
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 06 July 2018
Sec. Medicinal and Pharmaceutical Chemistry
Volume 6 - 2018 | https://doi.org/10.3389/fchem.2018.00246
 Mohammad Uzzal Hossain1
Mohammad Uzzal Hossain1 Chaman Ara Keya2Keshob Chandra Das3
Chaman Ara Keya2Keshob Chandra Das3 Abu Hashem4Taimur Md. Omar5
Abu Hashem4Taimur Md. Omar5 Md. Arif Khan6S. M. Rakib-Uz-Zaman7Md. Salimullah3*
Md. Arif Khan6S. M. Rakib-Uz-Zaman7Md. Salimullah3*An outbreak of West Nile Virus (WNV) like the recent Ebola can be more epidemic and fatal to public health throughout the world. WNV possesses utmost threat as no vaccine or drug is currently available for its treatment except mosquito control. The current study applied the combined approach of immunoinformatics and pharmacoinformatics to design potential epitope-based vaccines and drug candidates against WNV. By analyzing the whole proteome of 2994 proteins, the WNV envelope glycoprotein was selected as a therapeutic target based on its highest antigenicity. After proper assessment “KSFLVHREW” and “ITPSAPSYT” were found to be the most potential T and B-cell epitopes, respectively. Besides, we have designed and validated four novel drugs from a known WNV inhibitor, AP30451 by adopting computational approaches. Toxicity assessment and drug score confirmed the effectiveness of these drug candidates. This in silico research might greatly facilitate the wet lab experiments to develop vaccine and drug against WNV.
West Nile Virus (WNV) belongs to Flaviviridae family which shows the close association with dengue virus. WNV is transmitted by mosquito and causes febrile illness upon infection that further develops encephalitis including flaccid paralysis and cognitive dysfunction (Ceausu et al., 1997; Petersen and Marfin, 2002; Sejvar et al., 2003). Birds are the main target of WNV encephalitis but other vertebrates including humans are also at the edge of high risk. WNV fever and encephalitis have become prevalent throughout the world including the Middle East, Europe, Africa and America since the mid-1990s (Hubalek and Halouzka, 1999; Lanciotti et al., 1999; Petersen et al., 2003; Dauphin et al., 2004; Hayes et al., 2005; Mattar et al., 2005; Deardorff et al., 2006; Komar and Clark, 2006). The symptoms of WNV are more prevalent in elderly and immuno-compromised person although patients might be affected in any stages of age. The survey reported that fatality rate of most recent WNV outbreak is about 4–14% but it might be increased from 10 to 19% in hospitalized cases (Mattar et al., 2005). However, despite WNV's worldwide distribution and epidemic nature, currently, no effective medication is available that can prevent or cure this infection. Therefore, the patients with WNV infection were likely to have supportive treatment such as mosquito net usage, intensive nursing care, painkiller administration etc. (Hayes and Gubler, 2006; Division of Vector-Borne Infectious Disease, 2007).
The enveloped virion (50 nm) of WNV is comprised of RNA genome surrounded by a lipid bilayer membrane. The secondary structure of envelope protein (E) plays a vital role in translation, RNA synthesis, and packaging (Shi et al., 1996; Khromykh et al., 2001; Granwehr et al., 2004; Busch et al., 2005; Friebe and Harris, 2019). The envelope glycoproteins (E) intercede viral attachment and their entry by membrane fusion (Kanai et al., 2006). The domain of E protein is important for eliciting neutralizing antibodies as it is exposed on the viral surface. The envelope E glycoprotein is the foremost antigen which is also the essential target for vaccine development (Roehrig, 2003). The envelope (E) protein selected for drug design plays a crucial role in translation, RNA synthesis and packaging. We have selected a potential inhibitor AP30451 (experimentally validated from 80000 chemical compounds) which inhibits the translation and replication of WNV. The compound AP30451 inhibited translation of WNV mRNA and blocked WNV replication upon inhibitor screening of WNV replicon and Enzyme-linked Immunosorbent Assay (ELISA) of several cell types (Noueiry et al., 2007). And we have designed some inhibitor compounds from parent molecules AP30451. As E protein is essential for translation, it is, therefore, to be selected an inhibitor AP30451 which will ultimately inhibit the translation mechanism of WNV. The structural (capsid, envelope, and pre-membrane) and nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins were found during translation of viral RNA (Lindenbach et al., 2007). Receptor interaction, membrane fusion, and virion assembly are regulated by the envelope (E) protein. The conformation of E protein during virion assembly and premature fusion of it during virus exocytosis to the cell surface are both stabilized by the pre-membrane (prM) (Westaway et al., 2002). prM is cleaved to M (infectious virions that are released by exocytosis) via a furin mediated pathway upon the transportation through the trans-Golgi network (Guirakhoo et al., 1991; Elshuber et al., 2003).
The approach of vaccinomics which is integrated with bioinformatics tools has been better choice for the recent development of new vaccines (Flower, 2008; Poland et al., 2009). To emphasize this, here we propose an integrated approach, relying on vaccine development and drug prediction against WNV. Vaccinomics already demonstrated its potentials against some life-threatening diseases such as multiple sclerosis (Bourdette et al., 2005) malaria (López et al., 2001) and tumors (Knutson et al., 2001) through the development of the vaccine. An ideal vaccine that prompts immunity should essentially be able to eliminate the risk of re-infection as well as to elicit a specific immunological response (Atanas and Irini, 2013). In this perspective, immune dominant B-Cell and T-cell epitopes mapping is the prime concern of immunoinformatics based research to reduce expenses and valuable time for the development of vaccines. The chronic symptoms which may last for months to years might require being treated with the vaccine and drug simultaneously. Moreover, a mutation could shrink the effectiveness of the vaccine/drug candidates against the viral disease. Therefore, concurrent development of vaccine and drug candidates against viral disease might be the interest of the researchers and clinicians. To emphasize this, we have added both vaccine and drug therapy approach in the same study. Still, vaccination might be ineffective due to the emergence of a sudden outbreak of WNV. Post-therapeutics or drugs could be the best option until the new vaccine is developed. Here, we employ an integrated approach relying on both vaccine and drug development against WNV. Computer -aided screening techniques and docking simulation could significantly contribute to determine suitable inhibitors to the target proteins and to find out exclusive binding sites of potential target proteins respectively. Inhibitor molecules for dengue virus, envelope protein have recently been successfully identified based on this assumption methodology (Yennamalli et al., 2009). The approach of bioinformatics in the field of vaccine and drug discovery has already prolifically performed against some virus and bacteria (Sharmin and Islam, 2014; Hasan et al., 2015; Khan et al., 2015; Oany et al., 2015; Hossain et al., 2016a,b,c). Therefore, we have analyzed WNV entire proteome and identified its potential immunogenic regions. Further, computational methods have been performed for the prediction of inhibitor molecules against the active site of the target protein. This in silico research has been steered to design effective peptide vaccine and some novel drugs for the better medication system that could lead the possible treatment as well as prevention of WNV infection.
All the protein sequences of human West Nile virus available in UniProt Knowledge Base (UniProtKB) database (http://www.uniprot.org) (Apweiler et al., 2004; The UniProt Consortium, 2014) were retrieved and then stored in FASTA format for the analysis of immunogenic properties.
To identify the most potent antigenic protein, the antigenic value of each protein was assigned using an online prediction server, Vaxijen v2.0 (Doytchinova and Flower, 2007). Protein having highest antigenic value was considered as the most potent antigenic protein. Default parameter of this server was used for this identification.
MODELLER 9v11 (Šali et al., 1995) through HHpred (Söding, 2005; Söding et al., 2005) was used to predict the 3D structure of envelope glycoprotein of WNV. The model assessment tools Anolea (Melo et al., 1997), ProCheck (Laskowski et al., 1993), and Verify3D (Eisenberg et al., 1997) were employed to ensure the model quality.
NetCTL 1.2 (http://www.cbs.dtu.dk/services/NetCTL/) server was utilized for enlisting the interacted Cytotoxic T-lymphocyte (CTL) epitopes. The candidate epitopes were predicted based on the MHC class I supertypes (A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, and B62) and peptide binding (Larsen et al., 2005). The threshold value was set to 0.5 by which we could assess our findings more decisively to generate more epitopes. The best candidates for vaccine development were chosen for further analysis based on a combined score of class I binding, transporter of antigenic peptides (TAP) transport efficiency and proteasomal cleavage prediction.
Stabilized Matrix Method (SMM) (Peters and Sette, 2005; Tenzer et al., 2005) was implemented for the prediction of the suitable epitope from MHC II molecules.
The predicted epitopes were employed in Immune Epitope Database (IEDB) analysis resource to measure the epitope conservancy level within the all protein sequences of WNV (Bui et al., 2007).
We have employed IEDB population coverage tool (Bui et al., 2007) to calculate the population coverage of epitopes.
The allergenicity of proposed epitopes was assessed by a cross-reactive allergen prediction program, AllerHunter. Prediction of allergen and non-allergen epitopes were determined by AllerHunter with high sensitivity and specificity (Liao and Noble, 2003; Muh et al., 2009).
We have utilized PEP-FOLD server (Thevenet et al., 2012) to predict the 3D structure of the most prospective epitope represented with a 9-mer peptide sequence “KSFLVHREW” to analyse the binding affinity with Human Leukocyte Antigens (HLAs). The server proposed five 3D structures of corresponding epitope but only one was selected for the interaction with HLAs.
Docking experiments were performed by Autodock Vina (Trott and Olson, 2010) to reveal out the binding affinity between predicted epitopes and HLA molecules. The HLA-B*35:01 (3LKN) molecule was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) (Berman et al., 2000) and further subjected to Discovery studio (Van Joolingen et al., 2005) to remove the NP418 epitope which was complexed with HLA molecule. In order to compare with the previous binding affinity NP418 epitope and prepared HLA-B*35:01 was also performed in docking study. Moreover, 1D5M crystal structure was also retrieved to perform docking study with the selected epitope for Major Histocompatibility Complex II (MHC-II) molecule binding affinity.
To initiate an immune response, B cell epitope plays a vital role by interacting with B lymphocytes (Nair et al., 2002). IEDB tools were utilized to confirm the antigenic properties of B-cell epitope such as Kolaskar and Tongaonkar antigenicity scale (Kolaskar and Tongaonkar, 1990), Emini surface accessibility prediction (Emini et al., 1985), Karplus and Schulz flexibility prediction (Karplus and Schulz, 1985), Bepipred linear epitope prediction analysis (Larsen et al., 2006) and Chou and Fasman beta-turn prediction analysis (Chou and Fasman, 1978; Rini et al., 1992).
The selected epitopes were analyzed to evaluate the hydrophobic properties by using the Parker hydrophilicity prediction tool (Parker et al., 1986) of Immune Epitope Database (IEDB) in which default threshold of 3.448 was used.
The binding site of the constructed model was identified by using the meta-pocket server 2.0 (Bingding, 2009) and Discovery studio (Van Joolingen et al., 2005). The predicted binding site obtained by these servers could be employed for the discovery of effective drugs against envelope glycoprotein.
To convey noteworthy insight on drug-protein interactions active site analysis was carried out by using Computed Atlas of Surface Topography of proteins (CASTp) server (Dundas et al., 2006). The active binding sites were identified based on the structural similarity between template and model.
National Center for Biotechnology Information, NCBI (http://www.ncbi.nlm.nih.gov), Public Medline, Pubmed (www.ncbi.nlm.nih.gov/pubmed) and Google Scholar (http://scholar.google.com/) were searched to find out the inhibitor those are validated with the lab experiment. Therefore, the AP30451 compound was selected for our study as a control compound. Replication dependent luciferase expression and Adenosine Triphospate (ATP) quantification assay of AP30451 were previously performed and reported to assess its antiviral activity and cytotoxic effect on WNV (Noueiry et al., 2007). For the advancement of drug nature of this known inhibitor molecule, novel drug molecules were generated by employing ACD/Chemsketch (Fan et al., 2002) and Molinspiration (Mishra and Raghava, 2011). Thereafter, 2D structure data files (SDF) were stored as Mol file for further analysis.
The Mol file of AP30451 and designed molecules were converted to Protein Data Bank (PDB) file by using Open Bable (OL Boyle et al., 2011). Afterward, the 3D structure optimization of these compounds was accomplished by the ACD/Chemsketch (Fan et al., 2002).
For homology modeling and designed structures, an accurate alignment is an indispensable requirement to abate the impairment of residues during structure prediction. We have calculated the relative binding free energies for these complexes using the Yet Another Scientific Artificial Reality Application (YASARA) server (Sippl, 1993; Vriend and Sander, 1993; Kuszewski et al., 1997; Krieger et al., 2009).
Some sets of QSAR prediction tools Click2drug (http://www.click2drug.org/), Zinc database (http://zinc.docking.org/), Osiris property explorer (Sander, 2001), Molinspiration (Mishra and Raghava, 2011), and AcTor (Judson et al., 2008) were scrutinized to explore the structural properties of drugs.
The energy minimized homology model and the designed analogs with control compound were then performed in Autodock 4.2 (Duan et al., 2003; Morris et al., 2009) and Autodock vina (Trott and Olson, 2010) for docking experiments in which water molecules were omitted but added the polar hydrogen in built model. In docking experiment with autodock vina, a pdbqt file was generated by following grid box parameter (size 48 × 48 × 78 points and center with −14.359 × −6.818 × −39.272, and grid spacing 0.5°A as well). Besides, kollman united atom charges, genetic algorithm (LGA) and population size 150 were utilized in autodock 4.2 for docking studies. These docking experiments were preferred to generate the intermolecular energy, internal energy, of ligand and torsional free binding energy between built model and inhibitors. The molecular visualization of protein-ligands were analyzed by PyMol (Daniel and Bert, 2010), Yasara (Krieger et al., 2009), RasMol (Sayle and Milner-White, 1995), and Discovery studio (Van Joolingen et al., 2005).
The Osiris property explorer (Sander, 2001), Molinspiration (Mishra and Raghava, 2011), ACToR (Aggregated Computational Toxicology Resource) (Judson et al., 2008), ACD/I-Lab (https://ilab.acdlabs.com/iLab2/) and admetSAR (absorption, distribution, metabolism, excretion, and toxicity Structure-Activity Relationship database) (Cheng et al., 2012) were utilized for the calculation of ADME (Absorption, Distribution, Metabolism, Excretion) pharmacological properties and toxic profile. These properties confer the pharmacological activity of the compounds as drug candidates as well as their mode of action within the tissues.
The outline of the current study was shown in Figure 1.
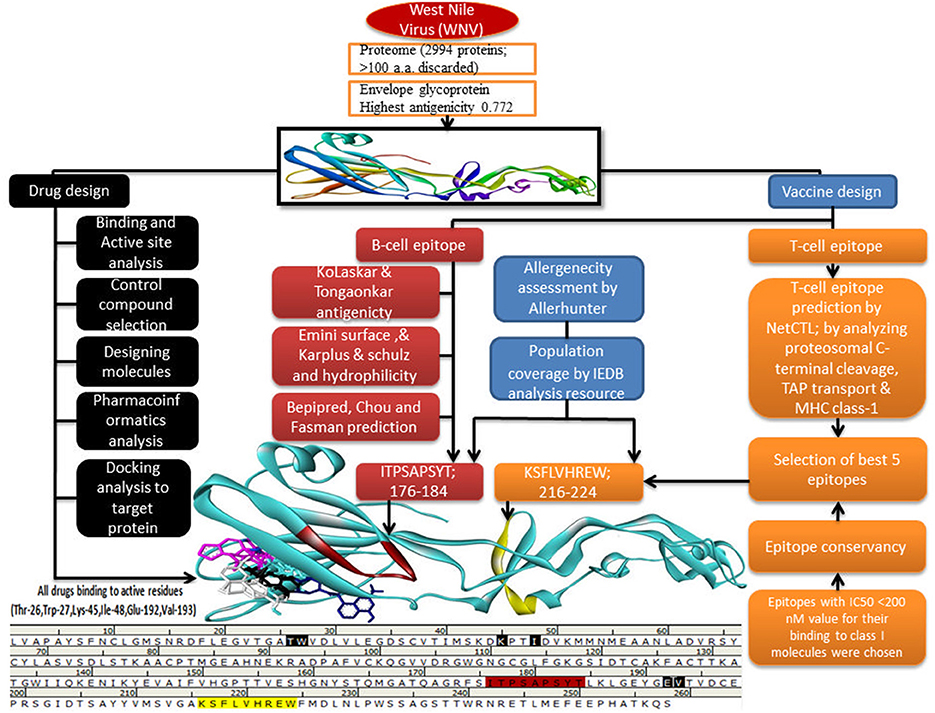
Figure 1. Graphical outline of the current study.
The proteome of WNV composed of 2994 proteins including envelope glycoprotein (758), Genome polyprotein (978), Polyprotein (1345), NS3, NS5 etc. was retrieved as FASTA format and saved in Excel sheet Supplementary Excel File 1. Non-structural proteins and proteins having less than 100 amino acid sequences were excluded from this study. Proteins were most unlikely to prioritize essential proteins when they are less than 100 amino acids (Kumar et al., 2010; Haag et al., 2012). These mini proteins (>100 a.a) have significant role in numerous biological phenomenon and regulatory purposes (Wang et al., 2008). But these mini proteins were deleted from the remaining set of proteins as they are less likely to correspond to the essential therapeutics candidate. In addition to this, the larger amino acid sequence has the possibility to be categorized as promising vaccine/drug targets (Kumar et al., 2010; Haag et al., 2012) Analysis of all the retrieved protein sequences in the VaxiJen server revealed the most antigenic protein as the envelope glycoprotein (UniProtKB id: F1CFF2) with highest prediction score of 0.772 Supplementary Excel File 1.
The 3D structure of the highest antigenic envelope glycoprotein was built by employing Modeller 9v11 Supplementary Figure S1. The PSI blast that identified 3P54_A template gives the best coverage of the selected protein. The Ramachandran plot was analyzed to evaluate the quality of structure and it showed 92.9% residues in most favored regions Supplementary Figure S2 and Supplementary Table S1. VERIFY3D and ANOELA also ensured an appropriate 3D structure of Envelope glycoprotein shown in Supplementary Figures S3, S4.
Five (5) Cytotoxic T-Cell epitopes (CTL) epitopes namely LADVRSYCY, TTVESHGNY, SGIDTSAYY, REWFMDLNL, and KSFLVHREW were selected on the basis of their higher NetCTL combinatorial score Table 1. The MHC-1 molecules were selected followed by half-maximal inhibitory concentration (IC50) value < 200 nm which assures the higher binding capability of selected epitopes to MHC-1 molecules Table 1. The 9-mer epitopes KSFLVHREW and SGIDTSAYY demonstrated the affinity for highest 13 MHC-I molecules Table 1. The conservancy of epitopes from IEDB analysis resource is the essential criteria to induce effective immunogenicity. In our analysis, epitope REWFMDLNL and KSFLVHREW were found with high conservation rate (>82%) whereas epitopes SGIDTSAYY, LADVRSYCY, and TTVESHGNY showed moderate to low conservation rate (5–33%) Table 1. We have also predicted MHC-II molecules of the “KSFLVHREW” as it showed higher conservancy and capability to interact with maximum interacting MHC-I molecules Table 2.

Table 1. 5 Potential T-cell epitopes with properties.

Table 2. MHC–II molecules from the selected peptide epitope.
Population coverage measures how many MHC-I molecules prone to respond to predicted epitopes among the people (in percentage) living in a given area. The highest population coverage 81% of epitopes from IEDB was observed in Germany whereas Mali showed the lowest (52.6%) Figure 2. Therefore, at least more than half of the world's population could be covered by these epitopes.
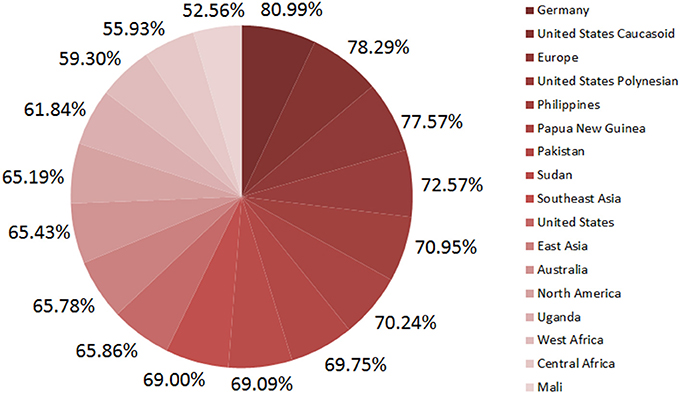
Figure 2. Population coverage of predicted epitopes.
AllerHunter was utilized to predict the sequence-based allergenicity of the epitopes. The analysis with 91.6% sensitivity and 89.3% specificity revealed non-allergenicity of the query epitopes.
AutoDock Vina generated binding mode between “KSFLVHREW” and selected HLA molecules. The binding energy between selected epitope and HLA-B*35:01 showed −7.5 kcal/mol which appeared very much similar to influenza NP418 epitope's binding energy −7.6 kcal/mol Figure 3A. The binding energy −7.1 kcal/mol of HLA-DR with the desired epitope was also promising Figure 3B.
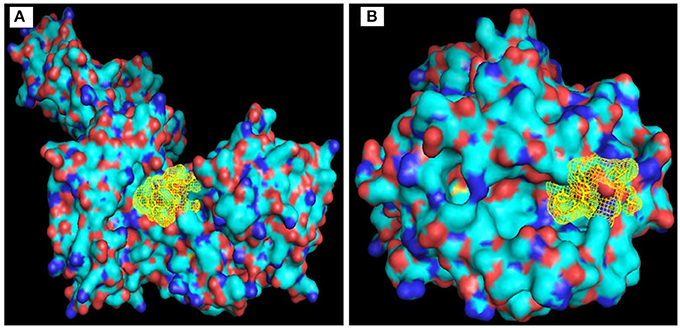
Figure 3. Visualization of binding of KSLVHREW epitope with (A) MHC-I (HLA B*35:01); and (B) MHC-II (HLA-DR).
We have implemented some sets of authentic bioinformatics tools to recognize the potential B-cell epitopes. The conserved region were assessed based on physico-chemical properties by Kolaskar and Tongaonkar antigenicity prediction tool. The tool predicted an average antigenic propensity value 1.058 in the scale of maximum value 1.240 and minimum value 0.920. The threshold for antigenicity in the conserved region was 1.00 whereas all values >1.00 from the result considered as potential antigenic determinants. We identified an epitope that meets the threshold criteria with the potentiality to initiate B cell response Figure 4A. Among others, we have found a region of amino acid residues (176–184) as surface accessibility to be urged for potential B cell epitope Figure 4B. The antigenic region of a protein generally contains beta turns which are usually surface accessible and exhibit hydrophilic nature. The region 107–115, 176–184, and 230–238 were reported as the β-turns region from Chou and Fasman Beta-turn prediction Figure 4C. It is speculated that the flexibility of the peptide has a strong correlation with antigenicity (Doytchinova and Flower, 2007). Karplus and Schulz flexibility prediction tool suggested the region of 176–184 as most flexible Figure 4D. Finally, we implemented the Bepipred linear epitope prediction tool to identify the region which could be predicted as the linear B-cell epitope Figures 4E, 5. To confirm the hydrophilic nature of a peptide region, Parker Hydrophilicity was also employed in the region. To analyze the all data from B-cell epitope prediction tools, the conserved sequence “ITPSAPSYT” of 176–184 region was shown to be able to prompt the immunity as B-cell epitope.
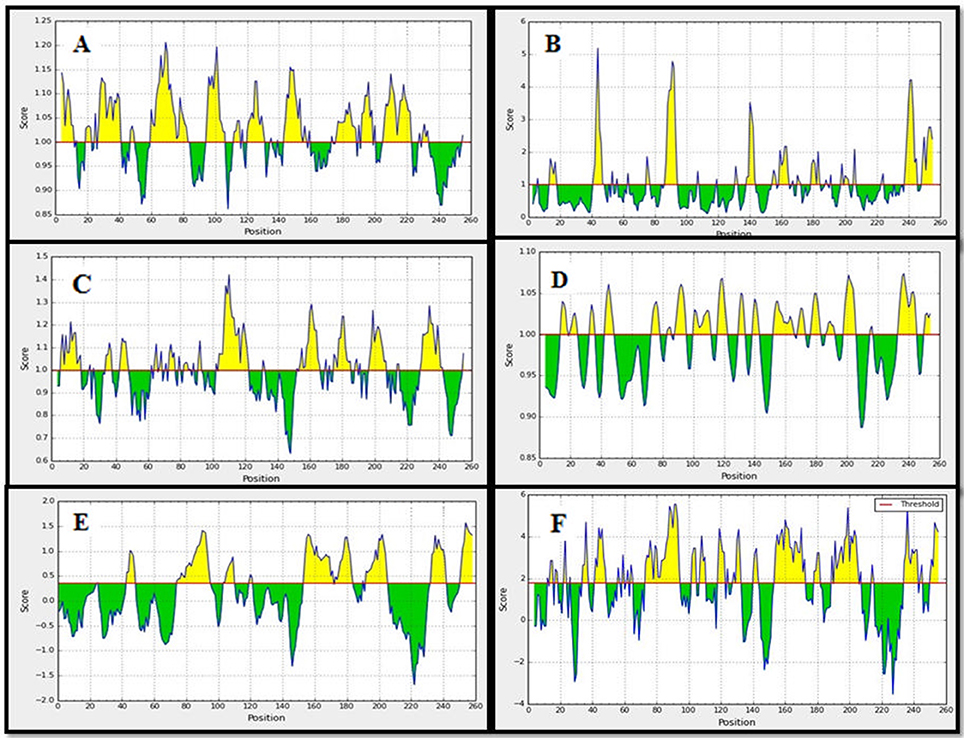
Figure 4. Prediction of B-cell antigenic properties for most antigenic conserved region. Epitope ITPSAPSYT (176–184) showed all the antigenic criteria to be predicted as B-cell epitope. (A) Kolaskar and Tongaonkar antigenicity prediction. (B) Emini surface accessibility prediction. (C) Chou and Fasman beta-turn prediction. (D) Karplus and Schulz flexibility prediction. (E) Bepipred linear epitope prediction. (F) Parker hydrophilicity prediction. The x-axis and y-axis represent the sequence position and corresponding antigenic properties score, respectively. The threshold level is 1.0 for most of the properties except for (E) (0.40) and (F) (0.1772). The regions having antigenic properties are shown in yellow color above the threshold value.
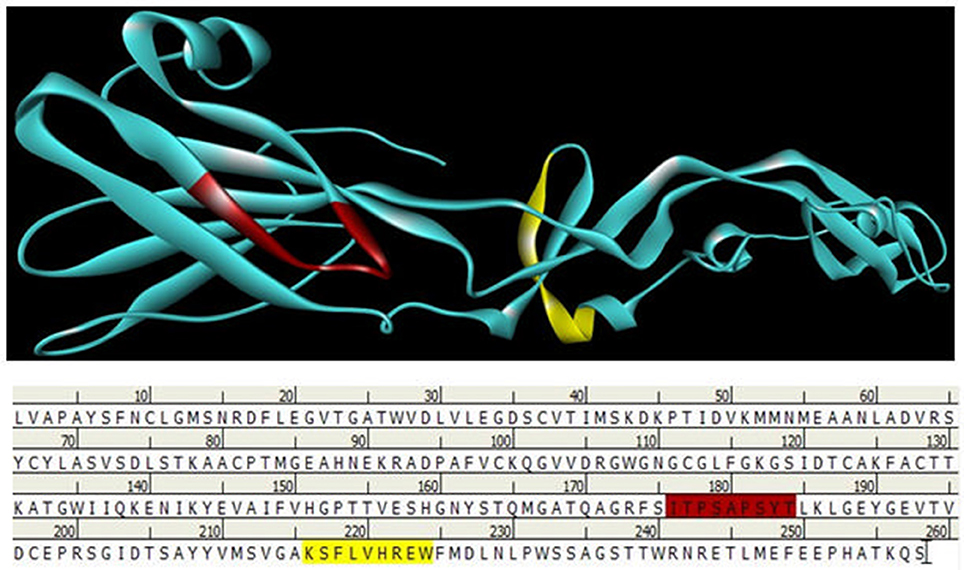
Figure 5. B and T cell epitopes depiction of envelope glycoprotein. Here red color indicates B cell epitope in 176–184 regions and Yellow color indicates T cell epitope is 216–224 regions.
We have found six possible binding sites of the modeled 3D protein structure Figure 6A. Then, the CASTp server identified the active site of the envelope glycoprotein. This gives an important prediction about the interacting sites on protein with the ligand molecules Figure 6B.
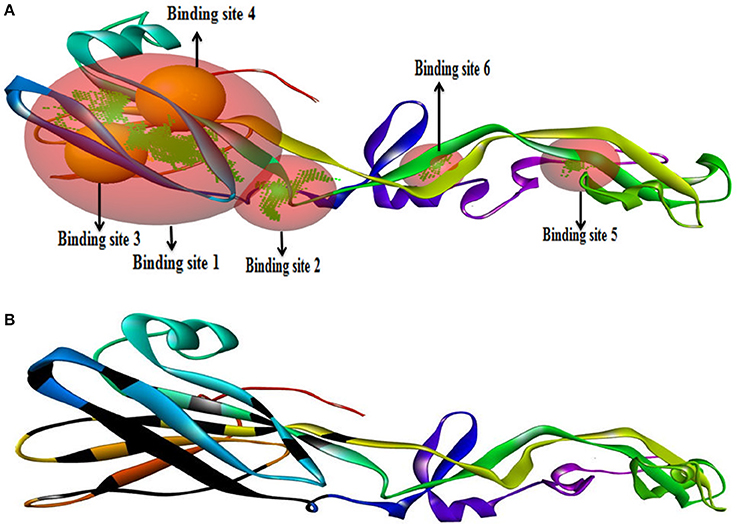
Figure 6. Binding site of envelope glycoprotein (A) Active site amino acid residues of envelope glycoprotein. (B) The active site residues indicated with black color within envelope glycoprotein of WNV.
The structural defect such as physical realism, stereochemistry, side-chain accurateness, and gap alignment might arise during homology modeling and designing molecules. Therefore, YASARA program was used for the minimization of these structural features which could offer the proper structural stability by yielding the energy (START vs. END) of the constructed model and designed inhibitors Figure 7. The energy comparison between the End energy (−123404.0 kJ/mol) and START energy (−53191.7 kJ/mol) of constructed model confirmed the accurateness of structural features. Besides, the energy comparison of designed molecule 1 (START −465.5 KJ/mol to END −924.0 KJ/mol), designed molecule 2 (START −164.6 KJ/mol to END −847.6 KJ/mol), designed molecule 3 (START −588.7 KJ/mol to END −1325.3 KJ/mol), and designed molecule 4 (START −60.7 KJ/mol to END −765.0 KJ/mol) ensures the structural stability Figure 8.
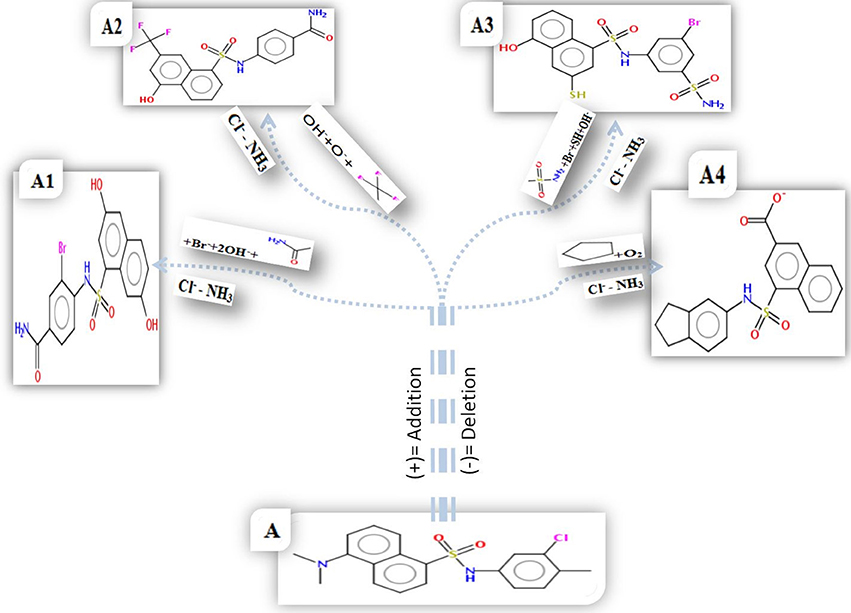
Figure 7. 2D structure of parent compound AP30451 and design molecules. (A) Parent compound AP30451. Here, Cl− and NH3were deleted from the original compounds to generate the novel compounds as WNV inhibitors. (A1) Br− and 2OH− were added in designed molecule-1. (A2) OH−, O− and CF3 added in design molecule-2. (A3) Br−, OH−, SH etc. were added in designed molecule-3. (A4) O2 and a ring structure added in designed molecule-4.
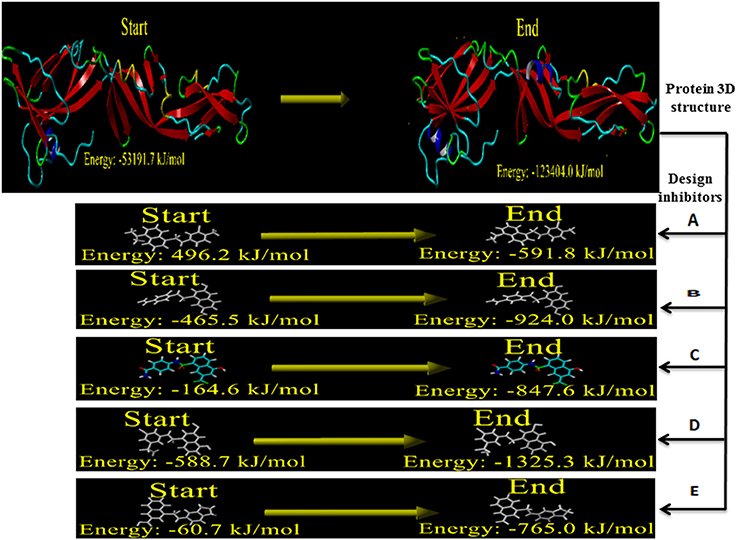
Figure 8. Energy minimization of built model of envelope glycoprotein and designed inhibitors.
To analyze the protein-drug interactions we have performed the molecular docking experiment with two docking software, namely Autodock 4.2 and Autodock Vina of MGL 1.5.6. The docking energy of AutoDock Vina (AP30451: −5.9 KJ/mol, designed molecule 1: −8.0 KJ/mol, designed molecule 2: −8.7 KJ/mol, designed molecule 3: −7.7 KJ/mol and designed molecule 4: −8.9 KJ/mol) and AutoDock 4.2 (AP30451: −4.93 KJ/mol, designed molecule 1: −7.89 KJ/mol, designed molecule 2: −7.67 KJ/mol, designed molecule 3: −6.99 KJ/mol and designed molecule 4: −7.03 KJ/mol) confirmed that they possess strong binding affinity with the target into the binding site Table 3 and Figure 9. Importantly, the interacting amino acid residues from the binding site were also found as active residues Figure 10. The most common interacting amino acid residues were found viz Thr 26, Trp 27, and Val 193 from control and all the inhibitor molecules Table 4 and Figure 11.
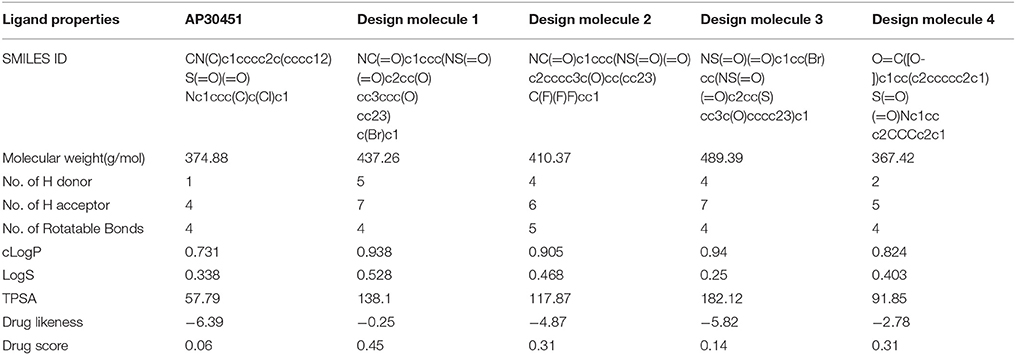
Table 3. QSAR properties of control drug and proposed WNV inhibitors.
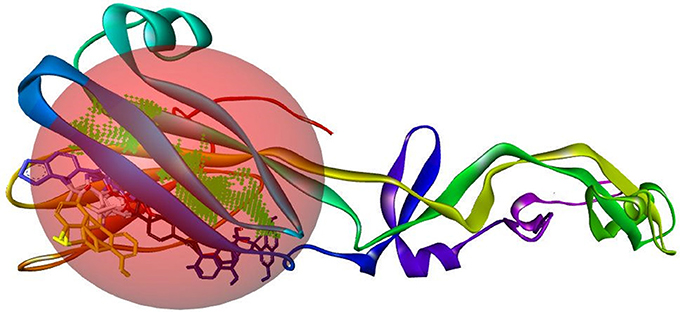
Figure 9. All the designed inhibitors bind in the binding site 1 of the envelopeglycoprotein along with AP30451. The color code of the inhibitors: AP30451 (White); designed molecule 1 (Blue); designed molecule 2 (Yellow); designed molecule 3 (red); designed molecule 4 (Indigo).
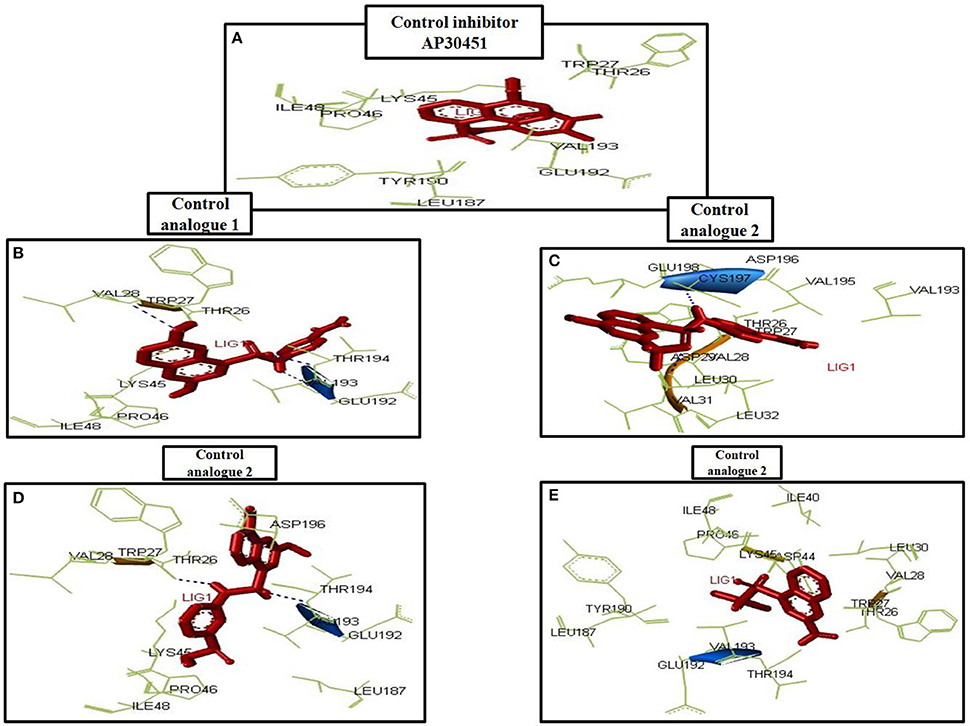
Figure 10. Amino acid interaction with inhibitors. (A) AP30451, (B) Designed molecule 1, (C) design molecule 2, (D) Designed molecule 3, (E) Designed molecule 4.
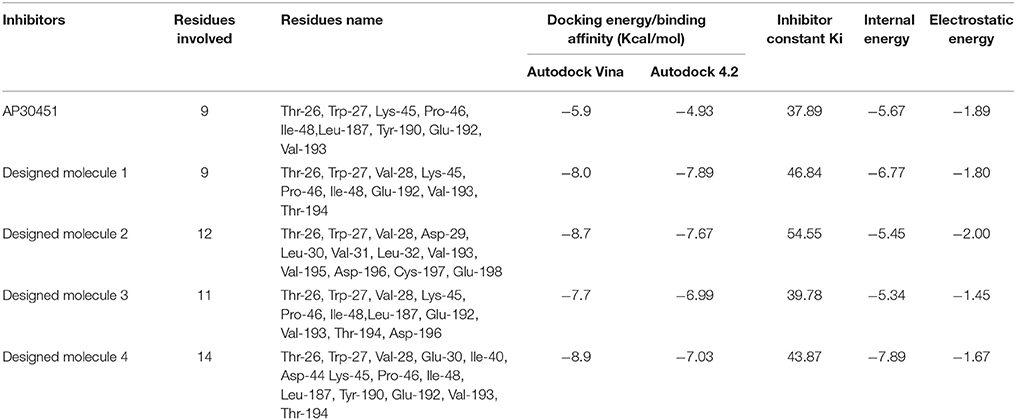
Table 4. Docking result of Designed inhibitors.
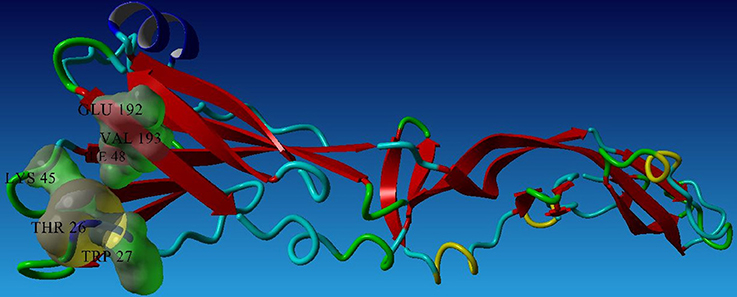
Figure 11. Most common interacting residues of envelope glycoprotein.
The important criteria for establishing a new drug is to analyse the structural activity relationship and pharmacoinformatics analysis. We have exploited some bioinformatics tools (Described in Materials and Methods section) for the analysis of druggable properties which upsurge these designed inhibitors as potential drug candidates. We also analyzed ADME properties (Absorption, Distribution, Metabolism, Excretion) including Human Intestinal Absorption (HIA), Skin permeability, Blood-Brain Barrier distribution (LogBB), Caco-2 cell permeability, Volume of distribution, CYP450 2C9 substrate and inhibitor etc. The outcomes from these analyses meet the criteria for the new drug discovery. The overall toxicity (mutagenic, irritative, reproductive toxicity, and carcinogenic) of designed drugs were satisfactory than the mother compound AP30451. The drug-likeness and oral bioavailability confirmed that designed molecule 1 (drug score 0.45) could be the best choice for the potential drug among all the designed inhibitors along with AP30451 Figure 12 and Table 5.
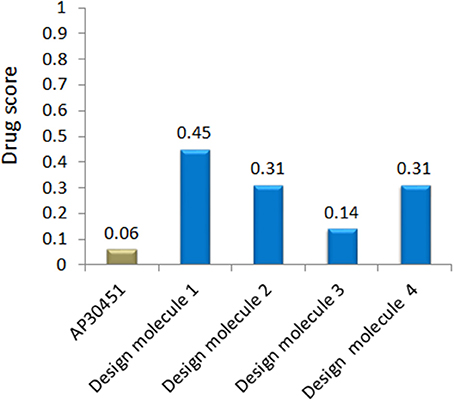
Figure 12. Drug score of WNV inhibitor.
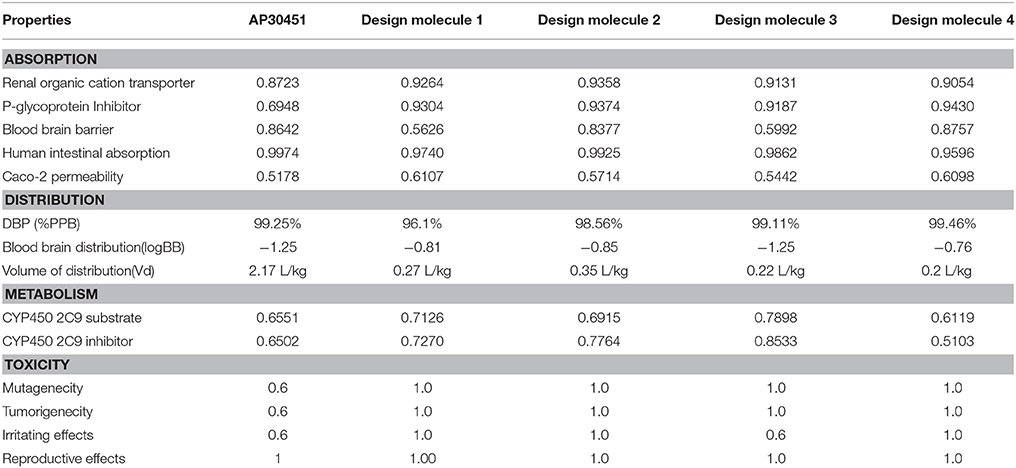
Table 5. ADME properties of designed inhibitors.
The underlying factors of recent emerging and re-emerging diseases include the microbial agent, human host and human environment (Morens and Fauci, 2013). Alarmingly, these sorts of diseases are now-a-days not confined to the developing countries. The infection of WNV is one of the emerging diseases and fatalities in humans and livestock around the globe. A few WNV vaccines in the pipeline provided a proof of concept for veterinary use. Still efforts on the development of effective therapeutics for human remain unsuccessful partly due to wet lab approaches which couldn't allow the faster method. However, the recent development of immunopharmacology tools together with the understanding of structure and function of WNV proteins could provide a faster and stronger platform for designing and developing vaccines/drugs as potential therapeutics. In the current study, we have been able to retrieve, store and utilize a lot of information about genomics and proteomics of WNV virus made available by the sequence-based technology which in turn allowed us the identification of epitopes from antigenic protein. The designing of epitope-based peptide vaccine against WNV achieved through the identification of a neutralizing epitope using similar bioinformatics tools applied for the recently recommended epitope-based vaccines such as dengue, chikungunya etc (Lapelosa et al., 2009; Chakraborty et al., 2010; Islam et al., 2012; Hasan et al., 2013; Hossain et al., 2018).
B-cell immunity is given the priority to design vaccine but T-cell was also shown to induce strong immune response (Van Joolingen et al., 2005). Also, T cell immune response is long-lasting immunity as foreign particles can avoid the effect of memory produced by an immune system. To suggest both B-cell and T-cell epitopes from the most antigenic protein we have used different bioinformatics tools to confer the immunity in WNV. A well-conserved T cell epitopes among the envelope glycoprotein sequences of WNV were considered as strong and potent. Our predicted human non-allergen T-cell epitope “KSFLVHREW” is expected to be effective as peptide vaccine as well as provide the protection broadly against various strains due to its higher binding affinity to interact highest numbers (13) of HLA and very high conservancy (82.46%) respectively Tables 1, 2, and Figure 6. This higher binding affinity of this epitope to MHC allele was confirmed by docking analysis by using a specific allele Figure 4. HLA alleles responsible for exposing affinity to KSFLVHREW were investigated for population coverage in WNV endemic regions. The significant level of population coverage was achieved for European, Asian and American population Figure 3. According to these results, it is obvious that this non-allergen vaccine would be effective for a vast population throughout a wide geographical region Figure 3. Through rigorous analysis of several characteristics such as the presence of antigenicity, beta-turns, flexibility, accessibility, linearity and hydrophilicity, this study also predicted, identified and confirmed a potent B-cell epitope “ITPSAPSYT” from WNV envelope glycoprotein Figures 5, 6. In addition to an effective vaccine, a universal drug is required for the minimization or complete elimination of chronic symptoms caused by the infection which may last for months to years. Further, sometimes the action of vaccine might not be effective due to acquired mutational change. Then the treatment with drugs could be the best choice against WNV. Nevertheless, vaccination won't be effective in the worst case of sudden WNV outbreak. Therefore, we have also identified the active sites of the highest antigenic protein of WNV for proposing some novel inhibitors alongside with pre-therapeutic vaccine design. Drug designing against WNV was based on the highest antigenic protein from its proteome, the envelope glycoprotein. Before designing the inhibitor, we have predicted the 3D model of the target glycoprotein based on a PSI blast identified model template 3P54_A and confirmed its superior quality Supplementary Figures S1–S4. We have designed four WNV inhibitors from a control WNV inhibitor AP30451 which upon structural stability analysis indicated their candidature as drugs Figures 7, 8. It is clearly apparent that all the designed molecules are the most potent inhibitors as they have lowest docking energy indicating their higher binding affinity to the envelope glycoprotein. Further, they interacted with active pocket amino acid Thr-26, Trp-27, and Val-193 of envelope glycoprotein Tables 3, 4 and Figures 9, 10. Besides, the mode of action of designed and control inhibitors was found similar when they were assessed for their pharmacological properties Table 5. Poor metabolism or toxic effect might urge to fail a drug compound in phase III clinical trials (Ju et al., 2011). The fundamental characteristics of the establishment of drugs are to measure their pharmacophore properties. The advancement of bioinformatics software could be employed to predict these properties before the wet lab confirmation (Ekins et al., 2007). Among the ADMET properties, all the designed molecules showed similar human intestinal absorption rate, higher metabolic rate, no sign to cross blood-brain barrier and balanced volume of drug distribution into the body. Furthermore, the toxic level of these compounds seems to be standard than the parent compound. Also the higher drug likeness of our designed compounds 1(0.45), 2(0.31), 3(0.14), and 4(0.41) over the experimentally validated control compounds AP30451 (0.05) support their effectiveness as inhibitory agents of the envelope glycoprotein of WNV Figure 12 and Table 5.
However, before the practical application of these predicted epitopes, experimental trials are necessary. For the inhibitors, the suggested compound has to be synthesized to test its efficacy in animal model experiments.
Vaccination and drug administration protect human from serious illness and complications against the diseases. Therefore, we have employed bioinformatics approach to look for an effective method of preventing viral disease. The current study suggests that both T and B cell epitope might uphold the progressive stage of novel peptide vaccine discovery to possible treatment against WNV. In addition, inhibitors are designed against WNV envelope glycoprotein as post-therapy which could be suggested to experimental validation for the development of drugs. The in silico results confirmed that the predicted epitopes could elicit the immune response and designed drugs also could assist as therapeutic agents against WNV infection.
MS: Conceived, designed, and guided the study, analyzed the data, drafting the manuscript and performed critical revision; CK: Guided the study, acquisition, and analyzed the data, helped in drafting the manuscript; KD and AH: Guided the study, analyzed the data, and helped in drafting the manuscript. TO, MK, and SR Helped in Bioinformatics analysis and helped in drafting the manuscript. MH: Helped to design the study, performed bioinformatics analysis, drafted, and developed the manuscript and performed critical revision. All authors have approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Authors are grateful to Ministry of Science and Technology (MOST), Bangladesh for its extended support to the National Institute of Biotechnology (NIB) during this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00246/full#supplementary-material
WNV, West Nile Virus; ALL, Acute Lymphoblastic Leukemia; RCSB, Research Collaboratory for Structural Bioinformatics; CADD, Computer Aided Drug Design; PDB, Protein data bank; 3D, 3 dimensional; LogS, Logarithm of solubility; ADME/Tox, Absorption, Distribution, Metabolism, Excretion, Toxicity; TPSA, The polar surface area; cLogP, Logarithm of partition coefficient; HIA, Human Intestinal Absorption HIA; Vd, Volume of Distribution; QSAR, Quantitative structural activity relationship; UniPortKB, UniProt Knowledge Base; IC50, half-maximal Inhibitory Concentration; CTL, Cytotoxic T-lymphocyte; TAP, Transport Associated Proteins; IEDB, Immune Epitope Database; MHC, Major Histocompatibility Complex.
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2004). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119. doi: 10.1093/nar/gkh131
Atanas, P., and Irini, D. (2013). T-cell epitope vaccine design by immunoinformatics. Open Biol. 3:120139. doi: 10.1098/rsob.120139
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Bingding, H. (2009). metaPocket: a meta approach to improve protein ligand binding site prediction. OMICS 13, 325–330 doi: 10.1089/omi.2009.0045
Bourdette, D. N., Edmonds, E., Smith, C., Bowen, J. D., Guttmann, C. R., Nagy, Z. P., et al. (2005). A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult. Scler. 11, 552–561. doi: 10.1191/1352458505ms1225oa
Bui, H. H., Sidney, J., Li, W., Fusseder, N., and Sette, A. (2007). Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics 8:361. doi: 10.1186/1471-2105-8-361
Busch, M. P. S., Caglioti, E. F., Robertson, J. D., McAuley, L. H., Tobler, H. K., and Linnen, J. M. (2005). Screening the blood supply for West Nile virus RNA by nucleic acid amplification testing. N. Engl. J. Med. 353, 460–467. doi: 10.1056/NEJMoa044029
Ceausu, E. S., Erscoiu, P., Calistru, D., Ispas, O., Dorobat, M., Homos, C., et al. (1997). Clinical manifestations in the West Nile virus outbreak. Rom. J. Virol. 48, 3–11.
Chakraborty, S., Chakravorty, R., and Ahmed, M. (2010). A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 10, 235–246. doi: 10.3233/ISB-2010-0435
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., et al. (2012). admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 52, 3099–3105. doi: 10.1021/ci300367a
Chou, P. Y., and Fasman, G. D. (1978). Empirical predictions of protein conformation. Annu. Rev. Biochem. 47, 251–276. doi: 10.1146/annurev.bi.47.070178.001343
Daniel, S., and Bert, L. d. G. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24, 417–422. doi: 10.1007/s10822-010-9352-6
Dauphin, G. S., Zientara, H. Z., and Murgue, B. (2004). West Nile: worldwide current situation in animals and humans. Comp. Immunol. Microbiol. Infect. Dis. 27, 343–355. doi: 10.1016/j.cimid.2004.03.009
Deardorff, E. J., Estrada-Franco, A. C., Brault, R., Navarro-Lopez, A., Campomanes-Cortes, P., Paz-Ramirez, M., et al. (2006). Introductions of West Nile virus strains to Mexico. Emerg. Infect. Dis. 12, 314–318. doi: 10.3201/eid1202.050871
Division of Vector-Borne Infectious Disease (2007). West Nile Virus Homepage. Centers for Disease Control and Prevention. Available online at: http://www.cdc.gov/ncidod/dvbid/westnile
Doytchinova, I. A., and Flower, D. R. (2007). VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4. doi: 10.1186/1471-2105-8-4
Duan, Y., Wu, C., Chowdhury, S., Lee, M. C., Xiong, G., Zhang, W., et al. (2003). A pointcharge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. doi: 10.1002/jcc.10349
Dundas, J., Ouyang, Z., Tseng, J., Binkowski, A., Turpaz, Y., and Liang, J. (2006). CASTp, computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, 116–118. doi: 10.1093/nar/gkl282
Eisenberg, D., Luthy, R., and Bowie, J. U. (1997). VERIFY3D, assessment of protein models with three-dimensional profiles. Meth. Enzymol. 277, 396–404. doi: 10.1016/S0076-6879(97)77022-8
Ekins, S., Mestres, J., and Testa, B. (2007). In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol. 152, 9–20. doi: 10.1038/sj.bjp.0707305
Elshuber, S., Allison, S. L., Heinz, F. X., and Mandl, C. W. (2003). Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borneencephalitis virus. J. Gen. Virol. 84, 183–191. doi: 10.1099/vir.0.18723-0
Emini, E. A., Hughes, J. V., Perlow, D. S., and Boger, J. (1985). Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 55, 836–839.
Fan, Y.-M., Cheng, M.-S., and Cheng, Y.-X. (2002). The reformation in our organic chemistry classes caused by the excellent software ACD/ChemSketch. J. Guang. Univ. 6:012.
Friebe, P., and Harris, E. (2019). Interplay of RNA elements in the dengue virus 5'and 3' ends required for viral RNA replication. J. Virol. 84, 6103–6118. doi: 10.1128/JVI.02042-09
Granwehr, B. P., Lillibridge, K. M., Higgs, P. W., Mason, J. F., Aronson, G. A. C., and Barrett, A. D. (2004). West Nile virus: where are we now? Lancet Infect. Dis. 4, 547–556. doi: 10.1016/S1473-3099(04)01128-4
Guirakhoo, F., Heinz, F. X., Mandl, C. W., Holzmann, H., and Kunz, C. (1991). Fusion activity of flaviviruses: comparison of mature and immature (prMcontaining) tick-borne encephalitis virions. J. Gen. Virol. 72(Pt. 6), 1323–1329. doi: 10.1099/0022-1317-72-6-1323
Haag, N. L., Velk, K. K., and Wu, C. (2012). In silico identification of drug targets in methicillin/multidrug-resistant Staphylococcusaureus. Int. J. Adv. Life Sci. 4, 21–32.
Hasan, M. A., Hossain, M., and Alam, M. J. (2013). A computational assay to design an epitope-based Peptide vaccine against Saint Louis encephalitis virus. Bioinform. Biol. Insights 7, 347–355. doi: 10.4137/BBI.S13402
Hasan, M. A., Khan, M. A., Datta, A., Mazumder, M. H. H., and Hossain, M. U. (2015). A comprehensive immunoinformatics and target site study revealed the corner-stone towards Chikungunya virus treatment. Mol. Immunol. 65, 189–204. doi: 10.1016/j.molimm.2014.12.013
Hayes, E. B., and Gubler, D. J. (2006). West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Ann. Rev. Med. 57, 181–194. doi: 10.1146/annurev.med.57.121304.131418
Hayes, E. B., Komar, N., Nasci, R. S., Montgomery, S. P., O'Leary, D. R., and Campbell, G. L. (2005). Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 11, 1167–1173. doi: 10.3201/eid1108.050289a
Hossain, M. U., Khan, M. A., Hashem, A., Islam, M. M., Morshed, M. N., Keya, C. A., et al. (2016a). Finding potential therapeutic targets against Shigella flexneri through proteome exploration. Front. Microbiol. 7:1817. doi: 10.3389/fmicb.2016.01817
Hossain, M. U., Khan, M. A., Rakib-Uz-Zaman, S. M., Ali, M. T., Islam, M. S., Keya, C. A., et al. (2016b). Treating diabetes mellitus: pharmacophore based designing of potential drugs from Gymnema sylvestre against insulin receptor protein. BioMed Res. Int. 2016:3187647. doi: 10.1155/2016/3187647
Hossain, M. U., Oany, A. R., Ahmed, S. A. I., Hasan, M. A., Khan, M. A., and Siddikey, M. A. A. (2016c). Identification of potential inhibitor and enzyme-inhibitor complex on trypanothione reductase to control Chagas disease. Comput. Biol. Chem. 65, 29–36 doi: 10.1016/j.compbiolchem.2016.10.002
Hossain, M. U., Omar, T. M., Oany, A. R., Kibria, K. M. K., Shibly, A. Z., Moniruzzaman, M., et al. (2018). Design of peptide-based epitope vaccine and further binding site scrutiny led to groundswell in drug discovery against Lassa virus. 3 Biotech. 8, 81. doi: 10.1007/s13205-018-1106-5
Hubalek, Z., and Halouzka, J. (1999). West Nile fever: a reemerging mosquitoborne viral disease in Europe. Emerg. Infect. Dis. 5, 643–650. doi: 10.3201/eid0505.990505
Islam, R., Sakib, M. S., and Zaman, A. (2012). A computational assay to design an epitope-based peptide vaccine against chikungunya virus. Future Virol. 7, 1029–1042. doi: 10.2217/fvl.12.95
Ju, S., Tardiff, D. F., Han, H., Divya, K., Zhong, Q., Maquat, L. E., et al. (2011). PLoS Biol. 9:e1001052. doi: 10.1371/journal.pbio.1001052
Judson, R. S., Richard, A., Dix, D., Houck, K., Elloumi, F., Martin, M., et al. (2008). ACToR—aggregated computational toxicology resource. Toxicol. Appl. Pharmacol. 233, 7–13. doi: 10.1016/j.taap.2007.12.037
Kanai, R., Kar, K., Anthony, K., Gould, L. H., Ledizet, M., Fikrig, E., et al. (2006). Crystal structure of West Nile Virus envelope glycoprotein reveals viral surface epitopes. J. Virol. 80, 11000–11008. doi: 10.1128/JVI.01735-06
Karplus, P. A., and Schulz, G. E. (1985). Prediction of chain flexibility in proteins. Naturwissenschaften 72, 212–213. doi: 10.1007/BF01195768
Khan, M. A., Hossain, M. U., Rakib-Uz-Zaman, S. M., and Morshed, M. N. (2015). Epitope -based peptide vaccine design and target site depiction against Ebola viruses: an immunoinformatics study. Scand. J. Immunol. 82, 25–34 doi: 10.1111/sji.12302
Khromykh, A. A., Meka, H., Guyatt, K. J., and Westaway, E. G. (2001). Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 75, 6719–6728. doi: 10.1128/JVI.75.14.6719-6728.2001
Knutson, K. L., Schiffman, K., and Disis, M. L. (2001). Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J. Clin. Invest. 107, 477–484. doi: 10.1172/JCI11752
Kolaskar, A. S., and Tongaonkar, P. C. (1990). A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1276, 172–174. doi: 10.1016/0014-5793(90)80535-Q
Komar, N., and Clark, G. G. (2006). West Nile virus activity in Latin America and the Caribbean. Rev. Panam. SaludPublica 19, 112–117. doi: 10.1590/S1020-49892006000200006
Krieger, E., Joo, K., Lee, J., Raman, S., Thompson, J., Tyka, M., et al. (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77(Suppl. 9), 114–122. doi: 10.1002/prot.22570
Kumar, G. S., Sarita, S., Kumar, G. M., Pant, K. K., and Seth, P. K. (2010). Definition of potential targets in Mycoplasma Pneumoniae through subtractive genome analysis. J. Antivir. Antiretrovir. 2, 038–041. doi: 10.4172/jaa.1000020
Kuszewski, J., Gronenborn, A. M., and Clore, G. M. (1997). Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J. Magn. Reson. 125, 171–177. doi: 10.1006/jmre.1997.1116
Lanciotti, R. S., Roehrig, J. T., Deubel, V., Smith, M., Parker, K., Steele, B., et al. (1999). Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286, 2333–2337. doi: 10.1126/science.286.5448.2333
Lapelosa, M., Gallicchio, E., Arnold, G. F., Arnold, E., and Levy, R. M. (2009). In silico vaccine design based on molecular simulations of rhinovirus chimeras presenting HIV-1 gp41 epitopes. J. Mol. Biol. 385, 675–691. doi: 10.1016/j.jmb.2008.10.089
Larsen, J. E., Lund, O., and Nielsen, M. (2006). Improved method for predicting linear B-cell epitopes. Immunome Res. 2:2. doi: 10.1186/1745-7580-2-2
Larsen, M. V., Lundegaard, C., Lamberth, K., Buus, S., Brunak, S., Lund, O., et al. (2005). An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 35, 2295–2303. doi: 10.1002/eji.200425811
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291. doi: 10.1107/S0021889892009944
Liao, L., and Noble, W. S. (2003). Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput. Biol. 10, 857–868. doi: 10.1089/106652703322756113
Lindenbach, B. D., Thiel, H. J., and Rice, C. M. (2007). “Flaviviridae: the virus and their replication,” in Fields Virology, 5th Edn. Vol. 1, eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Lippincott-Raven), 1101.
López, J. A., Weilenman, C., Audran, R., Roggero, M. A., Bonelo, A., Tiercy, J. M., et al. (2001). A synthetic malaria vaccine elicits a potent CD8+ and CD4+ T lymphocyte immune response in humans. Implications for vaccination strategies. Eur. J. Immunol. 31, 1989–1998. doi: 10.1002/1521-4141(200107)31:7<1989::AID-IMMU1989>3.0.CO;2-M
Mattar, S. E., Edwards, J., Laguado, M., Gonzalez, J., and Komar, N. N. (2005). West Nile virus antibodies in Colombian horses. Emerg. Infect. Dis. 11, 1497–1498. doi: 10.3201/eid1109.050426
Melo, F., Devos, D., Depiereux, E., and Feytmans, E. (1997). ANOLEA: a www server to assess protein structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 187–190.
Mishra, N. K., and Raghava, G. P. (2011). Prediction of specificity and cross-reactivity of kinase inhibitors. Lett. Drug Des. Discov. 8, 223–228 doi: 10.2174/157018011794578204
Morens, D. M., and Fauci, A. S. (2013). Emerging infectious diseases: threats to human health and global stability. PLoS Pathog. 9:e1003467. doi: 10.1371/journal.ppat.1003467
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., and Belew, R. K. (2009). Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 30, 2785–2791. doi: 10.1002/jcc.21256
Muh, H. C., Tong, J. C., and Tammi, M. T. (2009). AllerHunter: a SVM-pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS ONE 4:e5861. doi: 10.1371/journal.pone.0005861
Nair, D. T., Singh, K., Siddiqui, Z., Nayak, B. P., Rao, K. V., and Salunke, D. M. (2002). Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J. Immunol. 168, 2371–2382. doi: 10.4049/jimmunol.168.5.2371
Noueiry, A. O., Olivo, P. D., Slomczynska, U., Zhou, Y., Buscher, B., Geiss, B., et al. (2007). Identification of novel small-molecule inhibitors of West Nile virus infection. J. Virol. 81, 11992–12004. doi: 10.1128/JVI.01358-07
Oany, A. R., Ahmad, S. A. I., Hossain, M. U., and Jyoti, T. P. (2015). Highly conserved antigenic epitope regions in RNA dependent RNA polymerase-L of Crimean-Congo haemorrhagic fever virus: insights about novel vaccine. Adv. Appl. Bioinform. Chem. 8, 1–10. doi: 10.2147/AABC.S75250
OL Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: an open chemical toolbox. J. Cheminform. 3:33. doi: 10.1186/1758-2946-3-33
Parker, J. M., Guo, D., and Hodges, R. S. (1986). New hydrophilicity scale derived from highperformance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 23:25.
Peters, B., and Sette, A. (2005). Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132. doi: 10.1186/1471-2105-6-132
Petersen, L. R., and Marfin, A. A. (2002). West Nile virus: a primer for the clinician. Ann. Intern. Med. 137, 173–179. doi: 10.7326/0003-4819-137-3-200208060-00009
Petersen, L. R., Marfin, A. A., and Gubler, D. J. (2003). West Nile virus. JAMA 290, 524–528. doi: 10.1001/jama.290.4.524
Poland, G. A., Ovsyannikova, I. G., and Jacobson, R. M. (2009). Application of pharmacogenomics to vaccines. Pharmacogenomics 10, 837–852. doi: 10.2217/pgs.09.25
Rini, J. M., Schulze-Gahmen, U., and Wilson, I. A. (1992). Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 255, 959–965. doi: 10.1126/science.1546293
Roehrig, J. T. (2003). Antigenic structure of flavivirus proteins. Adv. Virus Res. 59, 141–176. doi: 10.1016/S0065-3527(03)59005-4
Šali, A., Potterton, L., Yuan, F., van Vlijmen, H., and Karplus, M. (1995). Evaluation of comparative protein modelling by MODELLER. Proteins 23, 318–326. doi: 10.1002/prot.340230306
Sander, T. (2001). OSIRIS property explorer. Organic Chemistry Portal. Available online at: http://www.organic-chemistry.org/prog/peo
Sayle, R. A., and Milner-White, E. J. (1995). RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 20, 374–376. doi: 10.1016/S0968-0004(00)89080-5
Sejvar, J. J., Haddad, M. B., Tierney, B. C., Campbell, G. L., Marfin, A. A., Van Gerpen, J. A., et al. (2003). Neurologic manifestations and outcome of West Nile virus infection. JAMA 290, 511–515. doi: 10.1001/jama.290.4.511
Sharmin, R., and Islam, A. B. M. M. K. (2014). A highly conserved WDYPKCDRA epitope in the RNA directed RNA polymerase of human coronaviruses can be used as epitope-based universal vaccine design. BMC Bioinformatics 15:161. doi: 10.1186/1471-2105-15-161
Shi, P. Y., Brinton, M. A., Veal, J. M., Zhong, Y. Y., and Wilson, W. D. (1996). Evidence for the existence of a pseudoknot structure at the 3' terminus of the flavivirus genomic RNA. Biochemistry 35, 4222–4230.
Sippl, M. J. (1993). Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362. doi: 10.1002/prot.340170404
Söding, J. (2005). Protein homology detection by HMM–HMM comparison. Bioinformatics 21, 951–960. doi: 10.1093/bioinformatics/bti125
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248. doi: 10.1093/nar/gki408
Tenzer, S., Peters, B., Bulik, S., Schoor, O., Lemmel, C., Schatz, M. M., et al. (2005). Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 62, 1025–1037. doi: 10.1007/s00018-005-4528-2
The UniProt Consortium (2014). Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198. doi: 10.1093/nar/gkt1140
Thevenet, P., Shen, Y., Maupetit, J., Guyon, F., Derreumaux, P., and Tufféry, P. (2012). PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 40, W288–W293. doi: 10.1093/nar/gks419
Trott, O., and Olson, A. J. (2010). AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461. doi: 10.1002/jcc.21334
Van Joolingen, W. R., Jong, T. D., Lazonder, A. W., Savelsbergh, E. R., and Manlove, S. (2005). Co-Lab: research and development of an online learning environment for collaborative scientific discovery learning. Comput. Human Behav. 21, 671–688. doi: 10.1016/j.chb.2004.10.039
Vriend, G., and Sander, C. (1993). Quality control of protein models: directional atomic contact analysis. J. Appl. Cryst. 26, 47–60. doi: 10.1107/S0021889892008240
Wang, F., Xiao, J., Pan, L., Yang, M., Zhang, G., Jin, S., et al. (2008). A systematic survey of mini-proteins in bacteria and archaea. PLoS ONE 3:e4027. doi: 10.1371/journal.pone.0004027
Westaway, E. G., Mackenzie, J. M., and Khromykh, A. A. (2002). Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 267, 323–351. doi: 10.1007/978-3-642-59403-8_16
Keywords: West Nile Virus, immunoinformatics, pharmacoinformatics, vaccine design, drug design
Citation: Hossain MU, Keya CA, Das KC, Hashem A, Omar TM, Khan MA, Rakib-Uz-Zaman SM and Salimullah M (2018) An Immunopharmacoinformatics Approach in Development of Vaccine and Drug Candidates for West Nile Virus. Front. Chem. 6:246. doi: 10.3389/fchem.2018.00246
Received: 27 July 2017; Accepted: 08 June 2018;
Published: 06 July 2018.
Edited by:
Rajeev K. Singla, Netaji Subhas Institute of Technology, IndiaReviewed by:
Dharmendra Kumar Yadav, Gachon University of Medicine and Science, South KoreaCopyright © 2018 Hossain, Keya, Das, Hashem, Omar, Khan, Rakib-Uz-Zaman and Salimullah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Md. Salimullah, c2FsaW0yOTY5QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.