 Haochang Hu
Haochang Hu Shaoyi Lin1,2
Shaoyi Lin1,2- 1School of Medicine, Ningbo University, Ningbo, China
- 2Department of Cardiology, Ningbo City First Hospital, Ningbo, China
Transcription factor 21 (TCF21) is specific for mesoderm and is expressed in the embryos’ mesenchymal derived tissues, such as the epicardium. It plays a vital role in regulating cell differentiation and cell fate specificity through epithelial-mesenchymal transformation during cardiac development. For instance, TCF21 could promote cardiac fibroblast development and inhibit vascular smooth muscle cells (VSMCs) differentiation of epicardial cells. Recent large-scale genome-wide association studies have identified a mass of loci associated with coronary heart disease (CHD). There is mounting evidence that TCF21 polymorphism might confer genetic susceptibility to CHD. However, the molecular mechanisms of TCF21 in heart development and CHD remain fundamentally problematic. In this review, we are committed to providing a detailed introduction of the biological roles of TCF21 in epicardial fate determination and the development of CHD.
Introduction
The helix-loop-helix (HLH) family of transcription factors possesses highly conserved bipartite domains for DNA binding and protein-protein interactions, which is pivotal in various developmental processes, covering myogenesis, neurogenesis, and hematopoiesis (Jiang and Yang, 2018). The HLH family members are divided into seven families according to tissue distribution, dimerization ability, and DNA binding specificity (Massari and Murre, 2000). Transcription factor 21 (TCF21, 6417bp)—also known as Pod1, bHLHa23, or Capsulin—encodes a transcription factor belonging to the cell-type-specific class II basic helix-loop-helix (bHLH) family at chromosome 6q23.2 (Jiang and Yang, 2018). Moreover, TCF21 is specific for mesoderm and is expressed in the embryos’ mesenchymal derived tissues, such as the epicardium, lung, intestine, gonad, and kidney (Plotkin and Mudunuri, 2008). It regulates the cell differentiation during embryonic development, such as coronary vasculature (Hidai et al., 1998). The ChIP-Seq studies in primary cultured human coronary vascular smooth muscle cells (VSMCs) have identified several TCF21 target genes, such as smooth muscle contraction, growth factor binding and matrix interaction, which are involved in the processes associated with coronary heart disease (CHD) pathophysiology (Sazonova et al., 2015). A wealth of evidence confirms that TCF21 takes an active role in VSMC phenotypic modulation in CHD. In this review, we endeavor to provide a detailed introduction of the biological roles of TCF21 in epicardial fate determination and the development of CHD.
The Origin of Epicardial Cells
The proepicardial cells, a mesoderm-derived cell cluster, are highly conserved in vertebrates including Xenopus, zebrafish, mice and even human (Cao and Cao, 2018), which could migrate to the myocardial surface, adhere and form the epicardium during embryogenesis (Tandon et al., 2016). Once the original epicardium forms, a portion of the epicardial cells could enter the subepicardial domain and produce epicardium-derived cells (EPDCs) by undergoing epithelial-mesenchymal transition (EMT) (Lie-Venema et al., 2007). During the EMT, epithelial cells downregulate their epithelial gene expression, lose their unique characteristics, and activate the mesenchymal genes to increase cell viability and motility, including aggressiveness (Li et al., 2018). Recent studies have confirmed that EMT could be affected by several signaling pathways in epicardial cells, including but not limited to transforming growth factorβ (TGF-β), retinoic acid (RA), platelet-derived growth factor (PDGF), and extra-cellular matrix (ECM) components (Cao et al., 2020). EPDCs are highly plastic and have been reported to differentiate into myocardial fibroblasts, VSMCs, and pericytes (Gittenberger-de Groot et al., 2010). Lineage tracing studies also confirmed that the VSMCs originated from the epicardial cells (Majesky, 2007).
TCF21 and Epicardial Cells
The Role of TCF21 in Epicardial Cells During Heart Development
Studies in different animal models have delivered several molecular signatures of proepicardial cells, including the conserved transcription factors [TCF21, Wilms tumor 1(WT1), and T-box factor 18 (Tb×18)] (Cao et al., 2020). The study of Xenopus embryos showed that proepicardial cells could migrate to the heart, retain their precursor cell characteristics and impair maturity in the absence of TCF21 (Tandon et al., 2013). It suggested that the morphological defect in epicardial integrity might be caused by the absence of TCF21 through regulating the specification and maturation of the proepicardial cells at earlier stages of epicardial development (Figure 1).
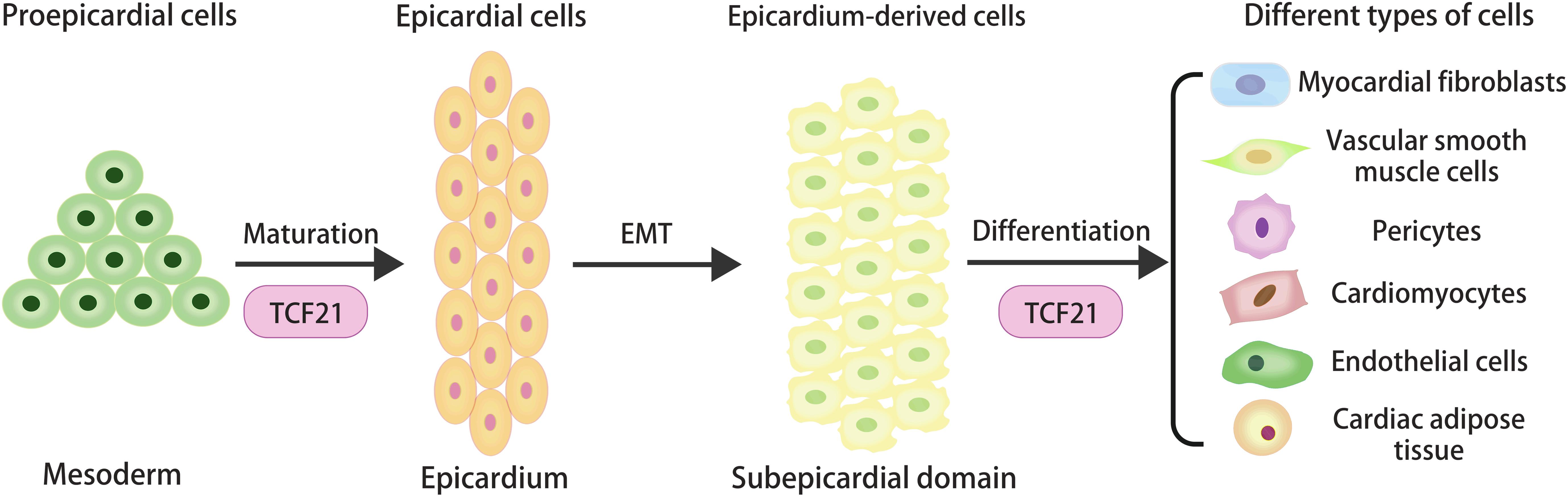
Figure 1. The origin and cellular contributions of epicardial cells during heart development, and the regulatory role of TCF21 in the specification and maturation of proepicardial cells and the determination of epicardial cell fate.
Previously, we recognized that TCF21 could promote cardiac fibroblast development and inhibit VSMC differentiation of EPDCs. Asha et al. found most epicardial cells expressing TCF21 were lumped in the myocardial fibroblast lineage before the initiation of EMT (Acharya et al., 2012). It was concluded from the study that a TCF21-deficient heart could not undergo EMT and form cardiac fibroblasts, which confirmed the unique role of TCF21 in epicardial EMT and differentiation (Acharya et al., 2012). The in vivo experiment showed that loss of TCF21 in mice resulted in a decrease of myocardial fibroblasts due to the premature differentiation of EPDCs into VSMCs through RA signals (Braitsch et al., 2012). Together, these data support the thesis that TCF21 plays an indispensable role in the development of myocardial fibroblasts.
With the development of single-cell sequencing technology, new insights into the differentiation of epicardial cells have been obtained. The near-term single-cell analysis of an epicardium model derived from human pluripotent stem cells showed that the different epicardial subsets were defined by high levels of transcription factors BNC1 or TCF21. In the TCF21 population, TCF21high cells could differentiate into myocardial fibroblasts and VSMCs, while TCF21low cells were mainly limited to VSMCs (Gambardella et al., 2019). These data revealed the complex regulatory effect of TCF21 on fundamental cells fate decisions during heart development. Further studies on the accurate regulatory mechanism of TCF21 in epicardial differentiation should be performed to better explain its role in normal heart development.
The Role of TCF21 in Myocardial Regeneration
After myocardial infarction, millions of myocardial cells die, which are replaced by fibrotic scar tissue. Regeneration and fibrosis are two important healing processes after heart injury. In response to cardiac damage, epicardial cells can be activated to migrate to the injured myocardium and differentiate into the desired cell lines (especially the cardiac fibroblasts) to support the regeneration of heart tissue (Limana et al., 2007; Bos et al., 2014; Cao et al., 2016). In addition to myocardial fibroblasts, VSMCs, and pericytes, epicardial cells also could give rise to cardiomyocytes, endothelial cells and cardiac adipose tissue during development (Zhou et al., 2008; Zangi et al., 2017). If true, activated epicardial cells can be harnessed to exploit their potential for differentiation to repair injured hearts. However, more evidence needs to be accumulated to prove these assumptions.
Genetic fate-mapping used by Kazu et al. showed that TCF21+ epicardial cells of zebrafish might differentiate into perivascular cells, but not to cardiomyocytes during heart regeneration (Kikuchi et al., 2011). In mice with myocardial infarction, TCF21 lineage-traced cells could transform into periostin-expressing myofibroblasts, which is necessary for adaptive healing and fibrosis in the heart (Kanisicak et al., 2016). In addition, the survival rate of myocardial infarction decreases with the reduction of periostin-expressing myofibroblasts, suggesting that fibrosis is an effective mechanism for repairing myocardium. In the past, fibrosis was deemed detrimental to the regeneration process, and it was thought that excessive fibrosis accelerates the progression of diseases. However, fibrosis is transient in zebrafish and fibrosis regression caused by fibroblast inactivation is concomitant with the regrowth of myocardial wall. Sanchez-Iranzo et al., 2018 found that cardiac fibroblasts in zebrafish not only contribute to the fibrosis response after injury but are also necessary for the proliferation of cardiomyocytes during regeneration. All the information on zebrafish’s myocardial regeneration provides a theoretical basis for regenerative medicine. Though regenerative medicine is regarded as a promising treatment for heart disease, many obstacles will have to be overcome before it can be used clinically.
TCF21 and the Development of Coronary Heart Disease
Coronary Heart Disease
According to the 2017 National Health Interview Survey, the age-adjusted prevalence of cardiovascular diseases (CVD) was 10.6% (11.0% for Whites, 9.7% for Blacks, 7.4% for Hispanics and 6.1% for Asians) (Virani et al., 2020). Coronary heart disease (CHD), the most common type of heart disease, is featured on the coronary stenosis caused by atherosclerotic plaque (Li et al., 2020), leading to the ischemia and apoptosis of cardiac cells (Firoozi et al., 2020). Acute myocardial infarction (AMI), the most severe consequences of CHD with high mortality, results from prolonged ischemia and irreversible necrosis of the heart muscle (Liu et al., 2009). Therefore, there is a pressing need to elucidate the pathogenesis of CHD and develop an effective therapy for patients with CHD.
As the basic pathological change of CHD, atherosclerosis affects the structure and function of three layers of the coronary artery vessel wall (Wang et al., 2017). Atherosclerotic plaque exists in the intimal space of coronary arteries, between the inner endothelial cell layer and the middle layer of the contractile VSMCs (Truskey, 2010). It is formed by absorbing circulating lipoproteins, recruiting inflammatory cells, and transferring the dedifferentiated VSMCs into the lesion to form a fibrous cap (Nurnberg et al., 2015). In the sections below, we highlight our current understanding of the influence of coronary VSMCs on CHD.
Phenotypic Modulation of VSMCs
Blood vessels are composed of three layers: the tunica intima, tunica media, and tunica adventitia (Truskey, 2010), with each layer having its unique histological, biological and functional characteristics. The tunica intima, the layer closest to blood, is a continuous lining formed by endothelial cells between the blood and tissues. The tunica media mainly consists of VSMCs and extracellular matrix (circular elastic fibers, collagen I and proteoglycans), which play an important role in maintaining tension and supporting vessels. The tunica adventitia is a loose connective tissue mainly comprising fibroblasts. Myocardial fibroblasts are the main non-myocyte cells in the heart and are critical in maintaining normal cardiac function (Souders et al., 2009).
As an important metabolic and endocrine organ, VSMCs are involved in many physiological and pathological processes through its proliferation, migration, and synthesis of extracellular matrix (Frismantiene et al., 2018). During vascular development, VSMCs secrete major extracellular proteins to keep the unique mechanical properties of vessel walls. Unlike conventional differentiated cells, the phenotypes of VSMCs can undergo reversible changes in different developmental stages, during the progression of diseases or as a response to injuries. The state of VSMCs can be switched between the “differentiated” contractile phenotype and the “dedifferentiated” synthetic phenotype, a process known as phenotypic transformation (Alexander and Owens, 2012). This extensive plasticity of VSMCs gives them the ability to participate in vascular repair.
Indeed, VSMCs principally exhibits the contractile phenotype with low proliferation and synthetic activity, which could regulate blood pressure, blood vessel diameter, and blood flow distribution (Owens et al., 2004), however, the synthetic phenotype loses its contractile function with the low expression of the contractile protein and gains synthetic, migratory, and proliferative properties. Although the phenotypic reversibility is important for normal homeostasis, VSMCs are vulnerable to physiological and non-physiological stimuli that could cause adverse phenotypic switching. A large number of studies have shown that the phenotypic transformation of VSMCs has made a great contribution to various major diseases such as atherosclerosis, asthma, hypertension, and cancer (Alexander and Owens, 2012).
VSMC Phenotypic Modulation and CHD
Phenotypic modulation of VSMCs is a physiological response to vascular injury. The lineage tracing studies have revealed that about 80 percent of the cells in atherosclerotic plaque lack traditional VSMC marks but about half of these cells originate from the VSMCs (Shankman et al., 2015; Cherepanova et al., 2016). Technical advances in the combination of single-cell RNA-sequencing and lineage tracing have demonstrated multiple dedifferentiated SMC phenotypes in atherosclerosis (Wirka et al., 2019). However, the question of whether VSMC phenotypic transformation in atherosclerosis is protective or deteriorative for the risk of CHD remains unsettled.
On the one hand, under the stimulation of proatherogenic factors, the contractile VSMCs could de-differentiate and be transformed into the macrophage-like inflammatory cells, which could secrete a large number of ECM and inflammatory factors, promoting plaque rupture and acute CHD events (Milutinovic et al., 2020). So, blocking the proinflammatory changes of the VSMC phenotype is of great importance for clinical treatment.
James et al. found that the gene expression associated with the contraction phenotype of VSMCs decreased significantly, while the expression levels of macrophage markers increased after cholesterol loading (Rong et al., 2003). However, MerTK and FcgR1 (CD64), two key marker genes commonly associated with mature macrophages, were not up-regulated in the macrophage-like cells. Cholesterol loading of VSMCs converted them to a macrophage-like state by downregulating the miRNA-143/145-myocardin axis (Vengrenyuk et al., 2015). Notably, these cells were classified as macrophages based largely on single cell-surface protein markers (Pan and Reilly, 2019). However, their transcriptome and functional properties suggested that they act differently from classical macrophages in the formation of atherosclerosis.
On the other hand, VSMCs transition to fibroblast-like cells may enhance the protection of the fibrous cap and reduce CHD events. Notably, the number of VSMCs in the fiber caps was negatively correlated with plaque stability. A thick fiber cap contains plentiful VSMCs surrounding the necrotic core to stabilize the fragile plaque. As the disease progresses, the plaque may rupture violently due to the reduced number of VSMCs in the fibrous cap, leading to arterial thrombosis and acute coronary events (Pan and Reilly, 2019).
In the present study, the single-cell RNA sequencing method was used to characterize the transcriptome phenotype of modulated VSMCs in mice and human atherosclerosis lesions. Though SMC-derived cells could be involved in exocytosis as “non-specialized” phagocytes, it does not have a macrophage-like transcriptional phenotype in vivo. Wirka et al. (2019) found that the modulated VSMCs transform into unique fibroblast-like cells, rather than into a classical macrophage phenotype in the ApoE–/– mouse model. Furthermore, the accumulation of VSMCs in the fibrous cap is monoclonal or oligoclonal, which plays a healing role in disease (Orlandi and Bennett, 2010). Therefore, we have reasons to speculate that the VSMCs phenotypic modulation seems to be restorative in atherosclerosis, rather than the primary driver of plaque formation (Bennett et al., 2016).
TCF21 and VSMC Phenotypic Modulation in CHD
Genome-wide association studies (GWAS) have identified over 160 loci associated with CHD (Wong et al., 2019). There is mounting evidence that TCF21 serves as the causal gene for CHD and that high expression of TCF21 is related to reduced CHD risk in human CHD-related tissues (Wirka et al., 2019). Besides, TCF21 was expressed in human coronary VSMCs but not in aortic SMCs or endothelial cells (Nurnberg et al., 2015). The rs12190287 located within the 3′-untranslated region (3′-UTR) of TCF21 was related to the TCF21 gene expression, which was identified as an expression quantitative trait locus (eQTL) (Schunkert et al., 2011). Race-based research suggested that the SNP of rs12190287 in TCF21 might be an independent genetic risk factor for CHD in Chinese and European patients (Wang et al., 2014; Wirtwein et al., 2017) but not in Japanese patients (Dechamethakun et al., 2014). Another CSMC eQTL variant located in the TCF21 promoter region, rs2327429, was identified as a methylation quantitative trait locus for TCF21 expression, suggesting that methylation regulation is a molecular trait that may mediate the risk of CHD (Gutierrez-Arcelus et al., 2013; Liu et al., 2018). A meta-analysis of two GWAS comprising 1,515 CHD cases and 5,019 controls showed that TCF21 rs12524865 conferred predisposition to CHD in the Chinese and Europeans (Lu et al., 2012). Though significant progress has been made in mapping the genetic contribution to CHD, the molecular mechanisms of TCF21 in CHD remain fundamentally problematic.
It is generally acknowledged that the TCF21 target gene relevant to CHD is enriched in the processes of vascular wall biology (Sazonova et al., 2015). It was observed that VSMC-specific knockout of TCF21 in mice inhibited VSMC phenotypic modulation (Wirka et al., 2019), and the absence of TCF21 results in the decreased fibroblasts in the lesion and the instability of plaque, suggesting that TCF21 functions as a protective factor by promoting VSMC transition to fibroblast-like cells in the lesion and fibrous cap (Figure 2). Regretfully, there are still fundamental questions related to the mechanisms by which it performs this role.
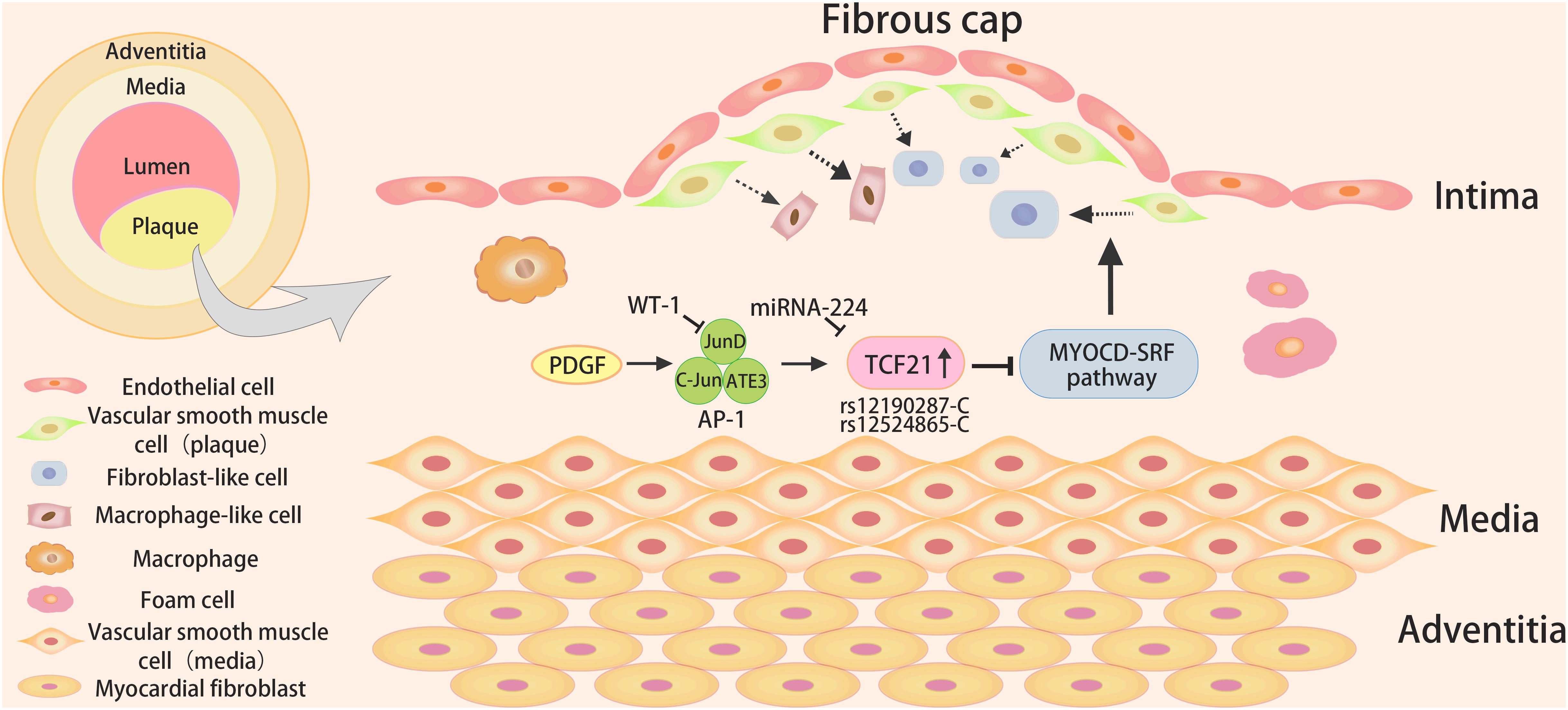
Figure 2. The role and signaling pathways of TCF21 in coronary heart disease. The TCF21 risk alleles (rs12190287-C or rs12524865-C) might result in increased TCF21 expression upon activation of PDGF signaling, and WT1could inhibit AP-1 mediated transcription and c-Jun mediated rs12190287 activation, leading to reduced expression of TCF21. In addition, TCF21 could block the MYOCD-SRF pathway to inhibit the differentiation of VSMCs in atherosclerotic plaque.
The Upstream Pathways of TCF21 in VSMC Phenotypic Modulation
Platelet-derived growth factor (PDGF) signaling mediated by PDGFRβ has been certified as having a major role in EMT, epicardial fate determination, VSMCs migration and proliferation, and atherosclerosis (Smith et al., 2011). The increased PDGF signaling promoted the formation of aggressive plaque in the thoracic aorta and coronary arteries (He et al., 2015). PDGF is a potent inducer of the transformation of contractile VSMCs to secreted phenotype (Raines and Ross, 1993; Owens et al., 2004). Clint et al. found that individuals carrying the TCF21 risk alleles (rs12190287-C or rs12524865-C) appeared to exhibit increased TCF21 expression upon activation of PDGF signaling in coronary VSMCs (Miller et al., 2013). It could increase enrichment of active histone modifications and form an open chromatin conformation, which binds to the atypical AP-1 element, likely including JunD, c-Jun, and ATF3, leading to altering TCF21 expression (Miller et al., 2013). Besides, WT1 as a transcriptional repressor, could inhibit AP-1 mediated transcription and c-Jun mediated TCF21 rs12190287 activation in vitro (Miller et al., 2013).
Studies using allelic imbalance sequencing have shown that SNPs could regulate miRNA-mediated repression by altering complementary miRNA binding sites (Kim and Bartel, 2009). The later studies by Clint et al. reported that the major risk C variants of TCF21 rs12190287 preferentially combines with miRNA-224 on account of the altered secondary structure of RNA, ultimately resulting in the downregulation of TCF21 expression (Miller et al., 2014). Furthermore, PDGF signals and TGF-β1 signals are the potential upstream mediators of miRNA-224 to direct the expression of allele-specific TCF21 rs12190287.
Taken together, these two mechanisms happen to explain the two pathways for the alteration of allele-specific gene expression (Mar et al., 2011). A transcriptional regulatory mechanism has been explored that the increased rs12190287 risk C allele could bring about elevated TCF21 expression mediated by PDGF signaling pathways. What is more, the C allele at rs12190287 interacts with miR-224 to reduce the expression of TCF21 through the miRNA mechanism.
The Downstream Pathways of TCF21 in VSMC Phenotypic Modulation
Myocardin (MYOCD) is an effective transcriptional co-activator in VSMCs that could bind to the DNA-binding transcription factor serum response factor (SRF) to form a complex and regulate the basic expression program of the VSMC gene (Wang et al., 2001). In addition, MYOCD serves as a major regulator of VSMC differentiation, which could facilitate the expression of VSMC differentiation markers. Manabu et al. used the Assay for Transposase-Accessible Chromatin using sequencing (ATAC-Seq) and ChIP-PCR methods to illuminate that TCF21, co-localized with SRF in the open chromatin region of VSMCs, could inhibit the binding of MYOCD in the human coronary VSMCs at the SRF enhancer (Nagao et al., 2020). Subsequent studies provide evidence for the speculation that TCF21 might disrupt the MYOCD-SRF transcriptional complex and transcriptional regulation of VSMC genes through direct interaction with MYOCD.
Except for the above pathway, Sylvia et al. found that TCF21 could bind to the VSMC marker ACTA2 locus and blocks its expression, indicating a directly inhibiting effect of TCF21 on some VSMC genes (Nurnberg et al., 2015). Also, the downregulated TCF21 expression induced by CXCL12 could decrease the promoter activity of ABCA1 and ABCA1 expression, thus inhibiting ABCA1-dependent cholesterol efflux from macrophages and promoting atherosclerosis (Gao et al., 2019).
It is important to consider that the role of TCF21 in VSMCs phenotypic modulation is regulated by multiple downstream pathways. Additional studies with human vascular disease samples should be conducted to better assess the exact mechanism in vivo.
TCF21 and Other Cardiovascular Disease
Hypertension is the pathogeny of hypertensive heart disease and an important risk factor for CHD. Large-scale association studies showed that TCF21 rs12190287 was a susceptibility locus for hypertension in the Japanese population (Fujimaki et al., 2015). A significant correlation was found between the polymorphism of TCF21 rs76987554 and hypertension (Liang et al., 2017). Previous studies have shown that TCF21 could reduce the cyclin-dependent kinase inhibitor P21 expression in MG63 cells, which is essential for VSMCs proliferation (Funato et al., 2003; Bond et al., 2006), and microvascular stenosis caused by abnormal VSMCs proliferation may play a pivotal part in the occurrence and development of hypertension (Feihl et al., 2008), suggesting that TCF21 might modulate blood pressure through p21-dependent microvascular remodeling. Caitlin et al. found that in the advanced stages of the disease, the predominant expression of TCF21 occurred in perivascular fibrosis caused by pressure overload in the adult mice (Braitsch et al., 2013). Therefore, we inferred that TCF21 might contribute to the genetic susceptibility to CHD by affecting the predisposition to hypertension.
Congenital heart disease is an occasional, isolated, non-syndromic defect that may be the result of an interaction between genetic and environmental factors (van der Linde et al., 2011). Ventricular septal defect (VSD) is one of the most common types of congenital heart disease. Recent research has shown that allele G at variant rs12190287 is a contributing risk factor for VSD in the Chinese population (Yang et al., 2017). Together, all these data indicated that TCF21 might confer genetic susceptibility to cardiovascular disease.
Conclusion and Prospects
In conclusion, the abovementioned studies demonstrated that TCF21 was engaged in the specification and maturation of proepicardial cells and the fate determination of epicardial cells during cardiomyogenesis. And TCF21 could activate the protective role of VSMC phenotypic modulation via the complex upstream and downstream signaling pathways in CHD. Both PDGF signaling pathway and miRNA mechanism might be potential mechanisms for the regulation of TCF21 expression. Moreover, TCF21 could modulate VSMCs response to vascular stress and injury by antagonizing the MYOCD-SRF pathway. Further investigation should be undertaken to explore whether and how TCF21 interacts with other factors to influence VSMC phenotypic modulation.
Recent evidence has confirmed the momentous role of TCF21 in epicardial cell differentiation, and it has been found that TCF21 rs12190287 polymorphism is associated with VSD, a congenital heart disease. Therefore, we speculated that TCF21 might play a regulatory role in heart development and related diseases. The study of the corresponding mechanism principle will be of great interest in the future.
The single-cell sequencing studies have shown that VSMCs transitioned almost exclusively to fibroblast-like cells, but not to macrophage-like cells in animal models with 16 weeks of disease progression (Wirka et al., 2019). Furthermore, the pro-differentiation related factors, such as SMAD3 and TGFβ1 could increase the risk of CHD (Miller et al., 2016; Iyer et al., 2018), which reinforces the evidence in reverse. If this is the case, it might clarify the mainly anti-atherosclerotic effect of VSMCs, thus providing strong evidence for treatment. Based on the flaws in current research, such as short observation time and defect in the detection method, a combination of complementary methods should be used to verify the atheroprotective role.
Author Contributions
XC, SL, and HH contributed to the conception and design of manuscript. HH, XC, and SW contributed to the writing and final approval of the submitted revision.
Funding
The research was supported by the grants from: National Natural Science Foundation of China (81900441), Natural Science Foundation of Zhejiang Province (LQ20H020001, LQ19H020010, LY19H020003), and Natural Science Foundation of Ningbo (2018A610418, 2017A610209).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acharya, A., Baek, S. T., Huang, G., Eskiocak, B., Goetsch, S., Sung, C. Y., et al. (2012). The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139, 2139–2149. doi: 10.1242/dev.079970
Alexander, M. R., and Owens, G. K. (2012). Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 74, 13–40. doi: 10.1146/annurev-physiol-012110-142315
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118, 692–702. doi: 10.1161/CIRCRESAHA.115.306361
Bond, M., Sala-Newby, G. B., Wu, Y. J., and Newby, A. C. (2006). Biphasic effect of p21Cip1 on smooth muscle cell proliferation: role of PI 3-kinase and Skp2-mediated degradation. Cardiovasc. Res. 69, 198–206. doi: 10.1016/j.cardiores.2005.08.020
Bos, D. J., van Raalten, T. R., Oranje, B., Smits, A. R., Kobussen, N. A., Belle, J., et al. (2014). Developmental differences in higher-order resting-state networks in autism spectrum disorder. Neuroimage Clin. 4, 820–827. doi: 10.1016/j.nicl.2014.05.007
Braitsch, C. M., Combs, M. D., Quaggin, S. E., and Yutzey, K. E. (2012). Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 368, 345–357. doi: 10.1016/j.ydbio.2012.06.002
Braitsch, C. M., Kanisicak, O., van Berlo, J. H., Molkentin, J. D., and Yutzey, K. E. (2013). Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J. Mol. Cell. Cardiol. 65, 108–119. doi: 10.1016/j.yjmcc.2013.10.005
Cao, J., Navis, A., Cox, B. D., Dickson, A. L., Gemberling, M., Karra, R., et al. (2016). Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232–243. doi: 10.1242/dev.130534
Cao, Y., and Cao, J. (2018). Covering and re-covering the heart: development and regeneration of the epicardium. J. Cardiovasc. Dev. Dis. 6:3 doi: 10.3390/jcdd6010003
Cao, Y., Duca, S., and Cao, J. (2020). Epicardium in heart development. Cold Spring Harb. Perspect. Biol. 12:a037192. doi: 10.1101/cshperspect.a037192
Cherepanova, O. A., Gomez, D., Shankman, L. S., Swiatlowska, P., Williams, J., Sarmento, O. F., et al. (2016). Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 22, 657–665. doi: 10.1038/nm.4109
Dechamethakun, S., Ikeda, S., Arai, T., Sato, N., Sawabe, M., and Muramatsu, M. (2014). Associations between the CDKN2A/B, ADTRP and PDGFD polymorphisms and the development of coronary atherosclerosis in Japanese patients. J. Atheroscler. Thromb. 21, 680–690. doi: 10.5551/jat.22640
Feihl, F., Liaudet, L., Levy, B. I., and Waeber, B. (2008). Hypertension and microvascular remodelling. Cardiovasc. Res. 78, 274–285. doi: 10.1093/cvr/cvn022
Firoozi, S., Pahlavan, S., Ghanian, M. H., Rabbani, S., Tavakol, S., Barekat, M., et al. (2020). A cell-free SDKP-conjugated self-assembling peptide hydrogel sufficient for improvement of myocardial infarction. Biomolecules 10:205. doi: 10.3390/biom10020205
Frismantiene, A., Philippova, M., Erne, P., and Resink, T. J. (2018). Cadherins in vascular smooth muscle cell (patho)biology: quid nos scimus? Cell Signal. 45, 23–42. doi: 10.1016/j.cellsig.2018.01.023
Fujimaki, T., Oguri, M., Horibe, H., Kato, K., Matsuoka, R., Abe, S., et al. (2015). Association of a transcription factor 21 gene polymorphism with hypertension. Biomed. Rep. 3, 118–122. doi: 10.3892/br.2014.371
Funato, N., Ohyama, K., Kuroda, T., and Nakamura, M. (2003). Basic helix-loop-helix transcription factor epicardin/capsulin/Pod-1 suppresses differentiation by negative regulation of transcription. J. Biol. Chem. 278, 7486–7493. doi: 10.1074/jbc.M212248200
Gambardella, L., McManus, S. A., Moignard, V., Sebukhan, D., Delaune, A., Andrews, S., et al. (2019). BNC1 regulates cell heterogeneity in human pluripotent stem cell-derived epicardium. Development 146:dev174441. doi: 10.1242/dev.174441
Gao, J. H., He, L. H., Yu, X. H., Zhao, Z. W., Wang, G., Zou, J., et al. (2019). CXCL12 promotes atherosclerosis by downregulating ABCA1 expression via the CXCR4/GSK3beta/beta-catenin(T120)/TCF21 pathway. J. Lipid Res. 60, 2020–2033. doi: 10.1194/jlr.RA119000100
Gittenberger-de Groot, A. C., Winter, E. M., and Poelmann, R. E. (2010). Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 14, 1056–1060. doi: 10.1111/j.1582-4934.2010.01077.x
Gutierrez-Arcelus, M., Lappalainen, T., Montgomery, S. B., Buil, A., Ongen, H., Yurovsky, A., et al. (2013). Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2:e00523. doi: 10.7554/eLife.00523
He, C., Medley, S. C., Hu, T., Hinsdale, M. E., Lupu, F., Virmani, R., et al. (2015). PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 6:7770. doi: 10.1038/ncomms8770
Hidai, H., Bardales, R., Goodwin, R., Quertermous, T., and Quertermous, E. E. (1998). Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech. Dev. 73, 33–43. doi: 10.1016/s0925-4773(98)00031-8
Iyer, D., Zhao, Q., Wirka, R., Naravane, A., Nguyen, T., Liu, B., et al. (2018). Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 14:e1007681. doi: 10.1371/journal.pgen.1007681
Jiang, X., and Yang, Z. (2018). Multiple biological functions of transcription factor 21 in the development of various cancers. Onco Targets Ther. 11, 3533–3539. doi: 10.2147/OTT.S164033
Kanisicak, O., Khalil, H., Ivey, M. J., Karch, J., Maliken, B. D., Correll, R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7:12260. doi: 10.1038/ncomms12260
Kikuchi, K., Gupta, V., Wang, J., Holdway, J. E., Wills, A. A., Fang, Y., et al. (2011). tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 138, 2895–2902. doi: 10.1242/dev.067041
Kim, J., and Bartel, D. P. (2009). Allelic imbalance sequencing reveals that single-nucleotide polymorphisms frequently alter microRNA-directed repression. Nat. Biotechnol. 27, 472–477. doi: 10.1038/nbt.1540
Li, J., Li, S. H., Wu, J., Weisel, R. D., Yao, A., Stanford, W. L., et al. (2018). Young bone marrow Sca-1 cells rejuvenate the aged heart by promoting epithelial-to-mesenchymal transition. Theranostics 8, 1766–1781. doi: 10.7150/thno.22788
Li, X., Wu, N., Ji, H., Huang, Y., Hu, H., Li, J., et al. (2020). A male-specific association between AGTR1 hypermethylation and coronary heart disease. Bosn J. Basic Med. Sci. 20, 31–36. doi: 10.17305/bjbms.2019.4321
Liang, J., Le, T. H., Edwards, D. R. V., Tayo, B. O., Gaulton, K. J., Smith, J. A., et al. (2017). Single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLoS Genet. 13:e1006728. doi: 10.1371/journal.pgen.1006728
Lie-Venema, H., van den Akker, N. M., Bax, N. A., Winter, E. M., Maas, S., Kekarainen, T., et al. (2007). Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Sci. World J. 7, 1777–1798. doi: 10.1100/tsw.2007.294
Limana, F., Zacheo, A., Mocini, D., Mangoni, A., Borsellino, G., Diamantini, A., et al. (2007). Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ. Res. 101, 1255–1265. doi: 10.1161/CIRCRESAHA.107.150755
Liu, B., Pjanic, M., Wang, T., Nguyen, T., Gloudemans, M., Rao, A., et al. (2018). Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am. J. Hum. Genet. 103, 377–388. doi: 10.1016/j.ajhg.2018.08.001
Liu, X., Wang, X., Shen, Y., Wu, L., Ruan, X., Lindpaintner, K., et al. (2009). The functional variant rs1048990 in PSMA6 is associated with susceptibility to myocardial infarction in a Chinese population. Atherosclerosis 206, 199–203. doi: 10.1016/j.atherosclerosis.2009.02.004
Lu, X., Wang, L., Chen, S., He, L., Yang, X., Shi, Y., et al. (2012). Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat. Genet. 44, 890–894. doi: 10.1038/ng.2337
Majesky, M. W. (2007). Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb. Vasc. Biol. 27, 1248–1258. doi: 10.1161/ATVBAHA.107.141069
Mar, J. C., Matigian, N. A., Mackay-Sim, A., Mellick, G. D., Sue, C. M., Silburn, P. A., et al. (2011). Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 7:e1002207. doi: 10.1371/journal.pgen.1002207
Massari, M. E., and Murre, C. (2000). Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 20, 429–440. doi: 10.1128/mcb.20.2.429-440.2000
Miller, C. L., Anderson, D. R., Kundu, R. K., Raiesdana, A., Nurnberg, S. T., Diaz, R., et al. (2013). Disease-related growth factor and embryonic signaling pathways modulate an enhancer of TCF21 expression at the 6q23.2 coronary heart disease locus. PLoS Genet. 9:e1003652. doi: 10.1371/journal.pgen.1003652
Miller, C. L., Haas, U., Diaz, R., Leeper, N. J., Kundu, R. K., Patlolla, B., et al. (2014). Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS Genet. 10:e1004263. doi: 10.1371/journal.pgen.1004263
Miller, C. L., Pjanic, M., Wang, T., Nguyen, T., Cohain, A., Lee, J. D., et al. (2016). Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 7:12092. doi: 10.1038/ncomms12092
Milutinovic, A., Suput, D., and Zorc-Pleskovic, R. (2020). Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: an updated review. Bosn J. Basic Med. Sci. 20, 21–30. doi: 10.17305/bjbms.2019.4320
Nagao, M., Lyu, Q., Zhao, Q., Wirka, R. C., Bagga, J., Nguyen, T., et al. (2020). Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ. Res. 126, 517–529. doi: 10.1161/CIRCRESAHA.119.315968
Nurnberg, S. T., Cheng, K., Raiesdana, A., Kundu, R., Miller, C. L., Kim, J. B., et al. (2015). Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells That Contribute to the Fibrous Cap. PLoS Genet. 11:e1005155. doi: 10.1371/journal.pgen.1005155
Orlandi, A., and Bennett, M. (2010). Progenitor cell-derived smooth muscle cells in vascular disease. Biochem. Pharmacol. 79, 1706–1713. doi: 10.1016/j.bcp.2010.01.027
Owens, G. K., Kumar, M. S., and Wamhoff, B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801. doi: 10.1152/physrev.00041.2003
Pan, H., and Reilly, M. P. (2019). A protective smooth muscle cell transition in atherosclerosis. Nat. Med. 25, 1194–1195. doi: 10.1038/s41591-019-0541-0
Plotkin, M., and Mudunuri, V. (2008). Pod1 induces myofibroblast differentiation in mesenchymal progenitor cells from mouse kidney. J. Cell. Biochem. 103, 675–690. doi: 10.1002/jcb.21441
Raines, E. W., and Ross, R. (1993). Smooth muscle cells and the pathogenesis of the lesions of atherosclerosis. Br. Heart J. 69(1 Suppl.), S30–S37. doi: 10.1136/hrt.69.1_suppl.s30
Rong, J. X., Shapiro, M., Trogan, E., and Fisher, E. A. (2003). Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. U.S.A. 100, 13531–13536. doi: 10.1073/pnas.1735526100
Sanchez-Iranzo, H., Galardi-Castilla, M., Sanz-Morejon, A., Gonzalez-Rosa, J. M., Costa, R., Ernst, A., et al. (2018). Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl. Acad. Sci. U.S.A. 115, 4188–4193. doi: 10.1073/pnas.1716713115
Sazonova, O., Zhao, Y., Nurnberg, S., Miller, C., Pjanic, M., Castano, V. G., et al. (2015). Characterization of TCF21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 11:e1005202. doi: 10.1371/journal.pgen.1005202
Schunkert, H., Konig, I. R., Kathiresan, S., Reilly, M. P., Assimes, T. L., Holm, H., et al. (2011). Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338. doi: 10.1038/ng.784
Shankman, L. S., Gomez, D., Cherepanova, O. A., Salmon, M., Alencar, G. F., Haskins, R. M., et al. (2015). KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 21, 628–637. doi: 10.1038/nm.3866
Smith, C. L., Baek, S. T., Sung, C. Y., and Tallquist, M. D. (2011). Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 108, e15–e26. doi: 10.1161/CIRCRESAHA.110.235531
Souders, C. A., Bowers, S. L., and Baudino, T. A. (2009). Cardiac fibroblast: the renaissance cell. Circ. Res. 105, 1164–1176. doi: 10.1161/CIRCRESAHA.109.209809
Tandon, P., Miteva, Y. V., Kuchenbrod, L. M., Cristea, I. M., and Conlon, F. L. (2013). Tcf21 regulates the specification and maturation of proepicardial cells. Development 140, 2409–2421. doi: 10.1242/dev.093385
Tandon, P., Wilczewski, C. M., Williams, C. E., and Conlon, F. L. (2016). The Lhx9-integrin pathway is essential for positioning of the proepicardial organ. Development 143, 831–840. doi: 10.1242/dev.129551
Truskey, G. A. (2010). Endothelial cell vascular smooth muscle cell co-culture assay for high throughput screening assays for discovery of anti-angiogenesis agents and other therapeutic molecules. Int. J. High Throughput Screen 2010, 171–181. doi: 10.2147/IJHTS.S13459
van der Linde, D., Konings, E. E., Slager, M. A., Witsenburg, M., Helbing, W. A., Takkenberg, J. J., et al. (2011). Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247. doi: 10.1016/j.jacc.2011.08.025
Vengrenyuk, Y., Nishi, H., Long, X., Ouimet, M., Savji, N., Martinez, F. O., et al. (2015). Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb. Vasc. Biol. 35, 535–546. doi: 10.1161/ATVBAHA.114.304029
Virani, S. S., Alonso, A., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2020). Heart disease and stroke statistics-2020 update: a report from the american heart association. Circulation 141, e139–e596. doi: 10.1161/CIR.0000000000000757
Wang, D., Chang, P. S., Wang, Z., Sutherland, L., Richardson, J. A., Small, E., et al. (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862. doi: 10.1016/s0092-8674(01)00404-4
Wang, D., Wang, Z., Zhang, L., and Wang, Y. (2017). Roles of cells from the arterial vessel wall in atherosclerosis. Mediat. Inflamm. 2017:8135934. doi: 10.1155/2017/8135934
Wang, Y., Wang, L., Liu, X., Zhang, Y., Yu, L., Zhang, F., et al. (2014). Genetic variants associated with myocardial infarction and the risk factors in Chinese population. PLoS One 9:e86332. doi: 10.1371/journal.pone.0086332
Wirka, R. C., Wagh, D., Paik, D. T., Pjanic, M., Nguyen, T., Miller, C. L., et al. (2019). Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 25, 1280–1289. doi: 10.1038/s41591-019-0512-5
Wirtwein, M., Melander, O., Sjogren, M., Hoffmann, M., Narkiewicz, K., Gruchala, M., et al. (2017). Relationship between selected DNA polymorphisms and coronary artery disease complications. Int. J. Cardiol. 228, 814–820. doi: 10.1016/j.ijcard.2016.11.060
Wong, D., Turner, A. W., and Miller, C. L. (2019). Genetic insights into smooth muscle cell contributions to coronary artery disease. Arterioscler Thromb. Vasc. Biol. 39, 1006–1017. doi: 10.1161/ATVBAHA.119.312141
Yang, L., Gao, X., Luo, H., Huang, Q., Su, D., Tan, X., et al. (2017). TCF21 rs12190287 polymorphisms are associated with ventricular septal defects in a chinese population. Genet. Test Mol. Biomarkers 21, 312–315. doi: 10.1089/gtmb.2016.0324
Zangi, L., Oliveira, M. S., Ye, L. Y., Ma, Q., Sultana, N., Hadas, Y., et al. (2017). Insulin-like growth factor 1 receptor-dependent pathway drives epicardial adipose tissue formation after myocardial injury. Circulation 135, 59–72. doi: 10.1161/CIRCULATIONAHA.116.022064
Keywords: transcription factor 21, development, epicardial cells differentiation, vascular smooth muscle cells, coronary heart disease
Citation: Hu H, Lin S, Wang S and Chen X (2020) The Role of Transcription Factor 21 in Epicardial Cell Differentiation and the Development of Coronary Heart Disease. Front. Cell Dev. Biol. 8:457. doi: 10.3389/fcell.2020.00457
Received: 14 March 2020; Accepted: 18 May 2020;
Published: 05 June 2020.
Edited by:
Eleonora Napoli, University of California, Davis, United StatesReviewed by:
Dimitris Beis, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceSandeep Kumar, Emory University, United States
Copyright © 2020 Hu, Lin, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuangshuang Wang, d2FuZ3NzMTAyM0AxMjYuY29t; Xiaomin Chen, Y2h4bWluQGhvdG1haWwuY29t; Y3htZG9jdG9yQDEyNi5jb20=