 Owen J. Curnow
Owen J. Curnow Douglas R. MacFarlane2*
Douglas R. MacFarlane2*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 10 December 2018
Sec. Green and Sustainable Chemistry
Volume 6 - 2018 | https://doi.org/10.3389/fchem.2018.00603
This article is part of the Research Topic Ionic Liquids: Properties and Applications View all 30 articles
A series of solvent-stabilized ionic liquid fluorides were prepared, [EMIM]F.nCH3COOH n = 1. 0, 1.6, 2.1, 2.4, and 3.2, either via exchange from the chloride salt using KF or AgF, or by neutralization of the hydroxide salt using HF. Azeotrope drying was used to remove water. Their viscosity, conductivity and density properties were determined. A diethanol solvate of the triaminocyclopropenium salt [C3(NPr2)3]F was found to be stable and its viscosity, conductivity and density properties were also determined. The monoethanol solvate, however, was found to be unstable with trace water present. Intramolecular stabilization of fluoride was achieved by using OH functionalized cations: [C3(NEt2)2N(CH2CH2OH)2]F, [C3(N(CH2CH2OH)2)3]F, and [Me3NCH2CH2OH]F.H2O, The first of these is an ionic liquid at ambient temperature and has a TGA mass loss onset at 175°C, indicating a useful range of liquid state stability.
Research in Ionic Liquids (ILs) has explored a vast variety of anion types, revealing a wide range of properties and applications. Halide anion based ILs were some of the first known, especially those based on iodide due to the generally lower melting points with increasing size and polarizability in Group 17. Generally, fluoride salts are high melting and therefore have attracted little recent attention in this field. On the other hand, fluoride anions have the potential to make ionic liquids with interesting properties due to the reactivity of fluoride (Maiti et al., 2008; Mallik and Siepmann, 2010). Indeed, fluoride salts, such as tetrabutylammonium fluoride (TBAF), have many uses as fluorinating reagents and for the removal of silicon-based protecting groups. The more “naked” (higher donor ability) the fluoride anion is, the higher the reactivity (Christie and Jenkins, 2003). However, this reactivity causes difficulties with organic fluoride salts often being difficult to use, either because they become unstable due to the reactive fluoride (Sun and DiMagno, 2005) or they are insoluble in aprotic solvents.
Triaminocyclopropenium (TAC) cations have been considered as counter ions to fluoride for two reasons: firstly, there are no acidic protons, and secondly, TAC cations have a high energy HOMO that leads to weak cation-anion interactions and good ionic liquid properties (Weiss et al., 1995a,b; Butchard et al., 2006; Curnow et al., 2011, 2012; Wallace et al., 2015). Cations with acidic protons will easily be deprotonated by fluoride, which becomes a much stronger base as it becomes less solvated. This is predicted to occur in imidazolium fluoride salts: ab initio calculations on 1-ethyl-3-methylimidazolium fluoride, [EMIM]F, show that covalent bonding between the fluorine atom and a proton extracted from the 2 position of the imdazolium cation is preferred over the ionic form. The result is HF and a stable Arduengo carbene (Turner et al., 2003; Maiti et al., 2008). As a result, imidazolium-based ionic liquid fluorides have only been reported with solvated fluoride anions (Swatloski et al., 2003; Maiti et al., 2008). TAC cations might be more resistant to Hofmann elimination, which causes the decomposition of tetraalkylammonium fluorides. On the other hand, TAC cations are sensitive to strong nucleophiles, such as hydroxide, and possibly fluoride itself. For this reason, even if triaminocyclopropenium salts proved to be stable to fluoride, any kind of naked fluoride ionic liquid would be highly moisture sensitive.
A number of solvate salts of fluorides are known, for example the t-butanol solvate of tetrabutylammonium fluoride, TBAF(tBuOH)4, which is a fluoride solvate that has been investigated as an anhydrous source of fluoride (Kim et al., 2008). However, due to the symmetric shape of the ions and t-butanol, and the non-dispersed charges, it is a solid.
Larson (Larson and McMahon, 1983) carried out gas phase measurements of the binding energy of fluoride with a variety of Brönsted and Lewis acids. The binding energy of the ethanol to fluoride hydrogen bond was found to be 132 kJ mol−1, a strong hydrogen bond that is not much weaker than that measured for hydrogen fluoride to fluoride (bifluoride) at 161 kJ mol−1. Other measurements have suggested this was low and found a bond energy of 191 kJ mol−1 (Wenthold and Squires, 1995). The strong hydrogen bond suggests that the alcohol solvate can be thought of as a discrete species. Fluorohydrogenate ([F(HF)x]−) anions in ionic liquids are considered as a discrete species, and have received considerable attention in the literature due to high conductivities and low viscosities (Hagiwara et al., 1999, 2003).
In this paper, we report on TAC fluoride salts with inter- and intra-molecular alcohol solvate interactions. However, the solvate molecules do not need to be limited to alcohols, they just need to be good hydrogen bond donors. So, prompted by earlier work on dimeric and higher order oligomeric acetic acid anions (Johansson et al., 2008), acetic acid was also investigated as a solvate molecule with imidazolium fluoride salts. Both the [AcOHF]− and [(AcOH)2F]− species, shown below, have been observed previously by NMR at sub-ambient temperature (Golubev et al., 2004).
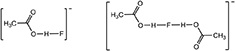
All operations were performed using standard Schlenk techniques with a dinitrogen atmosphere in order to reduce exposure to water. 1H-, 13C{1H}-NMR spectra were collected on a Varian Unity-300 operating at 300 and 75 MHz, respectively, or on a Varian INOVA-500 operating at 500 and 126 MHz, respectively, in CDCl3 or D2O, referenced to residual solvent peaks or TMS. 19F-NMR spectra were collected on a Varian Unity-300 operating at 282 MHz, in CDCl3 or D2O, referenced to C6H5F or CFCl3. Electrospray mass spectrometry was carried out on a Micromass LCT, with samples dissolved in acetonitrile. Water contents were determined by Karl Fischer titration using a Metrohm 831 KF coulometer. Chloride contents were determined using a AutolabEco Chemie, with associated GPES software, under a dinitrogen atmosphere. The electrodes were either a glassy carbon (3 mm diameter) or platinum (1 mm diameter) working electrode, a platinum wire counter electrode and a silver reference electrode. Fluoride, Ag+ and K+ were determined similarly using ISEs. Mircoanalysis was performed by Campbell Microanalytical Laboratory, Dunedin. [C3(NPr2)3]Cl (Curnow et al., 2011) and (Et2N)2C3O (Curnow et al., 2012) were prepared by previously-published methods. Other reagents were used as obtained commercially.
Differential Scanning Calorimetry (DSC) was carried out on a TA Q100 DSC and a Perkin-Elmer DSC 8000. The DSC 8000 was calibrated with indium (156.60°C) and cyclohexane (−87.0 and 6.5°C). Three heating and cooling cycles were carried out with data collected from a repeatable second cycle. A scan rate of 10°C min−1 was used, over a temperature range of at least −100 to 50°C, with a greater range used as required. Transition temperatures are reported as the onset temperature. Enthalpy for a transition was found by integrating the peak area, with entropy then calculated using ΔS = ΔH/T. Sample size was ~5–10 mg. Density measurements were carried out on an Anton Parr DMA 5000 instrument, an oscillating U-tube density meter, from 20 to 90°C in 10°C steps. All imidazolium fluoride acetic acid mixtures were measured on an Anton Parr AMVn automated microviscometer. The other samples were measured on a Brookfield DV-II+ Pro cone and plate viscometer. Samples that were measured using the cone and plate viscometer were examined for non-Newtonian behavior, although this was only over a limited shear rate range. All samples were measured from 20 (or above the melting point if solid) to 80 or 90°C, and performed sealed or under a dinitrogen gas flow. Conductivities were measured by AC impedance spectroscopy on a Solatron SI 1296 frequency response analyser, at ranges up to 0.01 Hz to 10 MHz. Measurements were carried out with a dip cell probe containing two platinum wires covered in glass. The resistance was identified using a Nyquist Plot, and conductivity then calculated using κ = l/AR, where l/A is the cell constant, which was determined using 0.01 mol L−1 KCl solution at 25°C. All samples were measured from 20 (or above the melting point if solid) to 80 or 90°C, and performed sealed or under a dinitrogen gas flow. Thermal Gravimetric Analysis (TGA) was carried out on a TA Q600 SDT (simultaneous DSC-TGA), using platinum pans. Temperature calibration carried out using zinc (419.6°C), and heat flow using a sapphire standard. Pans were cleaned after each TGA experiment by heating at 500°C under an air atmosphere, and if required cleaned with nitric acid. Sample size was ~10 mg.
Tris(diethanolamino)cyclopropenium chloride, [C3(N(C2H4OH)2)3]Cl (1)
Diethanolamine, HN(C2H4OH)2, (97.1 g, 0.925 mol) was mixed with CH2Cl2 (250 mL), and the suspension was cooled to 0°C. Pentachlorocyclopropane, C3Cl5H (24.7 g, 0.115 mol) was added dropwise and the solution stirred at ambient temperature overnight. The mixture was then heated to reflux for 24 h. Dichloromethane was then removed in vacuo. The mixture was dissolved into ethanol (150 mL) and molecular sieves (13X, 10 Å pore size) were added slowly until the solution was completely absorbed into the sieves. The crude product was extracted from the molecular sieves by soaking in ethanol (100 mL for 3 h followed by 100 mL for 19 h). This was repeated using methanol (2 × 100 mL), but with overnight soakings. Alcohol from the four fractions was removed in vacuo, yielding a pale yellow viscous liquid (24 g). All fractions were ~90% tris(diethanolamino)cyclopropenium chloride, with 10% [H2N(C2H4OH)2]Cl, as determined by NMR. The molecular sieves were washed with excess water to remove [H2N(C2H4OH)2]Cl, and the process was repeated to ultimately yield a pale yellow, extremely viscous liquid (7.0 g, 16%). 1H NMR (D2O, 300MHz): δ 3.81 (t, J = 5.3 Hz, 12H, NCH2), 3.61 ppm (t, J = 5.3 Hz, 12H, NCH2CH2). ES+ m/z 348.2158 (100%, M+).
Bis(diethylamino)diethanolaminocyclopropenium iodide, [C3(NEt2)2N(C2H4OH)2]I (2)
The cyclopropenone (Et2N)2C3O (7.68 g, 39.2 mmol) was dried using ethanol azeotropes (5 × 50 mL). Iodoethane (97.5 g, 625 mmol) was added and the solution heated to reflux for 18 h. After cooling to ambient temperature, HN(C2H4OH)2 (8.44 g, 80.3 mmol) was added with stirring for 48 h. Excess iodoethane was removed in vacuo and water (100 mL) added. Cyclopropenone was extracted with dichloromethane (100 mL and 50 mL). The product was then back-extracted with water from this dichloromethane solution (2 × 100 mL) and recombined with the first water layer. The final product was then extracted with CH2Cl2 (3 × 100 mL) to yield a yellow liquid (3.50 g, 22%). 1H NMR (300 MHz, D2O): 3.73 (t, J = 5.3 Hz, 4H, NCH2CH2OH), 3.47 (t, J = 5.3 Hz, 4H, NCH2CH2OH), 3.37 (q, J = 7.0 Hz, 8H, NCH2CH3), 1.17 (t, J = 7.2 Hz, 12H, NCH2CH3). 13C NMR (75 MHz, D2O, methanol reference): 117.89 (C1), 117.26 (C2), 59.46 (NCH2CH2OH), 54.38 (NCH2CH2OH), 47.20 (NCH2CH3), 13.98 (NCH2CH3).
Tris(diethanolamino)cyclopropenium fluoride, [C3(N(C2H4OH)2)3]F (3) via HF
Tris(diethanolamino)cyclopropenium chloride (1) (7.0 g, 19 mmol) was dissolved in water (250 mL) and Ag2O (7.0 g, 30 mmol) was added and stirred for 3 h in a blacked out flask. Water was partially (200 mL) removed in vacuo. AgCl was removed by filtration through Celite before the solution was neutralized to pH 7.5 with aqueous HF. Complete drying was achieved with additions (3 × 150 mL, 3 × 50 mL) of ethanol and isopropanol and removal of the solvent in vacuo. This was followed by extended drying under high vacuum with stirring and mild heat (40°C) to give a colorless solid (5.63 g, 84%). 1H NMR as for [C3(N(C2H4OH)2)3]Cl. 19F NMR (282 MHz, D2O, C6H5F): 122.05 (s). 13C NMR (75 MHz, D2O, acetonitrile reference): 118.18 (C3), 59.38 (NCH2CH2OH), 54.29 (NCH2CH2OH).
Tris(diethanolamino)cyclopropenium fluoride, [C3(N(C2H4OH)2)3]F (3) via AgF
Compound 1, [C3(C2H4OH)3]Cl (3.0 g, 7.8 mmol) was dissolved in water (200 mL) and AgF (1.30 g, 10.2 mmol) added with stirring. AgCl was removed by filtration through Celite. Excess AgF was removed by removal of solvent in vacuo, followed by the addition of alcohol solvent (50 mL, 5 × ethanol then 5 × isopropanol), after each addition the solution was filtered through Celite and the solvent removed in vacuo. This yielded a light yellow, extremely viscous liquid (2.3 g, 80%). Found: C, 48.82; H, 8.09; N, 11.06%. Calc. for C15H30N3O6F: C, 49.04; H, 8.23; N, 11.44%. Cl− content: 1,600 ppm.
Bis(diethylamino)diethanolaminocyclopropenium fluoride diethanol, [C3(NEt2)2N(C2H4OH)2]F (4)
Compound 2, [C3(NEt2)2N(C2H4OH)2]I (3.50 g, 8.52 mmol) was stirred in water (250 mL) in a blacked-out flask to which Ag2O (3.0 g, 13 mmol) was added with stirring for 3 h. AgI was removed by filtration through Celite before the solution was neutralized to pH 7.5 with aqueous HF. The solution was reduced in volume to 20 mL in vacuo, with drying being completed with ethanol azeotrope drying (3 × 200 mL). This yielded the product as a viscous yellow liquid (2.5 g, 96%). 19F NMR (282 MHz, D2O, C6H5F reference): 121.94 (s).
Tris(dipropylamino)cyclopropenium fluoride diethanol, [C3(NPr2)3]F.1.9EtOH (5.1.9EtOH)
Tris(dipropylamino)cyclopropenium chloride, [C3(NPr2)3]Cl (10.6 g, 28.5 mmol) was stirred in water (450 mL), in a blacked-out flask, to which Ag2O (6.70 g, 28.9 mmol) was added and then stirred for 3 h. AgCl was removed by filtering through Celite, before the solution was neutralized to pH 7.1 with aqueous HF. The solution was reduced in volume to 50 mL in vacuo, with drying being completed with ethanol azeotrope drying (3 × 500 mL), which yielded the product as an orange liquid (12.4 g, 97%). 1H NMR (300 MHz, CDCl3): 3.68 (q, J = 7.0 Hz, 3.81H, CH3CH2OH), 3.27 (t, J = 7.9 Hz, 12H, NCH2CH2CH3), 1.67 (m, 12H, NCH2CH2CH3), 1.21 (t, J = 7.0 Hz, 5.00H, CH3CH2OH), 0.94 (t, J = 7.3 Hz, 18H, NCH2CH2CH3). 19F NMR (282 MHz, D2O, C6H5F): 122.03 (s). Found: C, 61.95; H, 12.48; N, 8.77%. Calc. for C21H42N3F.1.9EtOH.2.09H2O: C, 61.95; H, 12.07; N, 8.74%. H2O content: 2,200 ppm. F− content: 44,300 ppm, calculated: 42,900 ppm.
1-Ethyl-3-methylimidazolium fluoride diethanol, [EMIM]F.1.8EtOH (6.1.8EtOH)
To [EMIM]Cl (33.0 g, 225 mmol) in ethanol (500 mL) was added KF (30 g, 520 mmol) suspended in ethanol (250 mL). After stirring for 1 h, the solid was removed by filtration. Two further additions of KF (35 and 25 g), with stirring (1 h) and removal, were made. The solvent was reduced in volume in vacuo, and acetonitrile (250 mL) was added and the solution cooled. A precipitate formed, and was filtered off. Ethanol was added to the filtrate, and excess solvent removed in vacuo. Cl− content: 3,120 ppm. To the [EMIM]F ethanol solvate (52 g) in water (200 mL) was added AgF (550 mg, 4.34 mmol) in water (100 mL). AgCl precipitate was removed by filtration. The solvent volume was reduced in vacuo with drying completed using ethanol azeotropes. The product was a pale yellow liquid (41.9 g, 87%) with 1.83 ethanol solvate molecules per fluoride. 1H NMR (400 MHz, D2O): 8.63 (s, 1H, CH), 7.40 (d, J = 2.0 Hz, 1H, CH), 7.33 (d, J = 2.0 Hz, 1H, CH), 4.14 (q, J = 7.5 Hz, 2H, CH2CH3), 3.80 (s, 3H, CH3), 3.56 (q, J = 7.3 Hz, 3.6H, CH3CH2OH), 1.41 (t, J = 7.5 Hz, 3H, CH2CH3), 1.09 (t, J = 7.3 Hz, 5.4H, CH3CH2OH). 19F NMR (377 MHz, D2O, CFCl3): −121.22 (s). Cl− content: 69 ppm. K+ content: < 100 ppm. Ag+ content: 821 ppm.
1-Ethyl-3-methylimidazolium fluoride acetic acid, [EMIM]F.1.0AcOH (6.1.0AcOH)
Acetic acid (1.77 g, 29.4 mmol) was added to [EMIM]F.1.8EtOH (6.31 g, 29.4 mmol). The mixture was dried with isopropanol (2 × 50 mL) and ethanol (50 mL) azeotropes. Additional acetic acid (0.111 g, 1.85 mmol) was then added. The product was a pale yellow liquid (5.04 g, 90%). 1H NMR (400 MHz, D2O): 8.63 (s, 1H, CH), 7.41 (s, 1H, CH), 7.35 (s, 1H, CH), 4.15 (q, J = 7.3 Hz, 2H, CH2CH3), 3.82 (s, 3H, CH3), 1.94 (s, 3H, CH3CO2HF), 1.43 (t, J = 7.3 Hz, 3H, CH2CH3). 19F NMR (377 MHz, D2O): −128.87 (s, CH3CO2HF), −142.77 (m, F2H−), 27.1:1 ratio. H2O content: 1330 ppm.
1-Ethyl-3-methylimidazolium fluoride acetic acid, [EMIM]F.1.6AcOH (6.1.6AcOH)
Acetic acid (3.00 g, 50.0 mmol) was added to [EMIM]F.1.8EtOH (6.25 g, 29.2 mmol). The mixture was dried with an ethanol (50 mL) azeotrope, followed by drying for 5 h in vacuo. The product was a pale yellow liquid (6.50 g, 99%). 1H NMR (400 MHz, D2O): 8.63 (s, 1H, CH), 7.41 (s, 1H, CH), 7.35 (s, 1H, CH), 4.15 (q, J = 7.5 Hz, 2H, CH2CH3), 3.82 (s, 3H, CH3), 1.95 (s, 4.8H, CH3CO2HF), 1.42 (t, 3JHH = 7.5 Hz, 3H, CH2CH3). 19F NMR (377 MHz, D2O): −128.88 (s, CH3CO2HF), −142.49 (m, F2H−), 24.0:1 ratio. H2O content: 1240 ppm.
1-Ethyl-3-methylimidazolium fluoride acetic acid, [EMIM]F.2.1AcOH (6.2.1AcOH)
Acetic acid (3.58 g, 59.6 mmol) was added to [EMIM]F.1.8EtOH (6.39 g, 29.8 mmol). The mixture was dried with ethanol (3 × 50 mL) azeotropes. Additional acetic acid (0.535 g, 8.91 mmol) was then added. The product was a pale yellow liquid (7.28 g, 96%). 1H NMR (400 MHz, D2O): 8.62 (s, 1H, CH), 7.40 (s, 1H, CH), 7.33 (s, 1H, CH), 4.14 (q, J = 7.3 Hz, 2H, CH2CH3), 3.80 (s, 3H, CH3), 1.95 (s, 6.3H, CH3CO2HF), 1.41 (t, J = 7.3 Hz, 3H, CH2CH3). 19F NMR (377 MHz, D2O): −128.81 (s, CH3CO2HF), −142.48 (m, F2H−), 28.4:1 ratio. H2O content: 1,030 ppm.
1-Ethyl-3-methylimidazolium fluoride acetic acid, [EMIM]F.2.4AcOH (6.2.4AcOH)
Acetic acid (4.09 g, 68.1 mmol) was added to [EMIM]F.1.8EtOH (5.83 g, 27.2 mmol). The mixture was dried with ethanol (2 × 50 mL) azeotropes. Additional acetic acid (0.495 g, 8.24 mmol) was then added. The product was a pale yellow liquid (7.48 g, 99%). 1H NMR (400 MHz, D2O): 8.60 (s, 1H, CH), 7.39 (s, 1H, CH), 7.32 (s, 1H, CH), 4.13 (q, 3JHH = 7.3 Hz, 2H, CH2CH3), 3.79 (s, 3H, CH3), 1.95 (s, 7.2H, CH3CO2HF), 1.39 (t, 3JHH = 7.3 Hz, 3H, CH2CH3). 19F NMR (377 MHz, D2O): −128.75 (s, CH3CO2HF), −142.46 (m, F2H−), 16.1:1 ratio. H2O content: 1,580 ppm.
1-Ethyl-3-methylimidazolium fluoride acetic acid, [EMIM]F.3.2AcOH (6.3.2AcOH)
Acetic acid (3.76 g, 62.6 mmol) was added to [EMIM]F.1.8EtOH (4.47 g, 20.8 mmol). The mixture was dried with ethanol (2 × 50 mL) azeotropes. Additional acetic acid (0.825 g, 13.7 mmol) was then added. The product was a pale yellow liquid (5.20 g, 77%). 1H NMR (400 MHz, D2O): 8.62 (s, 1H, CH), 7.40 (s, 1H, CH), 7.34 (s, 1H, CH), 4.14 (q, 3JHH = 7.3 Hz, 2H, CH2CH3), 3.81 (s, 3H, CH3), 1.98 (s, 9.6H, CH3CO2HF), 1.42 (t, 3JHH = 7.3 Hz, 3H, CH2CH3). 19F NMR (377 MHz, D2O): −128.80 (s, CH3CO2HF), −142.57 (m, F2H−), 20.4:1 ratio. H2O content: 900 ppm.
(2-hydroxyethyl)trimethylammonium fluoride hydrate, choline fluoride hydrate (7.H2O)
Choline chloride (10.2 g, 73.2 mmol) was dissolved into water (200 mL) in a blacked out flask. Ag2O (12.7 g, 54.8 mmol) was added and the solution stirred for 4 h. AgCl was removed by filtration through Celite and the filtrate was neutralized to pH 7.5 with aqueous HF. Water was removed by drying in vacuo at 40°C for 72 h to yield a white solid (9.89 g, 91%). Found: C, 40.34; H, 11.24; N, 9.03%. Calc. for C5H14NOF.1.43H2O: C, 40.34; H, 11.41; N, 9.41%.
In this work, three classes of cations were studied for the preparation of fluoride ionic liquids: the tris(dialkylamino)cyclopropenium cation [C3(NPr2)3]+, the imidazolium cation [EMIM]+, and hydroxyl-functionalized salts. Two hydroxy-functionalized TAC salts were prepared: Treatment of C3Cl5H with HN(CH2CH2OH)2 yielded the hexaol TAC product [C3(N(CH2CH2OH)2)3]Cl (1) whereas a diol TAC salt was prepared by treatment of the alkoxydiamino TAC salt [C3(NEt2)2OEt]I (generated by addition of alkylation of the cyclopropenone (Et2N)2C3O with ethyliodide) with HN(CH2CH2OH)2 to give [C3(NEt2)2N(CH2CH2OH)2]I (2) (Scheme 1). Separation of the hexaol 1 from the excess starting amine and product ammonium salt proved challenging but was eventually achieved by trapping the smaller amine/ammonium salt in Zeolite 13X, which has a 10 Å pore size.
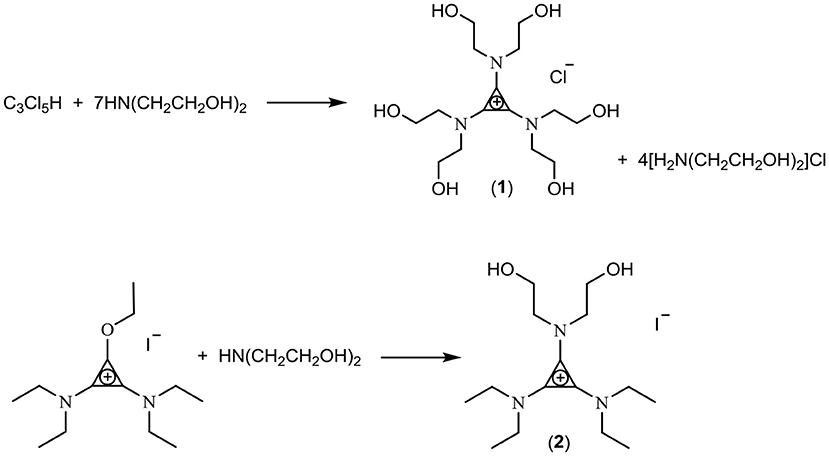
Scheme 1. Synthesis of hydroxyl-functionalized TAC salts.
Three methods were investigated for the conversion of chloride salts to fluoride salts: anion metathesis with AgF, neutralization of hydroxide with HF acid, and metathesis with KF in ethanol.
Anion metathesis with AgF from the chloride salt is the most direct route (Maiti et al., 2008; Vitz et al., 2009): addition of AgF to an aqueous solution of a chloride IL precipitates AgCl. This method was used for one synthesis of [C3(N(C2H4OH)2)3]F (3) and resulted in a final chloride content of 1,600 ppm, ~3% of total anions.
A common and readily-accessible source of fluoride is HF. Since HF is a weak acid, direct metathesis with chloride is difficult, however, the chloride salt can be converted first to a hydroxide salt, which is then neutralized with HF. It should also be noted that TAC ILs are sensitive to concentrated or heated hydroxide solutions with the conversion of TAC cations to cyclopropenones. There were two methods trialed for the conversion of chloride to hydroxide anions: firstly, one using silver oxide and the other using sodium isopropoxide.
The exchange of chloride to hydroxide using Ag2O (Scheme 2) involves mixing Ag2O with an aqueous solution of the chloride salt in a blacked-out container. Using this method, chloride contents of 500 ppm were obtained. Solutions of 3, [C3(NEt2)2N(C2H4OH)2]F (4) and [C3(NPr2)3]F (5) were synthesized using this method.
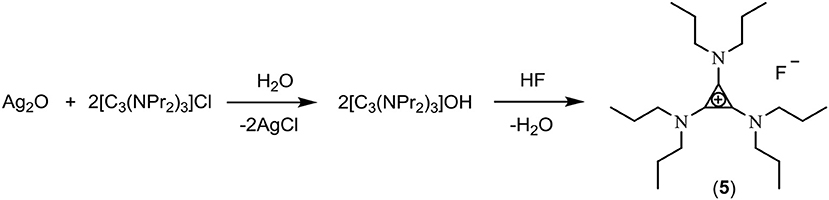
Scheme 2. Exchange using silver oxide.
The exchange of chloride to hydroxide via isopropoxide (Scheme 3) involves mixing isopropanol solutions of sodium isopropoxide and the chloride salt, which precipitates NaCl, leaving the cations in solution with isopropoxide. When this isopropoxide salt is added to water, hydroxide anions are formed and neutralization with aqueous HF then generates the fluoride salt. In the case of 5, the chloride content was 8%, which would significantly affect physical properties.
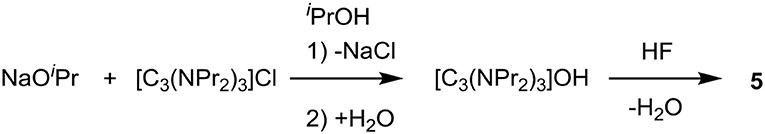
Scheme 3. Exchange using sodium isopropoxide.
The final method used for the exchange of chloride to fluoride was the use of KF in ethanol (Scheme 4; Dermeik and Sasson, 1989). This method is similar to that of the sodium isopropoxide exchange of chloride to hydroxide. It involves partially dissolving potassium fluoride in ethanol, followed by the addition of the chloride salt. The solubility ratio of potassium fluoride to potassium chloride is 25, therefore, due to the lower solubility of potassium chloride, this precipitates out. The ethanol was then removed in vacuo, and acetonitrile was added. After cooling, a precipitate of potassium fluoride (and possibly residual chloride) was removed. Ethanol was added and acetonitrile removed in vacuo to yield the ethanol solvate. This method was used to synthesize the ethanol solvate of [EMIM]F (6), and after carrying out this exchange three times, the chloride content was 3,100 ppm, ~2.4% of total anions, and the potassium ion concentration was 220 ppm, ~0.15% of [EMIM]+. Due to the chloride concentration being higher than preferred, a further step of adding silver fluoride was carried out. The amount of silver fluoride that was added was calculated to lower chloride to < 100 ppm, without having excess silver present. The final chloride concentration was 70 ppm.
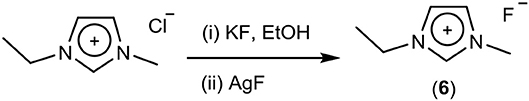
Scheme 4. Synthesis of fluoride salts using KF in ethanol.
Once a solution of fluoride ionic liquid is obtained, the solvent needs to be removed, and this is a critical and delicate step. As the fluoride anion becomes less solvated, its basicity increases, which leads to decomposition. We will now discuss a variety of drying techniques that were trialed.
An aqueous solution of [C3(NPr2)3]F was dried on a rotary evaporator at 40°C. This reduced the water content to ~20%, about four water molecules per TAC fluoride. When the temperature or vacuum is increased more water is removed, however, due to the heat and the increasing basicity of the fluoride, the salt decomposed to bis(dipropylamino)cyclopropenone and [C3(NPr2)3]HF2 (Scheme 5). Freeze drying was also attempted, the water content dropped to 1.8% but there was 25% decomposition to cyclopropenone.
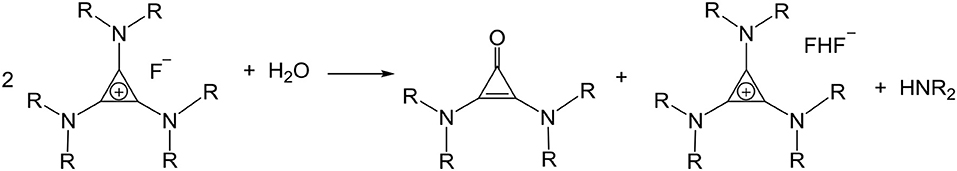
Scheme 5. Decomposition of [C3(NPr2)3]F.
Heterogeneous azeotrope drying using toluene has been reported previously (Zhao et al., 2008), however, with the fluoride salts this still leads to the TAC fluoride salt being concentrated in a wet phase, and resulted in 30% cyclopropenone. Therefore, homogeneous azeotropes were investigated whereby water is removed before the ionic liquid is concentrated. Both ethanol and isopropanol will form lower boiling homogeneous azeotropes with water, and both were used successfully. Due to the potential of the ionic salt to break the azeotrope, a large excess of ethanol or isopropanol is used. Isopropanol has the advantage of having a higher percentage of water in the azeotrope, so water can be removed with less additional solvent.
Azeotrope drying was used to synthesize 3, 4.1.9EtOH, 5.1.9EtOH, and 6.1.8EtOH. During the synthesis of 5.1.9EtOH, repeated additions of dry ethanol produced an ionic liquid fluoride with a water content of 2,200 ppm without cyclopropenone forming. The ethanol solvate could not easily be removed, because the strong hydrogen bonds between the alcohol solvate and fluoride anion provide stability to the IL fluoride.
Using another azeotrope, between benzene and ethanol, further ethanol can be removed from 5.1.9EtOH to form a monoethanol solvate: one addition of dried benzene to a sample of the diethanol solvate leaves 1.36 ethanol molecules per fluoride. After two more additions, this drops to 1.1 ethanol molecule per fluoride. However, significant amounts (17%) of bis(dipropylamino)cyclopropenone were detected in this product.
A range of acetic acid solvates of an imidazolium fluoride were synthesized with ~1, 1.5, 2, 2.5, and 3 equivalents of acetic acid per fluoride anion. The acetic acid solvates were synthesized from the ethanol solvate 6.1.8EtOH. Acetic acid was added and the mixture dried using ethanol or isopropanol azeotropes under reduced pressure with only small amounts of acetic acid being lost. Extra acetic acid was then added to give the desired HOAc/F− ratio. 1H-NMR was used to determine the precise ratios of acetic acid to [EMIM]+, which were found to be 1.0, 1.6, 2.1, 2.4, and 3.2.
The 13C and 19F NMR spectra also provide evidence for the formation of fluoride hydrogen-bonded anions. The 19F NMR of 5.1.9EtOH in D2O shows a peak at −122.03 ppm and 6.1.8EtOH at −121.22 ppm, which is the shift for sodium fluoride in D2O. However, for the various acetic acid mixtures, the 19F NMR peak is around −128.8 ppm. This compares to the shift for [FHF]−, at −142 ppm, hence suggesting that [AcOHF]− is persisting, or at least a significant contribution, in the D2O solution. A similar observation is made by examining the chemical shift of the methyl protons of the acetic acid. In D2O, acetic acid is observed at 2.08 ppm and the acetate ion at 1.90 ppm (Gottlieb et al., 1997), whereas for the fluoride acetic acid mixtures the chemical shifts are between 1.94 and 1.98 ppm. A small signal for [FHF]− was also observed due to some dissociation of the fluoride solvate with the acetic acid then generating some H+, which can hydrogen bond to two fluorides.
The viscosities of 5.1.9EtOH and the [EMIM]F-acetic acid mixtures were measured over the temperature range 20–50°C (Table 1). Plots of viscosity and conductivity vs. temperature are provided in the Supplementary Information. The viscosity of 5.1.9EtOH is lower than with DCA or anions (Walst et al., 2015) whereas the [EMIM]F acetic acid solvates are similar to the [EMIM]DCA salt. This is due to the acetic acid or ethanol being a co-solvent with much lower viscosity. While it is not possible to directly compare the viscosities of the acetic acid and ethanol solvates, due to the different cations and different number of solvate molecules, it is seems likely that the viscosity of the ethanol solvates is lower than the acetic acid solvates if the cation and number of solvate molecules is the same. Note that the viscosity of [C3(NPr2)3]NTf2 (Table 1) is approximately five times greater than that of [EMIM]NTf2 (Walst et al., 2015), so an increase by a factor of about 2.2 from the EMIM acetic acid solvates to 5.EtOH is small.
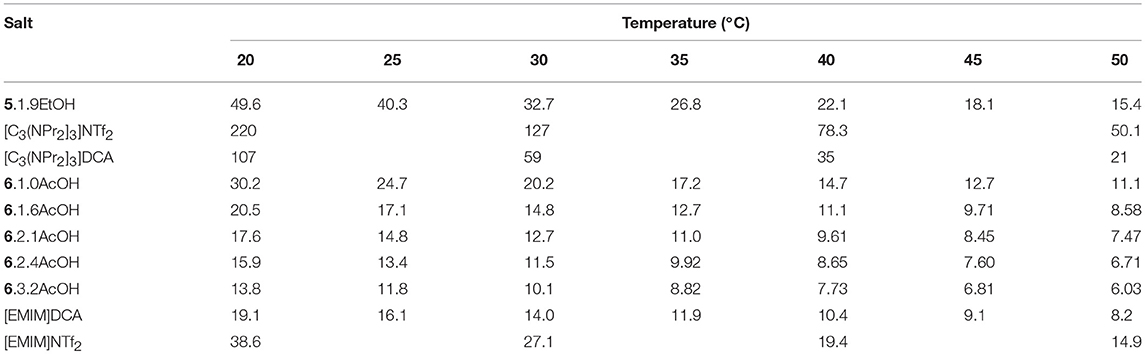
Table 1. Viscosity data for fluoride solvates (mPa s).
The temperature dependence (Table 2) was modeled with the VFT and Arrhenius equations (see Supplementary Information). D values (B/T0) of between 12.1 and 14.3 were found for the acetic acid solvates and 10.1 for 5.1.9EtOH, suggesting that these solvates are slightly less fragile than other ionic liquids with the same cations. The activation energies are between 22 and 31 kJ mol−1.
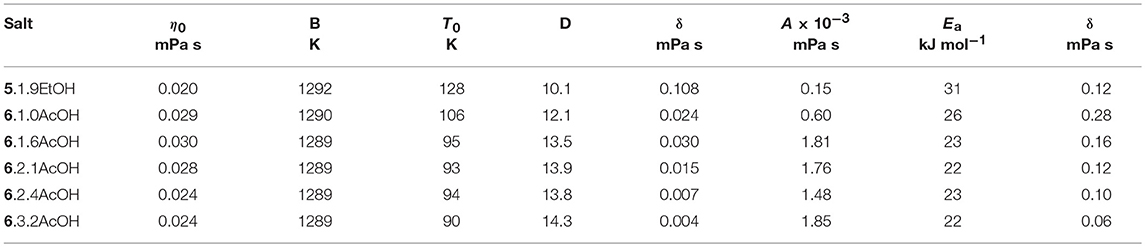
Table 2. Fitting parameters for temperature dependence of viscosity.
Addition of acetic acid decreases the viscosity, for example, from 30.2 with 1.0AcOH to 13.8 mPa s with 3.2AcOH at 20°C. This is due to the acetic acid being a lower viscosity liquid and indicates the presence of increasing amounts of free HOAc.
The conductivities were measured over a temperature range of 20–40°C (Table 3). The conductivity of 5.1.9EtOH is higher than the corresponding salt whereas the [EMIM]F acetic acid solvates have lower conductivity than the [EMIM]DCA ionic liquid. This is a reflection of the trends seen with the viscosity measurements. As with viscosity, it is not possible to directly compare their viscosities of the acetic acid and ethanol solvates, due to the different cations and different number of solvate molecules, it is probable that the conductivity of ethanol solvates is higher than the acetic acid solvates if the cation and number of solvate molecules is the same. The conductivity of the ethanol solvate is lower due to the large [C3(NPr2)3]+ cation, which causes higher viscosity and lowers the number of charge carriers in a given volume.
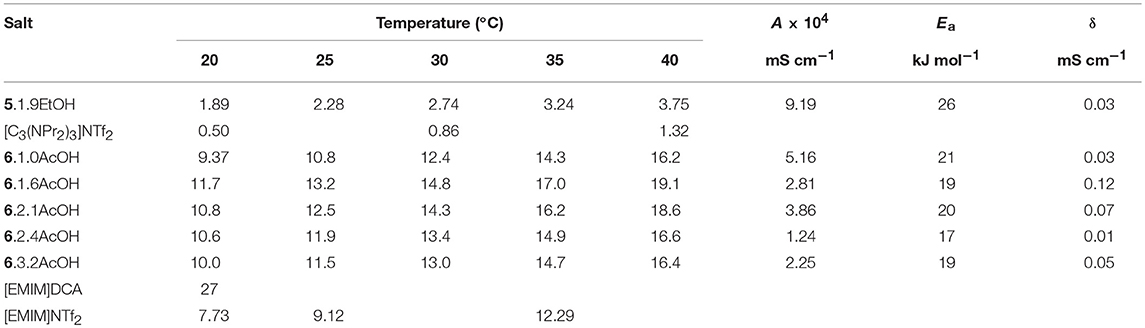
Table 3. Conductivity data and fitting parameters for fluoride solvates (mS cm−1).
The temperature dependence of the conductivity (Table 3) was modeled with the Arrhenius equation (see Supplementary Information). The activation energies were between 17 and 26 kJ mol−1. Due to the very limited temperature range and limited number of data points, the fitting to the VFT equation did not produce sensible results, so they are not reported here.
As the amount of acetic acid (Figure 1) increases, the conductivity increases from 9.4 mS cm−1 for [C2MIM]F.1.0AcOH to 11.7 mS cm−1 for [C2MIM]F.1.6AcOH, it then decreases to 10.0 mS cm−1 for [C2MIM]F.3.2AcOH as further acetic acid is added. Initially, the molar conductivity increases, consistent with decreasing viscosity, but further acetic acid decreases the concentration of ions.
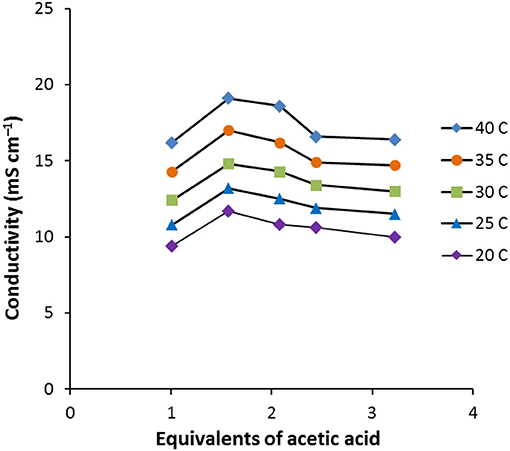
Figure 1. Conductivity vs. equivalents of acetic acid for 6.nAcOH (n = 1.0, 1.6, 2.1, 2.4, 3.2).
The densities for the [EMIM]F acetic acid mixtures were measured from 20 to 50°C (Table 4). As expected, density decreases as the relative concentration of acetic acid increases due to the lower density of acetic acid, and the density is linearly dependent on temperature (Table 5).
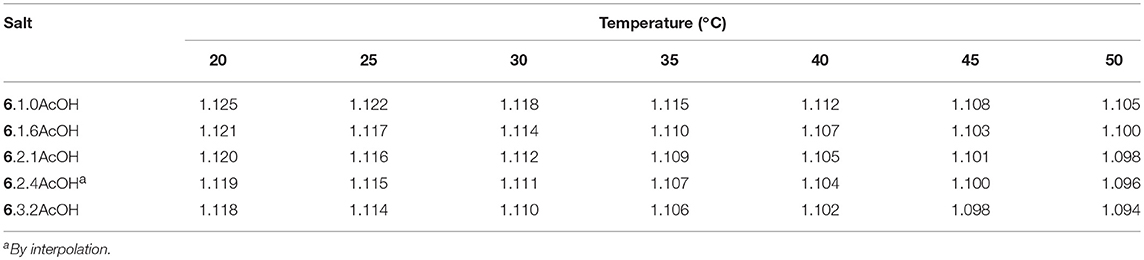
Table 4. Density data for imidazolium fluoride solvates (g mL−1).
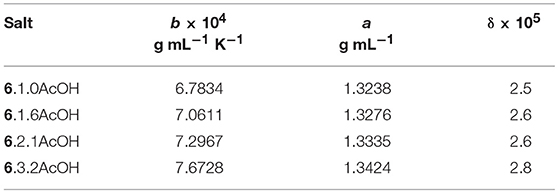
Table 5. Fitting parameters for temperature dependence of density, ρ = a – bT.
The ionicity of the fluoride solvates was assessed using a Walden plot (Figure 2; Xu et al., 2003). To complete the set of calculations, the density of 5.1.9EtOH was approximated by that of [C3(NPr2)3]DCA. The Walden product (= σ.η) and the deviation from ideal ionic behavior in the Walden plot, ΔW, are given in Table 6. The Walden products for the fluoride solvates are between 0.40 and 0.48 S cm2 P mol−1 and deviations from the ideal ionic behavior are between 0.32 and 0.40. Hence, all of the fluoride solvate ionic liquids can be classified as good ionic liquids in terms of the ionicity (Xu et al., 2003), with some degree of ion association or correlations evident suggesting some residual specific interaction between the cation and the anion solvate. There is a slight increase in ΔW as the amount of acetic acid increases. As a comparison, 6.2.3HF has a higher Walden product of 0.76 and a lower ΔW of 0.12 (Hagiwara et al., 2003); in that case, there is the potential for a contribution from proton hopping, which increases molar conductivity.
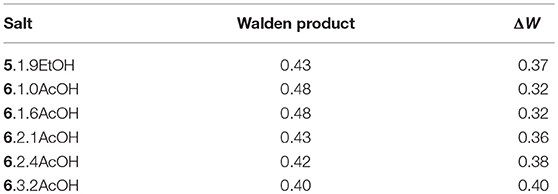
Table 6. Walden product and deviation from ideal at 20°C.
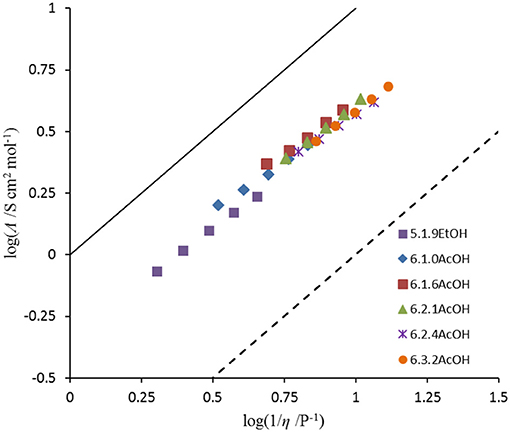
Figure 2. Walden plot of fluoride solvates.
Thermally stable ionic liquids would be desirable as they could be used in applications at elevated temperatures, however, the reactivity of fluoride and the volatility of acetic acid and ethanol are expected to hinder stability. The TGA of the TAC fluoride ethanol solvate 5.1.9EtOH (Figure 3) shows steady weight loss from ~50°C. Initially, this is likely to be loss of ethanol, which will lead to the decomposition of the cation.
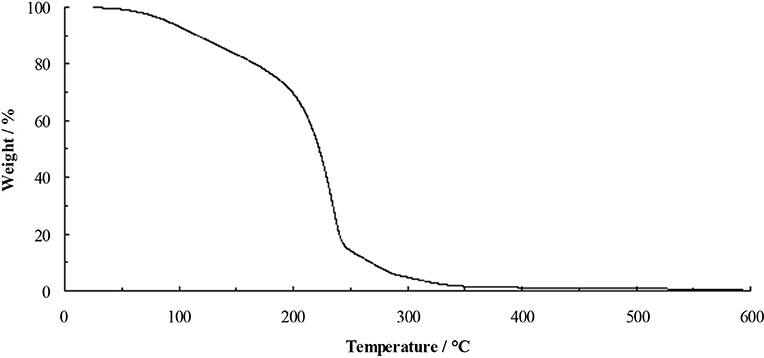
Figure 3. TGA of 5.1.9EtOH at 10°C min−1.
The TGA of the dihydroxyl derivative 4 (Figure 4) shows a profile similar to that of [C3(N(C2H4OH)2)3]F, with little weight loss before onset of decomposition at 175°C. This clearly indicates that hydroxyl functional groups do stabilize the fluoride anion. Salt 4 is a highly-viscous yellow liquid at ambient temperature, however, it is very sensitive to trace water and much care needs to be taken when drying this material.
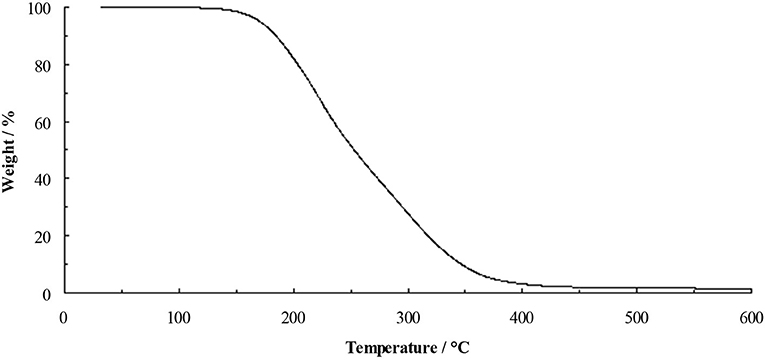
Figure 4. TGA of [C3(NEt2)2N(C2H4OH)2]F (4) at 10°C min−1.
The TGA of the hexahydroxyl derivative 3 shows an increase in stability compared to 4 with onset of decomposition at 191°C (Supplementary Information). The onset temperature is, as expected, much lower than other ionic liquids with non-coordinating anions such as , which can exceed 400°C. Potentially useful is the observation of the melting point at 87°C, which indicates a stable ionic liquid range at elevated temperatures. It is notably less sensitive to moisture during the drying process compared to 4. A sample of (2-hydroxyethyl)trimethylammonium (choline) fluoride hydrate (7.H2O) was prepared from the chloride salt using silver oxide and aqueous HF. The TGA shows good thermal stability compared to the ethanol solvate of 5, with an onset temperature of 139°C (Supplementary Information). However, this salt is a solid and no melting point is observed prior to decomposition.
The thermal behavior of 6.1.6AcOH and 6.3.2AcOH were examined using DSC: glass transitions were observed at −92 and −95°C, respectively, however, no melting points were observed.
Ionic liquid fluorides were synthesized by exchange of chloride to fluoride using one of three methods: anion metathesis with silver fluoride, anion metathesis using aqueous silver oxide followed by neutralization with HF acid or anion metathesis with potassium fluoride in ethanol. The fluoride salts were then isolated using homogeneous azetrope drying.
Whereas, naked fluoride salts are not stable in pure form, fluoride solvates with ethanol and acetic acid were found to be much more stable, probably due to strong hydrogen bond formation. Increasing amounts of acetic acid solvate decreases the viscosity and density, while the conductivity peaked at 6.1.6AcOH.
The low decomposition temperature of 5.1.9EtOH (starting at 50°C) led to the rational design of ionic liquids containing hydroxyl-functionalized cations; 3 and 4. These liquids demonstrate improved thermal stability with higher decomposition temperatures of 175 and 191°C, respectively, consistent with stabilization by intramolecular hydroxyl-fluoride bonding, leading to ionic liquids that are stable as the unsolvated ionic liquid fluorides.
OC conceived of the project and directed the TAC work. DM conceived of the acetic acid solvate concept and directed this work. KW carried out the experimental work. All authors made critical contributions to the report, gave significant input into the report writing process and analysis of data herein.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Some of this work appears in the Ph.D. thesis of KW (Walst, 2012). DM is grateful for the support of his Australian Laureate Fellowship from the Australian Research Council.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00603/full#supplementary-material
Butchard, J. R., Curnow, O. J., Garrett, D. J., and Maclagan, R. G. (2006). Structure of a discrete dichloride hexahydrate cube as a tris(diisopropylamino)cyclopropenium salt. Angew. Chem. Int. Ed. 45, 7550–7553. doi: 10.1002/anie.200603547
Christie, K. O., and Jenkins, H. D. B. (2003). Quantitative measure for the “nakedness” of fluoride ion sources. J. Am. Chem. Soc. 125, 9457–9461. doi: 10.1021/ja035675r
Curnow, O. J., Holmes, M. T., Ratten, L. C., Walst, K. J., and Yunis, R. (2012). A facile route to functionalised, protic and chiral ionic liquids based on the triaminocyclopropenium cation. RSC Adv. 2, 10794–10797. doi: 10.1039/c2ra22078d
Curnow, O. J., MacFarlane, D. R., and Walst, K. J. (2011). Triaminocyclopropenium salts as ionic liquids. Chem. Commun. 47, 10248–10250. doi: 10.1039/c1cc13979g
Dermeik, S., and Sasson, Y. (1989). Synthesis of quaternary ammonium fluoride salts by a solid-liquid halogen exchange process in protic solvents. J. Org. Chem. 54, 4827–4829. doi: 10.1021/jo00281a024
Golubev, N. S., Tolstoy, P. M., Smirnov, S. N., Denisov, G. S., and Limbach, H.-H. (2004). Low-temperature NMR spectra of fluoride–acetic acid hydrogen-bonded complexes in aprotic polar environment. J. Mol. Struct. 700, 3–12. doi: 10.1016/j.molstruc.2004.01.024
Gottlieb, H. E., Kotlyar, V., and Nudelman, A. (1997). NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515. doi: 10.1021/jo971176v
Hagiwara, R., Hirashige, T., Tsuda, T., and Ito, Y. (1999). Acidic 1-ethyl-3-methylimidazolium fluoride: a new room temperature ionic liquid. J. Fluorine Chem. 99, 1–3. doi: 10.1016/S0022-1139(99)00111-6
Hagiwara, R., Matsumoto, K., Nakamori, Y., Tsuda, T., Ito, Y., Matsumoto, H., et al. (2003). Physicochemical properties of 1,3-Dialkylimidazolium fluorohydrogenate room-temperature molten salts. J. Electrochem. Soc. 150, D195–D199. doi: 10.1149/1.1621414
Johansson, K. M., Izgorodin, E. I., Forsyth, M., MacFarlane, D. R., and Seddon, K. R. (2008). Protic ionic liquids based on the dimeric and oligomeric anions: [(AcO)xHx−1]−. Phys. Chem. Chem. Phys. 10, 2972–2978. doi: 10.1039/b801405a
Kim, D. W., Jeong, H.-J., Lim, S. T., and Sohn, M.-H. (2008). Tetrabutylammonium tetra(tert-butyl alcohol)-coordinated fluoride as a facile fluoride source. Angew. Chem. Int. Ed. 47, 8404–8406. doi: 10.1002/anie.200803150
Larson, J. W., and McMahon, T. B. (1983). Strong hydrogen bonding in gas-phase anions. An ion cyclotron resonance determination of fluoride binding energetics to Broensted acids from gas-phase fluoride exchange equilibrium measurements. J. Am. Chem. Soc. 105, 2944–2950. doi: 10.1021/ja00348a003
Maiti, A., Pagoria, P. F., Gash, A. E., Han, T. Y., Orme, C. A., Gee, R. H., et al. (2008). Solvent screening for a hard-to-dissolve molecular crystal. Phys. Chem. Chem. Phys. 10, 5050–5056. doi: 10.1039/B805169K
Mallik, B. S., and Siepmann, J. I. (2010). Thermodynamic, structural and transport properties of tetramethyl ammonium fluoride: first principles molecular dynamics simulations of an unusual ionic liquid. J. Phys. Chem. B 114, 12577–12584. doi: 10.1021/jp104261h
Sun, H., and DiMagno, S. G. (2005). Anhydrous tetrabutylammonium fluoride. J. Am. Chem. Soc. 127, 2050–2051. doi: 10.1021/ja0440497
Swatloski, R. P., Holbrey, J. D., and Rogers, R. D. (2003). Ionic liquids are not always green: hydrolysis of 1-butyl-3-methylimidazolium hexafluorophosphate. Green Chem. 5, 361–363. doi: 10.1039/b304400a
Turner, E. A., Pye, C. C., and Singer, R. D. (2003). Use of ab initio calculations toward the rational design of room temperature ionic liquids. J. Phys. Chem. A 107, 2277–2288. doi: 10.1021/jp021694w
Vitz, J., Erdmenger, T., Haensch, C., and Schubert, U. S. (2009). Extended dissolution studies of cellulose in imidazolium based ionic liquids. Green Chem. 11, 417–424. doi: 10.1039/b818061j
Wallace, A. J., Jayasinghe, C. D., Polson, M. I. J., Curnow, O. J., and Crittenden, D. L. (2015). Cyclopropenium cations break the rules of attraction to form closely bound dimers. J. Am. Chem. Soc. 137, 15528–15532. doi: 10.1021/jacs.5b10388
Walst, K. J. (2012). Synthesis and Characterization of Triaminocyclopropenium Ionic Liquids. Ph.D. thesis, University of Canterbury, Christchurch.
Walst, K. J., Yunis, R., Bayley, P. M., MacFarlane, D. R., Ward, C. J., Wang, R., et al. (2015). Synthesis and physical properties of tris(dialkylamino)cyclopropenium bistriflamide ionic liquids. RSC Adv. 5, 39565–39579. doi: 10.1039/C5RA05254H
Weiss, R., Brenner, T., Hampel, F., and Wolski, A. (1995a). The consequences of an electrostatic “forced marriage” between two electron-rich particles: strained ion pairs. Angew. Chem. Int. Ed. Engl. 34, 439–441. doi: 10.1002/anie.199504391
Weiss, R., Rechinger, M., Hampel, F., and Wolski, A. (1995b). Stable 1:1 adducts from iodoacetylenes and iodide ions: ion pair strain as an additional driving force? Angew. Chem. Int. Ed. Engl. 34, 441–443. doi: 10.1002/anie.199504411
Wenthold, P. G., and Squires, R. R. (1995). Bond dissociation energies of and . A gas-phase experimental and G2 theoretical study. J. Phys. Chem. 99, 2002–2005. doi: 10.1021/j100007a034
Xu, W., Cooper, E. I., and Angell, C. A. (2003). Ionic liquids: ion mobilities, glass temperatures, and fragilities. J. Phys. Chem. B 107, 6170–6178. doi: 10.1021/jp0275894
Keywords: ionic liquids, fluoride, imidazolium, cyclopropenium, salts, solvate
Citation: Curnow OJ, MacFarlane DR and Walst KJ (2018) Fluoride Ionic Liquids in Salts of Ethylmethylimidazolium and Substituted Cyclopropenium Cation Families. Front. Chem. 6:603. doi: 10.3389/fchem.2018.00603
Received: 07 August 2018; Accepted: 23 November 2018;
Published: 10 December 2018.
Edited by:
Jason B. Harper, University of New South Wales, AustraliaReviewed by:
Luke Henderson, Deakin University, AustraliaCopyright © 2018 Curnow, MacFarlane and Walst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Owen J. Curnow, b3dlbi5jdXJub3dAY2FudGVyYnVyeS5hYy5ueg==
Douglas R. MacFarlane, ZG91Z2xhcy5tYWNmYXJsYW5lQG1vbmFzaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.