 Tania Neva1†
Tania Neva1† Thais Carmona2†
Thais Carmona2† Juan M. Benito1Cédric Przybylski3
Juan M. Benito1Cédric Przybylski3 Carmen Ortiz Mellet4*
Carmen Ortiz Mellet4* Francisco Mendicuti2*
Francisco Mendicuti2* José M. García Fernández1*
José M. García Fernández1*- 1Instituto de Investigaciones Químicas (IIQ), CSIC - University of Sevilla, Sevilla, Spain
- 2Department of Analytical Chemistry, Physical Chemistry and Chemical Engineering, Faculty of Chemistry, University of Alcalá, Alcalá de Henares, Madrid, Spain
- 3Sorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, IPCM, Paris, France
- 4Department of Organic Chemistry, Faculty of Chemistry, University of Seville, Seville, Spain
The presence of a doubly-linked naphthylene clip at the O-2I and O-3II positions in the secondary ring of β-cyclodextrin (βCD) derivatives promoted their self-assembly into head-to-head supramolecular dimers in which the aromatic modules act either as cavity extension walls (if the naphthalene moiety is 1,8-disubstituted) or as folding screens that separate the individual βCD units (if 2,3-disubstituted). Dimer architecture is governed by the conformational properties of the monomer constituents, as determined by NMR, fluorescence, circular dichroism, and computational techniques. In a second supramolecular organization level, the topology of the assembly directs host-guest interactions and, reciprocally, guest inclusion impacts the stability of the supramolecular edifice. Thus, inclusion of adamantane carboxylate, a well-known βCD cavity-fitting guest, was found to either preserve the dimeric arrangement, leading to multicomponent species, or elicit dimer disruption. The ensemble of results highlights the potential of the approach to program self-organization and external stimuli responsiveness of CD devices in a controlled manner while keeping full diastereomeric purity.
Introduction
The incorporation of functional moieties into pre-existing cage molecules and macrocycles represents a powerful strategy to tailor their architectural, inclusion, self-assembling, stimulus responsiveness and biorecognition properties (Sansone and Casnati, 2013; Shetty et al., 2015; Gropp et al., 2017; Liu et al., 2017; Matsuoka and Nabeshima, 2018). A range of sophisticated molecular machines with programmed supramolecular behaviors have been designed by implementing this strategy, with applications in the fields of sensors, electronic or optical devices, natural ligand mimics, artificial catalysts or (bio) molecular carriers, among others (Jiménez Blanco et al., 2017; Webber and Langer, 2017; Yin et al., 2017; Guo et al., 2018; van Dijk et al., 2018; Yu et al., 2018; Zeng et al., 2018). The natural cyclooligosaccharides of the cyclodextrin family are paradigmatic examples in this context (Crini, 2014). The circular arrangement of α-(1 → 4)-linked D-glucopyranosyl units in the commercially available hexa-, hepta-, and octameric representatives (α, β, and γCD, respectively) draws a hollow truncated-cone frame with a hydrophobic interior that can host complementary guests in a size-conditional basis (Simões et al., 2015; Ryzhakov et al., 2016; Álvarez-Lorenzo et al., 2017; García-Moreno et al., 2017; Carreño et al., 2018). The volume constraint can be overcome by the incorporation of shaping and recognition modules leading to cavity extension, cooperativity effects or self-assembly, allowing several levels of organization to be implemented. The design of non-viral gene delivery systems (Ortiz Mellet et al., 2011; Gallego-Yerga et al., 2015; Evenou et al., 2018; Hong et al., 2018), antitoxins (Díaz-Moscoso et al., 2011; Joshi et al., 2011), nanocontainers (Gallego-Yerga et al., 2014b, 2017; Varan et al., 2017; Engel et al., 2018) and actuators (Smiljanic et al., 2006; Tan et al., 2014; Yang et al., 2014) highlights the potential of this tactic.
Appending aromatic walls at the CD rims has proven particularly beneficial to promote self-association and reinforce interactions with third species in a predictable manner (Benkovics et al., 2017; Zhu et al., 2017; Zhang et al., 2018), eventually enabling additional non-inclusion or interfacial complexing modes (McNally et al., 2009; de Jesus et al., 2012). Most reported examples of CD-aromatic conjugates focus on single position-linked derivatives that keep considerable mobility, which limits accurate three-dimensional definition, however (Gamieldien et al., 2010). Covalently bridging CD building blocks with aromatic tethers has been advantageously exploited for preorganization purposes in some cases (Carmona et al., 2015; Sun et al., 2015; Gallego-Yerga et al., 2018). Alternatively, prototypes featuring a two-point connected aromatic component at either the primary (Yang et al., 2008; Le Gac et al., 2016; Ménand et al., 2016, 2018; Yan et al., 2017) or the secondary rim (Balbuena et al., 2007, 2013) in a monomeric CD derivative have been reported, allowing unprecedented control over the topological and supramolecular attributes. The later architecture is particularly appealing: an aromatic component attached to the wider entrance of the inner cavity can act as gatekeeper, guest selector and/or aggregation promoter, the impact in the recognition abilities depending on the conformational bias (González-Álvarez et al., 2011, 2013). Thus, “hinge-type” o-xylylene segments connecting the vicinal secondary O-2 and O-3 positions in the same glucopyranosyl unit (e.g., 1) adopt a cap-like orientation that hinders inclusion and elicits instead the formation of supramolecular head-to-head (HH) dimers in which the individual constituents are closely packed (González-Álvarez et al., 2008; Mayordomo et al., 2013; Gallego-Yerga et al., 2014a). Differently, “clip-type” positional isomers in which the o-xylylene group intertwines the O-2I and O-3II positions in consecutive monosaccharide units (e.g., 2) preferentially take semi-open arrangements due to the reduced flexibility at the cyclic benzylic area. As a result, dimeric species with higher interfacial distances that readily dissociate in the presence of a suitable CD cavity-fitting guest are formed, which can be exploited for the spatiotemporal control of the recognition properties (Neva et al., 2018). Interestingly, the supramolecular abilities were drastically influenced by the type of joint between the aromatic and the cyclooligosaccaride parts: replacing the o-xylylene into a m-xylylene moiety in the clip-type geometry (e.g., 3) led to a fully open topology that abolished self-recognition and favored instead non-inclusion complexation of planar guests (Figure 1). It was anticipated that non-covalent interactions in this family of CD conjugates could be further tuned by modifying the nature of the aromatic clip, providing a flexible scheme to dynamically mold different self-assembling and host-guest fitting modes. The incorporation of naphthalene modules is particularly appealing at this respect: several relative orientations between the cyclooligosaccharide macroring and the aromatic surface can be preselected as a function of the naphthalene substitution pattern without altering the structure and inclusion capabilities of the CD cavity. We conceived that this concept could be implemented in the design of monomeric prototypes with the ability to form supramolecular dimers in which the CD cavities can be either interconnected, forming a single compartment, or separated by the aromatic walls. To probe this hypothesis, the new 1,8-disubstituted or 2,3-disubstituted naphthylene-equipped βCD isomers 4 and 5 (Figure 2) have been synthesized and their conformational, self-assembling and inclusion properties have been monitored by fluorescence spectroscopy, circular dichroism, NMR, MS, and computational techniques. The results underline the potential of strategies based on precise synthesis to drive aggregation of macromolecular entities and program their responsiveness to molecular recognition events, a critical aspect in the construction of functional devices.
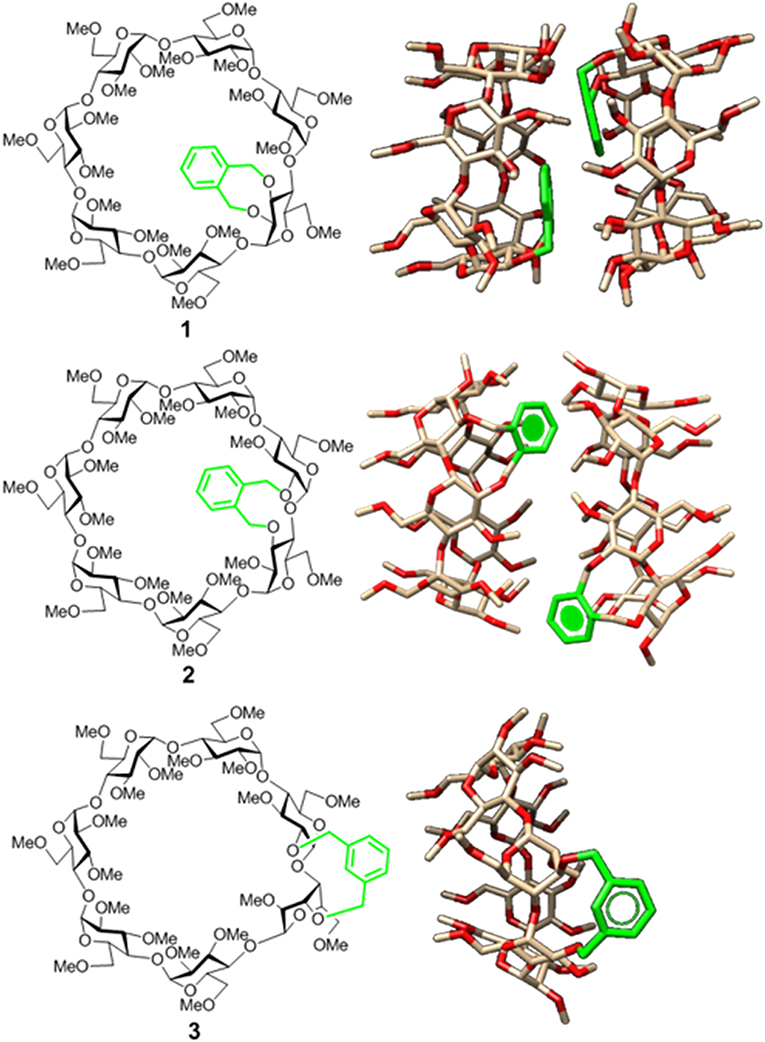
Figure 1. Structures of the doubly-linked isomeric βCD-xylylene conjugates 1–3 (Left) and 3D molecular models of the corresponding minima binding energy dimer (for 1 and 2) or monomer species (for 3) present in water solution (Right). Note that the aromatic ring (in green) orientation, relative to the CD cavity, shifts from cap-like to semi-open to fully open on moving from 1 to 3 (González-Álvarez et al., 2013; Neva et al., 2018).
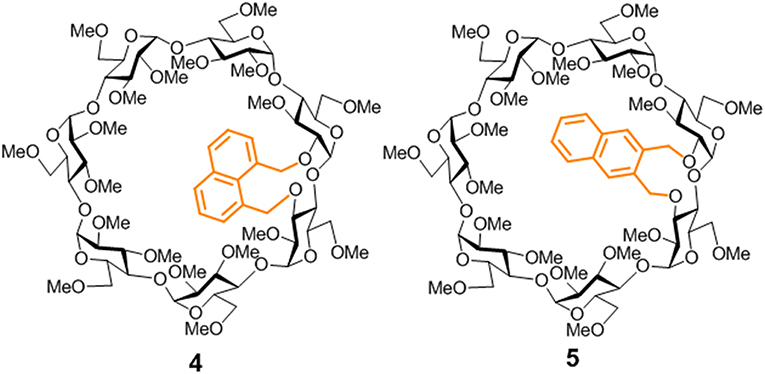
Figure 2. Structures of the new βCD conjugates equipped with naphthalene clips prepared in this work.
Experimental and Computational Methods
Synthesis and Compound Characterization
Reagents and solvents were purchased from commercial sources and used without further purification. 1H (13C) NMR spectra were recorded at 600, 500 and 400 (150.8, 125.7, and 100.6) MHz. 2D COSY, 1D and 2D TOCSY, NOESY, ROESY, HSQC, and HMBC experiments were used to assist assignments. Thin-layer chromatography (TLC) was carried out on aluminum sheets coated with Kieselgel 60 F245 (E. Merck), with visualization by UV light and by charring with 10% H2SO4. Flash column chromatography was carried out on silica gel (230–400 mesh). Elemental analyses were performed at the Institute for Chemical Research (Sevilla, Spain). The target cyclic ether conjugates 4 and 5 were readily obtained by alkylation of the diol precursor 7, obtained from 6 by regioselective bis-demethylation with diisobutylaluminiun hydride (DIBAL·H) (Roizel et al., 2002; Xiao et al., 2010), with ether 1,8- or 2,3-bis(bromomethyl)naphthalene (Kemp et al., 1980) (Figure 3; see the Supplementary Data and Supplementary Figures S1–S16 for experimental details and full characterization data).
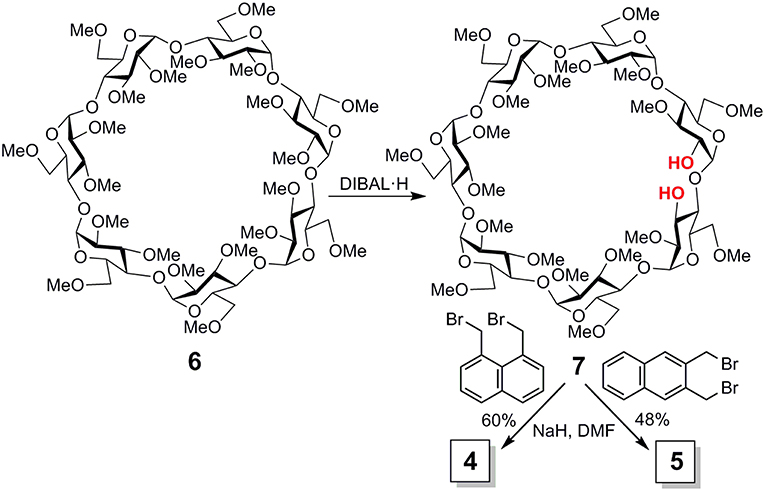
Figure 3. Synthesis of compounds 4 and 5.
Mass Spectrometry
The electrospray ionization-high resolution mass spectrometry (ESIHRMS) experiments were performed using a LTQ-Orbitrap XL ETD instrument from Thermo Scientific operated in positive ionization mode with a spray voltage at +3.6 kV. Samples were prepared at 20 μg·mL−1 in methanol/water (20:80 v/v). Once prepared, the fresh solutions were continuously infused at 3 μL·min−1 using a 250-μL syringe. The applied voltages were 45 and 120 V for the ion transfer capillary and the tube lens, respectively. The ion transfer capillary was held at 275°C. The resolution level was set to 30,000 (m/z = 400) for all studies, while the m/z range was set to m/z 200–2,000 in the profile mode and in the normal mass ranges. The spectra were analyzed using XCalibur 2.0.7 acquisition software (Thermo Scientific) without smoothing and background subtraction. The automatic gain control (AGC) allowed accumulation up to 2 × 105 ions. The maximum injection time was set to 500 ms using 1 μ scan count.
Fluorescence Spectroscopy
Steady-state fluorescence measurements were performed by using a high sensitivity spectrofluorimeter, the SLM 8100C Aminco, equipped with a cooled photomultiplier and a double (single) monochromator in the excitation (emission) path. Excitation and emission slit widths were selected at 8-nm for both channels. Polarizers were set at the magic angle conditions. The fluorescence decay measurements were achieved on a Time Correlated Single Photon Counting (TCSPC) FL900 Edinburgh Instruments Spectrometer. A NanoLed (Horiba), which emits short repetitive optical pulses at 296 nm, was used as a light source to excite the naphthyl groups. The system was equipped with two concave grating monochromators at both the excitation and emission paths and a red sensitive photomultiplier also immersed in a Peltier cooled housing. No polarizers were used. Data acquisition was carried out by using a multichannel time detector and a time window width of 200 ns with a total of 10,000 counts at the intensity maximum. The instrumental response function was regularly obtained by measuring the scattering of a Ludox solution. The cells housing temperature for both instruments were thermostatically controlled by using a Pt-100 probe with digital temperature processors. Right angle geometry and cylindrical quartz 2-mm inner path cells of 120 μL capacity were used for most of the experiments. Fluorescence intensities due to the inner effect were corrected (Icorr) according to
Where Aex and Aem are the absorption at the wavelength of excitation and emission, respectively.
However, for some of the checking typical rectangular (square cross-section) quartz 1-cm path cells were used.
Decay intensity profiles were fitted to a sum of exponential decay functions as
by the iterative deconvolution method (O'Connor et al., 1979). The average lifetime of a multiple-exponential decay function was then defined as,
where Ai is the pre-exponential factor of the component with a lifetime τi of the multi-exponential function intensity decay.
The fractional contribution fi of each decay time to the steady-state intensity, which represents the fraction of total fluorescence intensity I of the i-component at the wavelengths of observation, is given by
and the intensity weighted average lifetime <τ> from a dilute solution of a pair of emitting species, 1 and 2, that do not interact during the excited state lifetime can be obtained as,
From the fluorescence depolarization measurements, the anisotropy r is typically defined as:
where Ixy is the intensity of the emission that is measured when the excitation polarizer is in position x (V for vertical, H for horizontal), the emission polarizer is in position y, and the G factor (= IHV/IHH) corrects for any depolarization produced by the optical system (Lakowicz, 2008).
For a single isolated excited chromophore which is dynamically quenched by a quencher Q, the τ/τ0 ratio (with/without Q) is related with [Q] by the linear Stern-Volmer equation. For more complicated systems, the Stern-Volmer representations of <τ>/<τ0> are linear at the lowest [Q] region.
Circular Dichroism
Circular dichroism spectra were obtained by using a JASCO-715 spectropolarimeter. Recorded spectra were the average of three scans taken at a speed of 50 nm min−1 with a time response of 0.125 s. The sensitivity and resolution were fixed at 20 mdeg and 0.5 nm, respectively. Measurements were performed in 1 cm or 0.1 cm path quartz cells at 25°C.
Molecular Mechanics (MM) and Molecular Dynamics (MD) Simulations
All calculations were performed with Sybyl X2.0 (SYBYL- X 2.0; Tripos Associates; ed. St. Louis, Missouri, USA, 2012) and the Tripos force field (Clark et al., 1989). A relative permittivity ε = 3.5 (ε = 1) was used in the vacuum (in the presence of water). Charges for βCD derivatives were obtained by MOPAC (Frisch et al., 2004). The starting modified βCD derivatives were built with the macroring in the non-distorted form (ϕ = 0°, ψ = −3°, τ = 121.7° and side chain χ angles in the trans conformation) (Pozuelo et al., 1996) and the naphthyl substituent for 4 and 5 in the most probable conformations for the chain that links the naphthalene moiety to a glucopyranose unit of the βCD macroring. The selected conformations were those of the minima potential energies obtained by placement of the four torsional angles that describe the rotation around C(3)-O-CH2-Car(1) and C(2)-O-CH2-Car(8) ether bonds according to the procedures described elsewhere (González-Álvarez et al., 2011). Optimizations were carried out by the simplex algorithm, and the conjugate gradient was used as a termination method with gradients of 0.2 (0.5) kcal·mol−1Å−1 for the calculations carried out in vacuum (water) (Brunel et al., 1975; Press et al., 1988). Non-bonded cut-off distances were set at 12 Å. The Molecular Silverware algorithm (MS) and periodic boundary conditions (PBC) were used for system solvation when water was used as a solvent (Blanco, 1991).
The methods used for the conformational study of the monomeric 4 and 5 and for their dimer formation were similar to those described previously for 1 (González-Álvarez et al., 2008) and 2 or 3 (Neva et al., 2018). 2-ns MD simulations in the vacuum were performed at several temperatures ranging from 250 to 600 K on initial 4 and 5 conformations.
Emulation of dimerization processes were performed starting from the optimized conformations of 4 and 5. A single head-to-head orientation (HH) for the CD-to-CD approaching, in agreement with experimental findings, was considered (see Supplementary Figure S30 for the coordinate systems describing dimerization). Critical analysis of the structures generated by scanning the θ [O(4)-o-o'-O(4′)] dihedral angle in the −180 to +180o range (initially at 30° intervals and 10o later) and the y coordinate (oo' distance) from 2 to 0.7 nm (0.05 nm intervals) in the presence of water (fixed the ε [o-o'-O(4′)] angle at 90o) followed by optimization (gradient 1.5 kcal·mol−1Å−1) provided the most favorable θ angle for approaching. Supplementary Figure S29 depicts an example for dimerization of 5. Once θ was fixed, the dimerization was emulated by approaching along the y coordinate, in 0.05 nm steps, from 2 to 0.7 nm (Supplementary Figure S30). This was also done in the presence of water (MS and PBC). Every structure was optimized (1.5 kcal · mol−1Å−1) and saved for further analysis. Minima binding energy (MBE) structures for dimers were optimized once again (gradient 0.5 kcal · mol−1Å−1) and used as the starting conformations for the 1.0 ns MD simulations following the same strategy described earlier (González-Álvarez et al., 2008; Gonzalez-Álvarez et al., 2009b; González-Álvarez et al., 2011; Neva et al., 2018).
For computing complexation of 4-dimer with sodium adamantane-1-carboxylate (AC), we started from the most stable HH MBE 4-dimer structure. The anionic AC guest with the most favorable orientation (Supplementary Figure S41) was approached along the y coordinate from 2 to −2 nm (0.05 nm intervals) in the presence of water, in a way that is similar to the dimerization processes. Each structure generated and optimized in the presence of water was analyzed. In a similar manner, anionic AC complexation with 5 monomer (MBE structure) was computed.
NMR Titration Experiments
Association constants (Ka) for the inclusion complexes were determined in phosphate-buffered D2O (pD 7.4, 0.1 M) at 298 K by measuring the chemical shift variations in the 1H NMR spectra (500 MHz) of a solution of the CD derivatives 4 or 5 in the absence and in the presence of increasing amounts of AC. Typically, a stock solution of CD (ca. 0.5–1 mM) was prepared. A 500 μL aliquot of this solution was transferred to a NMR tube and the initial NMR spectrum was recorded. Then, a solution of AC (15–20 mM) was prepared in the stock CD solution (it is critically important to maintain a constant host concentration, pD and ionic strength throughout the titration experiment). Aliquots of the guest solution were sequentially added to the NMR tube and the corresponding spectrum was recorded after equilibration (Supplementary Figures S37, S38). Additions and measurements were continued until 90–100% complexation had been achieved. The chemical shifts of selected host resonances obtained at approximately ca. 15 different host-guest concentration ratios were plotted against the concentration of CD and used in an iterative least-squares fitting procedure using a 1:1 stoichiometry binding model (Bisson et al., 1998, 2000). Errors are estimated in ±15%.
Results and Discussion
Synthesis and Characterizations
Imposing distance restrictions in CDs through covalent bonds can lead to significant alterations in their primary structure, usually by provoking conformational changes in one or several of the monosaccharide building blocks, which may substantially perturb the inclusion capabilities (Fujita et al., 1988; Fukudome et al., 2007b; Álvarez-Dorta et al., 2015, 2016; Immel et al., 2001; Fukudome et al., 2007; León et al., 2018). Compounds 4 and 5 were purposely conceived to avoid this and keep the toroidal βCD cavity characteristic of βCD undistorted, so that any effect in their supramolecular properties can be ascribed to the presence of the naphthalene component, devoid from additional effects on the βCD macroring topology. According to the available X-ray data for permethylated βCD 6 (Caira et al., 2004), the through-space distance between the O-2I and O-3II positions in the cyclooligosaccharide backbone is 3.4 Å, which can be accommodated by benzylic positions in either 1,8- (for 4) or 2,3-disubstituted (for 5) naphthylene clips (separation distance <4.1 Å) without provoking any significant steric strain. Indeed, alkylation of the βCD diol precursor 6 with 1,8- or 2,3-bis(bromomethyl)naphthalene, conducted in dry N,N-dimethylformamide (DMF) in the presence of sodium hydride (NaH), afforded the target cyclic naphthylene ethers 4 and 5, respectively, as single products, as seen from thin layer chromatography (TLC) and mass spectrometry (MS) monitoring of the crude reaction mixtures (Figure 3). No side-products resulting from cross-linking or single-point etherification reactions were detected, pointing to a fast ring-closure once the bis(bromomethyl)naphthalene tether first reacts at either O-2I or O-3II. Analytically pure samples of 4 and 5, were isolated in 56 and 48%, respectively, after column chromatography on silica gel using 100:1 → 20:1 EtOAc/EtOH as eluent.
The 1H and 13C NMR data for 4 and 5 confirmed the presence of the dual-bridged joint involving consecutive glucopyranosyl subunits, with all the pyranoid rings in the ground state 4C1 chair conformation, in agreement with full preservation of the basket-shaped architecture. Selective irradiation experiments (1D TOCSY, Supplementary Figures S7, S15 for 4 and 5 in the Supplementary Materials, respectively) featured 3JH, H vicinal coupling constants in the 9–10 Hz range for the non-anomeric sugar protons (4.1–3.1 ppm), indicative of all-axial arrangements for the H-2, H-3, H-4, and H-5 protons about the six-membered pyranoid rings. The anomeric H-1 protons (5.32–5.14 ppm) exhibited coupling constant values in the 3.8–4.1 Hz, in agreement with the α-configuration. ROESY spectra further evidenced the expected intra-monosaccharide H-3—H-5 and inter-monosaccharide H-1—H-4 spatial correlations characteristic of undistorted cyclodextrin derivatives (Supplementary Figures S3, S10 for 4 and 5, respectively, in the Supplementary Materials). In the case of 4, the 1,8-dimethylnaphthylene clip (1H NMR resonances in the 8.0–7.5 ppm region) draws a 12-membered macrocycle with a five-carbon segment flattened region that imposes considerable rigidity, which is reflected in a broad dispersion of the benzylic proton resonances. The 2,3-dimethylnaphthylene clip in 5 (1H NMR resonances in the 7.9–7.5 ppm region), in which the flattened segment in the bridging area is reduced to four-carbon centers, is expected to keep substantially higher flexibility. Indeed, the corresponding proton NMR spectrum closely resembled the spectrum of the o-xylylenated βCD derivative 2 (Supplementary Figure S16).
Self-Assembling
The 2,3-naphthylene-equipped βCD derivative 5, but not the 1,8-naphthylene positional isomer 4, exhibited a very apparent negative solubility coefficient in water: when heated, clear (25°C) solutions became immediately turbid, returning to the limpid original state upon cooling. A negative water solubility coefficient is a characteristic feature of methylated cyclodextrin derivatives bearing aromatic appendages and can be ascribed to reversible dimerization processes (Filippone et al., 2002): in the dimer state, the hydrophobic aromatic moiety is shielded from the bulk, facilitating solvation and preventing unspecific clusterization at low temperatures, whereas heating results in dimer disruption, uncovering of the aromatic ring and precipitation. The dissimilar behavior of 4 and 5 was, however, intriguing. In principle, it could be interpreted in terms of differences in the propensity of 4 and 5 to dimerize. Notwithstanding, the high resolution electrospray ionization (HRESI)-mass spectra of 4 and 5, recorded from aqueous solutions, both exhibited prominent pseudomolecular peaks for the dimer species (Supplementary Figure S17). The data can be concealed assuming that the aromatic surface in the more rigid derivative 4 is similarly exposed to the solvent in the monomer and dimer entities, possibly acting as cavity extension module, whereas in 5-dimer the naphthalene walls are probably sandwiched between the two βCD constituents, in agreement with our starting hypothesis.
The possibility to drive the architecture of the CD dimer species by changing the type of joint to the naphthylene clip was further substantiated by fluorescence spectroscopy. Figure 4 shows the emission spectra for water solutions of 4 and 5 at increasing concentrations (from 0 to ~0.2 mM) at 25°C upon excitation of the naphthalene chromophore (295 nm). The spectrum for 4 exhibits a single band located at ~340 nm and a shoulder at ~380 nm, whereas the spectrum for 5 shows a double band with two maxima placed at ~330 and ~340 nm and a shoulder at ~350 nm. The low intensity shoulders located at longer wavelengths suggest the presence of intermolecular aromatic excimers, that is, the formation of short-lived dimeric complexes in an electronically excited state (Saigusa and Lim, 1996; Shirai et al., 2016). Conclusive confirmation of dimer formation was obtained from the analysis of the corresponding fluorescence decay profiles obtained from time-resolved fluorescence measurements, selecting the maximum of emission (340 nm in both βCD derivatives 4 and 5). The data were fitted to three-exponential decay functions at any concentration and temperature: a short-lived component (<0.5 ns) ascribed to the inherent scattering of the cylindrical cuvettes (not taken into account in the analysis), a second intermediate component in the 2–9 ns (for 4) or 3–12 ns (for 5) range that was attributed to the free dissociated monomer and a third slowest component in the 10–15 ns (for 4) or 20–60 ns (for 5), whose contribution increased with concentration and decreased with temperature, which was attributed to the dimeric entity (4-dimer or 5-dimer). In full agreement, the lifetime averages (<τ>) of the excited electronic states (obtained from Equation S3) increased with the concentration of 4 or 5 (Figure 5), a consequence of the increase of the fraction of the dimer species in the corresponding monomer/dimer equilibria.
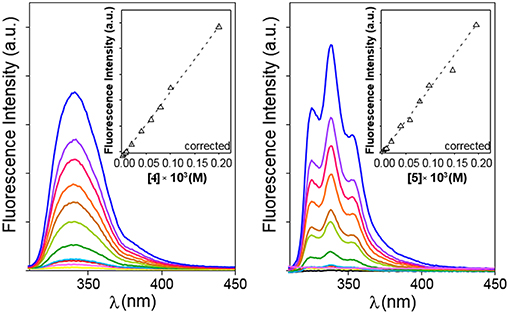
Figure 4. Emission spectra for 4 (Left) and 5 (Right) in water in the 0 to ~0.2 mM range of concentrations (yellow to blue solid lines) at 25°C. Variation of the corrected fluorescence intensity (Equation S1) with compound concentration measured at the intensity maxima are superimposed (dashed lines).
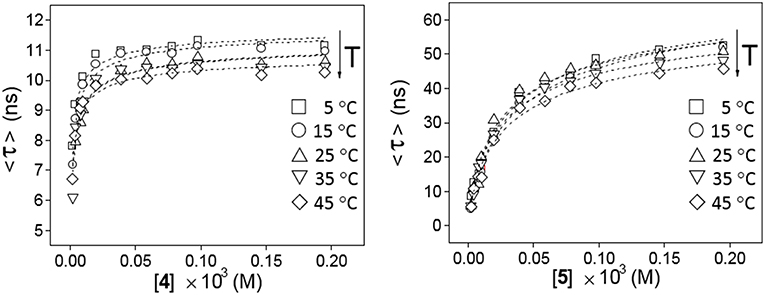
Figure 5. Variation of the weighted average lifetime <τ> with the concentration of 4 (Left) or 5 (Right) in water in the 5–45°C temperature range.
Plots of the corrected fluorescence intensity (Icorr, Equation 1 in the Experimental section), measured as the area under the emission spectra, against the concentration of 4 and 5 in water at 25°C (Figure 4 inserts) were linear in the whole range of concentrations used, meaning that the fluorescence quantum yield ϕ, i.e., the ratio of the number of photons emitted to the number of photons absorbed, does not change upon dimerization (ϕdimer ≅ ϕmonomer; see Equation 8 in the Experimental section). In other words, the fluorescence intensity is not sensitive to the association process of 4 or 5. Under this assumption, the dimerization isotherms fit the experimental data to Equation (9) (Experimental section). From them, the respective dimerization constants (KD), as well as the lifetimes for the monomeric (τ0 = τmonomer) and dimeric (τ∞ = τdimer) forms of both dimethylnaphthylene-clipped βCD derivatives 4 and 5, were derived. Table 1 collects the ensemble of data for the whole range of temperatures. Remarkably, the propensity to form dimers is about 2-to-3 orders of magnitude higher as compared with the previously reported o-xylylene clipped βCD derivative 2 (Neva et al., 2018). As expected from the shape of the isotherm plots in Figure 5, where a plateau is reached at much lower concentrations for 4 than for 5, the KD values for 4-dimer formation (4.1–8.0 · 105 M−1) were found to be above one order of magnitude higher as compared with the corresponding values for 5-dimer formation (1.4–2.2 · 104 M−1). On the other hand, the significantly larger average lifetimes at infinite concentration (ascribed to the dimer form) for 5 (67.5–79.8 ns) than for 4 (11.0–11.9 ns) point to considerable differences in the structure of the corresponding dimers, reinforcing the notion that the naphthylene moiety is probably better shielded from the solvent in 5-dimer than in 4-dimer.
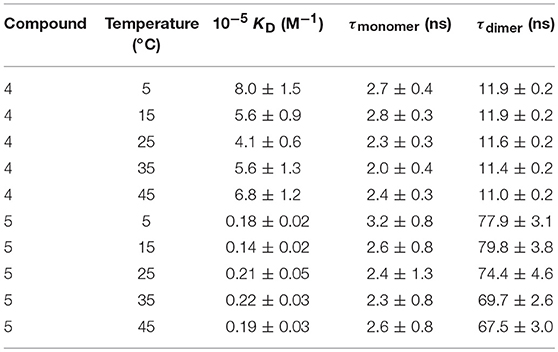
Table 1. Dimerization constant (KD) values and lifetime of the electronic excited state for the monomer (τmonomer) and dimer (τdimer) species obtained from analysis of decay profiles at different concentrations and temperatures for CDs 4 an 5.
From the van't Hoff linear representations shown in Figure 6, the ΔH and ΔS thermodynamic parameters associated to the dimerization process were found to be ΔH = −2.5 ± 5.1 kJ·mol−1, ΔS = +102.7 ± 17.3 J·K−1mol−1 for 4 and ΔH = +4.5 ± 3.6 kJ·mol−1, ΔS = +97.3 ± 12.2 J·K−1mol−1 for 5. Van der Waals or electrostatics favorable intermolecular interactions are usually characterized by relatively large ΔH < 0 values, whereas hydrophobic forces, which tend to reduce the hydrophobic surface of non-polar moieties, are instead accompanied by near zero or slightly positive ΔH values (Southall et al., 2002; Meyer et al., 2006; Kronberg, 2016). The experimental ΔH data are therefore consistent with a major role of the dimethylnaphthylene appended group in the dimerization process. The rather large and favorable ΔS values accompanying monomer-to-dimer transformation for 4 or 5 are also consistent with hydrophobic interactions as the driving force. They can be ascribed to a substantial loss of solvating shells during dimerization due to the large size of the naphthylene moiety, which overcomes the intrinsic entropy decrease due to the loss of degrees of freedom accompanying the association of monomeric entities. The preponderant role of hydrophobic forces was further confirmed by conducting fluorescent decay measurements for solutions of 4 and 5 in solvents of decreasing dielectric constant (water, water-methanol mixtures, methanol and ethanol; Supplementary Figures S18, S19): a steep decrease in <τ> values was observed, which correlates with an increase in the proportion of the shorter lifetime monomer component at the expenses of the dimer. For instance, whereas a 1.5 · 10−4 M solution of 4 in water at 25°C contains 90% of 4-dimer, in ethanol the dimer proportion drops to 40%.
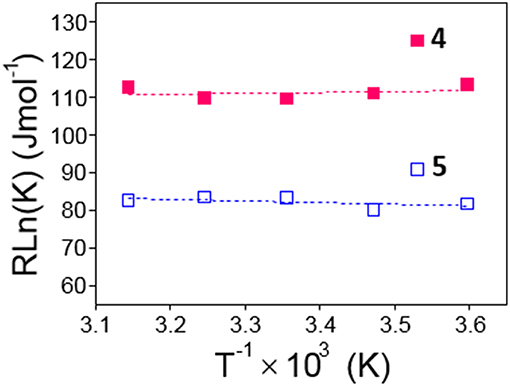
Figure 6. Van't Hoff linear representations for the dimerization processes of 4 and 5 obtained from the association constants collected in Table 1.
Structure and Conformation
In order to obtain information on the topology of the 1,8- and 2,3-dimethylnaphthylene βCD conjugates 4 and 5 and on the changes that may occur in the location of the naphthyl chromophore upon dimer formation, fluorescence quenching experiments using diacetyl as the quencher (q) were next conducted. Measurements were carried out at 25°C in water for two different concentrations of 4 (0.02 and 0.2 mM, meaning 4-dimer molar fractions of 0.69 and 0.88, respectively, for KD = 4.1·105 M−1; Table 1) or 5 (0.03 and 0.2 mM, meaning 5-dimer molar fractions of 0.25 and 0.53, respectively, for KD = 0.21·105 M−1; Table 1). From the Stern–Volmer (<τ>q=0/< τ > vs. [q]) plots (Lakowicz, 2008) (Supplementary Figure S24), which were linear in the range of quencher concentrations used (0 to 8 mM), the Stern-Volmer constants (Ksv) and bimolecular quenching constants (kq) were obtained (Table 2). For species bearing the same chromophore unit, kq values provides a qualitative assessment of the differences in the accessibility to the quencher: the lower the kq value, the less accessible is the chromophore. Changes in the concentration of 4 did not affect kq, suggesting that the 1,8-naphthylene group was similarly exposed in the monomer and in the dimer (kq = 1.4 ·109 M−1s−1). Differently, the kq value for water solutions of 5 decrease by about 15% on going from the lower (kq = 0.6 · 109 M−1s−1) to the higher concentration (kq = 0.5 · 109 M−1s−1), indicating that the 2,3-naphthylene moiety is less accessible to the quencher in 5-dimer as compared to the monomer form. The data could be interpreted as the shielding of the aromatic clip between the macrorings of two βCD units upon dimerization in the case of 5 but not in the case of 4, which points out to substantial differences in the architecture of 4-dimer and 5-dimer. Additionally, the lower kq values for 5 than for 4 whatever the concentration indicates that the 2,3-naphthylene moiety in 5 is less exposed to the quencher than the 1,8-naphthylene module in 4, both in monomer and dimer form, which further advocates for strong differences in the orientation of the aromatic appendage relative to the cyclooligosaccharide scaffold.

Table 2. Stern-Volmer plot parameters for the quenching 4 and 5 by diacetyl of at different concentrations at 25°C.
The presence of the naphthylene chromophore in 4 and 5 enables circular dichroism as a useful technique for conformational analyses: βCD is a chiral molecule; although it does not absorb above 200 nm, a chromophore appended to βCD (or included in the cavity) exhibits induced circular dichroism (ICD) (Superchio et al., 2004; Berova et al., 2007). Most interestingly, circular dichroism also provides precious information on intermolecular interactions and the structure of supramolecular assemblies: when two (or more) chromophores are positioned close in space and adopt a proper (chiral) mutual orientation, the interactions between their electronic transition dipoles result in large rotational strengths, often surpassing those connected with the perturbations on each chromophore exerted by the chiral non-chromophoric frame. The most significant case arises when chromophores with strong electric-dipole allowed transitions couple to each other (exciton coupling; EC) (Pescitelli et al., 2011).
It has been established that the sign of ICD for a chromophore inside the βCD cavity is positive when the electron transition dipole moment is parallel to the axis of CD while the one perpendicular to the axis is negative (Harata and Hisashi, 1975; Shimizu et al., 1979, 1981, 1982). This rule has been extended to derive a general law that the sign of ICD is reversed when the guest moves from inside of the cavity to outside and the direction of the transition is fixed, and it changes at the angle between the transition moment and the CD axis 54.7° (Kodaka and Fukaya, 1989; Kodaka, 1991, 1993). The major UV transitions of the naphthyl group are 1Bb (~230 nm), 1La (~275 nm), and 1Lb (~320 nm), and they do not change substantially with substitution (Kodaka, 1998). In full agreement, the ordinary absorption spectra for 4 or 5 in water exhibit two intense main bands whose maxima are placed at ~220 or ~227 nm and ~285 or ~287 nm and a rather weak shoulder located at ~315 or ~339 nm, which were ascribed to the 1Bb, 1La, and 1Lb electronic transitions, respectively (Figure 7, top panels). Here we follow the broadly accepted Platt's notation, where the electronic state is designated by A, B, C, …or K, L, M, … in accordance with its total momentum number Q (or total ring quantum number) and the superscript “1” indicates a singlet state, whereas the subscripts “a” and “b” denote the location of the nodes of the wave function: with a and b, the nodes are indicated to be present on C–C bonds and on carbon atoms, respectively (Platt, 1949; Michl, 1978). The electron transition dipole moment of the short- and the high-wavelengths 1Bb and 1Lb transitions are nearly parallel and perpendicular, respectively, to the long axis of naphthalene, while that of the 1La transition is tilted about 10–40° from the short axis (Kodaka, 1998; González-Álvarez et al., 2009a) (Figure 7, bottom panels). In the next paragraphs, the assignment of the ICD bands for compounds 4 and 5 conforms to these literature reports.
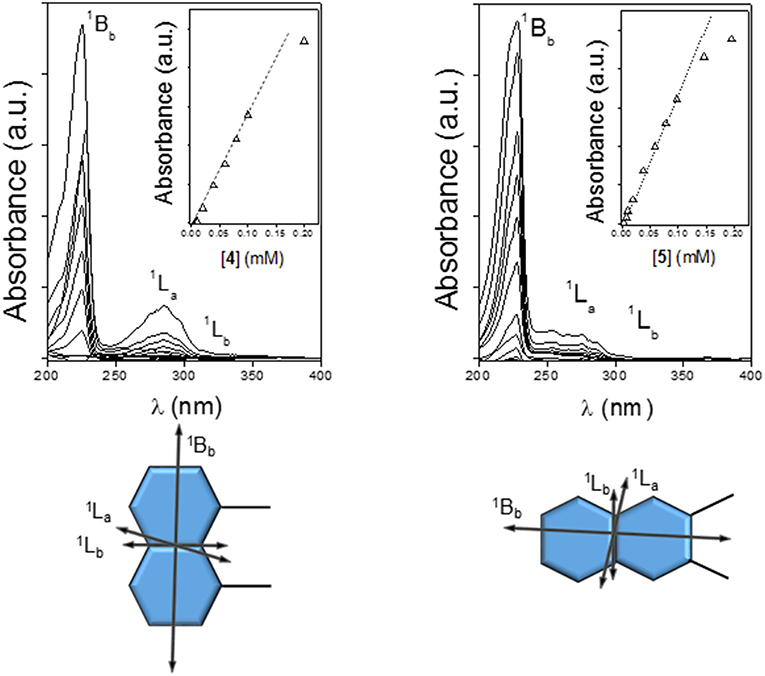
Figure 7. Absorption spectra for 4 (Top Left) and 5 (Top Right) in water at 25°C, with the corresponding absorbance vs. concentration plots (inserts) and directions of the electronic transition moments for the 1,8- (Bottom Left) and 2,3-naphthylene (Bottom Right) appended groups.
The circular dichroism spectrum (expressed as molar ellipticity; [θ]) of a 0.11 mM solution of 4 in water (4-dimer molar fraction 0.84; Figure 8, left panel) exhibits EC in the electronic 1Bb transition band, which represents a strong evidence for the presence of aggregates facing the naphthylene-bearing βCD secondary rims (head-to-head; HH dimers), likely stabilized through the mutual interaction between the aromatic groups. In addition, the 1La and 1Lb transitions provide negative and slightly positive bands, respectively. The intensities of all the three bands (expressed as ellipticity; θ, mdeg) in the ICD spectrum increased with the concentration of 4 (Figure 8, middle and right panels). At the lower concentrations used (0.67 · 10−4 and 0.022 · 10−4 M for the 0.1- and 1 cm path cell, respectively) the 4-dimer molar fractions were ~0.81 and 0.37, respectively, which means that the ICD signals at the highest concentration were mostly due to the dimer species. Negative and slightly positive 1La and 1Lb bands then agree with the respective transition moments adopting a nearly parallel (for 1La) and forming an angle slightly larger than 54.7° (for 1Lb) with respect to the 7-fold macroring axis. These geometrical requirements could be fulfilled for a fully open conformation of the 1,8-dimethylnaphthylene-βCD conjugate where the naphthylene group, slightly rotated about his short axis, essentially prolongs the βCD walls.
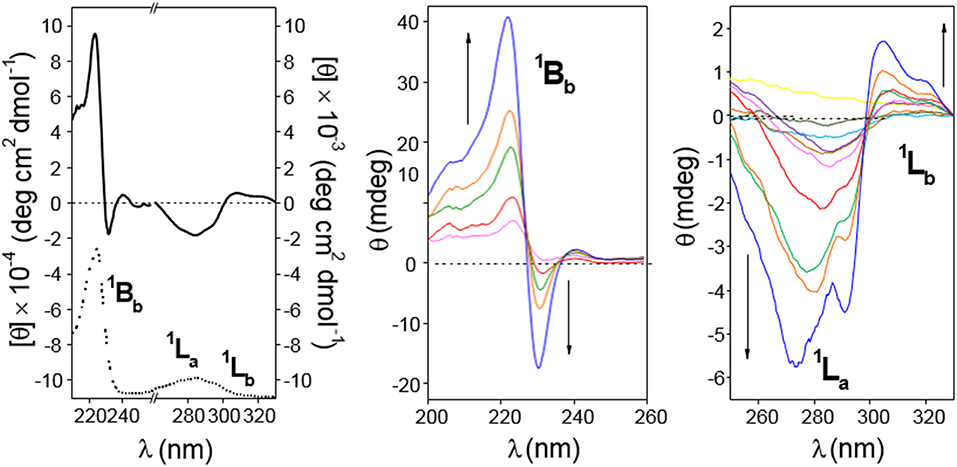
Figure 8. (Left) 210–330 nm range circular dichroism spectrum (solid line; as molar ellipticity [θ]), and absorption spectra (dotted line; arbitrary units) for 4 (0.11 mM in water). (Middle and Right) Circular dichroism spectra (as ellipticity; θ) for 4 in the 200–260 nm (from magenta to blue lines: 0.67, 1.12, 1.68 2.24, and 3.36 · 10−4 M in water; cell path 0.1 cm) and in the 260–330 nm range (from yellow to blue lines: 0.022 · 10−4 to 3.4 · 10−4 M in water; cell path 1 cm). All spectra were recorded at 25°C.
The ICD spectra of a 0.3 mM solution of 5 in water (5-dimer molar fraction ~0.59; Figure 9, left panel) likewise exhibited EC in the 1Bb band, with peaks of similar intensity but opposite sign as compared with 4, consistent with an HH dimer. This EC was still visible at lower concentrations down to 0.17 mM (5-dimer molar fraction ~0.51; Figure 10, right panel). The 1La and 1Lb transition bands were both positive and rather weak, especially the last one that was hardly visible. The 1La sign agrees with a naphthyl moiety placed outside the βCD cavity in a near perpendicular orientation relative to the macroring 7-fold axis, whereas the almost zero intensity of the 1Lb band is indicative of an angle of around 54.7° between the associated transition moment and the βCD main axis. This scenario would agree with a half-open conformation with a slightly long-axis rotated naphthylene group to provide the appropriate angles.
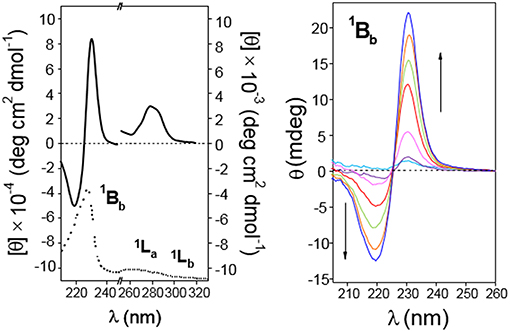
Figure 9. Circular dichroism spectra of aqueous solutions of CD 5. (Left) Represents molar ellipticity ([θ], solid lines) and pure absorption (dotted lines) of a 0.30 mM solution in the 210–330 nm range. (Right) Represents the ellipticity (θ) at variable concentration (from light blue to blue lines: 0.17, 0.26, 0.54, 1.09, 1.63, and 2.73 · 10−4 M) in the 200–260 nm (path 0.1 cm). All spectra recorded at 25°C.
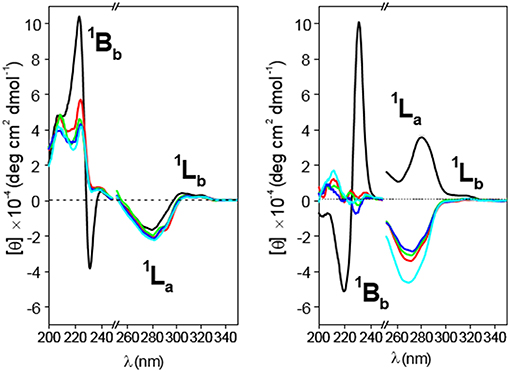
Figure 10. Circular dichroism spectra for 4 (Left) and 5 (Right) solutions in water (black line), 1:1 (v/v) water/methanol (red line), methanol (green line), ethanol (deep blue line), and propanol (light blue line) at 25°C. Concentrations were fixed at 0.19 and 0.20 mM for 4 and 5, respectively.
The occurrence of EC in both 4-dimer and 5-dimer, ascribable to HH species, had previously been detected in CD derivatives having a 1,8-naphthylene substituent attached at O-2I/O-3I in the same glucopyranosyl residue (as the xylylene moiety in 1, Figure 1). Only in the case of the γCD representative head-to-tail (HT) arrangements were found to predominate (González-Álvarez et al., 2013); for the αCD and βCD representatives the HH-dimer was the structure that better fitted most of the experimental and computational findings, as in the present work (González-Álvarez et al., 2011, 2013).
Circular dichroism spectra of 4 and 5 in solvents with decreasing polarity (water, 50:50 methanol/water, ethanol, propanol; Figure 10) led to the disappearance of the dual EC signal, which becomes a positive 1Bb band, in agreement with HH-dimer dissociation. This is an expected consequence of the weakening of hydrophobic attractive interactions and represents an additional evidence of the hydrophobic effect-driven nature of the dimerization process. Decreasing the medium polarity also makes the 1La band for 4 slightly more negative, whereas the 1Lb band still remains weakly positive (Figure 10, left panel). Taken together, positive 1Bb and negative 1La bands agree with a conformation for the monomer form of 4 where the naphthyl group is located outside the cavity with the 1Bb and 1La transitions moments nearly perpendicular and parallel, respectively, with respect to the βCD main axis, which essentially matches the situation encountered in 4-dimer. It can be concluded that self-assembly of 4 into dimers proceeds without any significant conformational change in the constitutive monomers. Sharply differently, dimer dissociation of compound 5 upon decreasing solvent polarity entails sign inversion of the 1La band from positive to negative, the 1Lb band remaining almost negligible (Figure 10, right panel). This trend points to a conformational change involving a twist of the 2,3-dimethylnaphthylene clip when going from the dimer to the monomer state and vice versa: in the monomer form the aromatic clip portion orients the 1La transition moment nearly parallel to the macroring axis while keeping a half-open disposition with respect to the βCD scaffold. Dimerization of 5 in water implies therefore a fitting process characterized by rotations about the benzyl and ether-type bonds to optimally dispose the naphthalene surfaces. Variable temperature 1H NMR spectra of 4 and 5 (MeOD) as well as 1H NMR spectra recorded in solvents with varying polarity (MeOD and 4:1 MeOD-D2O) further supported these notions (Supplementary Figures S20–S23).
Molecular mechanics (MM) and molecular dynamics (MD) simulations for 4 and 5 reproduced the topological features inferred from the ICD experimental data (see Experimental section for a detailed description of the computational protocols). Thus, the probability distributions for the torsional angles, obtained from the analysis of the 2-ns MD trajectories in the vacuum at different temperatures (using the most stable structures calculated by MM as the starting conditions; Supplementary Figure S25), predominantly afforded near fully-open and half-open conformations for the monomeric forms of 4 and 5, respectively (Figure 11, top panel). As expected, the ϕi and ψi angles for the bonds describing rotation about the bonds involving the glycosidic oxygens, C-1i–O and O— C-4i+1 (i = 1–7), adopted values typical for skewing states (0° ± 60°) from trans conformations in an undistorted βCD framework. Nevertheless, some ψi torsional angles could occasionally visit a cis state (Supplementary Figures S26, S27). The distributions of the four torsional angles χ describing the rotation around the bonds that link the appended aromatic moieties to the macroring (see Supplementary Figures S26, S27) hardly changed from their initial positions, indicating that the orientation of the naphthylene group remains confined in a relatively limited conformational space. The distributions of the distances between the center of mass of the naphthylene moiety and the βCD macroring for 4 and 5 (Supplementary Figure S28) at increasing temperatures further evidenced a significantly larger flapping amplitude for the latter, in agreement with the higher flexibility of the 2,3- vs. the 1,8-dimethylnaphthylene clip. MM data were also consistent with the preferred HH approaching of 4 or 5 upon self-assembly, yielding minimum binding energy (MBE) dimer aggregates featuring naphthylene—naphthylene distances fulfilling the requirements for the exciton coupling observed in the ICD spectra in water (Figure 11, middle panel). Van der Waals attractive interactions appeared as the most important contribution to the binding energy in both cases (Figure 11, bottom panel). The MBE structures were optimized again and used as starting structures for MD simulations. The corresponding 1-ns trajectories (Supplementary Figures S31, S32) next confirmed the stability of the dimer species. Most interestingly, the data support the initially theorized differences in the inner space topology of the assemblies: 4-dimer exhibits an extended tubular cavity in which the 1,8-dimethylnaphthylene moieties behave as cavity extension modules whereas in 5-dimer the 2,3-dimethylnaphthylene surfaces mutually act as partition elements between the two βCD cavities, resembling the situation previously encountered for the homologous o-xylylene derivative 2 (Neva et al., 2018).
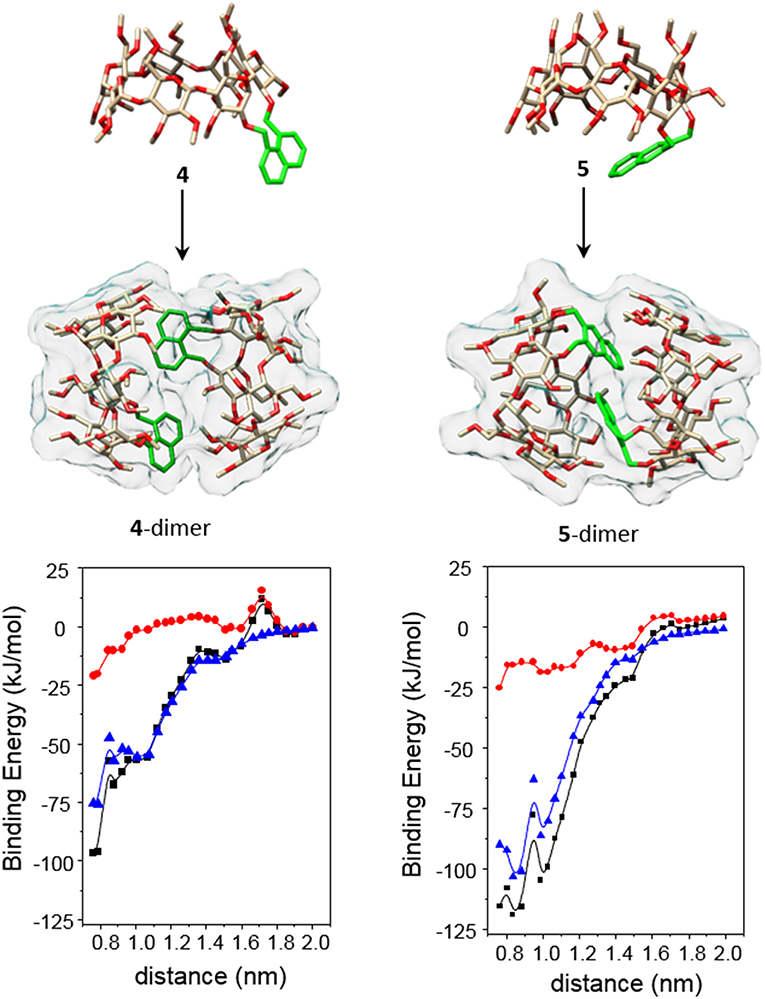
Figure 11. The (Top and Middle) represent 3D-views of the optimized (MM) structures of 4 and 5 (oxygen in red, carbon in tan; H atoms have been removed and the aromatic ring is highlighted in green for the sake of clarity) and minima binding energy (MBE) structures of the corresponding HH-dimers. The (Bottom) contains the changes of the total interaction energies (black) as well as the electrostatics (red) and van der Waals (blue) contributions, obtained after step by step approaching along the y axis defined by the center of mass of the glycosidic oxygens (see the Experimental section and the Supplementary Materials for details).
Inclusion Properties and the Impact in Dimer Stability
The revealed conformational differences between 4 and 5, both in the monomer and dimer states, let presume that their abilities to form inclusion complexes in water solution will quite differ. It is interesting to speculate that a guest molecule fitting in the βCD cavity will probably have a stronger impact in the orientation of the relatively flexible 2,3-naphthylene segment in 5 as compared to the more rigid 1,8-naphthylene appendage in 4, which at its turn would affect the corresponding monomer/dimer equilibrium. The close relationship between conformational properties and dimer stability offers then an opportunity for the spatiotemporal control of the aggregation/dissociation process through host-guest supramolecular chemistry. As a preliminary indication, the HRESI-mass spectrum of a water solution of 4 containing an excess of adamantane-1-carboxylate (AC), a preferred host for βCD derivatives that provides large association constants (Aoyagi et al., 1997; Kuwabara et al., 1999; Liu et al., 2004; Song et al., 2005), showed a relatively intense pseudomolecular peak at m/z 3331.5490, compatible with the AC guest trapped in the intact 4-dimer, whereas the 1:1 5:AC complex (m/z 1777.8173) was preferred in the case of 5, supporting that AC inclusion in the βCD cavity results in 5-dimer disruption (Supplementary Figure S33). Fluorescence spectroscopy and circular dichroism further reinforced this notion. Thus, sequential addition of AC aliquots to a water solution of 4 (AC:4 molar ratio from 0 to ~2) did not provoke significant changes in the average lifetime of the electronic excited states <τ> (Figure 12, left panel), whereas a similar experiment with 5 led to an important decrease in <τ>, meaning an increase in the proportion of the monomer (shorter lifetime) at the expenses of the dimer form. Analogously, the intensity of the 1Bb, 1La, and 1Lb bands in the corresponding ICD spectra of 4 were essentially unaltered in the presence of AC, whereas in the case of 5 monotonically decreased with AC concentration (Supplementary Figures S34, S35). Notably, adding a large excess of AC to 5 in water additionally triggered a drastic change in the sign of the 1La band from positive to negative, matching the effect observed upon decreasing the solvent polarity (Figure 12, right panel, and Supplementary Figure S36). This result implies that AC inclusion in the cavity of 5 leads to a reorientation of the 2,3-dimethylnaphthylene clip to meet the disposition encountered in the monomer, strongly supporting a guest-promoted conformational switch mechanism at the origin of the AC-induced dimer dissociation.
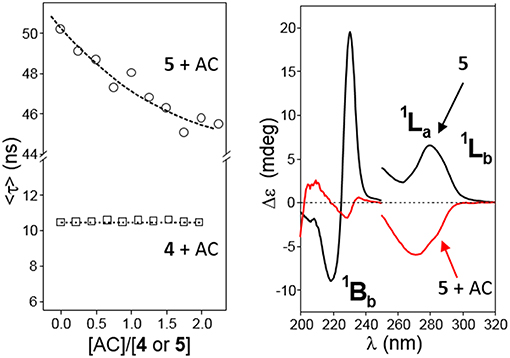
Figure 12. (Left) Plots of the average lifetime (<τ>) variations for water solutions of 4 (□) or 5 (◦, 1.36 · 10−4 M) at increasing AC concentrations at 25°C. (Right) Circular dichroism spectra of 5 (0.15 mM in water) in the absence (black line) and in the presence (red line) of a large excess of AC ([AC]/[5] = 86) at 25°C.
1H NMR titration experiments in buffered aqueous media at 25°C confirmed the formation of supramolecular complexes between AC and 4 or 5. In agreement with fluorescence and ICD data, the chemical shift variations of selected proton resonances of 4 (0.5 mM, meaning a 4-dimer molar fraction > 90%) induced by increasing proportions of AC afforded binding isotherms that fitted well to a 1:1 host:guest binding model between 4-dimer and the adamantane carboxylate guest, i.e., a 2:1 stoichiometry based on 4-monomer (Supplementary Figure S37). Least square fitting yielded an apparent association constant Ka = 1.3 ± 0.2 · 104 M−1 for the equilibrium 4-dimer + AC ⇆ 4-dimer:AC. In the case of compound 5, a similar analysis returned an apparent Ka = 1.1 ± 0.4 · 103 M−1 for a 1:1 complex between the monomer form of 5 and the AC guest, although this value is probably underestimated given that formation of the 5:AC inclusion complex requires 5-dimer disassembly (Supplementary Figure S38). For comparison, the Ka value for the 1:1 inclusion complexes of AC with permethylated βCD, which does not form dimers in solution, or with the o-xylylene derivative 2, which forms an HH-dimer in water solution that dissociates upon inclusion complex formation, were 1262 ± 130 M−1 and 5489 ± 1353 M−1, respectively (Neva et al., 2018).
Despite of the huge amount of information on the complexes between βCD and adamantane derivatives accumulated over more than 50 years, the orientation of the guest in the cyclooligosaccharide cavity continues to be a subject of debate. Negatively charged hydrophilic substituents, such as the carboxylate group in AC, are not included in the hydrophobic receptacle, but it is unclear whether they protrude from the wider (type I) or the narrower rim (type II complex) (Rüdiger et al., 1996; Gómez-Biagi et al., 2008; Krishnan et al., 2012). A recent study unequivocally demonstrates that both types of complexes coexist in approximately equal amounts in the case of the inclusion complex between AC and per-O-methylated βCD (Schönbeck, 2018), but the preferred guest orientation may well-depend on the modification of the βCD platform. A comparative analysis of NOE (or ROE) contacts between the α, β and γ protons in AC and the inside located H-3 and H-5 CD protons provides an indication on this issue (Figure 13, top panel). In our case, the asymmetric nature of the macroring in both 4 and 5 and the extensive signal overlapping precludes a precise quantification. Yet, a qualitative assessment of the NOESY spectra for the corresponding AC complexes was consistent with a largely predominant type II arrangement for the 1,8-dimethylnaphthylene derivative 4 (strong H-5/H-α and H-3/H-γ contacts, Supplementary Figure S39) and the presence of a much higher proportion of type I complex in the case of the 2,3-dimethylnaphthylene positional isomer 5 (H-3/H-α and H-5/H-γ spatial correlations were additionally present). In principle, access to the βCD cavity through the secondary rim is prevented in the dimers, guiding AC entrance through the primary face to give a type II orientation. In the case of 4-dimer, the resulting ternary species remains stable (Figure 13, middle panel). In contrast, AC inclusion in 5-dimer probably weakens the intermolecular naphthalene-naphthalene interactions by affecting the orientation of the aromatic clip (note that NOE contacts between aromatic and adamantane protons are also detected; Supplementary Figure S40), leading to dissociation into a 1:1 5-monomer:AC complex and a free host molecule. Inclusion through the secondary face becomes thus feasible, enabling equilibration between type I and type II 5:AC complexes (Figure 13, bottom panel).
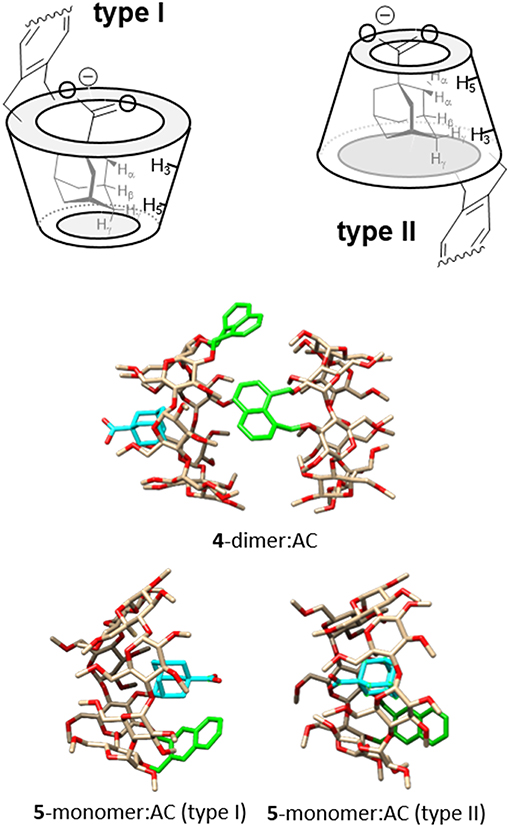
Figure 13. (Top) Schematic representation of type I and type II complexes between AC and βCD derivatives bearing aromatic clips, with indication of the expected diagnostic NOE contacts. (Middle) MBE structure of the ternary complex formed by 4-dimer and AC. (Bottom) MBE structures of the type I (left) and type II (right) 5-dimer:AC complexes obtained by approaching the AC guest to the CD host by the primary rim, to give a type I complex (left), or by the secondary rim, to give a type II complex (right). In the CD partners, oxygens are colored in red, carbons in tan; H atoms have been removed and the aromatic ring is highlighted in green; in the AC partner, the carbon atoms are highlighted in cyan for the sake of clarity.
Conclusion
We successfully synthesized two naphthylene-clipped β-cyclodextrin regioisomers with the ability to self-assemble into head-to-head supramolecular dimers in water solution, the aromatic appendage being either 1,8- (4) or 2,3-doubly-linked (5) to consecutive O-2I and O-3II positions in the secondary rim of the CD core through benzyl ether-type bridges. Such structural alteration translated into significant differences in the topological properties and stability of the dimer species as supported by MS, NMR, fluorescence and circular dichroism. Thus, 4-dimer adopts a very stable tubular-like arrangement in which the aromatic walls, in an open conformation, essentially extent the CD secondary faces of the monomer components leaving the primary rims exposed, whilst in 5-dimer the naphthalene platforms enjoy higher flexibility and take quasi-capped dispositions, acting as folding screens that separate the individual CD units. These conformational and architectural dissimilarities further condition the inclusion properties: 4-dimer can host the adamantane carboxylate guest to form a 4-dimer:AC stable ternary species whereas the same AC guest triggers 5-dimer disruption. Interestingly, the synthetic approach is compatible with the incorporation of functional elements at the primary positions of the cyclooligosaccharide, e.g., cationic centers to modulate the reactivity of guests confined in the CD cavity (Wei et al., 2018; Yi et al., 2018) or to mediate nucleic acid condensation (Aranda et al., 2013; Martínez et al., 2013; Jiménez Blanco et al., 2016; Martínez-Negro et al., 2017; Przybylski et al., 2018). The possibility of implementing different supramolecular organization levels (e.g., clusterization and/or inclusion and/or spatiotemporal controlled dissociation) with aromatic clips offers then a valuable tool to program the behavior of monodisperse CD-based devices in biological environments for (bio)molecular encapsulation and controlled release. It is worth mentioning that, although AC was just a model guest in this study, the adamantane moiety is present in many drugs (Wanka et al., 2013; de la Mata et al., 2015; Spilovska et al., 2016) and delivery systems (Rodríguez-Lavado et al., 2014; Štimac et al., 2017) that could be used in combination with customized analogs of 4 or 5 for biomedically oriented applications. Work in this direction is currently sought in our laboratories.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was supported by the Spanish Ministerio de Economía y Competitividad (contract numbers CTQ2015-64425-C2-1-R, CTQ2016-80600-P and SAF2016-76083-R), the Junta de Andalucía (contract number FQM2012-1467), the UAH (CCGP2017-EXP/027), and the European Regional Development Funds (FEDER and FSE). TN acknowledges the Junta de Andalucía for a doctoral fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Technical assistance from the research support services of the University of Seville (CITIUS) is acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00072/full#supplementary-material
References
Álvarez-Dorta, D., León, E. I., Kennedy, A. R., Martín, A., Pérez-Martín, A., and Suárez, E. (2016). Radical-mediated C–H functionalization: a strategy for access to modified cyclodextrins. J. Org. Chem. 81, 11766–11787. doi: 10.1021/acs.joc.6b02241
Álvarez-Dorta, D., León, E. I., Kennedy, A. R., Martín, A., Pérez-Martín, I., and Suárez, E. (2015). Easy access to modified cyclodextrins by an intramolecular radical approach. Angew. Chem. Int. Ed. 54, 3674–3678. doi: 10.1002/anie/201412300
Álvarez-Lorenzo, C., García-González, C. A., and Concheiro, A. (2017). Cyclodextrins as versatile building blocks for regenerative medicine. J. Control. Release 268, 269–281. doi: 10.1016/j.jconrel.2017.10.038
Aoyagi, T., Nakamura, A., Ikeda, H., Ikeda, T., Mihara, H., and Ueno, A. (1997). Alizarin Yellow-modified? β-cyclodextrin as a guest-responsive absorption change sensor. Anal. Chem. 69, 659–663. doi: 10.1021/ac960727z
Aranda, C., Urbiola, K., Méndez Ardoy, A., García Fernández, J. M., Ortiz Mellet, C., and Tros de Ilarduya, C. (2013). Targeted gene delivery by new folate–polycationic amphiphilic cyclodextrin–DNA nanocomplexes in vitro and in vivo. Eur. J. Pharm. Bipharm. 85, 390–397. doi: 10.1016/j.ejpb.2013.06.011
Balbuena, P., Gonçalves-Pereira, R., Jiménez Blanco, J. L., García-Moreno, M. I., Lesur, D., Ortiz Mellet, C., et al. (2013). o-Xylylene protecting group in carbohydrate chemistry: application to the regioselective protection of a single vic-diol segment in cyclodextrins. J. Org. Chem. 78, 1390–1403. doi: 10.1021/jo302178f
Balbuena, P., Lesur, D., González-Álvarez, M. J., Mendicuti, F., Ortiz Mellet, C., and García Fernández, J. M. (2007). One-pot regioselective synthesis of 2I,3I-O-(o-xylylene)-capped cyclomaltooligosaccharides: tailoring the topology and supramolecular properties of cyclodextrins. Chem. Commun. 3270–3272. doi: 10.1039/B705644C
Benkovics, G., Perez-Lloreta, M., Afonso, D., Darcsi, A., Béni, S., Fenyvesi, et al. (2017). A multifunctional β-cyclodextrin-conjugate photodelivering nitric oxide with fluorescence reporting. Int. J. Pharm. 531, 614–620. doi: 10.1016/j.ijpharm.2017.05.023
Berova, N., Di Bari, L., and Pescitelli, G. (2007). Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 36, 914– 931. doi: 10.1039/B515476F
Bisson, A. P., Carver, F. J., Eggleston, D. S., Haltiwanger, R. C., Hunter, C. A., Livingstone, D. L., et al. (2000). Synthesis and recognition properties of aromatic amide oligomers: molecular zippers. J. Am. Chem. Soc. 122, 8856–8868. doi: 10.1021/ja0012671
Bisson, A. P., Hunter, C. A., Morales, J. C., and Young, K. (1998). Cooperative interactions in a ternary mixture. Chem. Eur. J. 4, 845–851.
Blanco, M. (1991). Molecular silverware. I. General solutions to excluded volume constrained problems. J. Comput. Chem. 12, 237–247
Brunel, Y., Faucher, H., Gagnaire, D., and Rassat, A. (1975). Programme de minimisation de l'energie empirique d'une molecule par une methode simpliciale. Tetrahedron 31, 1075–1091. doi: 10.1016/0040-4020(75)80129-3
Caira, M. R., Bourne, S. A., Mhlongo, W. T., and Dean, P. M. (2004). New crystalline forms of permethylated β-cyclodextrin. Chem. Commun. 2004, 2216–2217. doi: 10.1039/B408660K
Carmona, T., Martina, K., Rinaldi, L., Boffa, L., Cravotto, G., and Mendicuti, F. (2015). Predicting self-assembly and structure in diluted aqueous solutions of modified mono- and bis-β-cyclodextrins that contain naphthoxy chromophore groups. New J. Chem. 39, 1714–1724. doi: 10.1039/C4NJ01556H
Carreño, A., Rodríguez, L., Páez-Hernández, D., Martin-Trasanco, R., Zúñiga, C., Oyarzún, D. P., et al. (2018). Two new fluorinated phenol derivatives pyridine Schiff bases: synthesis, spectral, theoretical characterization, Inclusion in epichlorohydrin-β-cyclodextrin polymer, and antifungal effect. Front. Chem. 6:312. doi: 10.3389/fchem.2018.00312
Clark, M., Cramer, R. D. III., and Van Opdenbosch, N. (1989). Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 10, 982–1012. doi: 10.1002/jcc.540100804
Crini, G. (2014). Review: a history of cyclodextrins. Chem. Rev. 114, 10940–10975. doi: 10.1021/cr500081p
de Jesus, M. B., Fraceto, L. F., Martini, M. F., Pickholza, M., Ferreira, C. V., and de Paula, E. (2012). Non-inclusion complexes between riboflavin and cyclodextrins. J. Pharm. Pharmacol. 64, 832–842. doi: 10.1111/j.2042-7158.2012.01492.x
de la Mata, M., Cotán, D., Oropesa-Avila, M., Garrido-Maraver, J., Cordero, M. D., Villanueva Paz, M., et al. (2015). Pharmacological chaperones and coenzyme Q10 treatment improves mutant β-glucocerebrosidase activity and mitocondrial function in neuronopathic forms of Gaucher disease. Sci. Rep. 5:10903. doi: 10.1038/srep10903
Díaz-Moscoso, A., Méndez-Ardoy, A., Ortega-Caballero, F., Benito, J. M., Ortiz Mellet, C., Defaye, J., et al. (2011). Chem. Med. Chem. 6, 181–192. doi: 10.1002/cmdc.201000419
Engel, S., Möller, N., and Ravoo, B. J. (2018). Stimulus-responsive assembly of nanoparticles using host–guest interactions of cyclodextrins. Chem. Eur. J. 24, 4741–4748. doi: 10.1002/chem.201705540
Evenou, P., Rossignol, J., Pembouong, G., Gothland, A., Colesnic, D., Barbeyron, R., et al. (2018). Bridging β-cyclodextrin prevents self-inclusion, promotes supramolecular polymerization, and promotes cooperative interaction with nucleic acids. Angew. Chem. Int. Ed. 57, 7753–7758. doi: 10.1002/anie.201802550
Filippone, S., Heimann, F., and Rassat, A. A. (2002). A highly water-soluble 2:1 β-cyclodextrin–fullerene conjugate. Chem. Commun. 1508–1509. doi: 10.1039/B202410A
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2004). Gaussian 03, Revision C.02. Wallingford, CT: Gaussian, Inc.
Fujita, K., Yamamura, H., Imoto, T., and Tabushi, I. (1988). Preparation of 3A,6A-anhydro-β-cyclodextrin and its Taka amylolysis. Chem. Lett. 17, 543–546. doi: 10.1246/cl.1988.543
Fukudome, M., Shimosaki, K., Koga, K., Yuan, D.-Q., and Fujita, K. (2007). Selective synthesis and ester cleavage property of 3A,2B-anhydro-3B-deoxy-3B-thio-β-cyclodextrin. Tetrahedron Lett. 48, 7493–7497. doi: 10.1016/j.tetlet.2007.08.062
Fukudome, M., Yoshikawa, K., Koga, K., Yuan, D.-Q., and Fujita, K. (2007b). Selective modification of beta-cyclodextrin: an unexpected tandem reaction enables the cross-linking of C2(A) and C2(B) via a sulfur atom. Chem. Commun. 3157–3159. doi: 10.1039/b703943c
Gallego-Yerga, L., Benito, J. M., Blanco-Fernández, L., Martínez-Negro, M., Vélaz, I., Aicart, E., et al. (2018). Plasmid-templated control of DNA–cyclodextrin nanoparticle morphology through molecular vector design for effective gene delivery. Chem. Eur. J. 24, 3825–3835. doi: 10.1002/chem.201705723
Gallego-Yerga, L., Blanco-Fernández, L., Urbiola, K., Carmona, T., Marcelo, G., Benito, J. M., et al. (2015). Host–guest-mediated DNA templation of polycationic supramolecules for hierarchical nanocondensation and the delivery of gene material. Chem. Eur. J. 21, 12093–12104. doi: 10.1002/chem.201501678
Gallego-Yerga, L., González-Álvarez, M. J., Mayordomo, N., Santoyo-González, F., Benito, J. M., Ortiz Mellet, C., et al. (2014a). Dynamic self-assembly of polycationic clusters based on cyclodextrins for pH-sensitive DNA nanocondensation and delivery by component design, Chem. Eur. J. 20, 6622–6627. doi: 10.1002/chem.201402026
Gallego-Yerga, L., Lomazzi, M., Sansone, F., Ortiz Mellet, C., Casnati, A., and Garcia Fernandez, J. M. (2014b). Glycoligand-targeted core-shell nanospheres with tunable drug release profiles from calixarene-cyclodextrin heterodimers. Chem. Commun. 50, 7440–7443. doi: 10.1039/c4cc02703e
Gallego-Yerga, L., Posadas, I., de la Torre, C., Ruiz-Almansa, J., Sansone, F., Ortiz Mellet, C., et al. (2017). Docetaxel-loaded nanoparticles assembled from β-cyclodextrin/calixarene giant surfactants: physicochemical properties and cytotoxic effect in prostate cancer and glioblastoma cells. Front. Pharmacol. 8:249. doi: 10.3389/fphar.2017.00249
Gamieldien, M. R., Maestre, I., Jaime, C., and Naidoo, K. J. (2010). Optimal configurations of “capped” β-cyclodextrin dimers in water maximise hydrophobic association. ChemPhysChem 11, 452–459. doi: 10.1002/cphc.200900541
García-Moreno, M. A., de la Mata, M., Sánchez-Fernández, E. M., Benito, J. M., Díaz-Quintana, A., Fustero, S., et al. (2017). Fluorinated chaperone–β-cyclodextrin formulations for β-glucocerebrosidase activity enhancement in neuronopathic Gaucher disease. J. Med. Chem. 60, 1829–1842. doi: 10.1021/acs.jmedchem.6b01550
Gómez-Biagi, R. F., Jagt, R. B. C., and Nitz, M. (2008). Remarkably stable inclusion complexes with heptakis-[6-deoxy-6-(2-aminoethylsulfanyl)]-β-cyclodextrin. Org. Biomol. Chem., 6, 4622–4626. doi: 10.1039/B813999G
González-Álvarez, M. J., Balbuena, P., Ortiz Mellet, C., García Fernández, J. M., and Mendicuti, F. (2008). Study of the conformational and self-aggregation properties of 2I,3I-O-(o-xylylene)-per-O-Me-α- and -β-cyclodextrins by fluorescence and molecular modeling. J. Phys. Chem. B 112, 13717–13729. doi: 10.1021/jp077670c
González-Álvarez, M. J., Benito, J. M., García Fernández, J. M., Ortiz Mellet, C., and Mendicuti, F. (2013). Influence of the macroring size on the self-association thermodynamics of cyclodextrins with a double-linked naphthalene at the secondary face. J. Phys. Chem. B 117, 5472–5485. doi: 10.1021/jp400784t
González-Álvarez, M. J., Marino, A., and Mendicuti, F. (2009a). Fluorescence, induced circular dichroism and molecular mechanics of 1-methyl naphthalenecarboxylate complexes with 2-hydroxypropyl cyclodextrins. J. Fluoresc. 19, 449–462. doi: 10.1007/s10895-008-0432-0
González-Álvarez, M. J., Méndez-Ardoy, A., Benito, J. M., García Fernández, J. M., and Mendicuti, F. (2011). Self-association of a naphthalene-capped-β-cyclodextrin through cooperative strong hydrophobic interactions. J. Photochem. Photobiol. A 223, 25–36. doi: 10.1016/j.jphotochem.2011.07.013
Gonzalez-Álvarez, M. J., Vicente, J., Ortiz Mellet, C., García Fernández, J. M., and Mendicuti, F. (2009b). Thermodynamics of the dimer formation of 2I,3I-O-(o-xylylene)-per-O-Me-γ-cyclodextrin: fluorescence, molecular mechanics and molecular dynamics. J. Fluoresc. 19, 975–988. doi: 10.1007/s10895-009-0497-4
Gropp, C., Quigley, B. L., and Diederich, F. (2017). Molecular recognition with resorcin[4]arene cavitands: switching, halogen-bonded capsules, and enantioselective complexation. J. Am. Chem. Soc. 140, 2705–2717. doi: 10.1021/jacs.7b12894
Guo, S., Song, Y., He, Y., Hu, X.-Y., and Wang, L. (2018). Highly efficient artificial light-harvesting systems constructed in aqueous solution based on supramolecular self-assembly. Angew. Chem. Int. Ed. 57, 3163–3167. doi: 10.1002/anie.201800175
Harata, K., and Hisashi, U. (1975). The circular dichroism spectra of the β-cyclodextrin complex with naphthalene derivatives. Bull. Chem. Soc. Jpn. 48:375. doi: 10.1246/bcsj.48.375
Hong, S. J., Ahn, M. H., Sangshetti, J., Choung, P. H., and Arote, R. B. (2018). Sugar-based gene delivery systems: current knowledge and new perspectives. Carbohydr. Polym. 181, 1180–1193. doi: 10.1016/j.carbpol.2017.11.105
Immel, S., Fujita, K., Fukudome, M., and Bolte, M. (2001). Two stereoisomeric 3I,2II-anhydro-α-cyclodextrins: a molecular dynamics and crystallographic study. Carbohydr. Res. 336, 297–308. doi: 10.1016/S0008-6215(01)00267-1
Jiménez Blanco, J. L., Benito, J. M., Ortiz Mellet, C., and García Fernández, J. M. (2017). Molecular nanoparticle-based gene delivery systems. J. Drug Deliv. Sci. Technol. 42, 18–37. doi: 10.1016/j.jddst.2017.03.012
Jiménez Blanco, J. L., Ortega-Caballero, F., Blanco-Fernández, L., Carmona, T., Marcelo, G., Martínez-Negro, et al. (2016). Trehalose-based Janus cyclooligosaccharides: the “click” synthesis and DNA-directed assembly into pH-sensitive transfectious nanoparticles. Chem. Commun. 52, 10117–10120. doi: 10.1039/c6cc04791b
Joshi, A., Kate, S., Poon, V., Mondal, D., Boggara, M. B., Saraph, A., et al. (2011). Structure-based design of a heptavalent anthrax toxin inhibitor. Biomacromolecules 12, 791–796. doi: 10.1021/bm101396u
Kemp, W., Storie, l,. T., and Tullozh, C. D. (1980). Synthesis of potentially basic hydrocarbons by sulphur extrusion and/or bis-Wittig reactions. Two syntheses of benz[5,6]indeno[2,1-a]phenalene and a new synthesis of dibenzo[de,mn]naphthacene (zethrene). J. Chem. Soc., Perkin Trans. 1, 2812–2817. doi: 10.1039/P19800002812
Kodaka, M. (1991). Sign of circular dichroism induced by β-cyclodextrin. J. Phys. Chem. 95, 2110–2112. doi: 10.1021/j100159a005
Kodaka, M. (1993). A general rule for circular dichroism induced by a chiral macrocycle. J. Am. Chem. Soc. 115, 3702–3705. doi: 10.1021/ja00062a040
Kodaka, M. (1998). Application of a general rule to induced circular dichroism of naphthalene derivatives complexed with cyclodextrins. J. Phys. Chem. A, 102, 8101– 8103. doi: 10.1021/jp983286r
Kodaka, M., and Fukaya, T. (1989). Induced circular dichroism spectrum of α-cyclodextrin complex with heptylviologen. Bull. Chem. Soc. Jpn. 62, 1154–1157. doi: 10.1246/bcsj.62.1154
Krishnan, R., Rakhi, A. M., and Gopidas, K. R. (2012). Study of β-cyclodextrin–Pyromellitic diimide complexation. Conformational analysis of binary and ternary complex structures by induced circular dichroism and 2D NMR spectroscopies. J. Phys. Chem. C 116, 25004–25014. doi: 10.1021/jp309788z
Kronberg, B. (2016). The hydrophobic effect. Curr. Opin. Colloid Interface Sci. 22:14. doi: 10.1016/j.cocis.2016.02.001
Kuwabara, T., Nakajima, H., Nanasawa, M., and Ueno, A. (1999). Color change indicators for molecules using methyl red-modified cyclodextrins. Anal. Chem. 71, 2844–2849. doi: 10.1021/ac9814041
Lakowicz, J. R. (2008). Principles of Fluorescence Spectroscopy, 3rd Edn. New York, NY: Springer, 280.
Le Gac, S., Boitrel, B., Sollogoub, M., and Ménand, M. (2016). Protonated hexaphyrin–cyclodextrin hybrids: molecular recognition tuned by a kinetic-to-thermodynamic topological adaptation. Chem. Commun. 52, 9347–9350. doi: 10.1039/C6CC04276G
León, E. I., Martín, A., Pèrez-Martín, I., and Suárez, E. (2018). Reductive radical cascades triggered by alkoxyl radicals in the β-cyclodextrin framework. Org. Lett. 20, 3385–3389. doi: 10.1021/acs.orglett.8b01308
Liu, Y., Zhao, Y.-L., Zhang, H.-Y., Fan, Z., Wen, G.-D., and Ding, F. (2004). Spectrophotometric study of inclusion complexation of aliphatic alcohols by β-cyclodextrins with azobenzene tether. J. Phys. Chem. B 108, 8836–8843. doi: 10.1021/jp0380024
Liu, Z., Nalluri, S. K. M., and Stoddart, J. F. (2017). Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes. Chem. Soc. Rev. 46, 2459–2478. doi: 10.1039/C7CS00185A
Martínez, A., Bienvenu, C., Jiménez Blanco, J. L., Vierling, P., Ortiz Mellet, C., García Fernández, J. M., et al. (2013). Amphiphilic oligoethyleneimine–β-cyclodextrin “click” clusters for enhanced DNA delivery. J. Org. Chem. 78, 8143–8148. doi: 10.1021/jo400993y
Martínez-Negro, M., Caracciolo, G., Palchetti, S., Pozzi, D., Capriottic, A. L., Cavaliere, C., et al. (2017). Biophysics and protein corona analysis of Janus cyclodextrin-DNA nanocomplexes. Efficient cellular transfection on cancer cells. Biochim. Biophys. Acta Gen. Sub. 1861, 1737–1749. doi: 10.1016/j.bbagen.2017.03.010
Matsuoka, R., and Nabeshima, T. (2018). Functional supramolecular architectures of dipyrrin complexes. Front. Chem. 6:349. doi: 10.3389/fchem.2018.00349
Mayordomo, N., González-Álvarez, M. J., Gallego Yerga, L., Ortiz Mellet, C., García Fernández, J. M., and Mendicuti, F. (2013). Competitive processes of a chromophore modified α-cyclodextrin in the presence of a fluorescence polarity sensitive probe. J. Photochem. Photobiol. A 256, 42–51. doi: 10.1016/j.jphotochem.2013.02.007
McNally, A., Forster, R. J., and Keyes, T. E. (2009). Interfacial supramolecular cyclodextrin-fullerene assemblies: host reorientation and guest stabilization. Phys. Chem. Chem. Phys. 11, 848–856. doi: 10.1039/B810772F
Ménand, M., Sollogoub, M., Boitrel, B., and Le Gac, S. (2016). Hexaphyrin-cyclodextrin hybrids: a nest for switchable aromaticity, asymmetric confinement, and isomorphic fluxionality. Angew. Chem. Int. Ed. 55, 297–301. doi: 10.1002/anie.201508009
Ménand, M., Sollogoub, M., Boitrel, B., and Le Gac, S. (2018). Cyclodextrin-sandwiched hexaphyrin hybrids: side-to-side cavity coupling switched by a temperature- and redox-responsive central device. Chem. Eur. J. 24, 5804–5812. doi: 10.1002/chem.201705958
Meyer, E. E., Rosenberg, K. J., and Israelachvili, J. (2006). Recent progress in understanding hydrophobic interactions. Proc. Natl. Acad. Sci. U.S.A 103, 15739–15746. doi: 10.1073/pnas.0606422103
Michl, J. (1978). Magnetic circular dichroism of cyclic π-electron systems. 1. Algebraic solution of the perimeter model for the A and B terms of high-symmetry systems with a (4N + 2)-electron [n]annulene perimeter. J. Am. Chem. Soc. 100, 6801–6811. doi: 10.1021/ja00490a001
Neva, T., Carmona, T., Benito, J. M., Przybylski, C., Ortiz Mellet, C., Mendicuti, F., et al. (2018). Xylylene clips for the topology-guided control of the inclusion and self-assembling properties of cyclodextrins. J. Org. Chem. 83, 5588–5597. doi: 10.1021/acs.joc.8b00602
O'Connor, D. V., Ware, W. R., and Andre, J. C. (1979). Deconvolution of fluorescence decay curves. A critical comparison of techniques. J. Phys. Chem. 83, 1333–1343. doi: 10.1021/j100473a019
Ortiz Mellet, C., García Fernández, and J. M., and Benito, J. M. (2011). Cyclodextrin-based gene delivery systems. Chem. Soc. Rev. 2011, 40, 1586–1608. doi: 10.1039/c0cs00019a
Pescitelli, G., Di Bari, L., and Berova, N. (2011). Conformational aspects in the studies of organic compounds by electronic circular dichroism. Chem. Soc. Rev. 40, 4603– 4625. doi: 10.1039/C1CS15036G
Platt, J. R. (1949). Classification of spectra of cata-condensed hydrocarbons. J. Chem. Phys. 17, 484–495. doi: 10.1063/1.1747293
Pozuelo, J., Madrid, J. M., Mendicuti, F., and Mattice, W. L. (1996). Inclusion complexes of chain molecules with cycloamyloses. 1. Conformational analysis of the isolated cycloamyloses using molecular dynamics simulations. Comput. Theor. Polym. Sci. 6, 125–134
Press, W. H., Flannery, B. P., Teukolski, S. A., and Vetterling, W. T. (1988). Numerical Recipes: The Art of Scientific Computing. Cambridge: Cambridge University Press.
Przybylski, C., Benito, J. M., Bonnet, V., Ortiz Mellet, C., and García Fernández, J. M. (2018). Revealing cooperative binding of polycationic cyclodextrins with DNA oligomers by capillary electrophoresis coupled to mass spectrometry. Anal. Chim. Acta 1002, 70–81. doi: 10.1016/j.aca.2017.11.034
Rodríguez-Lavado, J., de la Mata, M., Jiménez Blanco, J. L., García-Moreno, M. I., Benito, J. M., Díaz-Quintana, A., et al. (2014). Targeted delivery of pharmacological chaperones for Gaucher disease to macrophages by a mannosylated cyclodextrin carrier. Org. Biomol. Chem. 12, 2289–2301. doi: 10.1039/c3ob42530d
Roizel, B., Baltaze, J. P., and Sinaý, P. (2002). Diisobutylaluminum-promoted secondary rim selective de-O-methylation of permethylated cyclodextrins. Tetrahedron Lett. 43, 2371–2373. doi: 10.1016/S0040-4039(02)00274-5
Rüdiger, V., Eliseev, A., Simova, S., Schneider, H.-J., Blandamer, M. J., Cullis, P. M., et al. (1996). Conformational, calorimetric and NMR spectroscopic studies on inclusion complexes of cyclodextrins with substituted phenyl and adamantane derivatives. J. Chem. Soc., Perkin Trans. 2, 2119–2120. doi: 10.1039/P29960002119
Ryzhakov, A., Thi, T. D., Stappaerts, J., Bertoletti, L., Kimpe, K., Sá Couto, A. R., et al. (2016). Self-assembly of cyclodextrins and their complexes in aqueous solutions. J. Pharm. Sci. 105, 2556–2569. doi: 10.1016/j.xphs.2016.01.019
Saigusa, H., and Lim, E. C. (1996). Excimer formation in van der Waals dimers and clusters of aromatic molecules. Acc. Chem. Res. 29, 171–178. doi: 10.1021/ar950169v
Sansone, F., and Casnati, A. (2013). Multivalent glycocalixarenes for recognition of biological macromolecules: glycocalyx mimics capable of multitasking. Chem. Soc. Rev. 42, 4623–4639. doi: 10.1039/C2CS35437C
Schönbeck, C. (2018). Charge determines guest orientation: a combined NMR and molecular dynamics study of β-cyclodextrins and adamantane derivatives. J. Phys. Chem. B, 122, 4821–4827. doi: 10.1021/acs.jpcb.8b02579
Shetty, D., Khedkar, J. K., Park, K. M., and Kim, K. (2015). Can we beat the biotin-avidin pair?: cucurbit[7]uril-based ultrahigh affinity host–guest complexes and their applications. Chem. Soc. Rev. 44, 8747–8761. doi: 10.1039/C5CS00631G
Shimizu, H., Kaito, A., and Hatano, M. (1979). Induced circular dichroism of β-cyclodextrin complexes with substituted benzenes. Bull. Chem. Soc. Jpn. 52:2678. doi: 10.1246/bcsj.52.2678
Shimizu, H., Kaito, A., and Hatano, M. (1981). Induced circular dichroism of β-cyclodextrin complexes with o-, m-, and p-disubstituted benzenes. Bull. Chem. Soc. Jpn. 54, 513–515. doi: 10.1246/bcsj.54.513
Shimizu, H., Kaito, A., and Hatano, M. (1982). Induced circular dichroism of β-cyclodextrin complexes with azanaphthalenes - polarization directions of the π → π transitions in azanaphthalenes. J. Am. Chem. Soc. 104, 7059–7065. doi: 10.1021/ja00389a029
Shirai, S., Kurashige, Y., and Yanai, T. (2016). Computational evidence of inversion of 1La and 1Lb-derived excited states in naphthalene excimer formation from ab Initio multireference theory with large active space: DMRG-CASPT2 Study. J. Chem. Theory Comput. 12, 2366–2372. doi: 10.1021/acs.jctc.6b00210
Simões, S. M. N., Rey-Rico, A., Concheiro, A., and Alvarez-Lorenzo, C. (2015). Supramolecular cyclodextrin-based drug nanocarriers. Chem. Commun. 51, 6275–6289. doi: 10.1039/C4CC10388B
Smiljanic, N., Moreau, V., Yockot, D., Benito, J. M., García Fernández, J. M., and Djedaïni-Pilard, F. (2006). Supramolecular control of oligosaccharide-protein interactions: switchable and tunable ligands for concanavalin A based on β-cyclodextrin. Angew. Chem. Int. Ed. 45, 5465–5468. doi: 10.1002/anie.200601123
Song, Y., Chen, Y., and Liu, Y. (2005). Switchable fluorescence behaviors of pyronine Y at different pH values upon complexation with biquinolino-bridged bis(β-cyclodextrin). J. Photochem. Photobiol. A 173, 328–333. doi: 10.1016/j.jphotochem.2005.04.012
Southall, N. T., Dill, K. A., and Haymet, A. D. J. (2002). A view of the hydrophobic Effect. J. Phys. Chem. B 106, 521–533. doi: 10.1021/jp015514e
Spilovska, K., Zemek, F., Korabecny, J., Nepovimova, E., Soukup, O., Windisch, M., et al. (2016). Adamantane – a lead structure for drugs in clinical practice. Curr. Med. Chem. 23, 3245–3266. doi: 10.2174/0929867323666160525114026
Štimac, A., Šekutor, M., Mlinarić-Majerski, K., Frkanec, L., and Frkanec, R. (2017). Adamantane in drug delivery systems and surface recognition. Molecules 22, 297. doi: 10.3390/molecules22020297
Sun, H.-L., Chen, Y., Zhao, J., and Liu, Y. (2015). Photocontrolled reversible conversion of nanotube and nanoparticle mediated by β-Cyclodextrin dimers. Angew. Chem. Int. Ed. 54, 9376–9380. doi: 10.10002/anie.201503614
Superchio, S., Giorgio, E., and Rosini, C. (2004). Structural determinations by circular dichroism spectra analysis using coupled oscillator methods: an update of the applications of the DeVoe polarizability model. Chirality 16, 422–451. doi: 10.1002/chir.20056
Tan, S., Ladewig, K., Fu, Q., Blencowe, A., and Qiao, G. G. (2014). Cyclodextrin-based supramolecular assemblies and hydrogels: recent advances and future perspectives Macromol. Rapid Commun. 35, 1166–1184. doi: 10.1002/marc.201400080
van Dijk, L., Tilby, M. l. J., Szpera, R., Smith, O. A., Bunce, H. A. P., and Fletcher, S. P. (2018). Molecular machines for catalysis. Nat. Rev. Chem. 2, 0117. doi: 10.1038/s41570-018-0117
Varan, G., Varan, C., Erdogar, N., Hincal, A. A., and Bilensoy, E. (2017). Amphiphilic cyclodextrin nanoparticles. Int. J. Pharm. 531, 457–469. doi: 10.1016/j.ijpharm.2017.06.010
Wanka, L., Iqbal, K., and Schreiner, P. R. (2013). The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem. Rev. 113, 3516–3604. doi: 10.1021/cr100264t
Webber, M. J., and Langer, R. (2017). Drug delivery by supramolecular design. Chem. Soc. Rev. 46, 6600–6620. doi: 10.1039/c7cs00391a
Wei, X., Wu, W., Matsushita, R., Yan, Z., Zhou, D., Chruma, J. J., et al. (2018). Supramolecular photochirogenesis driven by higher-order complexation: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate to slipped cyclodimers via a 2:2 complex with β-cyclodextrin. J. Am. Chem. Soc. 140, 3959–3974. doi: 10.1021/jacs.7b12085
Xiao, S., Yang, M., Sinaý, P., Blériot, Y., Sollogoub, M., and Zhang, Y. (2010). Diisobutylaluminium Hydride (DIBAL-H) Promoted secondary rim regioselective demethylations of permethylated β-cyclodextrin: a mechanistic proposal. Eur. J. Org. Chem. 1510–1516. doi: 10.1002/ejoc.200901230
Yan, Z., Huang, Q., Liang, W., Yu, X., Zhou, D., Wu, W., et al. (2017). Enantiodifferentiation in the photoisomerization of (Z,Z)-1,3-cyclooctadiene in the cavity of γ-cyclodextrin–curcubit[6]uril-wheeled [4]rotaxanes with an encapsulated photosensitizer. Org. Lett. 19, 898–901. doi: 10.1021/acs.orglett.7b00057
Yang, C., Mori, T., and Inoue, Y. (2008). Supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by capped γ-cyclodextrins: critical control of enantioselectivity by cap rigidity. J. Org. Chem. 73, 5786–5794. doi: 10.1021/jo800533y
Yang, Y.-W., Sun, Y.-L., and Song, N. (2014). Switchable host–guest systems on surfaces. Acc. Chem. Res. 47, 1950–1960. doi: 10.1021/ar500022f
Yi, J., Liang, W., Wei, X., Yao, J., Yan, Z., Su, D., et al. (2018). Switched enantioselectivity by solvent components and temperature in photocyclodimerization of 2-anthracenecarboxylate with 6A,6X-diguanidio-γ-cyclodextrins. Chin. Chem. Lett. 29, 87–90. doi: 10.1016/j.cclet.2017.05.004
Yin, G.-Z., Zhang, W.-B., and Cheng, S. Z. D. (2017). Giant molecules: where chemistry, physics, and bio-science meet. Sci. China: Chem. 60, 338–352. doi: 10.1007/s11426-016-0436-x
Yu, G., Yung, B. C., Zhou, Z., Mao, Z., and Chen, X. (2018). Artificial molecular machines in nanotheranostics. ACS Nano 12, 7–12. doi: 10.1021/acsnano.7b07851
Zeng, M., Chen, K., Tan, J., Zhang, J., and Wei, Y. (2018). A Supramolecular catalyst self-assembled from polyoxometalates and cationic pillar[5]arenes for the room temperature oxidation of aldehydes. Front. Chem. 6:457. doi: 10.3389/fchem.2018.00457
Zhang, Q.-W., Elemans, J. A. A. W., White, P. B., and Nolte, R. J. M. (2018). A manganese porphyrin–α-cyclodextrin conjugate as an artificial enzyme for the catalytic epoxidation of polybutadiene, Chem. Commun. 54, 5586–5589. doi: 10.1039/C8CC02320D
Keywords: cyclodextrins, self-assembly, supramolecular chemistry, naphthalene, host-guest chemistry, fluorescence, circular dichroism
Citation: Neva T, Carmona T, Benito JM, Przybylski C, Ortiz Mellet C, Mendicuti F and García Fernández JM (2019) Dynamic Control of the Self-Assembling Properties of Cyclodextrins by the Interplay of Aromatic and Host-Guest Interactions. Front. Chem. 7:72. doi: 10.3389/fchem.2019.00072
Received: 03 November 2018; Accepted: 28 January 2019;
Published: 25 February 2019.
Edited by:
Leyong Wang, Nanjing University, ChinaReviewed by:
Cheng Yang, Sichuan University, ChinaLihua Yuan, Sichuan University, China
Dong-Sheng Guo, Nankai University, China
Copyright © 2019 Neva, Carmona, Benito, Przybylski, Ortiz Mellet, Mendicuti and García Fernández. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Ortiz Mellet, bWVsbGV0QHVzLmVz
Francisco Mendicuti, ZnJhbmNpc2NvLm1lbmRpY3V0aUB1YWguZXM=
José M. García Fernández, am9nYXJjaWFAaWlxLmNzaWMuZXM=
†These authors have contributed equally to this work