 Yanfei Yang
Yanfei Yang Yang Yang
Yang Yang Jing Yang
Jing Yang Xia Zhao1*
Xia Zhao1* Xiawei Wei
Xiawei Wei- 1Department of Gynecology and Obstetrics, Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, West China Second Hospital, Sichuan University, Chengdu, China
- 2Laboratory of Aging Research and Cancer Drug Target, State Key Laboratory of Biotherapy and Cancer Center, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
Ovarian cancer is one of the leading causes of death in patients with gynecological malignancy. Despite optimal cytoreductive surgery and platinum-based chemotherapy, ovarian cancer disseminates and relapses frequently, with poor prognosis. Hence, it is urgent to find new targeted therapies for ovarian cancer. Recently, the tumor microenvironment has been reported to play a vital role in the tumorigenesis of ovarian cancer, especially with discoveries from genome-, transcriptome- and proteome-wide studies; thus tumor microenvironment may present potential therapeutic target for ovarian cancer. Here, we review the interactions between the tumor microenvironment and ovarian cancer and various therapies targeting the tumor environment.
Highlights
– The tumor microenvironment plays important roles in the progression of ovarian cancer.
– Current “-omic” technology is revealing the molecular landscape of ovarian cancer tumor microenvironment, facilitating future therapeutic strategy.
– Ovarian cancer therapies targeting tumor microenvironment is rapidly developing, targets mainly focusing on cancer-associated fibroblasts, tumor-associated macrophages, angiogenesis and immune checkpoint blockade.
Introduction
Ovarian cancer is one of the leading causes of common, lethal gynecologic malignancy (Cortez et al., 2018). In 2018 worldwide, there were an estimated 295,414 cases and 184,799 deaths from ovarian cancer (Bray et al., 2018). Because of the lack of an early diagnosis method and the absence of specific early warning symptoms, patients with ovarian cancer are usually diagnosed at an advanced stage and have a poor prognosis (Scarlett and Conejo-Garcia, 2012).
Based on histological origin, ovarian tumors can be categorized into epithelial, germ cell, sex cord, or stromal tumors (Jayson et al., 2014). Around 90% of primary ovarian tumors are of epithelial origin (Colombo et al., 2010; Ledermann et al., 2013), so we mainly focus on evidence of epithelial ovarian cancer in this review. The World Health Organization (WHO) classified epithelial ovarian cancer (EOC) into the following types: serous, mucinous, endometrioid, clear cell, transitional cell, mixed epithelial, undifferentiated, and unclassified (Ledermann et al., 2013). According to architectural features, EOC is also classified into 3 grades by the International Federation of Gynecology and Obstetrics (FIGO) system (Colombo et al., 2010); in serous EOC, FIGO grade 1 is defined as low-grade while FIGO grade 2 and 3 are combined as high-grade (Bodurka et al., 2012). The classification with histosubtypes and grades are with prognostic significance (Ledermann et al., 2013).
The current standardized treatment for ovarian cancer is optimal cytoreductive surgery plus platinum-based chemotherapy with the carboplatin-paclitaxel regimen (Bolton et al., 2012). However, with the development of chemotherapy-resistant and refractory diseases, the sensitivity of chemotherapy has decreased (Lim and Ledger, 2016). Therefore, the long-term survival rate for ovarian cancer has decreased, and the recurrence rate has increased (Lim and Ledger, 2016). Hennessy et al. (2009) reported that despite benefiting from first-line therapy, 75% of patients with advanced ovarian cancer (stage III or IV) have tumor relapse at a median of 15 months from diagnosis. Moreover, for patients with early-stage disease (stage I or II), the long-term survival rates (>10 years) are 80–95%. In contrast, patients with advanced disease (stage III or IV) had a 10–30% long-term survival rate. Therefore, there is an urgent need to find new targeted therapies to improve the treatment efficacy of ovarian cancer. In recent years, the tumor microenvironment (TME) has been reported to play a vital role in the tumorigenesis of ovarian cancer and is considered a possible therapeutic target for ovarian cancer. Here, we review the interactions between the TME and ovarian cancer and various therapies targeting the tumor environment.
TME in Ovarian Cancer
The TME comprises (1) the extracellular matrix (ECM), which consists of chemokines, inflammatory cytokines, integrins, matrix metalloproteinases (MMPs) and other secreted molecules, and (2) stromal cells, including cancer cells, cancer stem cells, pericytes, cancer-associated fibroblasts (CAFs), endothelial cells (ECs) and immune cells (Figure 1) (Hanahan and Weinberg, 2011). In this part, we reviewed current findings on the impact of some key components above on ovarian cancer progression.
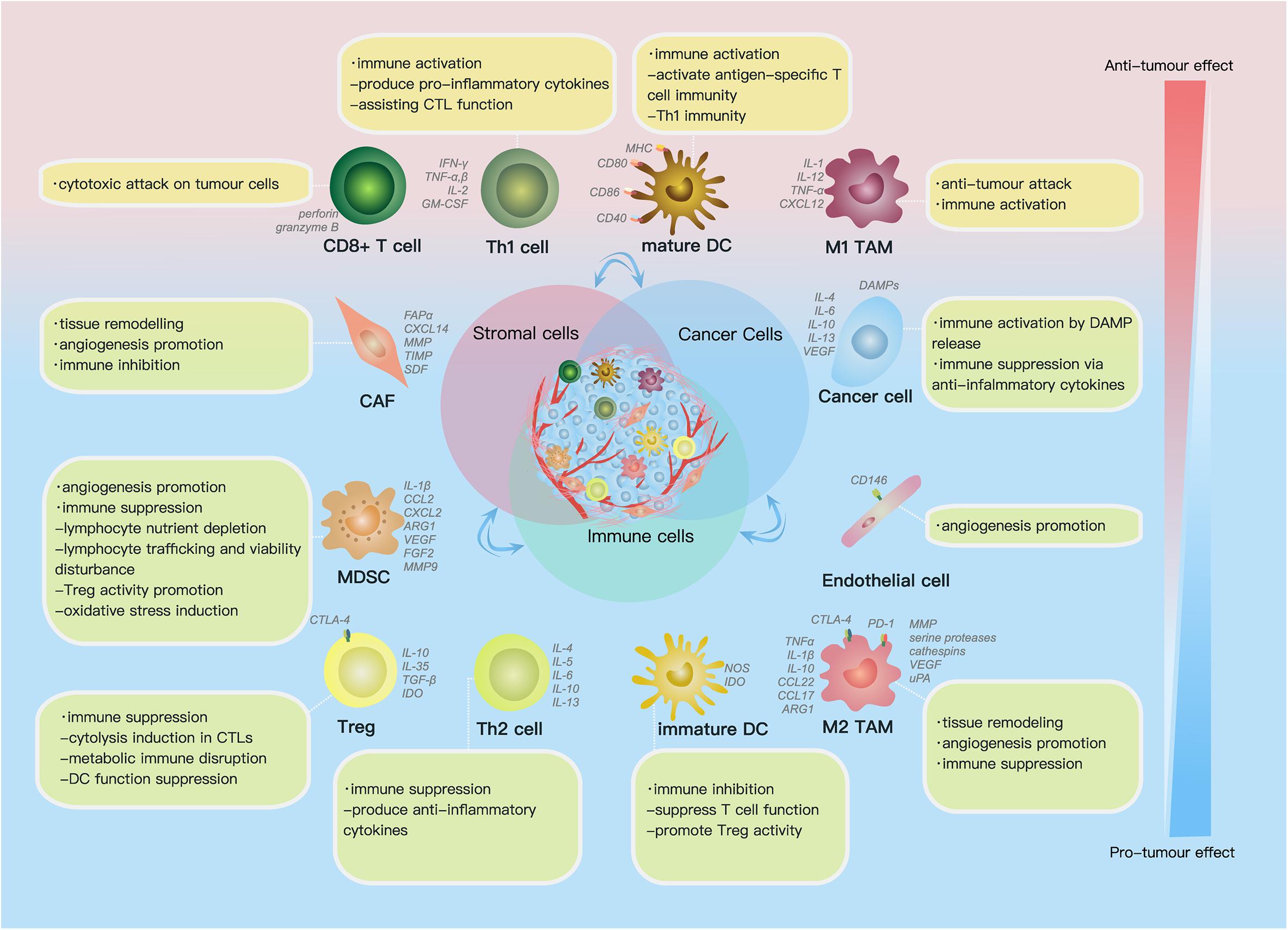
Figure 1. Cell components and functions in the tumor microenvironment (TME). Cell components in the TME can be classified into cancer cells, immune cells and stromal cells; these cells actively interact with each other by molecules they secrete [including cytokines, chemokines, damage-associated molecular patterns (DAMPs) etc.] and receptors they express, such as histocompatibility complex class (MHC) molecules, programmed cell death protein 1 (PD-1), etc., forming an evolving microenvironment. On the continuous spectrum from anti-tumor to pro-tumor effect, different cell components can locate at distinct positions, and the same group of cells may also be re-polarized depending on signals in the TME. The progression or regression of a single tumor site depends on the overall effect of the complex cellular and molecular regulating network in the TME.
Cancer-Associated Fibroblasts
Fibroblasts, which differentiate from mesenchymal-derived cells, are part of the TME (Ishii et al., 2016). They produce various MMPs, tissue inhibitor of metalloproteinases (TIMPs) and most of the proteins comprising the ECM, such as collagens, fibronectin and laminin (Kalluri and Zeisberg, 2006; Erdogan and Webb, 2017). These fibroblasts in the tumor milieu are also called “CAFs.” Additionally, CAFs can transdifferentiate from other cells, such as pericytes, epithelial cells and ECs, via exposure to platelet-derived growth factor (PDGF), tumor-derived transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), MMPs and reactive oxygen species (ROS) (Cai et al., 2012; Yu Y. et al., 2014; Denton et al., 2018).
CAFs are known to promote tumor progression via various mechanisms. CAFs can enhance tumor cell proliferation, invasion and migration. Sjoberg et al. (2016) showed that CAFs highly expressed CXCL14, which was an important factor in promoting cancer growth. CAFs also express the fibroblast activation protein α (FAP). Yang’s study indicated that FAPα enhanced the migration and invasion ability of HO-8910PM cells (a highly metastatic ovarian cancer cell line) Additionally, FAPα increased HO-8910PM cell proliferation.
CAFs promote immune inhibition and angiogenesis. Givel et al. (2018) found that CAFs increase the infiltration of FOXP3+ regulatory T lymphocytes (Tregs) at the tumor site, which exerts immune suppression effect in the tumor milieu. Additionally, in Orimo’s research, CAFs have high expression of stromal cell-derived factor-1 (SDF-1). Released SDF-1 promotes angiogenesis and tumor proliferation in a paracrine fashion (Orimo et al., 2005).
CAFs also increase platinum resistance and accelerate recurrence. Fauceglia’s study showed that CAFs expressed the FAP α. By analyzing 338 EOC tissues, they found that the overexpression of FAP acted as a hallmark for platinum resistance. Additionally, patients with FAP+ stroma had a shortened recurrence compared to that of patients with FAP- stroma (Mhawech-Fauceglia et al., 2015).
Several studies have indicated that CAFs was a biomarker of poor prognosis in ovarian cancer (Yang et al., 2013; Mhawech-Fauceglia et al., 2015; Zhao et al., 2017; Givel et al., 2018). In Givel’s study of CAFs in high-grade serous ovarian cancers (HGSOC), the results showed that the expression of CXCL12β and the infiltration of CAF-S1 (a subtype of CAFs) implied a dismal prognosis (Givel et al., 2018). Despite that accumulating studies have demonstrated the pro-tumor progression impact of CAFs, it is worthy of notifying that there are different subtypes of CAFs with heterogenous function status. Recently, Hussain et al. (2020) found 2 CAF subsets distinguished by the FAP expression level. The FAP-high CAF subtype, instead of the FAP-low subtype, was found to aggressively enhance tumor progression and negatively influence patient outcomes, which shed light on therapeutic strategies involving CAF modulation to consider CAF status in patient selection.
CAFs are a crucial cell population in the tumor microenvironment. CAFs promote the proliferation, invasion and migration of cancer cells and stimulate angiogenesis by coordinating with other cells. A deeper understanding of CAFs is needed to better understand how CAFs affect the tumor microenvironment.
Endothelial Cells
Endothelial cells (ECs) are components of the TME. Lining the vessels, ECs are crucial for transporting oxygen and nutrients and are closely associated with angiogenesis (Carmeliet and Jain, 2000; Hanahan and Weinberg, 2011). As we all know, angiogenesis is a complicated process accommodated by angiogenesis activators and inhibitors. Angiogenesis activators include VEGF, FGF-2, PDGF, TGFα and TGFβ, TNF-α, prostaglandin E2 and Interleukin 8 (IL-8). The angiogenesis inhibitors contain angiopoietin (Angs), Thrombospondin 1 (TSP-1) and endostatin. Moreover, the signal-transducing network of endothelial cells is associated with VEGF, FGF and Angs signals (Cross and Claesson-Welsh, 2001; Ahmed and Bicknell, 2009).
VEGF is a protein family consisting of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E and PLGF (placental growth factor). It is regulated by the ischaemia/hypoxia-induced genes (HIFs), epidermal growth factor (EGF) and PDGF (Semenza, 2000). There are three receptors for VEGF: VEGF receptors 1 (VEGFR1), VEGFR2 and VEGFR3. VEGFR1 and VEGFR2 are mainly expressed on ECs and are receptors for VEGF-A (Hagberg et al., 2010). Additionally, VEGFR1 is also a receptor for VEGF-B and PLGF. VEGFR3 is a receptor for VEGF-C and VEGF-D. Neuropilins (NRP1 and NRP2) are coreceptors for the VEGF family. With the help of NRP1, the binding affinity between VEGF-A and PLGF and VEGFR2 increases (Ferrara and Adamis, 2016). Similarly, with the effect of NRP2, VEGF-C, and VEGF-D have increased binding affinity with VEGFR-3. It is well known that the VEGF family is implicated in the adjustment of angiogenesis and lymphangiogenesis (Tammela and Alitalo, 2010; Apte et al., 2019). Among them, VEGF-A is crucial for angiogenesis, while VEGF-C and VEGF-D regulate lymphangiogenesis (Tammela and Alitalo, 2010; Apte et al., 2019).
Angs also a protein family consisting of Ang-1, Ang-2, Ang-3, and Ang-4. Through combined with the receptors of Angs “TIEs,” they perform different functions in angiogenesis. Ang-1 and -4 can bind to TIE2 and stimulate the tyrosine phosphorylation of TIE2. On the contrary, Ang-2 and -3 can competitive combined TIE2 without stimulating tyrosine phosphorylation, which stopping the signal transduction of angiogenesis (Sallinen et al., 2010; Lin et al., 2011; Sallinen et al., 2014). However, other study indicated that with Ang-1, Ang-2 block the TIE2 signaling, while Ang-2 induce the TIE2 signaling without Ang-1 (Yuan et al., 2009). Additionally, Ang-1 promotes the maturation and stabilization of vessel, while Ang-2 destabilize the stabilized vessel (Tse et al., 2003).
VEGF indicate poor clinical outcomes (Wimberger et al., 2014; Shen et al., 2017; Sopo et al., 2019). Wimberger et al. found that by Kaplan-Meier analyses, VEGFR1 expression was closely related to decreased overall survival (OS) and progression-free survival (PFS) (Wimberger et al., 2014). Sopo’ study discovered that VEGFR1, VEGF-A and VEGF-D were highly expressed in omental metastases compared to expression in primary ovarian epithelial tumors. Interestingly, patients with low VEGF-A expression were more likely to have a poor prognosis. Patients with high VEGF-C expression were related to a short PFS (Sopo et al., 2019).
The CD146 expression in the membrane of ECs promotes the migration of ECs and angiogenesis. CD146 is an endothelial biomarker and the extracellular domain of CD146 directly interacts with VEGFR2. Yan’s study demonstrated that CD146 can promote angiogenesis (Yan et al., 2003). Subsequently, Jiang’s report indicates CD146 promotes the migration of ECs and the formation of microvasculature by enhancing VEGFR2 phosphorylation and downstream signaling (AKT/p38 MAPKs/NF-κB) activation (Jiang et al., 2012). Interestingly, Zhou’s research indicated that the gene and protein levels of CD146 and VEGFRA were increased in patients with EOC compared to those of non-cancer patients (Zhou et al., 2019).
Enhanced Angs expression increase relapse and decrease survival time (Sallinen et al., 2010, 2014; Lin et al., 2011). Sallinen et al. observed that patients with ovarian carcinoma had higher Ang2 levels compared to those of patients with benign ovarian tumors. Furthermore, by analyzing the Kaplan–Meier curves, they found that increased Ang-2 levels (>2.7 ng/ml) were a biomarker for poor recurrence-free survival (Sallinen et al., 2010). Subsequently, they discovered that the expression levels of Ang-1 and Ang-2 were 26 and 44%, respectively, higher in women with ovarian cancer than in normal women. Increased Ang-2 expression was significantly related to advanced stage and grade of cancer and relapse of ovarian cancer. Additionally, elevated Ang-2 expression is a predictor of poor OS and short PFS (Sallinen et al., 2014).
As an important part of the tumor microenvironment, ECs are closely related to angiogenesis. VEGF and Angs are crucial regulators in angiogenesis. Both VEGF and Angs are associated with poor clinical outcomes, which provide possible targets for treatment.
Immune Cells
Immune cells include macrophages, dendritic cells (DCs), neutrophils, mast cells, myeloid-derived suppressor cells (MDSCs) and lymphocytes (Hanahan and Weinberg, 2011). They play significant roles both in tumor progression and tumor suppression, participating in evolving processes of tumorigenesis, metastasis, and angiogenesis by producing various signaling molecules, such as EGF, VEGF, MMP-9, IFNs, ILs, etc.
Macrophages
Macrophages are an essential population of immune cells that participate in inflammation and tumourigenesis (Grivennikov et al., 2010). Among them, macrophages residing in tumors are termed as tumor-associated macrophages (TAMs). TAMs can derive from resident macrophages or infiltrating macrophages from bone marrow monocytes circulating in the blood (Ghosn et al., 2010).
Depending on stimuli in the TME, TAMs can present two main phenotypes: the anti-tumor M1 macrophages and pro-tumor M2 macrophages (Sica et al., 2008; Grivennikov et al., 2010; Qian and Pollard, 2010; Sica, 2010; Gupta et al., 2018). When stimulated with interferon-gamma (IFN-γ), bacterial lipopolysaccharide (LPS) and granulocyte-macrophage-colony-stimulating factor (GM-CSF), monocytes differentiated into M1 macrophages, which can secrete IL-1, IL-12, TNFα and CXCL12 (Sica et al., 2008; Ramanathan and Jagannathan, 2014). M1 macrophages possess cytotoxicity, tumor suppression and immune-stimulation functions (Galdiero et al., 2013).
When stimulated with cytokines, including IL-4, IL-10, and IL-13, monocytes differentiated into M2 macrophages (Leyva-Illades et al., 2012; van Dalen et al., 2018). In the immune escape stage, the tumor macroenvironment maintains immunosuppression due to the secretion of many growth factors and cytokines, such as IL-4 and IL-13, by cancer cells. The immunosuppressive state accelerates monocytes to M2 macrophages; M2 macrophages, in turn, can promote tumor growth (Gordon, 2003; Roy and Li, 2016).
In ovarian cancer, TAMs are predominantly M2 macrophages, associating with tumor invasion, angiogenesis, metastatic disease and early recurrence (Pollard, 2004; Reinartz et al., 2014; Yin et al., 2016). They produce and secrete cytokines, which have immunosuppressive effects, such as IL-1R decoy, IL-10, CCL17 and CCL22 (Gordon, 2003). Via several mechanisms, they suppress adaptive immunity (Li et al., 2007; Noy and Pollard, 2014). Firstly, M2 macrophages can inhibit the proliferation of T cells and accelerate the immunosuppression of Treg cell transport to tumors by producing the chemokine CCL22 (Li et al., 2007). Secondly, M2 macrophages express the ligand receptors for CTLA-4 and PD-1. The activation of PD-1 and CTLA-4 inhibits cytotoxic function and regulates the cell cycle of T cells (Noy and Pollard, 2014). Then, M2 macrophages can also inhibit the activation of T cells through the depletion of L-arginine, which plays an essential role in T cell function (Galdiero et al., 2013). Arginase I (ARG1), a hallmark of M2 macrophages, is an L-arginine processing enzyme. In the TME, ARG1 decomposes L-arginine into L-ornithine and urea. The depletion of L-arginine suppresses the re-expression of the CD3 ζ chain, which is internalized by antigen stimulation and T cell receptor (TCR) signaling (Rodriguez et al., 2004).
Aside from immune suppression, M2 macrophages also take part in tissue repair, ECM remodeling and angiogenesis, which are processes involved in tumor progression as well (Mantovani et al., 2002; Coffelt et al., 2010; Ruffell et al., 2012; Finkernagel et al., 2016; Roy and Li, 2016). They can restructure ECM and regulate ECM components by degrading ECM via producing MMPs, serine proteases and cathepsins (Ruffell et al., 2012), which may facilitate tumor cell migration, invasion and metastasis. Additionally, they can secrete VEGF-A, which is an angiogenic factor, and produce proangiogenic cytokines, such as IL-1β, TNFα and uPA (urokinase-type plasminogen activator) (Roy and Li, 2016). In M2 macrophages, there is a subtype expressing TIE2, a tyrosine kinase receptor. The TIE2 macrophages are involved in angiogenesis (Fagiani and Christofori, 2013). These TIE2 macrophages recruited by CCL3, CCL5, CCL8, and TIE2-ligand Ang 2 are considered the most important reason for tumor vascularization because the deficiency of this cell type restricts the angiogenic switch (Ngambenjawong et al., 2017).
TAMs are plastic. The simple dichotomy of M1/M2 macrophages cannot account for the complexity of TAM heterogeneity (Ostuni et al., 2015). Transcriptome analysis uncovered a spectrum model of TAMs (Xue et al., 2014). M1 and M2 macrophages can be regarded as two ends of a continuum with wide ranges of functional states (Mantovani et al., 2002; Ostuni et al., 2015); the sub-populations of TAMs in between the two ends can share features of both M1 and M2 types (Qian and Pollard, 2010). For example, recently Singhal et al. (2019) found that TAMs could co-express M1/M2 markers, together with T cell co-inhibitory and co-stimulatory receptors.
The dynamic nature of the TME cellular environment gives a basis for the plasticity of TAMs. Macrophages present reversible changes in their functional phenotypes and distribution in response to different microenvironmental stimuli, including various cytokines and locally derived molecules, which are tissue- and tumor-specific (Stout et al., 2005; Okabe and Medzhitov, 2014; Ostuni et al., 2015; Kim and Bae, 2016). Therefore, in different histotypes of tumors (Zhang et al., 2014; Cassetta et al., 2019) and different microregions of the same tumor (Mantovani et al., 2002; Kim and Bae, 2016; Yang M. et al., 2018), there can be TAMs with different extent of infiltration and functional status.
In ovarian cancer, Zhang et al. (2014) found the density and the cancer islet/stroma ratio of TAMs vary among serous, mucinous, endometrioid, clear cell and undifferentiated histotypes. In the stroma and lumina of a small number of patient ovarian tumor samples, limited frequencies of iNOS expressing TAMs were found, which were thought to be cytotoxic (Klimp et al., 2001); in contrast, in the malignant ascites of ovarian cancer, abundant TAMs can be found, which are primarily M2-like with pro-tumor capacity (Gupta et al., 2018). As the tumor grows, stimuli in the TME alter, resulting in changes in TAM infiltration and polarization in a tumor progression level-dependent manner. In ovarian cancer studies, TAM and M2 macrophage density were found to increase as cancer stage and ascites volume increased or as lymphatic invasion appeared (Zhang et al., 2014; Ke et al., 2016; Yuan et al., 2017; Gupta et al., 2018); contrarily, M1/M2 ratio decreased as cancer stage increased (Zhang et al., 2014).
Despite expressing similar markers, TAMs may not always have similar functional implications. In colon cancer study, TAMs expressing PD-1 presented weakened phagocytic potency, associating with reduced survival (Gordon et al., 2017), while in early lung cancer study the PD-1+ TAMs did not affect tumor-specific T cell attack against tumor (Singhal et al., 2019). This indicates the necessity of future studies focusing on TAM functional status in the context of tumor tissue types and stages of the disease; this is especially true with ovarian cancer as it has many histotypes and high heterogeneity.
Several studies revealed the prognostic value of TAMs in ovarian cancer. The M1/M2 and M2/TAM ratio have been reported to be positively associated with PFS and OS, while the overall TAM density in ovarian tumors indicated no prognostic significance (Lan et al., 2013; Zhang et al., 2014; Yuan et al., 2017). M2 density in the ascites or tumor samples is associated with reduced relapse-free survival (Reinartz et al., 2014) and PFS (Lan et al., 2013; Yuan et al., 2017). However, there is a controversy in the relationship between M2 density alone and OS: Lan et al. (2013) reported a negative association between the two factors, while Zhang et al. (2014) found no significant relevance. This may be due to the difference of included tumor histotypes.
Dendritic Cells
Dendritic cells (DCs) capture endogenous or exogenous antigens, process them, and present the antigenic peptides to other immune cells (Banchereau et al., 2000), acting as a bridge connecting the innate and the adaptive immune system (Timmerman and Levy, 1999; Riboldi et al., 2005). There are two main subtypes of DCs: the conventional DC (cDC) that is specialized in antigen presentation, and the plasmacytoid DC (pDC) that produces IFN upon antigen stimulation aside from activating lymphocytes and other myeloid cells (Labidi-Galy et al., 2011; Vu Manh et al., 2015). cDCs comprise 5–10% of myeloid cells in most tumors; pDCs are rare in mouse tumors but found in most human tumors (Tang et al., 2017).
DCs play key roles in anti-tumor immunity because it is indispensable for T cell immune responses against tumors (Casey et al., 2015). DCs are responsible for tumor antigen recognition, which is the initiating event of the tumor-specific adaptive immune response. In both the animal ovarian cancer model and human HGSOC patients, DCs can sense damage-associated molecular patterns (DAMPs) released from dead cancer cells, such as double-stranded DNA (dsDNA) fragments and calreticulin, an endoplasmic reticulum (ER) chaperone, eliciting Th1 polarized immunity (Ding et al., 2018; Kasikova et al., 2019).
After capturing antigens, DCs present peptides processed from those antigens to CD4+ and CD8+ T cells via major histocompatibility complex class II (MHC II) and MHC I molecules respectively, which subsequently initiate a series of T cell activity (Dudek et al., 2013; Sabado et al., 2017). This process has been reported to be significant for tumor development prevention (MacKie et al., 2003; Galon et al., 2006).
Besides T cell activation, DCs are also crucial for the augmentation of cytotoxic T lymphocytes (CTLs) population in the TME. It is reported that intratumoral cDCs are responsible for intratumoral CTL proliferation both in vivo and in vitro (Diao et al., 2011), and they are the only group of phagocytosing tumor myeloid cells that can stimulate CD8+ T cell proliferation (Broz et al., 2014). As the major determinant of success in tumor deterrent, from the immune aspect (Budhu et al., 2010), is to increase the functional tumor-infiltrated CTL population, the significance of cDCs in the TME for anti-tumor responses is self-evident.
Effective T cell activation by DCs require DC maturation, a process happens after DC exposing to antigen, characterized by increased membrane expression of MHC and co-stimulatory molecules (CD80, CD86, CD40) (Bol et al., 2016; Bhatia et al., 2019), alteration of chemokine receptors to favor DC lymph node (LN) migration (Drakes and Stiff, 2018); mature DCs produce cytokines that favor Th1 (anti-tumor) immunity. Truxova et al. (2018) found in cohorts of HGSOC patients that tumor-infiltrated mature LAMP+ DCs is robustly associated with Th1 immune responses, clinically favorable cytotoxic activities in the TME and favorable OS.
The process of DC maturation can be hampered by multiple factors, leaving DC immatured, potentially developing into a tolerogenic status and promote immune tolerance (Dhodapkar et al., 2001). Immature DCs express low levels of co-stimulatory molecules and cytokines and mount limited immune activities (Drakes and Stiff, 2018). Factors that lead to DC dysfunction, including the inhibition of DC maturation, involve the immune-modulating molecules in the TME, such as IL-6, IL-10 and VEGF, tumor-derived soluble mediators and exosomes, the activation of oncogene STAT3 in DCs, the ER stress response, and the abnormal intracellular lipid accumulation (Cubillos-Ruiz et al., 2015; Tang et al., 2017; DeVito et al., 2019). These factors suppress DC functions by reducing the expression of co-stimulatory molecules and the secretion of pro-inflammatory cytokines, inhibiting DC lymph node chemotaxis, dampening DC differentiation, inducing tolerogenic phenotypes on DCs and shortening the lifespan of DCs (Tang et al., 2017).
Tolerogenic DCs suppresses anti-tumor immunity via several mechanisms. First, they produce less pro-inflammatory cytokines and induce immune suppressive cytokines. Labidi-Galy et al. (2011) found in a cohort of 44 ovarian cancer patients that intra-tumoural tolerogenic pDCs secreted fewer IFN-α, TNF-α, IL-6, macrophage inflammatory protein-1β and CCL5, while induced IL-10 from CD4+ T cells, promoting immune tolerance in these patients. Second, they harbor enzymes negatively regulating T effector cell functions, such as nitric oxide synthase (NOS) and Indoleamine 2,3-Dioxygenase (IDO) (Casey et al., 2015). IDO is an enzyme catalyzing tryptophan degradation, capable of suppressing tumor-infiltrated lymphocyte proliferation, promoting Treg differentiation, inducing T cell anergy, and promoting tumor angiogenesis as well as metastasis (Munn et al., 2005; Tanizaki et al., 2014; Munn and Mellor, 2016). In EOC patients, there was significantly increased frequency of IDO+ DCs in tumor draining LN compared to the normal donor LN; besides, in vitro study revealed IDO significantly inhibited proliferation of tumor-associated lymphocytes derived from EOC patients (Qian et al., 2009).
Many factors are affecting the actual DC functions and behaviors, which are with high plasticity, contributing to either pro-tumor or anti-tumor effect. Tumor expressing molecules are associated with mature DC infiltration. Recently, MacGregor et al. (2019a) found higher surface expression of B7-H4, a B7 family molecule, was correlated with higher mature DC (CD11c+HLA-DRhigh) infiltration in EOC patient samples, which may be associated with increased expression of CXCL17, a monocyte and DC chemoattractant in those tumors. This group have also found that tumour-to-stroma ratio (TSR), which represents the percentage of malignant cell component relative to the stroma in the tumor tissue, have an impact on infiltrated DC phenotype: high TSR was associated with elevated PD-L1 expression on mature DCs (CD11c+HLA-DRhigh) infiltrating in ovarian tumor tissue (MacGregor et al., 2019b).
DC functions can be regulated by their interactions with the proximal milieu, so different locations of DCs may result in different function. Labidi-Galy et al. (2011) discovered that in ovarian cancer patients, tumor pDCs produced less pro-inflammatory cytokines than pDCs from ascites or peripheral blood.
Also, DC performance can vary by different tumor development stage. In an ovarian cancer mouse model, at the early stage, tumor growth was prevented by infiltrating DCs and DC depletion at this stage accelerated tumor expansion; at the advanced stage, however, DCs become immunosuppressive in the TME, abrogating enduring activity of anti-tumor T cells, and DC depletion at this stage significantly delayed disease progression (Scarlett et al., 2012). Similarly, in a mouse model of ovarian cancer, Krempski et al. (2011) also found progressively gained immunosuppressive phenotype of infiltrating DCs as the tumor progressed over time, represented by gradually increased PD-1 expression.
More studies are favored in the future to reveal facts on how DCs functions are regulated, thereby providing clues for therapeutic strategies in maintaining their anti-tumor potential.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid cells that co-express the myeloid surface markers GR-1 and CD11b (Atretkhany and Drutskaya, 2016). MDSCs consist of three phenotypes: PMN-MDSC, M-MDSC and a small group of cells that have myeloid colony-forming activity, including myeloid progenitors and precursors (Gabrilovich, 2017). PMN-MDSCs are similar to neutrophils in phenotype and morphology and represent over 80% of MDSCs, while M-MDSCs are similar to monocyte (Gabrilovich, 2017). Studies have confirmed that MDSCs promote tumor progression by various mechanisms. First, MDSCs are implicated in immune suppression (Ostrand-Rosenberg and Fenselau, 2018). Despite their involvement in the inhibition of many cells in the immune system, MDSCs mainly target T cells. We summarized the mechanisms involved in immune suppression. (1) MDSCs accelerate lymphocyte nutrient depletion (Rodriguez et al., 2004; Srivastava et al., 2010). Both L-arginine and L-cysteine are essential amino acids that are important for T cell activation and function. MDSCs produce ARG1 and depletion of L-arginine through an ARG1-dependent manner (Rodriguez et al., 2004). MDSCs also sequester L-cysteine (Srivastava et al., 2010). Therefore, the amount of ζ-chain in the TCR complex is downregulated, and the proliferation of antigen-activated T cells is suppressed. (2) MDSCs disturb lymphocyte trafficking and viability (Hanson et al., 2009; Sakuishi et al., 2011). Galectin 9, which is expressed in MDSCs, binds to TIM3 on lymphocytes, which induces the apoptosis of T cells (Sakuishi et al., 2011). Similarly, MDSCs express ADAM17, which can decrease the L-selectin level on T cells and limit T cell recruitment in lymph nodes (Hanson et al., 2009). (3) MDSCs promote Treg cell activation and expansion (Gabrilovich et al., 2012). MDSCs stimulate CD4+ T cells to translate into induced Treg (iTreg) cells and expand natural Treg (nTreg) cells. These processes are associated with CD40-CD40L interactions, IFN-γ, IL-10, and TGFβ. (4) MDSCs stimulate the generation of oxidative stress. Oxidative stress is linked to ROS and RNS (reactive nitrogen species) (Gabrilovich, 2017). Superoxide reacts with NO and generates PNT (peroxynitrite), which nitrates T-cell receptors and limits the response of antigen-MHC complexes, thus suppressing T cells directly. PNT also nitrates T-cell-specific chemokines, which decreases the combination of antigenic peptides to MHC and limits the migration of T cells (Molon et al., 2011).
Moreover, MDSCs facilitate neovascularization through different mechanisms. Hypoxia in tumors induces MDSCs to produce VEGF, FGF2 and MMP9. Interestingly, the activation of STAT3 in MDSCs also stimulates neovascularization through IL-1β, CCL2 and CXCL2 release (Bruno et al., 2019). Additionally, these factors stimulate invasion and metastasis by producing MMPs (Ostrand-Rosenberg and Fenselau, 2018).
MDSC is an important part of the tumor microenvironment. MDSC promote tumor progression by regulating immune suppression and facilitating neovascularization. Moreover, the different tumor microenvironment is related to different functions and differentiation of MDSC. Nevertheless, the mechanism is still not clear.
Lymphocytes
Lymphocytes, a major component of the TME, include B lymphocytes and T lymphocytes and mediate innate and adaptive immunity, respectively (Sadelain et al., 2017). B lymphocytes accelerate tumor progression by producing protumorigenic cytokines and regulating the Th1: Th2 ratio (Quail and Joyce, 2013). T lymphocytes, a major component of the TME, are crucial for adaptive immunity (Sadelain et al., 2017). T cells develop in the thymus. Before encountering the initial antigen, T cells are regarded as naïve (TN) cells. After antigen encounter, naïve (TN) cells are activated and start differentiation (Smith-Garvin et al., 2009). They proliferate rapidly and release inflammatory cytotoxic granules and cytokines, which activate the immune response. According to the cytokine environment, T cells differentiate into various subsets (Wang M. et al., 2017).
Due to the exclusive expression of CD4 or CD8 markers, mature T cells are categorized into CD3+CD4+, CD3+CD8+ T cells and CD4+ Treg cells (Kishton et al., 2017). CD3+CD4+ T cells are also called helper T cells (Th cells) and regulate immune responses by releasing cytokines that promote or inhibit inflammation (Joyce and Pollard, 2009). CD3+CD4+ T cells can be divided into Th1 and Th2 cells. Among them, Th1 cells produce and release pro-inflammatory cytokines and assist CD3+CD8+ T cells in tumor rejection. Therefore, Th1 cells are antitumorigenic. However, Th2 cells release anti-inflammatory cytokines and promote tumor progression (Joyce and Pollard, 2009; Quail and Joyce, 2013). CD3+CD8+ T cells, called cytotoxic T lymphocytes (CTLs), produce inflammatory cytokines and cell lytic molecules such as perforin and granzyme, which specifically recognize and destroy pathogen-infected or malignant cells (Joyce and Pollard, 2009; Zhang and Bevan, 2011).
Treg cells (CD4+CD25+Foxp3+) also play a crucial role in the immune response (Patsoukis et al., 2016). During development in the thymus, Treg cells universally express Foxp3, representing 5–10% of CD4+ T cells. When responding to TCR and TGF-β, Treg cells show suppression. Treg cells protect hosts against autoimmune diseases through inhibiting self and autoreactive cells (de Aquino et al., 2015). Additionally, Treg cells play a tumourigenic role mainly through immunosuppression monitoring (Lindau et al., 2013). Treg cells regulate the immune response through four mechanisms (Vignali et al., 2008; Facciabene et al., 2012): (1) Secreting immunosuppressive molecules. Treg cells suppress effector T cell functions by secreting cytokines such as IL-10, IL-35, and TGFβ. Additionally, IL-10 and TGFβ are reported as key mediators that limit antitumor immunity and promote tumor progression (Facciabene et al., 2012). Interestingly, these cytokines not only inhibit the function of effector cells but also promote DC polarization to tolerogenic phenotypes. Additionally, Treg cells secrete VEGF, which is also an immunosuppressive molecule (Vignali et al., 2008). Through VEGF, Treg cells exert inhibition and regulate the differentiation of DCs. (2) Cytolysis. Treg cells induce the apoptosis of effector cells by secreting granzyme B and perforin (Vignali et al., 2008). (3) Metabolic disruption. Several mechanisms have been reported for the metabolic disruption regulated by Treg cells. However, it is still controversial. Treg cells deplete the local level of IL-2, which causes effector cells to starve and results in the apoptosis of effector cells. Moreover, with the expression of CD73 and CD39, Treg cells catalyze ATP to adenosine, which inhibits the function of effector T cells (Deaglio et al., 2007; Vignali et al., 2008). (4) Modulation of DC maturation and function. CTLA-4 (cytotoxic T-lymphocyte antigen 4) is expressed on Treg cells, and CD80 and CD86 are expressed on DCs. Treg cells induce DCs through CTLA4–CD80/CD86 interactions, which induces the release of IDO (indoleamine 2,3-dioxygenase). IDO expression depletes essential tryptophan and inhibits the function of effector T cells (Fallarino et al., 2003). Furthermore, Treg cells suppress the function of DCs by depleting costimulatory molecules and inhibiting LAG3 (lymphocyte-activation gene 3) binds to MHC class II molecules (Vignali et al., 2008). It has been reported that TLR (Toll-like receptor), GITR (glucocorticoid-induced TNF receptor), CTLA-4, and FR (folate receptor) directly or indirectly regulate the function of Treg cells (Pasare and Medzhitov, 2003; Callahan et al., 2010; de Aquino et al., 2015). TLR activation decreases the suppressive effect of Treg cells partially through IL-6. GITR, a costimulatory molecule, has a high expression level on Treg cells (Pasare and Medzhitov, 2003). Treatment with anti-GITR mAb downregulates the inhibition of Treg cells. Similarly, CTLA-4 and FR4 are expressed on Treg cells. When blocking CTLA-4 or deleting FR4, the inhibition of Treg cells decreased and active Treg cells were depleted (Callahan et al., 2010).
Several researches implied that lymphocytes were correlated with clinical outcomes in ovarian cancer. Plasma cell and B cell infiltration impacted the prognosis of ovarian cancer. CD138 and CD20 are markers for plasma cells and mature B cells, respectively. Lundgren et al. (2016) found that patients with high expression of CD138 and CD20 were related to advanced tumor grade. Additionally, the Kaplan–Meier analysis suggested that high expression of CD138 was linked to worse OS and OCSS (ovarian cancer-specific survival).
Tumor-infiltrating T cells are associated with clinical outcomes in ovarian cancer (Zhang et al., 2003). Through evaluating 186 frozen tissue samples from patients with advanced ovarian cancer, Zhang‘study demonstrated that the 5-year OS rate was higher in patients whose tumors had T cell infiltration compared to survival of patients whose tumors did not have T cell infiltration. They also confirmed that intratumor T cells were significantly associated with delayed relapse (Zhang et al., 2003).
CD8+ T lymphocyte infiltration extended survival time. In Hamanishi’s research, they demonstrated that patients with CD8+ T lymphocyte infiltration had prolonged PFS and OS (Hamanishi et al., 2007). Similarly, Sato et al. (2005) noticed that patients with high percentages of CD8+ T cells had a greater survival rate than that of patients with low percentages (55 months vs. 26 months). Clarke et al. (2009) also reported that in patients with advanced stage, CD8+ T lymphocyte infiltration was linked to increased PFS, OS and disease-specific survival. Interestingly, Ye et al. (2014) observed that CD137, a TNFR-family member, is expressed on both CD4+ and CD8+ T lymphocytes. Patients with CD137 expression had improved survival in ovarian cancer.
In contrast, Treg cell infiltration indicated poor clinical outcomes. In Curiel’s study, they evaluated 104 women with ovarian carcinoma and found that patients with advanced disease stage had a higher percentage of CD4+CD25+FOXP3+ Treg cells. Furthermore, Treg cells in the tumor sites were linked with decreased survival and a high death hazard (Curiel et al., 2004).
Lymphocytes play important roles in both innate immune responses and adaptive immune responses. Different lymphocytes have different functions. In ovarian cancer, specific lymphocytes infiltration is directly related to patient prognosis. At present, the mechanism of lymphocyte regulation is not completely understood, thereby deserving further investigation.
Novel Molecular Discoveries in Ovarian TME
Despite the advances of immunostaining technology that has provided important biological features in the ovarian cancer TME, it is still underpowered to adequately detail the multi-variant cellular and molecular interactions in it.
In the past decade, enormous technical development has been realized in sensitive detection and accurate quantification of genomic, transcriptomic, and proteomic features. The “-omic” techniques give scientists a scope from a higher dimension to reveal the TME molecular landscape as a whole. With information from this landscape, multilevel analysis has shed light on the heterogeneous nature of ovarian cancer, the complex and dynamic molecular events in the evolving TME, facilitating the advancement and validation of biomarkers for disease diagnosis, prognosis, treatment targets and treatment response prediction (Figure 2).
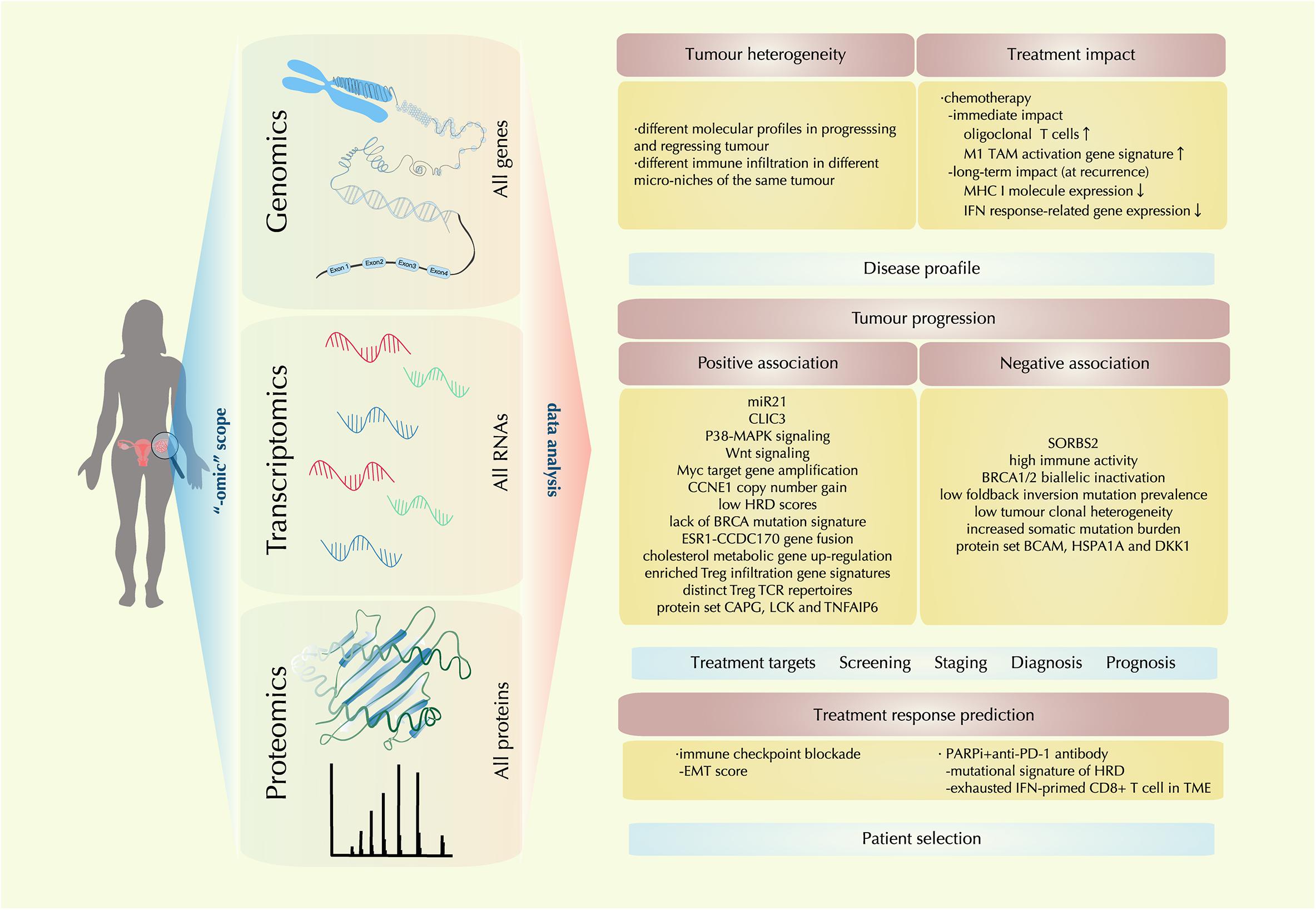
Figure 2. Recent molecular findings by “-omic technology” in the ovarian cancer tumor microenvironment (TME). With the tools of genomic, transcriptomic, proteomic technology, a comprehensive multi-level landscape of ovarian cancer TME is revealed. With data are drawn from the “-omic scope,” the molecular profile of ovarian cancer is further identified in terms of tumor heterogeneity and treatment impact; new discoveries on factors that are either positively or negatively associated with tumor progression are also identified, providing clues for treatment target exploration and novel biomarker designing for disease screening, staging, diagnosis, and prognosis; also, correlations between certain therapeutic regimens and ovarian cancer TME profiles are investigated, providing implication for precision medicine by precise patient selection.
Here we review the latest evidence provided by “-omic” technology, revealing the molecular characteristics of ovarian cancer TME in disease development and treatment intervention.
TME Heterogeneity in Ovarian Cancer
Ovarian cancer is a kind of highly heterogenous tumor; the diversified TME of ovarian cancer is one manifestation for this fact. With immunogenomic approaches, Jiménez-Sánchez et al. (2017) observed changes of different metastatic tumor sites of an HGSOC patient who received multiple times of chemotherapy. They found both progressing and regressing metastases during treatment in the same patient, characterized by immune cell exclusion and T cell infiltration with the oligoclonal expansion of specific subsets respectively. The progressing tumor presented with molecular patterns of immune suppression associated with Wnt signaling and higher HLA mutation and neoepitope loads while regressing sites showed patterns of immune activation with the expression of HLA, IFN-γ, CXCL9 etc. and enriched TCR signaling (Jiménez-Sánchez et al., 2017). By similar approach, recently the same group further discovered co-existence of both immune-cell-excluded and inflammatory microenvironment in the same tumor sites of the same patients with HGSOC, indicating ubiquitous variation in immune cell infiltration even in the micro-niches of the same tumor entity (Jiménez-Sánchez et al., 2020).
TME Molecular Factors Positively Associated With Tumor Progression
By -omic studies, there is a growing body of evidence unveiling the tumor driving signaling, interactions among cell components and immune profiles in the ovarian cancer TME. Comprehensive analysis of TME cell components, genomic alterations and gene expression revealed that amplification of Myc target genes and Wnt signaling were associated with impaired immune cell infiltration (Jiménez-Sánchez et al., 2020). Using next-generation sequencing technology, Au Yeung et al. (2016) identified high levels of microRNA-21 (miR21) from the exosomes secreted by cancer-associated adipocytes (CAAs) and CAFs, which later were transferred to ovarian cancer cells, suppressing cancer cell apoptosis and conferring chemoresistance. In order to investigate Treg antigen specificity, Ahmadzadeh et al. (2019) adopted TCR β chain deep sequencing in Tregs from multiple tumor samples, including ovarian cancer and discovered that TCR repertoires were distinct from conventional T cells, displaying tumor- and neoantigen-specific reactivity. This finding suggested Tregs clonally expand in an antigen-selective manner in the TME.
Recently, the interplay between metabolism and tumor progression as well as immune suppression among players in the TME has been reported with -omic tools. By phosphoproteomic techniques, Curtis et al. (2019) identified increased activation of the p38-MAPK pathway in CAFs that are co-cultured with ovarian cancer cells and verified that this activation lay the premise for glycogen mobilization in cancer cells, which as an energy source fueled metastatic tumor growth. Another proteomic study identified the central metabolic regulator of CAF, the methyltransferase nicotinamide N-methyltransferase (NNMT) being a prominent signature in metastatic stroma tissue of HGSOC patients, which is necessary for the differentiation of competent CAF phenotype, supporting cancer cell migration and proliferation (Eckert et al., 2019). In a mouse model of metastatic ovarian cancer, Goossens et al. (2019) used Gene Ontology (GO) enrichment analysis and found that at a later time after tumor inoculation (day 21), there was an up-regulation of cholesterol metabolic gene clusters in TAMs, resulting in membrane-cholesterol efflux and depletion of lipid rafts from TAMs, leading to IL-4-mediated immune-suppressive TAM reprogramming. All these discoveries provide novel therapeutic targets that could facilitate the current treatment strategy.
There are increasing -omic studies showing an association between TME cellular and molecular profile and patient clinical features. Barnett et al. (2010) earlier have found increased Treg infiltration, represented by several enriched immunologic pathway gene signatures, was associated with higher grade and advanced stage in serous ovarian cancer. From secretome analysis of fibroblasts and cancer cells, Hernandez-Fernaud et al. (2017) identified abundance chloride intracellular channel protein 3 (CLIC3) in the TME of aggressive ovarian cancers, which correlates with poor clinical outcome. Aside from single biomarkers, -omic research enables scientists to discover a correlation between a set of molecular patterns and patient prognosis. With proteomic technology, Finkernagel et al. (2019) identified 779 proteins in the ascites of HGSOC patients and identified protein marker sets to predict patient survival, with CAPG, LCK and TNFAIP6 as the core type 2 signature that has 91.2% correctness in identifying short-term survivors. Another multi-level -omic study with HGSOC primary tumor samples, discovered the association between short-term survival and copy number gain of CCNE1, lack of BRCA mutation signature, low homologous recombination deficiency scores, and the presence of ESR1-CCDC170 gene fusion (Yang S. Y. C. et al., 2018). One study with HGSOC metastatic samples revealed expression pattern of 22 matrisome genes and thereby generated a “matrix index” with their expression; this index was significantly correlated with Treg and Th2 cell signatures and can identify the patient group with shorter OS (Pearce et al., 2018).
TME Molecular Factors Negatively Associated With Tumor Progression
Together with novel findings of tumor promoting factors, there are also tumor-suppressing molecular patterns uncovered by recent -omic studies. In a study concentrating on RNA binding proteins (RBPs), the authors referred to published literature as well as oncogenic databases and conducted functional verification studies; they identified the sorbin and SH3 domain containing 2 (SORBS2) out of a pool of RBPs, as a suppressor of metastatic colonization of ovarian cancer, which exerted tumor suppressive function by dampening cancer invasiveness and repolarizing MDSCs and TAMs (Zhao L. et al., 2018). Another study investigated the correlation between tumor-infiltrating lymphocytes (TILs) and malignant diversity in HGSOC samples, where the authors found epithelial CD8+ TILs were negatively associated with tumor clonal heterogeneity, suggesting neoantigen-specific depletion of tumor clones and spatial antigen-specific T cell tracking of tumors (Zhang et al., 2018).
Molecular signatures correlating with good clinical prognosis have been reported as well. In Finkernagel et al.’s (2019) ascites proteomic study mentioned above, authors reported markers of BCAM, HSPA1A, and DKK1 as the core type 1 signature with 90.9% of correctness in the identification of long-term survivors. Similarly, in Yang S. Y. C. et al. (2018) study with primary HGSOC samples, they reported increased somatic mutation burden, BRCA1/2 biallelic inactivation, and enriched infiltration of activated as well as memory T cells in long-term survivors. Lastly, in Zhang et al.’s (2018) multi-level study with HGSOC tumor samples, they discovered the combinatorial prognostic value of high immune activity and low mutation prevalence of foldback inversions, which lead to best clinical outcomes.
Detailed mechanistic studies are needed in the future to determine the way of targeting the tumor suppressing molecular patterns that can potentiate therapeutic strategy.
Treatment Impact and Response Prediction in Ovarian Cancer TME
The tumouricidal treatment causes cell death, incurring multiple changes in the TME. After neoadjuvant chemotherapy (NACT), researchers adopted immunogenic analysis in HGSOC tumors and found increased NK cell infiltration and oligoclonal expansion of T cells, suggesting chemotherapy can potentiate immunogenicity of the primary tumor (Jiménez-Sánchez et al., 2020). Another gene expression analysis revealed ovarian cancer patients treated with paclitaxel had an enriched gene signature linked to M1 TAM activation (Wanderley et al., 2018). However, this immune activation property of chemotherapy may just exist in the early stage right after the treatment; when bulky tumor cells are eradicated, the chemo-resistant cancer stem cells (CSCs) remain and subsequently bring about recurrence. The positively selected CSCs by chemotherapy showed altered lipid metabolism signatures, resulting in accumulation of lactate, which acidifies ascites, leading to T cell dysfunction and Treg polarization (Ahmed et al., 2018). By comparing the gene expression profile of ascites-derived tumor cells from treatment naive (CN) and recurrent (CR) ovarian cancer patients, Ahmed and colleagues found massively reduced MHC I molecule (HLA-C and -B) expression and IFN response-related gene expression, including IFIT2, TMEM173 and MX2 in CR patients, suggesting an immuno-compromised ascites TME in CR after chemotherapy. Hence, CSCs could become a key target in treatment exploitation for CR patients (Ahmed et al., 2018). More comparative studies between CN and CR patients are needed in the future for the discovery of clues that can overcome treatment resistance and targets that can further improve the current regimens.
Due to the heterogeneous nature of ovarian cancer, responses of treatment targeting different tumor driving molecules vary among individuals. With “-omic” tools, the identification of tumor biomarkers that are associated with exceptional clinical response or resistance has become possible. Mak et al. (2016) conducted a genomic and proteomic analysis of EMT signatures in multiple cancers, including ovarian cancer; they identified a set of 77 EMT-related genes, generating an EMT score according to their expression signature; they found EMT score was positively correlated with expression levels of immune checkpoint genes, implicating predictive value of EMT score in treatment response of immune checkpoint blockade. Another study investigated immunogenomic profile in predicting combination treatment response of a PARP inhibitor (PARPi) and an anti-PD-1 antibody in ovarian cancer patients; the authors found two determinants associated with a positive response: mutational signature of defective homologous recombination DNA repair and exhausted CD8+ T cell primed by IFN in the TME. These findings are important for the patient selection of certain treatments, paving the way for future precision medicine.
In conclusion, more mechanistic and phenotypic investigation is required to decipher the roles of certain patterns of molecular alteration in disease development and treatment intervention, so as to facilitate the deployment of more individualized and molecularly informed treatments for ovarian cancer patients.
Therapeutic Strategies Targeting the TME
Therapies Targeting CAFs
Several therapies are targeting CAFs in ovarian cancer: (1) direct deletion FAP+ fibroblasts; (2) reverting the activated CAFs into a quiescent state; (3) targeting CAF-specific pathways (Chen and Song, 2019; Barrett and Puré, 2020; Truffi et al., 2020).
Direct Deletion FAP+ Fibroblasts
It is well known that CAF has phenotypic heterogeneity. Activated CAFs can selectively express a variety of different biomarkers in specific TMEs environments, such as alpha-SMA FAP, S100A4 and PDGFR (Barrett and Puré, 2020). FAP is a serine protease, which regulates the recruitment, proliferation and differentiation of myofibroblasts. FAP is an important surface marker in CAFs, which exists in more than 90% of CAFs (Chen and Song, 2019). FAP+ cells cannot only promote tumor progression but also block immunotherapy by producing ECM and direct signaling pathways (Puré and Blomberg, 2018). Studies have shown that inhibition of FAP can reduce the infiltration of CAF (Santos et al., 2009). Therefore, targeted therapy for FAP on CAF was proposed.
In Paulette’s research, they examined the gene expression of FAP in high-grade serous EOCs and found that a higher FAP expression in tumor tissue than normal control. They also reported that patients with high FAP expression showed poor OS. Then they blocked the FAP via a FAP-specific siRNA, the results demonstrated that the proliferation of cells was reduced 9–13% (Mhawech-Fauceglia et al., 2015). Thereby, downregulated FAP+ fibroblasts could reduce the proliferation of tumor cells and might be a new treatment for ovarian cancer.
Reverting the Activated CAFs Into a Quiescent State
There are two main states of CAFs: quiescent state and active state. In general, most CAFs are in a quiescent state and have low proliferative and metabolic capacity (Hansen et al., 2016; Chen and Song, 2019). However, once the homeostasis is broken, CAFs is activated in order to return to the quiescent state (Hansen et al., 2016). In the tumor microenvironment, not only cancer cells can enhance the activation of CAFs, but some cytokines can also activate CAFs (Chen and Song, 2019). Recent years, some scientists target therapy of CAF by restoring the activation state of tumorigenic CAFs to a static state (Rossmann et al., 1967; Froeling et al., 2011). But it has not been used in ovarian cancer.
Targeting CAFs Associated Signal Molecules
Cancer-associated fibroblasts regulate immune cells and ECM via a series of signal molecules. CAFs regulate the function of the myeloid cell and T cell (Barrett and Puré, 2020). CAFs decrease the number of MDSCs through inhibiting CXCL12/CXCR4 signal pathway and promote the differentiation of myeloid cells into DCs via stimulating IL6/STAT3 signal pathway (Gok Yavuz et al., 2019; Truffi et al., 2020). Besides, CAFs inhibit T cells through increase the expression of PD-L1/2, while CAFs activate T cells via stimulating the production of IL-6 (Barnas et al., 2010; Cho et al., 2011). CAFs can active the ECM through secreting growth factors (such as VEGF) and cytokines (such as TGF-β, IL-6 and IL-10) (Kohlhapp et al., 2015).
Recently, several therapies targeting CAFs associated signal molecules have been developed, including TGF-β inhibitors, PDGF inhibitor, Hedgehog inhibitors, FAK inhibitors and IL-6 inhibitors.
TGF-β1 is a cytokine produced by CAFs that plays a significant role in promoting tumorigenesis (Fabregat et al., 2014). A-83-01 is a TGF-β inhibitor. In Yamamura’s research, they found that in vitro TGF-β1 treatment stimulated HM-1 cell motility, invasion and adhesion. However, A-83-01 could counteract the effect of TGF-β1. Interestingly, in vivo, they found that mice treated with A-83-01 had a longer survival time than that of the control group (Yamamura et al., 2012). Similarly, in Gao’s study, they investigated the tumor-suppressive activity of LY2109761, a TGF-β type I (TβRI) and type II (TβRII) kinase. The results demonstrated that LY2109761 augmented ovarian cancer cell apoptosis. Moreover, combined cisplatin with LY2109761 enhanced the lethal effect of cisplatin in normal and cisplatin-resistant ovarian cancer cells. Furthermore, in vivo treatment with cisplatin and LY2109761 reduced the tumor volume in a cisplatin-resistant ovarian cancer model. Therefore, they confirmed that LY2109761 increases the antitumor activity of cisplatin (Gao et al., 2015).
PDGF is a factor that can stimulate other cell trans-differentiation for CAFs. In Matei’s report, they found that PDGFR was expressed in 39% of ovarian cancers. When ovarian cancer cells were treated with imatinib (a PDGFR inhibitor), cells were arrested in the G0–G1 phase. Therefore, they confirmed that imatinib could suppress the proliferation of ovarian cancer cells (Matei et al., 2004). Imatinib also showed its clinical activity on platinum-resistant ovarian carcinoma. In Matei’s study, in patients treated with imatinib mesylate and docetaxel, they found that 1 patient had a complete response and 4 patients had partial responses (ORR: 21.7%) (Matei et al., 2008). Overall, the inhibition of PDGF could suppress tumor proliferation.
Targeting Hedgehog, FAK and IL-6 is also an effective treatment for ovarian cancer. IPI-126 is a Hedgehog inhibitor. When treated with IPI-126, Hh signaling was suppressed, thereby inhibiting the proliferation of serous ovarian cancer (McCann et al., 2011). VS-6063, a FAK inhibitor, suppressed the phosphorylation of FAK. VS-6063 increase the chemosensitivity in taxane-resistant ovarian cancer cells, thus decrease the tumor load (Kang et al., 2013). Tocilizumab is a monoclonal antibody against IL-6R. In a phase I clinical trial, the results showed that patients treated with tocilizumab increase the serum IL-6 and soluble IL-6R. Moreover, increased sIL-6 indicated a longer survival time. Therefore, tocilizumab prolongs survival time in recurrent ovarian cancer (Dijkgraaf et al., 2015).
Cancer-associated fibroblasts is an important component of the tumor microenvironment, which play a pro-tumor function in the process of cancer development, making it a hot spot of targeted therapy. Nevertheless, targeted CAFs therapy still has challenges. Some studies suggest that directly targeted killing of CAF may be a way to reduce CAF infiltration in tumors. But due to the lack of specific cell surface markers, it is hard to precisely target CAFs. The reversal of the functional state of CAF provides a new idea for the development of new anticancer therapies. At present, limiting the function of CAF by targeting stromal CAF signals and effectors has become an important supplement to tumor therapy, but further mechanism and function studies are still needed.
Anti-angiogenesis Therapy
VEGF is the most typical activator of angiogenesis. Currently, anti-angiogenesis therapy is divided into three types (Cortez et al., 2018; Hironaka, 2019): (1) inhibiting VEGF; (2) inhibiting its receptor, VEGFR; and (3) inhibiting Angs (Table 1).
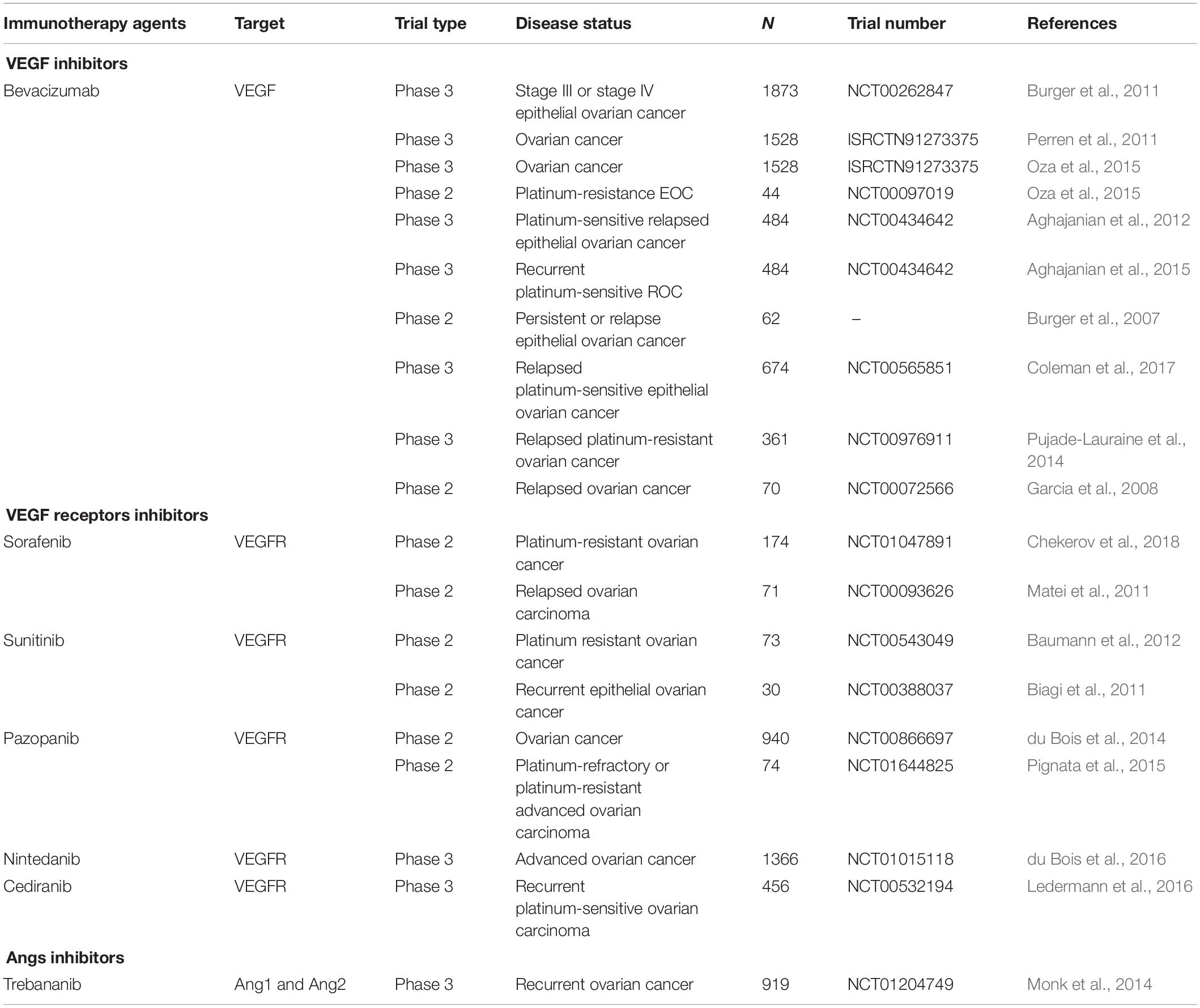
Table 1. Clinical trials of therapies that target angiogenesis in ovarian cancer.
Anti-VEGF
Bevacizumab is a humanized VEGF monoclonal antibody. It can inhibit ECs proliferation and activation by binding and inactivating VEGF (Eskander and Randall, 2011). Studies have reported that bevacizumab treatment of murine ovarian cancer models could suppress tumor proliferation and prolong survival time (Hu et al., 2002; Monk et al., 2006; Huynh et al., 2007; Mabuchi et al., 2008). Currently, using bevacizumab alone or combined with other therapeutics has had successful outcomes. Bevacizumab was quickly used in the clinic due to its amazing therapeutic effects in animal models. Bevacizumab, as the first FDA-approved anti-angiogenesis antibody, was applied to the treatment of ovarian cancer. Burger et al. (2011) designed a phase 3 trial (GOG-218) to explore the antitumor efficiency of bevacizumab in ovarian cancer. They enrolled 1873 women with newly diagnosed stage III or IV EOC. They randomly divided patients into three groups: control treatment, bevacizumab-initiation treatment, and bevacizumab-throughout treatment. The results demonstrated that the median PFS with the control treatment was 10.3 months, while with bevacizumab-initiation treatment and bevacizumab-throughout treatment, it was 11.2 and 14.1 months, respectively. Additionally, the median OS in the control treatment, bevacizumab-initiation treatment and bevacizumab-throughout treatment was 39.3, 38.7, and 39.7 months, respectively. Thereby, ovarian cancer patients treated with bevacizumab was supposed to have a longer PFS.
Subsequently, the ICON7 trial reported the similarity results in ovarian cancer patients. In the ICON7 trial, they enrolled 1528 patients with ovarian carcinoma and randomly allocated them into the standard-therapy group and bevacizumab group. The data indicated that when the follow-up was 19.4 months, patients in the bevacizumab group had a longer median PFS (restricted mean) than that of the standard-therapy group (21.8 months vs. 20.3 months). Moreover, when prolonging the follow-up to 42 months, the PFS (restricted mean) was 24.1 months in the bevacizumab group and 22.4 months in the standard-therapy group. Additionally, patients in the bevacizumab group had a median OS of 36.6 months compared with a median survival of 28.8 months in the standard-therapy group (Perren et al., 2011). Then, Oza et al. (2015) reported the final OS results of ICON7. They found no significant difference in restricted mean OS between the two groups (45.5 months in the bevacizumab group versus 44.6 months in the standard-therapy group). However, for patients with high risk, the restricted mean OS was longer in the bevacizumab group compared to that in the standard-therapy group (39.9 months in the bevacizumab group vs 34.5 months in the standard-therapy group). For patients with non-high risk, there was no significant difference in the restricted mean OS between the bevacizumab group and the standard-therapy group (49.7 months vs. 48.4 months). Overall, both the GOG-218 and ICON7 trial proved that chemotherapy combined with bevacizumab could not prolong the overall survival in ovarian cancer. However, high risk patients treated with chemotherapy plus bevacizumab show an OS benefit compared to those treated chemotherapy alone.
Bevacizumab also showed antitumor activity in platinum-resistant EOC. Cannistra et al. (2007) designed a phase 2 trial for bevacizumab in patients with platinum-resistant EOC or peritoneal serous cancer. They enrolled 44 patients and treated them with intravenous bevacizumab 15 mg/kg every 3 weeks. In the research, seven patients had a partial response. Patients had a median PFS of 4.4 months. So in platinum-resistant cancer, bevacizumab also had antitumor activity.
Bevacizumab also has antitumor activity in recurrent ovarian cancer. Aghajanian et al. (2012) reported a phase 3 clinical trial (OCEANS) in platinum-sensitive relapsed ovarian cancer. In total, 484 patients were enrolled and randomly assigned to the bevacizumab arm (n = 242) or placebo arm (n = 242). All patients were treated with carboplatin. The results showed that patients in the bevacizumab arm had a longer PFS compared to that of patients in the placebo arm (12.4 months vs. 8.4 months, respectively). Additionally, patients in the bevacizumab arm also showed an enhanced objective response rate (78.5% versus 57.4%) and duration of response (10.4 months versus 7.4 months) compared with those of patients in the placebo arm. They also analyzed the OS of the two arms. However, the results showed no significant difference in OS between the bevacizumab arm and the placebo arm (33.6 months in the bevacizumab arm vs 32.9 months in the placebo arm) (Aghajanian et al., 2015). Later, Coleman et al. (2017) reported a phase III clinical trial of bevacizumab combined with chemotherapy in patients with relapsed platinum-sensitive EOC. The data showed that the median OS was significantly longer in the chemotherapy plus bevacizumab group compared to that of the chemotherapy group (42.2 months in the chemotherapy plus bevacizumab group versus 37.3 months in the chemotherapy group). Additionally, the median PFS in patients with chemotherapy was 10.4 months, while in chemotherapy plus bevacizumab, the median PFS was 13.8 months (Coleman et al., 2017). Above studies implied that bevacizumab could improve the PFS and median OS in recurrent platinum-sensitive recurrent ovarian cancer, while it had no significant effect for OS.
There have also been studies using bevacizumab-treated relapsed platinum-resistant ovarian cancer. In a phase III clinical trial (AURELA) the results demonstrated that patients in the bevacizumab with chemotherapy group had a significantly increased PFS rate than that of the chemotherapy group. The median PFS in the bevacizumab with chemotherapy group and the chemotherapy group was 6.7 and 3.4 months, respectively. However, there was no meaningful OS increase in the bevacizumab with chemotherapy group. Therefore, they thought that the combination of bevacizumab with chemotherapy could improve the PFS in patients with relapsed platinum-resistant ovarian cancer (Pujade-Lauraine et al., 2014).
Bevacizumab combined with cyclophosphamide also showed its effect in recurrent ovarian cancer. In a phase 2 clinical trial, seventy patients with relapsed ovarian carcinoma were treated intravenously with bevacizumab and cyclophosphamide. The results showed that 17 patients had a partial response. Moreover, patients had a median progression time of 7.2 months and a median survival time of 16.9 months (Garcia et al., 2008).
VEGF plays a crucial role in angiogenesis. Nowadays, due to the pro-tumor effect in tumor progress, targeting VEGF became an attractive therapeutic method. As the first FDA-approved anti-angiogenesis antibody, bevacizumab was applied to the treatment of cancer. Studies demonstrated that bevacizumab combined with chemotherapy increases the PFS in newly prognosis ovarian cancer, platinum-resist ovarian cancer and recurrent ovarian cancer. Nevertheless, bevacizumab added to chemotherapy could not enhance the OS. Further investigated was needed.
Inhibitors of VEGF Receptors
Sorafenib (also called BAY 43-9006) is a double inhibitor of VEGF and RAF kinase that can promote tumor angiogenesis by targeting RTKs and the PAF/MEK/ERK pathway (Wilhelm et al., 2004). Chekerov et al. (2018) reported a randomized, double-blind, placebo-controlled, phase II study of the effect of sorafenib in combination with topotecan in women with platinum-resistant ovarian carcinoma. They randomly assigned patients into the topotecan combined sorafenib (oral 400 mg bid) group (n = 85) of the topotecan plus placebo group (n = 89). The results showed that patients in the topotecan plus sorafenib group had improved PFS compared with that of the topotecan plus placebo group (6.7 months vs. 4.4 months). There were the same results for OS (17.1 months in the topotecan plus sorafenib group vs. 10.1 months in the topotecan plus placebo group). Subsequently, Matei et al. (2011) described a phase 2 clinical trial of sorafenib in relapsed ovarian carcinoma. They treated patients with oral 400 mg sorafenib bid. The results suggested that only 2 women had partial response and 20 patients had stable disease. In brief, sorafenib could increase the survival time in ovarian cancer.
Sunitinib is a multi-targeted tyrosine kinase inhibitor that acts through targeting PDGF receptor (PDGFRs), VEGFR-1-2-3, KIT (a stem cell factor) and Flt3 (a tyrosine protein receptor) (Bauerschlag et al., 2010). Baumann et al. (2012) reported a phase II trial of sunitinib in patients with platinum-resistant ovarian cancer. Patients were divided into a non-continuous sunitinib group and a continuous sunitinib group. Six patients in the non-continuous group and two patients in the continuous group had a complete response or partial response. Additionally, the median PFS times for the non-continuous group and continuous group were 4.8 and 2.9 months, respectively. The median OS was 13.6 months vs. 13.7 months for the non-continuous group and continuous group. Additionally, Biagi et al. (2011) reported the effect of sunitinib in relapsed ovarian cancer. In patients who received oral sunitinib treatment, there was a median PFS of 4.1 months. Hence, using sunitinib in recurrent ovarian cancer patients could increase the survival time.
Pazopanib was an anti-angiogenic drug that targets VEGF receptor, FGF receptors (FGFRs) 1–3, and PDGFRs α and β. A phase 2 enrolled 940 patients with ovarian cancer and randomly divided into a pazopanib group (800 mg once daily for 24 months) (n = 472) and a placebo group (n = 478). The results revealed that patients in the pazopanib group had an improved median PFS compared with that of the placebo group (mean PFS 17.9 months versus 12.3 months). Conversely, there was no difference in OS between the two groups. Additionally, Pignata et al. (2015) reported a phase 2 trial to assess the effect of combined pazopanib and paclitaxel in patients with platinum-refractory or platinum-resistant advanced ovarian carcinoma. They enrolled 74 patients who were randomly assigned to the paclitaxel and pazopanib group (n = 37) or the paclitaxel group (n = 37). The data indicated that patients in the paclitaxel and pazopanib group had significantly longer PFS than that of patients in the paclitaxel group (mean PFS 6.35 vs 3.49 months). The median OS was 19.1 months in the paclitaxel and pazopanib group and 13.7 months in the paclitaxel group. Above researches indicated that pazopanib treatment had an improve PFS in ovarian cancer.
Nintedanib is also an oral angiokinase inhibitor, which can inhibit the effect of VEGFR1-3, FGRs 1-3 and PDGFRs α and β. A phase 3 trial was performed to elucidate the effect of nintedanib combined with first-line chemotherapy in advanced ovarian cancer. Of the 1366 patients enrolled, 911 were assigned to the nintedanib group, and 455 were assigned to the placebo group. The data suggested the mean PFS was 17.2 months in the nintedanib group compared with 16.6 months in the placebo group. Therefore, they suggested that the combination of nintedanib and first-line chemotherapy could significantly increase the PFS in advanced ovarian cancer.
Cediranib is a VEGF receptor (VEGFR1-3) inhibitor. In a randomized phase 3 trial (ICON6) the investigators randomly assigned 456 patients with relapsed platinum-sensitive ovarian carcinoma into three groups: group A received placebo with chemotherapy and then placebo maintenance, group B received cediranib 20 mg with chemotherapy and then placebo maintenance, and group C received cediranib once daily with chemotherapy and then cediranib once daily maintenance. The results showed that 90% of patients (410 of 456) had disease progression, including 96% of patients in group A (113 of 118), 90% of patients in group B (156 of 174) and 86% of patients in group C (141 of 164). Moreover, patients in group C had a longer median PFS compared to that of groups A and B (mean PFS = 8.7, 9.9, and 11.0 months in groups A, B, and C, respectively) (Ledermann et al., 2016). Thus, they considered cediranib with chemotherapy could significantly improve the PFS in patients with relapsed ovarian cancer.
VEGFR inhibition is an important component of anti-angiogenic therapy. Several VEGFR inhibitors have been introduced into clinical studies. The research results suggest that treatment of VEGFR inhibitors could improve the PFS in ovarian cancer. These provide a novel therapeutic option for ovarian cancer.
Angs Inhibitor
Ang1 and Ang2, expressed on ECs, were connected with the TIE2 receptor. They could mediate vascular remodeling by a signaling pathway, which was different from the VEGF pathway. Trebananib (also called AMG386) is a peptide-Fc. Through binding with Ang1 and Ang2, it prevents the connection of the TIE2 receptor and Angs. Thus, trebananib showed antitumor activity in ovarian cancer. A double blind phase 3 study detected the antitumor activity of trebananib in patients with recurrent ovarian cancer. A total of 919 patients were enrolled, and they were randomly divided into the trebananib group (n = 461) and the placebo group (n = 458). In the placebo group, patients were treated with intravenous paclitaxel 80 mg/m2 and placebo weekly. The results showed that patients with trebananib treatment had a significantly longer median PFS compared to patients with placebo treatment (7.2 months compared to 5.4 months). However, there were no significant differences between the trebananib group and the placebo group in the OS analysis (17.3 months in the paclitaxel group and 19.0 months in the placebo group) (Monk et al., 2014).
Trebananib inhibited Angs 1 and 2 and improved the PFS in ovarian cancer. However, the role of Angs inhibitors in recurrent ovarian cancer remains to be further studied.
TAM-Targeted Antitumor Strategies
Tumor-associated macrophages are a crucial part of the TME. It is known that TAMs have a significant association with the proliferation, invasion, migration and clinical outcomes of ovarian carcinoma. In recent years, significant progress has been made in research on TAM-targeted strategies. Based on previous research, TAM-targeted strategies can be divided into four types: (1) suppressing macrophage recruitment; (2) inhibiting TAM survival; (3) increasing the tumouricidal activity of M1 macrophages; and (4) limiting the tumor-promoting activity of M2 macrophages (Tang et al., 2013; Cassetta and Pollard, 2018).
Suppressing Macrophage Recruitment
Various chemokines and cytokines promote macrophage recruitment to tumor tissues, such as C-C motif chemokine ligand 2 (CCL2), macrophage colony-stimulating factor (GM-CSF), VEGF, CXCL-12 and hypoxia-inducible factors (HIFs) (Tang et al., 2013; Cassetta and Pollard, 2018). Thus, regulating the relevant chemoattractants is a promising approach for suppressing macrophage recruitment and tumor therapy.
CCL2 (also called monocyte chemotactic protein-1 [MCP-1]) is a member of the MCP chemokine family that is produced by tumor cells and stromal cells such as myeloid cells, ECs and fibroblasts and acts as a chemoattractant for T cells, NK cells and monocytes (Ueno et al., 2000; Conti and Rollins, 2004). CCR2 is a receptor of CCL2, including CCR2A and CCR2B. Among them, CCA2B is the main isoform of CCR2 that is highly expressed on NK cells and monocytes. CCR2A is expressed on smooth muscle cells and a portion of monocytes. It has been reported that CCL2-CCR2 signaling is involved in tumor metastasis (Lim et al., 2016). In the initial stage of metastasis, tumor cells breakdown ECM and travel to blood vessels. During this stage, CCL2 guides the migration of cancer cells through linking with CCR2. In addition, CCL2 promotes the migration of cancer cells by inducing the expression of MMP2 as well as MMP9 (Tang and Tsai, 2012). Then, cancer cells invade into blood vessels for metastatic dissemination, which requires TAMs. CCL2 promotes cancer cell intravasation and extravasation because it is a chemoattractant for TAMs. Interestingly, CCL2-CCR2 signaling stimulates angiogenic switching via recruiting myeloid cells and suppresses immune-mediated killing by recruiting MDSCs (Huang et al., 2007; Low-Marchelli et al., 2013).
CCL2 was highly expressed on paclitaxel-resistant ovarian cancer cells and showed an antitumor effect in ovarian cancer. Moisan et al. reported C1142 (a mouse CCL2 inhibitor) combined with carboplatin in the treatment of ovarian cancer mouse model could improve the efficacy of carboplatin (Moisan et al., 2014). Additionally, Sandhu et al. (2013) reported a phase I trial investigating the effect of carlumab in solid tumors. They enrolled forty-four patients in total, including eight ovarian cancer patients. All of them received different doses of carlumab (also called CNTO 888), which is a human anti-CCL2 monoclonal antibody. The results showed that patients with advanced ovarian carcinoma achieved a more than 50% decrease in CA125 and achieved 10.5 months of stabilized disease.
M-CSF (also called CSF-1) is also a chemokine for macrophages and is secreted by a variety of stromal cells and epithelial cells (Wyckoff et al., 2004). In addition, its receptor, named M-CSFR, CSF-1R or CD115, is a receptor tyrosine kinase and is restricted to mononuclear phagocytes (Bonelli et al., 2018). It is known that the binding of CSF-1 and CSF-1R regulates the differentiation, function and survival of macrophages through inducing tyrosine kinase (TK)–mediated autophosphorylation in the cytoplasm and the production of intracellular cascade signals (Chitu and Stanley, 2006; Hume and MacDonald, 2012). Hence, the CSF-1/CSF-1R axis can be blocked by anti-CSF antibodies, anti-CSF-1R antibodies and some molecule inhibitors that suppress the function of tyrosine kinases (Hume and MacDonald, 2012; Ries et al., 2015).
Studies implied that CSF-1R inhibitors suppressed the proliferation and metastasis in ovarian cancer via blocking macrophages. Moughon et al. (2015) reported using CSF-1R inhibitors in advanced ovarian carcinoma. They found that during the late stage of ovarian cancer, mice treated with GW2580, a CSF-1R kinase inhibitor, had markedly reduced ascites volume and infiltration of M2 macrophages. Thus, they thought that CSF-1R inhibitors decreased the ascites volume by blocking macrophages. Subsequently, Yu et al. (2018) reported the antitumor effect of CSF-1R inhibitors in cisplatin-resistant ovarian cancer. They found that CSF-1R was highly expressed in cisplatin-resistant SKOV3 and CaoV-3 cells (human ovarian cancer cell lines). When cancer cells were treated with pexidartinib, a CSF-1R inhibitor, with or without cisplatin, the combination of pexidartinib and cisplatin significantly inhibited ovarian cancer cell proliferation and induced apoptosis in cancer cells. They also found that the combination of pexidartinib and cisplatin treatment more efficiently suppressed tumor growth compared to using cisplatin alone in mouse models. Thus, they confirmed that CSF-1R inhibitors could inhibit the proliferation of cisplatin-resistant ovarian cancer. Recently, Lu and Meng (2019) reported the function of BLZ945 in ovarian cancer. They established an ovarian cancer model and treated them with docetaxel with or without BLZ945, which is an inhibitor of the CSF1CSF1R pathway. The data showed that both the docetaxel group and BLZ945 group had decreased tumor growth. Similarly, the docetaxel plus BLZ945 group had a significant decrease in tumor growth compared with that of the docetaxel or BLZ945 alone group. Additionally, docetaxel increased TAM infiltration, while the BLZ945 group showed decreased TAM abundance. They also found that the BLZ945 group had decreased VEGF and MMP9 expression levels, which were closely related to metastasis. Correspondingly, the DTX and BLZ945 combination group had significantly deregulated VEGF and MMP9. Moreover, the docetaxel plus BLZ945 group had less lung metastasis than those observed in the other groups. Therefore, they confirmed that BLZ945 inhibited the proliferation and metastasis of ovarian cancer by suppressing TAMs.
CCL2, M-CSF, VEGF, CXcl-12, and HIFs are molecules that can improve macrophage recruitment. As mentioned above, targeted related chemical attractants enhance the anti-tumor activity of ovarian cancer. Therefore, studies suggested that inhibition of macrophage recruitment can be used as a complement to treatment strategies.
Inhibiting TAM Survival
As a part of TAM-targeted strategies for cancer, the inhibition of TAM survival may be realized by attenuated bacteria, immunotoxin-conjugated mAbs and chemical reagents that induce macrophage apoptosis or by activating immune cells such as T lymphocytes to kill TAMs.
Bisphosphonate is an important drug for depleting macrophages. Kobayashi et al. reported that incubating SKOV3 and OVCAR5 cells (human ovarian cancer cell lines) with alendronate a second-generation bisphosphonate, they found that alendronate exerted concentration-dependent growth inhibition on these cells. Then, they treated the mogp-TAg transgenic mouse with alendronate and discovered a significant decrease in tumor mass in the reproductive tract. Additionally, mice that received alendronate treatment tolerated it well and showed no influence on body weight. Therefore, they suggested that alendronate inhibited the proliferation of ovarian cancer in vivo and in vitro (Kobayashi et al., 2017).
Apart from inducing macrophage apoptosis, TAMs can be suppressed by the activation of an adapted immune response, especially cytotoxic T lymphocytes. However, we did not find studies that applied the inhibition of the adapted immune response to ovarian cancer treatment.
Increasing the Tumouricidal Activity of M1 Macrophages
In the TME, immunosuppressive M2 macrophages comprise mainly TAMs (Ren et al., 2014). However, due to the plasticity of polarization, TAMs still maintain the potential to repolarize from tumor-promoting M2 macrophages to tumor-resisting M1 macrophages (Biswas and Mantovani, 2010). As mentioned before, the polarization of macrophages relies on cytokines. When exposed to IFN-γ, LPS, GM-CSF and IL-12, macrophages mainly polarize to M1 macrophages. In contrast, when exposed to IL-4, IL-10, and IL-13, macrophages polarize to M2 macrophages. Therefore, regulating these cytokines could contribute to TAM-targeted antitumor strategies.
It was reported that the NF-κB signaling pathway was associated with TAM polarization (Hagemann et al., 2009; Biswas and Lewis, 2010; Mancino and Lawrence, 2010; Tang et al., 2013).
The NF-κB family includes five transcription factors: RelA/p65, RelB, c-Rel, NF-κB1 (precursor proteins p50/p105) and NF-κB2 (p100/p52) (Hagemann et al., 2009). Several agents, such as TLR (Toll-like receptors) agonists, anti-IL-10R mAb and anti-CD40 mAb, can activate NF-κB through classical or non-classical pathways (Mantovani et al., 2004; Sica and Bronte, 2007; Mancino and Lawrence, 2010). In the NF-κB signaling pathway, NF-κB modulates many crucial genes in macrophages and many tumor-promoting genes, such as VEGF, IL-6, TNF-α, and COX2 (Hagemann et al., 2009; Biswas and Lewis, 2010). In addition, the inactivation of NF-κB mediates polarization of TAMs to immunosuppressive M2 macrophages, while NF-κB reactivation adjusts TAMs to tumouricidal M1 macrophages (Biswas and Lewis, 2010). The NF-κB signaling pathway also regulates the development and proliferation of T and B lymphocytes (Jost and Ruland, 2007), modules angiogenesis and plays a vital role in tumorigenesis (Pramanik et al., 2018).
Recently, a TLR-7 agonist, which can activate NF-κB, was applied to cancer treatment. Geller et al. (2010) reported a clinical trial on the antitumor activity of a TLR-7 agonist in relapsed ovarian, cervix and breast cancer. They enrolled fifteen patients, including ten patients with recurrent ovarian cancer. All of the patients received 852A (a TLR-7 agonist). The results showed that only one patient with stage IIIc serous ovarian cancer had stable disease after treatment with 24 doses of 852A, while she did not continue the trial because of disease progression.
IL-12 is also a factor that can enhance macrophage polarization to M1 macrophages. IL-12 can also promote Th1 response, which polarizes macrophage to M1 macrophages (Biswas et al., 2012; Lu, 2017; Shapouri-Moghaddam et al., 2018). Silver et al. (1999) reported that in an animal model of ovarian cancer, mice treated with IL-12 had decreased tumor growth and even tumor regression. Therefore, they suggested that IL-12 had an antitumor effect on ovarian cancer. Subsequently, a phase I trial reported the treatment of relapsed chemotherapy-resistant ovarian cancer with the IL-12 plasmid/lipopolymer complex. The results showed that 31% of the patients treated with IL-12 treatment had stable disease, while 69% of patients had progressive disease. In addition, patients with high IL-12 treatment had longer survival than that of patients with low IL-12 treatment (Anwer et al., 2010). Similarly, Anwer et al. (2013) reported that 17% of patients with platinum-sensitive relapsed ovarian cancer treated with IL-12 had completed response, 33% had a partial response, 42% had stable disease, and 8% had progressive disease. Later, Alvarez et al. (2014) reported that treated platinum-resistant relapsed ovarian cancer patients with EGEN-001 (an IL-12-based immunotherapeutic agent) no patients had complete or partial responses, 35% (7 of 16 patients) had stable disease, and 45% (9 of 16 patients) had progressive disease. Moreover, the median OS and PFS of EGEN-001-treated patients were 9.17 and 2.89 months, respectively. Therefore, they suggested the limited activity of EGEN-001 in platinum-resistant relapsed ovarian cancer.
Tumor-associated macrophages have the plasticity for polarization, which means that TAMs could repolarization from tumor promoting M2-type to tumor-killing M1-type. Several molecular are reported associated with TAM re-polarization, such as NF-κB and IL-12. Recent years, several researches implied that targeted NF-κB and IL-12 enhance macrophage polarization to M1 macrophages in ovarian cancer. However, the specific signaling pathways are not fully clear. Hence, further studies are needed.
Limiting the Tumor-Promoting Activity of M2 Macrophages
Apart from the above TAM-targeted antitumor strategies, limiting the tumor-promoting activity of M2 macrophages is also a promising strategy. STAT3, a member of the STATs (Signal Transducers and Activators of Transcription) family, is generally inactivated and located in the cytoplasm (Laudisi et al., 2018). It can be activated by various receptors, including cytokines such as IL-6 and IL-10, as well as IL-11 and growth factors such as VEGF, EGF and FGF (Chai et al., 2016; Furtek et al., 2016). Once binding to their ligand, the conformation of the receptors changes, which promotes signal propagation and leads to the activation of JAKs. Then, activated JAKs, including JAK1 and JAK2, induce the phosphorylation of STAT3 at Tyr705 and lead to the translocation of activated STAT3 dimers to the nucleus, where it binds to DNA and enhances gene transcription (Yu H. et al., 2014). It has been reported that STAT3 could modulate tumorigenesis by regulating associated gene expression. For instance, STAT3 regulates the expression of c-Myc and cyclin D1, which are linked to the cell cycle (Luwor et al., 2013). STAT3 also modulates angiogenesis via the gene expression of VEGF and IL-8 and regulates migration by MMP-2 and MMP-9 gene expression (Zhang et al., 2010). More recently, studies found that STAT3 is associated with the polarization of TAMs (Tang et al., 2013; Wang et al., 2018). Additionally, the inhibition of STAT3 decreased TAM polarization to M2 macrophages (Fujiwara et al., 2011).
HO-3867 is an inhibitor of STAT3 and has a significant antitumor effect on ovarian cancer (Selvendiran et al., 2010; Tierney et al., 2012; Rath et al., 2014; Tang et al., 2015; Bixel et al., 2017; Saini et al., 2017; Yoshikawa et al., 2018). Selvendiran et al. (2010) found that HO-3867 induced the apoptosis of A2780 cells through activating caspase-3 and caspase-8 and promoted G2/M cell-cycle arrest via regulating cell-cycle regulatory molecules such as cyclin, p21, p27, p53 and cdk2. Additionally, they observed a dose-dependent reduction of tumor volume in mice with ovarian cancer. Then, Rath et al. (2014) reported that HO-3867 reduced tumor growth in a chemotherapy-resistant ovarian cancer model in a dose-dependent manner. Subsequently, Saini et al. (2017) found that HO-3867 inhibited tumor size and tumor metastasis in ovarian cancer.
WP1066 is also a STAT3 inhibitor with antitumor activity. Tang et al. (2015) found that WP1066 markedly suppressed the clonogenicity and invasion activity of SKOV3 and SKOV3/DDP cells (a cisplatin-resistant ovarian cancer cell line). However, it increased the apoptosis of SKOV3 and SKOV3/DDP cells. When treated with WP1066 and cisplatin in combination, the inhibition of proliferation and apoptosis increased compared to that of cisplatin alone. Thus, they suggested that WP1066 inhibited proliferation and metastasis and increased the apoptosis and chemosensitivity of ovarian cancer cells.
M2 macrophages show tumor-promoting activity. Above studies provide that STAT3 is involved in increased M2 macrophages. The inhibition of STAT3 decreased the proliferation and metastasis of cancer cells.
In summary, TAMs-targeted treatment strategies include suppressing macrophage recruitment and TAM survival and increasing the transformation of M2 macrophages to M1 macrophages. TAMs participate in tumor progression through fairly complex mechanisms. However, our understanding of TAMs and its interaction with the tumor microenvironment are not deep enough. Therefore, more researches are needed to facilitate the development and clinical application of TAM-targeted antitumor strategies.
Immune Checkpoint Inhibitors
Immune checkpoints play important roles in modulating T cell function in the TME (Zhao and Subramanian, 2017). Immune checkpoint therapy limits inhibitory pathways in T cells, thereby enhancing antitumor immune responses (Sharma and Allison, 2015). Hence, it changes cancer treatment. Among them, CTLA-4 and PD1/PD-L1 are important immune checkpoints for ovarian cancer (Mittica et al., 2016; Bose, 2017) (Table 2).

Table 2. Immune checkpoint inhibitors in ovarian cancer.
Anti-CTLA-4 Antibody
CTLA-4 is a transmembrane glycoprotein that is mainly expressed on activated T cells, such as regulatory T cells and memory T cells (Tarhini and Iqbal, 2010). It is widely known that CTLA-4 inhibits the T lymphocyte-mediated antitumor immune response by intrinsic and extrinsic cell pathways (Tarhini and Iqbal, 2010; Brunner-Weinzierl and Rudd, 2018). The intrinsic cell pathways include suppressing cytokine receptor signaling and protein translation, activating ubiquitin ligases and recruiting phosphatases (Tarhini and Iqbal, 2010). In contrast, in the extrinsic cell pathways, CTLA-4 competitively binds to members of the B7 family and transmits inhibitory signals, thereby reducing the activation of T cells (Egen et al., 2002; Zhao Y. et al., 2018). In addition, CTLA-4 produces a reversing signal via B7 and induces the secretion of IDO, resulting in the decomposition of tryptophan and inhibition of the proliferation of T cells (Boasso et al., 2005).
Anti-CTLA-4 antibodies have immune suppression by limiting CTLA-4 to bind with members of the B7 family, thereby suppressing the recognition of antigen-specific T cells. Hodi et al. (2003) reported a trial investigating the activity of MDX-CTLA-4 (a CTLA-4 inhibitor, also called ipilimumab) in patients with metastatic ovarian cancer. One patient with ovarian carcinoma had a stable CA-125 level 1 month after antibody injection. Meanwhile, she experienced a reduction in ascites and pain. The other patient had a 43% decrease in CA-125 level in the initial 2 months. Subsequently, Hodi et al. (2008) treated ovarian cancer patients with ipilimumab. The data showed that one patient had obvious antitumor effects. There is also a phase II clinical trial underway studying the antitumor effect of ipilimumab in patients with relapsed platinum-sensitive ovarian carcinoma.
Anti-PD1/PD-L1 Antibodies
PD-1 (also called CD279) is a type I transmembrane protein that is expressed on a variety of immune cells, including activated T cells and B cells, NK cells, monocytes and DCs (Frydenlund and Mahalingam, 2017). PD-L1 and PD-L2 are receptors for PD-1, which all belong to the B7 family (Moy et al., 2017). Among them, PD-L1 shows a wide range of expression on hematopoietic cells and non-hematopoietic cells, while PD-L2 is only expressed on APCs (antigen presenting cells) (Moy et al., 2017). PD-L1 and PD-L2 can be induced by extrinsic proinflammatory signals, such as TNF-α, IFN-γ, ILs and GM-CSF (Wang X. et al., 2017; Zhang et al., 2017; Mimura et al., 2018). Apart from this, PD-L1 is also induced by intrinsic signaling pathways, including the PI3K-AKT pathway and the JAK/STAT pathway (Jiang et al., 2019). It is known that PD-1 has an immune-suppressive ability through binding with PD-1 receptors (Brunner-Weinzierl and Rudd, 2018). After binding with PD-1 receptors, PD-1 stimulates intracellular signaling pathways and suppresses immune cell activation, which inhibits the production of cytokines and antibodies from immune cells, which in turn exhausts the immune cells and maintains immune system homeostasis (McDermott and Atkins, 2013). Additionally, PD-1 can not only inhibit T lymphocyte activation and accelerate T lymphocyte apoptosis but can also be regulated by molecules in the TME, such as TGF-β, IL-7, IL-15, IL-21 and IFN-α (Kinter et al., 2008; Terawaki et al., 2011; Rekik et al., 2015; Jiang et al., 2019). PD-1 inhibitors, including anti-PD-1 antibodies and anti-PD-L1 antibodies, reverse the suppression of antigen-specific T cells through blocking PD-1 or PD-1 ligands (Hamanishi et al., 2016).
As precision medicine develops, patient selection tends to be a social economic trend, which also goes for PD-L1/PD-1 blockade therapy. In ovarian cancer, the expression of PD-L1 on DCs and macrophages correlates with clinical efficacy of PD-L1 and PD-1 blockade, suggesting that the APC PD-L1 expression may be a predicting factor of therapeutic benefit and patient selection indicator (Lin et al., 2018).
Anti-PD-1 antibodies
Nivolumab is an anti-PD-1 monoclonal antibody that inhibits the combination of PD-1 ligands. There was a clinical trial exploring the effect of Nivolumab in patients with platinum-resistant ovarian cancer. It divided twenty patients with platinum-resistant ovarian cancer into a high-dose cohort and a low-dose cohort. The results showed that in the low-dose group, one patient had a partial response, and four patients had stable disease. However, in the high-dose group, two patients had a complete response, and two patients had stable disease. Additionally, for all 20 patients, the median PFS time and median overall time were 3.5 and 20.0 months, respectively (Hamanishi et al., 2015).
Pembrolizumab is also a humanized anti-PD-1 monoclonal antibody. Andrea and his colleagues reported that treated advanced ovarian cancer patients with pembrolizumab, One patient with complete response, two patients with partial response, seven patients with stable disease and sixteen patients with disease progression. The median OS and PFS were 13.8 and 1.9 months (Varga et al., 2019), respectively.
Anti-PD-L1 antibodies
BMS-936559 is a humanized high affinity anti-PD-L1 monoclonal antibody that blocks PD-L1 binding to PD-1 and CD80. Brahmer et al. (2012) reported a clinical trial regarding the activity of BMS-936559 in advanced cancer. A total of 200 patients were enrolled, 17 of whom were ovarian cancer patients. The results demonstrated that at a 10 mg/kg dose, 1 ovarian cancer patient achieved a partial response, and 3 ovarian cancer patients achieved stable disease. Avelumab, a human anti-PD-L1 antibody, could specifically bind to PD-L1 and block the links with PD-1 (Heery et al., 2017). Disis et al. (2019) described a cohort study on the effect of avelumab in refractory or relapsed ovarian cancer. They enrolled 125 patients and treated them with 10 mg/kg avelumab every 2 weeks. The results demonstrated that 12 patients had an objective response. Among them, 1 patient had a complete response. Additionally, the median PFS and OS were 2.6 and 11.2 months, respectively.
Immune checkpoints are an important part of maintaining self-tolerance. Immune checkpoint therapy enhancing antitumor immune responses via suppressing inhibitory pathways in T cells. Currently researched suggest that immune checkpoint inhibition is a potential way to activate the immune response. But further exploration is needed to improve the cure rate.
Conclusion
The current standardized treatment for ovarian cancer is optimal cytoreductive surgery plus platinum-based chemotherapy with the carboplatin–paclitaxel regimen (Bolton et al., 2012). Nevertheless, due to chemotherapy-resistant and refractory diseases, the sensitivity of chemotherapy decreases, thereby decreasing the long-term survival rate and increasing the recurrence rate (Lim and Ledger, 2016). Currently, the TME is regarded as a possible therapeutic target for ovarian cancer. In this review, we summarized new targeted therapies at the interface between ovarian cancer and the TME (Figure 3): Several ways are adopted to target ovarian TME. CAFs targeting therapy includes direct deletion FAP+ fibroblasts, reverting the activated CAFs into a quiescent state, and targeting CAF-specific pathways; Anti-angiogenesis is an important TME targeting therapeutic strategy. Since VEGF is the most typical activator of angiogenesis, anti-angiogenesis therapy is divided into three types: anti-VEGF, inhibitors of VEGF receptors and Angs inhibitors. Among them, bevacizumab, the first FDA-approved anti-angiogenesis antibody, plays a crucial role in anti-angiogenesis therapy for ovarian cancer. TAM-targeted antitumor strategies also have drawn much research attention. Currently, the present TAM-targeted therapeutics consist of (i) suppressing macrophage recruitment (such as CCL2 inhibitors and CSF-1R inhibitors); (ii) inhibiting TAM survival (e.g., bisphosphonates); (iii) increasing the tumouricidal activity of M1 macrophages (for example, agonists of the NF-κB signaling pathway such as TLR-7 agonist and other agents such as IL-12); and (iv) limiting the tumor-promoting activity of M2 macrophages (inhibitor of STAT3). Finally, increasing evidence of the therapeutic effect of immune checkpoint inhibitors in ovarian cancer has been reported. Immune checkpoint inhibitors limit inhibitory pathways in T cells, thereby enhancing antitumor immune responses. In particular, CTLA-4 and PD1/PD-L1 are important immune checkpoints for ovarian cancer. We also summarized the ongoing clinical trials targeting ovarian cancer tumor microenvironment (Table 3). Although new therapeutic approaches targeting the TME could not cure ovarian cancer, they showed the potential to control the development of ovarian cancer. Besides, with the development of “-omic” technology, scientists are unveiling a more detailed “territory” of ovarian cancer TME; the data drawn from this area is certain to facilitate novel therapy exploration, with the expectation to bring breakthrough discoveries to this deadly disease. We believe that TEM-targeted strategies should be applied in ovarian cancer as a valuable adjuvant therapy.
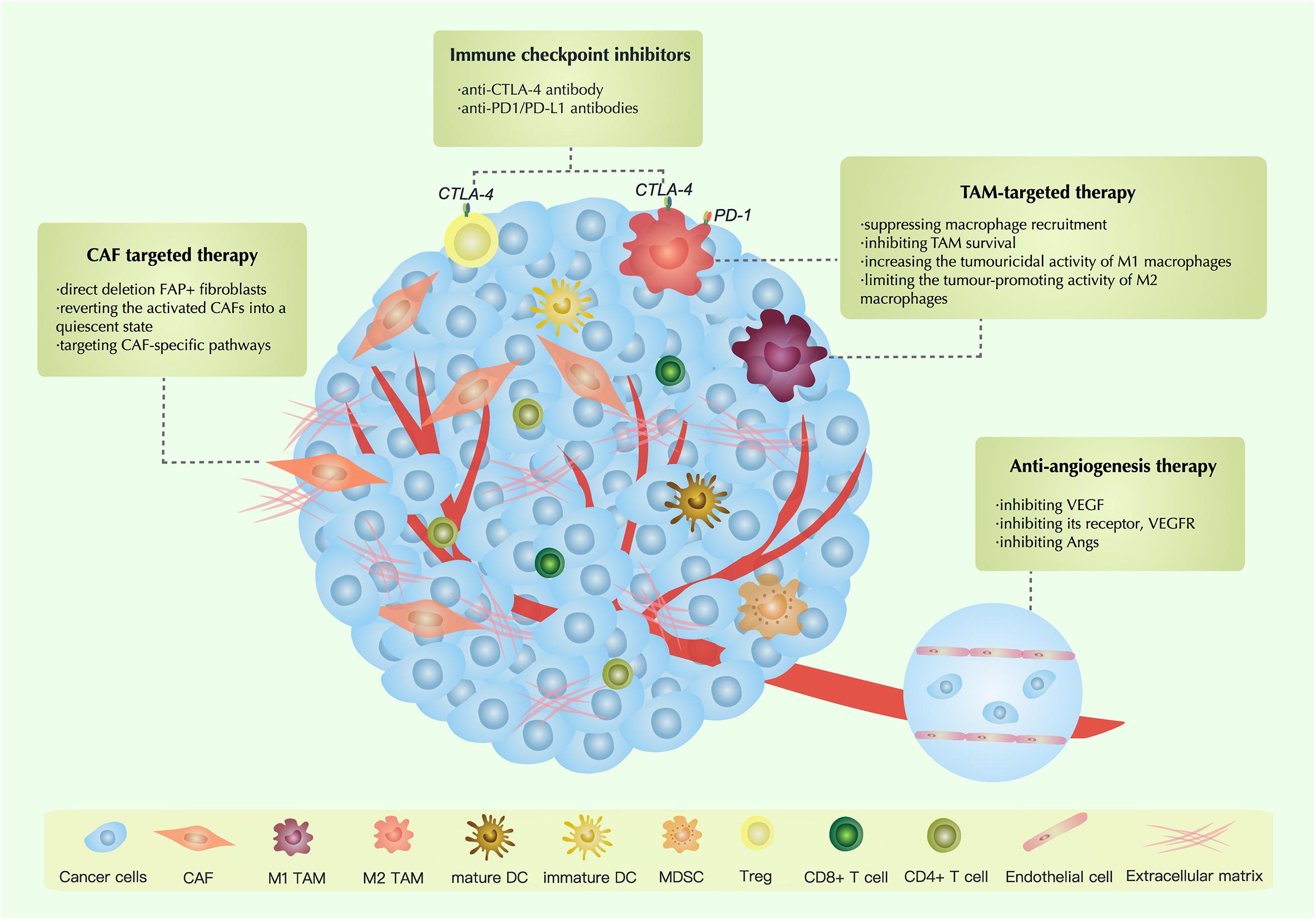
Figure 3. Tumor microenvironment related therapeutic strategies in ovarian cancer. The graph shows multiple strategies targeted the tumor microenvironment in ovarian cancer. Among them, several strategies are currently in clinical use, while others are at different phases of clinical development. CAFs: Cancer-associated fibroblasts; TGF-β: Transforming growth factor-β; TAMs: Tumor-associated macrophages; CCL2: chemokine (C-C motif) ligand 2; CSF-1R: colony stimulating factor-1 (CSF1) receptor; VEGF: Vascular endothelial growth factors; VEGFR: Vascular endothelial growth factor receptor.
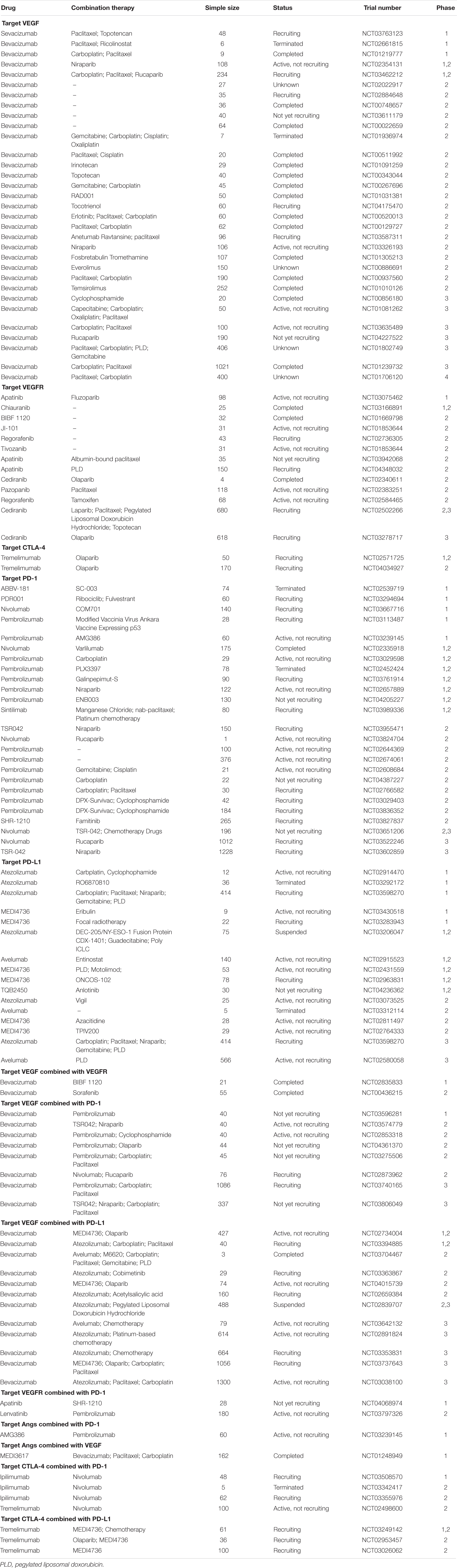
Table 3. Ongoing clinical trials targeting ovarian cancer tumor microenvironment (TME).
Author Contributions
YFY and JY drafted the manuscript. YY designed all figures and made substantial revision to the original manuscript. XZ and XW checked and modified the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key Research Projects of Science and Technology Department Foundation of Sichuan Province (2017SZ0002), a Pilot demonstration project for the transfer and transformation of new drug innovation results (2018ZX09201018-013), the Innovative research group projects (81821002), and the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aghajanian, C., Blank, S. V., Goff, B. A., Judson, P. L., Teneriello, M. G., Husain, A., et al. (2012). OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 30, 2039–2045. doi: 10.1200/jco.2012.42.0505
Aghajanian, C., Goff, B., Nycum, L. R., Wang, Y. V., Husain, A., and Blank, S. V. (2015). Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 139, 10–16. doi: 10.1016/j.ygyno.2015.08.004
Ahmadzadeh, M., Pasetto, A., Jia, L., Deniger, D. C., Stevanović, S., Robbins, P. F., et al. (2019). Tumor-infiltrating human CD4((+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol. 4:eaao4310. doi: 10.1126/sciimmunol.aao4310
Ahmed, N., Escalona, R., Leung, D., Chan, E., and Kannourakis, G. (2018). Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 53, 265–281. doi: 10.1016/j.semcancer.2018.10.002
Ahmed, Z., and Bicknell, R. (2009). Angiogenic signalling pathways. Methods Mol. Biol. 467, 3–24. doi: 10.1007/978-1-59745-241-0_1
Alvarez, R. D., Sill, M. W., Davidson, S. A., Muller, C. Y., Bender, D. P., DeBernardo, R. L., et al. (2014). A phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: a gynecologic oncology group study. Gynecol. Oncol. 133, 433–438. doi: 10.1016/j.ygyno.2014.03.571
Anwer, K., Barnes, M. N., Fewell, J., Lewis, D. H., and Alvarez, R. D. (2010). Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther. 17, 360–369. doi: 10.1038/gt.2009.159
Anwer, K., Kelly, F. J., Chu, C., Fewell, J. G., Lewis, D., and Alvarez, R. D. (2013). Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 131, 169–173. doi: 10.1016/j.ygyno.2013.07.081
Apte, R. S., Chen, D. S., and Ferrara, N. (2019). VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264. doi: 10.1016/j.cell.2019.01.021
Atretkhany, K. N., and Drutskaya, M. S. (2016). Myeloid-Derived Suppressor Cells and Proinflammatory Cytokines as Targets for Cancer Therapy. Biochemistry 81, 1274–1283. doi: 10.1134/s0006297916110055
Au Yeung, C. L., Co, N. N., Tsuruga, T., Yeung, T. L., Kwan, S. Y., Leung, C. S., et al. (2016). Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 7:11150. doi: 10.1038/ncomms11150
Banchereau, J., Briere, F., Caux, C., Davoust, J., Lebecque, S., Liu, Y. J., et al. (2000). Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811. doi: 10.1146/annurev.immunol.18.1.767
Barnas, J. L., Simpson-Abelson, M. R., Brooks, S. P., Kelleher, R. J. Jr., and Bankert, R. B. (2010). Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J. Immunol. 185, 2681–2692. doi: 10.4049/jimmunol.1000896
Barnett, J. C., Bean, S. M., Whitaker, R. S., Kondoh, E., Baba, T., Fujii, S., et al. (2010). Ovarian cancer tumor infiltrating T-regulatory (Treg) cells are associated with a metastatic phenotype. Gynecol. Oncol. 116, 556–562. doi: 10.1016/j.ygyno.2009.11.020
Barrett, R., and Puré, E. (2020). Cancer-associated fibroblasts: key determinants of tumor immunity and immunotherapy. Curr. Opin. Immunol. 64, 80–87. doi: 10.1016/j.coi.2020.03.004
Bauerschlag, D. O., Schem, C., Tiwari, S., Egberts, J. H., Weigel, M. T., Kalthoff, H., et al. (2010). Sunitinib (SU11248) inhibits growth of human ovarian cancer in xenografted mice. Anticancer. Res. 30, 3355–3360.
Baumann, K. H., du Bois, A., Meier, W., Rau, J., Wimberger, P., Sehouli, J., et al. (2012). A phase II trial (AGO 2.11) in platinum-resistant ovarian cancer: a randomized multicenter trial with sunitinib (SU11248) to evaluate dosage, schedule, tolerability, toxicity and effectiveness of a multitargeted receptor tyrosine kinase inhibitor monotherapy. Ann. Oncol. 23, 2265–2271. doi: 10.1093/annonc/mds003
Bhatia, R., Gautam, S. K., Cannon, A., Thompson, C., Hall, B. R., Aithal, A., et al. (2019). Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev. 38, 223–236. doi: 10.1007/s10555-018-09775-0
Biagi, J. J., Oza, A. M., Chalchal, H. I., Grimshaw, R., Ellard, S. L., Lee, U., et al. (2011). A phase II study of sunitinib in patients with recurrent epithelial ovarian and primary peritoneal carcinoma: an NCIC Clinical Trials Group Study. Ann. Oncol. 22, 335–340. doi: 10.1093/annonc/mdq357
Biswas, S. K., Chittezhath, M., Shalova, I. N., and Lim, J. Y. (2012). Macrophage polarization and plasticity in health and disease. Immunol. Res. 53, 11–24. doi: 10.1007/s12026-012-8291-9
Biswas, S. K., and Lewis, C. E. (2010). NF-kappaB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 88, 877–884. doi: 10.1189/jlb.0310153
Biswas, S. K., and Mantovani, A. (2010). Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11, 889–896. doi: 10.1038/ni.1937
Bixel, K., Saini, U., Kumar Bid, H., Fowler, J., Riley, M., Wanner, R., et al. (2017). Targeting STAT3 by HO3867 induces apoptosis in ovarian clear cell carcinoma. Int. J. Cancer 141, 1856–1866. doi: 10.1002/ijc.30847
Boasso, A., Herbeuval, J. P., Hardy, A. W., Winkler, C., and Shearer, G. M. (2005). Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4(+ T cells. Blood 105, 1574–1581. doi: 10.1182/blood-2004-06-2089
Bodurka, D. C., Deavers, M. T., Tian, C., Sun, C. C., Malpica, A., Coleman, R. L., et al. (2012). Reclassification of serous ovarian carcinoma by a 2-tier system: a Gynecologic Oncology Group Study. Cancer 118, 3087–3094. doi: 10.1002/cncr.26618
Bol, K. F., Schreibelt, G., Gerritsen, W. R., de Vries, I. J., and Figdor, C. G. (2016). Dendritic cell-based immunotherapy: state of the art and beyond. Clin. Cancer Res. 22, 1897–1906. doi: 10.1158/1078-0432.Ccr-15-1399
Bolton, K. L., Chenevix-Trench, G., Goh, C., Sadetzki, S., Ramus, S. J., Karlan, B. Y., et al. (2012). Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 307, 382–390. doi: 10.1001/jama.2012.20
Bonelli, S., Geeraerts, X., Bolli, E., Keirsse, J., Kiss, M., Pombo Antunes, A. R., et al. (2018). Beyond the M-CSF receptor - novel therapeutic targets in tumor-associated macrophages. FEBS J. 285, 777–787. doi: 10.1111/febs.14202
Bose, C. K. (2017). Immune checkpoint blockers and ovarian cancer. Indian J. Med. Paediatr. Oncol. 38, 182–189. doi: 10.4103/ijmpo.ijmpo_73_16
Brahmer, J. R., Tykodi, S. S., Chow, L. Q., Hwu, W. J., Topalian, S. L., Hwu, P., et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465. doi: 10.1056/NEJMoa1200694
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Broz, M. L., Binnewies, M., Boldajipour, B., Nelson, A. E., Pollack, J. L., Erle, D. J., et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. doi: 10.1016/j.ccell.2014.09.007
Brunner-Weinzierl, M. C., and Rudd, C. E. (2018). CTLA-4 and PD-1 Control of T-cell motility and migration: implications for tumor immunotherapy. Front. Immunol. 9:2737. doi: 10.3389/fimmu.2018.02737
Bruno, A., Mortara, L., Baci, D., Noonan, D. M., and Albini, A. (2019). Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front. Immunol. 10:771. doi: 10.3389/fimmu.2019.00771
Budhu, S., Loike, J. D., Pandolfi, A., Han, S., Catalano, G., Constantinescu, A., et al. (2010). CD8(+ T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. J. Exp. Med. 207, 223–235. doi: 10.1084/jem.20091279
Burger, R. A., Brady, M. F., Bookman, M. A., Fleming, G. F., Monk, B. J., Huang, H., et al. (2011). Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 365, 2473–2483. doi: 10.1056/NEJMoa1104390
Burger, R. A., Sill, M. W., Monk, B. J., Greer, B. E., and Sorosky, J. I. (2007). Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J. Clin. Oncol. 25, 5165–5171. doi: 10.1200/jco.2007.11.5345
Cai, J., Tang, H., Xu, L., Wang, X., Yang, C., Ruan, S., et al. (2012). Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 33, 20–29. doi: 10.1093/carcin/bgr230
Callahan, M. K., Wolchok, J. D., and Allison, J. P. (2010). Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin. Oncol. 37, 473–484. doi: 10.1053/j.seminoncol.2010.09.001
Cannistra, S. A., Matulonis, U. A., Penson, R. T., Hambleton, J., Dupont, J., Mackey, H., et al. (2007). Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J. Clin. Oncol. 25, 5180–5186. doi: 10.1200/jco.2007.12.0782
Carmeliet, P., and Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407, 249–257. doi: 10.1038/35025220
Casey, S. C., Amedei, A., Aquilano, K., Azmi, A. S., Benencia, F., Bhakta, D., et al. (2015). Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 35, S199–S223. doi: 10.1016/j.semcancer.2015.02.007
Cassetta, L., Fragkogianni, S., Sims, A. H., Swierczak, A., Forrester, L. M., Zhang, H., et al. (2019). Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 35, 588-602.e510. doi: 10.1016/j.ccell.2019.02.009
Cassetta, L., and Pollard, J. W. (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904. doi: 10.1038/nrd.2018.169
Chai, E. Z., Shanmugam, M. K., Arfuso, F., Dharmarajan, A., Wang, C., Kumar, A. P., et al. (2016). Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol. Ther. 162, 86–97. doi: 10.1016/j.pharmthera.2015.10.004
Chekerov, R., Hilpert, F., Mahner, S., El-Balat, A., Harter, P., De Gregorio, N., et al. (2018). Sorafenib plus topotecan versus placebo plus topotecan for platinum-resistant ovarian cancer (TRIAS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 19, 1247–1258. doi: 10.1016/s1470-2045(18)30372-3
Chen, X., and Song, E. (2019). Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 18, 99–115. doi: 10.1038/s41573-018-0004-1
Chitu, V., and Stanley, E. R. (2006). Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 18, 39–48. doi: 10.1016/j.coi.2005.11.006
Cho, Y. A., Yoon, H. J., Lee, J. I., Hong, S. P., and Hong, S. D. (2011). Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncol. 47, 1148–1153. doi: 10.1016/j.oraloncology.2011.08.007
Clarke, B., Tinker, A. V., Lee, C. H., Subramanian, S., van de Rijn, M., Turbin, D., et al. (2009). Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 22, 393–402. doi: 10.1038/modpathol.2008.191
Coffelt, S. B., Tal, A. O., Scholz, A., De Palma, M., Patel, S., Urbich, C., et al. (2010). Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 70, 5270–5280. doi: 10.1158/0008-5472.Can-10-0012
Coleman, R. L., Brady, M. F., Herzog, T. J., Sabbatini, P., Armstrong, D. K., Walker, J. L., et al. (2017). Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 18, 779–791. doi: 10.1016/s1470-2045(17)30279-6
Colombo, N., Peiretti, M., Parma, G., Lapresa, M., Mancari, R., Carinelli, S., et al. (2010). Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 21, (Suppl. 5), v23–v30. doi: 10.1093/annonc/mdq244
Conti, I., and Rollins, B. J. (2004). CCL2 (monocyte chemoattractant protein-1) and cancer. Semin. Cancer Biol. 14, 149–154. doi: 10.1016/j.semcancer.2003.10.009
Cortez, A. J., Tudrej, P., Kujawa, K. A., and Lisowska, K. M. (2018). Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 81, 17–38. doi: 10.1007/s00280-017-3501-8
Cross, M. J., and Claesson-Welsh, L. (2001). FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 22, 201–207.
Cubillos-Ruiz, J. R., Silberman, P. C., Rutkowski, M. R., Chopra, S., Perales-Puchalt, A., Song, M., et al. (2015). ER Stress Sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538. doi: 10.1016/j.cell.2015.05.025
Curiel, T. J., Coukos, G., Zou, L., Alvarez, X., Cheng, P., Mottram, P., et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949. doi: 10.1038/nm1093
Curtis, M., Kenny, H. A., Ashcroft, B., Mukherjee, A., Johnson, A., Zhang, Y., et al. (2019). Fibroblasts mobilize tumor cell glycogen to promote proliferation and metastasis. Cell Metab. 29, 141-155.e149. doi: 10.1016/j.cmet.2018.08.007
de Aquino, M. T., Malhotra, A., Mishra, M. K., and Shanker, A. (2015). Challenges and future perspectives of T cell immunotherapy in cancer. Immunol. Lett. 166, 117–133. doi: 10.1016/j.imlet.2015.05.018
Deaglio, S., Dwyer, K. M., Gao, W., Friedman, D., Usheva, A., Erat, A., et al. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265. doi: 10.1084/jem.20062512
Denton, A. E., Roberts, E. W., and Fearon, D. T. (2018). Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 1060, 99–114. doi: 10.1007/978-3-319-78127-3_6
DeVito, N. C., Plebanek, M. P., Theivanthiran, B., and Hanks, B. A. (2019). Role of tumor-mediated dendritic cell tolerization in immune evasion. Front. Immunol. 10:2876. doi: 10.3389/fimmu.2019.02876
Dhodapkar, M. V., Steinman, R. M., Krasovsky, J., Munz, C., and Bhardwaj, N. (2001). Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 193, 233–238. doi: 10.1084/jem.193.2.233
Diao, J., Zhao, J., Winter, E., and Cattral, M. S. (2011). Tumors suppress in situ proliferation of cytotoxic T cells by promoting differentiation of Gr-1((+) conventional dendritic cells through IL-6. J. Immunol. 186, 5058–5067. doi: 10.4049/jimmunol.1004125
Dijkgraaf, E. M., Santegoets, S. J., Reyners, A. K., Goedemans, R., Wouters, M. C., Kenter, G. G., et al. (2015). A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-(α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 26, 2141–2149. doi: 10.1093/annonc/mdv309
Ding, L., Kim, H. J., Wang, Q., Kearns, M., Jiang, T., Ohlson, C. E., et al. (2018). PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 25, 2972-2980.e2975. doi: 10.1016/j.celrep.2018.11.054
Disis, M. L., Taylor, M. H., Kelly, K., Beck, J. T., Gordon, M., Moore, K. M., et al. (2019). Efficacy and safety of Avelumab for patients with recurrent or refractory ovarian cancer: phase 1b Results From the JAVELIN solid tumor trial. JAMA Oncol. 5, 393–401. doi: 10.1001/jamaoncol.2018.6258
Drakes, M. L., and Stiff, P. J. (2018). Regulation of ovarian cancer prognosis by immune cells in the tumor microenvironment. Cancers 10:302. doi: 10.3390/cancers10090302
du Bois, A., Floquet, A., Kim, J. W., Rau, J., del Campo, J. M., Friedlander, M., et al. (2014). Incorporation of pazopanib in maintenance therapy of ovarian cancer. J. Clin. Oncol. 32, 3374–3382. doi: 10.1200/jco.2014.55.7348
du Bois, A., Kristensen, G., Ray-Coquard, I., Reuss, A., Pignata, S., Colombo, N., et al. (2016). Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 17, 78–89. doi: 10.1016/s1470-2045(15)00366-6
Dudek, A. M., Martin, S., Garg, A. D., and Agostinis, P. (2013). Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front. Immunol. 4:438. doi: 10.3389/fimmu.2013.00438
Eckert, M. A., Coscia, F., Chryplewicz, A., Chang, J. W., Hernandez, K. M., Pan, S., et al. (2019). Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728. doi: 10.1038/s41586-019-1173-8
Egen, J. G., Kuhns, M. S., and Allison, J. P. (2002). CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 3, 611–618. doi: 10.1038/ni0702-611
Erdogan, B., and Webb, D. J. (2017). Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 45, 229–236. doi: 10.1042/bst20160387
Eskander, R. N., and Randall, L. M. (2011). Bevacizumab in the treatment of ovarian cancer. Biologics 5, 1–5. doi: 10.2147/btt.S13071
Fabregat, I., Fernando, J., Mainez, J., and Sancho, P. (2014). TGF-beta signaling in cancer treatment. Curr. Pharm. Des. 20, 2934–2947.
Facciabene, A., Motz, G. T., and Coukos, G. (2012). T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 72, 2162–2171. doi: 10.1158/0008-5472.Can-11-3687
Fagiani, E., and Christofori, G. (2013). Angiopoietins in angiogenesis. Cancer Lett. 328, 18–26. doi: 10.1016/j.canlet.2012.08.018
Fallarino, F., Grohmann, U., Hwang, K. W., Orabona, C., Vacca, C., Bianchi, R., et al. (2003). Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212. doi: 10.1038/ni1003
Ferrara, N., and Adamis, A. P. (2016). Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 15, 385–403. doi: 10.1038/nrd.2015.17
Finkernagel, F., Reinartz, S., Lieber, S., Adhikary, T., Wortmann, A., Hoffmann, N., et al. (2016). The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget 7, 75339–75352. doi: 10.18632/oncotarget.12180
Finkernagel, F., Reinartz, S., Schuldner, M., Malz, A., Jansen, J. M., Wagner, U., et al. (2019). Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 9, 6601–6617. doi: 10.7150/thno.37549
Froeling, F. E., Feig, C., Chelala, C., Dobson, R., Mein, C. E., Tuveson, D. A., et al. (2011). Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-(β-catenin signaling to slow tumor progression. Gastroenterology 141, 1486-1497, 1497.e1481-1414. doi: 10.1053/j.gastro.2011.06.047
Frydenlund, N., and Mahalingam, M. (2017). PD-L1 and immune escape: insights from melanoma and other lineage-unrelated malignancies. Hum. Pathol. 66, 13–33. doi: 10.1016/j.humpath.2017.06.012
Fujiwara, Y., Komohara, Y., Kudo, R., Tsurushima, K., Ohnishi, K., Ikeda, T., et al. (2011). Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol. Rep. 26, 1533–1537. doi: 10.3892/or.2011.1454
Furtek, S. L., Backos, D. S., Matheson, C. J., and Reigan, P. (2016). Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 11, 308–318. doi: 10.1021/acschembio.5b00945
Gabrilovich, D. I. (2017). Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 5, 3–8. doi: 10.1158/2326-6066.Cir-16-0297
Gabrilovich, D. I., Ostrand-Rosenberg, S., and Bronte, V. (2012). Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268. doi: 10.1038/nri3175
Galdiero, M. R., Garlanda, C., Jaillon, S., Marone, G., and Mantovani, A. (2013). Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 228, 1404–1412. doi: 10.1002/jcp.24260
Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pagès, C., et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. doi: 10.1126/science.1129139
Gao, Y., Shan, N., Zhao, C., Wang, Y., Xu, F., Li, J., et al. (2015). LY2109761 enhances cisplatin antitumor activity in ovarian cancer cells. Int. J. Clin. Exp. Pathol. 8, 4923–4932.
Garcia, A. A., Hirte, H., Fleming, G., Yang, D., Tsao-Wei, D. D., Roman, L., et al. (2008). Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California. Chicago, and Princess Margaret Hospital phase II consortia. J. Clin. Oncol. 26, 76–82. doi: 10.1200/jco.2007.12.1939
Geller, M. A., Cooley, S., Argenta, P. A., Downs, L. S., Carson, L. F., Judson, P. L., et al. (2010). Toll-like receptor-7 agonist administered subcutaneously in a prolonged dosing schedule in heavily pretreated recurrent breast, ovarian, and cervix cancers. Cancer Immunol. Immunother. 59, 1877–1884. doi: 10.1007/s00262-010-0914-1
Ghosn, E. E., Cassado, A. A., Govoni, G. R., Fukuhara, T., Yang, Y., Monack, D. M., et al. (2010). Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. U.S.A. 107, 2568–2573. doi: 10.1073/pnas.0915000107
Givel, A. M., Kieffer, Y., Scholer-Dahirel, A., Sirven, P., Cardon, M., Pelon, F., et al. (2018). miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 9:1056. doi: 10.1038/s41467-018-03348-z
Gok Yavuz, B., Gunaydin, G., Gedik, M. E., Kosemehmetoglu, K., Karakoc, D., Ozgur, F., et al. (2019). Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1((+) TAMs. Sci. Rep. 9:3172. doi: 10.1038/s41598-019-39553-z
Goossens, P., Rodriguez-Vita, J., Etzerodt, A., Masse, M., Rastoin, O., Gouirand, V., et al. (2019). Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab 29, 1376-1389.e1374. doi: 10.1016/j.cmet.2019.02.016
Gordon, S. (2003). Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35. doi: 10.1038/nri978
Gordon, S. R., Maute, R. L., Dulken, B. W., Hutter, G., George, B. M., McCracken, M. N., et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. doi: 10.1038/nature22396
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Gupta, V., Yull, F., and Khabele, D. (2018). Bipolar tumor-associated macrophages in ovarian cancer as targets for therapy. Cancers 10:366. doi: 10.3390/cancers10100366
Hagberg, C. E., Falkevall, A., Wang, X., Larsson, E., Huusko, J., Nilsson, I., et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. doi: 10.1038/nature08945
Hagemann, T., Biswas, S. K., Lawrence, T., Sica, A., and Lewis, C. E. (2009). Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood 113, 3139–3146. doi: 10.1182/blood-2008-12-172825
Hamanishi, J., Mandai, M., Ikeda, T., Minami, M., Kawaguchi, A., Murayama, T., et al. (2015). Safety and Antitumor Activity of Anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 33, 4015–4022. doi: 10.1200/jco.2015.62.3397
Hamanishi, J., Mandai, M., Iwasaki, M., Okazaki, T., Tanaka, Y., Yamaguchi, K., et al. (2007). Programmed cell death 1 ligand 1 and tumor-infiltrating CD8(+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 3360–3365. doi: 10.1073/pnas.0611533104
Hamanishi, J., Mandai, M., and Konishi, I. (2016). Immune checkpoint inhibition in ovarian cancer. Int. Immunol. 28, 339–348. doi: 10.1093/intimm/dxw020
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hansen, J. M., Coleman, R. L., and Sood, A. K. (2016). Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 56, 131–143. doi: 10.1016/j.ejca.2015.12.016
Hanson, E. M., Clements, V. K., Sinha, P., Ilkovitch, D., and Ostrand-Rosenberg, S. (2009). Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4(+ and CD8(+ T cells. J. Immunol. 183, 937–944. doi: 10.4049/jimmunol.0804253
Heery, C. R., O’Sullivan-Coyne, G., Madan, R. A., Cordes, L., Rajan, A., Rauckhorst, M., et al. (2017). Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 18, 587–598. doi: 10.1016/s1470-2045(17)30239-5
Hennessy, B. T., Coleman, R. L., and Markman, M. (2009). Ovarian cancer. Lancet 374, 1371–1382. doi: 10.1016/s0140-6736(09)61338-6
Hernandez-Fernaud, J. R., Ruengeler, E., Casazza, A., Neilson, L. J., Pulleine, E., Santi, A., et al. (2017). Secreted CLIC3 drives cancer progression through its glutathione-dependent oxidoreductase activity. Nat. Commun. 8:14206. doi: 10.1038/ncomms14206
Hironaka, S. (2019). Anti-angiogenic therapies for gastric cancer. Asia Pac. J. Clin. Oncol. 15, 208–217. doi: 10.1111/ajco.13174
Hodi, F. S., Butler, M., Oble, D. A., Seiden, M. V., Haluska, F. G., Kruse, A., et al. (2008). Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. U.S.A. 105, 3005–3010. doi: 10.1073/pnas.0712237105
Hodi, F. S., Mihm, M. C., Soiffer, R. J., Haluska, F. G., Butler, M., Seiden, M. V., et al. (2003). Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. U.S.A. 100, 4712–4717. doi: 10.1073/pnas.0830997100
Hu, L., Hofmann, J., Zaloudek, C., Ferrara, N., Hamilton, T., and Jaffe, R. B. (2002). Vascular endothelial growth factor immunoneutralization plus Paclitaxel markedly reduces tumor burden and ascites in athymic mouse model of ovarian cancer. Am. J. Pathol. 161, 1917–1924. doi: 10.1016/s0002-9440(10)64467-7
Huang, B., Lei, Z., Zhao, J., Gong, W., Liu, J., Chen, Z., et al. (2007). CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 252, 86–92. doi: 10.1016/j.canlet.2006.12.012
Hume, D. A., and MacDonald, K. P. (2012). Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119, 1810–1820. doi: 10.1182/blood-2011-09-379214
Hussain, A., Voisin, V., Poon, S., Karamboulas, C., Bui, N. H. B., Meens, J., et al. (2020). Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J. Exp. Med. 217:e20191094. doi: 10.1084/jem.20191094
Huynh, H., Teo, C. C., and Soo, K. C. (2007). Bevacizumab and rapamycin inhibit tumor growth in peritoneal model of human ovarian cancer. Mol. Cancer Ther. 6, 2959–2966. doi: 10.1158/1535-7163.Mct-07-0237
Ishii, G., Ochiai, A., and Neri, S. (2016). Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 99(Pt B), 186–196. doi: 10.1016/j.addr.2015.07.007
Jayson, G. C., Kohn, E. C., Kitchener, H. C., and Ledermann, J. A. (2014). Ovarian cancer. Lancet 384, 1376–1388. doi: 10.1016/s0140-6736(13)62146-7
Jiang, T., Zhuang, J., Duan, H., Luo, Y., Zeng, Q., Fan, K., et al. (2012). CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 120, 2330–2339. doi: 10.1182/blood-2012-01-406108
Jiang, X., Wang, J., Deng, X., Xiong, F., Ge, J., Xiang, B., et al. (2019). Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 18, 10. doi: 10.1186/s12943-018-0928-4
Jiménez-Sánchez, A., Cybulska, P., Mager, K. L., Koplev, S., Cast, O., Couturier, D. L., et al. (2020). Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 52, 582–593. doi: 10.1038/s41588-020-0630-5
Jiménez-Sánchez, A., Memon, D., Pourpe, S., Veeraraghavan, H., Li, Y., Vargas, H. A., et al. (2017). Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 170, 927-938.e920. doi: 10.1016/j.cell.2017.07.025
Jost, P. J., and Ruland, J. (2007). Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood 109, 2700–2707. doi: 10.1182/blood-2006-07-025809
Joyce, J. A., and Pollard, J. W. (2009). Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9, 239–252. doi: 10.1038/nrc2618
Kalluri, R., and Zeisberg, M. (2006). Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. doi: 10.1038/nrc1877
Kang, Y., Hu, W., Ivan, C., Dalton, H. J., Miyake, T., Pecot, C. V., et al. (2013). Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 105, 1485–1495. doi: 10.1093/jnci/djt210
Kasikova, L., Hensler, M., Truxova, I., Skapa, P., Laco, J., Belicova, L., et al. (2019). Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 7:312. doi: 10.1186/s40425-019-0781-z
Ke, X., Zhang, S., Wu, M., Lou, J., Zhang, J., Xu, T., et al. (2016). Tumor-associated macrophages promote invasion via Toll-like receptors signaling in patients with ovarian cancer. Int. Immunopharmacol. 40, 184–195. doi: 10.1016/j.intimp.2016.08.029
Kim, J., and Bae, J. S. (2016). Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016:6058147. doi: 10.1155/2016/6058147
Kinter, A. L., Godbout, E. J., McNally, J. P., Sereti, I., Roby, G. A., O’Shea, M. A., et al. (2008). The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 181, 6738–6746. doi: 10.4049/jimmunol.181.10.6738
Kishton, R. J., Sukumar, M., and Restifo, N. P. (2017). Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109. doi: 10.1016/j.cmet.2017.06.016
Klimp, A. H., Hollema, H., Kempinga, C., van der Zee, A. G., de Vries, E. G., and Daemen, T. (2001). Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res. 61, 7305–7309.
Kobayashi, Y., Kashima, H., Rahmanto, Y. S., Banno, K., Yu, Y., Matoba, Y., et al. (2017). Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget 8, 72147–72156. doi: 10.18632/oncotarget.20046
Kohlhapp, F. J., Mitra, A. K., Lengyel, E., and Peter, M. E. (2015). MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34, 5857–5868. doi: 10.1038/onc.2015.89
Krempski, J., Karyampudi, L., Behrens, M. D., Erskine, C. L., Hartmann, L., Dong, H., et al. (2011). Tumor-infiltrating programmed death receptor-1(+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 186, 6905–6913. doi: 10.4049/jimmunol.1100274
Labidi-Galy, S. I., Sisirak, V., Meeus, P., Gobert, M., Treilleux, I., Bajard, A., et al. (2011). Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 71, 5423–5434. doi: 10.1158/0008-5472.Can-11-0367
Lan, C., Huang, X., Lin, S., Huang, H., Cai, Q., Wan, T., et al. (2013). Expression of M2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol. Cancer Res. Treat. 12, 259–267. doi: 10.7785/tcrt.2012.500312
Laudisi, F., Cherubini, F., Monteleone, G., and Stolfi, C. (2018). STAT3 interactors as potential therapeutic targets for cancer treatment. Int. J. Mol. Sci. 19:1787. doi: 10.3390/ijms19061787
Ledermann, J. A., Embleton, A. C., Raja, F., Perren, T. J., Jayson, G. C., Rustin, G. J. S., et al. (2016). Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 387, 1066–1074. doi: 10.1016/s0140-6736(15)01167-8
Ledermann, J. A., Raja, F. A., Fotopoulou, C., Gonzalez-Martin, A., Colombo, N., and Sessa, C. (2013). Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 24, (Suppl. 6), vi24–vi32. doi: 10.1093/annonc/mdt333
Leyva-Illades, D., McMillin, M., Quinn, M., and Demorrow, S. (2012). Cholangiocarcinoma pathogenesis: role of the tumor microenvironment. Transl. Gastrointest. Cancer 1, 71–80.
Li, H., Fan, X., and Houghton, J. (2007). Tumor microenvironment: the role of the tumor stroma in cancer. J. Cell. Biochem. 101, 805–815. doi: 10.1002/jcb.21159
Lim, H. J., and Ledger, W. (2016). Targeted therapy in ovarian cancer. Womens Health 12, 363–378. doi: 10.2217/whe.16.4
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N., and Muschel, R. J. (2016). Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710. doi: 10.18632/oncotarget.7376
Lin, H., Wei, S., Hurt, E. M., Green, M. D., Zhao, L., Vatan, L., et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 128, 805–815. doi: 10.1172/jci96113
Lin, Z., Liu, Y., Sun, Y., and He, X. (2011). Expression of Ets-1. Ang-2 and maspin in ovarian cancer and their role in tumor angiogenesis. J. Exp. Clin. Cancer Res. 30, 31. doi: 10.1186/1756-9966-30-31
Lindau, D., Gielen, P., Kroesen, M., Wesseling, P., and Adema, G. J. (2013). The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138, 105–115. doi: 10.1111/imm.12036
Low-Marchelli, J. M., Ardi, V. C., Vizcarra, E. A., van Rooijen, N., Quigley, J. P., and Yang, J. (2013). Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 73, 662–671. doi: 10.1158/0008-5472.Can-12-0653
Lu, X. (2017). Impact of IL-12 in Cancer. Curr. Cancer Drug. Targets 17, 682–697. doi: 10.2174/1568009617666170427102729
Lu, X., and Meng, T. (2019). Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology 224, 355–361. doi: 10.1016/j.imbio.2019.03.002
Lundgren, S., Berntsson, J., Nodin, B., Micke, P., and Jirstrom, K. (2016). Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J. Ovarian Res. 9:21. doi: 10.1186/s13048-016-0232-0
Luwor, R. B., Stylli, S. S., and Kaye, A. H. (2013). The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. 20, 907–911. doi: 10.1016/j.jocn.2013.03.006
Mabuchi, S., Terai, Y., Morishige, K., Tanabe-Kimura, A., Sasaki, H., Kanemura, M., et al. (2008). Maintenance treatment with bevacizumab prolongs survival in an in vivo ovarian cancer model. Clin. Cancer Res. 14, 7781–7789. doi: 10.1158/1078-0432.Ccr-08-0243
MacGregor, H. L., Garcia-Batres, C., Sayad, A., Elia, A., Berman, H. K., Toker, A., et al. (2019a). Tumor cell expression of B7-H4 correlates with higher frequencies of tumor-infiltrating APCs and higher CXCL17 expression in human epithelial ovarian cancer. Oncoimmunology 8:e1665460. doi: 10.1080/2162402x.2019.1665460
MacGregor, H. L., Sayad, A., Elia, A., Wang, B. X., Katz, S. R., Shaw, P. A., et al. (2019b). High expression of B7-H3 on stromal cells defines tumor and stromal compartments in epithelial ovarian cancer and is associated with limited immune activation. J. Immunother. Cancer 7:357. doi: 10.1186/s40425-019-0816-5
MacKie, R. M., Reid, R., and Junor, B. (2003). Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N. Engl. J. Med. 348, 567–568. doi: 10.1056/nejm200302063480620
Mak, M. P., Tong, P., Diao, L., Cardnell, R. J., Gibbons, D. L., William, W. N., et al. (2016). A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 22, 609–620. doi: 10.1158/1078-0432.Ccr-15-0876
Mancino, A., and Lawrence, T. (2010). Nuclear factor-kappaB and tumor-associated macrophages. Clin. Cancer Res. 16, 784–789. doi: 10.1158/1078-0432.Ccr-09-1015
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Mantovani, A., Sozzani, S., Locati, M., Allavena, P., and Sica, A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555. doi: 10.1016/s1471-4906(02)02302-5
Matei, D., Chang, D. D., and Jeng, M. H. (2004). Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and Akt inactivation. Clin. Cancer Res. 10, 681–690.
Matei, D., Emerson, R. E., Schilder, J., Menning, N., Baldridge, L. A., Johnson, C. S., et al. (2008). Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Cancer 113, 723–732. doi: 10.1002/cncr.23605
Matei, D., Sill, M. W., Lankes, H. A., DeGeest, K., Bristow, R. E., Mutch, D., et al. (2011). Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: a gynecologic oncology group trial. J. Clin. Oncol. 29, 69–75. doi: 10.1200/jco.2009.26.7856
McCann, C. K., Growdon, W. B., Kulkarni-Datar, K., Curley, M. D., Friel, A. M., Proctor, J. L., et al. (2011). Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PLoS One 6:e28077. doi: 10.1371/journal.pone.0028077
McDermott, D. F., and Atkins, M. B. (2013). PD-1 as a potential target in cancer therapy. Cancer Med. 2, 662–673. doi: 10.1002/cam4.106
Mhawech-Fauceglia, P., Yan, L., Sharifian, M., Ren, X., Liu, S., Kim, G., et al. (2015). Stromal Expression of Fibroblast Activation Protein Alpha (FAP) Predicts Platinum Resistance and Shorter Recurrence in patients with Epithelial Ovarian Cancer. Cancer Microenviron. 8, 23–31. doi: 10.1007/s12307-014-0153-7
Mimura, K., Teh, J. L., Okayama, H., Shiraishi, K., Kua, L. F., Koh, V., et al. (2018). PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 109, 43–53. doi: 10.1111/cas.13424
Mittica, G., Genta, S., Aglietta, M., and Valabrega, G. (2016). Immune checkpoint inhibitors: a new opportunity in the treatment of ovarian cancer? Int. J. Mol. Sci. 17:1169. doi: 10.3390/ijms17071169
Moisan, F., Francisco, E. B., Brozovic, A., Duran, G. E., Wang, Y. C., Chaturvedi, S., et al. (2014). Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 8, 1231–1239. doi: 10.1016/j.molonc.2014.03.016
Molon, B., Ugel, S., Del Pozzo, F., Soldani, C., Zilio, S., Avella, D., et al. (2011). Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 208, 1949–1962. doi: 10.1084/jem.20101956
Monk, B. J., Han, E., Josephs-Cowan, C. A., Pugmire, G., and Burger, R. A. (2006). Salvage bevacizumab (rhuMAB VEGF)-based therapy after multiple prior cytotoxic regimens in advanced refractory epithelial ovarian cancer. Gynecol. Oncol. 102, 140–144. doi: 10.1016/j.ygyno.2006.05.006
Monk, B. J., Poveda, A., Vergote, I., Raspagliesi, F., Fujiwara, K., Bae, D. S., et al. (2014). Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): a randomised, multicentre, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 15, 799–808. doi: 10.1016/s1470-2045(14)70244-x
Moughon, D. L., He, H., Schokrpur, S., Jiang, Z. K., Yaqoob, M., David, J., et al. (2015). Macrophage blockade using CSF1R inhibitors reverses the vascular leakage underlying malignant ascites in late-stage epithelial ovarian cancer. Cancer Res. 75, 4742–4752. doi: 10.1158/0008-5472.Can-14-3373
Moy, J. D., Moskovitz, J. M., and Ferris, R. L. (2017). Biological mechanisms of immune escape and implications for immunotherapy in head and neck squamous cell carcinoma. Eur. J. Cancer 76, 152–166. doi: 10.1016/j.ejca.2016.12.035
Munn, D. H., and Mellor, A. L. (2016). IDO in the tumor microenvironment: inflammation. counter-regulation, and tolerance. Trends Immunol. 37, 193–207. doi: 10.1016/j.it.2016.01.002
Munn, D. H., Sharma, M. D., Baban, B., Harding, H. P., Zhang, Y., Ron, D., et al. (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642. doi: 10.1016/j.immuni.2005.03.013
Ngambenjawong, C., Gustafson, H. H., and Pun, S. H. (2017). Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 114, 206–221. doi: 10.1016/j.addr.2017.04.010
Noy, R., and Pollard, J. W. (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61. doi: 10.1016/j.immuni.2014.06.010
Okabe, Y., and Medzhitov, R. (2014). Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844. doi: 10.1016/j.cell.2014.04.016
Orimo, A., Gupta, P. B., Sgroi, D. C., Arenzana-Seisdedos, F., Delaunay, T., Naeem, R., et al. (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348. doi: 10.1016/j.cell.2005.02.034
Ostrand-Rosenberg, S., and Fenselau, C. (2018). Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 200, 422–431. doi: 10.4049/jimmunol.1701019
Ostuni, R., Kratochvill, F., Murray, P. J., and Natoli, G. (2015). Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 36, 229–239. doi: 10.1016/j.it.2015.02.004
Oza, A. M., Cook, A. D., Pfisterer, J., Embleton, A., Ledermann, J. A., Pujade-Lauraine, E., et al. (2015). Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 16, 928–936. doi: 10.1016/s1470-2045(15)00086-8
Pasare, C., and Medzhitov, R. (2003). Toll pathway-dependent blockade of CD4(+CD25(+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036. doi: 10.1126/science.1078231
Patsoukis, N., Bardhan, K., Weaver, J., Herbel, C., Seth, P., Li, L., et al. (2016). The role of metabolic reprogramming in T cell fate and function. Curr. Trends Immunol. 17, 1–12.
Pearce, O. M. T., Delaine-Smith, R. M., Maniati, E., Nichols, S., Wang, J., Böhm, S., et al. (2018). Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov. 8, 304–319. doi: 10.1158/2159-8290.Cd-17-0284
Perren, T. J., Swart, A. M., Pfisterer, J., Ledermann, J. A., Pujade-Lauraine, E., Kristensen, G., et al. (2011). A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 365, 2484–2496. doi: 10.1056/NEJMoa1103799
Pignata, S., Lorusso, D., Scambia, G., Sambataro, D., Tamberi, S., Cinieri, S., et al. (2015). Pazopanib plus weekly paclitaxel versus weekly paclitaxel alone for platinum-resistant or platinum-refractory advanced ovarian cancer (MITO 11): a randomised, open-label, phase 2 trial. Lancet Oncol. 16, 561–568. doi: 10.1016/s1470-2045(15)70115-4
Pollard, J. W. (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78. doi: 10.1038/nrc1256
Pramanik, K. C., Makena, M. R., Bhowmick, K., and Pandey, M. K. (2018). Advancement of NF-kappaB signaling pathway: a novel target in pancreatic cancer. Int. J. Mol. Sci. 19:3890. doi: 10.3390/ijms19123890
Pujade-Lauraine, E., Hilpert, F., Weber, B., Reuss, A., Poveda, A., Kristensen, G., et al. (2014). Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J. Clin. Oncol. 32, 1302–1308. doi: 10.1200/jco.2013.51.4489
Puré, E., and Blomberg, R. (2018). Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene 37, 4343–4357. doi: 10.1038/s41388-018-0275-3
Qian, B. Z., and Pollard, J. W. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. doi: 10.1016/j.cell.2010.03.014
Qian, F., Villella, J., Wallace, P. K., Mhawech-Fauceglia, P., Tario, J. D. Jr., Andrews, C., et al. (2009). Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2,3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. 69, 5498–5504. doi: 10.1158/0008-5472.Can-08-2106
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Ramanathan, S., and Jagannathan, N. (2014). Tumor associated macrophage: a review on the phenotypes, traits and functions. Iran. J. Cancer Prev. 7, 1–8.
Rath, K. S., Naidu, S. K., Lata, P., Bid, H. K., Rivera, B. K., McCann, G. A., et al. (2014). HO-3867, a safe STAT3 inhibitor, is selectively cytotoxic to ovarian cancer. Cancer Res. 74, 2316–2327. doi: 10.1158/0008-5472.Can-13-2433
Reinartz, S., Schumann, T., Finkernagel, F., Wortmann, A., Jansen, J. M., Meissner, W., et al. (2014). Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 134, 32–42. doi: 10.1002/ijc.28335
Rekik, R., Belhadj Hmida, N., Ben Hmid, A., Zamali, I., Kammoun, N., and Ben Ahmed, M. (2015). PD-1 induction through TCR activation is partially regulated by endogenous TGF-beta. Cell Mol. Immunol. 12, 648–649. doi: 10.1038/cmi.2014.104
Ren, F., Fan, M., Mei, J., Wu, Y., Liu, C., Pu, Q., et al. (2014). Interferon-gamma and celecoxib inhibit lung-tumor growth through modulating M2/M1 macrophage ratio in the tumor microenvironment. Drug Des. Devel. Ther. 8, 1527–1538. doi: 10.2147/dddt.S66302
Riboldi, E., Musso, T., Moroni, E., Urbinati, C., Bernasconi, S., Rusnati, M., et al. (2005). Cutting edge: proangiogenic properties of alternatively activated dendritic cells. J. Immunol. 175, 2788–2792. doi: 10.4049/jimmunol.175.5.2788
Ries, C. H., Hoves, S., Cannarile, M. A., and Ruttinger, D. (2015). CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr. Opin. Pharmacol. 23, 45–51. doi: 10.1016/j.coph.2015.05.008
Rodriguez, P. C., Quiceno, D. G., Zabaleta, J., Ortiz, B., Zea, A. H., Piazuelo, M. B., et al. (2004). Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 64, 5839–5849. doi: 10.1158/0008-5472.Can-04-0465
Rossmann, P., Hornych, A., and Englis, M. (1967). [Histology and ultrastructure of crystalloid inclusions in the podocytes in paraproteinaemia]. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 344, 151–158.
Roy, A., and Li, S. D. (2016). Modifying the tumor microenvironment using nanoparticle therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 8, 891–908. doi: 10.1002/wnan.1406
Ruffell, B., Affara, N. I., and Coussens, L. M. (2012). Differential macrophage programming in the tumor microenvironment. Trends Immunol. 33, 119–126. doi: 10.1016/j.it.2011.12.001
Sabado, R. L., Balan, S., and Bhardwaj, N. (2017). Dendritic cell-based immunotherapy. Cell Res. 27, 74–95. doi: 10.1038/cr.2016.157
Sadelain, M., Riviere, I., and Riddell, S. (2017). Therapeutic T cell engineering. Nature 545, 423–431. doi: 10.1038/nature22395
Saini, U., Naidu, S., ElNaggar, A. C., Bid, H. K., Wallbillich, J. J., Bixel, K., et al. (2017). Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: a potential therapeutic target. Oncogene 36, 168–181. doi: 10.1038/onc.2016.197
Sakuishi, K., Jayaraman, P., Behar, S. M., Anderson, A. C., and Kuchroo, V. K. (2011). Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 32, 345–349. doi: 10.1016/j.it.2011.05.003
Sallinen, H., Heikura, T., Koponen, J., Kosma, V. M., Heinonen, S., Yla-Herttuala, S., et al. (2014). Serum angiopoietin-2 and soluble VEGFR-2 levels predict malignancy of ovarian neoplasm and poor prognosis in epithelial ovarian cancer. BMC Cancer 14:696. doi: 10.1186/1471-2407-14-696
Sallinen, H., Heikura, T., Laidinen, S., Kosma, V. M., Heinonen, S., Yla-Herttuala, S., et al. (2010). Preoperative angiopoietin-2 serum levels: a marker of malignant potential in ovarian neoplasms and poor prognosis in epithelial ovarian cancer. Int. J. Gynecol. Cancer 20, 1498–1505. doi: 10.1111/IGC.0b013e3181f936e3
Sandhu, S. K., Papadopoulos, K., Fong, P. C., Patnaik, A., Messiou, C., Olmos, D., et al. (2013). A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 71, 1041–1050. doi: 10.1007/s00280-013-2099-8
Santos, A. M., Jung, J., Aziz, N., Kissil, J. L., and Puré, E. (2009). Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 119, 3613–3625. doi: 10.1172/jci38988
Sato, E., Olson, S. H., Ahn, J., Bundy, B., Nishikawa, H., Qian, F., et al. (2005). Intraepithelial CD8(+ tumor-infiltrating lymphocytes and a high CD8(+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 18538–18543. doi: 10.1073/pnas.0509182102
Scarlett, U. K., and Conejo-Garcia, J. R. (2012). Modulating the tumor immune microenvironment as an ovarian cancer treatment strategy. Expert Rev. Obstet. Gynecol. 7, 413–419. doi: 10.1586/eog.12.41
Scarlett, U. K., Rutkowski, M. R., Rauwerdink, A. M., Fields, J., Escovar-Fadul, X., Baird, J., et al. (2012). Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 209, 495–506. doi: 10.1084/jem.20111413
Selvendiran, K., Tong, L., Bratasz, A., Kuppusamy, M. L., Ahmed, S., Ravi, Y., et al. (2010). Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 9, 1169–1179. doi: 10.1158/1535-7163.Mct-09-1207
Semenza, G. L. (2000). HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 88, 1474–1480. doi: 10.1152/jappl.2000.88.4.1474
Shapouri-Moghaddam, A., Mohammadian, S., Vazini, H., Taghadosi, M., Esmaeili, S. A., Mardani, F., et al. (2018). Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440. doi: 10.1002/jcp.26429
Sharma, P., and Allison, J. P. (2015). The future of immune checkpoint therapy. Science 348, 56–61. doi: 10.1126/science.aaa8172
Shen, W., Li, H. L., Liu, L., and Cheng, J. X. (2017). Expression levels of PTEN. HIF-1alpha, and VEGF as prognostic factors in ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 21, 2596–2603.
Sica, A. (2010). Role of tumour-associated macrophages in cancer-related inflammation. Exp. Oncol. 32, 153–158.
Sica, A., Allavena, P., and Mantovani, A. (2008). Cancer related inflammation: the macrophage connection. Cancer Lett. 267, 204–215. doi: 10.1016/j.canlet.2008.03.028
Sica, A., and Bronte, V. (2007). Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 117, 1155–1166. doi: 10.1172/jci31422
Silver, D. F., Hempling, R. E., Piver, M. S., and Repasky, E. A. (1999). Effects of IL-12 on human ovarian tumors engrafted into SCID mice. Gynecol. Oncol. 72, 154–160. doi: 10.1006/gyno.1998.5239
Singhal, S., Stadanlick, J., Annunziata, M. J., Rao, A. S., Bhojnagarwala, P. S., O’Brien, S., et al. (2019). Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci. Transl. Med. 11:eaat1500. doi: 10.1126/scitranslmed.aat1500
Sjoberg, E., Augsten, M., Bergh, J., Jirstrom, K., and Ostman, A. (2016). Expression of the chemokine CXCL14 in the tumour stroma is an independent marker of survival in breast cancer. Br. J. Cancer 114, 1117–1124. doi: 10.1038/bjc.2016.104
Smith-Garvin, J. E., Koretzky, G. A., and Jordan, M. S. (2009). T cell activation. Annu. Rev. Immunol. 27, 591–619. doi: 10.1146/annurev.immunol.021908.132706
Sopo, M., Anttila, M., Hamalainen, K., Kivela, A., Yla-Herttuala, S., Kosma, V. M., et al. (2019). Expression profiles of VEGF-A. VEGF-D and VEGFR1 are higher in distant metastases than in matched primary high grade epithelial ovarian cancer. BMC Cancer 19:584. doi: 10.1186/s12885-019-5757-3
Srivastava, M. K., Sinha, P., Clements, V. K., Rodriguez, P., and Ostrand-Rosenberg, S. (2010). Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 70, 68–77. doi: 10.1158/0008-5472.Can-09-2587
Stout, R. D., Jiang, C., Matta, B., Tietzel, I., Watkins, S. K., and Suttles, J. (2005). Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 175, 342–349. doi: 10.4049/jimmunol.175.1.342
Tammela, T., and Alitalo, K. (2010). Lymphangiogenesis: Molecular mechanisms and future promise. Cell 140, 460–476. doi: 10.1016/j.cell.2010.01.045
Tang, C. H., and Tsai, C. C. (2012). CCL2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-kappaB signaling pathway. Biochem. Pharmacol. 83, 335–344. doi: 10.1016/j.bcp.2011.11.013
Tang, M., Diao, J., and Cattral, M. S. (2017). Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell Mol. Life. Sci. 74, 761–776. doi: 10.1007/s00018-016-2317-8
Tang, X., Mo, C., Wang, Y., Wei, D., and Xiao, H. (2013). Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 138, 93–104. doi: 10.1111/imm.12023
Tang, Y. J., Sun, Z. L., Wu, W. G., Xing, J., He, Y. F., Xin, D. M., et al. (2015). Inhibitor of signal transducer and activator of transcription 3 (STAT3) suppresses ovarian cancer growth, migration and invasion and enhances the effect of cisplatin in vitro. Genet. Mol. Res. 14, 2450–2460. doi: 10.4238/2015.March.30.3
Tanizaki, Y., Kobayashi, A., Toujima, S., Shiro, M., Mizoguchi, M., Mabuchi, Y., et al. (2014). Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 105, 966–973. doi: 10.1111/cas.12445
Tarhini, A. A., and Iqbal, F. (2010). CTLA-4 blockade: therapeutic potential in cancer treatments. Oncol. Targets Ther. 3, 15–25.
Terawaki, S., Chikuma, S., Shibayama, S., Hayashi, T., Yoshida, T., Okazaki, T., et al. (2011). IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 186, 2772–2779. doi: 10.4049/jimmunol.1003208
Tierney, B. J., McCann, G. A., Cohn, D. E., Eisenhauer, E., Sudhakar, M., Kuppusamy, P., et al. (2012). HO-3867, a STAT3 inhibitor induces apoptosis by inactivation of STAT3 activity in BRCA1-mutated ovarian cancer cells. Cancer Biol. Ther. 13, 766–775. doi: 10.4161/cbt.20559
Timmerman, J. M., and Levy, R. (1999). Dendritic cell vaccines for cancer immunotherapy. Annu. Rev. Med. 50, 507–529. doi: 10.1146/annurev.med.50.1.507
Truffi, M., Sorrentino, L., and Corsi, F. (2020). Fibroblasts in the Tumor Microenvironment. Adv. Exp. Med. Biol. 1234, 15–29. doi: 10.1007/978-3-030-37184-5_2
Truxova, I., Kasikova, L., Hensler, M., Skapa, P., Laco, J., Pecen, L., et al. (2018). Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J. Immunother. Cancer 6:139. doi: 10.1186/s40425-018-0446-3
Tse, V., Xu, L., Yung, Y. C., Santarelli, J. G., Juan, D., Fabel, K., et al. (2003). The temporal-spatial expression of VEGF, angiopoietins-1 and 2, and Tie-2 during tumor angiogenesis and their functional correlation with tumor neovascular architecture. Neurol. Res. 25, 729–738. doi: 10.1179/016164103101202084
Ueno, T., Toi, M., Saji, H., Muta, M., Bando, H., Kuroi, K., et al. (2000). Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 6, 3282–3289.
van Dalen, F. J., van Stevendaal, M., Fennemann, F. L., Verdoes, M., and Ilina, O. (2018). Molecular repolarisation of tumour-associated macrophages. Molecules 24:9. doi: 10.3390/molecules24010009
Varga, A., Piha-Paul, S., Ott, P. A., Mehnert, J. M., Berton-Rigaud, D., Morosky, A., et al. (2019). Pembrolizumab in patients with programmed death ligand 1-positive advanced ovarian cancer: analysis of KEYNOTE-028. Gynecol. Oncol. 152, 243–250. doi: 10.1016/j.ygyno.2018.11.017
Vignali, D. A., Collison, L. W., and Workman, C. J. (2008). How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532. doi: 10.1038/nri2343
Vu Manh, T. P., Bertho, N., Hosmalin, A., Schwartz-Cornil, I., and Dalod, M. (2015). Investigating evolutionary conservation of dendritic cell subset identity and functions. Front. Immunol. 6:260. doi: 10.3389/fimmu.2015.00260
Wanderley, C. W., Colón, D. F., Luiz, J. P. M., Oliveira, F. F., Viacava, P. R., Leite, C. A., et al. (2018). Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 Profile in a TLR4-dependent manner. Cancer Res. 78, 5891–5900. doi: 10.1158/0008-5472.Can-17-3480
Wang, M., Zhao, J., Zhang, L., Wei, F., Lian, Y., Wu, Y., et al. (2017). Role of tumor microenvironment in tumorigenesis. J. Cancer 8, 761–773. doi: 10.7150/jca.17648
Wang, X., Yang, L., Huang, F., Zhang, Q., Liu, S., Ma, L., et al. (2017). Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 184, 7–14. doi: 10.1016/j.imlet.2017.02.006
Wang, Y., Shen, Y., Wang, S., Shen, Q., and Zhou, X. (2018). The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 415, 117–128. doi: 10.1016/j.canlet.2017.12.003
Wilhelm, S. M., Carter, C., Tang, L., Wilkie, D., McNabola, A., Rong, H., et al. (2004). BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109. doi: 10.1158/0008-5472.Can-04-1443
Wimberger, P., Chebouti, I., Kasimir-Bauer, S., Lachmann, R., Kuhlisch, E., Kimmig, R., et al. (2014). Explorative investigation of vascular endothelial growth factor receptor expression in primary ovarian cancer and its clinical relevance. Gynecol. Oncol. 133, 467–472. doi: 10.1016/j.ygyno.2014.03.574
Wyckoff, J., Wang, W., Lin, E. Y., Wang, Y., Pixley, F., Stanley, E. R., et al. (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022–7029. doi: 10.1158/0008-5472.Can-04-1449
Xue, J., Schmidt, S. V., Sander, J., Draffehn, A., Krebs, W., Quester, I., et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. doi: 10.1016/j.immuni.2014.01.006
Yamamura, S., Matsumura, N., Mandai, M., Huang, Z., Oura, T., Baba, T., et al. (2012). The activated transforming growth factor-beta signaling pathway in peritoneal metastases is a potential therapeutic target in ovarian cancer. Int. J. Cancer 130, 20–28. doi: 10.1002/ijc.25961
Yan, X., Lin, Y., Yang, D., Shen, Y., Yuan, M., Zhang, Z., et al. (2003). A novel anti-CD146 monoclonal antibody. AA98, inhibits angiogenesis and tumor growth. Blood 102, 184–191. doi: 10.1182/blood-2002-04-1004
Yang, M., McKay, D., Pollard, J. W., and Lewis, C. E. (2018). Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 78, 5492–5503. doi: 10.1158/0008-5472.CAN-18-1367
Yang, S. Y. C., Lheureux, S., Karakasis, K., Burnier, J. V., Bruce, J. P., Clouthier, D. L., et al. (2018). Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 10:81. doi: 10.1186/s13073-018-0590-x
Yang, W., Han, W., Ye, S., Liu, D., Wu, J., Liu, H., et al. (2013). Fibroblast activation protein-alpha promotes ovarian cancer cell proliferation and invasion via extracellular and intracellular signaling mechanisms. Exp. Mol. Pathol. 95, 105–110. doi: 10.1016/j.yexmp.2013.06.007
Ye, Q., Song, D. G., Poussin, M., Yamamoto, T., Best, A., Li, C., et al. (2014). CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 20, 44–55. doi: 10.1158/1078-0432.Ccr-13-0945
Yin, M., Li, X., Tan, S., Zhou, H. J., Ji, W., Bellone, S., et al. (2016). Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Invest. 126, 4157–4173. doi: 10.1172/jci87252
Yoshikawa, T., Miyamoto, M., Aoyama, T., Soyama, H., Goto, T., Hirata, J., et al. (2018). JAK2/STAT3 pathway as a therapeutic target in ovarian cancers. Oncol. Lett. 15, 5772–5780. doi: 10.3892/ol.2018.8028
Yu, H., Lee, H., Herrmann, A., Buettner, R., and Jove, R. (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. doi: 10.1038/nrc3818
Yu, Y., Xiao, C. H., Tan, L. D., Wang, Q. S., Li, X. Q., and Feng, Y. M. (2014). Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 110, 724–732. doi: 10.1038/bjc.2013.768
Yu, R., Jin, H., Jin, C., Huang, X., Lin, J., and Teng, Y. (2018). Inhibition of the CSF-1 receptor sensitizes ovarian cancer cells to cisplatin. Cell Biochem. Funct. 36, 80–87. doi: 10.1002/cbf.3319
Yuan, H. T., Khankin, E. V., Karumanchi, S. A., and Parikh, S. M. (2009). Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol. Cell. Biol. 29, 2011–2022. doi: 10.1128/mcb.01472-08
Yuan, X., Zhang, J., Li, D., Mao, Y., Mo, F., Du, W., et al. (2017). Prognostic significance of tumor-associated macrophages in ovarian cancer: a meta-analysis. Gynecol. Oncol. 147, 181–187. doi: 10.1016/j.ygyno.2017.07.007
Zhang, A. W., McPherson, A., Milne, K., Kroeger, D. R., Hamilton, P. T., Miranda, A., et al. (2018). Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 173, 1755-1769.e1722. doi: 10.1016/j.cell.2018.03.073
Zhang, L., Conejo-Garcia, J. R., Katsaros, D., Gimotty, P. A., Massobrio, M., Regnani, G., et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348, 203–213. doi: 10.1056/NEJMoa020177
Zhang, M., He, Y., Sun, X., Li, Q., Wang, W., Zhao, A., et al. (2014). A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian. Res. 7:19. doi: 10.1186/1757-2215-7-19
Zhang, N., and Bevan, M. J. (2011). CD8((+) T cells: foot soldiers of the immune system. Immunity 35, 161–168. doi: 10.1016/j.immuni.2011.07.010
Zhang, X., Yue, P., Fletcher, S., Zhao, W., Gunning, P. T., and Turkson, J. (2010). A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 79, 1398–1409. doi: 10.1016/j.bcp.2010.01.001
Zhang, X., Zeng, Y., Qu, Q., Zhu, J., Liu, Z., Ning, W., et al. (2017). PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 22, 1026–1033. doi: 10.1007/s10147-017-1161-7
Zhao, L., Ji, G., Le, X., Wang, C., Xu, L., Feng, M., et al. (2017). Long Noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 77, 1369–1382. doi: 10.1158/0008-5472.Can-16-1615
Zhao, L., Wang, W., Huang, S., Yang, Z., Xu, L., Yang, Q., et al. (2018). The RNA binding protein SORBS2 suppresses metastatic colonization of ovarian cancer by stabilizing tumor-suppressive immunomodulatory transcripts. Genome Biol. 19:35. doi: 10.1186/s13059-018-1412-6
Zhao, Y., Yang, W., Huang, Y., Cui, R., Li, X., and Li, B. (2018). Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol. Biochem. 47, 721–734. doi: 10.1159/000490025
Zhao, X., and Subramanian, S. (2017). Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res. 77, 817–822. doi: 10.1158/0008-5472.Can-16-2379
Keywords: ovarian cancer, tumor microenvironment, anti-angiogenesis therapy, tumor-associated macrophage-targeted strategies, immune checkpoint inhibitors
Citation: Yang Y, Yang Y, Yang J, Zhao X and Wei X (2020) Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 8:758. doi: 10.3389/fcell.2020.00758
Received: 02 April 2020; Accepted: 20 July 2020;
Published: 11 August 2020.
Edited by:
Cecilia Ana Suarez, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Esther Hoste, Ghent University, BelgiumPriyanka Gupta, University of Alabama at Birmingham, United States
Copyright © 2020 Yang, Yang, Yang, Zhao and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Zhao, eGlhLXpoYW9AMTI2LmNvbQ==; Xiawei Wei, eGlhd2Vpd2VpQHNjdS5lZHUuY24=; d2VpeGlhd2Vpc2N1QDEyNi5jb20=
†These authors have contributed equally to this work