 Noriko Funato1,2*
Noriko Funato1,2*- 1Department of Signal Gene Regulation, Tokyo Medical and Dental University, Tokyo, Japan
- 2Research Core, Tokyo Medical and Dental University, Tokyo, Japan
The synchondroses formed via endochondral ossification in the cranial base are an important growth center for the neurocranium. Abnormalities in the synchondroses affect cranial base elongation and the development of adjacent regions, including the craniofacial bones. In the central region of the cranial base, there are two synchondroses present—the intersphenoid synchondrosis and the spheno-occipital synchondrosis. These synchondroses consist of mirror image bipolar growth plates. The cross-talk of several signaling pathways, including the parathyroid hormone-like hormone (PTHLH)/parathyroid hormone-related protein (PTHrP), Indian hedgehog (Ihh), Wnt/β-catenin, and fibroblast growth factor (FGF) pathways, as well as regulation by cilium assembly and the transcription factors encoded by the RUNX2, SIX1, SIX2, SIX4, and TBX1 genes, play critical roles in synchondrosis development. Deletions or activation of these gene products in mice causes the abnormal ossification of cranial synchondrosis and skeletal elements. Gene disruption leads to both similar and markedly different abnormalities in the development of intersphenoid synchondrosis and spheno-occipital synchondrosis, as well as in the phenotypes of synchondroses and skeletal bones. This paper reviews the development of cranial synchondroses, along with its regulation by the signaling pathways and transcription factors, highlighting the differences between intersphenoid synchondrosis and spheno-occipital synchondrosis.
Introduction
In vertebrates, the cranial base lies below the brain and forms a central bone structure of the skull. Within the cranial base, synchondroses play a critical role in the longitudinal growth of the skull (McBratney-Owen et al., 2008; Wei et al., 2017). Precocious ossification and/or malformation of cranial synchondroses can induce the fusion of adjacent bones and subsequent cranium deformities, such as microcephaly and midface hypoplasia (Goldstein et al., 2014; Funato et al., 2020). In order to understand the nature of craniofacial development and related congenital anomalies, identifying the signaling molecules that regulate synchondrosis development is necessary.
Cartilaginous segments that persist between the ossification centers in the cranial base represent various synchondroses, such as the sphenoethmoidal synchondrosis, intersphenoid synchondrosis (ISS), spheno-occipital synchondrosis (SOS), and intraoccipital synchondrosis (Figure 1A). The ISS is located between the presphenoid and basisphenoid bones in the central region of the cranial base, while the SOS is located between the basisphenoid and basioccipital bones (Figures 1A,B). The medial line of the cranial base is originally composed of the hypophyseal, acrochordal, and parachordal cartilages (McBratney-Owen et al., 2008). Subsequently, the hypophyseal cartilage and acrochordal cartilage develop into the ISS and SOS, respectively (McBratney-Owen et al., 2008). The cartilage primordium of the ISS is derived from the neural crest, whereas the SOS has a more complex origin, wherein its cartilage primordium is derived from the neural crest as well as the cranial mesoderm (Figure 1C; McBratney-Owen et al., 2008). The SOS contributes to the embryonic and postnatal elongation of the cranial base, until its ossification between the ages of 16 and 18 years in humans, whereas complete ossification of the ISS occurs between 2 and 3 years of age (Madeline and Elster, 1995), suggesting that the role of the SOS, in particular, is important in the postnatal stage.
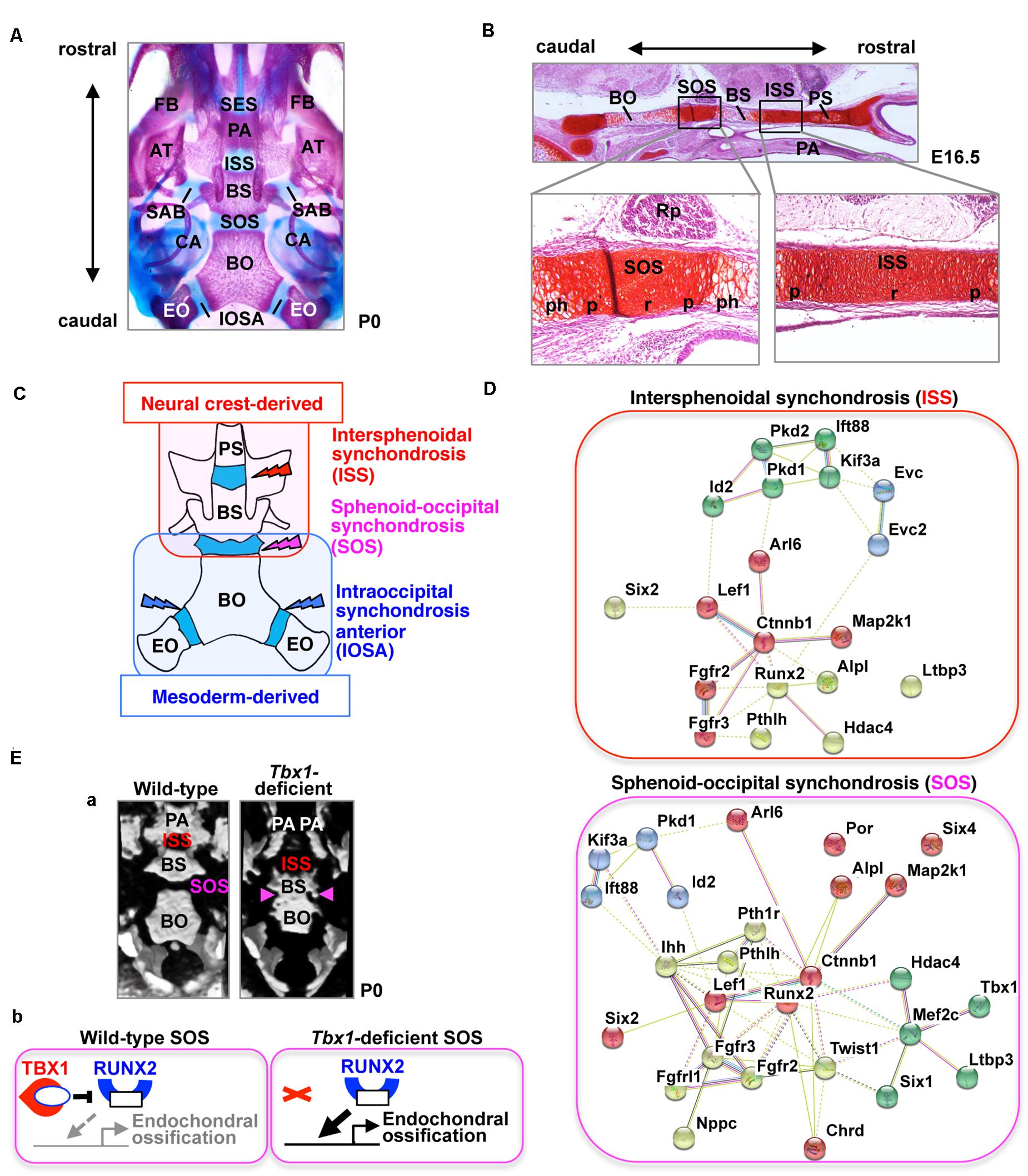
Figure 1. The cranial base and synchondroses. (A) Ventral view of bone staining of the mouse cranial base at postnatal day (P) 0. The middle line of the cranial base is formed by the presphenoid, basisphenoid (BS), and basioccipital (BO) bones. Between the mineralized bones, there are two cartilaginous synchondroses, the intersphenoid synchondrosis (ISS) and the spheno-occipital synchondrosis (SOS). Please note that the presphenoid bone is invisible because of the palatine process (PA). AT, ala temporalis (greater wing) of the basisphenoid bone; CA, canalicular part of auditory capsule; EO, exoccipital bone; FB, facial bone; IOSA, intraoccipital synchondrosis; SAB, synchondrosis, alar-basisphenoidalis; SES, spheno-ethmoidal synchondrosis. (B) Safranin-O staining of the mouse cranial base at embryonic day (E) 16.5. The presphenoid (PS), basisphenoid (BS), and basioccipital (BO) bones are separated by two synchondroses, the intersphenoid synchondrosis (ISS) and spheno-occipital synchondrosis (SOS). The synchondrosis is composed of bipolar growth plates with a central resting (r), proliferating (p), and prehypertrophic (ph) zones. PA, palate; Rp, Rathke’s pouch. (C) Schematic illustration of the tissue origins of the cranial base derived from the neural crest shown in red and those derived from the mesoderm in blue (McBratney-Owen et al., 2008). BS, basisphenoid bone; BO, basioccipital bone; EO, exoccipital bone; PS, presphenoid bone. (D) STRING protein-protein interaction network of mouse genes involved in abnormal synchondroses. The network was constructed using the STRING tool3, with mouse genes involved in abnormal synchondroses (Table 1) used as input. Different colors represent different kinds of evidence of connection between proteins. (E) (a) Skulls from wild-type and Tbx1-deficient mice at birth were analyzed by micro-computed tomography and are shown in a “bird’s eye view.” In Tbx1-deficient mice, the spheno-occipital synchondrosis (SOS) was completely mineralized (Funato et al., 2020). BO, basioccipital bone; BS, basisphenoid bone; ISS, intersphenoid synchondrosis; PA, palatine process. (b) A predicted model for TBX1-mediated regulation of endochondral ossification of SOS. By inhibiting the activity of RUNX2 and the expression of RUNX2 target genes, TBX1 negatively regulates chondrocyte differentiation as well as subsequent endochondral ossification in the SOS.
The cranial base is formed by endochondral ossification, which begins with the formation of a cartilage primordium from condensed mesenchymal cells (McBratney-Owen et al., 2008). Chondrocyte proliferation maintains the synchondroses and leads to elongation of the cranial base (Matsushita et al., 2009). Immature chondrocytes undergo hypertrophy and subsequent apoptosis, followed by the formation of ossification centers after the invasion of osteoblasts from the perichondrium (St-Jacques et al., 1999). Endochondral ossification of the cranial synchondroses is different from that of skeletal bones in several ways. The synchondrosis is composed of bipolar growth plates with resting, proliferating, pre-hypertrophic, and hypertrophic zones that produce growth in opposing directions, whereas long bones are composed of a unipolar growth plate (Wei et al., 2016). This review presents new insights on the signaling pathways and transcription factors involved in the regulation of synchondrosis development, highlighting the differences and similarities between synchondroses present in the cranial base.
Signaling Pathways in Synchondrosis Development
For the normal progression of the development of synchondroses, stringent regulation of chondrocyte differentiation in the cranial synchondroses is crucial. To find the relationship between genetic or molecular interaction networks in the synchondroses, genetically modified mice associated with abnormal synchondroses were investigated. Using the Mouse Genome Informatics1 database and PubMed2, 31 mouse genes with abnormal annotations in SOS and/or ISS were discovered (Table 1). These genes indicated that the regulation of synchondrosis development involves the interaction of several signaling pathways, including the parathyroid hormone-like hormone (PTHLH)/parathyroid hormone-related protein (PTHrP), Indian hedgehog (Ihh), Wnt/β-catenin, and fibroblast growth factor (FGF) pathways, as well as control by cilium assembly and by transcription factors encoded by specific genes (Figure 1D). This review focuses on the genes listed in Table 1.
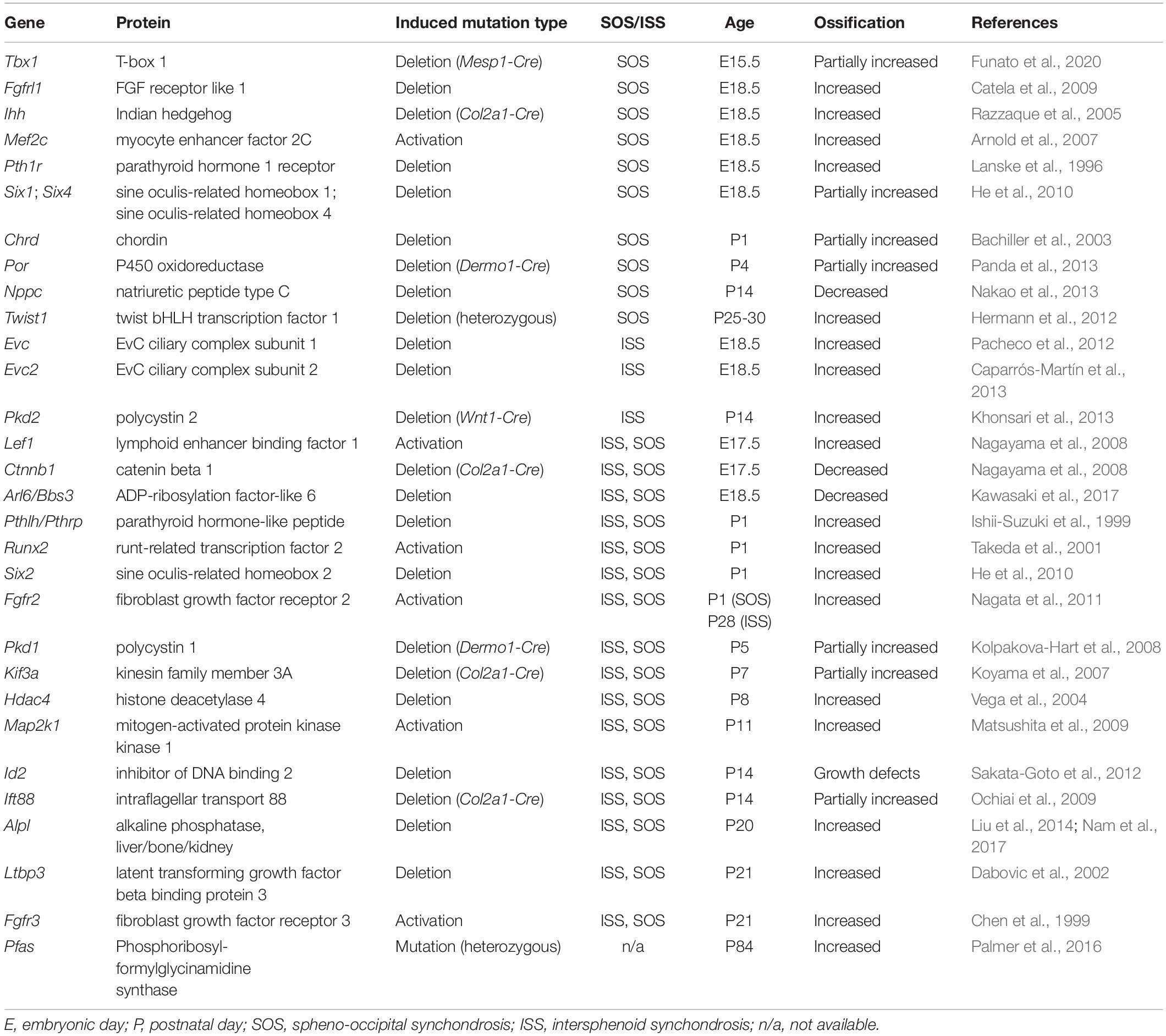
Table 1. Mouse genes involved in abnormal development of the cranial synchondroses.
Runt-Related Transcription Factor 2
Runt-Related Transcription Factor 2 (RUNX2), a gene implicated in cleidocranial dysplasia (Online Mendelian Inheritance in Man; OMIM #119600), is a crucial transcription factor of osteoblast and chondrocyte differentiation (Ducy et al., 1997; Komori et al., 1997; Yoshida et al., 2004). Skull radiography of patients with cleidocranial dysplasia caused by RUNX2 haploinsufficiency showed persistent synchondroses primarily associated with defective development of membranous bones (Kreiborg et al., 1999; Al Kaissi et al., 2013). Chondrocyte-specific constitutive Runx2 expression in mice has also been shown to induce precocious endochondral ossification in the cranial cartilage (Takeda et al., 2001).
RUNX2 and histone deacetylase 4 (HDAC4) are expressed in prehypertrophic and hypertrophic chondrocytes present in developing cartilages. HDAC4 regulates chondrocyte differentiation and endochondral bone formation by interacting with and inhibiting the activity of RUNX2 (Inada et al., 1999; Enomoto et al., 2000; Vega et al., 2004). Furthermore, Hdac4-deletion mice exhibit precocious endochondral ossification of cranial synchondrosis (Vega et al., 2004). Myocyte enhancer factor 2C (MEF2C) regulates a Runx2 enhancer in chondrocytes, and an activating form of MEF2C in mice causes precocious chondrocyte hypertrophy as well as ossification in SOS (Arnold et al., 2007). Runx2 expression has been detected in the cartilaginous condensation of the cranial cartilages at embryonic day 13.5 (Funato et al., 2020), yet ossification of synchondroses did not occur in the wild-type embryos. This time lag between Runx2 expression and execution of chondrocyte differentiation in the synchondroses implies that multiple layers of regulation are required in synchondrosis development and that HDAC4 and MEF2C could be the regulators involved in this process.
T-Box Transcription Factor Family 1
T-box Transcription Factor Family 1 (TBX1) is the candidate gene of DiGeorge (OMIM #188400), velocardiofacial (OMIM #192430), and conotruncal anomaly face (OMIM #217095) syndromes. Tbx1-deficient mice exhibit most features similar to the human syndromes, including microcephaly (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Funato et al., 2012, 2015). Mice lacking Chrd—which encodes chordin, i.e., an antagonist of bone morphogenetic proteins (BMPs)—exhibit recapitulating phenotypes of DiGeorge syndrome and Tbx1-deletion mice (Bachiller et al., 2003). Recently, we reported that TBX1 is a specific and essential regulator of chondrocyte differentiation and subsequent ossification at the SOS (Funato et al., 2020). By inhibiting the activity of RUNX2 and the expression of RUNX2 target genes, TBX1 negatively regulates chondrocyte differentiation and subsequent ossification in the SOS (Figure 1E).
FGF Pathway
The FGF receptor (FGFR) family is a subfamily of receptor tyrosinekinases. Dominant gain-of-function mutations of FGFR2 induce craniofacial dysmorphology in Apert (OMIM #101200), Crouzon (OMIM #123500), Pfeiffer (OMIM #101600), Jackson-Weiss (OMIM #123150), and Antley-Bixler (OMIM #207410) syndromes. Mice carrying the Fgfr2 mutation exhibit accelerated chondrocyte maturation, accompanied by precocious ossification in the SOS and ISS synchondroses, at birth and 4 week-old stage, respectively (Nagata et al., 2011). A patient with Antley-Bixler syndrome was also identified to be harboring a mutation in FGFRL1 (Rieckmann et al., 2009). Fgfrl1-deficient mice showed precocious ossification in the SOS at E18.5 (Catela et al., 2009). Homozygous mutations in the POR gene, encoding cytochrome P450 oxidoreductase, induce midface hypoplasia and craniosynostosis in Antley-Bixler syndrome, accompanied by genital anomalies and disordered steroidogenesis (OMIM #201750). Conditional deletion of Por in osteoprogenitors with Dermo1-Cre affects synchondrosis and long bone development in mice recapitulating Antley-Bixler syndrome (Panda et al., 2013). Although the craniofacial dysmorphology caused by POR mutations and by FGFR2 mutations overlap, the pathogenesis underlying the skeletal malformation in POR deficiency remains to be elucidated.
Gain-of-function mutation of FGFR3 is reported in most cases of achondroplasia (OMIM #100800) and Muenke syndrome (OMIM #602849), which are associated with craniofacial and skeletal abnormalities. Targeted mutations in Fgfr3 in mice carrying the equivalent human syndromes lead to decreased chondrocyte proliferation along with accelerated osteoblast differentiation, resulting in precocious ossification of synchondroses (Chen et al., 1999, 2001; Matsushita et al., 2009; Laurita et al., 2011). Additionally, chondrocyte-specific expression of constitutively active mitogen-activated protein kinase 1 (MAP2K1)/MEK1 causes precocious ossification of cranial synchondroses and effectively rescues the Fgfr3-deficient mouse phenotype (Matsushita et al., 2009).
PTHLH/PTHrP Pathway
PTHLH, also known as PTHrP, maintains chondrocyte proliferation in conjunction with parathyroid hormone 1 receptor (PTH1R). PTHLH/PTHrP impedes chondrocyte differentiation through the inhibition of Runx2 expression (Li et al., 2004). In Pthlh/Pthrp-deletion mice, chondrocyte differentiation is accelerated in both the SOS as well as the ISS (Ishii-Suzuki et al., 1999). Moreover, PTHLH/PTHrP promotes dephosphorylation and nuclear localization of HDAC4, subsequently inhibiting MEF2C transcription (Kozhemyakina et al., 2009). Pth1r-deletion mice are shown to exhibit abnormal neurocranium morphology due to excessive mineralization of synchondroses present between the basioccipital, exoccipital, and basisphenoid bones (Lanske et al., 1996).
Ihh Pathway
The Ihh pathway coordinates diverse aspects of bone morphogenesis via PTHLH/PTHrP-dependent and independent processes (St-Jacques et al., 1999). Ihh is expressed in the synchondroses within the prehypertrophic chondrocytes via RUNX2 regulation and promotes chondrocyte proliferation as well as differentiation (Young et al., 2006; Nagayama et al., 2008; Ushijima et al., 2014). In Ihh-deficient synchondroses, chondrocyte proliferation is decreased, and their differentiation is initially delayed (Razzaque et al., 2005; Young et al., 2006). Furthermore, conditional deletion of Ihh with Col2a1-Cre results in loss of the SOS at E18.5 (Razzaque et al., 2005).
Cilium Assembly
The hedgehog signaling pathway requires cilium assembly. Kinesin family member 3A (KIF3A) is an intraflagellar transport (IFT) motor protein essential for the formation of cilia (Huangfu et al., 2003). Conditional deletion of Kif3a with Col2a1-Cre results in precocious ossification of synchondroses, by disrupting the expression pattern of Ihh in synchondroses (Koyama et al., 2007). Conditional deletion of Ift88, which encodes IFT88/polaris, ultimately results in a deformed basicranium, along with precocious ossification of synchondroses due to disruption of the Ihh signaling pathway (Ochiai et al., 2009). Polycystin-1 and polycystin-2, which are encoded by Pkd1 and Pkd2, form a protein complex and localize to the primary cilium. Conditional deletion of Pkd1 with Dermo1-Cre exhibits a premature closure of both the ISS and SOS (Kolpakova-Hart et al., 2008), whereas conditional deletion of Pkd2 in neural crest with Wnt1-Cre exhibits abnormal ossification of neural crest-derived ISS (Khonsari et al., 2013).
EVC and EVC2 are the disease genes implicated in Ellis-van Creveld syndrome (OMIM #225500) as well as Weyers acrofacial dysostosis (OMIM #193530). EvC ciliary complex subunit 1 (EVC) and EVC2 localize at the base of chondrocyte cilia and function as positive regulators of Ihh-mediated bone development (Takeda et al., 2002; Ruiz-Perez et al., 2007; Caparrós-Martín et al., 2013). Both Evc- and Evc2-deficient mice exhibit precocious ossification of the ISS at E18.5 (Pacheco et al., 2012; Caparrós-Martín et al., 2013).
ADP-ribosylation factor-like 6, which is encoded by ARL6/BBS3, regulates intracellular traffic. Mutations in ARL6/BBS3 account for Bardet-Biedl syndrome-3 (OMIM #600151), which is characterized by retinal dystrophy, renal structural abnormalities, history of obesity, and skeletal abnormalities. Arl6/Bbs3-deficient mice are shown to exhibit hypomorphic cranial synchondroses at E18.5 (Kawasaki et al., 2017).
Wnt/β-Catenin Pathway
The Wnt/β-catenin and Ihh signaling pathways interact with one another to regulate the development of the endochondral bones (Mak et al., 2006). Conditional deletion of Ctnnb1, which encodes CTNNB1/β-catenin, with Col2a1-Cre results in abnormal bone formation (Day et al., 2005; Nagayama et al., 2008). β-catenin and T-cell factor/lymphoid enhancer factor 1 (TCF/LEF1) are transcriptional mediators of the Wnt/β-catenin signaling pathway that directly interact with the Ihh promoter in chondrocytes in vivo, suggesting that the Wnt/β-catenin signaling pathway regulates Ihh expression (Später et al., 2006). Cartilage overexpression of a constitutively active form of LEF1 causes accelerated chondrocyte hypertrophy, topographical disorganization, and excessive bone collar formation in the ISS and SOS (Nagayama et al., 2008). Interestingly, LEF1 is reported to interact with and consequently inhibit the activity of RUNX2 (Kahler and Westendorf, 2003), suggesting that LEF1 might regulate RUNX2 activity during the development of synchondroses.
SIX Homeobox Family
The sine oculis homeobox (SIX) family of transcription factors regulates the embryonic development of the ears and kidneys. Six2-deficient mice display precocious ossification of synchondroses at birth due to disruptions in chondrocyte differentiation, in conjunction with reduced proliferation and accelerated terminal differentiation of the cells (He et al., 2010). SIX1 is implicated in Branchiootic syndrome 3 (OMIM #608389) and deafness (OMIM #605192). Double knockout mice of Six1 and Six4 genes show a precocious partial ossification of the SOS at E18.5 (He et al., 2010).
Discussion
During synchondrosis development, the cross-talk between several signaling pathways, including PTHLH/PTHrP, FGF, Ihh, and Wnt/β-catenin, and control by cilium assembly and by transcription factors, play critical roles. Since the cranial abnormalities in female carriers of the P250R mutation in FGFR3 are more severe than those of the male carriers (Lajeunie et al., 1999), it would be interesting to study whether the onset and complete ossification of synchondroses vary based on gender in wild-type and genetically modified mice. Histological analysis of precocious ossification of synchondroses indicated that the deletion of the RUNX2 inhibitors HDAC4, MEF2C, and TBX1 in mice resulted in accelerated chondrocyte differentiation and, consequently, precocious endochondral ossification of cranial synchondroses (Vega et al., 2004; Arnold et al., 2007; Funato et al., 2020). Consistent with the precocious ossification of the synchondroses in these genetically modified mice, chondrogenic markers were ectopically expressed during synchondrosis formation. Since bone collar ossification occurs secondary to chondrocyte hypertrophy during endochondral bone formation (Chung et al., 2001; Arnold et al., 2007), precocious ossification of synchondroses in these genetically modified mice could occur when chondrocyte hypertrophy is accelerated. The accelerated chondrocyte hypertrophy may also result in a shortage of the reserves of resting and proliferating chondrocytes.
Phenotypic Differences Between SOS and ISS
The synchondrosis phenotype is different among genetically modified mice. Deletion of Pthlh/Pthrp or Six2 or overexpression of Runx2 in chondrocytes resulted in precocious ossification both in the ISS and the SOS. Precocious ossification is specific to the SOS in Tbx1-, Fgfrl1-, Ihh-, and Pth1r-deficient mice and Mef2c-superactivating mice, whereas it is specific to the ISS in Arl6/Bbs3-, Evc-, and Evc2-deficient mice (Table 1). Phenotypic differences among the synchondroses may be due to varying origins of the ISS and SOS (Figure 1C). The cartilage primordium of the ISS is derived from the neural crest, whereas the SOS has a more complex origin, comprising the cartilage primordium derived from the neural crest along with the cranial mesoderm (McBratney-Owen et al., 2008). TBX1 is a specific regulator of SOS development. Since TBX1 is expressed in the mesoderm-derived primordium cartilage of the SOS, differences in the expression pattern of TBX1 likely contribute to the discordant abnormalities between the ISS and SOS (Funato et al., 2020). A consequence of functional redundancy of family genes might also contribute to the same. In the synchondroses of Ihh-deficient mice, the hypertrophic chondrocytes in the ISS are more affected than those in the SOS (Young et al., 2006). The remnants of the notochord express Sonic hedgehog (Shh) near the primordium of the SOS but not in the ISS. Since Shh has a redundant interaction with Ihh (Zhang et al., 2001), Shh may induce the milder phenotype of the SOS than the ISS of Ihh-deficient mice (Young et al., 2006).
Phenotypic Differences Between the Growth Plate and Synchondroses
The growth plates of cranial synchondroses and long bones contribute to bone elongation as well as shaping of the mature bone via endochondral ossification. However, the growth plate of synchondrosis and the long bone are histologically, environmentally, and developmentally different in the following aspects: (1) the mirror image growth plates of synchondrosis produce longitudinal bone growth in bipolar directions, but the growth plate of long bones produces growth in unipolar direction; (2) the long bones are overlaid by articular synovial layers, which are absent in the synchondrosis; (3) the growth plate in developing long bones present the secondary ossification center, which is absent in the synchondrosis; (4) mechanical stress influences the growth of long bones (Sharir et al., 2011); and (5) the ISS originates from the neural crest, while the SOS has a complex unique contribution of both the neural crest and cranial mesoderm, and long bones are derived from mesoderm. Therefore, discordant abnormalities in the growth plates of the long bones and synchondroses are likely attributable to the differences in location-specific downstream signaling targets and the expression patterns of the signaling factors, which differ according to the unique origins and anatomical structures.
RUNX2, HDAC4, and MEF2C control endochondral ossification in the growth plates of both synchondroses and long bones (Takeda et al., 2001; Vega et al., 2004; Arnold et al., 2007). However, in other mutant mice, discordant abnormalities between long bones and synchondroses have been reported. Zinc finger transcriptional coregulator 521 (ZFP521), whose expression is regulated by PTHLH/PTHrP, associates with and antagonizes RUNX2 activity in chondrocytes via an HDAC4-dependent mechanism (Correa et al., 2010). Deletion of Zfp521 in chondrocytes does not affect the synchondrosis development; however, long bones appear to be hypomorphic (Correa et al., 2010). Deletion of Tbx1 results in precocious endochondral ossification of the SOS, but not in the skeletal cartilages despite TBX1 expression in immature chondrocytes (Funato et al., 2015, 2020).
In the synchondroses of Pthlh/Pthrp-deletion mice, chondrocyte differentiation is significantly accelerated compared with those chondrocytes present in long bones (Ishii-Suzuki et al., 1999). Ihh is expressed in prehypertrophic chondrocytes and stimulates Pthlh/Pthrp expression in periarticular chondrocytes in long bones. In the synchondrosis, an overlaid periarticular layer is absent, and the Ihh signaling relays cross-talks between Ihh-producing prehypertrophic chondrocytes and PTHLH/PTHrP-producing proliferating chondrocytes (Young et al., 2006). Since PTHLH/PTHrP is expressed in both the resting and the proliferating chondrocytes in the synchondroses and in the resting chondrocytes of long bones, varied distribution of PTHLH/PTHrP-expressing chondrocytes may contribute to the discordant phenotypes between the synchondrosis and long bones of Pthlh/Pthrp-deficient mice (Young et al., 2006; Nagayama et al., 2008).
Conclusion
Synchondroses are formed through endochondral ossification and play a critical role in the elongation of the basicranium. Deletions or activation of genes can cause the precocious ossification or hypoplasia of synchondroses, suggesting that stringent regulation of signaling pathways is crucial for proper synchondrosis development. The disruption of genes leads to both similar and distinctly different abnormalities in the development of the two synchondroses and also between the growth plates of synchondrosis and skeletal bones. Despite its importance, few studies have addressed the molecular mechanisms that regulate the endochondral ossification of synchondroses. It is important to fully elucidate the interaction of signaling pathways for the regulation of synchondrosis development. In addition, the detailed molecular mechanisms that mark the differences between the synchondroses and the skeletal bones should be deciphered. Hopefully, these insights from future studies will provide possible strategies for biologics-based therapies to treat synchondrosis anomalies.
Author Contributions
NF contributed to the conceptual idea, performed the database searches, analyzed the data, and wrote the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [20K09901]; and the Astellas Foundation for Research on Metabolic Disorders.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Editage (www.editage.com) for English language editing.
Footnotes
References
Al Kaissi, A., Ben Chehida, F., Kenis, V., Ganger, R., Radler, C., Hofstaetter, J. G., et al. (2013). Broad spectrum of skeletal malformation complex in patients with cleidocranial dysplasia syndrome: radiographic and tomographic study. Clin. Med. Insights Arthritis Musculoskelet. Disord. 6, 45–55. doi: 10.4137/CMAMD.S11933
Arnold, M. A., Kim, Y., Czubryt, M. P., Phan, D., McAnally, J., Qi, X., et al. (2007). transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 12, 377–389. doi: 10.1016/j.devcel.2007.02.004
Bachiller, D., Klingensmith, J., Shneyder, N., Tran, U., Anderson, R., Rossant, J., et al. (2003). The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development 130, 3567–3578. doi: 10.1242/dev.00581
Caparrós-Martín, J. A., Valencia, M., Reytor, E., Pacheco, M., Fernandez, M., Perez-Aytes, A., et al. (2013). The ciliary Evc/Evc2 complex interacts with smo and controls hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 22, 124–139. doi: 10.1093/hmg/dds409
Catela, C., Bilbao-Cortes, D., Slonimsky, E., Kratsios, P., Rosenthal, N., and Te Welscher, P. (2009). Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated in Fgfrl1 null mice. Dis. Model. Mech. 2, 283–294. doi: 10.1242/dmm.002287
Chen, L., Adar, R., Yang, X., Monsonego, E. O., Li, C., Hauschka, P. V., et al. (1999). Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J. Clin. Invest. 104, 1517–1525. doi: 10.1172/JCI6690
Chen, L., Li, C., Qiao, W., Xu, X., and Deng, C. (2001). A Ser365Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum. Mol. Genet. 10, 457–465. doi: 10.1093/hmg/10.5.457
Chung, U. I., Schipani, E., McMahon, A. P., and Kronenberg, H. M. (2001). Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Invest. 107, 295–304. doi: 10.1172/JCI11706
Correa, D., Hesse, E., Seriwatanachai, D., Kiviranta, R., Saito, H., Yamana, K., et al. (2010). Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev. Cell 19, 533–546. doi: 10.1016/j.devcel.2010.09.008
Dabovic, B., Chen, Y., Colarossi, C., Obata, H., Zambuto, L., Perle, M. A., et al. (2002). Bone Abnormalities in latent TGF-β binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-β bioavailability. J. Cell Biol. 156, 227–232. doi: 10.1083/jcb.200111080
Day, T. F., Guo, X., Garrett-Beal, L., and Yang, Y. (2005). Wnt/β-Catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750. doi: 10.1016/J.DEVCEL.2005.03.016
Ducy, P., Zhang, R., Geoffroy, V., Ridall, A. L., and Karsenty, G. (1997). Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754. doi: 10.1016/s0092-8674(00)80257-3
Enomoto, H., Enomoto-Iwamoto, M., Iwamoto, M., Nomura, S., Himeno, M., Kitamura, Y., et al. (2000). Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 275, 8695–8702. doi: 10.1074/jbc.275.12.8695
Funato, N., Nakamura, M., Richardson, J. A., Srivastava, D., and Yanagisawa, H. (2012). Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 21, 2524–2537. doi: 10.1093/hmg/dds071
Funato, N., Nakamura, M., Richardson, J. A., Srivastava, D., and Yanagisawa, H. (2015). Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 24, 424–435. doi: 10.1093/hmg/ddu458
Funato, N., Srivastava, D., Shibata, S., and Yanagisawa, H. (2020). TBX1 regulates chondrocyte maturation in the spheno-occipital synchondrosis. J. Dent. Res. 99, 1182–1191. doi: 10.1177/0022034520925080
Goldstein, J. A., Paliga, J. T., Wink, J. D., Bartlett, S. P., and Nah, H. D. (2014). Earlier evidence of spheno-occipital synchondrosis fusion correlates with severity of midface hypoplasia in patients with syndromic craniosynostosis. Plast. Reconstr. Surg. 134, 504–510. doi: 10.1097/PRS.0000000000000419
He, G., Tavella, S., Hanley, K. P., Self, M., Oliver, G., Grifone, R., et al. (2010). Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev. Biol. 344, 720–730. doi: 10.1016/j.ydbio.2010.05.509
Hermann, C. D., Lee, C. S. D., Gadepalli, S., Lawrence, K. A., Richards, M. A., Olivares-Navarrete, R., et al. (2012). Interrelationship of cranial suture fusion, basicranial development, and resynostosis following suturectomy in Twist1+/- mice, a murine model of Saethre-Chotzen syndrome. Calcif. Tissue Int. 91, 255–266. doi: 10.1007/s00223-012-9632-3
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., and Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. doi: 10.1038/nature02061
Inada, M., Yasui, T., Nomura, S., Miyake, S., Deguchi, K., Himeno, M., et al. (1999). Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 214, 279–290. doi: 10.1002/(sici)1097-0177(199904)214:4<279::aid-aja1>3.0.co;2-w
Ishii-Suzuki, M., Suda, N., Yamazaki, K., Kuroda, T., Senior, P. V., Beck, F., et al. (1999). Differential responses to parathyroid hormone-related protein (PTHrP) deficiency in the various craniofacial cartilages. Anat. Rec. 255, 452–457. doi: 10.1002/(sici)1097-0185(19990801)255:4<452::aid-ar10>3.0.co;2-e
Jerome, L. A., and Papaioannou, V. E. (2001). DiGeorge syndrome phenotype in mice mutant for the T-Box gene, Tbx1. Nat. Genet. 27, 286–291. doi: 10.1038/85845
Kahler, R. A., and Westendorf, J. J. (2003). Lymphoid enhancer factor-1 and β-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. J. Biol. Chem. 278, 11937–11944. doi: 10.1074/jbc.M211443200
Kawasaki, M., Izu, Y., Hayata, T., Ideno, H., Nifuji, A., Sheffield, V. C., et al. (2017). Bardet-Biedl syndrome 3 regulates the development of cranial base midline structures. Bone 101, 179–190. doi: 10.1016/j.bone.2016.02.017
Khonsari, R. H., Ohazama, A., Raouf, R., Kawasaki, M., Kawasaki, K., Porntaveetus, T., et al. (2013). Multiple postnatal craniofacial anomalies are characterized by conditional loss of polycystic kidney disease 2 (Pkd2). Hum. Mol. Genet. 22, 1873–1885. doi: 10.1093/hmg/ddt041
Kolpakova-Hart, E., McBratney-Owen, B., Hou, B., Fukai, N., Nicolae, C., Zhou, J., et al. (2008). Growth of cranial synchondroses and sutures requires polycystin-1. Dev. Biol. 321, 407–419. doi: 10.1016/j.ydbio.2008.07.005
Komori, T., Yagi, H., Nomura, S., Yamaguchi, A., Sasaki, K., Deguchi, K., et al. (1997). Targeted Disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764. doi: 10.1016/s0092-8674(00)80258-5
Koyama, E., Young, B., Nagayama, M., Shibukawa, Y., Enomoto-iwamoto, M., Iwamoto, M., et al. (2007). Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 2169, 2159–2169. doi: 10.1242/dev.001586
Kozhemyakina, E., Cohen, T., Yao, T. P., and Lassar, A. B. (2009). Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 Pathway. Mol. Cell. Biol. 29, 5751–5762. doi: 10.1128/mcb.00415-09
Kreiborg, S., Jensen, B. L., Larsen, P., Schleidt, D. T., and Darvann, T. (1999). Anomalies of craniofacial skeleton and teeth in cleidocranial dysplasia. J. Craniofac. Genet. Dev. Biol. 19, 75–79.
Lajeunie, E., El Ghouzzi, V., Le Merrer, M., Munnich, A., Bonaventure, J., and Renier, D. (1999). Sex related expressivity of the phenotype in coronal craniosynostosis caused by the recurrent P250R FGFR3 mutation. J. Med. Genet. 36, 9–13.
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., et al. (1996). PTH/PTHrP receptor in early development and indian hedgehog-regulated bone growth. Science 273, 663–666. doi: 10.1126/science.273.5275.663
Laurita, J., Koyama, E., Chin, B., Taylor, J. A., Lakin, G. E., Hankenson, K. D., et al. (2011). The Muenke syndrome mutation (FgfR3 P244R) causes cranial base shortening associated with growth plate dysfunction and premature perichondrial ossification in murine basicranial synchondroses. Dev. Dyn. 240, 2584–2596. doi: 10.1002/dvdy.22752
Li, T. F., Dong, Y., Ionescu, A. M., Rosier, R. N., Zuscik, M. J., Schwarz, E. M., et al. (2004). Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp. Cell Res. 299, 128–136. doi: 10.1016/j.yexcr.2004.05.025
Lindsay, E. A., Vitelli, F., Su, H., Morishima, M., Huynh, T., Pramparo, T., et al. (2001). Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410, 97–101. doi: 10.1038/35065105
Liu, J., Nam, H. K., Campbell, C., Gasque, K. C. S., Millán, J. L., and Hatch, N. E. (2014). Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl-/- mouse model of infantile hypophosphatasia. Bone 67, 81–94. doi: 10.1016/j.bone.2014.06.040
Madeline, L. A., and Elster, A. D. (1995). Postnatal development of the central skull base: normal variants. Radiology 196, 757–763. doi: 10.1148/radiology.196.3.7644640
Mak, K. K., Chen, M. H., Day, T. F., Chuang, P. T., and Yang, Y. (2006). Wnt/beta-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development 133, 3695–3707. doi: 10.1242/dev.02546
Matsushita, T., Wilcox, W. R., Chan, Y. Y., Kawanami, A., Bükülmez, H., Balmes, G., et al. (2009). FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum. Mol. Genet. 18, 227–240. doi: 10.1093/hmg/ddn339
McBratney-Owen, B., Iseki, S., Bamforth, S. D., Olsen, B. R., and Morriss-Kay, G. M. (2008). Development and tissue origins of the mammalian cranial base. Dev. Biol. 322, 121–132. doi: 10.1016/j.ydbio.2008.07.016
Nagata, M., Nuckolls, G. H., Wang, X., Shum, L., Seki, Y., Kawase, T., et al. (2011). The Primary site of the acrocephalic feature in Apert syndrome is a dwarf cranial base with accelerated chondrocytic differentiation due to aberrant activation of the FGFR2 signaling. Bone 48, 847–856. doi: 10.1016/j.bone.2010.11.014
Nagayama, M., Iwamoto, M., Hargett, A., Kamiya, N., Tamamura, Y., Young, B., et al. (2008). Wnt/β-catenin signaling regulates cranial base development and growth. J. Dent. Res. 87, 244–249. doi: 10.1177/154405910808700309
Nakao, K., Okubo, Y., Yasoda, A., Koyama, N., Osawa, K., Isobe, Y., et al. (2013). The effects of C-type natriuretic peptide on craniofacial skeletogenesis. J. Dent. Res. 92, 58–64. doi: 10.1177/0022034512466413
Nam, H. K., Sharma, M., Liu, J., and Hatch, N. E. (2017). Tissue nonspecific alkaline phosphatase (TNAP) regulates cranial base growth and synchondrosis maturation. Front. Physiol. 8:161. doi: 10.3389/fphys.2017.00161
Ochiai, T., Nagayama, M., Nakamura, T., Morrison, T., Pilchak, D., Kondo, N., et al. (2009). Roles of the primary cilium component polaris in synchondrosis development. J. Dent. Res. 88, 545–550. doi: 10.1177/0022034509337775
Pacheco, M., Valencia, M., Caparrós-Martín, J. A., Mulero, F., Goodship, J. A., and Ruiz-Perez, V. L. (2012). Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 50, 28–41. doi: 10.1016/j.bone.2011.08.025
Palmer, K., Fairfield, H., Borgeia, S., Curtain, M., Hassan, M. G., Dionne, L., et al. (2016). Discovery and characterization of spontaneous mouse models of craniofacial dysmorphology. Dev. Biol. 415, 216–227. doi: 10.1016/j.ydbio.2015.07.023
Panda, S. P., Guntur, A. R., Polusani, S. R., Fajardo, R. J., Gakunga, P. T., Roman, L. J., et al. (2013). Conditional deletion of cytochrome P450 reductase in osteoprogenitor cells affects long bone and skull development in mice recapitulating Antley-Bixler syndrome: role of a redox enzyme in development. PLoS One 8:75638. doi: 10.1371/journal.pone.0075638
Razzaque, M. S., Soegiarto, D. W., Chang, D., Long, F., and Lanske, B. (2005). Conditional deletion of indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 207, 453–461. doi: 10.1002/path.1870
Rieckmann, T., Zhuang, L., Flück, C. E., and Trueb, B. (2009). Characterization of the first FGFRL1 mutation identified in a craniosynostosis patient. Biochim. Biophys. Acta Mol. Basis Dis. 1792, 112–121. doi: 10.1016/j.bbadis.2008.11.006
Ruiz-Perez, V. L., Blair, H. J., Rodriguez-Andres, M. E., Blanco, M. J., Wilson, A., Liu, Y. N., et al. (2007). Evc Is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 134, 2903–2912. doi: 10.1242/dev.007542
Sakata-Goto, T., Takahashi, K., Kiso, H., Huang, B., Tsukamoto, H., Takemoto, M., et al. (2012). Id2 controls chondrogenesis acting downstream of BMP signaling during maxillary morphogenesis. Bone 50, 69–78. doi: 10.1016/j.bone.2011.09.049
Sharir, A., Stern, T., Rot, C., Shahar, R., and Zelzer, E. (2011). Muscle force regulates bone shaping for optimal load-bearing capacity during embryogenesis. Development 138, 3247–3259. doi: 10.1242/dev.063768
Später, D., Hill, T. P., O’Sullivan, R. J., Gruber, M., Conner, D. A., and Hartmann, C. (2006). Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development 133, 3039–3049. doi: 10.1242/dev.02471
St-Jacques, B., Hammerschmidt, M., and McMahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. doi: 10.1101/gad.13.16.2072
Takeda, H., Takami, M., Oguni, T., Tsuji, T., Yoneda, K., Sato, H., et al. (2002). Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc. Natl. Acad. Sci. U.S.A. 99, 10549–10554. doi: 10.1073/pnas.152337899
Takeda, S., Bonnamy, J. P., Owen, M. J., Ducy, P., and Karsenty, G. (2001). Continuous Expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15, 467–481. doi: 10.1101/gad.845101
Ushijima, T., Okazaki, K., Tsushima, H., Ishihara, K., Doi, T., and Iwamoto, Y. (2014). CCAAT/enhancer binding protein β regulates expression of indian hedgehog during chondrocytes differentiation. PLoS One 9:104547. doi: 10.1371/journal.pone.0104547
Vega, R. B., Matsuda, K., Oh, J., Barbosa, A. C., Yang, X., Meadows, E., et al. (2004). Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119, 555–566. doi: 10.1016/j.cell.2004.10.024
Wei, X., Hu, M., Mishina, Y., and Liu, F. (2016). Developmental regulation of the growth plate and cranial synchondrosis. J. Dent. Res. 95, 1221–1229. doi: 10.1177/0022034516651823
Wei, X., Thomas, N., Hatch, N. E., Hu, M., and Liu, F. (2017). Postnatal craniofacial skeletal development of female C57BL/6NCrl mice. Front. Physiol. 8:697. doi: 10.3389/fphys.2017.00697
Yoshida, C. A., Yamamoto, H., Fujita, T., Furuichi, T., Ito, K., Inoue, K., et al. (2004). Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of indian hedgehog. Genes Dev. 18, 952–963. doi: 10.1101/gad.1174704
Young, B., Minugh-Purvis, N., Shimo, T., St-Jacques, B., Iwamoto, M., Enomoto-Iwamoto, M., et al. (2006). Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev. Biol. 299, 272–282. doi: 10.1016/j.ydbio.2006.07.028
Keywords: cranial base, cartilage, mesoderm, neural crest, spheno-occipital synchondrosis, intersphenoid synchondrosis, RUNX2
Citation: Funato N (2020) New Insights Into Cranial Synchondrosis Development: A Mini Review. Front. Cell Dev. Biol. 8:706. doi: 10.3389/fcell.2020.00706
Received: 07 June 2020; Accepted: 13 July 2020;
Published: 11 August 2020.
Edited by:
Erika Kuchler, Universidade Positivo, BrazilReviewed by:
Karla Carpio Horta, University of São Paulo, BrazilSabrina Kathrin Schulze, University of Potsdam, Germany
Arthur Cunha, Rio de Janeiro State University, Brazil
Copyright © 2020 Funato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noriko Funato, noriko-funato@umin.ac.jp