 Qiang He
Qiang He Hui Liu2
Hui Liu2 Shuwen Deng
Shuwen Deng Xuan Jiang
Xuan Jiang Wei Lu
Wei Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 31 August 2020
Sec. Cell Death and Survival
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00830
Increasing evidence shows that, in addition to the classical function of protein processing and transport, the Golgi apparatus (GA) is also involved in apoptosis, one of the most common forms of cell death. The structure and the function of the GA is damaged during apoptosis. However, the specific effect of the GA on the apoptosis process is unclear; it may be involved in initiating or promoting apoptosis, or it may inhibit apoptosis. Golgi-related apoptosis is associated with a variety of neurological diseases including glioma, Alzheimer’s disease (AD), Parkinson’s disease (PD), and ischemic stroke. This review summarizes the changes and the possible mechanisms of Golgi structure and function during apoptosis. In addition, we also explore the possible mechanisms by which the GA regulates apoptosis and summarize the potential relationship between the Golgi and certain neurological diseases from the perspective of apoptosis. Elucidation of the interaction between the GA and apoptosis broadens our understanding of the pathological mechanisms of neurological diseases and provides new research directions for the treatment of these diseases. Therefore, we propose that the GA may be a potential therapeutic target for apoptosis-related neurological diseases.
In plants, invertebrates, and many protists, Golgi stacks are independent of each other and scattered throughout the cytoplasm. In vertebrates, Golgi mini-stacks are laterally fused into a twisted continuous ribbon structure that is maintained at the centrosome and paranuclear position by interaction with microtubules and dynein motors (Makhoul et al., 2018; Kulkarni-Gosavi et al., 2019). This difference obviously reveals that the higher complexity of the Golgi apparatus (GA) structure is required for more complex functions in vertebrates, especially mammals. The GA lies at the heart of the secretory pathway; proteins and lipids are sequentially modified and processed in the Golgi stacks to ultimately help target transports to their correct destination (Lowe, 2011). In addition to these classic functions, the GA is also involved in higher-order functions such as mitosis (Lowe et al., 2000), DNA repair (Farber-Katz et al., 2014), stress response (Li T. et al., 2016), autophagy (Chang et al., 2012), inflammation (Chen and Chen, 2018), and apoptosis (Chiu et al., 2002). Furthermore, the GA is an important signaling platform for many cascade signals originating from the plasma membrane or other organelles, including ubiquitin ligases, phospholipases, phosphatases, and many types of trimeric G proteins (Mayinger, 2011).
Apoptosis is an orderly autonomous death process controlled by genes. In this process, cells that are harmful to the body or unneeded will be removed (Peter, 2011). If infected cells are apoptotic before viral progeny are produced, virus replication will be limited (Nguyen and Blaho, 2006). In ischemic stroke, apoptosis is a vital pathway that mediates neuron death (Xu et al., 2019). Three main apoptotic pathways are known: receptor-mediated apoptotic pathways, mitochondria-mediated apoptotic pathways, and endoplasmic reticulum (ER)-mediated apoptotic pathways. These apoptotic pathways will eventually be integrated into the caspase cascade pathway, where the effector caspases will cleave proteins that maintain cell structure and metabolism. Then, the apoptotic cells will be engulfed and degraded by nearby phagocytes or other surrounding cells. In receptor-mediated apoptotic pathways, the most common death receptors on the plasma membrane surface [such as Fas and tumor necrosis factor receptors (TNFRs)] bind to the corresponding ligands to transmit apoptotic signals, thereby activating the caspase cascade pathway. In mitochondria-mediated apoptotic pathways, the mitochondrial permeability transition pore is irreversibly over-opened under various proapoptotic signals, which causes the release of cytochrome C (an active proapoptotic protein) from mitochondria into the cytosol to initiate the downstream apoptotic pathway. In ER-mediated apoptotic pathways, the most common apoptotic pathway (unfolded protein response, UPR) is primarily triggered by misfolded and unfolded proteins in the ER. In UPR, CHOP, and caspase 12 play key roles in apoptotic signal transmission (Elmore, 2007; Savitskaya and Onishchenko, 2015). In fact, according to the source of the apoptosis-stimulating signal, the mitochondria- and the ER-mediated apoptosis pathways can be collectively referred to as intrinsic apoptosis pathways, and the receptor-mediated apoptosis pathways can be referred to as extrinsic apoptotic pathways. The above-mentioned apoptotic pathways are regulated by the Bcl-2 family of proteins, which is divided into anti-apoptotic proteins (such as Bcl-2 and Bcl-xL) and proapoptotic proteins (such as Bak and Bax) (Popgeorgiev et al., 2018). In addition, p53 transcription factor activation promotes the expression of proapoptotic proteins such as Bax, Puma, and NoxA. In turn, these factors induce mitochondria-mediated apoptosis and subsequent caspase cascade activation (Savitskaya and Onishchenko, 2015).
The in-depth study of the apoptosis mechanism has led to an increasing number of technical means for detecting apoptosis. However, morphological observations play an important role in distinguishing apoptosis from other types of cell death. Of these, transmission electron microscopy is considered to be the gold standard for confirming apoptosis (White and Cinti, 2004). Apoptotic cell characteristics that can be observed by transmission electron microscopy include the following: (1) an electron-dense nucleus (marginalization in the early phase), (2) nuclear fragmentation, (3) an intact cell membrane even late in the cell disintegration phase, (4) disorganized cytoplasmic organelles, (5) large clear vacuoles, and (6) blebs at the cell surface. Cells will break into apoptotic bodies with intact plasma membranes that contain cytoplasmic organelles in the later stages of apoptosis (Elmore, 2007). Obviously, Golgi morphology also changes drastically during apoptosis. In normal cells viewed by transmission electron microscopy, the GA pattern is initially highly ordered, appearing as ribbon-like stacks in a vesicular structure. In cells treated with apoptosis inducers, the Golgi stacks become swollen, disordered, and lysed into vesicles and tubules (Snigirevskaia et al., 2014). Similarly, Golgi marker immunofluorescence shows that the GA pattern is dispersed and fragmented throughout the cytoplasm in apoptotic cells, unlike in normal cells where it accumulates in the perinuclear area in a typical semilunar shape (Bottone et al., 2013). While morphological alterations of the GA are common in various physiological or pathological conditions, the Golgi fragmentation mechanism in apoptosis is quite distinct (Figure 1).
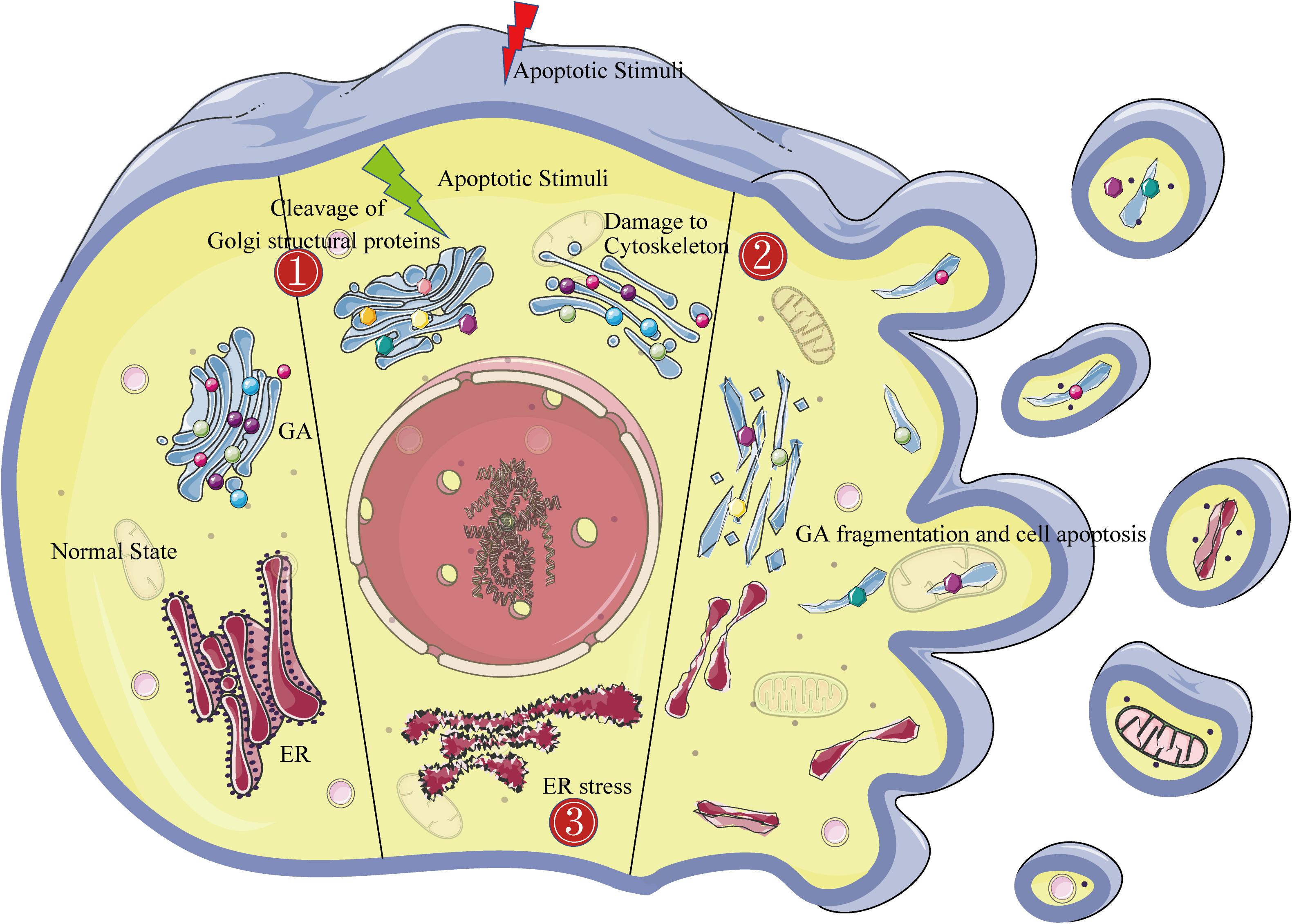
Figure 1. The Golgi fragmentation mechanism during apoptosis. The left side of the figure shows the morphological structure of the endoplasmic reticulum and the Golgi in normal cells. The middle of the figure shows the three mechanisms of Golgi fragmentation in apoptotic cells: ➀ activated caspases cleave Golgi structural proteins (represented by dots of various colors); ➁ the cytoskeleton is damaged (represented by blue bands); and ➂ endoplasmic reticulum stress. The right side of the figure shows the complete fragmentation of the Golgi apparatus at the end of apoptosis.
One of the characteristics of apoptosis is caspase cascade signaling activation including caspase 3, caspase 8, or caspase 9 (Savitskaya and Onishchenko, 2015). Activated caspases cleave important structural proteins and cause the destruction of organelles, such as the Golgi (Chiu et al., 2002), mitochondria (Yu et al., 2014), and nucleus (Al-Ghorbani et al., 2016). The currently known Golgi structural proteins that can be cleaved by activated caspases are described in the following subsections.
One of the most widely studied Golgi matrix proteins is GM130, which is anchored to the cis-Golgi. An important step in the biogenesis of the Golgi ribbon is to dynamically and continuously incorporate vesicles from the ER into the Golgi stacks (Marra et al., 2007). GM130 can specifically capture vesicles from the ER (Wong and Munro, 2014; Lowe, 2019). If GM130 is knocked out, this capture process will be ablated, resulting in the shortening of the Golgi cisternae and the disruption of the Golgi ribbon (Marra et al., 2007; Liu et al., 2017). Recent studies have shown that the methylation of the N-terminal arginine of GM130 plays an important role in maintaining the structure of the Golgi (Zhou et al., 2010). During Fas-induced apoptosis, GM130 decreases rapidly, accompanied by GA fragmentation (Walker et al., 2004). He et al. (2020) found that HSV-1 infection-induced endothelial cell apoptosis and Golgi fragmentation were significantly reduced after GM130 cleavage was inhibited by the pan-caspase inhibitor, Z-VAD. These results reveal that activated caspases cleave GM130, which might be involved in GA disruption during apoptosis.
Giantin, primarily localized to the Golgi cisternal rims, is the largest golgin in mammals (Linstedt et al., 1995). Koreishi et al. (2013) found that giantin plays a role in laterally linking the Golgi cisternae within the Golgi ribbon. Depletion of giantin with siRNA causes dispersion of the Golgi stacks. However, the exogenous expression of giantin in Drosophila S2 cells promotes Golgi stack clustering, similar to the Golgi ribbon in mammalian cells (Koreishi et al., 2013). During apoptosis, giantin is cleaved by caspase 2, caspase 3, and caspase 7, accompanied by the cessation of vesicular transport between the ER and the GA (Lowe et al., 2004). We speculate that cleavage of giantin by activated caspases could promote fragmentation of the Golgi.
P115 consists of an N-terminal globular head, a coiled-coil tail, and a short C-terminal acidic domain, often forming dimers which are found in the cis-Golgi (An et al., 2009). It was initially found that P115 mainly acts on intercisternal transport in the Golgi stacks (Waters et al., 1992). Both GM130 and giantin can bind to the C-terminal acidic tail of P115. Further experiments confirmed that P115, GM130, and giantin form a tethering complex, tethering coat protein complex I vesicles to the Golgi membranes (Sönnichsen et al., 1998; Beard et al., 2005). Mice lacking P115 exhibit destruction of the Golgi structure, revealing the importance of P115 in maintaining the integrity of the Golgi (Kim et al., 2012). A recent experiment found that P115 is cleaved by caspase 3 and caspase 8 during apoptosis (Chiu et al., 2002). Relative to that, in control cells expressing wild-type P115, cells expressing the cleavage-resistant form of P115 show delayed Golgi fragmentation during apoptosis. These experimental results revealed that Golgi fragmentation during apoptosis is partly due to P115 cleavage (Chiu et al., 2002).
GRASP65 is mainly targeted to the cis-Golgi and is a member of the Golgi reassembly and stacking protein (GRASP) family (Zhang and Wang, 2015). However, whether GRASP65 acts as a stacking factor is still controversial. This is because, in mammalian cells, there are contradictory reports on the integrity of the Golgi stack after the depletion of GRASP65 (Xiang and Wang, 2010; Rabouille and Linstedt, 2016). A less controversial role of GRASP65 is that it can promote formation of the Golgi ribbons in mammals. Jarvela et al. (2014) found that the inactivation of GRASP65 specifically causes the cis-side of the Golgi ribbons to be disconnected. In addition, Cheng et al. (2010) demonstrated that Golgi fragmentation is dependent on caspases and identified GRASP65 as a new substrate for caspase 3. The expression of the caspase-resistant form of GRASP65 partially inhibits Golgi fragmentation in apoptotic cells (Lane et al., 2002).
Golgin160 (also known as GOLGA3) is a coiled-coil protein enriched in the cis-Golgi cisternae (Hicks et al., 2006). Yadav et al. (2009, 2012) have proved that golgin160 can recruit the dynein microtubule motor protein to the GA, which is important for maintaining the positioning of the GA and the integrity of the Golgi ribbons. The loss of golgin160 in cultured cells results in Golgi positioning defect and generates dispersed ministacks (Yadav et al., 2009, 2012). During apoptosis, golgin160 is cleaved by caspase 2, caspase 3, and caspase 7 (Mancini et al., 2000). The inhibition of caspase-mediated golgin160 cleavage can prevent GA fragmentation and even apoptosis. The exogenous expression of a caspase-resistant mutant of golgin160 attenuates the kinetics of GA fragmentation, revealing that caspase-mediated cleavage of golgin160 is involved in inducing apoptotic changes in Golgi morphology (Mancini et al., 2000; Maag et al., 2005).
Golgin84 is a member of the golgin family, which is mainly presented on the Golgi cisternal rims (Gillingham and Munro, 2016). Satoh et al. (2003) found that golgin84 can stimulate Golgi stacking and increase the length of the Golgi stacks. In addition, it is generally believed that golgin84 plays an important role in tethering the intra-Golgi transport vesicles (Sohda et al., 2010; Lowe, 2019). In golgin84-knockdown cells, the Golgi ribbon breaks into ministacks, accompanied by the accumulation of some intra-Golgi transport vesicles (Sohda et al., 2010). During apoptosis triggered by Chlamydia trachomatis infection, activated caspases cleave golgin84, accompanied by GA fragmentation. However, inhibiting golgin84 proteolytic cleavage can prevent Golgi fragmentation (Heuer et al., 2009).
Syntaxin 5, located in the cis-Golgi cisternae and ER, is a member of the SNARE family (Hui et al., 1997; Linders et al., 2019). It specifically pairs with other homologous SNAREs to form the “SNARE complex,” driving the fusion of the vesicle membrane with the target membrane. It has been found that syntaxin 5 is involved in the vesicle fusion events of ER–Golgi, intra-Golgi, and early/recycling endosome to the trans-Golgi network trafficking (Dascher et al., 1994; Tai et al., 2004; Volchuk et al., 2004). RNA interference-mediated silencing of syntaxin 5 leads to fragmentation of the GA, revealing that syntaxin 5 is necessary for maintenance of the Golgi structure (Suga et al., 2005). During apoptosis, syntaxin 5 is cleaved by caspase 3 and caspase 7, which is accompanied by disorders in vesicular trafficking (Lowe et al., 2004). We speculate that cleavage of syntaxin 5 by activated caspases is involved in Golgi fragmentation during apoptosis.
Taken together, the aforementioned Golgi proteins are either related to vesicle tethering and fusion or to Golgi stacking and ribbon formation, which are involved in maintaining the structural integrity of the Golgi. Moreover, these proteins have been confirmed to be cleaved by activated caspases during apoptosis (Table 1). Therefore, we propose that, under different apoptotic stimuli, activated caspases cleave one or several Golgi structural proteins, resulting in fragmentation of the GA.
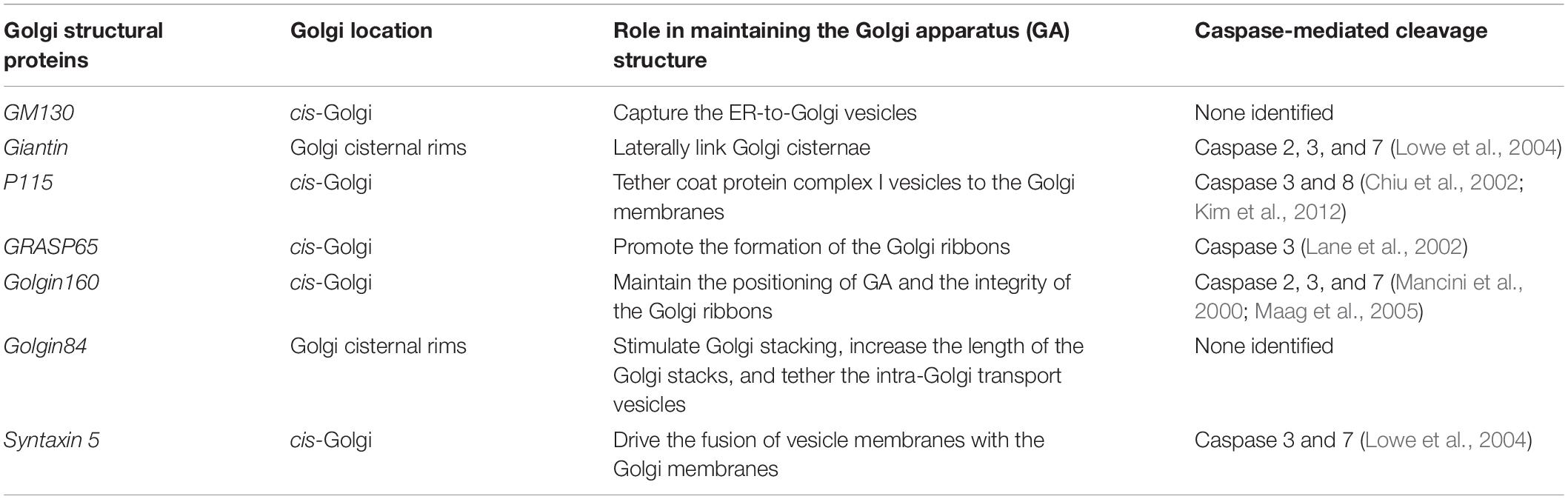
Table 1. Caspase cleavage of Golgi structural proteins associated with Golgi fragmentation during apoptosis.
The cytoskeleton mainly consists of microtubules and actin filaments that are essential for maintaining GA structure. It is generally believed that microtubules play a key role in generating and positioning the Golgi ribbon and that actin finely and synergistically regulates the Golgi architecture (Makhoul et al., 2018). Golgi-derived microtubules work in a search-and-capture manner to contact other nearby Golgi stacks. These Golgi stacks move along the microtubules toward the minus end and eventually gather near the perinuclear centrosome (Lowe, 2011). Tang et al. (2016) found that Mena interacts with GRASP65 to promote local actin polymerization, thereby promoting the interconnection between Golgi stacks to form the Golgi ribbon. In motor neurons, the overexpression of microtubule-destabilizing proteins stathmin 1 and stathmin 2 led to the defective polymerization of Golgi-derived microtubules and severe Golgi fragmentation; however, stathmin 1/2 knockdown or treatment with the microtubule-stabilizing drug taxol completely reverses this effect (Bellouze et al., 2016). Perforation/fragmentation and severe swelling of Golgi cisternae have been observed by transmission electron microscopy using actin-depolymerizing toxins (cytochalasin D, latrunculin B, mycalolide B, and Clostridium botulinum C2 toxin) (Lazaro-Dieguez et al., 2006). In addition, recent studies found that the formin homology 2 (FH2) domain protein 1 (FHDC1) enriched on cis-Golgi binds microtubules through a unique C-terminal domain and actin through its FH2 domain. Knockdown or overexpression of FHDC1 can cause the dispersal of the Golgi ribbon into mini-stacks, indicating the potential role of actin and microtubule Golgi networks in maintaining the Golgi ribbon structure (Copeland et al., 2016). Microtubules are reportedly depolymerized at apoptosis execution phase onset (Moss and Lane, 2006; Ndozangue-Touriguine et al., 2008). However, recent results indicate that the microtubule network reforms during the later stages of apoptosis and contributes to nuclear and cell fragmentation (Moss and Lane, 2006). Actin is similarly degraded during apoptosis (Coleman and Olson, 2002). Moreover, mounting evidence proves that the actin cytoskeleton is a sensor and mediator of apoptosis (Desouza et al., 2012). In addition, both tubulin and actin are cleaved by caspases (Fischer et al., 2003; Moss and Lane, 2006). Therefore, given the importance of the cytoskeleton to the GA, we speculate that cytoskeleton damage may contribute to GA fragmentation in apoptosis.
Endoplasmic reticulum and GA are closely related in morphology and function. The stability of the Golgi dynamic structure depends on efficient bidirectional vesicle transport with the ER (Kokubun et al., 2019). In a stacked Golgi, new cisternae would form at the cis face, pass through the stack, and then peel off from the trans face, carrying secretory cargo proteins forward. The vesicles of the ER would shuttle to the Golgi to form new cisternae to maintain stack integrity (Mollenhauer and Morré, 1991; Emr et al., 2009). The ER is an important organelle in eukaryotic cells that is mainly responsible for the synthesis, processing, and modification of intracellular proteins. However, unfolded and misfolded proteins can accumulate in the ER and lead to ER stress when cells are subjected to a variety of strong stimulating factors such as apoptosis inducers, toxin stimulation, or Ca2+ metabolic imbalance. When the stimulus persists or is too strong, ER stress activates the apoptotic pathway and disrupts the ER-to-GA transport (Short et al., 2007; Qi and Chen, 2019). The GA becomes disordered and diffused under treatment with the ER stress inducer thapsigargin (Nakagomi et al., 2008). In addition, mutant SOD1 inhibits the ER-to-GA transport of secreted proteins in neurons, thereby causing GA fragmentation (Atkin et al., 2014). Protein-disulfide isomerase Bax inhibitor 1 (BI-1), located on the ER, inhibits ER stress. Nakagomi et al. (2008) found that BI-1 overexpression in cortical neurons can partially inhibit the Golgi fragmentation induced by various insults, further revealing that ER stress plays an important role in regulating Golgi morphology. Based on these data, we propose that ER stress causes GA fragmentation to varying degrees by disrupting the ER-to-GA transport during ER stress-triggered apoptosis.
The GA is damaged by various pathways during apoptosis. However, increasing evidence shows that the subcellular organelle is involved in apoptosis signaling pathway regulation and that the GA is also involved (Hicks and Machamer, 2005; Sasaki and Yoshida, 2019). Studies have identified many components located in the GA that are related to apoptosis regulation, such as P115, GRASP65, and Bruce (Cheng et al., 2010; How and Shields, 2011; Tassi et al., 2012). A highly conserved Golgi anti-apoptotic protein (GAAP) located only in the GA has also been found (Carrara et al., 2017). The following sections focus on the related molecules and mechanisms of Golgi that promote or inhibit apoptosis signals (Figures 2, 3).
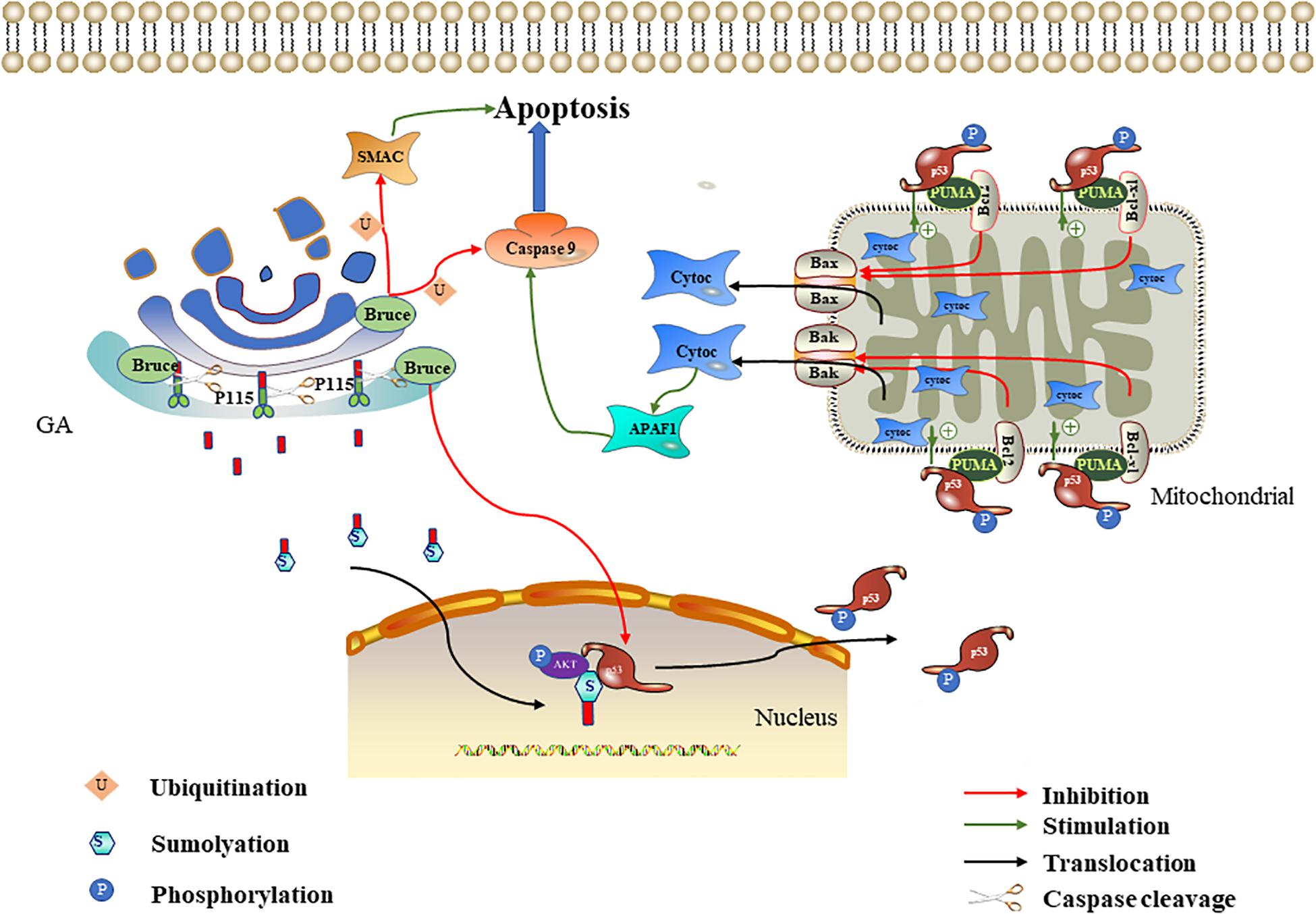
Figure 2. Effect of P115 and Bruce on apoptosis. The p115 caspase cleavage fragment promotes apoptosis mediated by the ERK/p53/p53 upregulated modulator of apoptosis (PUMA) pathways. The p115 caspase cleavage fragment enters the nucleus after being sumoylation and then acts as a scaffold to promote ERF-mediated p53 phosphorylation. The phosphorylated p53 upregulates PUMA. PUMA then relieves the Bcl-2/Bcl-xl inhibition of Bax/Bak, leading to mitochondrial cytochrome c release. Cytochrome c activates APAF1 and caspase 9 in succession to promote apoptosis. Bruce degrades caspase 9/SMAC through ubiquitination and downregulates p53 to inhibit apoptosis.
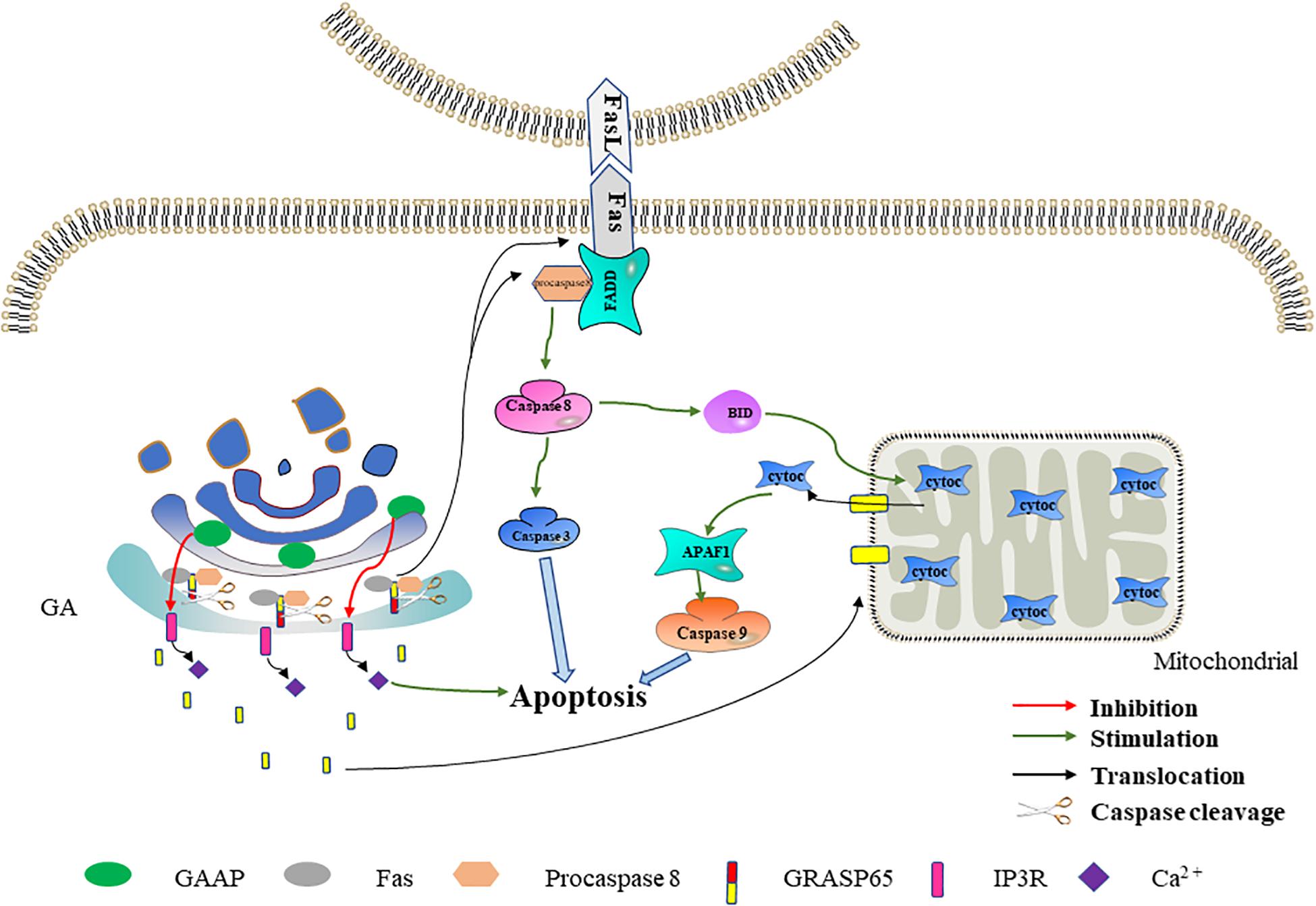
Figure 3. The effect of Golgi reassembly and stacking protein 65 (GRASP65) and Golgi anti-apoptotic protein (GAAP) on apoptosis. GRASP65 caspase cleavage promotes Fas-mediated apoptosis. After GRASP65 is cleaved by caspases, Golgi-tethered Fas and procaspase 8 are transported to the cell membrane, thereby further promoting apoptosis. In addition, the caspase cleavage fragment of GRASP65 will translocate to the mitochondria, thereby promoting the release of cytochrome c and promoting apoptosis. GAAP inhibits Ca2+ release into the cytoplasm by interacting with IP3R. The decreased Ca2+ in the cytoplasm inhibits apoptosis to a certain extent.
P115 is a 961-kDa Golgi peripheral membrane protein. It is mainly involved in vesicle transport between Golgi cisternae (Sapperstein et al., 1995). In Chiu et al. (2002) found that P115 was cleaved by caspase 3 and caspase 8 at residue TEKD757, resulting in a C-terminal fragment (CTF) containing 205 residues during apoptosis. CTF enters the nucleus through sumoylation (Mukherjee and Shields, 2009). The CTF in the nucleus serves as a scaffold to tether ERK and p53, thereby promoting p53 phosphorylation and activation by ERK (How and Shields, 2011). ERK is a mitogen-activated protein kinase, and its signaling cascade plays an important role in apoptosis (Li et al., 2014; da Cunha Jaeger et al., 2020). p53 is an important tumor suppressor gene, and it is often involved in the transcriptional regulation of key pro-apoptotic genes, such as p53 upregulated modulator of apoptosis (PUMA) and hdm2 (Khan et al., 2020). PUMA is a member of the Bcl-2 family that interacts with anti-apoptotic Bcl-2 family members (Bcl-xl and Bcl-2) and relieves the Bcl-2/Bcl-xl inhibition of Bax/Bak. When the Bax/Bak inhibition is lifted, they will translocate to the mitochondria and trigger the release of cytochrome C; this ultimately activates the caspase cascade signaling pathway and thereby induces apoptosis (Nakano and Vousden, 2001). How and Shields (2011) further proved that P115 CTF expression can promote p53 activation, thereby increasing PUMA expression. The above-mentioned literature indicates that, in the early stage of apoptosis, P115 located on the Golgi is partly cleaved to produce CTF. P115 CTF promotes the mitochondria-mediated apoptosis pathway through the ERK/p53/PUMA pathway, thereby amplifying the apoptotic signal and leading to the full activation of caspases and apoptosis (Hilton et al., 2014; Stokholm et al., 2016; Figure 2).
Bruce (also known as apollon) is a conserved peripheral membrane protein located on the trans-Golgi. Its amino and carboxyl termini contain the baculovirus-IAP repeat (BIR) and the ubiquitin-conjugating enzymes (UBC) domain, respectively (Hauser et al., 1998). Bruce is a member of the IAP family, so named for their ability to bind and inhibit caspases and other pro-apoptotic factors (Pohl and Jentsch, 2008; Vasudevan and Ryoo, 2015). In 293T and HeLa cells, overexpressed Bruce can inhibit apoptosis induced by various stimuli, including staurosporine and ultraviolet light (Bartke et al., 2004). Bruce can inhibit apoptosis in multiple ways, and its inhibitory effect mainly plays a role in the mitochondria-mediated apoptosis pathway. On the one hand, the BIR motif on Bruce binds to activated caspase 9 and SMAC, and then the UBC domain is used to induce the ubiquitination of the two, thereby inhibiting apoptosis. Because the mitochondria release pro-apoptotic proteins SMAC and cytochrome c after mitochondria-mediated apoptosis is triggered, cytochrome c combines with Apaf-1 and dATP to form the apoptosome (a multi-protein platform with a catalytic effect). It can recruit and activate caspase 9, leading to caspase cascade signaling pathway activation (Zhang, 2019; Li et al., 2020). Therefore, the Golgi can degrade caspase 9 and SMAC through Bruce to inhibit apoptosis. Interestingly, Bruce can also be cleaved by activated caspase 9 or degraded by SMAC through the ubiquitination pathway (Hao et al., 2004; Qiu and Goldberg, 2005). Therefore, the degree of apoptosis depends on the relative activities of Bruce, caspase 9, and SMAC. On the other hand, the Golgi can inhibit apoptosis by downregulating p53 through Bruce. Ren et al. (2005) found that the deletion of the C-terminal half of Bruce activates p53 in mice, which in turn upregulates bax and bak and leads to mitochondria-mediated apoptosis. Therefore, we speculate that, under normal physiological conditions, complete Bruce should inhibit p53 expression, thereby down-regulating bax and bak and inhibiting mitochondria-mediated apoptosis. In conclusion, the Golgi can degrade caspase 9/SMAC and downregulate p53 through Bruce to inhibit apoptosis (Figure 2).
Fas (also known as CD95) is a member of the TNFR superfamily, which transmits apoptotic signals by binding to Fas ligand (FasL) expressed in the cell membranes of other cells (Andera, 2009; Xie et al., 2019). The Fas–FasL system is a major signaling pathway for apoptosis induction. After binding to FasL, the Fas receptor interacts with the signal adapter, FADD, through its death domain (Thomas et al., 2004). FADD then recruits inactive procaspase 8 through a homologous motif. Fas, FADD, and procaspase 8 form a death-inducing signaling complex (Gomez-Angelats and Cidlowski, 2001). After the complex is formed, procaspase 8 drives its activation through self-cleavage. The activated caspase 8 directly activates the caspase cascade signaling pathway or cleaves BID to trigger the mitochondria-mediated apoptosis pathway to indirectly activate the caspase cascade signaling pathway, thereby leading to apoptosis (El Mchichi et al., 2007; Logue and Martin, 2008).
GRASP65 is a cis-Golgi protein that plays a role in Golgi structure, membrane trafficking, and cell signaling (Ahat et al., 2019a, b). In the early stage of apoptosis, cleavage of GRASP65 by caspase 3 may promote Fas-mediated apoptosis in one of two ways (Lane et al., 2002). In one method, the Golgi promote the translocation of Fas and procaspase 8 to the cell membrane by GRASP65 cleavage, thereby promoting Fas-mediated apoptosis. GRASP65 can regulate the transport of proteins containing C-terminal hydrophobic motifs on the GA through its tandem PDZ-type “GRASP” domains. The Fas C-terminus terminates with a hydrophobic leucine–valine motif (Ivanov et al., 2003). The interaction of GRASP65 with Fas and procaspase 8 has been detected in isolated Golgi components during hypoxia/reoxygenation-induced apoptosis (Wang et al., 2004). Therefore, we speculate that, in the early stage of apoptosis, GRASP65 tethers Fas and procaspase 8 to the GA through its PDZ domain. When GRASP65 is cleaved by caspase 3, Fas and procaspase 8 are transported to the cell membrane to promote apoptosis. In the other method, the GA releases the GRASP65 caspase fragment to the mitochondria, thereby promoting Fas-mediated apoptosis. Cheng et al. (2010) found that the C-terminus obtained by cleaving GRASP65 targets the mitochondria during Fas-mediated apoptosis. In addition, the GRASP65 caspase cleavage C-terminus fragment can make cells more sensitive to apoptosis induced by mitochondrial toxicants such as CCCP, antimycin A, and FasL (Cheng et al., 2010). These results reveal that, following the mitochondrial targeting of the caspase cleavage fragment released by GRASP65, mitochondrion permeability increases and thereby promotes cytochrome c release and apoptosis. Based on the above-mentioned data, we speculate that the Golgi may further promote Fas-mediated apoptosis through GRASP65 caspase cleavage (Figure 3).
A new anti-apoptotic protein has been isolated and identified in camel pox virus. It was named GAAP based on its intracellular localization and its first described function. GAAP has significant amino acid conservation with orthologs throughout eukaryotes, prokaryotes, and some orthopoxviruses, indicating a high degree of conserved functions (Gubser et al., 2007). In addition to its anti-apoptotic functions, GAAP also regulates the Ca2+ flux of the GA and ER and forms cation-selective channels. GAAP is widely expressed in various tissues of the human body (Gubser et al., 2007; de Mattia et al., 2009; Carrara et al., 2017).
Ca2+ is a ubiquitous intracellular messenger involved in both intrinsic and extrinsic apoptosis signaling pathways (Tiwari et al., 2017; Zhou et al., 2019). Under physiological conditions, Ca2+ in the cytoplasm is maintained at a low level, while subcellular organelles such as ER and GA store higher concentrations of Ca2+. Ca2+-induced signals are triggered by its transmembrane entry and/or release from the cell reservoir (mainly the ER and the GA) (Chung et al., 2017; Smaardijk et al., 2017). Ca2+ in the ER and the GA is mainly released through the IP3R (inositol-1,4,5-trisphosphate receptor) channel (Lin et al., 1999; Parys and Vervliet, 2020). The Ca2+ signals between Ca2+ storage organelles and mitochondria play an important role in promoting apoptosis (Marchi et al., 2018). Molecular and pharmacological methods that reduce cytoplasmic Ca2+ levels can protect cells from apoptosis, while conditions that increase cytoplasmic Ca2+ levels have the opposite effect (Ma et al., 1999; Nakamura et al., 2000; Pinton and Rizzuto, 2006). GAAP inhibits extracellular Ca2+ influx and reduces Ca2+ release from organelle reservoirs by interacting with IP3R, thereby reducing the increase in cytosolic Ca2+ flux induced by apoptotic stimuli (de Mattia et al., 2009). Therefore, we speculate that the Golgi may reduce the cytosolic Ca2+ flux and inhibit apoptosis through GAAP. In addition, Guo et al. (2018) found that GAAP in Arabidopsis interacts with IRE1, which initiates ER stress and thereby inhibits ER stress-induced apoptosis. The association between GAAP and ER stress in Arabidopsis provides a direction for future research on the human GAAP anti-apoptosis mechanism (Figure 3).
Apoptosis is an important pathological mechanism in many neurological diseases (Su et al., 2019), and the GA can perceive and transmit apoptotic signals through its own unique molecular mechanism (Machamer, 2015). More than 40% of GA-related genes known to be associated with disease affect the central or the peripheral nervous system, highlighting the importance of the GA in neurological diseases (Zappa et al., 2018). The following discussion explores the potential link between Golgi and certain neurological diseases from the perspective of apoptosis (Figure 4).
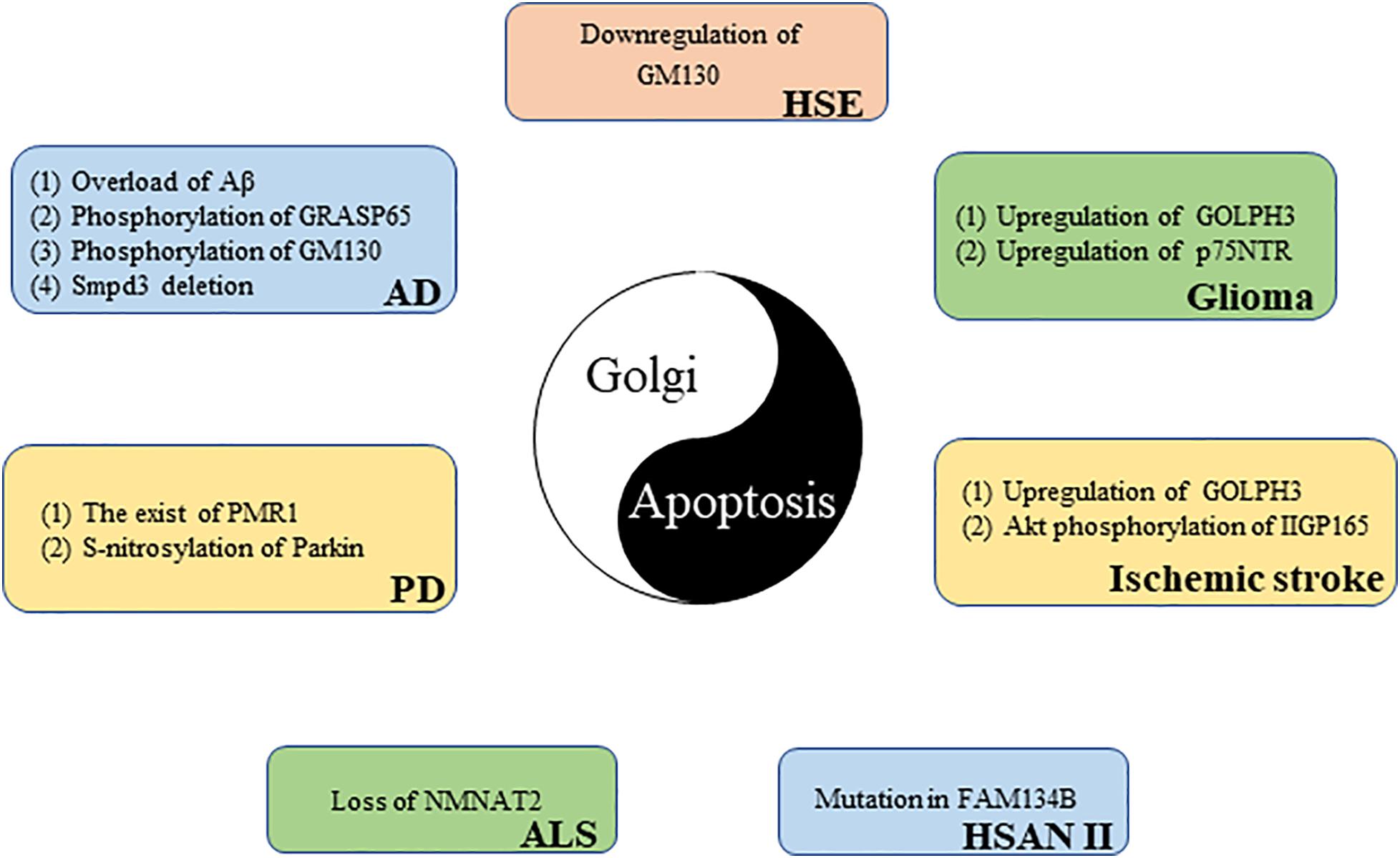
Figure 4. The link between the Golgi apparatus and neurological diseases from the perspective of apoptosis.
Neuroepithelial-derived tumors are collectively called gliomas; they account for 30% of all brain and central nervous system tumors and 80% of all malignant brain tumors (Goodenberger and Jenkins, 2012). GOLPH3, a member of the trans-Golgi network protein family, is the first example of an oncogene that functions in secretory trafficking at the GA (Buschman et al., 2015). GOLPH3 is highly expressed in glioma tissues, and its expression level is directly proportional to tumor malignancy. However, there is a negative correlation between tumor suppressor gene NDRG1 expression and GOLPH3 expression in glioma samples. GOLPH3 knockdown increases NDRG1 and triggers apoptosis in glioma cells. These results reveal that GOLPH3 causes glioma cells to escape apoptosis monitoring by downregulating NDRG1, which may promote glioma occurrence and development (Li X. et al., 2016). The poor prognosis of glioma patients is largely due to acquired chemotherapy resistance. Peng et al. (2018) found that GOLPH3 regulation sensitizes glioma cells to apoptotic stimuli induced by the chemotherapy drug temozolomide. The p75 neurotrophin receptor (p75NTR) is a glycoprotein belonging to the TNFR/nerve growth factor receptor family that is traditionally detected at the cell membrane (Wang et al., 2020). Recently, Giraud et al. (2011) found, using immunofluorescence microscopy, that a part of p75NTR is located in the GA of glioma cells. p75NTR retention in the GA prevents p75NTR expression on the cell surface. p75NTR can be sequestered in the GA; this sequestration is responsible for cell resistance to apoptosis and glioma formation (Giraud et al., 2011).
Alzheimer’s disease (AD) patients show a progressive decline in memory and cognitive function because AD leads to the gradual loss of neuronal function (Bondi et al., 2017). One of the main pathological features of AD is the abnormal production and deposition of amyloid beta peptide (Aβ). Aβ is mainly produced in the cis-Golgi (Choy et al., 2012). However, too much Aβ can cause GA fragmentation before cell death. Experiments revealed that Golgi fragmentation in AD is caused by GRASP65 phosphorylation, which is induced by Aβ-triggered cyclin-dependent kinase 5 (Cdk5) activation (Joshi et al., 2014). GM130 is also phosphorylated by Aβ-activated Cdk5, causing GA fragmentation (Sun et al., 2008). Golgi fragmentation, in turn, promotes Aβ production (Joshi et al., 2014). The aggregated Aβ neurotoxicity manifests as neuronal apoptosis, in which caspase 8, caspase 9, and caspase 3 are activated (Takada et al., 2020). These results reveal the importance of the Golgi in AD (Joshi et al., 2014). In addition, Stoffel et al. (2018) found that neutral sphingomyelinase, smpd3, expression in the central nervous system is restricted to the GA of neurons. In the brains of smpd3−/− mice, Golgi vesicular protein transport in neurons is inhibited, which leads to Aβ deposition, UPR, and apoptosis. Finally, smpd3−/− mice show a progressive cognitive decline similar to the clinical manifestations of familial and sporadic AD, indicating that smpd3 may be a susceptible gene for AD (Stoffel et al., 2018).
Amyotrophic lateral sclerosis (ALS) is a deadly neurodegenerative disease characterized by the progressive degeneration of motor neurons (Zhang et al., 2019). Nicotinamide adenine dinucleotide (NAD+) is an important cofactor for cell metabolism maintenance. By increasing NAD+ expression, survival time is moderately prolonged in ALS mice (Harlan et al., 2020). NMNAT2, a key enzyme responsible for NAD+ production, is mainly located in the GA (Milde et al., 2013). Harlan et al. (2020) found that the NMNAT2 expression is reduced in the spinal cord of ALS patients. The loss of NMNAT2 in neurons can cause GA fragmentation, further leading to neuronal apoptosis (Pottorf et al., 2018). These results suggest that NMNAT2 may be a potential target for the treatment of ALS.
Parkinson’s disease (PD) is a degenerative neurological disease commonly found in middle-aged and elderly people that it is characterized by a progressive loss of dopaminergic neurons (Surmeier, 2018). The main toxic effector of PD is α-synuclein, which destroys intracellular Ca2+ homeostasis and causes neuronal death. Buttner et al. (2013) demonstrated that the Ca2+/Mn2+ ATPase, PMR1, residing on the GA is a conserved mediator that can regulate α-synuclein-induced Ca2+ imbalance and cell apoptosis. In addition, parkin (the pathogenic gene for autosomal recessive juvenile PD) is partially located in the trans-Golgi network (Kubo et al., 2001). An increased expression of S-nitrosylated-parkin and p53 is simultaneously found in brain tissues of patients with PD. Sunico et al. (2013) further found that, in a mouse model of pesticide-induced PD, S-nitrosylation of parkin reduced its activity as a repressor of p53 gene expression, resulting in p53 upregulation, and p53 upregulation triggers neuronal apoptosis. These results suggest that p53-mediated neuronal apoptosis caused by S-nitrosylation of parkin is involved in the pathophysiology of PD.
Hereditary sensory and autonomic neuropathy type II (HSAN II) is a hereditary, degenerative, peripheral nervous system disease characterized by the progressive loss of peripheral sensory nerve function (Rotthier et al., 2012). Patients with HSAN II usually have progressive sensory and autonomic dysfunction that results in reduced sensitivity to pain, temperature, and touch (Arain and Chand, 2015). FAM134B is a newly identified cis-Golgi protein. The mutation of FAM134B directly causes HSAN II (Kurth et al., 2009; Falcao de Campos et al., 2019). Kurth et al. (2009) further found that FAM134B knockdown led to cis-Golgi compartment shrinkage that triggered the apoptosis of primary dorsal root ganglion neurons; this may be related to the neurotrophic factor transport disorder induced by FAM134B mutation. These results reveal that Golgi dysfunction is an important cause of neurogenetic diseases.
Herpes simplex virus encephalitis (HSE) caused by HSV-1 infection is the most common sporadic encephalitis worldwide (Piret and Boivin, 2020). DeBiasi et al. (2002) found apoptotic neurons and glial cells in the brain tissue sections of patients with acute HSE. The GA exhibit a disordered and diffused cytoplasmic distribution in HSV-1-infected primary neurons (Martin et al., 2017). In addition, blood–brain barrier disruption is an important pathological mechanism for HSE development (Liu et al., 2019). Recently, He et al. (2020) found that HSV-1-induced blood–brain barrier damage involves apoptosis that is associated with GM130-mediated Golgi stress. The expression of GM130 is downregulated and accompanied by GA fragmentation in brain microvascular endothelial cells infected with HSV-1. The downregulation of GM130 promotes caspase cascade pathway activation. Eventually, activated caspases downregulate the tight junction proteins occludin and claudin 5, which implies the disrupted barrier function of endothelial cells. Interestingly, GM130 downregulation is partially caspase dependent (He et al., 2020). Moreover, apoptosis can inhibit HSV-1 proliferation in cells to a certain extent (Nguyen and Blaho, 2006), which has a certain positive significance for HSE. In short, the interaction between GA and apoptosis is closely related to the pathological mechanism of HSE.
Stroke is the second most common cause of death worldwide, and ischemic stroke is the most common type of stroke (Wang et al., 2016). Cerebral ischemia–reperfusion injury is one of the important pathological mechanisms of ischemic stroke. P115 cleavage, Golgi fragmentation, and apoptosis have been observed in vitro in a cerebral ischemia–reperfusion model (Zhong et al., 2015). GOLPH3 is upregulated in a rat model of ischemic stroke (You et al., 2014). Li et al. further found that GOLPH3 not only acts as a Golgi stress sensor, owing to its rapid upregulation during oxidative stress, but also triggers Golgi stress and transmits a specific Golgi stress signal to downstream effectors. Stress-induced GOLPH3 upregulation leads to Golgi fragmentation and apoptosis (Li T. et al., 2016). Recently, Ran et al. (2007) discovered a 165-kDa protein induced by cerebral ischemia that was named ischemia-induced Golgi protein 165 (IIGP165) because of its localization in the GA. The Akt phosphorylation of IIGP165 prevents apoptotic cell death (Ran et al., 2007).
Multiple studies provide ample evidence that the structure and the function of the GA are damaged by various mechanisms during apoptosis. However, in the face of different apoptotic situations, the Golgi can inhibit or promote apoptosis through different Golgi-related proteins. Golgi-related apoptosis is involved in the pathological mechanisms of various neurological diseases. In the future, the Golgi may be a potential therapeutic target for apoptosis-related neurological diseases. However, the exact mechanisms and specific relationships between GA and apoptosis in numerous interactions must still be studied. Whether the Golgi is upstream or downstream of apoptosis in specific diseases should be studied. In addition, the crosstalk of apoptotic signals between the GA and the other subcellular organelles also requires investigation.
QH carried out the literature review and drafted the manuscript. HL, SD, and XC helped in drafting the manuscript. DL and XJ conceived, designed, and coordinated the study. WZ and WL contributed to and finalized the draft. All the authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (No. 81571181).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; BI-1, Bax inhibitor 1; BIR, baculovirus-IAP repeat; Cdk5, cyclin-dependent kinase 5; COPI, coat protein complex I; CTF, C-terminal fragment; ER, endoplasmic reticulum; FasL, Fas ligand; FH2, formin homology 2; FHDC1, formin homology 2 domain protein 1; GA, Golgi apparatus; GAAP, Golgi anti-apoptotic protein; HSAN II, hereditary sensory and autonomic neuropathy type II; HSE, Herpes simplex virus encephalitis; IIGP165, ischemia-induced Golgi protein 165; IP3R, inositol-1,4,5-trisphosphate receptor; NAD+, nicotinamide adenine dinucleotide; p75NTR, p75 neurotrophin receptor; PD, Parkinson’s disease; PUMA, p53 upregulated modulator of apoptosis; TNFR, tumor necrosis factor receptor; UBC, ubiquitin-conjugating enzymes; UPR, unfolded protein response.
Ahat, E., Li, J., and Wang, Y. (2019a). New insights into the Golgi stacking proteins. Front. Cell. Dev. Biol. 7:131. doi: 10.3389/fcell.2019.00131
Ahat, E., Xiang, Y., Zhang, X., Bekier, M. E. II, and Wang, Y. (2019b). GRASP depletion-mediated Golgi destruction decreases cell adhesion and migration via the reduction of alpha5beta1 integrin. Mol. Biol. Cell 30, 766–777. doi: 10.1091/mbc.E18-07-0462
Al-Ghorbani, M., Thirusangu, P., Gurupadaswamy, H. D., Girish, V., Shamanth Neralagundi, H. G., Prabhakar, B. T., et al. (2016). Synthesis and antiproliferative activity of benzophenone tagged pyridine analogues towards activation of caspase activated DNase mediated nuclear fragmentation in Dalton’s lymphoma. Bioorg. Chem. 65, 73–81. doi: 10.1016/j.bioorg.2016.02.001
An, Y., Chen, C. Y., Moyer, B., Rotkiewicz, P., Elsliger, M. A., Godzik, A., et al. (2009). Structural and functional analysis of the globular head domain of p115 provides insight into membrane tethering. J. Mol. Biol. 391, 26–41. doi: 10.1016/j.jmb.2009.04.062
Andera, L. (2009). Signaling activated by the death receptors of the TNFR family. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 153, 173–180. doi: 10.5507/bp.2009.029
Arain, F. M., and Chand, P. (2015). Hereditary sensory autonomic neuropathy II, a rare disease in a large Pakistani family. J. Pak. Med. Assoc. 65, 1128–1130.
Atkin, J. D., Farg, M. A., Soo, K. Y., Walker, A. K., Halloran, M., Turner, B. J., et al. (2014). Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 129, 190–204. doi: 10.1111/jnc.12493
Bartke, T., Pohl, C., Pyrowolakis, G., and Jentsch, S. (2004). Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol. Cell 14, 801–811. doi: 10.1016/j.molcel.2004.05.018
Beard, M., Satoh, A., Shorter, J., and Warren, G. (2005). A cryptic Rab1-binding site in the p115 tethering protein. J. Biol. Chem. 280, 25840–25848. doi: 10.1074/jbc.M503925200
Bellouze, S., Baillat, G., Buttigieg, D., de la Grange, P., Rabouille, C., and Haase, G. (2016). Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol. Neurodegener. 11:43. doi: 10.1186/s13024-016-0111-6
Bondi, M. W., Edmonds, E. C., and Salmon, D. P. (2017). Alzheimer’s disease: past, present, and future. J. Int. Neuropsychol. Soc. 23, 818–831. doi: 10.1017/s135561771700100x
Bottone, M. G., Santin, G., Aredia, F., Bernocchi, G., Pellicciari, C., and Scovassi, A. I. (2013). Morphological features of organelles during apoptosis: an overview. Cells 2, 294–305. doi: 10.3390/cells2020294
Buschman, M. D., Rahajeng, J., and Field, S. J. (2015). GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res. 75, 624–627. doi: 10.1158/0008-5472.can-14-3081
Buttner, S., Faes, L., Reichelt, W. N., Broeskamp, F., Habernig, L., Benke, S., et al. (2013). The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca(2+) levels to alpha-synuclein toxicity in Parkinson’s disease models. Cell Death Differ. 20, 465–477. doi: 10.1038/cdd.2012.142
Carrara, G., Parsons, M., Saraiva, N., and Smith, G. L. (2017). Golgi anti-apoptotic protein: a tale of camels, calcium, channels and cancer. Open Biol. 7:170045. doi: 10.1098/rsob.170045
Chang, S. H., Hong, S. H., Jiang, H. L., Minai-Tehrani, A., Yu, K. N., Lee, J. H., et al. (2012). GOLGA2/GM130, cis-Golgi matrix protein, is a novel target of anticancer gene therapy. Mol. Ther. 20, 2052–2063. doi: 10.1038/mt.2012.125
Chen, J., and Chen, Z. J. (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76. doi: 10.1038/s41586-018-0761-3
Cheng, J. P., Betin, V. M., Weir, H., Shelmani, G. M., Moss, D. K., and Lane, J. D. (2010). Caspase cleavage of the Golgi stacking factor GRASP65 is required for Fas/CD95-mediated apoptosis. Cell Death Dis. 1:e82. doi: 10.1038/cddis.2010.59
Chiu, R., Novikov, L., Mukherjee, S., and Shields, D. (2002). A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J. Cell Biol. 159, 637–648. doi: 10.1083/jcb.200208013
Choy, R. W., Cheng, Z., and Schekman, R. (2012). Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 109, E2077–E2082. doi: 10.1073/pnas.1208635109
Chung, W. Y., Jha, A., Ahuja, M., and Muallem, S. (2017). Ca(2+) influx at the ER/PM junctions. Cell Calcium 63, 29–32. doi: 10.1016/j.ceca.2017.02.009
Coleman, M. L., and Olson, M. F. (2002). Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death Differ. 9, 493–504. doi: 10.1038/sj.cdd.4400987
Copeland, S. J., Thurston, S. F., and Copeland, J. W. (2016). Actin- and microtubule-dependent regulation of Golgi morphology by FHDC1. Mol. Biol. Cell 27, 260–276. doi: 10.1091/mbc.E15-02-0070
da Cunha Jaeger, M., Ghisleni, E. C., Cardoso, P. S., Siniglaglia, M., Falcon, T., Brunetto, A. T., et al. (2020). HDAC and MAPK/ERK inhibitors cooperate to reduce viability and stemness in Medulloblastoma. J. Mol. Neurosci. 70, 981–992. doi: 10.1007/s12031-020-01505-y
Dascher, C., Matteson, J., and Balch, W. E. (1994). Syntaxin 5 regulates endoplasmic reticulum to Golgi transport. J. Biol. Chem. 269, 29363–29366.
de Mattia, F., Gubser, C., van Dommelen, M. M., Visch, H. J., Distelmaier, F., Postigo, A., et al. (2009). Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol. Biol. Cell 20, 3638–3645. doi: 10.1091/mbc.E09-05-0385
DeBiasi, R. L., Kleinschmidt-DeMasters, B. K., Richardson-Burns, S., and Tyler, K. L. (2002). Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J. Infect. Dis. 186, 1547–1557. doi: 10.1086/345375
Desouza, M., Gunning, P. W., and Stehn, J. R. (2012). The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture 2, 75–87. doi: 10.4161/bioa.20975
El Mchichi, B., Hadji, A., Vazquez, A., and Leca, G. (2007). p38 MAPK and MSK1 mediate caspase-8 activation in manganese-induced mitochondria-dependent cell death. Cell Death Differ. 14, 1826–1836. doi: 10.1038/sj.cdd.4402187
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Emr, S., Glick, B. S., Linstedt, A. D., Lippincott-Schwartz, J., Luini, A., Malhotra, V., et al. (2009). Journeys through the Golgi–taking stock in a new era. J. Cell Biol. 187, 449–453. doi: 10.1083/jcb.200909011
Falcao de Campos, C., Vidailhet, M., Toutain, A., de Becdelievre, A., Funalot, B., Bonello-Palot, N., et al. (2019). Hereditary sensory autonomic neuropathy type II: report of two novel mutations in the FAM134B gene. J. Peripher. Nerv. Syst. 24, 354–358. doi: 10.1111/jns.12352
Farber-Katz, S. E., Dippold, H. C., Buschman, M. D., Peterman, M. C., Xing, M., Noakes, C. J., et al. (2014). DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156, 413–427. doi: 10.1016/j.cell.2013.12.023
Fischer, U., Janicke, R. U., and Schulze-Osthoff, K. (2003). Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 10, 76–100. doi: 10.1038/sj.cdd.4401160
Gillingham, A. K., and Munro, S. (2016). Finding the Golgi: golgin coiled-coil proteins show the way. Trends Cell Biol. 26, 399–408. doi: 10.1016/j.tcb.2016.02.005
Giraud, S., Loum, E., Bessette, B., Mathonnet, M., and Lalloue, F. (2011). P75 neurotrophin receptor is sequestered in the Golgi apparatus of the U-87 MG human glioblastoma cell line. Int. J. Oncol. 38, 391–399. doi: 10.3892/ijo.2010.862
Gomez-Angelats, M., and Cidlowski, J. A. (2001). Protein kinase C regulates FADD recruitment and death-inducing signaling complex formation in Fas/CD95-induced apoptosis. J. Biol. Chem. 276, 44944–44952. doi: 10.1074/jbc.M104919200
Goodenberger, M. L., and Jenkins, R. B. (2012). Genetics of adult glioma. Cancer Genet. 205, 613–621. doi: 10.1016/j.cancergen.2012.10.009
Gubser, C., Bergamaschi, D., Hollinshead, M., Lu, X., van Kuppeveld, F. J., and Smith, G. L. (2007). A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog. 3:e17. doi: 10.1371/journal.ppat.0030017
Guo, K., Wang, W., Fan, W., Wang, Z., Zhu, M., Tang, X., et al. (2018). Arabidopsis GAAP1 and GAAP3 modulate the unfolded protein response and the onset of cell death in response to ER stress. Front. Plant Sci. 9:348. doi: 10.3389/fpls.2018.00348
Hao, Y., Sekine, K., Kawabata, A., Nakamura, H., Ishioka, T., Ohata, H., et al. (2004). Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 6, 849–860. doi: 10.1038/ncb1159
Harlan, B. A., Killoy, K. M., Pehar, M., Liu, L., Auwerx, J., and Vargas, M. R. (2020). Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol. 327:113219. doi: 10.1016/j.expneurol.2020.113219
Hauser, H.-P., Bardroff, M., Pyrowolakis, G., and Jentsch, S. (1998). A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 141, 1415–1422. doi: 10.1083/jcb.141.6.1415
He, Q., Liu, H., Huang, C., Wang, R., Luo, M., and Lu, W. (2020). Herpes simplex Virus 1-induced blood-brain barrier damage involves apoptosis associated with GM130-mediated golgi stress. Front. Mol. Neurosci. 13:2. doi: 10.3389/fnmol.2020.00002
Heuer, D., Rejman Lipinski, A., Machuy, N., Karlas, A., Wehrens, A., Siedler, F., et al. (2009). Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457, 731–735. doi: 10.1038/nature07578
Hicks, S. W., Horn, T. A., McCaffery, J. M., Zuckerman, D. M., and Machamer, C. E. (2006). Golgin-160 promotes cell surface expression of the beta-1 adrenergic receptor. Traffic 7, 1666–1677. doi: 10.1111/j.1600-0854.2006.00504.x
Hicks, S. W., and Machamer, C. E. (2005). Golgi structure in stress sensing and apoptosis. Biochim. Biophys. Acta 1744, 406–414. doi: 10.1016/j.bbamcr.2005.03.002
Hilton, D., Stephens, M., Kirk, L., Edwards, P., Potter, R., Zajicek, J., et al. (2014). Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 127, 235–241. doi: 10.1007/s00401-013-1214-6
How, P. C., and Shields, D. (2011). Tethering function of the caspase cleavage fragment of Golgi protein p115 promotes apoptosis via a p53-dependent pathway. J. Biol. Chem. 286, 8565–8576. doi: 10.1074/jbc.M110.175174
Hui, N., Nakamura, N., Sönnichsen, B., Shima, D., Nilsson, T., and Warren, G. (1997). An isoform of the Golgi t-SNARE, syntaxin 5, with an endoplasmic reticulum retrieval signal. Mol. Biol. Cell 8, 1777–1787. doi: 10.1091/mbc.8.9.1777
Ivanov, V. N., Lopez Bergami, P., Maulit, G., Sato, T. A., Sassoon, D., and Ronai, Z. (2003). FAP-1 association with Fas (Apo-1) inhibits Fas expression on the cell surface. Mol. Cell Biol. 23, 3623–3635. doi: 10.1128/mcb.23.10.3623-3635.2003
Jarvela, T., Linstedt, A. D., and Glick, B. S. (2014). Isoform-specific tethering links the Golgi ribbon to maintain compartmentalization. Mol. Biol. Cell 25, 133–144. doi: 10.1091/mbc.e13-07-0395
Joshi, G., Chi, Y., Huang, Z., and Wang, Y. (2014). Abeta-induced Golgi fragmentation in Alzheimer’s disease enhances Abeta production. Proc. Natl. Acad. Sci. U.S.A. 111, E1230–E1239. doi: 10.1073/pnas.1320192111
Khan, H., Reale, M., Ullah, H., Sureda, A., Tejada, S., Wang, Y., et al. (2020). Anti-cancer effects of polyphenols via targeting p53 signaling pathway: updates and future directions. Biotechnol. Adv. 38:107385. doi: 10.1016/j.biotechadv.2019.04.007
Kim, S., Hill, A., Warman, M. L., and Smits, P. (2012). Golgi disruption and early embryonic lethality in mice lacking USO1. PLoS One 7:e50530. doi: 10.1371/journal.pone.0050530
Kokubun, H., Jin, H., and Aoe, T. (2019). Pathogenic effects of impaired retrieval between the endoplasmic reticulum and Golgi complex. Int. J. Mol. Sci. 20:5614. doi: 10.3390/ijms20225614
Koreishi, M., Gniadek, T. J., Yu, S., Masuda, J., Honjo, Y., and Satoh, A. (2013). The golgin tether giantin regulates the secretory pathway by controlling stack organization within Golgi apparatus. PLoS One 8:e59821. doi: 10.1371/journal.pone.0059821
Kubo, S. I., Kitami, T., Noda, S., Shimura, H., Uchiyama, Y., Asakawa, S., et al. (2001). Parkin is associated with cellular vesicles. J. Neurochem. 78, 42–54. doi: 10.1046/j.1471-4159.2001.00364.x
Kulkarni-Gosavi, P., Makhoul, C., and Gleeson, P. A. (2019). Form and function of the Golgi apparatus: scaffolds, cytoskeleton and signalling. FEBS Lett. 593, 2289–2305. doi: 10.1002/1873-3468.13567
Kurth, I., Pamminger, T., Hennings, J. C., Soehendra, D., Huebner, A. K., Rotthier, A., et al. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 41, 1179–1181. doi: 10.1038/ng.464
Lane, J. D., Lucocq, J., Pryde, J., Barr, F. A., Woodman, P. G., Allan, V. J., et al. (2002). Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 156, 495–509. doi: 10.1083/jcb.200110007
Lazaro-Dieguez, F., Jimenez, N., Barth, H., Koster, A. J., Renau-Piqueras, J., Llopis, J. L., et al. (2006). Actin filaments are involved in the maintenance of Golgi cisternae morphology and intra-Golgi pH. Cell Motil. Cytoskeleton 63, 778–791. doi: 10.1002/cm.20161
Li, Q., Chen, M., Liu, H., Yang, L., Yang, T., and He, G. (2014). The dual role of ERK signaling in the apoptosis of neurons. Front. Biosci. (Landmark Ed.) 19, 1411–1417. doi: 10.2741/4291
Li, T., You, H., Mo, X., He, W., Tang, X., Jiang, Z., et al. (2016). GOLPH3 mediated Golgi stress response in modulating N2A cell death upon oxygen-glucose deprivation and reoxygenation injury. Mol. Neurobiol. 53, 1377–1385. doi: 10.1007/s12035-014-9083-0
Li, X., Li, M., Tian, X., Li, Q., Lu, Q., Jia, Q., et al. (2016). Golgi phosphoprotein 3 inhibits the apoptosis of human glioma cells in part by downregulating N-myc downstream regulated gene 1. Med. Sci. Monit. 22, 3535–3543. doi: 10.12659/msm.900349
Li, Z., Guo, D., Yin, X., Ding, S., Shen, M., Zhang, R., et al. (2020). Zinc oxide nanoparticles induce human multiple myeloma cell death via reactive oxygen species and Cyt-C/Apaf-1/Caspase-9/Caspase-3 signaling pathway in vitro. Biomed. Pharmacother. 122:109712. doi: 10.1016/j.biopha.2019.109712
Lin, P., Yao, Y., Hofmeister, R., Tsien, R. Y., and Farquhar, M. G. (1999). Overexpression of CALNUC (nucleobindin) increases agonist and thapsigargin releasable Ca2+ storage in the Golgi. J. Cell Biol. 145, 279–289. doi: 10.1083/jcb.145.2.279
Linders, P. T., Horst, C. V., Beest, M. T., and van den Bogaart, G. (2019). Stx5-mediated ER-Golgi transport in mammals and yeast. Cells 8:780. doi: 10.3390/cells8080780
Linstedt, A. D., Foguet, M., Renz, M., Seelig, H. P., Glick, B. S., and Hauri, H. P. (1995). A C-terminally-anchored Golgi protein is inserted into the endoplasmic reticulum and then transported to the Golgi apparatus. Proc. Natl. Acad. Sci. U.S.A. 92, 5102–5105. doi: 10.1073/pnas.92.11.5102
Liu, C., Mei, M., Li, Q., Roboti, P., Pang, Q., Ying, Z., et al. (2017). Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. U.S.A. 114, 346–351. doi: 10.1073/pnas.1608576114
Liu, H., Qiu, K., He, Q., Lei, Q., and Lu, W. (2019). Mechanisms of blood-brain barrier disruption in herpes simplex encephalitis. J. Neuroimmune Pharmacol. 14, 157–172. doi: 10.1007/s11481-018-9821-6
Logue, S. E., and Martin, S. J. (2008). Caspase activation cascades in apoptosis. Biochem. Soc. Transact. 36, 1–9. doi: 10.1042/bst0360001
Lowe, M. (2011). Structural organization of the Golgi apparatus. Curr. Opin. Cell Biol. 23, 85–93. doi: 10.1016/j.ceb.2010.10.004
Lowe, M. (2019). The physiological functions of the golgin vesicle tethering proteins. Front. Cell Dev. Biol. 7:94. doi: 10.3389/fcell.2019.00094
Lowe, M., Gonatas, N. K., and Warren, G. (2000). The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J. Cell Biol. 149, 341–356. doi: 10.1083/jcb.149.2.341
Lowe, M., Lane, J. D., Woodman, P. G., and Allan, V. J. (2004). Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J. Cell Sci. 117(Pt 7), 1139–1150. doi: 10.1242/jcs.00950
Ma, T. S., Mann, D. L., Lee, J. H., and Gallinghouse, G. J. (1999). SR compartment calcium and cell apoptosis in SERCA overexpression. Cell Calcium 26, 25–36. doi: 10.1054/ceca.1999.0049
Maag, R. S., Mancini, M., Rosen, A., and Machamer, C. E. (2005). Caspase-resistant Golgin-160 disrupts apoptosis induced by secretory pathway stress and ligation of death receptors. Mol. Biol. Cell 16, 3019–3027. doi: 10.1091/mbc.e04-11-0971
Machamer, C. E. (2015). The Golgi complex in stress and death. Front. Neurosci. 9:421. doi: 10.3389/fnins.2015.00421
Makhoul, C., Gosavi, P., and Gleeson, P. A. (2018). The Golgi architecture and cell sensing. Biochem. Soc. Trans. 46, 1063–1072. doi: 10.1042/BST20180323
Mancini, M., Machamer, C. E., Roy, S., Nicholson, D. W., Thornberry, N. A., Casciola-Rosen, L. A., et al. (2000). Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 149, 603–612. doi: 10.1083/jcb.149.3.603
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi: 10.1016/j.ceca.2017.05.003
Marra, P., Salvatore, L., Mironov, A. Jr., Di Campli, A., Di Tullio, G., Trucco, A., et al. (2007). The biogenesis of the Golgi ribbon: the roles of membrane input from the ER and of GM130. Mol. Biol. Cell 18, 1595–1608. doi: 10.1091/mbc.e06-10-0886
Martin, C., Leyton, L., Hott, M., Arancibia, Y., Spichiger, C., McNiven, M. A., et al. (2017). Herpes simplex virus type 1 neuronal infection perturbs Golgi Apparatus integrity through activation of Src Tyrosine Kinase and Dyn-2 GTPase. Front. Cell Infect. Microbiol. 7:371. doi: 10.3389/fcimb.2017.00371
Mayinger, P. (2011). Signaling at the Golgi. Cold Spring Harb. Perspect. Biol. 3:a005314. doi: 10.1101/cshperspect.a005314
Milde, S., Gilley, J., and Coleman, M. P. (2013). Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 11:e1001539. doi: 10.1371/journal.pbio.1001539
Mollenhauer, H. H., and Morré, D. J. (1991). Perspectives on Golgi apparatus form and function. J. Electron Microscopy Tech. 17, 2–14. doi: 10.1002/jemt.1060170103
Moss, D. K., and Lane, J. D. (2006). Microtubules: forgotten players in the apoptotic execution phase. Trends Cell Biol. 16, 330–338. doi: 10.1016/j.tcb.2006.05.005
Mukherjee, S., and Shields, D. (2009). Nuclear import is required for the pro-apoptotic function of the Golgi protein p115. J. Biol. Chem. 284, 1709–1717. doi: 10.1074/jbc.M807263200
Nakagomi, S., Barsoum, M. J., Bossy-Wetzel, E., Sutterlin, C., Malhotra, V., and Lipton, S. A. (2008). A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 29, 221–231. doi: 10.1016/j.nbd.2007.08.015
Nakamura, K., Bossy-Wetzel, E., Burns, K., Fadel, M. P., Lozyk, M., Goping, I. S., et al. (2000). Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150, 731–740. doi: 10.1083/jcb.150.4.731
Nakano, K., and Vousden, K. H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694. doi: 10.1016/s1097-2765(01)00214-3
Ndozangue-Touriguine, O., Hamelin, J., and Breard, J. (2008). Cytoskeleton and apoptosis. Biochem. Pharmacol. 76, 11–18. doi: 10.1016/j.bcp.2008.03.016
Nguyen, M. L., and Blaho, J. A. (2006). Apoptosis during herpes simplex virus infection. Adv. Virus Res. 69, 67–97. doi: 10.1016/s0065-3527(06)69002-7
Parys, J. B., and Vervliet, T. (2020). New insights in the IP3 receptor and its regulation. Adv. Exp. Med. Biol. 1131, 243–270. doi: 10.1007/978-3-030-12457-1_10
Peng, Y., He, X., Chen, H., Duan, H., Shao, B., Yang, F., et al. (2018). Inhibition of microRNA-299-5p sensitizes glioblastoma cells to temozolomide via the MAPK/ERK signaling pathway. Biosci. Rep. 38:BSR20181051. doi: 10.1042/BSR20181051
Peter, M. E. (2011). Programmed cell death: Apoptosis meets necrosis. Nature 471, 310–312. doi: 10.1038/471310a
Pinton, P., and Rizzuto, R. (2006). Bcl-2 and Ca 2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 13, 1409–1418. doi: 10.1038/sj.cdd.4401960
Piret, J., and Boivin, G. (2020). Immunomodulatory strategies in herpes simplex virus encephalitis. Clin. Microbiol. Rev. 33:e00105-19. doi: 10.1128/cmr.00105-19
Pohl, C., and Jentsch, S. (2008). “Regulation of apoptosis and cytokinesis by the anti-apoptotic E2/E3 ubiquitin-ligase BRUCE,” in Proceedings of the Ernst Schering Foundation Symposium Proceedings, eds S. Jentsch and B. Haendler (Berlin: Springer), 115–126. doi: 10.1007/2789_2008_104
Popgeorgiev, N., Jabbour, L., and Gillet, G. (2018). Subcellular localization and dynamics of the Bcl-2 family of proteins. Front. Cell Dev. Biol. 6:13. doi: 10.3389/fcell.2018.00013
Pottorf, T., Mann, A., Fross, S., Mansel, C., and Vohra, B. P. S. (2018). Nicotinamide Mononucleotide Adenylyltransferase 2 maintains neuronal structural integrity through the maintenance of golgi structure. Neurochem. Int. 121, 86–97. doi: 10.1016/j.neuint.2018.09.010
Qi, Z., and Chen, L. (2019). Endoplasmic reticulum stress and autophagy. Adv. Exp. Med. Biol. 1206, 167–177. doi: 10.1007/978-981-15-0602-4_8
Qiu, X. B., and Goldberg, A. L. (2005). The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J. Biol. Chem. 280, 174–182. doi: 10.1074/jbc.M411430200
Rabouille, C., and Linstedt, A. D. (2016). GRASP: a multitasking tether. Front. Cell Dev. Biol. 4:1. doi: 10.3389/fcell.2016.00001
Ran, R., Pan, R., Lu, A., Xu, H., Davis, R. R., and Sharp, F. R. (2007). A novel 165-kDa Golgin protein induced by brain ischemia and phosphorylated by Akt protects against apoptosis. Mol. Cell. Neurosci. 36, 392–407. doi: 10.1016/j.mcn.2007.07.014
Ren, J., Shi, M., Liu, R., Yang, Q.-H., Johnson, T., Skarnes, W. C., et al. (2005). The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc. Natl. Acad. Sci. U.S.A. 102, 565–570. doi: 10.1073/pnas.0408744102
Rotthier, A., Baets, J., Timmerman, V., and Janssens, K. (2012). Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 8:73. doi: 10.1038/nrneurol.2011.227
Sapperstein, S. K., Walter, D. M., Grosvenor, A. R., Heuser, J. E., and Waters, M. G. (1995). p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc. Natl. Acad. Sci. U.S.A. 92, 522–526. doi: 10.1073/pnas.92.2.522
Sasaki, K., and Yoshida, H. (2019). Golgi stress response and organelle zones. FEBS Lett. 593, 2330–2340. doi: 10.1002/1873-3468.13554
Satoh, A., Wang, Y., Malsam, J., Beard, M. B., and Warren, G. (2003). Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic 4, 153–161. doi: 10.1034/j.1600-0854.2003.00103.x
Savitskaya, M. A., and Onishchenko, G. E. (2015). Mechanisms of Apoptosis. Biochemistry (Mosc) 80, 1393–1405. doi: 10.1134/S0006297915110012
Short, D. M., Heron, I. D., Birse-Archbold, J. L., Kerr, L. E., Sharkey, J., and McCulloch, J. (2007). Apoptosis induced by staurosporine alters chaperone and endoplasmic reticulum proteins: identification by quantitative proteomics. Proteomics 7, 3085–3096. doi: 10.1002/pmic.200600964
Smaardijk, S., Chen, J., Wuytack, F., and Vangheluwe, P. (2017). SPCA2 couples Ca(2+) influx via Orai1 to Ca(2+) uptake into the Golgi/secretory pathway. Tissue Cell 49(2 Pt A), 141–149. doi: 10.1016/j.tice.2016.09.004
Snigirevskaia, E. S., Moshkov, A. V., Iurinskaia, V. E., Vereninov, A. A., and Komissarchik, I. (2014). [Ultrastructural and X-ray spectral analysis of cells U-937 during apoptosis process induced by hypertony]. Tsitologiia 56, 828–840.
Sohda, M., Misumi, Y., Yamamoto, A., Nakamura, N., Ogata, S., Sakisaka, S., et al. (2010). Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic 11, 1552–1566. doi: 10.1111/j.1600-0854.2010.01123.x
Sönnichsen, B., Lowe, M., Levine, T., Jämsä, E., Dirac-Svejstrup, B., and Warren, G. (1998). A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol. 140, 1013–1021. doi: 10.1083/jcb.140.5.1013
Stoffel, W., Jenke, B., Schmidt-Soltau, I., Binczek, E., Brodesser, S., and Hammels, I. (2018). SMPD3 deficiency perturbs neuronal proteostasis and causes progressive cognitive impairment. Cell Death Dis. 9:507. doi: 10.1038/s41419-018-0560-7
Stokholm, M. G., Danielsen, E. H., Hamilton-Dutoit, S. J., and Borghammer, P. (2016). Pathological a-synuclein in gastrointestinal tissues from prodromal P arkinson disease patients. Ann. Neurol. 79, 940–949. doi: 10.1002/ana.24648
Su, X. J., Huang, L., Qu, Y., and Mu, D. (2019). Progress in research on the role of Omi/HtrA2 in neurological diseases. Rev. Neurosci. 30, 279–287. doi: 10.1515/revneuro-2018-0004
Suga, K., Hattori, H., Saito, A., and Akagawa, K. (2005). RNA interference-mediated silencing of the syntaxin 5 gene induces Golgi fragmentation but capable of transporting vesicles. FEBS Lett. 579, 4226–4234. doi: 10.1016/j.febslet.2005.06.053
Sun, K. H., de Pablo, Y., Vincent, F., Johnson, E. O., Chavers, A. K., and Shah, K. (2008). Novel genetic tools reveal Cdk5’s major role in Golgi fragmentation in Alzheimer’s disease. Mol. Biol. Cell 19, 3052–3069. doi: 10.1091/mbc.E07-11-1106
Sunico, C. R., Nakamura, T., Rockenstein, E., Mante, M., Adame, A., Chan, S. F., et al. (2013). S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 8:29. doi: 10.1186/1750-1326-8-29
Surmeier, D. J. (2018). Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 285, 3657–3668. doi: 10.1111/febs.14607
Tai, G., Lu, L., Wang, T. L., Tang, B. L., Goud, B., Johannes, L., et al. (2004). Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the trans-Golgi network. Mol. Biol. Cell 15, 4011–4022. doi: 10.1091/mbc.e03-12-0876
Takada, E., Okubo, K., Yano, Y., Iida, K., Someda, M., Hirasawa, A., et al. (2020). Molecular mechanism of Apoptosis by amyloid beta-protein fibrils formed on neuronal cells. ACS Chem. Neurosci. 11, 796–805. doi: 10.1021/acschemneuro.0c00011
Tang, D., Zhang, X., Huang, S., Yuan, H., Li, J., and Wang, Y. (2016). Mena-GRASP65 interaction couples actin polymerization to Golgi ribbon linking. Mol. Biol. Cell 27, 137–152. doi: 10.1091/mbc.E15-09-0650
Tassi, E., Zanon, M., Vegetti, C., Molla, A., Bersani, I., Perotti, V., et al. (2012). Role of Apollon in human melanoma resistance to antitumor agents that activate the intrinsic or the extrinsic apoptosis pathways. Clin. Cancer Res. 18, 3316–3327. doi: 10.1158/1078-0432.ccr-11-2232
Thomas, L. R., Henson, A., Reed, J. C., Salsbury, F. R., and Thorburn, A. (2004). Direct binding of Fas-associated death domain (FADD) to the tumor necrosis factor-related apoptosis-inducing ligand receptor DR5 is regulated by the death effector domain of FADD. J. Biol. Chem. 279, 32780–32785. doi: 10.1074/jbc.M401680200
Tiwari, M., Prasad, S., Shrivastav, T. G., and Chaube, S. K. (2017). Calcium signaling during meiotic cell cycle regulation and Apoptosis in mammalian oocytes. J. Cell Physiol. 232, 976–981. doi: 10.1002/jcp.25670
Vasudevan, D., and Ryoo, H. D. (2015). Regulation of cell death by IAPs and their Antagonists. Curr. Top. Dev. Biol. 114, 185–208. doi: 10.1016/bs.ctdb.2015.07.026
Volchuk, A., Ravazzola, M., Perrelet, A., Eng, W. S., Di Liberto, M., Varlamov, O., et al. (2004). Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol. Biol. Cell 15, 1506–1518. doi: 10.1091/mbc.e03-08-0625
Walker, A., Ward, C., Sheldrake, T. A., Dransfield, I., Rossi, A. G., Pryde, J. G., et al. (2004). Golgi fragmentation during Fas-mediated apoptosis is associated with the rapid loss of GM130. Biochem. Biophys. Res. Commun. 316, 6–11. doi: 10.1016/j.bbrc.2004.02.015
Wang, H., Naghavi, M., Allen, C., Barber, R., Bhutta, Z., Carter, A., et al. (2016). GBD 2015 mortality and causes of death collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1459–1544.
Wang, X., Ma, W., Wang, T., Yang, J., Wu, Z., Liu, K., et al. (2020). BDNF-TrkB and proBDNF-p75NTR/sortilin signaling pathways are involved in mitochondria-mediated neuronal apoptosis in dorsal root ganglia after sciatic nerve transection. CNS Neurol. Disord. Drug Targets 19, 66–82. doi: 10.2174/1871527319666200117110056
Wang, X., Zhang, J., Kim, H. P., Wang, Y., Choi, A. M., and Ryter, S. W. (2004). Bcl-XL disrupts death-inducing signal complex formation in plasma membrane induced by hypoxia/reoxygenation. FASEB J. 18, 1826–1833. doi: 10.1096/fj.04-2047com
Waters, M. G., Clary, D. O., and Rothman, J. E. (1992). A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 118, 1015–1026. doi: 10.1083/jcb.118.5.1015
White, M. K., and Cinti, C. (2004). A morphologic approach to detect apoptosis based on electron microscopy. Methods Mol. Biol. 285, 105–111. doi: 10.1385/1-59259-822-6
Wong, M., and Munro, S. (2014). Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 346:1256898. doi: 10.1126/science.1256898
Xiang, Y., and Wang, Y. (2010). GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 188, 237–251. doi: 10.1083/jcb.200907132
Xie, J., Li, B., Yao, B., Zhang, P., Wang, L., Lu, H., et al. (2019). Transforming growth factor-beta1-regulated Fas/FasL pathway activation suppresses nucleus pulposus cell apoptosis in an inflammatory environment. Biosci. Rep. 40:BSR20191726. doi: 10.1042/bsr20191726
Xu, B., Qin, Y., Li, D., Cai, N., Wu, J., Jiang, L., et al. (2019). Inhibition of PDE4 protects neurons against oxygen-glucose deprivation-induced endoplasmic reticulum stress through activation of the Nrf-2/HO-1 pathway. Redox Biol. 28:101342. doi: 10.1016/j.redox.2019.101342
Yadav, S., Puri, S., and Linstedt, A. D. (2009). A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol. Biol. Cell 20, 1728–1736. doi: 10.1091/mbc.E08-10-1077
Yadav, S., Puthenveedu, M. A., and Linstedt, A. D. (2012). Golgin160 recruits the dynein motor to position the Golgi apparatus. Dev. Cell 23, 153–165. doi: 10.1016/j.devcel.2012.05.023
You, H., Li, T., Zhang, J., Lei, Q., Tao, X., Xie, P., et al. (2014). Reduction in ischemic cerebral infarction is mediated through golgi phosphoprotein 3 and Akt/mTOR signaling following salvianolate administration. Curr. Neurovasc. Res. 11, 107–113. doi: 10.2174/1567202611666140307124857
Yu, J., Nagasu, H., Murakami, T., Hoang, H., Broderick, L., Hoffman, H. M., et al. (2014). Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. U.S.A. 111, 15514–15519. doi: 10.1073/pnas.1414859111
Zappa, F., Failli, M., and De Matteis, M. A. (2018). The Golgi complex in disease and therapy. Curr. Opin. Cell Biol. 50, 102–116. doi: 10.1016/j.ceb.2018.03.005
Zhang, T. M. (2019). TRIAP1 inhibition activates the cytochrome c/Apaf-1/Caspase-9 signaling pathway to enhance human ovarian cancer sensitivity to cisplatin. Chemotherapy 64, 119–128. doi: 10.1159/000501633
Zhang, X., Chen, S., Lu, K., Wang, F., Deng, J., Xu, Z., et al. (2019). Verapamil ameliorates motor neuron degeneration and improves lifespan in the SOD1(G93A) mouse model of ALS by enhancing Autophagic flux. Aging Dis. 10, 1159–1173. doi: 10.14336/ad.2019.0228
Zhang, X., and Wang, Y. (2015). GRASPs in Golgi structure and function. Front. Cell Dev. Biol. 3:84. doi: 10.3389/fcell.2015.00084
Zhong, B., Hu, Z., Tan, J., Lu, T., Lei, Q., Chen, C., et al. (2015). Hsp20 protects against oxygen-glucose deprivation/reperfusion-induced Golgi fragmentation and apoptosis through Fas/FasL pathway. Oxid. Med. Cell. Longev. 2015:606934.
Zhou, Z., Sun, X., Zou, Z., Sun, L., Zhang, T., Guo, S., et al. (2010). PRMT5 regulates Golgi apparatus structure through methylation of the golgin GM130. Cell Res. 20, 1023–1033. doi: 10.1038/cr.2010.56
Zhou, Z., Zhou, B., Chen, H., Tang, X., and Wang, Y. (2019). Reactive oxygen species (ROS) and the calcium-(Ca(2+)) mediated extrinsic and intrinsic pathways underlying BDE-47-induced apoptosis in rainbow trout (Oncorhynchus mykiss) gonadal cells. Sci. Total Environ. 656, 778–788. doi: 10.1016/j.scitotenv.2018.11.306
Keywords: Golgi apparatus, apoptosis, caspases, neurological diseases, therapeutic target
Citation: He Q, Liu H, Deng S, Chen X, Li D, Jiang X, Zeng W and Lu W (2020) The Golgi Apparatus May Be a Potential Therapeutic Target for Apoptosis-Related Neurological Diseases. Front. Cell Dev. Biol. 8:830. doi: 10.3389/fcell.2020.00830
Received: 08 May 2020; Accepted: 04 August 2020;
Published: 31 August 2020.
Edited by:
Jiyan Zhang, Independent Researcher, Bejing, ChinaReviewed by:
Akihiko Nakano, RIKEN, JapanCopyright © 2020 He, Liu, Deng, Chen, Li, Jiang, Zeng and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenbo Zeng, emVuZ3diQHdoLmlvdi5jbg==; Wei Lu, bHV3ZWkwMzM4QGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.