 Nicolas Clere
Nicolas Clere Sarah Renault
Sarah Renault Isabelle Corre
Isabelle Corre- 1Micro and Nanomédecines Translationnelles, Université d’Angers, INSERM UMR U1066, CNRS 6021, Angers, France
- 2Sarcomes Osseux et Remodelage des Tissus Calcifiés, Université de Nantes, INSERM UMR U1238, Nantes, France
Cancer is one of the most important causes of morbidity and mortality worldwide. Tumor cells grow in a complex microenvironment constituted of immune, stromal, and vascular cells that supports growth, angiogenesis, and metastasis. Endothelial cells (ECs) are major components of the vascular microenvironment. These cells have been described for their plasticity and potential to transdifferentiate into mesenchymal cells through a process known as endothelial-to-mesenchymal transition (EndMT). This complex process is controlled by various factors, by which ECs convert into a phenotype characterized by mesenchymal protein expression and motile, contractile morphology. Initially described in normal heart development, EndMT is now identified in several pathologies, and especially in cancer. In this review, we highlight the process of EndMT in the context of cancer and we discuss it as an important adaptive process of the tumor microenvironment that favors tumor growth and dissemination but also resistance to treatment. Thus, we underline targeting of EndMT as a potential therapeutic strategy.
Introduction
The tumor microenvironment (TME) is a complex network of stromal fibroblastic, immune, and endothelial cells (ECs), embedded in a supportive extracellular matrix. It is now considered as an essential player in cancer biology, participating to tumor progression, metastatic dissemination, and immune surveillance of cancer cells but also to responses to therapies (Maman and Witz, 2018). Furthermore, in response to environmental and oncogenic signals that evolved continuously while tumor growths, the TME appears highly dynamic and plastic (Quail and Joyce, 2013). In this environment, ECs that line microvessels are the key masters of angiogenesis. Indeed, in response to tumor environmental cues, ECs promote the formation of new vessels through proliferation, migration, adhesion, and matrix digestion to support tumor progression and dissemination (Potente et al., 2011). But beside their role in angiogenesis, ECs have been characterized the last decade as capable of an important phenotypic plasticity (Dejana et al., 2017), illustrated by their ability to modify their endothelial phenotype toward a mesenchymal profile. This plasticity, named the endothelial-to-mesenchymal transition (EndMT), was initially described in cardiac embryonic development but has been also highlighted in several postnatal pathologies such as cardiac fibrosis, atherosclerosis, pulmonary hypertension, and vascular calcification (Li et al., 2018a). Endothelial-to-mesenchymal transition appears as essential in these inflammation-associated disorders and may be a key link between endothelial dysfunction and inflammation (Cho et al., 2018). In the context of cancer, the process of EndMT was identified in 2007 in melanoma and pancreatic tumor mouse model (Zeisberg et al., 2007) and critically involved in tumor progression. Since then, increasing evidence has reinforced the relevance of endothelial plasticity through EndMT in cancer biology. This review aims to describe the main features and roles of EndMT in cancer and to highlight its importance in shaping a tumor supportive microenvironment that favors tumor growth, metastatic dissemination, and resistance to treatment. Therefore, targeting EndMT as therapeutic strategy will also be discussed.
Description
The EndMT process is characterized as a transdifferentiation program where ECs lose their endothelial characteristics and gain mesenchymal features. This phenotypic switch is characterized by profound morphological, functional and molecular changes. Endothelial cells lose their cellular adhesion and delaminate, reorganize their cytoskeleton that converts their apico-basal polarity toward a front-end back polarity to form spindle-shape cells with enhanced properties of migration. This transition is accompanied with a marked decrease in endothelial markers such as VE-cadherin, CD31, Tie-1, Tie-2, and vWF conjugated to an increased expression of mesenchymal biomarkers such as α-SMA, SM22a, CD44, N-Cadherin, vimentin, COL I/III, and FSP-1 (S100A4) (Piera-Velazquez and Jimenez, 2019) (Figure 1). As described by Dejana et al. (2017), EndMT markers evolve from an early step with partial downregulation of endothelial markers and up-regulation of some early mesenchymal markers (α-SMA, SM22a, and FSP-1) to a later step, characterized by a decline of endothelial markers and up-regulation of mesenchymal markers such as matrix proteins and matrix metalloproteases (MMPs) (Fibronectin, COL I, and MMPs). Initially considered as a complete process of differentiation, EndMT might also be partial in pathophysiological context and in particular in cancer. Intermediate stages of differentiation in tumors-derived ECs have been identified (Xiao et al., 2015) and these tumor ECs display heterogeneity in their potentiality to undergo EndMT. This is consistent with the phenotypic heterogeneity of ECs in tumors (Maishi et al., 2019; Goveia et al., 2020), combined with the diversity of signals emanating from TME, as fine tuning of EndMT appears to be under the control of several factors such as transforming growth factor-β (TGF-β) and basic fibroblast growth factor (bFGF) (Xiao and Dudley, 2017). Moreover, partial EndMT has been assimilated as an initial step of endothelial sprouting in angiogenesis process (Welch-Reardon et al., 2015; Wang et al., 2017). Reversibility of EndMT is barely described in cancer, but a mesenchymal-to-endothelial transition was recently suggested as contributing to Kaposi sarcoma (Li et al., 2018b). Few in vitro studies suggest that EndMT reversibility may occur for a short period of time upon exposure to EndMT inducers (Rieder et al., 2011), while prolonged exposure forces mesenchymal differentiated cells to reach a no-return point toward endothelial features (Xiao et al., 2015).
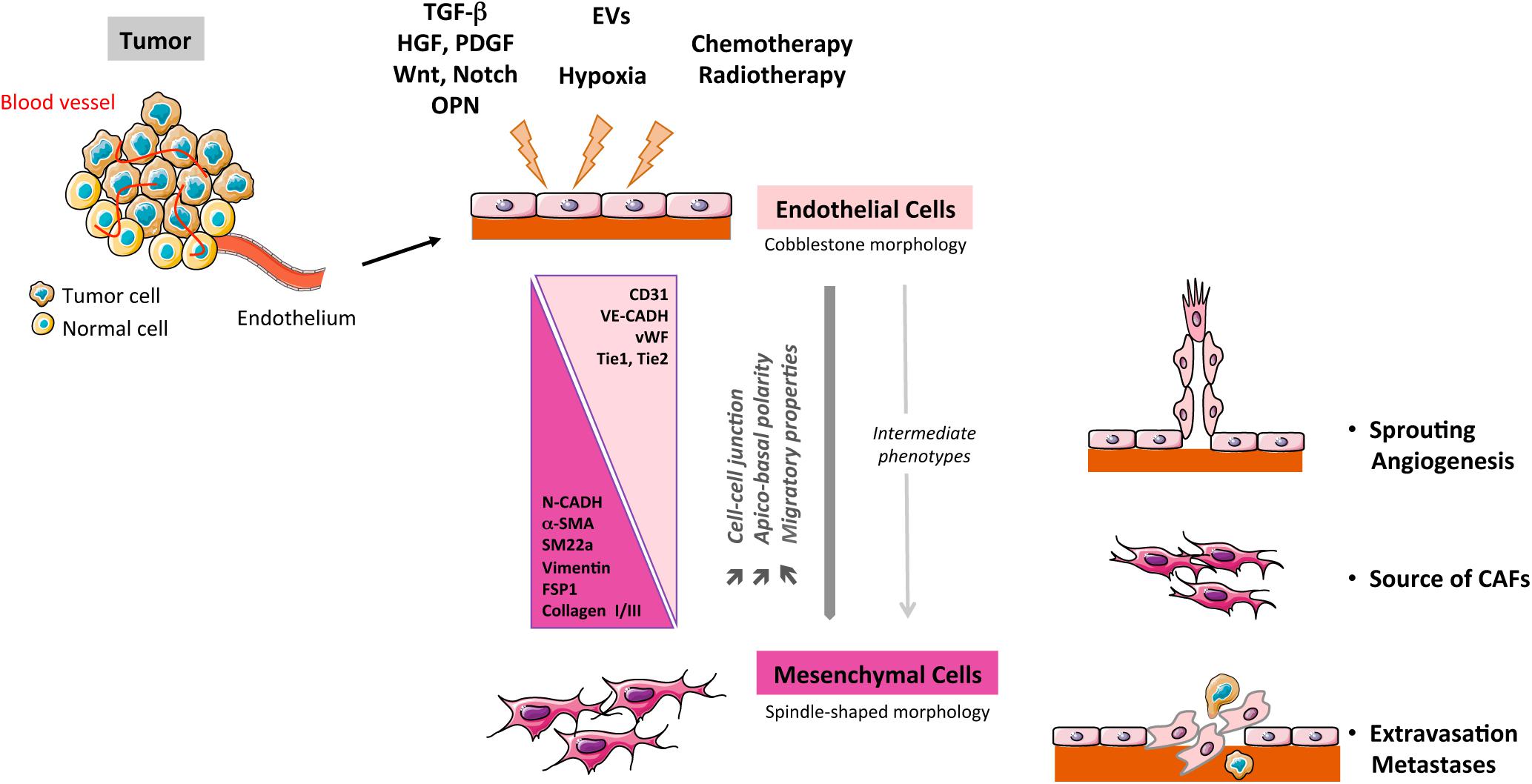
Figure 1. Schematic representation of the EndMT process in cancer. TGF-β, transforming growth factor-β; PDGF, platelet derived growth factor; HGF, hepatocyte growth factor; CAFs, cancer-associated fibroblasts; OPN, osteopontin; EVs: extracellular vesicles.
Identifying the process of EndMT in living animals remains challenging. This process is inherently transitional and progressive, rendering detection of mesenchymal cells originating from endothelium difficult unless cells have not terminated their full transition. Therefore, detecting EndMT may be quite often underestimated in vivo. Nevertheless, exploration of EndMT has been rendered possible in vivo through fate mapping approach in transgenic mice harboring EC-specific lineage tracing, with the limitation that endothelial promoters are rarely totally exclusive to endothelial lineage (Li et al., 2018a). The first in vivo evidence of EndMT in a tumor context was shown in subcutaneous melanoma model in Tie-2-Cre x R26TRosa Lox-stop-LacZ-Lox crossed mice (Zeisberg et al., 2007). Since then, fate-mapping tracing has allowed to characterize EndMT in models of lung (Choi et al., 2018), pancreas (Anderberg et al., 2013), and brain (Huang et al., 2016). More importantly, EndMT has now been discovered in tumor specimen of human patients in a number of cancers: colorectal carcinoma (Fan et al., 2018), pancreatic ductal adenocarcinoma (Fan et al., 2019), lung cancer (Choi et al., 2018), or glioblastoma (GBM) (Huang et al., 2016; Table 1).
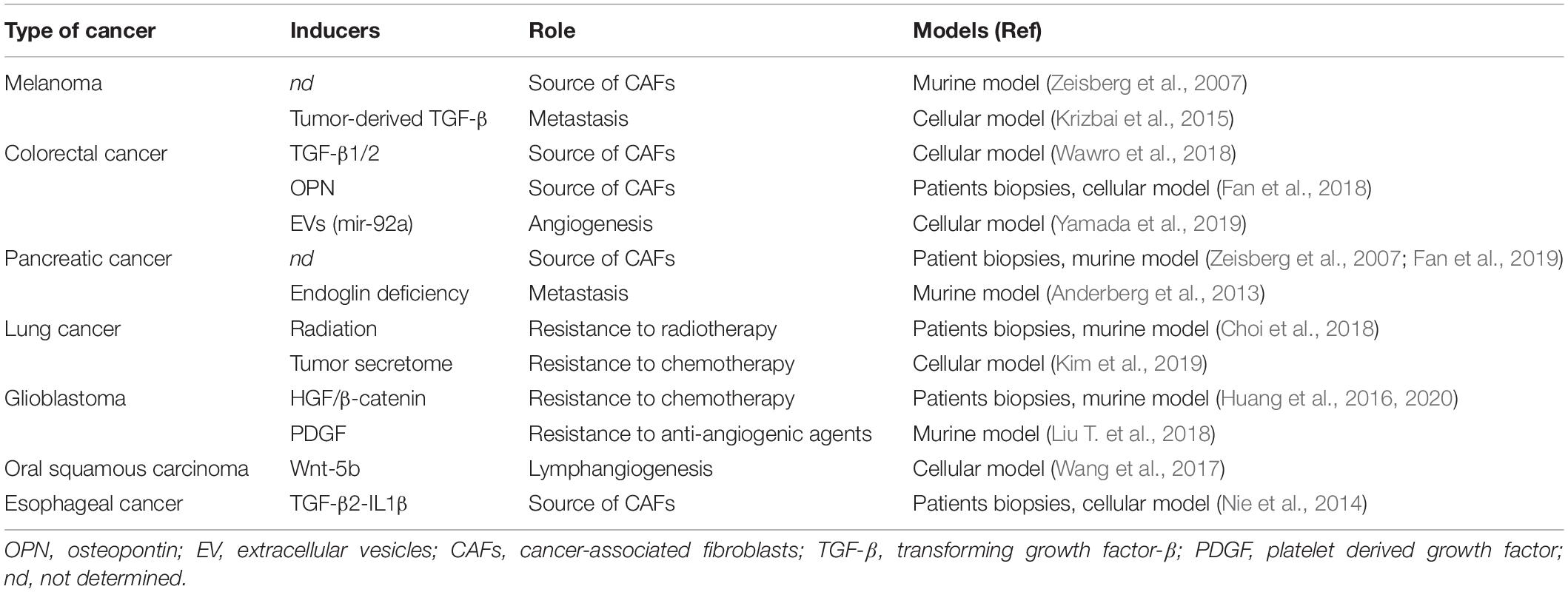
Table 1. Inducers and Roles of EndMT in different types of cancer.
Regulation of EndMT
Major Inducers in Cancer
Members of TGF-β family of proteins are considered today as the major inducers of EndMT, both in physiologic cardiac development (Garside et al., 2013) but also in cancer, as TGF-βs are soluble factors often found overexpressed in tumors (Principe et al., 2014). Therefore, TGF-β1 produced by melanoma tumor cells leads to EndMT (Krizbai et al., 2015). TGF-β2 secreted by tumor in synergy with Interleukin-1β was shown essential to induce EndMT in esophageal carcinoma (Nie et al., 2014). TGF-β2 is also required to promote EndMT in the context of invasive colon carcinoma, via a tubulin-β3 dependent mechanism (Wawro et al., 2018). Nevertheless, additional signals to TGF-βs may be required, as highlighted in a 3D spheroid model of lung cancer where EndMT does not rely on TGF-β (Kim et al., 2019). In fact, TGF-β signaling appears insufficient in promoting EndMT in GBM but requires the Notch signaling pathway (Marin-Ramos et al., 2019). Such a crosstalk between Notch and TGF-β participates to the formation of a transient mesenchymal/endothelial niche in a murine breast cancer xenograft model (Ghiabi et al., 2015). In cancer studies, additional soluble factors have been depicted as EndMT inducers. In brain tumor, Hepatocyte growth factor (HGF) (Huang et al., 2016) and platelet-derived growth factor (PDGF) (Liu T. et al., 2018), two growth factors secreted by tumor cells, appear critical to drive EndMT. The bone matrix protein, osteopontin (OPN), through its binding to integrin αvβ3, promotes EndMT through an epigenetic controlled-repression of VE-cadherin in colorectal cancer (Fan et al., 2018). The Wnt signaling pathway through β-catenin/LEF signaling has also be shown to promote EndMT in oral squamous carcinoma (Wang et al., 2017) and very recently in GBM (Huang et al., 2020).
Hypoxia is an important hallmark of the TME and plays a major role in promoting angiogenesis. Nevertheless, hypoxia has also been described as a potent inducer of EndMT, especially in pulmonary arterial hypertension (Suzuki et al., 2018) and in radiation-induced fibrosis (Choi et al., 2015), through hypoxia-inducible factor (HIF)-dependent pathways (Tang et al., 2018; Toullec et al., 2018). Alternatively, hypoxia-induced EndMT in primary and immortalized ECs was shown to involve RhoJ leading to HIF-dependent and trimethylated histone H3K4-dependent pathways (Liu L. et al., 2018). Furthermore, expression of TGF-β, the master inducer of EndMT, may also be regulated by hypoxia (Nishi et al., 2004; Hung et al., 2013). Endothelial-to-mesenchymal transition-relied transcription factors Snail (Xu et al., 2015) and Twist-1 (Mammoto et al., 2018) are also identified as targets of hypoxia. Although particularly relevant in tumors, hypoxia, and HIF signaling remain to be deeper explored in EndMT in cancer.
Responses to cancer treatment have important issues, leading to efficiency but often to resistance. Cancer therapies such as conventional radiotherapies (Mintet et al., 2015; Choi et al., 2018), high-dose radiation therapies (Banerjee et al., 2020) but also chemotherapies (Murugavel et al., 2018) are also currently identified as promoting EndMT in diverse type of tumors, with consequences in tumor response as described below.
Main Intracellular Signaling Modulators of EndMT
Transcription Factors
As mentioned, TGF-β induces EndMT in cancer and involves the Snail family of transcription factors such as Snail, Slug, Twist, and ZEB. These factors are potent transcriptional repressors, notably of endothelial markers VE-cadherin and CD31 (Piera-Velazquez and Jimenez, 2019). A direct regulation of Snail by TGF-β2 has been highlighted in mouse embryonic stem cell-derived ECs (Kokudo et al., 2008) and Medici et al. (2011) described Snail as a master transcription factor of EndMT, induced both by Smad-dependent and PI3K/p38 MAPK-dependent signaling pathways. Importantly, Snail has been identified as a direct target of HIF-1α in hypoxia-induced EndMT (Xu et al., 2015). Furthermore, hypoxia-induced EndMT involves TGF-β-induced phosphorylation of transcription factor Twist-1 (Mammoto et al., 2018) but also increased expression and nuclear translocation of Snail, Slug and Zeb1 (Doerr et al., 2016). TGF-β2 drives also EndMT through a Smad-dependent activation of the myocardin-related transcription factor-A (MRTF-A), in pancreatic MS-1 ECs (Mihira et al., 2012) while in an invasive colorectal cancer microenvironment, TGF-β2-induced EndMT involves MRTF-A and B transcription factors through Smad-independent RhoA pathway (Ciszewski et al., 2017).
Epigenetic Regulation
With EndMT characterized as a cellular transdifferentation where gene expression changes are necessary, epigenetics is now considered as a strong regulator of this process (Hulshoff et al., 2018). Furthermore, epigenetic regulation is also determinant for cellular differentiation during cancer pathologies (Petronis, 2010). The global histones methylation of lysine residues has been reported to be associated with EndMT. Thus, EZH2 (enhancer of zeste homolog 2), the major histone methyltransferase that processes the transcriptional repressive trimethylation on lysine 27 of histone H3 (H3K27me3), is down-expressed during EndMT induced by IL-1β and TGF-β2 (Maleszewska et al., 2015). This is associated with reduced H3K27me3 repressive marks at the promoter of the mesenchymal gene transgelin/SM22α. Recently, in colorectal cancer, EZH2 was identified as interacting with the transcription factor TCF12, helping this latter to transcriptionally repress VE-cadherin gene and thus facilitating EndMT (Fan et al., 2018). Additionally, Jumonji domain-containing protein 2B, JMJD2B, a demethylase of the repressive histone mark H3K9me3, is induced by EndMT-promoting stimuli and is associated to de-repression of genes in EndMT-related genes and pathways (Glaser et al., 2020). Description of EndMT in melanoma intratumoral ECs has recently shed the light on two transcription factors from the ETS family, ERG and Fli-1 (Nagai et al., 2018). These pivotal transcription factors promote expression of EC-specific genes but indirectly repress mesenchymal-related genes through epigenetic regulation by modifying histones methylation/acetylation marks. The dowregulation of ERG and Fli1 has been described in tumor-induced EndMT both in vitro and in clinical cancer patients (Nagai et al., 2018), pointing the alleviation of mesenchymal genes repression.
Accumulative evidence highlights that non-coding RNAs including miRNAs and lncRNAs are important epigenetic regulators of EndMT [for review, Hulshoff et al. (2018)]. Depending on their gene targets that mainly belonged to TGF signaling pathways, miRNAs may promote or repress the process of EndMT. The list of miRNAs regulating EndMT is constantly increasing in pathologies (cardiac, fibrosis, and pulmonary hypertension) (Kim, 2018). In malignant disorders, overexpression of miR-302c suppresses EndMT, therefore inhibiting hepatocarcinoma tumor growth (Zhu et al., 2014). A few of lncRNAs have been associated with EndMT. Metastasis-associated lung adenocarcinoma transcript 1 MALAT-1 facilitates EndMT through down-regulation of Smad3, TGFBR2-targeting miR-145 (Xiang et al., 2017) and recently, a combination of low expression of three lncRNAs (LOC340340, LOC101927256, MNX1-AS1) was proposed as an EndMT index in pancreas adenocarcinoma (Fan et al., 2019).
Metabolic Regulation
Activation of the EndMT program has been recently associated with profound metabolic alterations in ECs. Fatty acid oxidation (FAO) was shown to contribute to vessels sprouting by promoting, in angiogenic ECs, a de novo nucleotide synthesis for DNA replication for proliferation (Schoors et al., 2015; Eelen et al., 2018). Xiong and colleagues now demonstrate that inhibition of FAO, marked by a decreased expression of mitochondrial enzyme carnitine O-palmitoyltransferase 1 (CPT1) and by a fall of acetyl-CoA levels, is critical for TGF-β-induced EndMT (Xiong et al., 2018). Therefore, metabolic plasticity and metabolic transcriptome heterogeneity among normal and tumor ECs, as recently described (Rohlenova et al., 2020), have to be considered as important indicators of endothelial fate, with engagement to EndMT associated with a lower energy-producing metabolism of FAO inhibition.
Roles of EndMT in Cancer
Angiogenesis as a Partial EndMT
Angiogenesis is required for an optimal tumor progression (Folkman, 1971) and is initiated by the sprouting of ECs from existent vessels. Sprouting-leader endothelial tip cells lose their apical-basal polarity, weaken their cell-cell interaction, degrade extracellular matrix to become more motile, but never complete their transition as they do maintain some cell-to-cell interactions. Overall, this sprouting resumes EndMT features and may be compared to a partial EndMT (Potenta et al., 2008; Welch-Reardon et al., 2015). Moreover, the master EndMT transcription factors Slug and Snail are found expressed in tumor ECs in several carcinoma (Welch-Reardon et al., 2015), with Slug expression recently associated with angiogenesis and tumor metastasis (Gu et al., 2017). Recently, colon cancer cells, via extracellular vesicles, have been shown to induce a partial EndMT associated with angiogenesis, in a miR-92a-dependent inhibition of junctional Claudin-11 and up-regulation of Snail, and vimentin (Yamada et al., 2019). Partial EndMT, stimulated by tumor-secreted Wnt ligand, has also been involved in oral cancer lymphangiogenesis (Wang et al., 2017).
EndMT as a Source of Cancer-Associated Fibroblasts (CAFs)
CAFs are major components of the TME, contributing to tumorigenesis in multiple ways: cancer cell proliferation, migration, invasion, stemness, mainly through the secretion of cytokines, chemokines, extracellular vesicles, and matrix remodeling [for review, Bu et al. (2019)]. CAFs may originate from several cell types, namely resident fibroblasts, epithelial cells, mesenchymal stromal stem cell from bone marrow, and adipocytes. However, myofibroblastic/mesenchymal cells issued from ECs through EndMT represent also a unique source of CAFs, as initially described in mouse model (Zeisberg et al., 2007). The actual contribution of EndMT in CAFs production is still not fully characterized. Nevertheless, several studies indicate that EndMT-derived cells share functions with CAF-like cells, such as adhesion to collagen and wound healing (Wawro et al., 2018), production of VEGF in esophageal cancer (Nie et al., 2014) but also promotion of angiogenesis, tumor growth and stemness in colorectal cancer (Fan et al., 2018) or driving monocyte M2 polarization in pancreatic cancer (Fan et al., 2019).
EndMT and Metastasis
One major hallmark of EndMT is endothelial cytoskeletal reorganization through the Rho/ROCK signaling pathway (Mihira et al., 2012; Ciszewski et al., 2017), coupled to the loss of endothelial adhesion molecules (VE-cadherin, claudins). Therefore, EndMT contributes to disruption of the endothelial barrier that may favor the intra- and extravasation tumor cells. Indeed, melanoma cells increase their transendothelial migration when EndMT is induced by tumor secretome (Krizbai et al., 2015). To the same extend, hepatic and lung metastatic seeding is increased in pancreatic cancer mouse model upon endoglin deficiency-induced EndMT (Anderberg et al., 2013). Hence, EndMT by facilitating tumor transendothelial migration can be considered as an essential process for an optimal metastatic dissemination (Gasparics et al., 2016).
EndMT and Therapeutic Resistance
Vasculature is a double-edge sword in tumor, as it promotes tumor progression through angiogenesis but at the same time it assures delivery of drugs to tumor. Therefore, plasticity and dynamics of the endothelial compartment may impact response to therapies. The role of EndMT in resistance to therapies emerges as a novel field, both in the context of chemo- and radiotherapy. In fact, EndMT confers tumor radioresistance and promotes colorectal tumor growth tumor, by awaking cancer stem cells and stimulating polarization of M2 tumor-associated macrophages TAM (Choi et al., 2018). Resistance to chemotherapies cisplatin and gefitinib in a multicellular lung tumor spheroid model is alleviated when EndMT is reversed, implying EndMT as a resistance factor (Kim et al., 2019). Recently, a robust EndMT induced by HGF/c-Met was described in GBM, resulting in aberrant vasculature and in an increase of tumor cells chemoresistant to temozolomide (Huang et al., 2016), undoubtedly linked to drug delivery failure and hypoxia. The same group documented the induction of resistance of EndMT-issued cells to anti-angiogenic chemotherapies in this brain tumor. Indeed, PDGF-induced EndMT is associated with a decreased expression of VEGFR2, resulting in resistance to anti-VEGF treatment (Liu T. et al., 2018) whereas HGF/β-catenin-induced-EndMT leads to endothelial temozolomide resistance through up-regulation of drug pumping and efflux proteins ABBC1/Multidrug resistance associated protein (MRP-1; Huang et al., 2020). Furthermore, EndMT induces abnormal recruitment of pericytes (Choi et al., 2018) and generates pericyte-like cells abnormally covering the tumor vasculature (Anderberg et al., 2013). Such an aberrant coverage of pericytes is described as a signature of resistance to anti-VEGF anti-angiogenic therapy in two different cancer models [pancreas (Helfrich et al., 2010) and melanoma (Franco et al., 2011)]. Hence, EndMT appears to participate to establish a perivascular niche supportive for resistance in many ways: promotion of hypoxia, failure of drug delivery, vanishing of drug target (VEGFR2), and recruitment of pro-tumoral immune cells. Overall, EndMT has to be considered as a relevant factor of resistance.
Targeting of EndMT
In light of the overall pro-tumoral effect of EndMT, targeting of this process could represent a therapeutic avenue to treat cancer. Currently, several drugs have been tested as potential EndMT inhibitors. The mTOR inhibitor rapamycin is able to block EndMT, either by preventing EC migration and matrix degradation (Gao et al., 2011) or by lowering TGF-β, TNF-α and VEGF levels (Gonzalez-Mateo et al., 2015). Spironolactone, an aldosterone receptor-blocker, abrogates EndMT in a model of fibrosis, by blocking TGF-β and Notch signaling (Chen et al., 2015). Notably, a derived-form of temozolomide, the standard chemotherapy in GBM, can suppress EndMT in tumor-associated ECs via the same mechanisms of TGF-β and Notch signaling inhibition (Marin-Ramos et al., 2019), supporting the clinical value of this molecule.
An innovative adjuvant strategy could use molecules targeting metabolic pathways. As inhibition of fatty acid oxidation is described to potentiate EndMT (Xiong et al., 2018), one may consider activation of FAO to limit the EndMT process. Indeed, pharmacological approaches to increase FAO are available, mainly through the use of peroxisome proliferator-activated receptor PPAR agonists (Djouadi et al., 2005) or fatty acid synthase FASN inhibitors (Thupari et al., 2004) but are not explored yet in cancer.
Inhibiting kinase signaling engaged by recently identified EndMT-inducing factors such as PDGF or HGF (Huang et al., 2016; Liu T. et al., 2018) would be of interest. For example, Nintedanib, a tyrosine kinase inhibitor of PDGF, FGF, and VEGF, has been shown to ameliorate pulmonary hypertension by blocking EndMT (Tsutsumi et al., 2019) and inhibitors of the HGF receptor c-Met (e.g., carbozantinib) are at present intensively explored in Ewing sarcoma and osteosarcoma clinical trials (Italiano et al., 2020).
As TGF-β is currently considered as potent inducer of EndMT in cancer, targeting TGF-β could represent a potential therapeutic approach. Several TGF-β inhibitors have emerged, among which the small molecule inhibitor galunisertib (LY2157299) appears as one of the most advanced and promising molecules tested in two phase-II trials in pancreatic cancer (NCT01373164) or in hepatocellular carcinoma (NCT01246986). Despite not tested on tumor EndMT yet, galunisertib harbors a very safe toxicity profile, with no cardiac toxicity that was a limitation for the use of the first generation of TGF-β inhibitors tested in clinic (Kovacs et al., 2015). Monoclonal antibodies against TGF-β are now also available and tested in different pathologies. As such, fresolimumab, initially developed for the treatment of idiopathic pulmonary fibrosis, has been investigated in phase-II trials in renal cell carcinoma and melanoma, but also metastatic breast cancer (NCT01401062) (Formenti et al., 2019) where TGF-β blocking was also associated with an increase of the immune anti-tumor response. If the beneficial properties of fresolimumab were confirmed in impairing the process of EndMT, inhibiting the TGF-β pathway could be particularly efficient for the treatment of aggressive tumors with the targeting of two essential components of the TME.
Conclusion
Despite its beneficial role in embryo development, the process of EndMT may be also detrimental, notably in cancer where it contributes to tumor development. This induced plasticity of the endothelium has an important role in shaping the TME. Endothelial-to-mesenchymal transition participates to initial angiogenesis sprouting, reinforces the stromal fibroblastic microenvironment and remodels the vasculature to support tumor cell dissemination and metastasis. Moreover, it contributes to the resistance to cancer treatment, by affecting directly or indirectly the microenvironment. Thus, the process of EndMT regulates different stages of tumorigenesis from initiation, dissemination to response to treatments.
Exploring EndMT remains very challenging mainly due to its inherent dynamics and to the complexity of its mechanisms. Nevertheless, its full understanding still requires deep exploration. Importantly, the role of EndMT transitional populations still need to be grasped, as these intermediate populations could also be relevant in tumor biology.
In conclusion, EndMT can be considered as an important process that reinforces the TME plasticity and devotion to cancer cells. Therefore, modulation of EndMT could be envisaged as a complementary therapeutic arm to counteract tumor progression. Currently, several strategies to inhibit EndMT are considered and deserve exploration in cancer context.
Author Contributions
NC, SR, and IC contributed to the writing and editing of the manuscript. SR and IC elaborated the figure and table. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by “Région Pays de la Loire” for the TRENDOMOS project («2019-Pari Scientifique» Program), INSERM, CNRS and by the association La Ligue contre le cancer (Comities 22, 29, 37, 44, 49, and 56).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderberg, C., Cunha, S. I., Zhai, Z., Cortez, E., Pardali, E., Johnson, J. R., et al. (2013). Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J. Exp. Med. 210, 563–579. doi: 10.1084/jem.20120662
Banerjee, D., Barton, S. M., Grabham, P. W., Rumeld, A. L., Okochi, S., Street, C., et al. (2020). High-dose radiation increases notch1 in tumor vasculature. Int. J. Radiat. Oncol. Biol. Phys. 106, 857–866. doi: 10.1016/j.ijrobp.2019.11.010
Bu, L., Baba, H., Yoshida, N., Miyake, K., Yasuda, T., Uchihara, T., et al. (2019). Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 38, 4887–4901. doi: 10.1038/s41388-019-0765-y
Chen, X., Cai, J., Zhou, X., Chen, L., Gong, Y., Gao, Z., et al. (2015). Protective effect of spironolactone on endothelial-to-mesenchymal transition in HUVECs via notch pathway. Cell Physiol. Biochem. 36, 191–200. doi: 10.1159/000374063
Cho, J. G., Lee, A., Chang, W., Lee, M. S., and Kim, J. (2018). Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 9:294. doi: 10.3389/fimmu.2018.00294
Choi, S. H., Hong, Z. Y., Nam, J. K., Lee, H. J., Jang, J., Yoo, R. J., et al. (2015). A Hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 21, 3716–3726. doi: 10.1158/1078-0432.CCR-14-3193
Choi, S. H., Kim, A. R., Nam, J. K., Kim, J. M., Kim, J. Y., Seo, H. R., et al. (2018). Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6(+) cancer cell and macrophage polarization. Nat. Commun. 9:5108. doi: 10.1038/s41467-018-07470-w
Ciszewski, W. M., Sobierajska, K., Wawro, M. E., Klopocka, W., Chefczynska, N., Muzyczuk, A., et al. (2017). The ILK-MMP9-MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumor microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2283–2296. doi: 10.1016/j.bbamcr.2017.09.004
Dejana, E., Hirschi, K. K., and Simons, M. (2017). The molecular basis of endothelial cell plasticity. Nat. Commun. 8:14361. doi: 10.1038/ncomms14361
Djouadi, F., Aubey, F., Schlemmer, D., Ruiter, J. P., Wanders, R. J., Strauss, A. W., et al. (2005). Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 14, 2695–2703. doi: 10.1093/hmg/ddi303
Doerr, M., Morrison, J., Bergeron, L., Coomber, B. L., and Viloria-Petit, A. (2016). Differential effect of hypoxia on early endothelial-mesenchymal transition response to transforming growth beta isoforms 1 and 2. Microvasc. Res. 108, 48–63. doi: 10.1016/j.mvr.2016.08.001
Eelen, G., de Zeeuw, P., Treps, L., Harjes, U., Wong, B. W., and Carmeliet, P. (2018). Endothelial cell metabolism. Physiol. Rev. 98, 3–58. doi: 10.1152/physrev.00001.2017
Fan, C. S., Chen, L. L., Hsu, T. A., Chen, C. C., Chua, K. V., Li, C. P., et al. (2019). Endothelial-mesenchymal transition harnesses HSP90alpha-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 12:138. doi: 10.1186/s13045-019-0826-2
Fan, C. S., Chen, W. S., Chen, L. L., Chen, C. C., Hsu, Y. T., Chua, K. V., et al. (2018). Osteopontin-integrin engagement induces HIF-1alpha-TCF12-mediated endothelial-mesenchymal transition to exacerbate colorectal cancer. Oncotarget 9, 4998–5015. doi: 10.18632/oncotarget.23578
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186. doi: 10.1056/NEJM197111182852108
Formenti, S. C., Hawtin, R. E., Dixit, N., Evensen, E., Lee, P., Goldberg, J. D., et al. (2019). Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J. Immunother. Cancer 7:177. doi: 10.1186/s40425-019-0633-x
Franco, M., Roswall, P., Cortez, E., Hanahan, D., and Pietras, K. (2011). Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 118, 2906–2917. doi: 10.1182/blood-2011-01-331694
Gao, H., Zhang, J., Liu, T., and Shi, W. (2011). Rapamycin prevents endothelial cell migration by inhibiting the endothelial-to-mesenchymal transition and matrix metalloproteinase-2 and -9: an in vitro study. Mol. Vis. 17, 3406–3414.
Garside, V. C., Chang, A. C., Karsan, A., and Hoodless, P. A. (2013). Co-ordinating Notch, BMP, and TGF-beta signaling during heart valve development. Cell Mol. Life Sci. 70, 2899–2917. doi: 10.1007/s00018-012-1197-9
Gasparics, A., Rosivall, L., Krizbai, I. A., and Sebe, A. (2016). When the endothelium scores an own goal: endothelial cells actively augment metastatic extravasation through endothelial-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 310, H1055–H1063. doi: 10.1152/ajpheart.00042.2016
Ghiabi, P., Jiang, J., Pasquier, J., Maleki, M., Abu-Kaoud, N., Halabi, N., et al. (2015). Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 13:27. doi: 10.1186/s12967-015-0386-3
Glaser, S. F., Heumuller, A. W., Tombor, L., Hofmann, P., Muhly-Reinholz, M., Fischer, A., et al. (2020). The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 117, 4180–4187. doi: 10.1073/pnas.1913481117
Gonzalez-Mateo, G. T., Aguirre, A. R., Loureiro, J., Abensur, H., Sandoval, P., Sanchez-Tomero, J. A., et al. (2015). Rapamycin protects from type-i peritoneal membrane failure inhibiting the angiogenesis, lymphangiogenesis, and Endo-MT. Biomed. Res. Int. 2015, 989560. doi: 10.1155/2015/989560
Goveia, J., Rohlenova, K., Taverna, F., Treps, L., Conradi, L. C., Pircher, A., et al. (2020). An Integrated Gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37, 21–36. doi: 10.1016/j.ccell.2019.12.001
Gu, A., Jie, Y., Yao, Q., Zhang, Y., and Mingyan, E. (2017). Slug is associated with tumor metastasis and angiogenesis in ovarian cancer. Reprod. Sci. 24, 291–299. doi: 10.1177/1933719116654989
Helfrich, I., Scheffrahn, I., Bartling, S., Weis, J., von Felbert, V., Middleton, M., et al. (2010). Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 207, 491–503. doi: 10.1084/jem.20091846
Huang, M., Liu, T., Ma, P., Mitteer, R. A. Jr., and Zhang, Z. (2016). c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Invest. 126, 1801–1814. doi: 10.1172/JCI84876
Huang, M., Zhang, D., Wu, J. Y., Xing, K., Yeo, E., Li, C., et al. (2020). Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 12:532. doi: 10.1126/scitranslmed.aay7522
Hulshoff, M. S., Xu, X., Krenning, G., and Zeisberg, E. M. (2018). Epigenetic regulation of endothelial-to-mesenchymal transition in chronic heart Disease. Arterioscler. Thromb. Vasc. Biol. 38, 1986–1996. doi: 10.1161/ATVBAHA.118.311276
Hung, S. P., Yang, M. H., Tseng, K. F., and Lee, O. K. (2013). Hypoxia-induced secretion of TGF-beta1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant. 22, 1869–1882. doi: 10.3727/096368912X657954
Italiano, A., Mir, O., Mathoulin-Pelissier, S., Penel, N., Piperno-Neumann, S., Bompas, E., et al. (2020). Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 21, 446–455. doi: 10.1016/S1470-2045(19)30825-3
Kim, J. (2018). MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. BMB Rep. 51, 65–72. doi: 10.5483/bmbrep.2018.51.2.011
Kim, S. H., Song, Y., and Seo, H. R. (2019). GSK-3beta regulates the endothelial-to-mesenchymal transition via reciprocal crosstalk between NSCLC cells and HUVECs in multicellular tumor spheroid models. J. Exp. Clin. Cancer Res. 38:46. doi: 10.1186/s13046-019-1050-1
Kokudo, T., Suzuki, Y., Yoshimatsu, Y., Yamazaki, T., Watabe, T., and Miyazono, K. (2008). Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 121(Pt 20), 3317–3324. doi: 10.1242/jcs.028282
Kovacs, R. J., Maldonado, G., Azaro, A., Fernandez, M. S., Romero, F. L., Sepulveda-Sanchez, J. M., et al. (2015). Cardiac safety of TGF-beta receptor i kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc. Toxicol. 15, 309–323. doi: 10.1007/s12012-014-9297-4
Krizbai, I. A., Gasparics, A., Nagyoszi, P., Fazakas, C., Molnar, J., Wilhelm, I., et al. (2015). Endothelial-mesenchymal transition of brain endothelial cells: possible role during metastatic extravasation. PLoS One 10:e0119655. doi: 10.1371/journal.pone.0119655
Li, Y., Lui, K. O., and Zhou, B. (2018a). Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 15, 445–456. doi: 10.1038/s41569-018-0023-y
Li, Y., Zhong, C., Liu, D., Yu, W., Chen, W., Wang, Y., et al. (2018b). Evidence for kaposi sarcoma originating from mesenchymal stem cell through KSHV-induced mesenchymal-to-endothelial transition. Cancer Res. 78, 230–245. doi: 10.1158/0008-5472.CAN-17-1961
Liu, L., Chen, J., Sun, L., and Xu, Y. (2018). RhoJ promotes hypoxia induced endothelial-to-mesenchymal transition by activating WDR5 expression. J. Cell Biochem. 119, 3384–3393. doi: 10.1002/jcb.26505
Liu, T., Ma, W., Xu, H., Huang, M., Zhang, D., He, Z., et al. (2018). PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 9:3439. doi: 10.1038/s41467-018-05982-z
Maishi, N., Annan, D. A., Kikuchi, H., Hida, Y., and Hida, K. (2019). Tumor endothelial heterogeneity in cancer progression. Cancers 11:1511. doi: 10.3390/cancers11101511
Maleszewska, M., Gjaltema, R. A., Krenning, G., and Harmsen, M. C. (2015). Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22alpha expression in endothelial cells in response to interleukin-1beta and transforming growth factor-beta2. Cell Signal. 27, 1589–1596. doi: 10.1016/j.cellsig.2015.04.008
Maman, S., and Witz, I. P. (2018). A history of exploring cancer in context. Nat. Rev. Cancer 18, 359–376. doi: 10.1038/s41568-018-0006-7
Mammoto, T., Muyleart, M., Konduri, G. G., and Mammoto, A. (2018). Twist1 in hypoxia-induced pulmonary hypertension through transforming growth factor-beta-smad signaling. Am. J. Respir. Cell Mol. Biol. 58, 194–207. doi: 10.1165/rcmb.2016-0323OC
Marin-Ramos, N. I., Jhaveri, N., Thein, T. Z., Fayngor, R. A., Chen, T. C., and Hofman, F. M. (2019). NEO212, a conjugate of temozolomide and perillyl alcohol, blocks the endothelial-to-mesenchymal transition in tumor-associated brain endothelial cells in glioblastoma. Cancer Lett. 442, 170–180. doi: 10.1016/j.canlet.2018.10.034
Medici, D., Potenta, S., and Kalluri, R. (2011). Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 437, 515–520. doi: 10.1042/BJ20101500
Mihira, H., Suzuki, H. I., Akatsu, Y., Yoshimatsu, Y., Igarashi, T., Miyazono, K., et al. (2012). TGF-beta-induced mesenchymal transition of MS-1 endothelial cells requires Smad-dependent cooperative activation of Rho signals and MRTF-A. J. Biochem. 151, 145–156. doi: 10.1093/jb/mvr121
Mintet, E., Rannou, E., Buard, V., West, G., Guipaud, O., Tarlet, G., et al. (2015). identification of endothelial-to-mesenchymal transition as a potential participant in radiation Proctitis. Am. J. Pathol. 185, 2550–2562. doi: 10.1016/j.ajpath.2015.04.028
Murugavel, S., Bugyei-Twum, A., Matkar, P. N., Al-Mubarak, H., Chen, H. H., Adam, M., et al. (2018). Valproic acid induces endothelial-to-mesenchymal transition-like phenotypic switching. Front. Pharmacol. 9:737. doi: 10.3389/fphar.2018.00737
Nagai, N., Ohguchi, H., Nakaki, R., Matsumura, Y., Kanki, Y., Sakai, J., et al. (2018). Downregulation of ERG and FLI1 expression in endothelial cells triggers endothelial-to-mesenchymal transition. PLoS Genet. 14:e1007826. doi: 10.1371/journal.pgen.1007826
Nie, L., Lyros, O., Medda, R., Jovanovic, N., Schmidt, J. L., Otterson, M. F., et al. (2014). Endothelial-mesenchymal transition in normal human esophageal endothelial cells cocultured with esophageal adenocarcinoma cells: role of IL-1beta and TGF-beta2. Am. J. Physiol. Cell Physiol. 307, C859–C877. doi: 10.1152/ajpcell.00081.2014
Nishi, H., Nakada, T., Hokamura, M., Osakabe, Y., Itokazu, O., Huang, L. E., et al. (2004). Hypoxia-inducible factor-1 transactivates transforming growth factor-beta3 in trophoblast. Endocrinology 145, 4113–4118. doi: 10.1210/en.2003-1639
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727. doi: 10.1038/nature09230
Piera-Velazquez, S., and Jimenez, S. A. (2019). Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 99, 1281–1324. doi: 10.1152/physrev.00021.2018
Potenta, S., Zeisberg, E., and Kalluri, R. (2008). The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 99, 1375–1379. doi: 10.1038/sj.bjc.6604662
Potente, M., Gerhardt, H., and Carmeliet, P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887. doi: 10.1016/j.cell.2011.08.039
Principe, D. R., Doll, J. A., Bauer, J., Jung, B., Munshi, H. G., Bartholin, L., et al. (2014). TGF-beta: duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 106:djt369. doi: 10.1093/jnci/djt369
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Rieder, F., Kessler, S. P., West, G. A., Bhilocha, S., de la Motte, C., Sadler, T. M., et al. (2011). Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am. J. Pathol. 179, 2660–2673. doi: 10.1016/j.ajpath.2011.07.042
Rohlenova, K., Goveia, J., Garcia-Caballero, M., Subramanian, A., Kalucka, J., Treps, L., et al. (2020). Single-Cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. 31, 862–877. doi: 10.1016/j.cmet.2020.03.009
Schoors, S., Bruning, U., Missiaen, R., Queiroz, K. C., Borgers, G., Elia, I., et al. (2015). Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197. doi: 10.1038/nature14362
Suzuki, T., Carrier, E. J., Talati, M. H., Rathinasabapathy, A., Chen, X., Nishimura, R., et al. (2018). Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 314, L118–L126. doi: 10.1152/ajplung.00296.2017
Tang, H., Babicheva, A., McDermott, K. M., Gu, Y., Ayon, R. J., Song, S., et al. (2018). Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 314, L256–L275. doi: 10.1152/ajplung.00096.2017
Thupari, J. N., Kim, E. K., Moran, T. H., Ronnett, G. V., and Kuhajda, F. P. (2004). Chronic C75 treatment of diet-induced obese mice increases fat oxidation and reduces food intake to reduce adipose mass. Am. J. Physiol. Endocrinol. Metab. 287, E97–E104. doi: 10.1152/ajpendo.00261.2003
Toullec, A., Buard, V., Rannou, E., Tarlet, G., Guipaud, O., Robine, S., et al. (2018). HIF-1alpha deletion in the endothelium, but not in the epithelium, protects from radiation-induced enteritis. Cell Mol. Gastroenterol. Hepatol. 5, 15–30. doi: 10.1016/j.jcmgh.2017.08.001
Tsutsumi, T., Nagaoka, T., Yoshida, T., Wang, L., Kuriyama, S., Suzuki, Y., et al. (2019). Nintedanib ameliorates experimental pulmonary arterial hypertension via inhibition of endothelial mesenchymal transition and smooth muscle cell proliferation. PLoS One 14:e0214697. doi: 10.1371/journal.pone.0214697
Wang, S. H., Chang, J. S., Hsiao, J. R., Yen, Y. C., Jiang, S. S., Liu, S. H., et al. (2017). Tumour cell-derived WNT5B modulates in vitro lymphangiogenesis via induction of partial endothelial-mesenchymal transition of lymphatic endothelial cells. Oncogene 36, 1503–1515. doi: 10.1038/onc.2016.317
Wawro, M. E., Chojnacka, K., Wieczorek-Szukala, K., Sobierajska, K., and Niewiarowska, J. (2018). Invasive colon cancer cells induce transdifferentiation of endothelium to cancer-associated fibroblasts through microtubules enriched in tubulin-beta3. Int. J. Mol. Sci. 20:53. doi: 10.3390/ijms20010053
Welch-Reardon, K. M., Wu, N., and Hughes, C. C. (2015). A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 35, 303–308. doi: 10.1161/ATVBAHA.114.303220
Xiang, Y., Zhang, Y., Tang, Y., and Li, Q. (2017). MALAT1 modulates tgf-beta1-induced endothelial-to-mesenchymal transition through downregulation of miR-145. Cell Physiol. Biochem. 42, 357–372. doi: 10.1159/000477479
Xiao, L., and Dudley, A. C. (2017). Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 241, 25–35. doi: 10.1002/path.4814
Xiao, L., Kim, D. J., Davis, C. L., McCann, J. V., Dunleavey, J. M., Vanderlinden, A. K., et al. (2015). Tumor endothelial cells with distinct patterns of TGFbeta-driven endothelial-to-mesenchymal transition. Cancer Res. 75, 1244–1254. doi: 10.1158/0008-5472.CAN-14-1616
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q. S., Edmunds, L. R., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell 69, 689–698. doi: 10.1016/j.molcel.2018.01.010
Xu, X., Tan, X., Tampe, B., Sanchez, E., Zeisberg, M., and Zeisberg, E. M. (2015). Snail is a direct target of hypoxia-inducible factor 1alpha (HIF1alpha) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J. Biol. Chem. 290, 16653–16664. doi: 10.1074/jbc.M115.636944
Yamada, N. O., Heishima, K., Akao, Y., and Senda, T. (2019). Extracellular vesicles containing microRNA-92a-3p facilitate partial endothelial-mesenchymal transition and angiogenesis in endothelial cells. Int. J. Mol. Sci. 20:406. doi: 10.3390/ijms20184406
Zeisberg, E. M., Potenta, S., Xie, L., Zeisberg, M., and Kalluri, R. (2007). Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128. doi: 10.1158/0008-5472.CAN-07-3127
Keywords: endothelial, mesenchymal, plasticity, cancer, CAF, metastasis, resistance
Citation: Clere N, Renault S and Corre I (2020) Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 8:747. doi: 10.3389/fcell.2020.00747
Received: 05 June 2020; Accepted: 17 July 2020;
Published: 14 August 2020.
Edited by:
Lucas Treps, VIB-KU Leuven Center for Cancer Biology, BelgiumReviewed by:
Giorgio Stassi, University of Palermo, ItalyTze-Sing Huang, National Health Research Institutes, Taiwan
Copyright © 2020 Clere, Renault and Corre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Corre, aXNhYmVsbGUuY29ycmVAdW5pdi1uYW50ZXMuZnI=