 Ana F. Pina1,2,3
Ana F. Pina1,2,3 Diego O. Borges1,4
Diego O. Borges1,4 Maria João Meneses1,2
Maria João Meneses1,2 Patrícia Branco5,6
Patrícia Branco5,6 Rita Birne5,6
Rita Birne5,6 Antonio Vilasi7
Antonio Vilasi7 Maria Paula Macedo1,3,6*
Maria Paula Macedo1,3,6*- 1Centro de Estudos de Doenças Crónicas, NOVA Medical School/Faculdade de Ciências Médicas, Universidade Nova de Lisboa, Lisbon, Portugal
- 2ProRegeM Ph.D. Programme, NOVA Medical School/Faculdade de Ciências Médicas, Universidade Nova de Lisboa, Lisbon, Portugal
- 3Department of Medical Sciences, University of Aveiro, Aveiro, Portugal
- 4Molecular Biosciences Ph.D. Programme, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal
- 5Department of Nephrology, Centro Hospitalar Lisboa Ocidental, Lisbon, Portugal
- 6Portuguese Diabetes Association – Education and Research Center (APDP-ERC), Lisbon, Portugal
- 7Institute of Clinical Physiology – National Research Council, Reggio Calabria Unit1, Reggio Calabria, Italy
Kidney function in metabolism is often underestimated. Although the word “clearance” is associated to “degradation”, at nephron level, proper balance between what is truly degraded and what is redirected to de novo utilization is crucial for the maintenance of electrolytic and acid–basic balance and energy conservation. Insulin is probably one of the best examples of how diverse and heterogeneous kidney response can be. Kidney has a primary role in the degradation of insulin released in the bloodstream, but it is also incredibly susceptible to insulin action throughout the nephron. Fluctuations in insulin levels during fast and fed state add another layer of complexity in the understanding of kidney fine-tuning. This review aims at revisiting renal insulin actions and clearance and to address the association of kidney dysmetabolism with hyperinsulinemia and insulin resistance, both highly prevalent phenomena in modern society.
Introduction
Insulin is a vital hormone with several functions among which its central role in energy and glucose homeostasis is fundamental (Plum, 2006). Beyond the traditional crosstalk between the most recognized insulin-responsive organs (liver, adipose tissue, and skeletal muscle), kidney insulin action and resistance have recently been suggested as critical components of metabolic and dysmetabolic states (Artunc et al., 2016). The important role of insulin in the kidney is further corroborated by the increased prevalence of chronic kidney disease in subjects with type 2 diabetes mellitus (T2DM) and non-alcoholic fatty liver disease (NAFLD; Musso et al., 2014; Kim et al., 2018; Mok et al., 2019; Kiapidou et al., 2020). Furthermore, the kidney can adjust the levels of essential players of nutrient homeostasis such as glucose and insulin (Krebs, 1963; Chamberlain and Stimmler, 1967).
First reports of insulin actions in the kidney were originated in the 1950s (Farber et al., 1951). Since then, the knowledge acquired has dramatically increased, nonetheless much remains unveiled. It is now known that insulin impacts on tubular glucose reabsorption (Nakamura et al., 2015). In fact, a whole therapeutic class based on this mechanism was recently developed. Additionally, hyperinsulinemia driven by pancreatic hypersecretion and/or impairments in hepatic insulin clearance could explain changes in glomerular filtration rates and increased renal gluconeogenesis, which in turn causes fatty liver deposition and, as a direct consequence, may lead to dysglycemia (Bril et al., 2014; Naderpoor et al., 2017; Jung et al., 2018; Bergman et al., 2019).
Kidney is, in fact, the most critical organ responsible for insulin clearance after the liver (Elgee and Williams, 1954; Narahara et al., 1958); still, its relevance in the maintenance of proper insulinemia and insulin sensitivity is underestimated. The control of insulin clearance/action exerted by the kidney is complex. Furthermore, the diversity of insulin actions is only possible thanks to the differentiation of specific kidney segments in which insulin regulates different pathways. In this review, we will briefly describe insulin production and its main actions in the liver, adipose tissue, and skeletal muscle. Moreover, we will focus on kidney and on renal insulin actions as well as insulin clearance/degradation. Overall, this work aims at highlighting the critical role of kidney-insulin interplay in the development of dysmetabolism.
Overview of Insulin Biosynthesis and Non-Renal Action and Metabolism
The insulin gene (INS) is represented by only one copy in the human genome, and its transcription is mainly regulated by the same enhancers of other glucose-related genes (Andrali et al., 2008; Gao et al., 2014). In pancreas β-cell, INS is first transcribed as preproinsulin and, after cleavage and folding in the rough endoplasmic reticulum (ER), proinsulin is formed (Hutton, 1994). The conversion into insulin occurs after the removal of the c-peptide in the Golgi apparatus, with a final maturation achieved by c-terminal amino acid elimination (Steiner, 2004). Mature insulin is composed of A and B chains linked by two disulfide bonds, with a third bond within A chain (Mayer et al., 2007). For storage, insulin is complexed in hexamers with zinc, and an actin network is involved in the organization of mature insulin granules for first and second insulin secretion phases (Kalwat and Thurmond, 2013).
Insulin secretion is triggered by cytoplasmic increments in ATP levels, derived from glucose internalization and subsequent metabolism (Prentki and Matschinsky, 1987). This increase in ATP shuts down membrane ATP-sensitive K+ channels, depolarizing plasma membrane, and activating voltage-gated calcium channels (Detimary et al., 1998). Indeed, the resultant Ca2+ influx seems to be necessary for the fusion of insulin granules with the plasma membrane, ending up in its secretion (Hou et al., 2009). The first wave of insulin secretion is composed of primed granules that are in close proximity with plasma membrane (Olofsson et al., 2002). However, most of these granules remain more distant from the plasma membrane and are further boosted with newly synthesized insulin that will culminate in 75–95% of total insulin secretion in the second phase (Kou et al., 2014).
With the insulin pools ready to be released, insulin secretion does not occur in a constant manner, but in waves of 4–5 min interval (Song et al., 2000). The pulsatile secretion profile is considered primordial for proper insulin responsiveness, as constant release would result in down-regulation of insulin receptor (INSR) expression (Matveyenko et al., 2012). After binding and activation of its receptor, insulin, as an anabolic hormone, promotes increased biosynthesis of macromolecules further needed to maintain energetic balance during nutrient privation (Bedinger and Adams, 2015).
At cellular level, insulin mostly relies on the specific bind to its transmembrane receptor to perform its actions (Figure 1). Insulin receptor is a tyrosine kinase receptor with two extracellular α subunits and two transmembrane β subunits (Ye et al., 2017). Insulin binding to INSR produces an autophosphorylation that leads to the internalization of the complex and activation of downstream effectors. INSR substrate 1 (IRS1) and 2 (IRS2) are the main targets that intracellularly coordinate insulin action (Thirone et al., 2006; Kubota et al., 2016). In the liver, the activity of these proteins is differently controlled by insulin. Although both IRS1 and IRS2 are degraded after insulin activation, only IRS2 is transcriptionally repressed by insulin activation (Hirashima et al., 2003). Further transduction of insulin signaling occurs via phosphatidylinositol 3-kinase (PI3K)/AKT phosphorylation. AKT or Protein kinase B (PKB) are considered the main effectors of metabolic insulin actions (Taniguchi et al., 2006; Molinaro et al., 2019). On the other hand, AKT1 and 3 are the leading players of mitogenic effects of insulin by RAS/mitogen activated protein kinase (MAPK) cascade and crosstalk with growth hormones (Xu et al., 2006; Xu and Messina, 2009; Qiu et al., 2017).
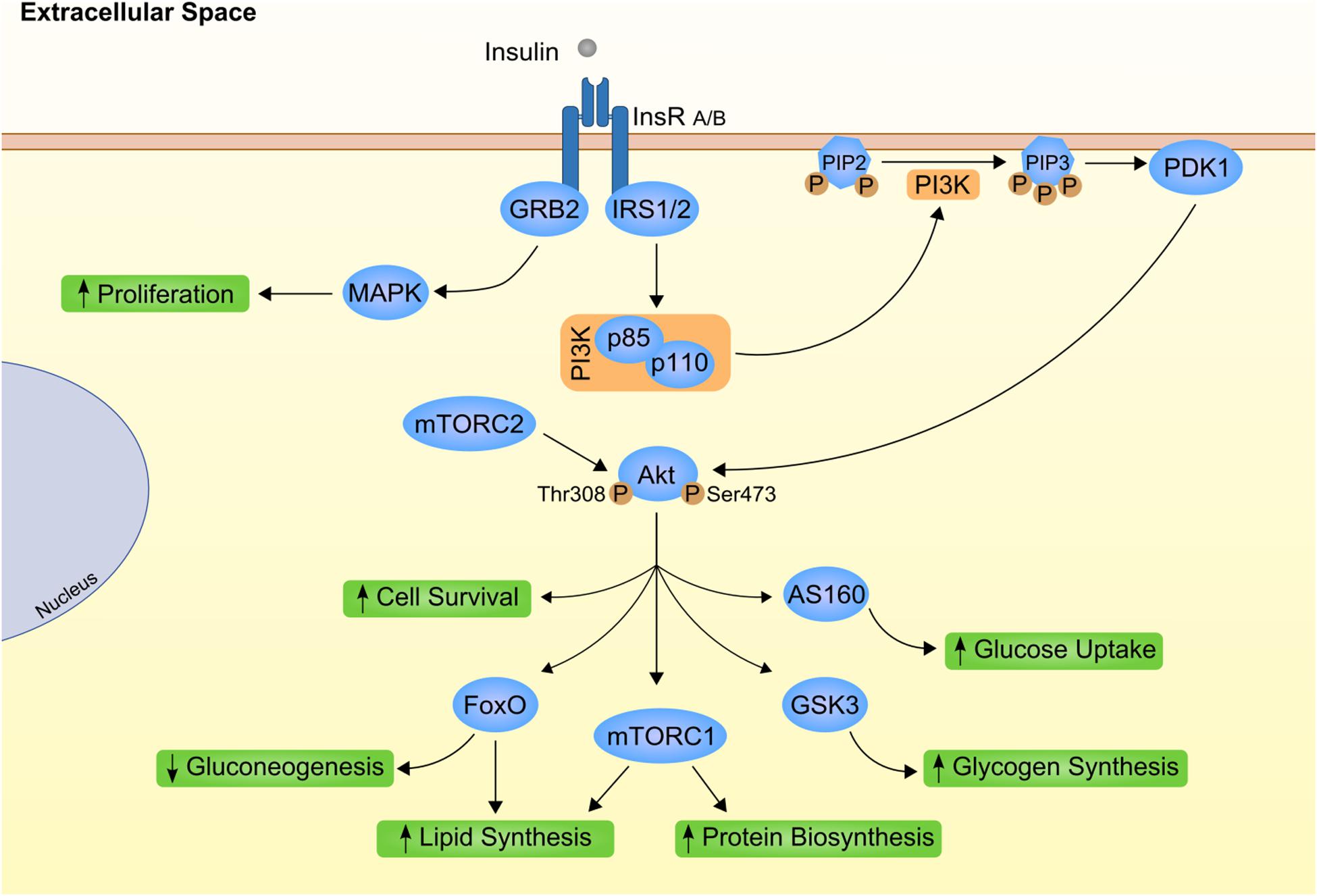
Figure 1. Canonical insulin signaling cascade. Insulin (gray circle) binds to its transmembrane kinase receptor at cell membrane and triggers insulin signaling cascade. Insulin receptor present two isoforms with distinct affinity for insulin already associated to differential insulin internalization (INSR A and INSR B; Calzi et al., 1997). Binding of insulin auto-phosphorylates INSR at Tyr-960. Further recruitment and phosphorylation of insulin receptor substrate (IRS) 1 and 2 will mostly result in subsequent activation of phosphoinositide-dependent kinase 1 (PDK1) through phosphoinositide-3 kinase (PI3K; Yamada et al., 2002). PDK1 is responsible for propagation of insulin signal to one of the most important downstream effectors, Akt. Importantly, Akt has two distinct phosphorylation sites, Thr308 activated by PDK1 (Alessi et al., 1997) and Ser473 phosphorylated by mammalian target of rapamycin complex (mTORC) 2 protein (Bayascas and Alessi, 2005). Finally, fully activated Akt can interact with different proteins, eliciting different effects as stimulation of glucose uptake and glycogen synthesis by AS160 and glycogen synthase kinase 3 (GSK3), respectively (Ng et al., 2008). On the other hand, INRS activation also promote growth factor receptor-bound protein 2 (GRB2) interaction with Shc proteins and activation of mitogen activated protein kinases (MAPK; Skolnik et al., 1993; Xu et al., 2006). This as part of the insulin-mediated proliferative stimuli. PIP2, phosphatidylinositol 4,5 biphosphate; PIP3, phosphatidylinositol 3,4,5 triphosphate.
Given the scope of this review, we will summarize insulin actions on non-renal target organs, but detailed information is available in recent reviews (Gancheva et al., 2018; Tokarz et al., 2018). The first organ to be reached by insulin is the liver, where 50–70% is removed at first pass (Duckworth et al., 1998). In hepatocytes, insulin signaling recruits forkhead box transcription factor 1 (FoxO1) to the cytoplasm, inhibiting the transcription of gluconeogenic enzymes (Ling et al., 2018). In such an abundance of glucose, insulin also stimulates the synthesis of glycogen by phosphorylation of glycogen synthase kinase 3 (GSK3), which leads to dephosphorylation and activation of glycogen synthase (McManus et al., 2005). After the replenishment of liver glycogen stores in the fed state, glucose starts to be degraded to pyruvate, which is the precursor of lipid biosynthesis via de novo lipogenesis (DNL; Sanders and Griffin, 2016). Herein, insulin signaling is also crucial for upregulation of sterol regulatory element binding protein 1c (SREPB1c), a transcription factor associated to further transcription of DNL (Oh et al., 2003; Roder et al., 2007; Krycer et al., 2010).
Only a fraction of the insulin that reaches the liver will get to the periphery, to perform its action at skeletal muscle, adipose tissue, and other insulin-sensitive organs. Skeletal muscle is the greater insulin-dependent “kidnapper” of glucose, it is responsible for ∼60 to 80% of whole-body glucose disposal (DeFronzo et al., 1976; Fernandes et al., 2011). Insulin signals to myocytes through classical signaling, promoting glucose uptake by the dynamic translocation of glucose transporter 4 (GLUT4; Zorzano et al., 1996). Glucose is here mainly used for glycolysis or stored as glycogen as already described for the liver.
During the fed state, a clear inhibitory effect in lipolysis and consequent blunt of free fatty acid (FFA) release is mediated by insulin acting on adipocytes (Dimitriadis et al., 2011). The resultant decrease in FFA availability reduces its hepatic uptake and fatty acid oxidation, changing the disposal of substrates that are mostly used for gluconeogenesis (Galgani et al., 2008). Not exclusively for liver, the effect of insulin in reducing adipose tissue FFA release is important for other organs that, at fed state, change energy source to glucose and recover glycogen stores contributing to normoglycemia (Petersen and Shulman, 2018). In this context, glucose uptake is also stimulated by insulin in the fed state at adipocytes. Furthermore, translocation of glucose transporters to the membrane of adipocytes was very well characterized in 3T3-L1 adipocytes by activation of the canonical PI3K/AKT2 pathway (Ng et al., 2008).
As mentioned above, all these organs expressing INSR are responsible for insulin clearance and local degradation. However, a significant part of insulin degradation is played in the liver (50–70%) by its specialized machinery of internalization and degradation (Najjar and Perdomo, 2019). In the case of internalization, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1) phosphorylation by insulin promotes rapidly receptor-mediated uptake (Poy et al., 2002). Carcinoembryonic antigen-related cell adhesion molecule 1 expression occurs preferentially in liver, and restoration of its function only at liver of null CEACAM-1 mice reverse the hyperinsulinemia and fatty liver deposition observed in global knockout CEACAM-1 model (Russo et al., 2017). Regarding degradation, insulin-degrading enzyme (IDE) is considered the most critical protease to cleave insulin intracellularly and is highly expressed in the liver (Kuo et al., 1993).
The amount of insulin that remains in circulation after liver first pass (30–50%) will be mostly degraded by the kidney (Elgee and Williams, 1954; Narahara et al., 1958). However, the kidney is not just an endpoint for insulin degradation, as insulin also acts throughout the entire nephron. Whereas hepatic insulin clearance is regaining attention as the very first alteration leading to hyperinsulinemia, compensatory mechanisms that could emerge from the kidney are not well known. Impairments in hepatic insulin clearance increase peripheral insulinemia, and very little is described regarding the exact effects of the increased amount of insulin that will further reach the kidney.
Kidney and Insulin Actions
Kidney works together with the liver for maintenance of optimal insulin levels. In humans, kidney removes 6–8 U of insulin per day by two major routes (Figure 2), that apparently have distinct preferential physiological goals: post-glomerular secretion (insulin signaling) and glomerular filtration (nutrient conservation and homeostasis) (Ritz et al., 2013). In the kidney, insulin acts at multiple sites along the nephron, from the glomerulus (Coward et al., 2005; Welsh et al., 2010; Mima et al., 2011; Lay and Coward, 2014) to the renal tubule (Tiwari et al., 2008, 2013; Li et al., 2013), to modulate different functions such as glomerular filtration, gluconeogenesis, renal sodium handling, among others.
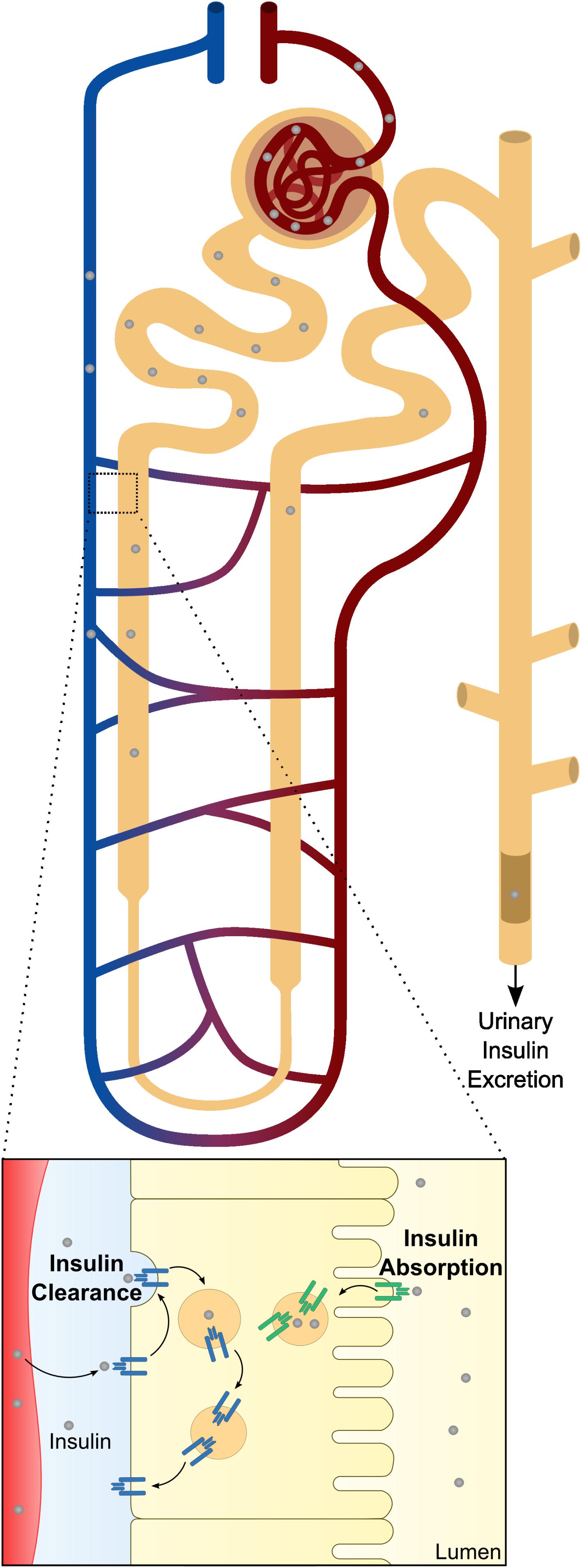
Figure 2. Schematic representation of renal insulin handling. Insulin is depicted in gray circles at different portions of the nephron. As insulin is a small molecule, it will be fully filtered by the glomerular system until it reaches the proximal tubule. At proximal tubule cells, almost all the filtered insulin will be absorbed at the luminal membrane. In physiological conditions, only a small percentage will be excreted in urine. Beyond glomerular filtration, insulin also raise from the perivenous capillaries. In a closer look to the proximal tubule cells is represented both mechanisms of insulin clearance. The increased levels of insulin receptor (INSR) and consequent increased uptake of insulin at the basolateral membrane is also depicted.
Many alterations in insulin associated mechanisms in the kidney are driven by insulin resistance (IR), leading to diabetic nephropathy if not reestablished (Svensson and Eriksson, 2006). In the Asiatic population, it was described that the prevalence of diabetic nephropathy with impaired estimated glomerular filtration rate (eGFR) (with or without albuminuria) was 31.6%, and the prevalence of albuminuria (with or without impaired eGFR) was 16.9 and 22.0%, respectively (Mok et al., 2019). Patients may already show diabetic kidney disease (DKD) at the time of T2DM diagnosis. Nevertheless, 10 years after the diagnosis of T2DM, low-level albuminuria is present in 24.9% of the patients and 5.3% already present macroalbuminuria (Adler et al., 2003).
Glomerular Insulin Actions
Podocytes are the primary constituent cell of the glomerulus, with their long finger-like projections to the glomerular capillaries at the glomerular basement membrane (GBM). These cells have intercellular junctions that form filtration barriers to help maintaining normal renal function. When damaged, podocytes lose their arrangement resulting in a reduction in barrier function. Indeed, one of the DKD features is the podocyte loss with consequent albuminuria (Pagtalunan et al., 1997; Wolf et al., 2005). Lately, it has been unveiled the role of insulin in podocytes (Figure 3); in other words, podocyte IR gives rise to the albuminuric feature.
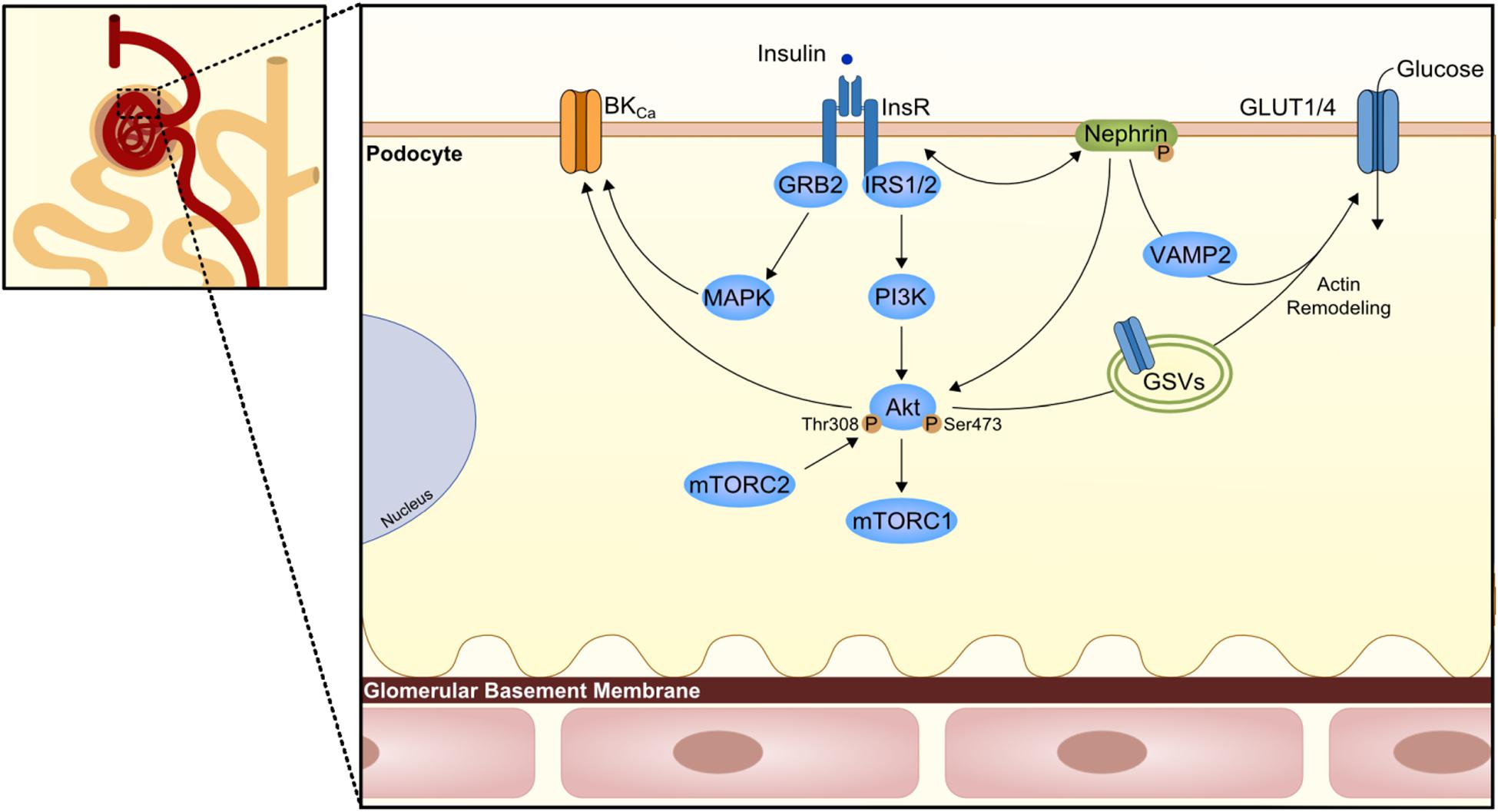
Figure 3. Podocyte insulin signaling. Podocytes are the first cells to interact with insulin at the nephron and express several proteins of the canonical insulin signaling pathway. However, here the podocyte-specific protein nephrin is known to have a role in the trafficking of glucose transporters (GLUT1 or GLUT4) to podocyte membrane and consequently promote glucose uptake. The trafficking seems to involve Vamp2 and actin remodeling. On the other branch of insulin signaling, an effect in the large-conductance Ca2+-activated K+ channels (BKCa) is also important for maintenance of podocyte integrity and proper glomerular filtration. GRB2, growth factor receptor-bound protein 2; GSV, GLUT storage vesicle; VAMP2, Vesicle-associated membrane protein 2.
Podocytes reveal to express proteins of the insulin signaling canonical pathway, namely the INSR, and both IRS1 and IRS2 (Coward et al., 2005; Santamaria et al., 2015), with IRS2 being the most prevalent one. Coward et al. (2005) depicted the effect of insulin in human podocytes, which results in glucose uptake not only through GLUT4 but also through glucose transporter 1 (GLUT1; Figure 3). As insulin promotes the translocation of GLUT4 to the membrane through the activation of PI3K-AKT2-PkB pathway, there is a remodeling of the cortical actin of the cytoskeleton with subsequent contraction (Welsh et al., 2010). In compliance, podocytes-specific deletion of INSR in mice revealed DKD features based on substantial albuminuria and histological features as podocyte foot structure loss and glomerulosclerosis (Welsh et al., 2010). AKT seems to play a central role as its phosphorylation appears to be severely affected in several models of both type 1 diabetes mellitus (T1DM; streptozotocin-induced T1DM) and T2DM (db/db mice, Zucker rats) (Tejada et al., 2008; Mima et al., 2011). Moreover, AKT isoform 2 deletion results in serious glomerular lesions in mice. This can lead to rapid disease progression, also associated with tubular dilatation and microalbuminuria (Canaud et al., 2013). Other relevant players that might contribute to podocyte IR is SH2-domain-containing inositol phosphatase 2 (SHIP2), a down regulator of the PI3K signaling pathway shown to be upregulated in the Zucker rats. Moreover, protein tyrosine-phosphatase 1B (PTP1B), a negative regulator of the INSR activity, or phosphatase and tensin homolog when increased, appears to also compromise the insulin signaling pathway (Mima et al., 2011; Garner et al., 2018).
Podocytes also present an insulin-dependent alternative pathway, the cyclic guanosine monophosphate(cGMP)-dependent protein kinase G (PKG), from which the PKG isoform I-alpha levels are increased in glomeruli of the hyperinsulinemic Zucker rats (Piwkowska et al., 2013). These high insulin levels increase glomerular barrier albumin permeability through a PKGI-reliant mechanism via the NAD(P)H-dependent generation of superoxide anion.
An important player in podocytes physiology is the protein nephrin, a podocyte-specific protein, which is responsible for the maintenance of the integrity of the filtration barrier. In fact, nephrin mutations are involved in severe nephrotic syndrome (Lenkkeri et al., 1999). Nephrin appears to play a most outstanding role in the trafficking of GLUT4 and GLUT1 by interacting with Vamp2 as well as by interacting with insulin-stimulated actin remodeling (Coward et al., 2007; Lay et al., 2017). Of interest, nephrin can also induce phosphorylation of p70S6K in a PI3K-dependent manner independently of INSR/AKT2 activation (Villarreal et al., 2016; Figure 3).
The development of glomerular IR is triggered by several factors: high glucose and/or insulin levels, increased FFAs levels, or an inflammatory milieu. Moreover, insulin signaling appears to be relevant for the adaptative ER stress response in DKD; this is the case for mice with podocyte-specific heterozygous INSR deletion (Madhusudhan et al., 2015). In support of this view, stable overexpression of INSR or knock-down of PTP1B was protective against ER stress (Garner et al., 2018). Podocyte mitochondria play an essential role in cellular metabolism. When dysfunctional as a result of reactive oxygen species production, mitochondria triggers apoptosis, which can also be observed along with a compromised IR state (Susztak et al., 2006). Certainly, the preservation or reestablishment of podocyte integrity is essential in the prevention of the onset and development of DKD.
Insulin Tubular Actions
In the kidney tubule, insulin has several roles: metabolism, electrolyte and acid-base regulation and absorption of filtered substances. However, the exact mechanisms by which insulin performs these distinct roles is not fully understood. Nonetheless, it seems that, at least some of them, are mediated by INSR, and can be explained by the recruitment of specific IRS, as recently shown by Nakamura et al. (2020) specifically for gluconeogenesis and sodium reabsorption regulation. Still, there are overlapping mediators in downstream pathways. In the following paragraphs we will summarize the most relevant and well-known insulin actions in the tubular segment.
Insulin receptor is present throughout the entire nephron (Butlen et al., 1988; Stechi et al., 1994), however, insulin binding capacity of luminal and basolateral membrane tubule cells is different. There is evidence showing same affinity of INSR in both membrane sides of the cell, nonetheless its abundance is asymmetrical (Hammerman, 1985). In fact, the binding capacity of the contraluminal compared to luminal membrane seems to be several times greater due to higher expression of INSR (Talor et al., 1982). Figure 4 summarizes insulin signaling in proximal tubule (PT), regarding its actions in both gluconeogenesis and sodium reabsorption. Additionally, insulin actions through INSR are thought to be different in the proximal and distal nephron regions. Tiwari et al. (2013) demonstrated, in a mouse model with deletion of INSR in the kidney tubule cells, that depending on the segment targeted with INSR deletion, there were different phenotypes further described. In case of decreased INSR at PT, animals had a mild diabetic phenotype, without increased IR when compared to control. These animals shown to have an higher activity of gluconeogenesis enzymes (Tiwari et al., 2013). On the other hand, in animals with the deletion of INSR targeted to distal parts of the tubule, elevated blood pressure and impaired sodium excretion was observed (Tiwari et al., 2008).
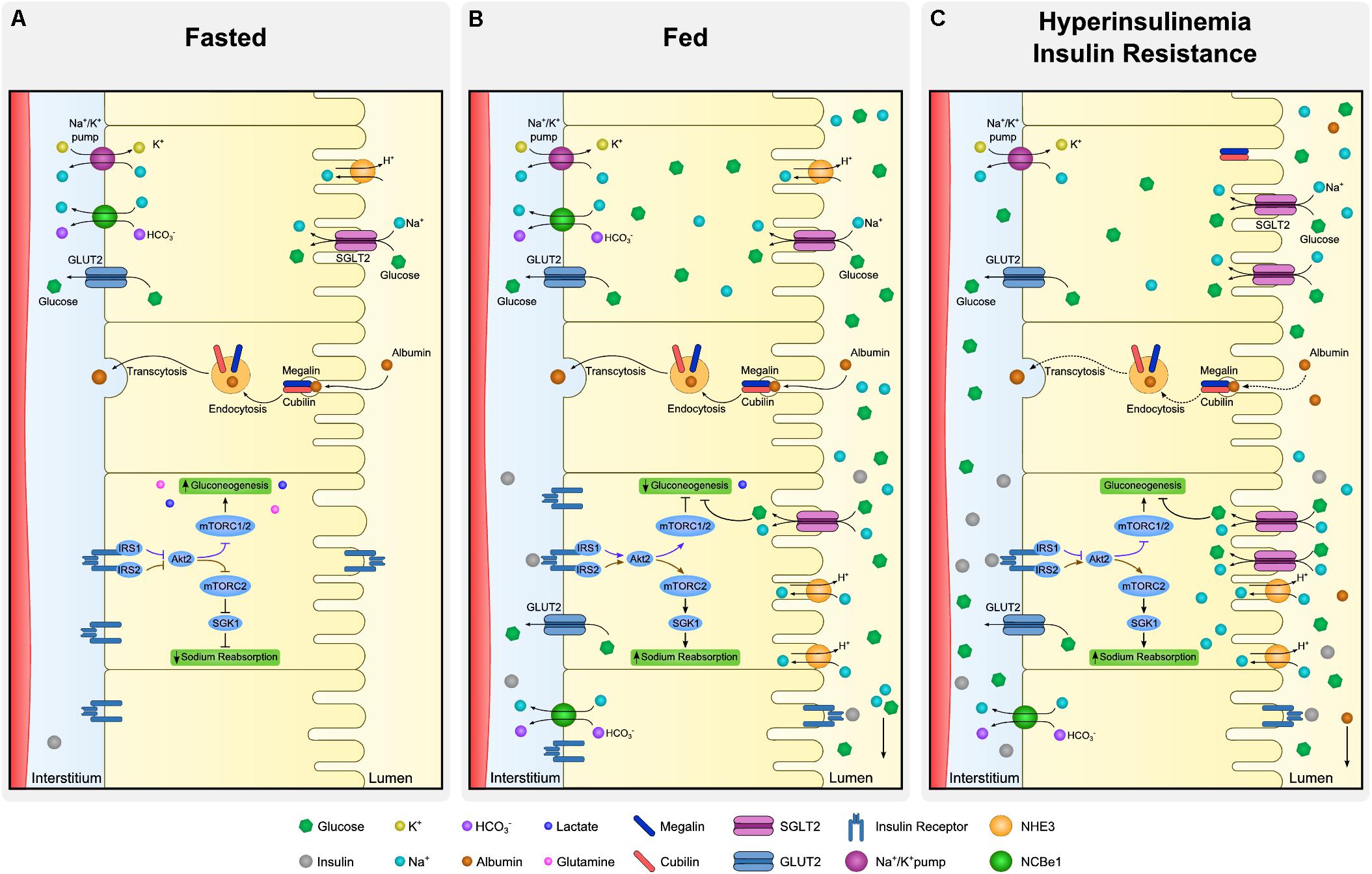
Figure 4. Dynamics of proximal tubule cells at fasting, fed and insulin resistant states. Proximal tubule cells are subjected to distinct microenvironments (lumen and interstitium) and the regulation of absorption and reabsorption of molecules is complex. Although all the described processes occur in every cell of the proximal tubule simultaneously, each specific process is illustrated in a different cell. At fasting (A), low levels of insulin allow expression of gluconeogenic enzymes whereas sodium reabsorption is downregulated. Expression of glucose transporter 2 (GLUT2) at basolateral membrane is mostly associated to glucose output and not to its uptake. Moreover, albumin absorption is performed by megalin and cubilin at luminal membrane and transcytosis allow albumin to be rerouted back to the organism. At fed state (B), increased availability of insulin and glucose promote drastic changes in proximal tubule dynamics. In the case of insulin, luminal uptake is mostly associated to degradation and basolateral to signaling activation. Insulin receptor (INSR) activation downregulates gluconeogenesis and increases sodium reabsorption by different proteins as type 3 Na-H exchanger (NHE-3) and sodium-glucose transport protein 2 (SGLT2). Together with sodium, SGLT2 also co-transport glucose from the lumen. Finally, hyperinsulinemia is linked to perturbations of proximal tubule cells in many aspects (C). As in many other organs, insulin signaling desensitization is associated to inefficient inhibition of gluconeogenesis contributing to maintenance of increased levels of glucose. Derangements at podocyte level increases filtration of albumin and overloads luminal capacity of reabsorption. Such impairment in albumin reabsorption culminates with albuminuria, frequent observed in hyperinsulinemic states.
Glucose Reabsorption
Glucose is reabsorbed by the PT cells from the kidney tubule lumen to the bloodstream (Figure 4A). In the kidney, GLUT2 is in the basolateral membrane and diffuses glucose out of the cell, contrary to the liver, where GLUT2 acts in glucose uptake. Sodium-glucose transporter proteins (SGLT) are responsible for glucose and sodium co-transport by the luminal membrane of kidney cells. Sodium-glucose co-transporter 2 (SGLT2) is a high-capacity/low affinity sodium-glucose cotransporter present in the apical membrane of proximal convoluted tubule cells (Vallon et al., 2011). These transporters are responsible for approximately 90% of filtered glucose reabsorption, and an important target for T2DM therapy (SGLT2 inhibitors; DeFronzo et al., 2012). The remaining 10% of glucose in the tubule is absorbed by SGLT1, a low capacity and high affinity sodium-glucose cotransporter (Wright et al., 2011). Kidney has a threshold for glucose excretion which, in healthy conditions, relates to a glycemic value around 180 mg/dl (Figure 4B). This threshold, however, can be altered in diabetes (Rave et al., 2006). It is not clear if SGLT2 glucose transport is or not directly dependent on insulin signaling (Ferrannini et al., 2020). Nonetheless, SGLT2 expression was shown to be upregulated by insulin on human cultured PT cells, in a dose-dependent manner (Nakamura et al., 2015). Therefore, in hyperinsulinemic states, frequently observed in prediabetes and T2DM, an excessive glucose absorption can be observed. Of notice, glucose is still highly absorbed by SGLT2 in IR states, suggesting that this mechanism is not affected by IR, though it is upregulated by hyperinsulinemia. In this case, a vicious cycle can happen where increased insulin levels drive an increase in glucose SGLT2 overexpression increasing glycemic levels which in turn will increment the insulin secretion (Figure 4C). Therefore, SGLT2 overexpression inhibition is a potential new target for highly prevalent hyperinsulinemia related conditions, namely some dysglycemic phenotypes and obesity. In this case, lowering insulin levels could be paramount to prevent SGLT2 overexpression. Thus, SGLT2 inhibitors might become a relevant therapeutic approach for hyperinsulinemia related conditions, other than T2DM, namely obesity and prediabetes.
Gluconeogenesis Regulation
Kidney has a major role in gluconeogenesis along with the liver and the intestine. Gluconeogenesis occurs mainly in PT cells essentially from lactate and glutamine (Figure 4A). Moreover, PT cells do not use glucose, as they get their energy mostly from fatty acid oxidation (Gerich et al., 1963). Insulin regulates gluconeogenesis in PT cells to meet the fluctuating needs of the body. While in the fasting state the kidney contributes in 40% to overall gluconeogenesis, in the post-absorptive state its contribution drops to 20% (Gerich et al., 1963; Figure 4B).
Nakamura et al. (2020) demonstrated that insulin directly inhibits gluconeogenesis in isolated PT through IRS1/AKT2/mTORC1/2, and that mTORC1 positively regulates insulin signaling (Nakamura et al., 2020). Specifically, in the fasting state, suppressed insulin signaling increases FoxO1 activity, increasing the expression of gluconeogenic genes, such as PEPCK and glucose-6-phosphatase. In parallel, decreased glucose reabsorption via SGLT2 on the luminal membrane downregulates the NADH/NAD+ ratio, activating sirtuin 1 and peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α), a coactivator of FoxO1. Therefore, these two mechanisms lead to enhanced gluconeogenesis. On the contrary, in the fed state, the increased insulin levels and glucose reabsorption result in the suppression of gluconeogenesis through downregulation of the previously mentioned gluconeogenic genes (Nakamura et al., 2015; Sasaki et al., 2017). Whereas glucose is reabsorbed by the luminal membrane, insulin interacts with the basolateral membrane, thus gluconeogenesis regulation results from the integration of signals from distinct cell microenvironment.
Sodium
Insulin impacts on the fine-tuning of several electrolytes by the kidney. Among them sodium handling is probably the best described (DeFronzo et al., 1975). However, the insulin role on sodium retention in normoglycemia is not completely established. Regulation of sodium absorption is paramount to maintain the extracellular volume in a physiological range. Kidney is the principal organ involved in sodium excretion, responding and adapting to sodium intake (Krekels et al., 2015). Additionally, sodium has a major role in driving electrochemical forces that support kidney primary role in fine-tuning body composition. Sodium reabsorption and excretion results from the integration of a complex network of sensors, neural-hormonal stimuli and hemodynamic and metabolic mechanisms (Frame and Wainford, 2017).
Contrary to glucose, sodium is absorbed along the nephron by distinct apical sodium transport proteins. Usually, approximately 65% of filtered sodium is reabsorbed in the PT along with water, mainly through type 3 Na-H exchanger (NHE-3) and SGLT proteins. In addition, the thick ascending limb is responsible for approximately 25% of reabsorption through sodium-potassium chloride cotransporter-2 (NKCC2). Finally, 5–10% of sodium is reabsorbed in the collecting duct by epithelial sodium channel (ENaC) and less than 10% is excreted in urine (Esteva-Font et al., 2012).
The association of insulin with sodium absorption was suggested almost a century ago (Atchley et al., 1933), nonetheless discrimination of insulin- and glucose-mediated effects has not been clarified (DeFronzo et al., 1976; Manhiani et al., 2011). It is still under debate if insulin has a causal effect on hypertension under normoglycemia. Nevertheless, it is known that insulin stimulates sodium absorption in all the tubule segments where it takes place (Kirchner, 1988; Friedberg et al., 1991; Ghezzi and Wright, 2012). In the PT, insulin regulates several sodium transporters, in both luminal (NHE-3, SGLT2) and basolateral membrane (Na/K-ATPase, NBCe1; Figure 4A; Gesek and Schoolwerth, 1991; Feraille et al., 1994; Ruiz et al., 1998). Recently, Nakamura et al. (2020) showed that, regarding sodium absorption in PT, insulin recruits an IRS (IRS2) different from the one orchestrating gluconeogenesis (IRS1). Insulin receptor substrate 2 acts through the AKT2/mTORC2 pathway (Nakamura et al., 2020; Figure 4A). In fact, mTORC2 is known to activate serum/glucocorticoid regulated kinase 1 (SGK1), that will then stimulate ENaC and NHE-3, increasing sodium reabsorption (Satoh et al., 2015). It has been suggested that, in healthy conditions, with rising insulin levels in fed state, IRS2 desensitize, suppressing sodium reabsorption at PT and increasing its delivery in the distal convoluted tubule (Figure 4B). Moreover, with IR, the desensitizing mechanism is abolished and therefore sodium will not reach the distal tubule (Ecelbarger, 2020; Figure 4C). Of notice, by recruiting distinct IRS, kidney cells can somehow dissociate pathways performing distinct functions.
Finally, it must be kept in mind that insulin can interact with intrarenal and systemic renin-angiotensin-aldosterone system in several ways (Muscogiuri et al., 2008) and therefore, indirectly interfere with sodium reabsorption in different mechanism out of the scope of this review.
Albumin Absorption
The luminal membrane of PT cells is primarily responsible for the reabsorption of proteins that are freely filtered in the glomerulus by receptor-mediated endocytosis (Figure 4A; Christensen and Gburek, 2004). This is the case of albumin reabsorption that can have an important role in energy conservation. It has been suggested that albumin endocytosis is a regulated process, dependent on membrane receptors megalin and cubilin (Christensen and Birn, 2001). More recent evidence suggest that insulin might also have a role in the regulation of tubular albumin absorption (Kumari et al., 2019).
Albuminuria is of major clinical relevance in diagnosis and follow-up of kidney disease including subjects with diabetes. Insulin resistance was found to be associated with decreased INSR expression in tubular cells in rat models (Wang et al., 2005). In these observations, Kumari et al. (2019) analyzed urine samples from mice with targeted deletion of INSR from the renal PT. These mice had an impaired uptake of albumin, without any glomerulopathy. They also demonstrated that in healthy humans, albumin absorption capacity and excretion vary from the fast to the fed state. Moreover, IR was associated with microalbuminuria even in normoglycemia as described in the RISC study (Pilz et al., 2014) and thus can be present regardless of diabetes diagnosis. Altogether, these evidences suggest that albuminuria might be an important marker of kidney tubular dysfunction and might reflect tubule cells IR (Figure 4C). These reinforces the kidney contribution to diabetes development and highlights insulin and albumin dynamics prior and regardless of the development of diabetes.
Kidney Insulin Clearance
In the systemic circulation, besides insulin metabolization by the liver, the kidney is the major site of insulin clearance (around 25%) (Elgee and Williams, 1954; Narahara et al., 1958); its action might be required to limit excessive insulin levels. Evidences supporting this theory started to rise in the middle of the 20th century (Zubrod et al., 1951; Elgee and Williams, 1954; Narahara et al., 1958; Ricketts and Wildberger, 1962). In a study from 1966, Beck et al. (1966) found that when insulin was injected intravenously in mice, it concentrated in liver and kidney; however, with higher insulin doses, raised insulin levels were found in rat’s kidney, while these levels were found to be reduced in liver. Nevertheless, this early study has some methodological limitations. Indeed, kidney insulin clearance remains constant in spite of insulinemia variations, but varies with creatinine clearance (Rubenstein et al., 1967). Globally, these evidences suggest that kidney insulin clearance is a non-saturable process, although dependent on glomerular filtration rate.
Insulin freely filtered in the glomerulus is absorbed by the lining cells of the PT (Figure 2). Upon entering the cell, insulin is transported through the luminal membrane into the PT cells and is degraded. Luminal insulin reabsorption limits urine insulin excretion, thus less than 2% of insulin reaches the urine in normal fasting conditions (Rubenstein and Spitz, 1968). Insulin is transported through the luminal membrane by a receptor-mediated endocytic mechanism (Rabkin et al., 1984). Endocytic internalization of insulin seems to be more related to insulin degradation than to insulin biological actions (Figure 2).
While glomerular filtration of insulin could not account for its total estimated renal extraction, a second mechanism was postulated. In this context, it was observed that a significant amount of insulin was cleared by the post-glomerular peritubular capillaries into the tubule cells (Chamberlain and Stimmler, 1967). In humans, this route represents around one-third of cleared insulin in the kidney, where it enters tubule cells not just by endocytosis, but also by INSR mediated uptake. In this case, through INSR binding, insulin signal to the kidneys’ tubular apparatus is crucial to maintain central physiologic functions, similarly to what happens in extra-renal tissues, namely regarding glucose homeostasis and blood pressure.
Insulin degrading activity has been observed at cytosol, lysosomes and mitochondria in addition to the membrane, indicating that it occurs in distinct cell sites. Degradation at the membrane level, however, seems to represent less than 2% of total degrading activity (Rabkin et al., 1984). Insulin can be initially hydrolyzed by an insulin protease followed by the action of plasma-membrane-associated or lysosomal proteases. This pathway can degrade insulin entering through both luminal and contraluminal membrane. In another possible pathway, endocytic vesicles containing insulin fuse with lysosomes. This pathway comprehends glutathione-insulin transhydrogenase (GIT) action, followed by hydrolysis of intact A and B chains by lysosomal proteases, and seems to need insulin internalization. It may act primarily on insulin delivered by luminal uptake and it is most active when supraphysiological levels of insulin are present (Rabkin et al., 1984).
Azizi et al. (2015) demonstrated that, in a culture of human adipose microvascular endothelial cells, insulin can go through microvascular endothelial cells by transcytosis (Azizi et al., 2015). Regarding insulin handling in the kidney tubule, Dahl et al. (1989) hypothesized that insulin molecules could also pass tubular cells by a retroendocytic pathway instead of being degraded. The authors demonstrated that cultured opossum kidney cells exhibited a retroendocytic pathway for insulin (Dahl et al., 1989). Using the same model, the authors later demonstrated that inhibition of insulin degradation diverted intact insulin from the degradative to the retroendocytic pathway (Dahl et al., 1990). Although captivating, especially regarding a potential contribution to hyperinsulinemic states, this hypothesis was not further explored.
Whether insulin clearance mechanisms attributed to other organs affects renal function and insulin clearance it is not clear. For example, the lack of CEACAM-1, a key protein enrolled in hepatic insulin clearance driving hyperinsulinemia, in the kidney leads to increased renin levels contributing to a potentiation of the RAS system and hypertension. These effects are exacerbated upon high fat diet exposure. Hence, the described CEACAM-1 renal effects can be due to the lack of its expression as well as the observed hyperinsulinemia (Huang et al., 2013; Li et al., 2015).
Despite early conflicting results, further studies showed that insulin is excreted in urine. However, in physiological conditions, it represents a minimal proportion of insulin filtered in the glomerulus. In health, a minor amount of insulin appears in the urine, as the majority is absorbed in PT. Tubule absorbing capacity of insulin does not saturate and thus the insulin fraction excreted in urine is constantly small, regardless of insulin levels. However, the amount of insulin excreted in urine varies physiologically (e.g., fasting and post-prandial) and in pathological conditions (obesity, diabetes) depending on the affected nephron region (e.g., glomerulopathy vs. tubulopathy) (Rubenstein and Spitz, 1968). Considering that insulin is internalized in the apical membrane by a receptor-mediated endocytic mechanism, the increased urinary insulin excretion might represent a tubular dysfunction. Subjects with tubulopathy show large amounts of insulin in urine approximating the amount that is filtered (Rabkin et al., 1984). Conversely, subjects with nephrotic syndrome show normal amounts of insulin in urine. When both glomerular and tubule lesion occur urine insulin excretion increase (Rabkin et al., 1984).
Clinical Insights on Insulin Dysregulation
Insulin resistance is a common feature in chronic kidney disease (CKD) patients, even in absence of diabetes (DeFronzo et al., 1981; Shinohara et al., 2002; Becker et al., 2005; Kobayashi et al., 2005; Landau et al., 2011), and it is a risk factor for CKD progression (Fox et al., 2004). Its prevalence in CKD ranges from 30 to 50%, and this mainly depends on the adopted method of measurement (Spoto et al., 2016). Insulin resistance can be detected at the very early stages, when eGFR is still within the normal range, suggesting a potential role in triggering CKD (Fliser et al., 1998). A large study based on the Atherosclerosis Risk in Communities (ARICs) cohort confirmed that CKD development increases in strict parallelism with the number of metabolic syndrome criteria measured in non-diabetic adults, and this relationship remains significant even after controlling for the development of diabetes and hypertension (Kurella et al., 2005). Insulin resistance has also been associated with prevalent CKD and rapid decline in renal function in elderly, non-diabetic, Asian individuals (Cheng et al., 2012), and with microalbuminuria in the general population (Mykkänen et al., 1998), and in patients with T1DM (Yip et al., 1993; Ekstrand et al., 1998) and T2DM (Groop et al., 1993), indicating that this relationship is independent of diabetes (Mykkänen et al., 1998; Chen et al., 2003, 2004). The proposed mechanism by which IR contributes to kidney damage involves the worsening of renal hemodynamics through activation of the sympathetic nervous system (Rowe et al., 1981), sodium retention, decreased Na+, K+-ATPase activity, and increased GFR (Gluba et al., 2013).
The etiology of IR in CKD is multifactorial, depending on classical and CKD-peculiar risk factors, such as physical inactivity, inflammation and oxidative stress, adipokine derangements, vitamin D deficiency, metabolic acidosis, anemia and microbial toxins (Spoto et al., 2016).
Long-term hemodialysis has a positive effect on IR (DeFronzo et al., 1978), but there is little clinical data regarding the effect of peritoneal dialysis.
In addition to being a risk factor for CKD onset and progression, IR is also involved in the increased cardiovascular (CV) risk in this population. IR may be responsible for high blood pressure via direct stimulation of renin–angiotensin–aldosterone system (Nickenig et al., 1998), activation of sympathetic system (Sowers et al., 2001) and downregulation of the natriuretic peptide system (Sarzani et al., 1999).
However, the association between IR and CV complications in CKD patients is still to be clarified, as well as the relationship between IR and all-cause and CV mortality.
A positive association between IR and all-cause mortality was found in smokers and physically inactive CKD patients (Xu et al., 2014) and in a small cohort of 170 Japanese, non-diabetic, dialysis patients (Shinohara et al., 2002), whereas association with CV mortality was found in a cohort of peritoneal patients. However, no association was found in the ULSAM cohort, including 3-4 stage CKD patients (Jia et al., 2014) and in a small study performed in peritoneal dialysis patients (Sánchez-Villanueva et al., 2013).
Even though the prognostic value of IR for death and CV events need to be clarified, the association with CKD is well established. Even in this case, however, if IR is responsible of the onset and progression of CKD, or if CKD is responsible for IR is still to be clarified. A possible answer to this question could derive from clinical trials aiming at assessing the effect of drugs used for IR treatment on kidney function.
Thiazolidinediones (TZDs) are a class of oral diabetic medications that increase insulin sensitivity by acting on PPARγ (Yki-Järvinen, 2004). The effect of TZDs on kidney function has been previously described in mice models (Fujii et al., 1997). Treatment with TZDs has been demonstrated to improve insulin sensitivity in patients with T2DM after a 3-month treatment, and to reduce albuminuria, the last effect likely mediated by the concurrent increase in serum adiponectin concentration (Miyazaki et al., 2007). These results were confirmed in a meta-analysis of 15 double-blind, randomized, clinical trials involving diabetic patients (Sarafidis et al., 2010) and in a large study involving 4351 diabetic patients (Lachin et al., 2011). Even though a meta-analysis reported an increase in cardiovascular mortality linked to the use of TZDs in dialysis patients (Nissen and Wolski, 2007), no definitive proof are available on the risk related to this medication in this population.
Another interesting class of hypoglycemic drugs with positive kidney outcomes are the SGLT2 inhibitors (SGLT2i) which inhibit glucose and sodium reabsorption in the PT (Ferrannini, 2017). These drugs have a renoprotective effect in patients with T2DM (Perkovic et al., 2019) independently of glycemic control (Cherney et al., 2017). The renal protective effect can also be attributed to altered hemodynamics, reduced inflammation and fibrosis as well as controlled blood pressure and weight loss (Williams et al., 2020). In rats treated with SGLT2i the glycemic improvement was accompanied by a decrease in insulin and lipid levels (Huang et al., 2020). Moreover, the actions of SGLT2i are associated with increased insulin sensitivity and decreased albuminuria (Cherney et al., 2017). Interestingly, Jaikumkao et al. observed that in an animal model of diet induced obesity characterized by IR and impaired renal function dapagliflozin treatment resulted in improved IR, renal function and renal insulin signaling (Jaikumkao et al., 2018).
Conclusion
Insulin is a hormone which acts not only on the most recognized insulin-responsive organs (liver, adipose tissue, and skeletal muscle), but also on the kidney. Moreover, the kidney has a primordial role in insulin clearance and may impact on insulin plasma levels. Whereas its main action is mainly related to homeostasis of glucose, including modulation of gluconeogenesis and lipolysis, in kidney the effects of insulin and IR change according to whether the target is in glomeruli or tubules. More specifically, if in glomerular podocytes insulin promotes glucose uptake, with an involvement in barrier permeability, in tubules it contributes to glucose reabsorption and gluconeogenesis regulation, and plays an important role in sodium homeostasis.
Importantly, insulin intervenes in albumin reabsorption at tubular level. Moreover, IR has been associated with microalbuminuria even in normoglycemia, and thus can be present regardless of diabetes diagnosis. These findings reinforce the kidney contribution to diabetes development and highlights insulin and albumin dynamics prior and regardless of the development of diabetes.
Insulin is cleared in the PT of kidney by two major routes, either by absorption of filtered insulin, or by post-glomerular capillary secretion, and only a minor amount appears to be excreted in urine. A decreased renal insulin clearance might lead to higher insulin levels, in the organ or systemically, favoring IR. Nonetheless, the impact of renal insulin clearance affection in the kidney or in insulin plasma levels still needs to be further unveiled.
It is clear now that kidney is not a mere target of insulin action, but insulin, more precisely IR, is also able to trigger CKD even in absence of diabetes. IR has been associated with prevalent CKD, rapid decline in renal function and microalbuminuria in the general population and in diabetic patients. In addition to being a risk factor for CKD onset and progression, IR is also involved in the increased cardiovascular risk in this population. However, if IR is responsible for the onset and progression of CKD, or if CKD is responsible for IR is still to be clarified. Preliminary confirmations come from clinical trials aiming at exploring the effect of TZDs, a class of oral diabetic medications, on kidney function. However, more focused studies, aiming also at testing the safety of these medications in CKD patients, are needed to better understand if treatment of IR may improve renal function in this population.
Author Contributions
AP and MPM conceptualized the study. All authors originally drafted the manuscript, reviewed, edited, and critically approved the final version of the manuscript.
Funding
This work was supported by “Fundação para a Ciência e a Tecnologia” – FCT to AP (PD/BD/136887/2018); MJM (PD/BD/114256/2016), MPM (PTDC/DTP-EPI/0207/2012), DOB e MPM (PTDC/BIM-MET/2115/2014); iNOVA4Health (UIDB/Multi/04462/2020), by the European Commission Marie Skłodowska-Curie Actions H2020 (grant agreements nos. 722619 and 734719), and by the Sociedade Portuguesa de Diabetologia.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADP, adenosine diphosphate; ATP, adenosine triphosphate; CEACAM-1, carcinoembryonic antigen-related cell adhesion molecule 1; cGMP, cyclic guanosine monophosphate; CKD, chronic kidney disease; CV, cardiovascular; DKD, diabetic kidney disease; DNL, de novo lipogenesis; eGFR, estimated glomerular filtration rate; ENaC, epithelial sodium channel; ER, endoplasmic reticulum; FFA, free fatty acid; FoxO1, forkhead box O1; GBM, glomerular basement membrane; GIT, glutathione-insulin transhydrogenase; GLUT1, glucose transporter 1; GLUT2, glucose transporter 2; GLUT4, glucose transporter 4; GRB2, growth factor receptor-bound protein 2; GSK3, glycogen synthase kinase 3; GSV, GLUT storage vesicle; IDE, insulin-degrading enzyme; INSR, insulin receptor; IR, insulin resistance; IRS1, insulin receptor substrate 1; IRS2, insulin receptor substrate 2; MAPK, mitogen activated protein kinase; mTOR, mammalian target of rapamycin; NAFLD, non-alcoholic fatty liver disease; NBCe1, sodium bicarbonate cotransporter 1; NHE-3, sodium/hydrogen exchanger 3; NKCC2, sodium-potassium chloride cotransporter-2; PGC1 α, peroxisome proliferator-activated receptor- γ coactivator 1 α; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5 biphosphate; PIP3, phosphatidylinositol 3,4,5 triphosphate; PKB, protein kinase B; PKG, cGMP-dependent protein kinase G; PKGI, cGMP-dependent protein kinase G isoform I; PPAR γ, peroxisome proliferator-activated receptor γ; PT, proximal tubule; PTEN, phosphatase and tensin homolog; PTP1B, protein tyrosine-phosphatase 1B; SGK1, serum/glucocorticoid regulated kinase 1; SGLT, sodium-glucose transport proteins; SGLT2, sodium-glucose co-transporter 2; SGLT2i, sodium-glucose co-transporter-2 inhibitors; SHIP2, SH2-domain-containing inositol phosphatase 2; SREBP-1c, sterol regulatory element binding protein-1c; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TZDs, thiazolidinediones; Vamp2, vesicle-associated membrane protein 2.
References
Adler, A. I., Stevens, R. J., Manley, S. E., Bilous, R. W., Cull, C. A., Holman, R. R., et al. (2003). Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 63, 225–232. doi: 10.1046/j.1523-1755.2003.00712.x
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R. J., Reese, C. B., et al. (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269. doi: 10.1016/S0960-9822(06)00122-9
Andrali, S. S., Sampley, M. L., Vanderford, N. L., and Özcan, S. (2008). Glucose regulation of insulin gene expression in pancreatic β-cells. Biochem. J. 415, 1–10. doi: 10.1042/BJ20081029
Artunc, F., Schleicher, E., Weigert, C., Fritsche, A., Stefan, N., and Häring, H. (2016). The impact of insulin resistance on the kidney and vasculature. Nat. Publ. Gr. 12, 721–737. doi: 10.1038/nrneph.2016.145
Atchley, D. W., Loeb, R. F., Richards, D. W., Benedict, E. M., and Driscoll, M. E. (1933). ON DIABETIC ACIDOSIS: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J. Clin. Invest. 12, 297–326. doi: 10.1172/JCI100504
Azizi, P. M., Zyla, R. E., Guan, S., Wang, C., Liu, J., Bolz, S. S., et al. (2015). Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol. Biol. Cell 26, 740–750. doi: 10.1091/mbc.E14-08-1307
Bayascas, J. R., and Alessi, D. R. (2005). Regulation of Akt/PKB Ser473 Phosphorylation. Mol. Cell 18, 143–145. doi: 10.1016/j.molcel.2005.03.020
Beck, L. V., Zaharko, D. S., Roberts, N., and King, C. (1966). On insulin I-131 metabolism in mice. Modifying effects of anti-insulin serum and of total insulin dosage. Diabetes 15, 336–341. doi: 10.2337/diab.15.5.336
Becker, B., Kronenberg, F., Kielstein, J. T., Haller, H., Morath, C., Ritz, E., et al. (2005). Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. J. Am. Soc. Nephrol. 16, 1091–1098. doi: 10.1681/ASN.2004090742
Bedinger, D. H., and Adams, S. H. (2015). Metabolic, anabolic, and mitogenic insulin responses: a tissue-specific perspective for insulin receptor activators. Mol. Cell. Endocrinol. 415, 143–156. doi: 10.1016/j.mce.2015.08.013
Bergman, R. N., Piccinini, F., Kabir, M., Kolka, C. M., and Ader, M. (2019). Hypothesis: role of reduced hepatic insulin clearance in the pathogenesis of type 2 diabetes. Diabetes Metab. Res. Rev. 68, 1709–1716. doi: 10.2337/db19-0098
Bril, F., Lomonaco, R., Orsak, B., Ortiz-Lopez, C., Webb, A., Tio, F., et al. (2014). Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 59, 2178–2187. doi: 10.1002/hep.26988
Butlen, D., Vadrot, S., Roseau, S., and Morel, F. (1988). Insulin receptors along the rat nephron: [125I] Insulin binding in microdissected glomeruli and tubules. Pflügers Arch. Eur. J. Physiol. 412, 604–612. doi: 10.1007/BF00583761
Calzi, S. L., Choice, C. V., and Najjar, S. M. (1997). Differential effect of pp120 on insulin endocytosis by two variant insulin receptor isoforms. Am. J. Physiol. Metab. 273, E801–E808. doi: 10.1152/ajpendo.1997.273.4.E801
Canaud, G., Bienaimé, F., Viau, A., Treins, C., Baron, W., Nguyen, C., et al. (2013). AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 19, 1288–1296. doi: 10.1038/nm.3313
Chamberlain, M. J., and Stimmler, L. (1967). The renal handling of insulin. J. Clin. Invest. 46, 911–919. doi: 10.1172/JCI105597
Chen, J., Muntner, P., Hamm, L. L., Fonseca, V., Batuman, V., Whelton, P. K., et al. (2003). Insulin resistance and risk of chronic kidney disease in nondiabetic US adults. J. Am. Soc. Nephrol. 14, 469–477. doi: 10.1097/01.ASN.0000046029.53933.09
Chen, J., Muntner, P., Hamm, L. L., Jones, D. W., Batuman, V., Fonseca, V., et al. (2004). The metabolic syndrome and chronic kidney disease in U.S. adults. Ann. Intern. Med. 140, 167–174. doi: 10.7326/0003-4819-140-3-200402030-00007
Cheng, H. T., Huang, J. W., Chiang, C. K., Yen, C. J., Hung, K. Y., and Wu, K. D. (2012). Metabolic syndrome and insulin resistance as risk factors for development of chronic kidney disease and rapid decline in renal function in elderly. J. Clin. Endocrinol. Metab. 97, 1268–1276. doi: 10.1210/jc.2011-2658
Cherney, D. Z. I., Zinman, B., Inzucchi, S. E., Koitka-Weber, A., Mattheus, M., von Eynatten, M., et al. (2017). Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: an exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 5, 610–621. doi: 10.1016/S2213-8587(17)30182-1
Christensen, E. I., and Birn, H. (2001). Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am. J. Physiol. Physiol. 280, F562–F573. doi: 10.1152/ajprenal.2001.280.4.F562
Christensen, E. I., and Gburek, J. (2004). Protein reabsorption in renal proximal tubule - Function and dysfunction in kidney pathophysiology. Pediatr. Nephrol. 19, 714–721. doi: 10.1007/s00467-004-1494-0
Coward, R. J. M., Welsh, G. I., Koziell, A., Hussain, S., Lennon, R., Ni, L., et al. (2007). Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes Metab. Res. Rev. 56, 1127–1135. doi: 10.2337/db06-0693
Coward, R. J. M., Welsh, G. I., Yang, J., Tasman, C., Lennon, R., Koziell, A., et al. (2005). The human glomerular podocyte is a novel target for insulin action. Diabetes Metab. Res. Rev. 54, 3095–3102. doi: 10.2337/diabetes.54.11.3095
Dahl, D. C., Tsao, T., Duckworth, W. C., Frank, B. H., and Rabkin, R. (1990). Effect of bacitracin on retroendocytosis and degradation of insulin in cultured kidney epithelial cell line. Diabetes Metab. Res. Rev 39, 1339–1346. doi: 10.2337/diab.39.11.1339
Dahl, D. C., Tsao, T., Duckworth, W. C., Mahoney, M. J., and Rabkin, R. (1989). Retroendocytosis of insulin in a cultured kidney epithelial cell line. Am. J. Physiol. Cell Physiol. 257, C190–C196. doi: 10.1152/ajpcell.1989.257.2.c190
DeFronzo, R. A., Alvestrand, A., Smith, D., Hendler, R., Hendler, E., and Wahren, J. (1981). Insulin resistance in uremia. J. Clin. Invest. 67, 563–568. doi: 10.1172/JCI110067
DeFronzo, R. A., Cooke, C. R., Andres, R., Faloona, G. R., and Davis, P. J. (1975). The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J. Clin. Invest. 55, 845–855. doi: 10.1172/JCI107996
DeFronzo, R. A., Davidson, J. A., and del Prato, S. (2012). The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes. Metab. 14, 5–14. doi: 10.1111/j.1463-1326.2011.01511.x
DeFronzo, R. A., Goldberg, M., and Agus, Z. S. (1976). The effects of glucose and insulin on renal electrolyte transport. J. Clin. Invest. 58, 83–90. doi: 10.1172/JCI108463
DeFronzo, R. A., Tobin, J. D., Rowe, J. W., and Andres, R. (1978). Glucose intolerance in uremia. Quantification of pancreatic beta cell sensitivity to glucose and tissue sensitivity to insulin. J. Clin. Invest. 62, 425–435. doi: 10.1172/JCI109144
Detimary, P., Gilon, P., and Henquin, J.-C. (1998). Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem. J. 333, 269–274. doi: 10.1042/bj3330269
Dimitriadis, G., Mitrou, P., Lambadiari, V., Maratou, E., and Raptis, S. A. (2011). Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 93, S52–S59. doi: 10.1016/S0168-8227(11)70014-6
Duckworth, W. C., Bennett, R. G., and Hamel, F. (1998). Insulin degradation: progress and potential. Endocr. Rev. 19, 608–624.
Ecelbarger, C. M. (2020). Refining insulin signaling in the proximal tubule at the level of the substrate. Kidney Int. 97, 256–258. doi: 10.1016/j.kint.2019.11.018
Ekstrand, A. V., Groop, P. H., and Grönhagen-Riska, C. (1998). Insulin resistance precedes microalbuminuria in patients with insulin-dependent diabetes mellitus. Nephrol. Dial. Transplant. 13, 3079–3083. doi: 10.1093/ndt/13.12.3079
Elgee, N. J., and Williams, R. H. (1954). Degradation of Insulin-I131 by Liver and Kidney in vivo. Exp. Biol. Med. 87, 352–355. doi: 10.3181/00379727-87-21380
Esteva-Font, C., Ballarin, J., and Fernández-Llama, P. (2012). Molecular biology of water and salt regulation in the kidney. Cell. Mol. Life Sci. 69, 683–695. doi: 10.1007/s00018-011-0858-4
Farber, S. J., Berger, E. Y., and Earle, D. P. (1951). Effect of diabetes and insulin on the maximum capacity of the renal tubules to reabsorb glucose. J. Clin. Invest. 30, 125–129. doi: 10.1172/JCI102424
Feraille, E., Carranza, M. L., Rousselot, M., and Favre, H. (1994). Insulin enhances sodium sensitivity of Na-K-ATPase in isolated rat proximal convoluted tubule. Am. J. Physiol. Ren. Fluid Electrolyte Physiol. 267, F55–F62. doi: 10.1152/ajprenal.1994.267.1.f55
Fernandes, A. B., Patarrão, R. S., Videira, P. A., and Macedo, M. P. (2011). Understanding postprandial glucose clearance by peripheral organs: the role of the hepatic parasympathetic system. J. Neuroendocrinol. 23, 1288–1295. doi: 10.1111/j.1365-2826.2011.02226.x
Ferrannini, E. (2017). Sodium-glucose co-transporters and their inhibition: clinical physiology. Cell Metab. 26, 27–38. doi: 10.1016/j.cmet.2017.04.011
Ferrannini, E., Pereira-Moreira, R., Seghieri, M., Rebelos, E., Souza, A. L., Chueire, V. B., et al. (2020). Insulin enhances renal glucose excretion: relation to insulin sensitivity and sodium-glucose cotransport. BMJ Open Diabetes Res. Care 8:e001178. doi: 10.1136/bmjdrc-2020-001178
Fliser, D., Pacini, G., Engelleiter, R., Kautzky-Willer, A., Prager, R., Franek, E., et al. (1998). Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int. 53, 1343–1347. doi: 10.1046/j.1523-1755.1998.00898.x
Fox, C. S., Larson, M. G., Leip, E. P., Culleton, B., Wilson, P. W. F., and Levy, D. (2004). Predictors of new-onset kidney disease in a community-based population. J. Am. Med. Assoc. 291, 844–850. doi: 10.1001/jama.291.7.844
Frame, A. A., and Wainford, R. D. (2017). Renal sodium handling and sodium sensitivity. Kidney Res. Clin. Pract. 36, 117–131. doi: 10.23876/j.krcp.2017.36.2.117
Friedberg, C. E., Van Buren, M., Bijlsma, J. A., and Koomans, H. A. (1991). Insulin increases sodium reabsorption in diluting segment in humans: evidence for indirect mediation through hypokalemia. Kidney Int. 40, 251–256. doi: 10.1038/ki.1991.207
Fujii, M., Takemura, R., Yamaguchi, M., Hasegawa, G., Shigeta, H., Nakano, K., et al. (1997). Troglitazone (CS-045) ameliorates albuminuria in streptozotocin-induced diabetic rats. Metabolism 46, 981–983. doi: 10.1016/S0026-0495(97)90264-X
Galgani, J. E., Moro, C., and Ravussin, E. (2008). Metabolic flexibility and insulin resistance. Am. J. Physiol. Metab. 295, E1009–E1017. doi: 10.1152/ajpendo.90558.2008
Gancheva, S., Jelenik, T., Álvarez-Hernández, E., and Roden, M. (2018). Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 98, 1371–1415. doi: 10.1152/physrev.00015.2017
Gao, T., McKenna, B., Li, C., Reichert, M., Nguyen, J., Singh, T., et al. (2014). Pdx1 Maintains β cell identity and function by repressing an α cell program. Cell Metab. 19, 259–271. doi: 10.1016/j.cmet.2013.12.002
Garner, K. L., Betin, V. M. S., Pinto, V., Graham, M., Abgueguen, E., Barnes, M., et al. (2018). Enhanced insulin receptor, but not PI3K, signalling protects podocytes from ER stress. Sci. Rep. 8:3902. doi: 10.1038/s41598-018-22233-9
Gerich, J. E., Meyer, C., Hans, W. J., and Stumvoll, M. (1963). Renal gluconeogenesis. Adv. Enzyme Regul. 1, 385–400. doi: 10.1016/0065-2571(63)90034-7
Gesek, F. A., and Schoolwerth, A. C. (1991). Insulin increases Na(+)-H+ exchange activity in proximal tubules from normotensive and hypertensive rats. Am. J. Physiol. Physiol. 260, F695–F703. doi: 10.1152/ajprenal.1991.260.5.F695
Ghezzi, C., and Wright, E. M. (2012). Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am. J. Physiol. Cell Physiol. 303, 348–354. doi: 10.1152/ajpcell.00115.2012
Gluba, A., Mikhailidis, D. P., Lip, G. Y. H., Hannam, S., Rysz, J., and Banach, M. (2013). Metabolic syndrome and renal disease. Int. J. Cardiol. 164, 141–150. doi: 10.1016/j.ijcard.2012.01.013
Groop, L., Ekstrand, A., Forsblom, C., Widén, E., Groop, P. H., Teppo, A. M., et al. (1993). Insulin resistance, hypertension and microalbuminuria in patients with Type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 36, 642–647. doi: 10.1007/BF00404074
Hammerman, M. R. (1985). Interaction of insulin with the renal proximal tubular cell. Am. J. Physiol. Physiol. 249, F1–F11. doi: 10.1152/ajprenal.1985.249.1.F1
Hirashima, Y., Tsuruzoe, K., Kodama, S., Igata, M., Toyonaga, T., Ueki, K., et al. (2003). Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J. Endocrinol. 179, 253–266. doi: 10.1677/joe.0.1790253
Hou, J. C., Min, L., and Pessin, J. E. (2009). “Chapter 16 insulin granule biogenesis, trafficking and exocytosis,” in Vitamins and Hormones, ed. G. Litwack (Amsterdam: Elsevier Inc), 473–506. doi: 10.1016/S0083-6729(08)00616-X
Huang, J., Ledford, K. J., Pitkin, W. B., Russo, L., Najjar, S. M., and Siragy, H. M. (2013). Targeted Deletion of Murine CEACAM 1 Activates PI3K-Akt signaling and contributes to the expression of (pro)renin receptor via CREB Family and NF-κB transcription factors. Hypertension 62, 317–323. doi: 10.1161/HYPERTENSIONAHA.113.01324
Huang, Z., Huang, L., Wang, C., Zhu, S., Qi, X., Chen, Y., et al. (2020). SUN-672 SGLT2 inhibitor reduces hyperinsulinemia and restores pulsatile growth hormone secretion in Obese MC4RKO mice. J. Endocr. Soc. 4:672. doi: 10.1210/jendso/bvaa046.1158
Hutton, J. C. (1994). Insulin secretory granule biogenesis and the proinsulin-processing endopeptidases. Diabetologia 37, S48–S56. doi: 10.1007/BF00400826
Jaikumkao, K., Pongchaidecha, A., Chueakula, N., Thongnak, L., Wanchai, K., Chatsudthipong, V., et al. (2018). Renal outcomes with sodium glucose cotransporter 2 (SGLT2) inhibitor, dapagliflozin, in obese insulin-resistant model. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2021–2033. doi: 10.1016/j.bbadis.2018.03.017
Jia, T., Huang, X., Qureshi, A. R., Xu, H., Ärnlöv, J., Lindholm, B., et al. (2014). Validation of insulin sensitivity surrogate indices and prediction of clinical outcomes in individuals with and without impaired renal function. Kidney Int. 86, 383–391. doi: 10.1038/ki.2014.1
Jung, S.-H., Jung, C.-H., Reaven, G. M., and Kim, S. H. (2018). Adapting to insulin resistance in obesity: role of insulin secretion and clearance. Diabetologia 61, 681–687. doi: 10.1007/s00125-017-4511-0
Kalwat, M. A., and Thurmond, D. C. (2013). Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med. 45:e37. doi: 10.1038/emm.2013.73
Kiapidou, S., Liava, C., Kalogirou, M., Akriviadis, E., and Sinakos, E. (2020). Chronic kidney disease in patients with non-alcoholic fatty liver disease: what the Hepatologist should know? Ann. Hepatol. 19, 134–144. doi: 10.1016/j.aohep.2019.07.013
Kim, K.-S., Park, S. W., Cho, Y.-W., and Kim, S.-K. (2018). Higher prevalence and progression rate of chronic kidney disease in elderly patients with Type 2 diabetes mellitus. Diabetes Metab. J. 42, 224–232. doi: 10.4093/dmj.2017.0065
Kirchner, K. A. (1988). Insulin increases loop segment chloride reabsorption in the euglycemic rat. Am. J. Physiol. Ren. Fluid Electrolyte Physiol. 255, F1206–F1213. doi: 10.1152/ajprenal.1988.255.6.f1206
Kobayashi, S., Maesato, K., Moriya, H., Ohtake, T., and Ikeda, T. (2005). Insulin resistance in patients with chronic kidney disease. Am. J. Kidney Dis. 2012:691369. doi: 10.1053/j.ajkd.2004.09.034
Kou, S.-W., Hu, Y.-H., and Wu, D.-A. (2014). The response of first and second phase insulin secretion in newly diagnosed type 2 Diabetes Mellitus. Int. J. Diabetes Clin. Diagnosis 1:103. doi: 10.15344/2394-1499/2014/103
Krekels, M., Kroon, A., and de Leeuw, P. (2015). Sodium intake as a modulator of kidney function. Curr. Hypertens. Rev. 11, 57–60. doi: 10.2174/1573402111666150530204512
Krycer, J. R., Sharpe, L. J., Luu, W., and Brown, A. J. (2010). The Akt–SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 21, 268–276. doi: 10.1016/j.tem.2010.01.001
Kubota, N., Kubota, T., Kajiwara, E., Iwamura, T., Kumagai, H., Watanabe, T., et al. (2016). Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat. Commun. 7:12977. doi: 10.1038/ncomms12977
Kumari, M., Sharma, R., Pandey, G., Ecelbarger, C. M., Mishra, P., and Tiwari, S. (2019). Deletion of insulin receptor in the proximal tubule and fasting augment albumin excretion. J. Cell. Biochem. 120, 10688–10696. doi: 10.1002/jcb.28359
Kuo, W. L., Montag, A. G., and Rosner, M. R. (1993). Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 132, 604–611. doi: 10.1210/endo.132.2.7678795
Kurella, M., Lo, J. C., and Chertow, G. M. (2005). Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J. Am. Soc. Nephrol. 16, 2134–2140. doi: 10.1681/ASN.2005010106
Lachin, J. M., Viberti, G., Zinman, B., Haffner, S. M., Aftring, R. P., Paul, G., et al. (2011). Renal function in type 2 diabetes with rosiglitazone, metformin, and glyburide monotherapy. Clin. J. Am. Soc. Nephrol. 6, 1032–1040. doi: 10.2215/CJN.09291010
Landau, M., Kurella-Tamura, M., Shlipak, M. G., Kanaya, A., Strotmeyer, E., Koster, A., et al. (2011). Correlates of insulin resistance in older individuals with and without kidney disease. Nephrol. Dial. Transplant. 26, 2814–2819. doi: 10.1093/ndt/gfq817
Lay, A., and Coward, R. J. (2014). Recent advances in our understanding of insulin signalling to the podocyte. Nephrol. Dial. Transplant. 29, 1127–1133. doi: 10.1093/ndt/gft471
Lay, A. C., Hurcombe, J. A., Betin, V. M. S., Barrington, F., Rollason, R., Ni, L., et al. (2017). Prolonged exposure of mouse and human podocytes to insulin induces insulin resistance through lysosomal and proteasomal degradation of the insulin receptor. Diabetologia 60, 2299–2311. doi: 10.1007/s00125-017-4394-0
Lenkkeri, U., Männikkö, M., McCready, P., Lamerdin, J., Gribouval, O., Niaudet, P., et al. (1999). Structure of the Gene for Congenital Nephrotic Syndrome of the Finnish Type (NPHS1) and Characterization of Mutations. Am. J. Hum. Genet. 64, 51–61. doi: 10.1086/302182
Li, C., Culver, S. A., Quadri, S., Ledford, K. L., Al-Share, Q. Y., Ghadieh, H. E., et al. (2015). High-fat diet amplifies renal renin angiotensin system expression, blood pressure elevation, and renal dysfunction caused by Ceacam1 null deletion. Am. J. Physiol. Metab. 309, E802–E810. doi: 10.1152/ajpendo.00158.2015
Li, L., Garikepati, R. M., Tsukerman, S., Kohan, D., Wade, J. B., Tiwari, S., et al. (2013). Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am. J. Physiol. Physiol. 304, F279–F288. doi: 10.1152/ajprenal.00161.2012
Ling, A. V., Gearing, M. E., Semova, I., Shin, D.-J., Clements, R., Lai, Z. W., et al. (2018). FoxO1 is required for most of the metabolic and hormonal perturbations produced by hepatic insulin receptor deletion in male mice. Endocrinology 159, 1253–1263. doi: 10.1210/en.2017-00870
Madhusudhan, T., Wang, H., Dong, W., Ghosh, S., Bock, F., Thangapandi, V. R., et al. (2015). Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat. Commun. 6:6496. doi: 10.1038/ncomms7496
Manhiani, M. M., Cormican, M. T., and Brands, M. W. (2011). Chronic sodium-retaining action of insulin in diabetic dogs. Am. J. Physiol. Ren. Physiol. 300, 957–965. doi: 10.1152/ajprenal.00395.2010
Matveyenko, A. V., Liuwantara, D., Gurlo, T., Kirakossian, D., Dalla Man, C., Cobelli, C., et al. (2012). Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes Metab. Res. Rev 61, 2269–2279. doi: 10.2337/db11-1462
Mayer, J. P., Zhang, F., and DiMarchi, R. D. (2007). Insulin structure and function. Biopolymers 88, 687–713. doi: 10.1002/bip.20734
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., et al. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583. doi: 10.1038/sj.emboj.7600633
Mima, A., Ohshiro, Y., Kitada, M., Matsumoto, M., Geraldes, P., Li, C., et al. (2011). Glomerular-specific protein kinase C-β-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 79, 883–896. doi: 10.1038/ki.2010.526
Miyazaki, Y., Cersosimo, E., Triplitt, C., and DeFronzo, R. A. (2007). Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 72, 1367–1373. doi: 10.1038/sj.ki.5002516
Mok, K. Y., Chan, P. F., Lai, L. K. P., Chow, K. L., and Chao, D. V. K. (2019). Prevalence of diabetic nephropathy among Chinese patients with type 2 diabetes mellitus and different categories of their estimated glomerular filtration rate based on the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation in primary car. J. Diabetes Metab. Disord. 18, 281–288. doi: 10.1007/s40200-018-00382-y
Molinaro, A., Becattini, B., Mazzoli, A., Bleve, A., Radici, L., Maxvall, I., et al. (2019). Insulin-Driven PI3K-AKT signaling in the hepatocyte is mediated by redundant PI3Kα and PI3Kβ activities and is promoted by RAS. Cell Metab. 29:1400-1409.e5. doi: 10.1016/j.cmet.2019.03.010
Muscogiuri, G., Chavez, A., Gastaldelli, A., Perego, L., Tripathy, D., Saad, M., et al. (2008). The crosstalk between insulin and renin-angiotensin-aldosterone signaling systems and its effect on glucose metabolism and diabetes prevention. Curr. Vasc. Pharmacol. 6, 301–312. doi: 10.2174/157016108785909715
Musso, G., Gambino, R., Tabibian, J. H., Ekstedt, M., Kechagias, S., Hamaguchi, M., et al. (2014). Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med. 11:e1001680. doi: 10.1371/journal.pmed.1001680
Mykkänen, L., Zaccaro, D. J., Wagenknecht, L. E., Robbins, D. C., Gabriel, M., and Haffner, S. M. (1998). Microalbuminuria is associated with insulin resistance in nondiabetic subjects: the insulin resistance atherosclerosis study. Diabetes Metab. Res. Rev. 47, 793–800. doi: 10.2337/diabetes.47.5.793
Naderpoor, N., Lyons, J. G., Mousa, A., Ranasinha, S., de Courten, M. P. J., Soldatos, G., et al. (2017). Higher glomerular filtration rate is related to insulin resistance but not to obesity in a predominantly obese non-diabetic cohort. Sci. Rep. 7:45522. doi: 10.1038/srep45522
Najjar, S. M., and Perdomo, G. (2019). Hepatic insulin clearance: mechanism and physiology. Physiology 34, 198–215. doi: 10.1152/physiol.00048.2018
Nakamura, M., Tsukada, H., Seki, G., Satoh, N., Mizuno, T., Fujii, W., et al. (2020). Insulin promotes sodium transport but suppresses gluconeogenesis via distinct cellular pathways in human and rat renal proximal tubules. Kidney Int. 97, 316–326. doi: 10.1016/j.kint.2019.08.021
Nakamura, N., Matsui, T., Ishibashi, Y., and Yamagishi, S. (2015). Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol. Metab. Syndr. 7:48. doi: 10.1186/s13098-015-0044-1
Narahara, H. T., Everett, N. B., Simmons, B. S., and Williams, R. H. (1958). Metabolism of Insulin-I 131 and Glucagon-I 131 in the Kidney of the Rat. Am. J. Physiol. Content 192, 227–231. doi: 10.1152/ajplegacy.1958.192.2.227
Ng, Y., Ramm, G., Lopez, J. A., and James, D. E. (2008). Rapid Activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3-L1 adipocytes. Cell Metab. 7, 348–356. doi: 10.1016/j.cmet.2008.02.008
Nickenig, G., Röling, J., Strehlow, K., Schnabel, P., and Böhm, M. (1998). Insulin induces upregulation of vascular receptor gene expression by posttranscriptional mechanisms. Circulation 98, 2453–2460. doi: 10.1161/01.CIR.98.22.2453
Nissen, S. E., and Wolski, K. (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 356, 2457–2471. doi: 10.1056/NEJMoa072761
Oh, S.-Y., Park, S.-K., Kim, J.-W., Ahn, Y.-H., Park, S.-W., and Kim, K.-S. (2003). Acetyl-CoA Carboxylase β gene is regulated by sterol regulatory element-binding protein-1 in liver. J. Biol. Chem. 278, 28410–28417. doi: 10.1074/jbc.M300553200
Olofsson, C. S., Göpel, S. O., Barg, S., Galvanovskis, J., Ma, X., Salehi, A., et al. (2002). Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflügers Arch. 444, 43–51. doi: 10.1007/s00424-002-0781-5
Pagtalunan, M. E., Miller, P. L., Jumping-Eagle, S., Nelson, R. G., Myers, B. D., Rennke, H. G., et al. (1997). Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest. 99, 342–348. doi: 10.1172/JCI119163
Perkovic, V., Jardine, M. J., Neal, B., Bompoint, S., Heerspink, H. J. L., Charytan, D. M., et al. (2019). Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 380, 2295–2306. doi: 10.1056/NEJMoa1811744
Petersen, M. C., and Shulman, G. I. (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 98, 2133–2223. doi: 10.1152/physrev.00063.2017
Pilz, S., Rutters, F., Nijpels, G., Stehouwer, C. D. A., HØjlund, K., Nolan, J. J., et al. (2014). Insulin sensitivity and albuminuria: the risc study. Diabetes Care 37, 1597–1603. doi: 10.2337/dc13-2573
Piwkowska, A., Rogacka, D., Kasztan, M., Angielski, S., and Jankowski, M. (2013). Insulin increases glomerular filtration barrier permeability through dimerization of protein kinase G type Iα subunits. Biochim. Biophys. Acta Mol. Basis Dis. 1832, 791–804. doi: 10.1016/j.bbadis.2013.02.011
Plum, L. (2006). Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 116, 1761–1766. doi: 10.1172/JCI29063
Poy, M. N., Yang, Y., Rezaei, K., Fernström, M. A., Lee, A. D., Kido, Y., et al. (2002). CEACAM1 regulates insulin clearance in liver. Nat. Genet. 30, 270–276. doi: 10.1038/ng840
Prentki, M., and Matschinsky, F. M. (1987). Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol. Rev. 67, 1185–1248. doi: 10.1152/physrev.1987.67.4.1185
Qiu, H., Yang, J.-K., and Chen, C. (2017). Influence of insulin on growth hormone secretion, level and growth hormone signalling. Sheng Li Xue Bao 69, 541–556. doi: 10.13294/j.aps.2017.0062
Rabkin, R., Ryan, M. P., and Duckworth, W. C. (1984). The renal metabolism of insulin. Diabetologia 27, 351–357. doi: 10.1007/BF00304849
Rave, K., Nosek, L., Posner, J., Heise, T., Roggen, K., and van Hoogdalem, E. J. (2006). Renal glucose excretion as a function of blood glucose concentration in subjects with type 2 diabetes - Results of a hyperglycaemic glucose clamp study. Nephrol. Dial. Transplant. 21, 2166–2171. doi: 10.1093/ndt/gfl175
Ricketts, H. T., and Wildberger, H. L. (1962). The effect of renal disease on the degradation of insulin. Trans. Am. Clin. Climatol. Assoc. 74, 102–108.
Ritz, E., Adamczak, M., and Wiecek, A. (2013). Carbohydrate Metabolism in Kidney Disease and Kidney Failure. Amsterdam: Elsevier Inc, doi: 10.1016/B978-0-12-391934-2.00002-3
Roder, K., Zhang, L., and Schweizer, M. (2007). SREBP-1c mediates the retinoid-dependent increase in fatty acid synthase promoter activity in HepG2. FEBS Lett. 581, 2715–2720. doi: 10.1016/j.febslet.2007.05.022
Rowe, J. W., Young, J. B., Minaker, K. L., Stevens, A. L., Pallotta, J., and Landsberg, L. (1981). Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes Metab. Res. Rev. 30, 219–225. doi: 10.2337/diab.30.3.219
Rubenstein, A. H., Lowy, C., Welborn, T. A., and Fraser, T. R. (1967). Urine insulin in normal subjects. Metabolism 16, 234–244. doi: 10.1016/0026-0495(67)90172-2
Rubenstein, A. H., and Spitz, I. (1968). Role of the kidney in insulin metabolism and excretion. Diabetes Metab. Res. Rev. 17, 161–169. doi: 10.2337/diab.17.3.161
Ruiz, O. S., Qiu, Y.-Y., Cardoso, L. R., and Arruda, J. A. L. (1998). Regulation of the renal Na–HCO3 cotransporter: IX. Modulation by insulin, epidermal growth factor and carbachol. Regul. Pept. 77, 155–161. doi: 10.1016/S0167-0115(98)00115-3
Russo, L., Muturi, H. T., Ghadieh, H. E., Ghanem, S. S., Bowman, T. A., Noh, H. L., et al. (2017). Liver-specific reconstitution of CEACAM1 reverses the metabolic abnormalities caused by its global deletion in male mice. Diabetologia 60, 2463–2474. doi: 10.1007/s00125-017-4432-y
Sánchez-Villanueva, R., Estrada, P., Del Peso, G., Grande, C., Díez, J. J., Iglesias, P., et al. (2013). Repeated analysis of estimated insulin resistance using the HOMAIR index in nondiabetic patients on peritoneal dialysis and its relationship with cardiovascular disease and mortality. Nefrologia 33, 85–92. doi: 10.3265/Nefrologia.pre2012.Nov.11430
Sanders, F. W. B., and Griffin, J. L. (2016). De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol. Rev. 91, 452–468. doi: 10.1111/brv.12178
Santamaria, B., Marquez, E., Lay, A., Carew, R. M., González-Rodríguez, Á, and Welsh, G. I et al. (2015). IRS2 and PTEN are key molecules in controlling insulin sensitivity in podocytes. Biochim. Biophys. Acta Mol. Cell Res. 1853, 3224–3234. doi: 10.1016/j.bbamcr.2015.09.020
Sarafidis, P. A., Stafylas, P. C., Georgianos, P. I., Saratzis, A. N., and Lasaridis, A. N. (2010). Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am. J. Kidney Dis. 55, 835–847. doi: 10.1053/j.ajkd.2009.11.013
Sarzani, R., Dessì-Fulgheri, P., Salvi, F., Serenelli, M., Spagnolo, D., Cola, G., et al. (1999). A novel promoter variant of the natriuretic peptide clearance receptor gene is associated with lower atrial natriuretic peptide and higher blood pressure in obese hypertensives. J. Hypertens. 17, 1301–1305. doi: 10.1097/00004872-199917090-00010
Sasaki, M., Sasako, T., Kubota, N., Sakurai, Y., Takamoto, I., Kubota, T., et al. (2017). Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes Metab. Res. Rev 66, 2339–2350. doi: 10.2337/db16-1602
Satoh, N., Nakamura, M., Suzuki, M., Suzuki, A., Seki, G., and Horita, S. (2015). Roles of Akt and SGK1 in the regulation of renal tubular transport. Biomed Res. Int. 2015, 1–8. doi: 10.1155/2015/971697
Shinohara, K., Shoji, T., Emoto, M., Tahara, H., Koyama, H., Ishimura, E., et al. (2002). Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. J. Am. Soc. Nephrol. 13, 1894–1900. doi: 10.1097/01.ASN.0000019900.87535.43
Skolnik, E. Y., Batzer, A., Li, N., Lee, C. H., Lowenstein, E., Mohammadi, M., et al. (1993). The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science 260, 1953–1955. doi: 10.1126/science.8316835
Song, S. H., McIntyre, S. S., Shah, H., Veldhuis, J. D., Hayes, P. C., and Butler, P. C. (2000). Direct measurement of pulsatile insulin secretion from the portal vein in human subjects 1. J. Clin. Endocrinol. Metab. 85, 4491–4499. doi: 10.1210/jcem.85.12.7043
Sowers, J. R., Epstein, M., and Frohlich, E. D. (2001). Diabetes, hypertension, and cardiovascular disease an update. Hypertension 37, 1053–1059. doi: 10.1161/01.HYP.37.4.1053
Spoto, B., Pisano, A., and Zoccali, C. (2016). Insulin resistance in chronic kidney disease: a systematic review. Am. J. Physiol. Renal Physiol. 311, F1087–F1108. doi: 10.1152/ajprenal.00340.2016
Stechi, L. A., De Carll, S., and Bartoll, E. (1994). In situ characterization of renal insulin receptors in the rat. J. Recept. Res. 14, 347–356. doi: 10.3109/10799899409101509
Steiner, D. F. (2004). The proinsulin c-peptide—a multirole model. Exp. Diabesity Res. 5, 7–14. doi: 10.1080/15438600490424389
Susztak, K., Raff, A. C., Schiffer, M., and Bottinger, E. P. (2006). Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes Metab. Res. Rev 55, 225–233. doi: 10.2337/diabetes.55.01.06.db05-0894
Svensson, M., and Eriksson, J. W. (2006). Insulin resistance in diabetic nephropathy — cause or consequence? Diabetes. Metab. Res. Rev. 22, 401–410. doi: 10.1002/dmrr.648
Talor, Z., Emmanouel, D. S., and Katz, A. I. (1982). Insulin binding and degradation by luminal and basolateral tubular membranes from rabbit kidney. J. Clin. Invest. 69, 1136–1146. doi: 10.1172/JCI110549
Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96. doi: 10.1038/nrm1837
Tejada, T., Catanuto, P., Ijaz, A., Santos, J. V., Xia, X., Sanchez, P., et al. (2008). Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 73, 1385–1393. doi: 10.1038/ki.2008.109
Thirone, A. C. P., Huang, C., and Klip, A. (2006). Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 17, 70–76. doi: 10.1016/j.tem.2006.01.005
Tiwari, S., Sharma, N., Gill, P. S., Igarashi, P., Kahn, C. R., Wade, J. B., et al. (2008). Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 6469–6474. doi: 10.1073/pnas.0711283105
Tiwari, S., Singh, R. S., Li, L., Tsukerman, S., Godbole, M., Pandey, G., et al. (2013). Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. J. Am. Soc. Nephrol. 24, 1209–1214. doi: 10.1681/ASN.2012060628
Tokarz, V. L., MacDonald, P. E., and Klip, A. (2018). The cell biology of systemic insulin function. J. Cell Biol. 217, 2273–2289. doi: 10.1083/jcb.201802095
Vallon, V., Platt, K. A., Cunard, R., Schroth, J., Whaley, J., Thomson, S. C., et al. (2011). SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 22, 104–112. doi: 10.1681/ASN.2010030246
Villarreal, R., Mitrofanova, A., Maiguel, D., Morales, X., Jeon, J., Grahammer, F., et al. (2016). Nephrin contributes to insulin secretion and affects mammalian target of rapamycin signaling independently of insulin receptor. J. Am. Soc. Nephrol. 27, 1029–1041. doi: 10.1681/ASN.2015020210
Wang, X. L., Lu, J. M., Pan, C. Y., Tian, H., and Li, C. L. (2005). A comparison of urinary albumin excretion rate and microalbuminuria in various glucose tolerance subjects. Diabet. Med. 22, 332–335. doi: 10.1111/j.1464-5491.2004.01408.x
Welsh, G. I., Hale, L. J., Eremina, V., Jeansson, M., Maezawa, Y., Lennon, R., et al. (2010). Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 12, 329–340. doi: 10.1016/j.cmet.2010.08.015
Williams, D. M., Nawaz, A., and Evans, M. (2020). Renal outcomes in type 2 diabetes: a review of cardiovascular and renal outcome trials. Diabetes Ther. 11, 369–386. doi: 10.1007/s13300-019-00747-3
Wolf, G., Chen, S., and Ziyadeh, F. N. (2005). From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes Metab. Res. Rev. 54, 1626–1634. doi: 10.2337/diabetes.54.6.1626
Wright, E. M., Loo, D. D. F., and Hirayama, B. A. (2011). Biology of Human Sodium Glucose Transporters. Physiol. Rev. 91, 733–794. doi: 10.1152/physrev.00055.2009
Xu, H., Huang, X., Ärnlöv, J., Cederholm, T., Stenvinkel, P., Lindholm, B., et al. (2014). Clinical correlates of insulin sensitivity and its association with mortality among men with CKD stages 3 and 4. Clin. J. Am. Soc. Nephrol. 9, 690–697. doi: 10.2215/CJN.05230513
Xu, J., Keeton, A. B., Franklin, J. L., Li, X., Venable, D. Y., Frank, S. J., et al. (2006). Insulin enhances growth hormone induction of the MEK/ERK signaling pathway. J. Biol. Chem. 281, 982–992. doi: 10.1074/jbc.M505484200
Xu, J., and Messina, J. L. (2009). “Crosstalk between growth hormone and insulin signaling,” in Vitamins and Hormones, Ed. G. Litwack (Amsterdam: Elsevier Inc), 125–153. doi: 10.1016/S0083-6729(08)00606-7
Yamada, T., Katagiri, H., Asano, T., Tsuru, M., Inukai, K., Ono, H., et al. (2002). Role of PDK1 in insulin-signaling pathway for glucose metabolism in 3T3-L1 adipocytes. Am. J. Physiol. Metab. 282, E1385–E1394. doi: 10.1152/ajpendo.00486.2001
Ye, L., Maji, S., Sanghera, N., Gopalasingam, P., Gorbunov, E., Tarasov, S., et al. (2017). Structure and dynamics of the insulin receptor: implications for receptor activation and drug discovery. Drug Discov. Today 22, 1092–1102. doi: 10.1016/j.drudis.2017.04.011
Yip, J., Mattock, M. B., Morocutti, A., Sethi, M., Trevisan, R., and Viberti, G. (1993). Insulin resistance in insulin-dependent diabetic patients with microalbuminuria. Lancet 342, 883–887. doi: 10.1016/0140-6736(93)91943-G
Yki-Järvinen, H. (2004). Thiazolidinediones. N. Engl. J. Med. 351, 1106–1118. doi: 10.1056/NEJMra041001
Zorzano, A., Munoz, P., Camps, M., Mora, C., Testar, X., and Palacin, M. (1996). Insulin-Induced Redistribution of GLUT4 Glucose Carriers in the Muscle Fiber: in Search of GLUT4 Trafficking Pathways. Diabetes 45, S70–S81. doi: 10.2337/diab.45.1.S70
Keywords: diabetic nephropathy, insulin, insulin resistance, insulin clearance, albuminuria
Citation: Pina AF, Borges DO, Meneses MJ, Branco P, Birne R, Vilasi A and Macedo MP (2020) Insulin: Trigger and Target of Renal Functions. Front. Cell Dev. Biol. 8:519. doi: 10.3389/fcell.2020.00519
Received: 18 March 2020; Accepted: 02 June 2020;
Published: 29 July 2020.
Edited by:
Samar Abd ElHafeez, Alexandria University, EgyptReviewed by:
Sonia Michael Najjar, Ohio University, United StatesKarem Salem, Fayoum University, Egypt
Copyright © 2020 Pina, Borges, Meneses, Branco, Birne, Vilasi and Macedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Paula Macedo, cGF1bGEubWFjZWRvQG5tcy51bmwucHQ=