molecular semiconductors: the role of Hartree–Fock exchange in charge-transfer processes. Chem. Phys. 331, 321–331. doi: 10.1016/j.chemphys.2006.11.002
Scharber, M. C., Wuhlbacher, D., Koppe, M., Denk, P., Waldauf, C., Heeger, A. J., et al. (2006). Design rules for donors in bulk-heterojunction solar cells-towards 10 % energy-conversion efficiency. Adv. Mater. 18, 789–794. doi: 10.1002/adma.200501717
Sharma, G. D., Zervaki, G. E., Angaridis, P. A., Kitsopoulos, T. N., and Coutsolelos, A. G. (2014). Triazine-bridged porphyrin triad as electron donor for solution-processed bulk hetero-junction organic solar cells. J. Phys. Chem. C 118, 5968–5977. doi: 10.1021/jp500090h
Shiau, S. Y., Chang, C. H., Chen, W. J., Wang, H. J., Jeng, R. J., and Lee, R. H. (2015). Star-shaped organic semiconductors with planar triazine core and diketopyrrolopyrrole branches for solution-processed small-molecule organic solar cells. Dyes. Pigments 115, 35–49. doi: 10.1016/j.dyepig.2014.12.007
Tawada, Y., Tsuneda, T., Yanagisawa, S., Yanai, T., and Hirao, K. (2004). A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 120, 8425–8433. doi: 10.1063/1.1688752
Vala, M., Krajčovič, J., Luňák, SJr., Ouzzane, I., Bouillon, J. P., and Weiter, M. (2014). HOMO and LUMO energy levels of N,N′-dinitrophenyl-substituted polar diketopyrrolopyrroles (DPPs). Dyes Pigments 106, 136–142. doi: 10.1016/j.dyepig.2014.03.005
Wang, J. L., Liu, K. K., Liu, S., Xiao, F., Chang, Z. F., Zheng, Y. Q., et al. (2017). Donor end-capped hexafluorinated oligomers for organic solar cells with 9.3% efficiency by engineering the position of π-bridge and sequence of two-step annealing. Chem. Mater. 29, 1036–1046. doi: 10.1021/acs.chemmater.6b03796
Wazzan, N., El-Shishtawy, R. M., and Irfan, A. (2018). DFT and TD–DFT calculations of the electronic structures and photophysical properties of newly designed pyrene-core arylamine derivatives as hole-transporting materials for perovskite solar cells. Theor. Chem. Acc. 137:9. doi: 10.1007/s00214-017-2183-y
Wu, J., Kan, Y. -H., Wu, Y., and Su, Z. -M. (2013). Computational design of host materials suitable for green-(deep) blue phosphors through effectively tuning the triplet energy while maintaining the ambipolar property. J. Phys. Chem. C 117, 8420–8428. doi: 10.1021/jp4008174
Xiao, S., Stuart, A. C., Liu, S., Zhou, H., and You, W. (2010). Conjugated polymer based on polycyclic aromatics for bulk heterojunction organic solar cells: a case study of quadrathienonaphthalene polymers with 2% efficiency. Adv. Funct. Mater. 20, 635–643. doi: 10.1002/adfm.200901407
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57. doi: 10.1016/j.cplett.2004.06.011
Yao, H., Ye, L., Zhang, H., Li, S., Zhang, S., and Hou, J. (2016). Molecular design of benzodithiophene-based organic photovoltaic materials. Chem. Rev. 116, 7397–7457. doi: 10.1021/acs.chemrev.6b00176
Zhang, G., and Musgrave, C. B. (2007). Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 111, 1554–1561. doi: 10.1021/jp061633o
Zhang, L., Pei, K., Yu, M., Huang, Y., Zhao, H., Zeng, M., et al. (2012). Theoretical investigations on donor-acceptor conjugated copolymers based on naphtha [1, 2-c: 5, 6-c] bis [1, 2, 5] thiadiazole for organic solar cell applications. J. Phys. Chem. C 116, 26154–26161. doi: 10.1021/jp306656c
Zhang, Y. M., Tan, H., Xiao, M. J., Bao, X. C., Tao, Q., and Wang, Y. F. (2014). D–A–Ar-type small molecules with enlarged π-system of phenanthrene at terminal for high-performance solution processed organic solar cells. Org. Electron. 15, 1173–1183. doi: 10.1016/j.orgel.2014.03.011
Zhang, Z., Zhou, Z., Hu, Q., Liu, F., Russell, T. P., and Zhu, X. (2017). 1, 3-bis (thieno [3, 4-b] thiophen-6-yl)-4 H-thieno [3, 4-c] pyrrole-4, 6 (5 H)-dione-Based small-molecule donor for efficient solution-processed solar cells. ACS Appl. Mater. Interfaces 9, 6213–6219. doi: 10.1021/acsami.6b14572
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241.
Keywords: star-shaped molecules, diketopyrrolopyrrole derivatives, optical and electronic properties, frontier molecular orbitals (FMOs), organic solar cells (OSCs)
Citation: Zhang X and Jin R (2019) Rational Design of Low-Band Gap Star-Shaped Molecules With 2,4,6-Triphenyl-1,3,5-triazine as Core and Diketopyrrolopyrrole Derivatives as Arms for Organic Solar Cells Applications. Front. Chem. 7:122. doi: 10.3389/fchem.2019.00122
Received: 04 December 2018; Accepted: 15 February 2019;
Published: 19 March 2019.
Edited by:
Doo Soo Chung, Seoul National University, South KoreaReviewed by:
Sungu Hwang, Pusan National University, South KoreaAhmad Irfan, King Khalid University, Saudi Arabia
Copyright © 2019 Zhang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruifa Jin, cnVpZmFqaW5AMTYzLmNvbQ==