 Marina I. Oliveira da Silva
Marina I. Oliveira da Silva Márcia A. Liz
Márcia A. Liz- 1Instituto de Ciências Biomédicas Abel Salazar (ICBAS), Universidade do Porto, Porto, Portugal
- 2Neurodegeneration Group, Instituto de Biologia Molecular e Celular (IBMC) and Nerve Regeneration Group, Instituto de Investigação e Inovação em Saúde (i3S), Universidade do Porto, Porto, Portugal
Alpha-Synuclein (αSyn), a protein highly enriched in neurons where it preferentially localizes at the pre-synapse, has been in the spotlight because its intraneuronal aggregation is a central phenomenon in Parkinson’s disease. However, the consequences of αSyn accumulation to neuronal function are not fully understood. Considering the crucial role of actin on synaptic function and the fact that dysregulation of this cytoskeleton component is emerging in neurodegenerative disorders, the impact of αSyn on actin is a critical point to be addressed. In this review we explore the link between αSyn and actin and its significance for physiology and pathology. We discuss the relevance of αSyn-actin interaction for synaptic function and highlight the actin-depolymerizing protein cofilin-1 as a key player on αSyn-induced actin dysfunction in Parkinson’s disease.
Introduction
In the last years, a large body of evidences point to the neuronal cytoskeleton damage as a major contributor to neurodegeneration (Eira et al., 2016). From the various cytoskeleton dysfunctions, alterations in microtubule (MT) stability are a main causative agent in several neurodegenerative disorders and include: (i) variations in the levels of tubulin post-translational modifications with a consequent impact on axonal transport (Dompierre et al., 2007; d’Ydewalle et al., 2011; Zhang et al., 2014; Qu et al., 2017; Magiera et al., 2018), and (ii) dysregulation of MT associated proteins, such as the key example of tau hyperphosphorylation. This modification promotes tau detachment from MTs, what either impacts on axonal transport (Alonso et al., 1997) or leads to recruitment of MT severing enzymes causing axon degeneration (Qiang et al., 2006). Concerning the actin cytoskeleton, dysregulation of this component causing neurodegeneration mostly derives from actin accumulations as the case of cofilin-actin rods (Minamide et al., 2000; Munsie and Truant, 2012).
In the case of Parkinson’s disease (PD), the most common synucleinopathy, characterized by the intraneuronal accumulation of aggregated αSyn, several reports demonstrated αSyn-induced alterations of the microtubule cytoskeleton dynamics and axonal transport defects (Carnwath et al., 2018; Prots et al., 2018). As αSyn is a protein that impacts on the synapse both physiologically and pathologically, an association between the protein and the actin cytoskeleton, a cell component crucial for synaptic function, has been suggested. This review will focus on the current knowledge on the interaction between αSyn and actin and actin-binding proteins (ABPs) concluding with a critical perspective of the implication of these interactions in health and disease. Addressing this topic will certainly contribute with new insights into αSyn-related pathology opening new lines of research targeting neurodegeneration.
Alpha-Synuclein: From Physiology to Pathology
αSyn was originally identified in the Torpedo electric organ and is one of the most abundant proteins in the brain (Maroteaux and Scheller, 1991). It belongs to the synuclein family of proteins, a group of small soluble proteins that transiently bind to neuronal membranes, which also includes β-Synuclein and γ-Synuclein (Clayton and George, 1998). αSyn is a 140 amino-acid protein containing three distinct motifs (Maroteaux et al., 1988): the N-terminal contains seven repeats of a 11-residue sequence (XKTKEGVXXXX) responsible for αSyn interaction with vesicles containing phospholipids, the central region contains a non-amyloid component (NAC) sequence which is relatively hydrophobic and prone to aggregation, and the intrinsically unstructured C-terminus region, responsible for multiple protein interactions (Emamzadeh, 2016). αSyn is a natively unfolded protein implying that it lacks a secondary organized structure (Weinreb et al., 1996), what enables the protein to adopt several conformations comprising an unstructured soluble cytosolic form, an α-helical membrane bound structure or a β-sheet-like prone to aggregate conformation (Burre et al., 2013).
αSyn is ubiquitously expressed but highly enriched in the nervous systems (Lavedan, 1998). In neurons, αSyn is enriched in the pre-synaptic terminals where it interacts with synaptic vesicles and participates in several steps of the vesicle cycle comprising trafficking, docking, fusion, and recycling after exocytosis, therefore playing a central role in the regulation of the synaptic transmission (Scott and Roy, 2012; Diao et al., 2013; Wang et al., 2014). Notably, synuclein proteins were proposed to have a major impact in the long-term function of synaptic transmission, as αβγ-Syn triple-knockout mice showed reduced SNARE (SNAP REceptor)-complex assembly, and presented neuropathological signs and shortened lifespan (Burre et al., 2010). Interestingly, although αSyn plays an important role at the pre-synapse, it is one of the last proteins to be targeted to the synapse during development, suggesting that its physiologic function is not focused on the synapse formation but more directed to its maintenance (Cheng et al., 2011). Although αSyn role at the synapse is the most well studied physiological function, a number of additional cellular localizations have been described, including mitochondria, nucleus, endoplasmic reticulum, Golgi complex, and cytoskeleton components, suggesting that the protein contributes to the regulation of innumerous cellular processes (Burre et al., 2018).
αSyn is a protein deeply associated with disease since it was described as the main component of Lewy Bodies (LBs), the pathological hallmark of synucleinopathies (Spillantini et al., 1997), a group of diseases caused by αSyn aggregation and pathology from which PD is the most common one. Although being a cytosolic protein, and its intra-neuronal accumulation resulting in neurodegeneration, it is now known that cell-to-cell transmission of αSyn aggregates also occurs contributing to the progression and propagation of the disease (Karpowicz et al., 2019). Further supporting αSyn link with pathology, familial forms of PD are related with duplication, triplications and point mutations (mainly A30P and A53T mutations) in the SNCA gene, encoding for αSyn, which increase the aggregation potential of the protein (Burre et al., 2018).
αSyn aggregated species were shown to induce neurotoxicity through several processes: (i) affecting membrane permeabilization of several cell components, including plasma membrane and endoplasmic reticulum (Colla et al., 2012), mitochondria (Parihar et al., 2009), and vesicle membranes (Volles and Lansbury, 2002; Burre et al., 2018); (ii) increasing reactive oxygen species (ROS) production (Parihar et al., 2009) and Ca2+ influx (Danzer et al., 2007); and (iii) disrupting protein synthesis machinery and degradation systems, namely the autophagy-lysosomal and the ubiquitin-proteasomal systems (Lindersson et al., 2004; Garcia-Esparcia et al., 2015). αSyn accumulation was also shown to negatively affect SNARE-complex assembly and disassembly impairing neurotransmitter release and leading to decreased neuronal excitability and synaptic firing, what culminates in synaptotoxicity (Vekrellis et al., 2011). In the same line, αSyn overexpression in neurons decreased spine density and impaired spine dynamics (Blumenstock et al., 2017). Interestingly, synaptic structure and function is highly dependent on actin dynamics, suggesting an impact of αSyn on that cytoskeleton component.
Actin Cytoskeleton: a Critical Component for Neuronal Function
The integrity of the cytoskeleton is crucial for neuronal maintenance and function and depends on a critical regulation of its components: actin filaments, microtubules and intermediate filaments. The actin cytoskeleton, the component of interest in this review, is composed of actin filaments (F-actin) that are formed by the association of globular actin (G-actin) to a growing polymer (Mitchison and Cramer, 1996). Actin monomers polymerize/depolymerize from the actin filament constituting the fundamental process driving actin dynamics (Carlier, 1998). There is a considerable number of ABPs regulating actin dynamics among which are: (i) nucleation factors (formins and Arp2/3) that promote the assembly of G-actin into filaments and the development of branched networks (Bugyi et al., 2006; Korobova and Svitkina, 2008); (ii) actin-monomer binding proteins (profilin) that provide new subunits to the filament enhancing the assembly of G-actin into F-actin (Mockrin and Korn, 1980); (iii) proteins that bundle (fascin) or crosslink (α-actinins and filamins) the actin filaments (Tseng et al., 2004); (iv) tropomyosins which are able to regulate actin dynamics through binding along F-actin and interaction with other ABPs or by direct contact of F-actin with different tropomyosin isoforms (Gunning et al., 2008; Schevzov et al., 2012); (v) spectrins that are localized at subcortical regions linking actin cytoskeletal meshwork to membrane receptors (Burridge et al., 1982); (vi) capping proteins (adducin) which bind to actin filaments blocking their growth (Matsuoka et al., 2000) and (vii) severing proteins (actin depolymerizing factor (ADF)/cofilin-1 and gelsolin) which control the rate of actin polymerization (Weeds et al., 1991; Andrianantoandro and Pollard, 2006).
In neurons, actin acquires several structural rearrangements that are differentially distributed in the cell. Lamellipodia is an actin structure characterized by a branched network of short actin filaments while filopodia is composed of long parallel bundles of actin filaments (Neukirchen and Bradke, 2011). Lamellipodia and filopodia are actin arrangements enriched in the more dynamic neuronal structures including the growth cone, dendrites and dendritic spines. In the growth cones, besides filopodia and lamellipodia, there is, in the transition zone, an actomyosin contractile structure named actin arcs which are perpendicular to F-actin and are suggested to interact with microtubules and allow them to invade the growth cone (Schaefer et al., 2002). In dendritic spines, actin is the core structure providing the architecture for the formation, stability, motility, and morphology of the spines (Basu and Lamprecht, 2018). Actin patches are structurally similar to lamellipodia and are found in the axons and dendrites (Korobova and Svitkina, 2010; Spillane et al., 2011). It is suggested that they promote locally actin filopodia formation, as these actin patches are not motile structures (Spillane et al., 2011). More recently, a component of the neuronal subcortical cytoskeleton was described, the actin rings (Xu et al., 2013). This structure is along the axon and dendrites and is composed by a ring of short actin filaments capped by adducin and distanced by ∼190 nm by spectrin (Xu et al., 2013). More recently, non-muscle myosin II was proposed to regulate the axonal actin ring contraction and expansion (Costa et al., 2020). While actin rings are considered to give mechanical support to neurons, there is a dynamic pool of actin in the structure of the axon, termed actin trails, which have been suggested to be composed of actin “hotspots” and provide for a flexible actin cytoskeleton network in the axon (Ganguly et al., 2015).
In mature neurons a dysregulation of the actin cytoskeleton has tremendous implications for spine density, morphology and function (Zhang and Benson, 2001). An emergent number of studies have also reported the abundance and relevance of actin and ABPs in the pre-synaptic terminals, where actin dynamics plays essential functions on the vesicle pool organization, synaptic vesicle mobilization and exocytosis and posterior endocytosis (Rust and Maritzen, 2015). The crucial roles of actin at the synapse turns it an important protein to study in the context of neurodegenerative disorders where synaptic dysfunction is a central causing agent.
αSyn-Actin Cytoskeleton Link: Impact on Neuronal Health and Disease
Evidences of αSyn Interaction With Actin and Actin-Binding Proteins
The most αSyn recognized physiological function is on the synaptic vesicle cycle, a process where actin plays a key role. This functional “proximity” between αSyn and actin boosted the research on identifying an interaction between the two proteins. This interaction was investigated mainly in in vitro assays, either with cell-free or with cell-line approaches, which although were critical for the demonstration of the interaction of αSyn with actin and actin-binding proteins (Zhou et al., 2004; Peng et al., 2005; Esposito et al., 2007; Sousa et al., 2009; Welander et al., 2011; Lee et al., 2012), lack the validation in primary neuronal cultures which would be a more relevant scenario. In this respect, in a cellular PD model using rotenone-treated dopaminergic neurons, it was observed an increase in the expression levels of F-actin and αSyn. However, a putative interaction between the two proteins was not explored in that context (Mattii et al., 2019). Further supporting an interaction, αSyn and actin co-immunoprecipitated in rat brain homogenates under physiological conditions (Sousa et al., 2009).
Additionally, proteomic studies demonstrated alterations on the expression levels of several ABPs in models of αSyn overexpression in D. melanogaster (Xun et al., 2007a,b) and C. elegans (Ichibangase et al., 2008). Moreover, gelsolin was found in LBs from PD and Dementia with Lewy Bodies (DLB) patients and was shown to have a positive effect on αSyn aggregation in the presence of high Ca2+ concentrations (Welander et al., 2011).
αSyn Impact on Actin Dynamics: Cofilin-1 Involvement
Based on the data demonstrating a putative interaction between αSyn and actin, it was investigated whether αSyn directly binds to actin modulating actin dynamics. In vitro cell-free assays showed that WT αSyn decreased the rate of polymerized actin, an effect suggested to occur due to the αSyn-mediated sequestration of the actin monomers. Interestingly, this effect was decreased in the presence of high concentrations of Ca2+, a scenario mimicking a stimulated state of neurons. In opposite, A30P aSyn, increased actin polymerization and stabilization of the actin filaments (Sousa et al., 2009). Validation of the impact of αSyn on actin dynamics was performed in studies with neuronal cell lines and primary cultures of hippocampal neurons, expressing either WT or A30P αSyn, which demonstrated that physiologically WT αSyn regulates actin dynamics, while the pathologic A30P aSyn disrupts the actin cytoskeleton (Sousa et al., 2009).
The proposed physiologic impact of WT αSyn on actin dynamics, and the fact that both αSyn and actin play a role on the synapse, raise the question of whether αSyn-actin interaction might regulate neurotransmitter homeostasis. Supporting this hypothesis, a report demonstrated a critical dependence of the interaction of αSyn with the actin cytoskeleton for the trafficking and transport activity of norepineprhine transporters (Jeannotte and Sidhu, 2008).
Concerning the pathological impact of αSyn on the actin cytoskeleton, further studies showed that the extracellular addition of high concentrations of WT or A30P αSyn to hippocampal neurons induced a stabilization of the actin cytoskeleton, by increasing the number of lamellipodia and filopodia and resistance to depolymerization, with the mutant protein having a more pronounced effect. These αSyn-induced actin alterations were mediated by the activation of the actin signaling pathway Rac1/PAK2/LIMK/cofilin-1 and to require GRP78, an endoplasmic reticulum chaperone present at the cell membrane of several cell types (Bellani et al., 2014). Cofilin-1 is an actin depolymerizing protein which activity is negatively regulated by phosphorylation in the Serine 3 residue, promoted by several kinases, including LIMK (Arber et al., 1998). As such, pathologic αSyn induced cofilin-1 phosphorylation leading to inactivation of its depolymerizing action and consequently to actin stabilization. Importantly, this outcome was recapitulated using fibroblasts from PD patients carrying multiplications of the SNCA gene (Bellani et al., 2014), which presented a threefold increase in the phospho-cofilin 1/cofilin 1 ratio and an increased number and thickness of actin stress fibers structures (Bellani et al., 2014).
Additional work with primary hippocampal neurons confirmed a negative effect of pathologic αSyn on actin dynamics, mediated by cofilin-1 inactivation, which impacted on neuronal functions such as axon elongation and migration (Tilve et al., 2015). Additionally, and also supporting αSyn-induced cofilin-1 inactivation, in a glaucoma animal model, consisting on elevated intraocular pressure which results in retinal neurodegeneration, the intravitreal injection of αSyn antibodies hampered neurodegeneration, an effect that was suggested to involve upregulation of cofilin-1 (Teister et al., 2017).
The presented studies claim that pathologic concentrations of αSyn induce cofilin-1 inactivation resulting on actin stabilization and neuronal dysfunction. However, cofilin-1 was also placed in the context of αSyn-induced neurodegeneration in a different scenario. In a study addressing the mechanisms of protein aggregates entry in cells, downregulation of cofilin-1 decreased αSyn aggregates entry, while both, the silencing of ROCK1 and the pharmacological inhibition of Rho, increased aggregate entry (Zhong et al., 2018). These observations suggested Rho-ROCK1-LIMK-Cofilin-1 pathway as a relevant signaling cascade triggering αSyn aggregates entry in the host cells (Zhong et al., 2018). Regardless of the context, it is remarkable the impact of αSyn in the actin cytoskeleton through cofilin-1.
αSyn-Induced Disruption of the Actin Cytoskeleton in in vivo Models of PD
In the previous sections the analysis of the effect of αSyn on the actin cytoskeleton was mainly derived from cell-based studies. It is important to understand what happens in vivo by using disease models. In this respect, a report using a PD Drosophila model based on the neuronal overexpression of αSyn, validated the pathologic impact of the protein on the actin cytoskeleton, as αSyn transgenic flies showed increased F-actin and the presence of rod-shaped actin-cofilin rich inclusions in whole-mounted brains (Ordonez et al., 2018). Rod structures were also observed in the brainstem region of a PD mouse model expressing the A53T mutant form of αSyn, and in the cingulate cortex region of a DLB patient (Ordonez et al., 2018). Following experiments in fly demonstrated that the disruption of the actin cytoskeleton induced by αSyn was mediated by its interaction with α-spectrin, and resulted on the mislocalization of the mitochondrial fission protein Drp1 and subsequent mitochondrial dysfunction (Ordonez et al., 2018). This study pointed to a critical interaction between αSyn and α-spectrin resulting in actin dysfunction which consequently affects mitochondria. Additionally, and although not highly explored, the study also shows the scenario of αSyn induction of cofilin-actin rods. These are structures formed upon localized cofilin-1 activation (by dephosphorylation), leading to its association to F-actin and promoting the formation of short actin filaments saturated with cofilin-1. Rod formation has been mainly studied in the context of Alzheimer’s disease (AD) and shown to have a tremendous impact in neurons causing synaptic dysfunction, blocking axonal transport, and exacerbating mitochondrial membrane potential loss what culminates in cognitive impairment (Bamburg and Bernstein, 2016). Considering the impact of cofilin-actin rods on neurodegeneration, the pathologic relevance of rod formation upon αSyn overexpression should be further addressed. In this respect, one study reported the presence of oxidized γ-Synuclein in cofilin-actin rods in the thalamus of mice subjected to traumatic brain injury (Surgucheva et al., 2014).
Discussion
The current revision summarizes the studies supporting the link between αSyn and the actin cytoskeleton. This is an important topic as while the interplay between αSyn and the microtubules was recently reviewed (Carnwath et al., 2018; Calogero et al., 2019), the link αSyn-actin was less explored. Considering the αSyn structural features responsible for the interaction with cytoskeleton components, it was described that αSyn is a functional microtubule-associated protein with its C-terminal region suggested to be responsible for the interaction with microtubules (Alim et al., 2004; Cartelli et al., 2016). In the case of αSyn interaction with actin, although it is implied by the available literature, no structural features underlying this interaction have been explored to date what deserves future investigation.
One important question raised by this review is whether physiologically αSyn, by regulating actin dynamics, impacts on the actin-derived functions on synaptic transmission. In this respect, while there are studies with αSyn KO mice showing decreased SNARE-complex assembly and changes in synaptic structure and size (Burre et al., 2010; Greten-Harrison et al., 2010), other reports showed no major defects in synaptic function in αSyn KO mice (Abeliovich et al., 2000; Chandra et al., 2004; Chadchankar and Yavich, 2011). Taking this into consideration, it would be important to explore neuronal actin dynamics and actin organization in synaptic structures in αSyn KO mice. This would clarify whether the αSyn-actin link acts on the physiology of the synapse, what could explain the impairment of synaptic activity in PD.
The most striking point of the present review is the highlight of cofilin-1 as a central player in αSyn-induced neuronal dysfunction. Cofilin-1 depolymerizing activity upon actin is crucial for synaptic function since the remodeling of the pre- and post-synapses intimately relies on actin dynamics (Pontrello et al., 2012). As such, we might hypothesize that the reported αSyn-activation of the actin signaling pathway Rac1/PAK2/LIMK/cofilin-1 via GRP78 (Bellani et al., 2014) which results on cofilin inactivation, and consequent blockage of actin dynamics (Sousa et al., 2009; Bellani et al., 2014) occurs at the pre-synapse, and might contribute for the synaptic dysfunction observed in the several synucleinopathies. An additional pathway by which pathogenic αSyn might cause synaptic dysfunction is via the induction of cofilin-actin rods formation (Cichon et al., 2012), which was observed in a PD Drosophila model (Ordonez et al., 2018). In AD, Aβ-induced formation of rods occurs through a pathway involving the cellular prion protein (PrPC) and NADPH oxidase (NOX) (Walsh et al., 2014), which results in the dysregulation of cofilin activity via oxidation and dephosphorylation, and consequent formation of cofilin-actin rods. Interestingly, αSyn was shown to interact with PrPC to induce synaptic dysfunction (Ferreira et al., 2017). This finding raises the question of whether, similarly to what occurs in AD, αSyn-induced rod formation is mediated through a PrPC-dependent pathway culminating on disruption of synaptic activity.
While the literature suggests that αSyn might impact on neuronal function, through modulation of cofilin-1 activity, is still unclear whether αSyn induces an activation or inactivation of the ABP. Interestingly, a similar scenario was seen in AD where cofilin-1 activity was shown to be regulated in multiple ways depending on the pathogenesis context (Kang and Woo, 2019). Importantly, inhibition of cofilin activity or expression was shown to have ameliorative effects in AD (Deng et al., 2016). In the case of αSyn-induced neurodegeneration, it will be important to analyze the phosphorylation status of cofilin-1 in different pathological scenarios and cellular contexts, as well as the impact of cofilin-actin rods formation for neuronal function. These studies will be critical to point cofilin-1 as novel therapeutic target to prevent neurodegeneration in synucleinopathies.
Another point that stands out from the studies reported in this review is that the few reported experiments in primary neurons were performed with cultures of hippocampal neurons (Sousa et al., 2009; Bellani et al., 2014; Tilve et al., 2015). Although this observation suggests that additional studies should be performed with primary cultures of dopaminergic neurons, the cell type mainly affected in typical motor PD, research on the impact of αSyn on actin in hippocampal neurons is also essential considering the symptomology of dementia which has been linked to Syn pathology in the hippocampus leading to neuronal dysfunction (Hall et al., 2014).
In summary, this review presents a critical perspective in the αSyn impact on the actin cytoskeleton. The literature here revised strongly suggests that αSyn interacts/modulates actin and ABPs what has consequences to pathophysiology, as summarized in Figure 1. Nevertheless, this topic requires further investigation what might be of extremely importance in the context of both health and disease.
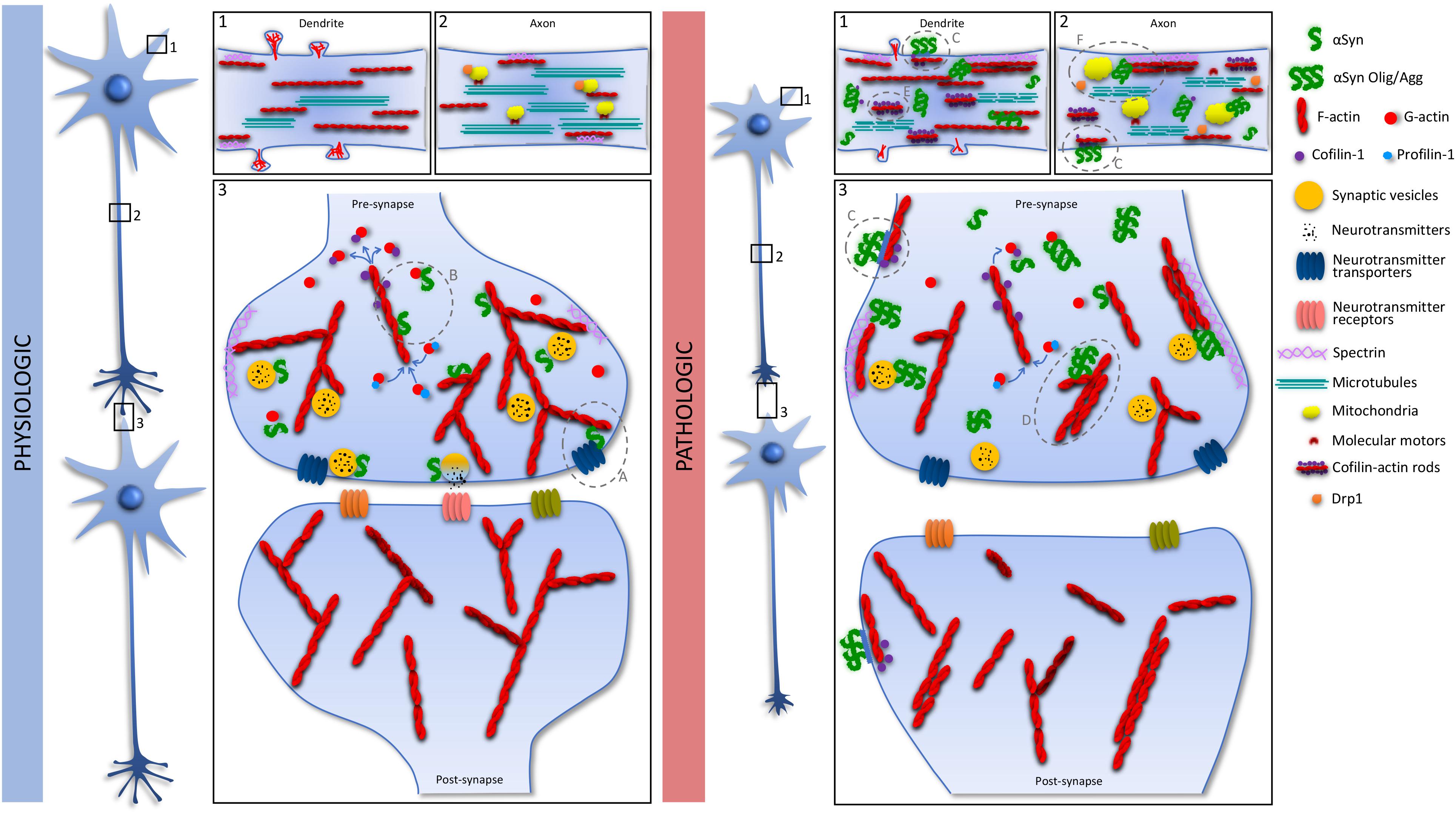
Figure 1. Schematic representation of the link between αSyn and actin. Under physiologic conditions αSyn is mainly concentrated at the pre-synapse and interacts with synaptic vesicles at the several stages of the vesicle cycle. At the pre-synapse αSyn-actin interaction was described as being crucial for the trafficking and transport activity of neurotransmitter transporters (Physiologic, Panel 3; A). Additionally, we suggest that the reported αSyn regulation of actin dynamics might contribute for the proper actin-derived functions at the pre-synapse (Physiologic, Panel 3; B). Under pathologic conditions, overexpression and misfolding of αSyn affects several cellular processes. At the cell membrane, extracellular misfolded αSyn uses a pathway culminating in cofilin-1 activation and actin remodeling to enter the cells (Pathologic, Panels 1–3; C). Intracellularly, synaptic dysfunction might occur due to the αSyn-induced stabilization of the actin cytoskeleton (Pathologic, Panel 3; D), through inactivation of cofilin-1 or αSyn-induced cofilin-actin rod formation (Pathologic, Panel 1; E), that also affects axonal transport. Additionally, αSyn interaction with spectrin (Pathologic, Panel 2; F), disrupts the actin cytoskeleton with consequent mislocalization of Drp1 and mitochondrial impairment.
Author Contributions
MO and ML conceived the structure and content and wrote the manuscript. MO produced the figure. Both authors contributed to the article and approved the submitted version.
Funding
The authors were supported by the FEDER – Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020 – Operacional Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT – Fundação Para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior in the framework of the project POCI-01-0145-FEDER-028336 (PTDC/MED-NEU/28336/2017). MO was a FCT fellow (SFRH/BD/118728/2016). ML was a FCT Investigator (IF/00902/2015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Alim, M. A., Ma, Q. L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., et al. (2004). Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers. Dis. 6, 435–442. doi: 10.3233/jad-2004-6412
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303. doi: 10.1073/pnas.94.1.298
Andrianantoandro, E., and Pollard, T. D. (2006). Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell. 24, 13–23. doi: 10.1016/j.molcel.2006.08.006
Arber, S., Barbayannis, F. A., Hanser, H., Schneider, C., Stanyon, C. A., Bernard, O., et al. (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805–809. doi: 10.1038/31729
Bamburg, J. R., and Bernstein, B. W. (2016). Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton 73, 477–497. doi: 10.1002/cm.21282
Basu, S., and Lamprecht, R. (2018). The role of actin cytoskeleton in dendritic spines in the maintenance of long-term memory. Front. Mol. Neurosci. 11:143. doi: 10.3389/fnmol.2018.00143
Bellani, S., Mescola, A., Ronzitti, G., Tsushima, H., Tilve, S., Canale, C., et al. (2014). GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 21, 1971–1983. doi: 10.1038/cdd.2014.111
Blumenstock, S., Rodrigues, E. F., Peters, F., Blazquez-Llorca, L., Schmidt, F., Giese, A., et al. (2017). Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol. Med. 9, 716–731. doi: 10.15252/emmm.201607305
Bugyi, B., Papp, G., Hild, G., Lorinczy, D., Nevalainen, E. M., Lappalainen, P., et al. (2006). Formins regulate actin filament flexibility through long range allosteric interactions. J. Biol. Chem. 281, 10727–10736. doi: 10.1074/jbc.M510252200
Burre, J., Sharma, M., and Sudhof, T. C. (2018). Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb. Perspect. Med. 8:a024091. doi: 10.1101/cshperspect.a024091
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Burre, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Sudhof, T. C. (2013). Properties of native brain alpha-synuclein. Nature 498, E4–E6. doi: 10.1038/nature12125
Burridge, K., Kelly, T., and Mangeat, P. (1982). Nonerythrocyte spectrins: actin-membrane attachment proteins occurring in many cell types. J. Cell Biol. 95(2 Pt 1), 478–486. doi: 10.1083/jcb.95.2.478
Calogero, A. M., Mazzetti, S., Pezzoli, G., and Cappelletti, G. (2019). Neuronal microtubules and proteins linked to Parkinson’s disease: a relevant interaction? Biol. Chem. 400, 1099–1112. doi: 10.1515/hsz-2019-0142
Carlier, M. F. (1998). Control of actin dynamics. Curr. Opin. Cell Biol. 10, 45–51. doi: 10.1016/s0955-0674(98)80085-9
Carnwath, T., Mohammed, R., and Tsiang, D. (2018). The direct and indirect effects of alpha-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 14, 1685–1695. doi: 10.2147/NDT.S166322
Cartelli, D., Aliverti, A., Barbiroli, A., Santambrogio, C., Ragg, E. M., Casagrande, F. V., et al. (2016). alpha-Synuclein is a novel microtubule dynamase. Sci. Rep. 6:33289. doi: 10.1038/srep33289
Chadchankar, H., and Yavich, L. (2011). Sub-regional differences and mechanisms of the short-term plasticity of dopamine overflow in striatum in mice lacking alpha-synuclein. Brain Res. 1423, 67–76. doi: 10.1016/j.brainres.2011.09.026
Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., et al. (2004). Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. U.S.A. 101, 14966–14971. doi: 10.1073/pnas.0406283101
Cheng, F., Vivacqua, G., and Yu, S. (2011). The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 42, 242–248. doi: 10.1016/j.jchemneu.2010.12.001
Cichon, J., Sun, C., Chen, B., Jiang, M., Chen, X. A., Sun, Y., et al. (2012). Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J. Biol. Chem. 287, 3919–3929. doi: 10.1074/jbc.M111.301911
Clayton, D. F., and George, J. M. (1998). The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254. doi: 10.1016/s0166-2236(97)01213-7
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M. K. (2012). Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012
Costa, A. R., Sousa, S. C., Pinto-Costa, R., Mateus, J. C., Lopes, C. D., Costa, A. C., et al. (2020). The membrane periodic skeleton is an actomyosin network that regulates axonal diameter and conduction. eLife 9:55471. doi: 10.7554/eLife.55471
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
Deng, Y., Wei, J., Cheng, J., Zhong, P., Xiong, Z., Liu, A., et al. (2016). Partial amelioration of synaptic and cognitive deficits by inhibiting cofilin dephosphorylation in an animal model of Alzheimer’s Disease. J. Alzheimers Dis. 53, 1419–1432. doi: 10.3233/JAD-160167
Diao, J., Burre, J., Vivona, S., Cipriano, D. J., Sharma, M., Kyoung, M., et al. (2013). Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2:e00592. doi: 10.7554/eLife.00592
Dompierre, J. P., Godin, J. D., Charrin, B. C., Cordelieres, F. P., King, S. J., Humbert, S., et al. (2007). Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007
d’Ydewalle, C., Krishnan, J., Chiheb, D. M., Van Damme, P., Irobi, J., Kozikowski, A. P., et al. (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974. doi: 10.1038/nm.2396
Eira, J., Silva, C. S., Sousa, M. M., and Liz, M. A. (2016). The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 141, 61–82. doi: 10.1016/j.pneurobio.2016.04.007
Emamzadeh, F. N. (2016). Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 21:29. doi: 10.4103/1735-1995.181989
Esposito, A., Dohm, C. P., Kermer, P., Bahr, M., and Wouters, F. S. (2007). alpha-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis. 26, 521–531. doi: 10.1016/j.nbd.2007.01.014
Ferreira, D. G., Temido-Ferreira, M., Vicente Miranda, H., Batalha, V. L., Coelho, J. E., Szego, E. M., et al. (2017). alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 20, 1569–1579. doi: 10.1038/nn.4648
Ganguly, A., Tang, Y., Wang, L., Ladt, K., Loi, J., Dargent, B., et al. (2015). A dynamic formin-dependent deep F-actin network in axons. J. Cell Biol. 210, 401–417. doi: 10.1083/jcb.201506110
Garcia-Esparcia, P., Hernandez-Ortega, K., Koneti, A., Gil, L., Delgado-Morales, R., Castano, E., et al. (2015). Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 3:76. doi: 10.1186/s40478-015-0257-4
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U.S.A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
Gunning, P., O’Neill, G., and Hardeman, E. (2008). Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 88, 1–35. doi: 10.1152/physrev.00001.2007
Hall, H., Reyes, S., Landeck, N., Bye, C., Leanza, G., Double, K., et al. (2014). Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 137(Pt 9), 2493–2508. doi: 10.1093/brain/awu193
Ichibangase, T., Saimaru, H., Takamura, N., Kuwahara, T., Koyama, A., Iwatsubo, T., et al. (2008). Proteomics of Caenorhabditis elegans over-expressing human alpha-synuclein analyzed by fluorogenic derivatization-liquid chromatography/tandem mass spectrometry: identification of actin and several ribosomal proteins as negative markers at early Parkinson’s disease stages. Biomed. Chromatogr. 22, 232–234. doi: 10.1002/bmc.931
Jeannotte, A. M., and Sidhu, A. (2008). Regulated interactions of the norepineprhine transporter by the actin and microtubule cytoskeletons. J. Neurochem. 105, 1668–1682. doi: 10.1111/j.1471-4159.2008.05258.x
Kang, D. E., and Woo, J. A. (2019). Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s Disease. J. Alzheimers Dis. 72, S131–S144. doi: 10.3233/JAD-190585
Karpowicz, R. J. Jr., Trojanowski, J. Q., and Lee, V. M. (2019). Transmission of alpha-synuclein seeds in neurodegenerative disease: recent developments. Lab. Invest. 99, 971–981. doi: 10.1038/s41374-019-0195-z
Korobova, F., and Svitkina, T. (2008). Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol. Biol. Cell 19, 1561–1574. doi: 10.1091/mbc.E07-09-0964
Korobova, F., and Svitkina, T. (2010). Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 21, 165–176. doi: 10.1091/mbc.E09-07-0596
Lee, H. J., Lee, K., and Im, H. (2012). alpha-Synuclein modulates neurite outgrowth by interacting with SPTBN1. Biochem. Biophys. Res. Commun. 424, 497–502. doi: 10.1016/j.bbrc.2012.06.143
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. doi: 10.1074/jbc.M306390200
Magiera, M. M., Bodakuntla, S., Ziak, J., Lacomme, S., Marques Sousa, P., Leboucher, S., et al. (2018). Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37:e0100440. doi: 10.15252/embj.2018100440
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/jneurosci.08-08-02804.1988
Maroteaux, L., and Scheller, R. H. (1991). The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res. Mol. Brain Res. 11, 335–343. doi: 10.1016/0169-328x(91)90043-w
Matsuoka, Y., Li, X., and Bennett, V. (2000). Adducin: structure, function and regulation. Cell Mol. Life. Sci. 57, 884–895. doi: 10.1007/PL00000731
Mattii, L., Pardini, C., Ippolito, C., Bianchi, F., Sabbatini, A. R. M., and Vaglini, F. (2019). Rho-inhibition and neuroprotective effect on rotenone-treated dopaminergic neurons in vitro. Neurotoxicology 72, 51–60. doi: 10.1016/j.neuro.2019.02.006
Minamide, L. S., Striegl, A. M., Boyle, J. A., Meberg, P. J., and Bamburg, J. R. (2000). Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2, 628–636. doi: 10.1038/35023579
Mitchison, T. J., and Cramer, L. P. (1996). Actin-based cell motility and cell locomotion. Cell 84, 371–379. doi: 10.1016/s0092-8674(00)81281-7
Mockrin, S. C., and Korn, E. D. (1980). Acanthamoeba profilin interacts with G-actin to increase the rate of exchange of actin-bound adenosine 5’-triphosphate. Biochemistry 19, 5359–5362. doi: 10.1021/bi00564a033
Munsie, L. N., and Truant, R. (2012). The role of the cofilin-actin rod stress response in neurodegenerative diseases uncovers potential new drug targets. Bioarchitecture 2, 204–208. doi: 10.4161/bioa.22549
Neukirchen, D., and Bradke, F. (2011). Neuronal polarization and the cytoskeleton. Semin. Cell Dev. Biol. 22, 825–833. doi: 10.1016/j.semcdb.2011.08.007
Ordonez, D. G., Lee, M. K., and Feany, M. B. (2018). alpha-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97, 108–124. doi: 10.1016/j.neuron.2017.11.036
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2009). Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int. J. Biochem. Cell Biol. 41, 2015–2024. doi: 10.1016/j.biocel.2009.05.008
Peng, X., Tehranian, R., Dietrich, P., Stefanis, L., and Perez, R. G. (2005). Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118(Pt 15), 3523–3530. doi: 10.1242/jcs.02481
Pontrello, C. G., Sun, M. Y., Lin, A., Fiacco, T. A., DeFea, K. A., and Ethell, I. M. (2012). Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc. Natl. Acad. Sci. U.S.A. 109, E442–E451. doi: 10.1073/pnas.1118803109
Prots, I., Grosch, J., Brazdis, R. M., Simmnacher, K., Veber, V., Havlicek, S., et al. (2018). alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. U.S.A. 115, 7813–7818. doi: 10.1073/pnas.1713129115
Qiang, L., Yu, W., Andreadis, A., Luo, M., and Baas, P. W. (2006). Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 26, 3120–3129. doi: 10.1523/JNEUROSCI.5392-05.2006
Qu, X., Yuan, F. N., Corona, C., Pasini, S., Pero, M. E., Gundersen, G. G., et al. (2017). Stabilization of dynamic microtubules by mDia1 drives Tau-dependent Abeta1-42 synaptotoxicity. J. Cell Biol. 216, 3161–3178. doi: 10.1083/jcb.201701045
Rust, M. B., and Maritzen, T. (2015). Relevance of presynaptic actin dynamics for synapse function and mouse behavior. Exp. Cell Res. 335, 165–171. doi: 10.1016/j.yexcr.2014.12.020
Schaefer, A. W., Kabir, N., and Forscher, P. (2002). Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J. Cell Biol. 158, 139–152. doi: 10.1083/jcb.200203038
Schevzov, G., Curthoys, N. M., Gunning, P. W., and Fath, T. (2012). Functional diversity of actin cytoskeleton in neurons and its regulation by tropomyosin. Int. Rev. Cell Mol. Biol. 298, 33–94. doi: 10.1016/B978-0-12-394309-5.00002-X
Scott, D., and Roy, S. (2012). alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012
Sousa, V. L., Bellani, S., Giannandrea, M., Yousuf, M., Valtorta, F., Meldolesi, J., et al. (2009). {alpha}-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 20, 3725–3739. doi: 10.1091/mbc.E08-03-0302
Spillane, M., Ketschek, A., Jones, S. L., Korobova, F., Marsick, B., Lanier, L., et al. (2011). The actin nucleating Arp2/3 complex contributes to the formation of axonal filopodia and branches through the regulation of actin patch precursors to filopodia. Dev. Neurobiol. 71, 747–758. doi: 10.1002/dneu.20907
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Surgucheva, I., He, S., Rich, M. C., Sharma, R., Ninkina, N. N., Stahel, P. F., et al. (2014). Role of synucleins in traumatic brain injury - an experimental in vitro and in vivo study in mice. Mol. Cell. Neurosci. 63, 114–123. doi: 10.1016/j.mcn.2014.10.005
Teister, J., Anders, F., Beck, S., Funke, S., von Pein, H., Prokosch, V., et al. (2017). Decelerated neurodegeneration after intravitreal injection of alpha-synuclein antibodies in a glaucoma animal model. Sci. Rep. 7:6260. doi: 10.1038/s41598-017-06702-1
Tilve, S., Difato, F., and Chieregatti, E. (2015). Cofilin 1 activation prevents the defects in axon elongation and guidance induced by extracellular alpha-synuclein. Sci. Rep. 5:16524. doi: 10.1038/srep16524
Tseng, Y., An, K. M., Esue, O., and Wirtz, D. (2004). The bimodal role of filamin in controlling the architecture and mechanics of F-actin networks. J. Biol. Chem. 279, 1819–1826. doi: 10.1074/jbc.M306090200
Vekrellis, K., Xilouri, M., Emmanouilidou, E., Rideout, H. J., and Stefanis, L. (2011). Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 10, 1015–1025. doi: 10.1016/S1474-4422(11)70213-7
Volles, M. J., and Lansbury, P. T. Jr. (2002). Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602. doi: 10.1021/bi0121353
Walsh, K. P., Minamide, L. S., Kane, S. J., Shaw, A. E., Brown, D. R., Pulford, B., et al. (2014). Amyloid-beta and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One 9:e95995. doi: 10.1371/journal.pone.0095995
Wang, L., Das, U., Scott, D. A., Tang, Y., McLean, P. J., and Roy, S. (2014). alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 24, 2319–2326. doi: 10.1016/j.cub.2014.08.027
Weeds, A. G., Gooch, J., Hawkins, M., Pope, B., and Way, M. (1991). Role of actin-binding proteins in cytoskeletal dynamics. Biochem. Soc. Trans. 19, 1016–1020. doi: 10.1042/bst0191016
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., and Lansbury, P. T. Jr. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. doi: 10.1021/bi961799n
Welander, H., Bontha, S. V., Nasstrom, T., Karlsson, M., Nikolajeff, F., Danzer, K., et al. (2011). Gelsolin co-occurs with Lewy bodies in vivo and accelerates alpha-synuclein aggregation in vitro. Biochem. Biophys. Res. Commun. 412, 32–38. doi: 10.1016/j.bbrc.2011.07.027
Xu, K., Zhong, G., and Zhuang, X. (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. doi: 10.1126/science.1232251
Xun, Z., Sowell, R. A., Kaufman, T. C., and Clemmer, D. E. (2007a). Lifetime proteomic profiling of an A30P alpha-synuclein Drosophila model of Parkinson’s disease. J. Proteome Res. 6, 3729–3738. doi: 10.1021/pr0700504
Xun, Z., Sowell, R. A., Kaufman, T. C., and Clemmer, D. E. (2007b). Protein expression in a Drosophila model of Parkinson’s disease. J. Proteome Res. 6, 348–357. doi: 10.1021/pr060488o
Zhang, L., Liu, C., Wu, J., Tao, J. J., Sui, X. L., Yao, Z. G., et al. (2014). Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 41, 1193–1205. doi: 10.3233/JAD-140066
Zhang, W., and Benson, D. L. (2001). Stages of synapse development defined by dependence on F-actin. J. Neurosci. 21, 5169–5181. doi: 10.1523/jneurosci.21-14-05169.2001
Zhong, Z., Grasso, L., Sibilla, C., Stevens, T. J., Barry, N., and Bertolotti, A. (2018). Prion-like protein aggregates exploit the RHO GTPase to cofilin-1 signaling pathway to enter cells. EMBO J. 37:822. doi: 10.15252/embj.201797822
Keywords: alpha-Synuclein, Parkinson’s disease, actin cytoskeleton, actin-binding proteins, cofilin-1
Citation: Oliveira da Silva MI and Liz MA (2020) Linking Alpha-Synuclein to the Actin Cytoskeleton: Consequences to Neuronal Function. Front. Cell Dev. Biol. 8:787. doi: 10.3389/fcell.2020.00787
Received: 21 April 2020; Accepted: 27 July 2020;
Published: 12 August 2020.
Edited by:
Beate Winner, University of Erlangen Nuremberg, GermanyReviewed by:
Christian Gonzalez-Billault, University of Chile, ChileGraziella Cappelletti, University of Milan, Italy
Copyright © 2020 Oliveira da Silva and Liz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Márcia A. Liz, maliz@ibmc.up.pt