 Marina Ferreira Batista
Marina Ferreira Batista Carlos Alcides Nájera
Carlos Alcides Nájera Isabela Meneghelli
Isabela Meneghelli Diana Bahia
Diana Bahia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 10 June 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00396
This article is part of the Research Topic Intracellular Infectious Diseases View all 12 articles
The trypanosomatid (protozoan) parasites Trypanosoma cruzi and Leishmania spp. are causative agents of Chagas disease and Leishmaniasis, respectively. They display high morphological plasticity, are capable of developing in both invertebrate and vertebrate hosts, and are the only trypanosomatids that can survive and multiply inside mammalian host cells. During internalization by host cells, these parasites are lodged in “parasitophorous vacuoles” (PVs) comprised of host cell endolysosomal system components. PVs effectively shelter parasites within the host cell. PV development and maturation (acidification, acquisition of membrane markers, and/or volumetric expansion) precede parasite escape from the vacuole and ultimately from the host cell, which are key determinants of infective burden and persistence. PV biogenesis varies, depending on trypanosomatid species, in terms of morphology (e.g., size), biochemical composition, and parasite-mediated processes that coopt host cell machinery. PVs play essential roles in the intracellular development (i.e., morphological differentiation and/or multiplication) of T. cruzi and Leishmania spp. They are of great research interest as potential gateways for drug delivery systems and other therapeutic strategies for suppression of parasite multiplication and control of the large spectrum of diseases caused by these trypanosomatids. This mini-review focuses on mechanisms of PV biogenesis, and processes whereby PVs of T. cruzi and Leishmania spp. promote parasite persistence within and dissemination among mammalian host cells.
Trypanosoma cruzi and Leishmania spp. are evolutionarily closely related trypanosomatid protozoan parasites and the causative agents of Chagas disease and leishmaniasis, respectively, (Chagas, 1909; Brener, 1997; Harmer et al., 2018). These are classified by WHO as neglected tropical diseases (NTDs), and collectively affect over 20 million people worldwide – mainly populations living in remote, poorly developed areas (WHO, 2020a, b). T. cruzi and Leishmania spp. have complex and distinctive life cycles, and both are transmitted by insect vectors (triatomine bugs and phlebotomine flies, respectively) to various mammalian species (including humans) that act as persistent hosts (Ashford, 2000; Zingales et al., 2012; Kaufer et al., 2017). Trypanosomatids display remarkable plasticity in adapting to distinctive host organisms and environments, and in adapting and developing resistance to the action of drugs intended to control infection, thus presenting a challenge to any therapeutic strategy (Genois et al., 2014; Reis-Cunha et al., 2015; Laffitte et al., 2016; Reis-Cunha et al., 2018; Kaufer et al., 2019). Such adaptability is based on morphological and biochemical changes. For example, T. cruzi has five morphologically distinct developmental forms: non-infectious multiplicative epimastigote and infectious less-replicative metacyclic trypomastigote forms that colonize the insect vector, an intracellular amastigote form that multiplies within mammalian host cells, and infectious extracellular amastigote and trypomastigote forms that enter the mammalian host bloodstream (Lima et al., 2010; Ferreira et al., 2012).
Leishmania spp. have two clearly defined developmental forms: promastigote forms that colonize insect vectors (divided into non-infectious procyclic and infectious metacyclic subgroups), and amastigote forms that multiply within mammalian host cells (primarily macrophages; Tomlinson et al., 1995; Kaufer et al., 2017). The host cell interior is thus an important developmental environment for trypanosomatids, and they have developed various strategies for entering host cells. For this purpose, T. cruzi can utilize “passive” pathways such as endocytosis (Mortara et al., 2005, 2008; de Souza et al., 2010; Fernandes et al., 2015), or “active” parasite-mediated invasive pathways such as actin depolymerization induced in either phagocytic or non-phagocytic cells by extracellular amastigotes (Mortara, 1991; Mortara et al., 2008; Caradonna and Burleigh, 2011; Fernandes et al., 2013, 2015) or induction of lysosome-mediated membrane repair that promotes internalization (Fernandes et al., 2011).
In contrast, entry of Leishmania spp. into host cells occurs mainly via endocytosis, primarily by “professional phagocytes” such as macrophages and neutrophils. Neutrophils act as “Trojan horses,” they deliver parasites into host cells after being phagocytosed by macrophages (Farah et al., 1975; Chang and Dwyer, 1976; Ribeiro-Gomes et al., 2004). In addition, Leishmania amazonensis was recently reported to enter non-phagocytic cells (e.g., fibroblasts) using a Ca2+-dependent mechanism similar to one utilized by T. cruzi, involving membrane damage and repair via lysosomal exocytosis (Fernandes et al., 2015; Cavalcante-Costa et al., 2019).
Following cell invasion, intracellular obligate parasites use various strategies to withstand host cell immune responses and persist in the host cell. One such strategy, commonly observed in medically relevant infectious diseases, is formation of pathogen-containing vacuoles within host cells after pathogen internalization. Among trypanosomatids, only T. cruzi and Leishmania spp. are intracellular parasites of mammals.
After entering host cells, they are sheltered within a type of vacuole termed “parasitophorous vacuoles” (PVs), an essential preliminary step in further intracellular development of the parasites. PVs act as filters for nutrients to the detriment of host cell immune response factors, creating a niche for parasite differentiation, and/or multiplication. PVs also function as intermediary chambers facilitating development of parasites until they reach the cell cytoplasm, where they multiply and then exit the host cell (the case for T. cruzi; Basu and Ray, 2005; Barrias et al., 2013 for reviews), or until they are transferred safely from cell to cell without contacting the extracellular milieu (the case for L. amazonensis; Real et al., 2014). This mini-review focuses on mechanisms of PV biogenesis, and processes whereby PVs of T. cruzi and Leishmania spp. promote parasite persistence within and dissemination among mammalian host cells.
Trypanosomatids, in their evolutionary history, were initially non-parasitic free-living organisms, as evidenced by their common ancestor Bodo saltans, and subsequently underwent selective pressure leading to development of the capacity to colonize host organisms, as either extracellular or intracellular parasites (Jaskowska et al., 2015). Ability to live inside host cells was a key evolutionary adaptation toward parasitism in mammalian hosts. For example, T. brucei (which is more closely related to T. cruzi than to Leishmania) does not require an intracellular/vacuolar environment in order to parasitize mammals, and exists exclusively as an extracellular parasite in the bloodstream of its mammalian host during its life cycle (Stevens et al., 1998; Hamilton et al., 2007). Adaptation to such an intracellular lifestyle has apparently involved genome reduction during the evolutionary history of many parasitic microorganisms (Casadevall, 2008). Genomes of the PV-forming intracellular parasites Leishmania spp. and T. cruzi are smaller than those of extracellular parasites such as T. brucei, indicating that PV biogenesis was a later adaptation that was beneficial to previously exclusively extracellular parasites (Jaskowska et al., 2015). T. cruzi likely appeared earlier than Leishmania spp. during trypanosomatid evolutionary history, which could account for the persistence of T. cruzi in mammalian host organisms as both extracellular (like T. brucei) and intracellular forms (like Leishmania spp.). From an evolutionary point of view, PV biogenesis may be a refined adaptation for parasitism that enhanced the fitness of trypanosomatid parasites involved in medically relevant pathologies.
Although both T. cruzi and Leishmania spp. depend on PV development to persist inside host cells, the former is sheltered transiently by PVs and then is released to host cell cytosol where it multiplies, whereas the latter is sheltered permanently by PVs throughout its intracellular life cycle, and multiplies within PVs (Fernandes et al., 2011; Fernandes and Andrews, 2012). Its release from these PVs to cytoplasm depends on an acid pH-dependent signaling event promoted by lysosome recruitment toward PV; this leads to parasite exit into cytosol, where they differentiate into replicative amastigote forms and multiply (Ley et al., 1990; Andrade and Andrews, 2004; Fernandes and Andrews, 2012). Leishmania spp., on the other hand, take advantage of PVs to differentiate into amastigotes and also to multiply. Analogously to the diverse spectrum of disease outcomes in leishmaniasis (Burza et al., 2018), PVs of Leishmania spp. display diverse morphologies depending on species: the great majority of Leishmania spp. (including L. major and L. donovani) develop in single compact PVs, whereas members of the L. mexicana complex (L. amazonensis, L. mexicana) multiply within larger PVs (Real and Mortara, 2012).
Thus, PVs are customized to fulfill the requirements of parasites for their intracellular development, as reflected by the construction of doubly infected, chimeric PVs, i.e., single pathogen-containing vacuoles that host different parasite species simultaneously. In a model system of chimeric vacuoles hosting L. amazonensis amastigotes (primo-infection) and L. major promastigotes (superinfection), the latter was unable to differentiate into amastigote form (Real et al., 2010). In another model of chimeric vacuoles using L. amazonensis large PVs as recipient vacuoles for T. cruzi, the latter differentiated into replication-competent amastigote forms not in cytosol but within phagolysosome-like L. amazonensis large PVs, indicating that trypomastigote-to-amastigote differentiation of T. cruzi occurs under the acidic pH of PVs and precedes the release from PV to cytosol (Pessoa et al., 2016). This finding is consistent with previous reports that PV alkalinization impairs parasite PV escape (Ley et al., 1990; Stecconi-Silva et al., 2003). Rather than being released from PVs (as T. cruzi does), Leishmania spp. remain associated with PVs even during cell-to-cell parasite spreading. L. amazonensis takes advantage of host macrophage apoptosis to transfer from macrophage to macrophage in vitro, and remains associated with host lysosomal components on its surface that trigger anti-inflammatory cytokine production by recipient non-apoptotic macrophages (Real et al., 2014). PVs thus provide an additional shelter from the extracellular milieu and immune system surveillance, and participate in the late intracellular life cycle of parasites; i.e., egress from host cells and reinfection of new ones.
Several parasite species utilize a strategy based on formation of a specifically designed, customized PV during the process of cell infection. Intracellular persistence of trypanosomatids depends on several host-related features. In contrast to various intracellular pathogens that interfere with phagosome maturation to avoid transport to lysosomes, Leishmania spp. and T. cruzi recruit lysosome markers during the process of infection, and need an acidic environment to maintain their intracellular life cycle. This strategy requires that the parasites remodel and subvert the host endolysosomal pathway to benefit themselves.
In cells infected with L. amazonensis, an exchange of biomolecules (e.g., lipids, proteins, and sialoglycoproteins) between cells and parasites was observed following contact. The PV was labeled with the same molecular markers of the parasite, indicating that in addition to host cell internalized components, there is a shedding of proteins from the intracellular parasites to PV (Henriques and De Souza, 2000). Henriques et al. (2003) confirmed the transfer of lipids by labeling macrophages with 32P and then exposing the cells to L. amazonensis. The main phospholipid component of PV was phosphatidylcholine. Changes were observed in PV protein composition in relation to time of infection and morphological form of the parasite (Henriques et al., 2003).
Gagnon et al. (2002) showed, based on observation of calnexin markers, that endoplasmic reticulum (ER) participates in formation of phagosomes in macrophages, by fusion with plasmatic membrane during early phagocytosis and its subsequent maturation. Entry of pathogens such as L. donovani into macrophages evidently required ER proteins such as calnexin and calreticulin, indicating participation of ER in the internalization process. Similarly, Canton et al. (2012) showed that ER elements are involved in PV formation in L. amazonensis, through action of SNARE protein.
Leishmania donovani also is able to upregulate Rab5a, an early endosome protein. The parasite retains Rab5a, along with its effector protein EEA1, in PVs, thereby forming and maintaining an early endosome compartment and delaying maturation (Verma et al., 2017). Such delay is observed in many other Leishmania species and is mediated by parasite surface components such as lipophosphoglycan (LPG; Lodge and Descoteaux, 2005), favoring differentiation into amastigote forms of promastigotes otherwise sensitive to the harsh environment of fully matured phagolysosomal vacuoles. In contrast, L. amazonensis acquires Rab5a and EEA1 soon after internalization but does not maintain these early endosome markers; rather, it rapidly acquires late endosome and lysosome markers (Courret et al., 2002; reviewed by Veras et al., 2019). For L. amazonensis (and possibly other members of the L. mexicana complex), PV maturation in terms of acquisition of lysosomal membrane markers and content is accompanied by a striking increase in PV volumetric size, which is dependent on host cell factors such as lysosomal traffic regulator LYST/Beige (Wilson et al., 2008), CD36 receptor (Okuda et al., 2016), and V-ATPase subunit d isoform 2 (ATP6V0d2; Pessoa et al., 2019). Lysosome marker recruitment and PV enlargement are impaired when the host cell lacks CD36 receptor, thereby impairing parasite multiplication as well (Okuda et al., 2016). In another mechanism possibly involving CD36, ATP6V0d2 knockdown depletes macrophage cholesterol and inhibits PV enlargement without impairing parasite multiplication (Pessoa et al., 2019). Increase of cholesterol level by addition of oxidized low-density lipoprotein (ox-LDL), of which CD36 is the receptor, results in PV enlargement and impaired parasite multiplication.
Initial parasite-host cell interaction leading to parasite internalization involves recognition of conserved parasite components, termed pathogen-associated molecular patterns (PAMPs), by host pattern recognition receptors (PRRs); the parasites then take advantage of adhesion to host cells to access safe intracellular environments (Bahia et al., 2018). Such interaction modifies cellular signaling pathways and thereby determines parasite fate. Activation of host signaling pathways [phosphatidylinositol-3-kinase/protein kinase C (PI3K/PKC) – mTOR pathway and endolysosomal pathway for T. cruzi; phagocytic pathway for Leishmania spp.] leads to parasite internalization and PV formation (Romano et al., 2009; Martins et al., 2011; Salassa and Romano, 2018; Ferreira et al., 2019).
Trypanosoma cruzi endocytic entry into non-professional phagocytic cells has been clearly shown to require lysosome recruitment (Meirelles et al., 1987; Carvalho and de Souza, 1989) and actin reorganization (Rosestolato et al., 2002). Metacyclic and culture-derived trypomastigote forms both depend on transient presence of Ca2+ during entry into cells driven by parasite surface molecules (gp82- MT/oligopeptidase B-CDT; Yoshida, 2006). Intact microtubule machinery is essential for T. cruzi internalization (Rodriguez et al., 1996; Rosestolato et al., 2002). During T. cruzi infection, microtubules play a role in directing lysosomes to PVs, which act as sites of microtubule organization (Tyler et al., 2005).
PV establishment in T. cruzi is directly related to its mechanism of entry into cells. Internalization of T. cruzi in non-phagocytic cells clearly depends on early lysosomal exocytosis to parasite infection sites (Tardieux et al., 1992). Following parasite-induced plasma membrane damage, membrane repair is stimulated via lysosomal exocytosis, and T. cruzi takes advantage of this process to enter the cell (Fernandes et al., 2011). Woolsey et al. (2003) demonstrated that trypomastigotes can also utilize a different invasion route which is dependent on phosphatidylinositol 3-phosphate (PIP-3) plasma membrane-associated molecules in non-phagocytic cells. Only 20% of the analyzed parasite population presented early endosomal markers (e.g., EEA1), in contrast to the 50% of the population that invaded via class I phosphatidylinositol-3-kinases (PI3K)-mediated PI3-P accumulation. These findings indicate that the main endocytic internalization pathway of T. cruzi in non-professional phagocytic cells does not require lysosomal exocytosis, contrary to previously proposed mechanisms.
Parasitophorous vacuole features formed during these two processes are distinctive. In the lysosome-dependent pathway (early lysosome fusion), Ca2+-dependent exocytosis of lysosomes is activated and lysosomes fuse with nascent PVs (Tardieux et al., 1992; Fernandes et al., 2011). In contrast, in the lysosome-independent pathway (late lysosome fusion), parasite entry occurs through membrane invagination resulting from PIP3 accumulation, and lysosome markers are acquired only during PV maturation (Woolsey et al., 2003).
In HeLa cells, extracellular amastigotes induce PI3K pathway to promote rearrangement of actin cytoskeleton and their own phagocytosis (Ferreira et al., 2019). This process likely interferes with PV formation, although the mechanism remains unclear.
Martins et al. (2011) showed that both metacyclic and culture-derived trypomastigotes elicit lysosome recruitment during invasion by activating autophagy-related signaling pathways. Parasite surface glycoprotein gp82 and host actin remodeling are required for induction of lysosome recruitment to plasma membrane during metacyclic invasion through activation of PI3K/PKC-mTOR pathway (Martins et al., 2011). In contrast, invasion of culture-derived trypomastigotes depends on activation of autophagy-related proteins ATG5 and Beclin, independently of mTOR pathway (Romano et al., 2009; Figure 1).
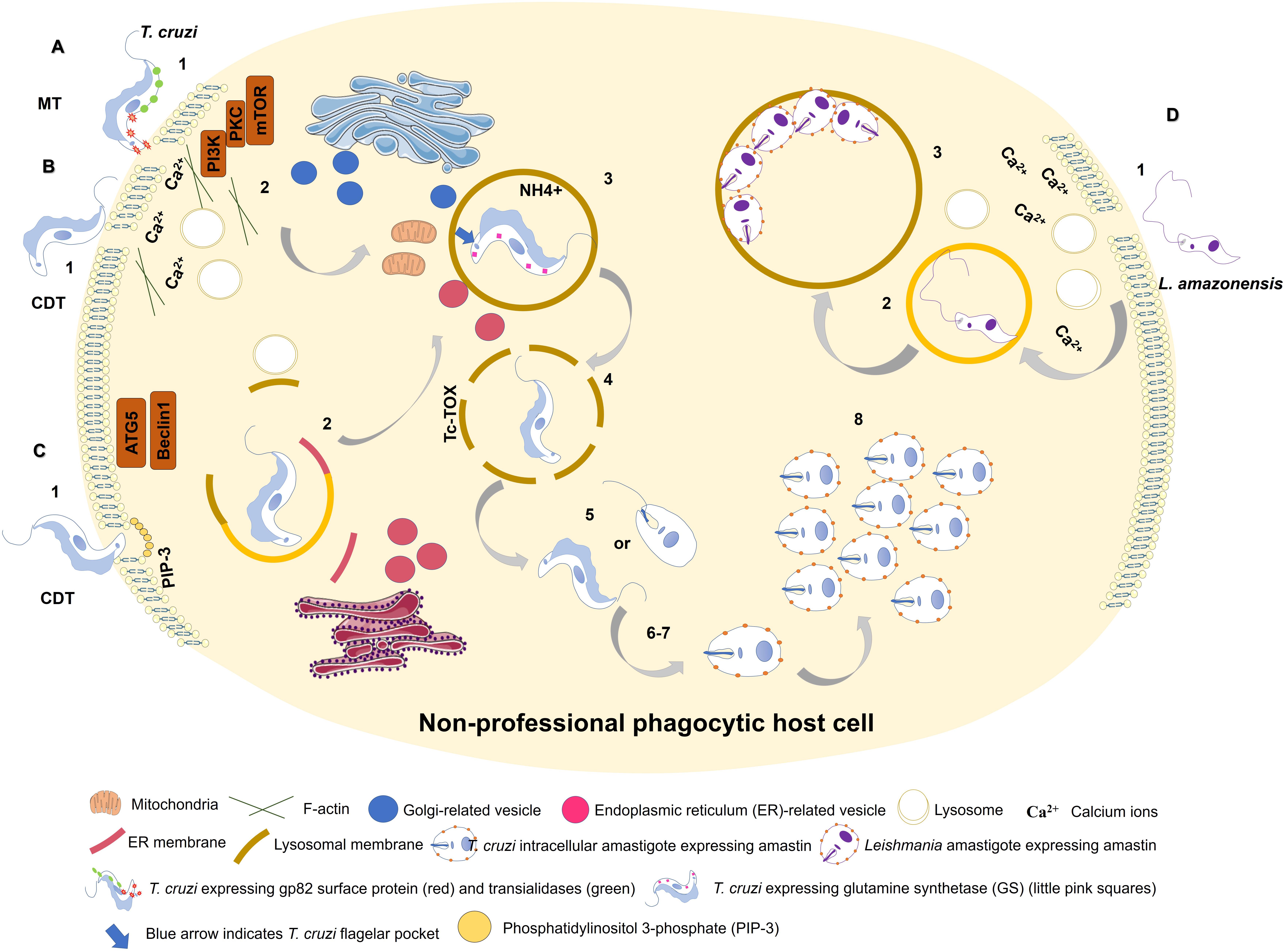
Figure 1. Entry of Trypanosoma cruzi metacyclic trypomastigote (MT) forms into non-professional phagocytic cell (A). 1: MT entry requires parasite surface protein gp82 and recruitment of lysosomes to the infection site in a Ca2+-dependent manner. 2: MT invasion activates PI3K/PKC-mTOR signaling and F-actin disruption. 3: Acidic parasitophorous vacuole is formed as T. cruzi begins internalization. Mitochondria are localized near flagellar pocket (blue arrow). T. cruzi vacuolar closure and maturation/acidification involve continuous communication with ER/Golgi vesicles. Glutamine synthetase (GS) controls pH by regulating NH4+ vacuolar content (small pink squares). 4: T. cruzi vacuolar degradation. 5: T. cruzi vacuolar escape into cytoplasm. MT form remains intact, or an intermediate form between MT and intracellular amastigote (IA) is present. 6: T. cruzi extravacuolar differentiation into IA forms. 7: Free T. cruzi IA in cytoplasm. 8: T. cruzi IA cytoplasmic replication. T. cruzi culture-derived trypomastigote (CDT) invasion by lysosome-dependent pathway (early lysosome fusion; B). 1: CDT induces membrane damage. 2: CDT requires parasite oligopeptidase B surface protein and recruitment of lysosomes to the infection site in a Ca2+-dependent manner. F-actin disruption also plays a role in CDT invasion. 3-8: Similar to (A). T. cruzi culture-derived trypomastigote (CDT) invasion by lysosome-independent pathway (late lysosome fusion; C). 1: CDT invades by membrane invagination resulting in a PI3K-dependent PIP3 accumulation. In deprivation of nutrients host cell increases CDT internalization through ATG5/Beclin1 pathway. 2: lysosome markers are acquired only during PV maturation. ER membrane is donated to PV during its membrane construction. 3-8: Similar to (A). Model of L. amazonensis non-phagocytic cell internalization as proposed by Cavalcante-Costa et al. (2019); D). 1: L. amazonensis induces membrane damage and Ca2+ recruitment during invasion process. 2: L. amazonensis vacuolar formation and maturation. 3: L. amazonensis multiplication into large vacuoles.
Host autophagy processes, in addition to regulating invasion, have been shown to affect PV maturation (Ghartey-Kwansah et al., 2020). In L. donovani, both canonical and non-canonical autophagy are triggered, at different infection time points (Pitale et al., 2019). Salassa and Romano (2018) suggested that autophagy is involved in PV maturation in T. cruzi infection, based on observed recruitment of LC3 (an autophagosome marker) to PV. Confirmation of this idea will require elucidation of the process involved. e.g., canonical autophagy, xenophagy, or LC3-associated phagocytosis (Salassa and Romano, 2018).
Intracellular parasites exploit host membrane resources and organelles to promote PV maintenance and maturation, in order to complete their life cycle. Reignault et al. (2019) observed formation of PVs during the first 2 h (not later times) of internalization by peritoneal macrophages of T. cruzi amastigotes and trypomastigotes. Electron microscopic and 3D reconstruction techniques indicated that during biogenesis of T. cruzi PVs, ER and lysosomes act as membrane donors for generation of PVs. Morphological changes were observed in cellular distribution of Golgi complex and mitochondria during PV biogenesis; these organelles moved from the perinuclear region to the PV vicinity. No membrane fusion with Golgi complex or mitochondria was observed; however, it is conceivable that both organelles function in synchrony with PV development, in view of their proximity. In the context of PV development, the observed exchange of membranes between parasite and PV suggest occurrence of emergent signaling pathways between parasite and host cell, and indicate involvement of the host actin cytoskeleton, which surrounds the PV from its biogenesis until its rupture (Reignault et al., 2014, 2019).
Lysosome-mediated PV acidification is necessary for both PV maturation and trypomastigote-to-amastigote differentiation; it enables proper functioning of parasite-derived Tc-TOX protein (de Souza et al., 2010). Carvalho and de Souza (1989) were the first to suggest a T. cruzi mechanism of PV dissolution. Stecconi-Silva et al. (2003) demonstrated later that Tc-TOX induces formation of pores to degrade PV membrane, thereby promoting parasite release into host cell cytoplasm.
Intracellular parasites are able to persist and overcome host cell defense mechanisms through a variety of strategies, e.g., secreting proteins, hijacking host proteins, and recruiting host proteins/structures. Certain parasite-derived factors have been implicated in subversion of host cell functions.
Lipophosphoglycan, a glycoconjugated virulence factor on Leishmania spp. surfaces, enhances parasite survival by targeting host defense proteins. During L. donovani phagocytosis, LPG delays PV maturation by inducing F-actin depolymerization around the PV site, resulting in inhibition of vesicular trafficking holding (e.g., lack of LAMP-1 marker; Winberg et al., 2009), and of recruitment to PV of protein kinase Cα (PKCα), a kinase involved in F-actin degradation and regulation of PV development (Holm et al., 2001). GP63, another virulence factor, is a metalloprotease surface protein that interferes with PV acidification. da Silva Vieira et al. (2019) reported that GP63 and LPG expression varies in L. braziliensis, and that these PV effects are strain-specific (da Silva Vieira et al., 2019).
Certain host ER- and Golgi-localized N-ethylmaleimide-sensitive-factor attachment protein receptors (SNAREs) are related to endosome/lysosome fusion and are coopted by trypanosomatids for PV biogenesis (Ndjamen et al., 2010; Canton and Kima, 2012; Cueto et al., 2017). Vacuoles containing either L. donovani or L. pifanoi recruit endoplasmic markers such as calnexin and SNARE Sec22b to PV formation sites during phagocytosis (Ndjamen et al., 2010). L. amazonensis engages SNAREs (syntaxin-5, Sec22b) that play important roles in PV development; syntaxin-5 inhibition blocked PV enlargement and reduced parasite burden (Canton and Kima, 2012), and Sec22b and syntaxin-5 participated in trafficking of parasite-derived molecules in host cells (Canton and Kima, 2012; Arango Duque et al., 2019).
Leishmania spp. metacyclic promastigotes utilize the host classic exocytic pathway (ER/Golgi complex) to deliver GP63 and LPG from PVs toward the extracellular milieu, thereby affecting parasite persistence (Arango Duque et al., 2019). Casgrain et al. (2016) observed that L. mexicana GP63 cleaved VAMP3 and VAMP8, two endocytic SNAREs associated with phagosome biogenesis and function, and helped maintain parasite intracellular development to allow PV expansion. Matte et al. (2016) reported that cleavage of VAMP8 by L. major GP63 was associated with inhibition of LC3 recruitment to phagosomes during LC3-associated phagocytosis (LAP). This pathway is typically activated by external particles already contained within a single-membraned phagosome or endosome, and leads to deposition of LC3 on the cytosolic side of the phagosome, thereby promoting more rapid fusion with lysosomes (Evans et al., 2018). Disruption resulting from VAMP8 cleavage impairs host cell antimicrobial machinery (Matte et al., 2016).
In T. cruzi, two SNAREs (VAMP3, VAMP7) are recruited to PVs at different times during PV development: VAMP3 appears only in early phases following parasite internalization, whereas VAMP7 is readily recruited and maintained throughout PV maturation and maintenance, and is essential for parasite invasion and lysosome-PV fusion events (Cueto et al., 2017; Figure 2).
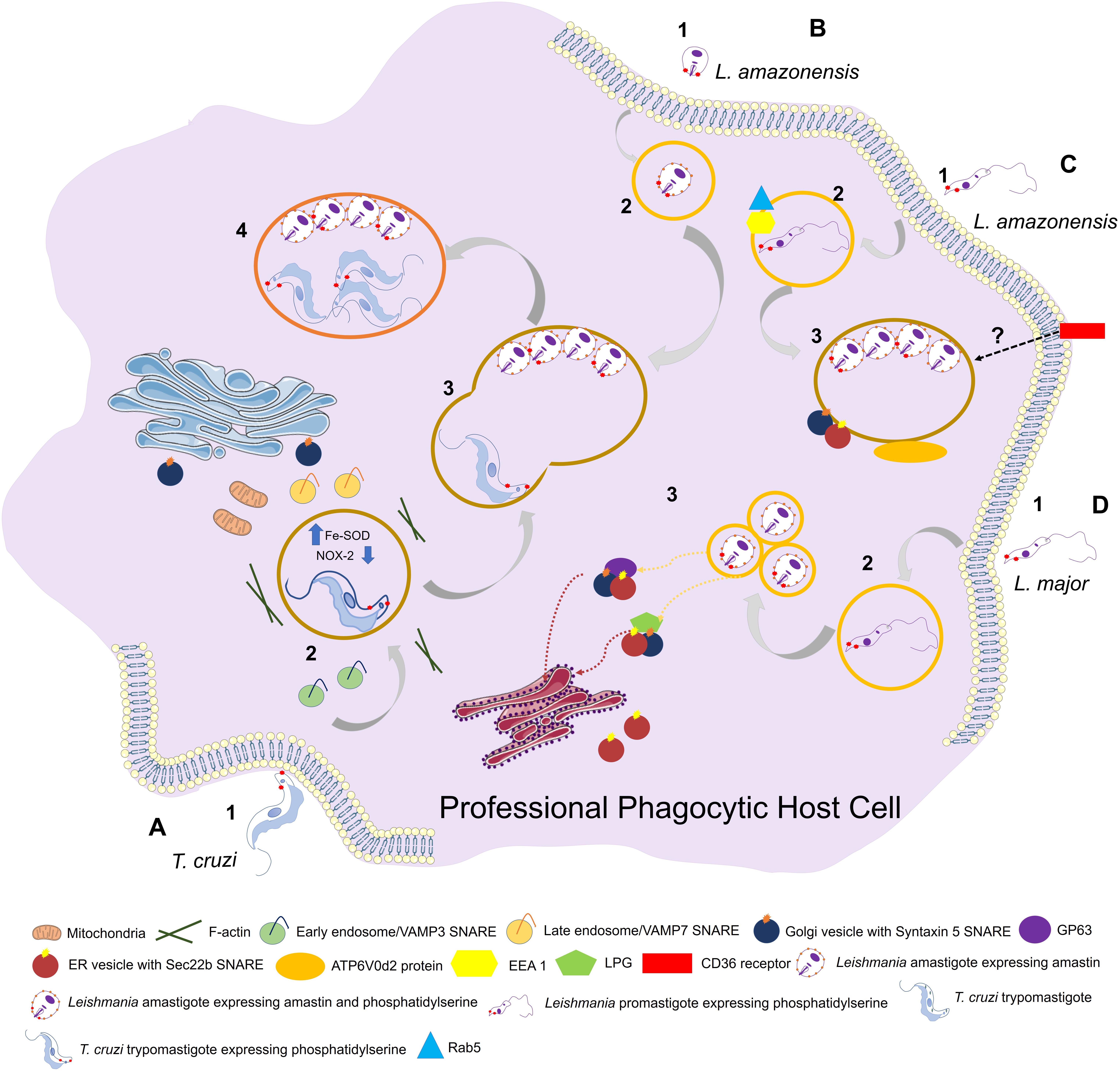
Figure 2. Entry of trypanosomatids in a professional phagocytic cell. (A) 1: Uptake of Trypanosoma cruzi trypomastigote by phagocytosis. This process can be facilitated through expression of phosphatidylserine (PS) in parasite surface. VAMP3 (SNARE) recruitment to entry site in early-phase infection. 2: T. cruzi vacuolar closure and maturation. VAMP7 (SNARE) is recruited and maintained during PV maturation. Parasite uses Fe-SOD to reduce the amount of O2– produced by host (NOX-2) inside phagosome. PS exposition creates a permissive state to parasite survival. (B) 1: Uptake of L. amazonensis amastigote by phagocytosis. 2: L. amazonensis vacuolar formation and maturation. 3: L. amazonensis and T. cruzi vacuolar fusion. 4: Chimeric vacuole formation with L. amazonensis amastigotes and T. cruzi trypomastigotes. (C) 1: Uptake of L. amazonensis promastigote by phagocytosis. 2: L. amazonensis vacuolar formation and maturation. Rab5 and EEA1 endosome markers are acquired after internalization. 3: L. amazonensis multiplication into large vacuoles. ATP6V0d2 subunit of V-ATPase participates in cholesterol influx, an essential process in PV maintenance. Syntaxin 15 and Sec22b are SNAREs that play important roles in PV development. (D) 1: Uptake of L. major by phagocytosis. 2: L. major vacuolar formation and maturation. The parasite is developed in a single compact PV. 3: L. major multiplication into tight individual vacuoles. Parasite virulence factors GP63 and LPG are transferred to host cell (ER/ERGIC; “ER-Golgi intermediate compartment”) by SNAREs. Both promastigotes and amastigotes of L. amazonesis and L. major can present PS in its surface with the same functions as in T. cruzi facilitating parasite phagocytosis and, once the parasite is inside the cell, aiding in its survival.
Amastin proteins, a family of stage-specific parasite surface factors first described in T. cruzi and showing upregulated expression in amastigote forms (Teixeira et al., 1994; Cruz et al., 2012), are reportedly involved in parasite intracellular survival and PV biogenesis (Cruz et al., 2012; de Paiva et al., 2015). The major amastin subfamily in Leishmania spp., termed δ-amastins, includes (~42 isoforms. Genome analysis of L. amazonensis showed that amastin subfamilies are species-specific, and show correlations with disease outcomes and PV volumetric size (Real et al., 2013). In L. braziliensis, δ-amastin knockdown reduced parasite PV attachment, thereby inhibiting multiplication and release into cytosol both in vitro and in vivo (Teixeira et al., 1994; de Paiva et al., 2015). T. cruzi has a smaller number of δ-amastin gene copies (12 in total) than do Leishmania spp. δ-amastin superexpression in T. cruzi in vitro was correlated with rapid differentiation of culture-derived trypomastigotes into intracellular amastigote forms, and with host cell egress. Overexpression of δ-amastin in T. cruzi extracellular amastigotes in vivo led to earlier parasite tropism (relative to wild-type) toward livers of infected mice (Cruz et al., 2012). Thus, parasite-derived membrane factors display a wide variety of functions depending on trypanosomatid species, ranging from parasite extracellular morphological development and parasite tropism in host organisms, to intracellular multiplication and PV biogenesis.
Another family of proteins, the trans-sialidases (TSs), also play important roles in parasite-PV interactions. Freire-de-Lima et al. (2015) showed that T. cruzi TSs help the parasite salvage host cell sialic acid for its own benefit. Besides their well-documented involvement in adhesion, invasion, and immune modulation, TSs facilitate parasite escape from PVs to cytosol, but the mechanism for this is unclear (Freire-de-Lima et al., 2015, 2017; da Fonseca et al., 2019). T. cruzi culture-derived trypomastigotes, in comparison with metacyclic forms, express higher TS levels and escape earlier from PVs throughout their intracellular life cycle, suggesting a link between TSs and PV escape. In infected cells lacking surface sialic acid and lysosome membranes, TS-overexpressing metacyclic forms and culture-derived trypomastigotes with high native TS expression show similar PV escape kinetics (Rubin-de-Celis et al., 2006, Rubin-de-Celis and Schenkman, 2012).
Crispim et al. (2018) showed that the ATP-dependent enzyme glutamine synthetase (GS) is associated with PV evasion in T. cruzi. GS regulates the level of metabolites derived from amino acid consumption by converting accumulated NH4+ and glutamate into glutamine. Blocking of GS by methionine sulfoximine (MS) inhibited trypomastigote PV escape into cytoplasm in vitro. T. cruzi PV escape is associated with acidic pH environment, and GS therefore may regulate intravacuolar NH4+ content and acidification (Crispim et al., 2018). Another enzyme associated with parasite survival within PVs is cytosolic iron superoxide dismutase (Fe-SOD), an O2– catabolizing enzyme. In T. cruzi, Fe-SOD reduces the amount of O2– produced by host NADPH oxidase (NOX-2) inside phagosomes, thereby counteracting host cell oxidative stress involved in defense against intracellular pathogens. Fe-SOD overexpression in parasites in vivo resulted in increased parasitemia and parasite burden in infected mice (Martínez et al., 2019).
Last but not least, recognition of phosphatidylserine (PS) on the membrane surface of apoptotic cells (apoptosis) is necessary for its elimination by phagocytes (endocytosis) without causing inflammation. A reproduction of this mechanism, termed “apoptotic mimicry,” is used by some intracellular parasites, including T. cruzi (trypomastigote forms) and Leishmania spp. (promastigote and amastigote forms), during the invasion process and maintenance of infection. In classical mimicry, the parasite expresses PS in order to be phagocytosed by macrophages, and a permissive state is created by decrease of NO production through induction of cytokines (e.g., TGF-β1; anti-inflammatory cytokine) and synthesis of IL-10, allowing the parasite to survive inside the host cell. In contrast, in non-classical mimicry the host cell expresses PS during the infection process. Leishmania spp., for example, initially colonizes a neutrophil and then induces its PS expression in order to be phagocytosed by a macrophage – the ideal host for the parasite. The amount of PS expressed determines the infection capacity of the parasite, and depends on an intrinsic pressure upon the host’s immune system (De Freitas Balanco et al., 2001; DaMatta et al., 2007; El-Hani et al., 2012; Wanderley et al., 2020).
Trypanosoma cruzi and Leishmania spp. are trypanosomatid pathogens that depend on customized PV niches during the infection process. PV development mechanisms are complex, and vary among parasite species and strains. During evolution, mammalian host cells have adapted to resist invasion of intracellular parasites by establishment of hostile intracellular environments. Development of more efficient therapeutic strategies against Chagas disease and leishmaniasis will require better understanding of the PV processes related to such structural biogenesis, and the mechanisms whereby parasite factors subvert host cell responses.
MB, CN, IM, and DB conceived and wrote the manuscript.
This study was supported by funding from FAPEMIG (PPM-00604-16), CNPq (404182/2016-0), and CAPES. DB is a recipient of a CNPq fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Dr. Fernando Real (Institut Cochin, INSERM U1016, Paris, France) for critical revision of the manuscript, and to Dr. S. Anderson for English editing.
Andrade, L. O., and Andrews, N. W. (2004). Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200, 1135–1143. doi: 10.1084/jem.20041408
Arango Duque, G., Jardim, A., Gagnon, E., Fukuda, M., and Descoteaux, A. (2019). The host cell secretory pathway mediates the export of Leishmania virulence factors out of the parasitophorous vacuole. PLoS Pathog. 15:e1007982. doi: 10.1371/journal.ppat.1007982
Ashford, R. W. (2000). The leishmaniasis as emerging and reemerging zoonoses. Int. J. Parasitol. 30, 1269–1281. doi: 10.1016/B978-0-12-803265-7.00007-5
Bahia, D., Satoskar, A. R., and Dussurget, O. (2018). Editorial: cell signaling in host-pathogen interactions: the host point of view. Front. Immunol. 9:221. doi: 10.3389/fimmu.2018.00221
Barrias, E. S., de Carvalho, T. M. U., and De Souza, W. (2013). Trypanosoma cruzi: entry into mammalian host cells and parasitophorous vacuole formation. Front. Immunol. 4:186. doi: 10.3389/fimmu.2013.00186
Basu, M. K., and Ray, M. (2005). Macrophage and Leishmania: an unacceptable coexistence. Crit. Rev. Microbiol. 31, 145–154. doi: 10.1080/1040841059100510
Brener, Z. (1997). “Trypanosoma cruzi: morfologia e ciclo evolutivo,” in Clínica e Terapêutica da Doença de Chagas: uma Abordagem Prática Para o Clínico Geral, eds J. C. P. Dias and J. R. Coura (Rio de Janeiro: FIOCRUZ), 24–31.
Canton, J., and Kima, P. E. (2012). Targeting host syntaxin-5 preferentially blocks Leishmania parasitophorous vacuole development in infected cells and limits experimental Leishmania infections. Am. J. Pathol. 181, 1348–1355. doi: 10.1016/j.ajpath.2012.06.041
Canton, J., Ndjamen, B., Hatsuzawa, K., and Kima, P. E. (2012). Disruption of the fusion of Leishmania parasitophorous vacuoles with ER vesicles results in the control of the infection. Cell. Microbiol. 14, 937–948. doi: 10.1111/j.1462-5822.2012.01767.x
Caradonna, K. L., and Burleigh, B. A. (2011). Mechanisms of host cell invasion by Trypanosoma cruzi. Adv. Parasitol. 76, 33–61. doi: 10.1016/B978-0-12-385895-5.00002-5
Carvalho, T. M. U., and de Souza, W. (1989). Early events related with the behaviour of Trypanosoma cruzi within an endocytic vacuole in mouse peritoneal macrophages. Cell Struct. Funct. 14, 383–392. doi: 10.1247/csf.14.383
Casadevall, A. (2008). Evolution of intracellular pathogens. Annu. Rev. Microbiol. 62, 19–33. doi: 10.1146/annurev.micro.61.080706.093305
Casgrain, P. A., Martel, C., McMaster, W. R., Mottram, J. C., Olivier, M., and Descoteaux, A. (2016). Cysteine peptidase B regulates Leishmania mexicana virulence through the modulation of GP63 expression. PLoS Pathog. 12:e1005658. doi: 10.1371/journal.ppat.1005658
Cavalcante-Costa, V. S., Costa-Reginaldo, M., Queiroz-Oliveira, T., Oliveira, A. C. S., Couto, N. F., Dos Anjos, D. O., et al. (2019). Leishmania amazonensis hijacks host cell lysosomes involved in plasma membrane repair to induce invasion in fibroblasts. J. Cell Sci. 132:jcs226183. doi: 10.1242/jcs.226183
Chagas, C. (1909). Nova tripanozomiase humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi. Gen., n. Spp., ajente etiolojico de nova entidade morbida do homem. Mem. Inst. Oswaldo Cruz 1, 159–218. doi: 10.1590/S0074-02761909000200008
Chang, K. P., and Dwyer, D. M. (1976). Multiplication of a human parasite (Leishmania donovani) in phagolysosomes of hamster macrophages in vitro. Science 193, 678–680. doi: 10.1126/science.948742
Courret, N., Fréhel, C., Gouhier, N., Pouchelet, M., Prina, E., Roux, P., et al. (2002). Biogenesis of Leishmania-harbouring parasitophorous vacuoles following phagocytosis of the metacyclic promastigote or amastigote stages of the parasites. J. Cell Sci. 115, 2303–2316.
Crispim, M., Damasceno, F. S., Hernández, A., Barisón, M. J., Sauter, I. P., Pavani, R. S., et al. (2018). The glutamine synthetase of Trypanosoma cruzi is required for its resistance to ammonium accumulation and evasion of the parasitophorous vacuole during host-cell infection. PLoS Negl. Trop. Dis. 12:e0006170. doi: 10.1371/journal.pntd.0006170
Cruz, M. C., Souza-Melo, N., da Silva, C. V., DaRocha, W. D., Bahia, D., Araújo, P. R., et al. (2012). Trypanosoma cruzi: role of δ-amastin on extracellular amastigote cell invasion and differentiation. PLoS One 7:e51804. doi: 10.1371/journal.pone.0051804
Cueto, J. A., Vanrell, M. C., Salassa, B. N., Nola, S., Galli, T., Colombo, M. I., et al. (2017). Soluble N-ethylmaleimide-sensitive factor attachment protein receptors required during Trypanosoma cruzi parasitophorous vacuole development. Cell. Microbiol. 19:e12713. doi: 10.1111/cmi.12713
da Fonseca, L. M., da Costa, K. M., Chaves, V. S., Freire-de-Lima, C. G., Morrot, A., Mendonça-Previato, L., et al. (2019). Theft and reception of host cell’s sialic acid: dynamics of Trypanosoma cruzi trans-sialidases and mucin-like molecules on chagas’ disease immunomodulation. Front. Immunol. 10:164. doi: 10.3389/fimmu.2019.00164
da Silva Vieira, T. S., Arango Duque, G., Ory, K., Gontijo, C. M., Soares, R. P., and Descoteaux, A. (2019). Leishmania braziliensis: strain-specific modulation of phagosome maturation. Front. Cell. Infect. Microbiol. 9:319. doi: 10.3389/fcimb.2019.00319
DaMatta, R. A., Seabra, S. H., Deolindo, P., Arnholdt, A. C. V., Manhães, L., Goldenberg, S., et al. (2007). Trypanosoma cruzi exposes phosphatidylserine as an evasion mechanism. FEMS Microbiol. Lett. 266, 29–33. doi: 10.1111/j.1574-6968.2006.00495.x
De Freitas Balanco, J. M., Costa Moreira, M. E., Bonomo, A., Bozza, P. T., Amarante-Mendes, G., Pirmez, C., et al. (2001). Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr. Biol. 11, 1870–1873. doi: 10.1016/S0960-9822(01)00563-2
de Paiva, R. M. C., Grazielle-Silva, V., Cardoso, M. S., Nakagaki, B. N., Mendonça-Neto, R. P., Canavaci, A. M. C., et al. (2015). Amastin knockdown in Leishmania braziliensis affects parasite-macrophage interaction and results in impaired viability of intracellular amastigotes. PLoS Pathog. 11:e1005296. doi: 10.1371/journal.ppat.1005296
de Souza, W., de Carvalho, T. M. U., and Barrias, E. S. (2010). Review on Trypanosoma cruzi: host cell interaction. Int. J. Cell Biol. 2010:295394. doi: 10.1155/2010/295394
El-Hani, C. N., Borges, V. M., Wanderley, J. L. M., and Barcinski, M. A. (2012). Apoptosis and apoptotic mimicry in Leishmania: an evolutionary perspective. Front. Cell. Infect. Microbiol. 2:96. doi: 10.3389/fcimb.2012.00096
Evans, R. J., Sundaramurthy, V., and Frickel, E. M. (2018). The interplay of host autophagy and eukaryotic pathogens. Front. Cell Dev. Biol. 6:118. doi: 10.3389/fcell.2018.00118
Farah, F. S., Samra, S. A., and Nuwayri, N. S. (1975). The role of the macrophage in cutaneous Leishmaniasis. Immunol. Commun. 29, 755–764.
Fernandes, M. C., and Andrews, N. W. (2012). Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol. Rev. 36, 734–747. doi: 10.1111/j.1574-6976.2012.00333.x
Fernandes, M. C., Corrotte, M., Miguel, D. C., Tam, C., and Andrews, N. W. (2015). The exocyst is required for trypanosome invasion and the repair of mechanical plasma membrane wounds. J. Cell Sci. 128, 27–32. doi: 10.1242/jcs.150573
Fernandes, M. C., Cortez, M., Flannery, A. R., Tam, C., Mortara, R. A., and Andrews, N. W. (2011). Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J. Exp. Med. 208, 909–921. doi: 10.1084/jem.20102518
Fernandes, M. C., Flannery, A. R., Andrews, N. W., and Mortara, R. A. (2013). Extracellular amastigotes of Trypanosoma cruzi are potent inducers of phagocytosis in mammalian cells. Cell. Microbiol. 15, 977–991. doi: 10.1111/cmi.12090
Ferreira, B. L., Ferreira, É. R., Bonfim-Melo, A., Mortara, R. A., and Bahia, D. (2019). Trypanosoma cruzi extracellular amastigotes selectively trigger the PI3K/Akt and Erk pathways during HeLa cell invasion. Microbes Infect. 21, 485–489. doi: 10.1016/j.micinf.2019.06.003
Ferreira, E. R., Bonfim-Melo, A., Mortara, R. A., and Bahia, D. (2012). Trypanosoma cruzi extracellular amastigotes and host cell signaling: more pieces to the puzzle. Front. Immunol. 30:363. doi: 10.3389/fimmu.2012.00363
Freire-de-Lima, L., Fonseca, L. M., Oeltmann, T., Mendonca-Previato, L., and Previato, J. O. (2015). The trans-sialidase, the major Trypanosoma cruzi virulence factor: three decades of studies. Glycobiology 25, 1142–1149. doi: 10.1093/glycob/cwv057
Freire-de-Lima, L., Gentile, L. B., da Fonseca, L. M., da Costa, K. M., Lemos, J. S., Jacques, L. R., et al. (2017). Role of inactive and active Trypanosoma cruzi trans-sialidases on T cell homing and secretion of inflammatory cytokines. Front. Microbiol. 8:1307. doi: 10.3389/fmicb.2017.01307
Gagnon, E., Duclos, S., Rondeau, C., Chevet, E., Cameron, P. H., Steele-Mortimer, O., et al. (2002). Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 110, 119–131.
Genois, M., Paquet, E. R., Laffitte, M. N., Maity, R., Rodrigue, A., Ouellette, M., and Masson, J. (2014). DNA repair pathways in trypanosomatids: from DNA repair to drug resistance. Microbiol. Mol. Biol. Rev. 78, 40–73. doi: 10.1128/MMBR.00045-13
Ghartey-Kwansah, G., Aboagye, B., Adu-Nti, F., Opoku, Y. K., and Abu, E. K. (2020). Clearing or subverting the enemy: role of autophagy in protozoan infections. Life Sci. 247:117453. doi: 10.1016/j.lfs.2020.117453
Hamilton, P. B., Gibson, W. C., and Stevens, J. R. (2007). Patterns of co-evolution between trypanosomes and their hosts deduced from ribosomal RNA and protein-coding gene phylogenies. Mol. Phylogenet. Evol. 44, 15–25. doi: 10.1016/j.ympev.2007.03.023
Harmer, J., Yurchenko, V., Nenarokova, A., Lukeš, J., and Ginger, M. L. (2018). Farming, slaving and enslavement: histories of endosymbioses during kinetoplastid evolution. Parasitology 145, 1311–1323. doi: 10.1017/S0031182018000781
Henriques, C., Atella, G. C., Bonilha, V. L., and De Souza, W. (2003). Biochemical analysis of proteins and lipids found in parasitophorous vacuoles containing Leishmania amazonensis. Parasitol. Res. 89, 123–133. doi: 10.1007/s00436-002-0728-y
Henriques, C., and De Souza, W. (2000). Redistribution of plasma-membrane surface molecules during formation of the Leishmania amazonensis-containing parasitophorous vacuole. Parasitol. Res. 86, 215–225. doi: 10.1007/s004360050034
Holm, A., Tejle, K., Magnusson, K. E., Descoteaux, A., and Rasmusson, B. (2001). Leishmania donovani lipophosphoglycan causes periphagosomal actin accumulation: correlation with impaired translocation of PKCα and defective phagosome maturation. Cell. Microbiol. 3, 439–447. doi: 10.1046/j.1462-5822.2001.00127.x
Jaskowska, E., Butler, C., Preston, G., and Kelly, S. (2015). Phytomonas: trypanosomatids adapted to plant environments. PLoS Pathog. 11:e1004484. doi: 10.1371/journal.ppat.1004484
Kaufer, A., Ellis, J., Stark, D., and Barratt, J. (2017). The evolution of trypanosomatid taxonomy. Parasit. Vectors 10:287.
Kaufer, A., Stark, D., and Ellis, J. (2019). Evolutionary insight into the trypanosomatidae using alignment-free phylogenomics of the kinetoplast. Pathogens 8:157. doi: 10.3390/pathogens8030157
Laffitte, M. N., Leprohon, P., Papadopoulou, B., and Ouelletteb, M. (2016). Plasticity of the Leishmania genome leading to gene copy number variations and drug resistance. F1000Res. 5:2350. doi: 10.12688/f1000research.9218.1
Ley, V., Robbins, E. S., Nussenzweig, V., and Andrews, N. W. (1990). The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the PH of acidic compartments. J. Exp. Med. 171, 401–413. doi: 10.1084/jem.171.2.401
Lima, F. M., Oliveira, P., Mortara, R. A., Silveira, J. F., and Bahia, D. (2010). The challenge of Chagas’ disease: has the human pathogen, Trypanosoma cruzi, learned how to modulate signaling events to subvert host cells? New Biotechnol. 27, 837–843. doi: 10.1016/j.nbt.2010.02.003
Lodge, R., and Descoteaux, A. (2005). Modulation of phagolysosome biogenesis by the lipophosphoglycan of Leishmania. Clin. Immunol. 114, 256–265. doi: 10.1016/j.clim.2004.07.018
Martínez, A., Prolo, C., Estrada, D., Rios, N., Alvarez, M. N., Piñeyro, M. D., et al. (2019). Cytosolic Fe-superoxide dismutase safeguards Trypanosoma cruzi from macrophage-derived superoxide radical. Proc. Natl. Acad. Sci. U.S.A. 116, 8879–8888. doi: 10.1073/pnas.1821487116
Martins, R. M., Alves, R. M., Macedo, S., and Yoshida, N. (2011). Starvation and rapamycin differentially regulate host cell lysosome exocytosis and invasion by Trypanosoma cruzi metacyclic forms. Cell. Microbiol. 13, 943–954. doi: 10.1111/j.1462-5822.2011.01590.x
Matte, C., Casgrain, P. A., Séguin, O., Moradin, N., Hong, W. J., and Descoteaux, A. (2016). Leishmania major promastigotes evade LC3-associated phagocytosis through the action of GP63. PLoS Pathog. 12:e1005690. doi: 10.1371/journal.ppat.1005690
Meirelles, M. N. L., Araujo Jorge, T. C., de Souza, W., Moreira, A. L., and Barbosa, H. S. (1987). Trypanosoma cruzi: phagolysosomal fusion after invasion into non professional phagocytic cells. Cell Struct. Funct. 12, 387–393. doi: 10.1247/csf.12.387
Mortara, R. A. (1991). Trypanosoma cruzi: amastigotes and trypomastigotes interact with different structures on the surface of HeLa cells. Exp. Parasitol. 14, 1–14. doi: 10.1016/0014-4894(91)90002-e
Mortara, R. A., Andreoli, W. K., Fernandes, M. C. D. C., da Silva, C. V., Fernandes, A. B., L’Abbate, C., et al. (2008). Host cell actin remodeling in response to Trypanosoma cruzi: trypomastigote versus amastigote entry. Subcell. Biochem. 47, 92–100. doi: 10.1007/978-0-387-78267-6_7
Mortara, R. A., Andreoli, W. K., Taniwaki, N. N., Fernandes, A. B., da Silva, C. V., Fernandes, M. C. D. C., et al. (2005). Mammalian cell invasion and intracellular trafficking by Trypanosoma cruzi infective forms. An. Acad. Bras. Ciênc. 77, 77–94. doi: 10.1590/s0001-37652005000100006
Ndjamen, B., Kang, B., Hatsuzawa, K., and Kima, P. E. (2010). Leishmania parasitophorous vacuoles interact continuously with the host cell’s endoplasmic reticulum; parasitophorous vacuoles are hybrid compartments. Cell. Microbiol. 12, 1480–1494. doi: 10.1038/jid.2014.371
Okuda, K., Tong, M., Dempsey, B., Moor, K. J., Gazzinelli, R. T., and Silverman, N. (2016). Leishmania amazonensis engages CD36 to drive parasitophorous vacuole maturation. PLoS Pathog. 12:e1005669. doi: 10.1371/journal.ppat.1005669
Pessoa, C. C., Ferreira, E. R., Bayer-Santos, E., Rabinovitch, M., Mortara, R. A., and Real, F. (2016). Trypanosoma cruzi differentiates and multiplies within chimeric parasitophorous vacuoles in macrophages coinfected with Leishmania amazonensis. Infect. Immun. 84, 1603–1614. doi: 10.1128/IAI.01470-15
Pessoa, C. C., Reis, L. C., Ramos-Sanchez, E. M., Orikaza, C. M., Cortez, C., de Castro Levatti, E. V., et al. (2019). ATP6V0d2 controls Leishmania parasitophorous vacuole biogenesis via cholesterol homeostasis. PLoS Pathog. 15:e1007834. doi: 10.1371/journal.ppat.1007834
Pitale, D. M., Gendalur, N. S., Descoteaux, A., and Shaha, C. (2019). Leishmania donovani induces autophagy in human blood–derived neutrophils. J. Immunol. 202, 1163–1175. doi: 10.4049/jimmunol.1801053
Real, F., Florentino, P. T. V., Reis, L. C., Ramos-Sanchez, E. M., Veras, P. S. T., Goto, H., et al. (2014). Cell-to-cell transfer of Leishmania amazonensis amastigotes is mediated by immunomodulatory LAMP-rich parasitophorous extrusions. Cell. Microbiol. 16, 1549–1564. doi: 10.1111/cmi.12311
Real, F., and Mortara, R. A. (2012). The diverse and dynamic nature of Leishmania parasitophorous vacuoles studied by multidimensional imaging. PLoS Negl. Trop. Dis. 6:e1518. doi: 10.1371/journal.pntd.0001518
Real, F., Mortara, R. A., and Rabinovitch, M. (2010). Fusion between Leishmania amazonensis and Leishmania major parasitophorous vacuoles: live imaging of coinfected macrophages. PLoS Negl. Trop. Dis. 4:e905. doi: 10.1371/journal.pntd.0000905
Real, F., Vidal, R. O., Carazzolle, M. F., Mondego, J. M. C., Costa, G. G. L., Herai, R. H., et al. (2013). The genome sequence of Leishmania (Leishmania) amazonensis: functional annotation and extended analysis of gene models. DNA Res. 20, 567–581. doi: 10.1093/dnares/dst031
Reignault, L. C., Alcantara, C. L., Barrias, E. S., and de Souza, W. (2019). 3D reconstruction of Trypanosoma cruzi-macrophage interaction shows the recruitment of host cell organelles towards parasitophorous vacuoles during its biogenesis. J. Struct. Biol. 205, 133–146. doi: 10.1016/j.jsb.2018.12.010
Reignault, L. C., Barrias, E. S., Soares Medeiros, L. C., De Souza, W., and De Carvalho, T. M. U. (2014). Structures containing galectin-3 are recruited to the parasitophorous vacuole containing Trypanosoma cruzi in mouse peritoneal macrophages. Parasitol. Res. 113, 2323–2333. doi: 10.1007/s00436-014-3887-8
Reis-Cunha, J. L., Rodrigues-Luiz, G. F., Valdivia, H. O., Baptista, R. P., Mendes, T. A. O., de Morais, G. L., et al. (2015). Chromosomal copy number variation reveals differential levels of genomic plasticity in distinct Trypanosoma cruzi strains. BMC Genomics 16:499. doi: 10.1186/s12864-015-1680-4
Reis-Cunha, J. L., Valdivia, H. O., and Bartholomeu, D. C. (2018). Gene and chromosomal copy number variations as an adaptive mechanism towards a parasitic lifestyle in trypanosomatids. Curr. Genomics 19, 87–97. doi: 10.2174/1389202918666170911161311
Ribeiro-Gomes, F. L., Otero, A. C., Gomes, N. A., Moniz-de-Souza, M. C. A., Cysne-Finkelstein, L., Arnholdt, A. C., et al. (2004). Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462. doi: 10.4049/jimmunol.172.7.4454
Rodriguez, A., Samoff, E., Rioult, M. G., Chung, A., and Andrews, N. W. (1996). Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J. Cell Biol. 134, 349–362. doi: 10.1083/jcb.134.2.349
Romano, P. S., Arboit, M. A., Vázquez, C. L., and Colombo, M. I. (2009). The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 5, 6–18. doi: 10.4161/auto.5.1.7160
Rosestolato, C. T. F., Dutra, J. M. F., de Souza, W., and Carvalho, T. M. U. (2002). Participation of host cell actin filaments during interaction of trypomastigote forms of Trypanosoma Cruzi with host cells. Cell Struct. Funct. 27, 91–98. doi: 10.1247/csf.27.91
Rubin-de-Celis, S. S., and Schenkman, S. (2012). T rypanosoma cruzi trans-sialidase as a multifunctional enzyme in Chagas’ disease. Cell. Microb. 14, 1522–1530. doi: 10.1111/j.1462-5822.2012.01831.x
Rubin-de-Celis, S. S. C., Uemura, H., Yoshida, N., and Schenkman, S. (2006). Expression of trypomastigote trans-sialidase in metacyclic forms of Trypanosoma cruzi increases parasite escape from its parasitophorous vacuole. Cell. Microbiol. 8, 1888–1898. doi: 10.1111/j.1462-5822.2006.00755.x
Salassa, B. N., and Romano, P. S. (2018). Autophagy: a necessary process during the Trypanosoma cruzi life-cycle. Virulence 10, 460–469. doi: 10.1080/21505594.2018.1543517
Stecconi-Silva, R. B., Andreoli, W. K., and Mortara, R. A. (2003). Parameters affecting cellular invasion and escape from the parasitophorous vacuole by different infective forms of Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 98, 953–958. doi: 10.1590/s0074-02762003000700016
Stevens, J., Noyes, H., and Gibson, W. (1998). The Evolution of trypanosomes infecting humans and primates. Mem. Inst. Oswaldo Cruz 93, 669–676. doi: 10.1590/S0074-02761998000500019
Tardieux, I., Webster, P., Ravesloot, J., Boron, W., Lunn, J. A., Heuser, J. E., et al. (1992). Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71, 1117–1130. doi: 10.1016/s0092-8674(05)80061-3
Teixeira, S. M. R., Russell, D. G., Kirchhoffnll, L. V., and Donelson, J. E. (1994). A differentially expressed gene family encoding ‘amastin’, a surface protein of Trypanosoma cruzi amastigotes. J. Biol. Chem. 269, 20509–20516.
Tomlinson, S., Frevert, U., Vandekerckhove, F., and Nussenzweig, V. (1995). The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low PH. Parasitology 110, 547–554. doi: 10.1017/S0031182000065264
Tyler, K. M., Luxton, G. W. G., Applewhite, D. A., Murphy, S. C., and Engman, D. M. (2005). Responsive microtubule dynamics promote cell invasion by Trypanosoma cruzi. Cell. Microbiol. 7, 1579–1591. doi: 10.1111/j.1462-5822.2005.00576.x
Veras, P. S. T., de Menezes, J. P. B., and Dias, B. R. S. (2019). Deciphering the role played by autophagy in Leishmania infection. Front. Immunol. 10:2523. doi: 10.3389/fimmu.2019.02523
Verma, J. K., Rastogi, R., and Mukhopadhyay, A. (2017). Leishmania donovani resides in modified early endosomes by upregulating Rab5a expression via the downregulation of MiR-494. PLoS Pathog. 13:e1006459. doi: 10.1371/journal.ppat.1006459
Wanderley, J. L. M., Damatta, R. A., and Barcinski, M. A. (2020). Apoptotic mimicry as a strategy for the establishment of parasitic infections: parasite- and host-derived phosphatidylserine as key molecule. Cell Commun. Signal. 18:10. doi: 10.1186/s12964-019-0482-8
WHO (2020a). Chagas Disease (American Trypanosomiasis). Available online at: https://www.who.int/chagas/epidemiology/en/ (accessed January 16, 2020).
WHO (2020b). Leishmaniasis. Available online at: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed January 16, 2020).
Wilson, J., Huynh, C., Kennedy, K. A., Ward, D. M., Kaplan, J., Aderem, A., and Andrews, N. W. (2008). Control of parasitophorous vacuole expansion by LYST/Beige restricts the intracellular growth of Leishmania amazonensis. PLoS pathog. 4:e1000179. doi: 10.1371/journal.ppat.1000179
Winberg, M. E., Holm, A., Sarndahl, E., Vinet, A. F., Descoteaux, A., Magnusson, K. E., et al. (2009). Leishmania donovani lipophosphoglycan inhibits phagosomal maturation via action on membrane rafts. Microbes Infect. 11, 215–222. doi: 10.1016/j.micinf.2008.11.007
Woolsey, A. M., Sunwoo, L., Petersen, C. A., Brachmann, S. M., Cantley, L. C., and Burleigh, A. (2003). Novel Pl 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J. Cell Sci. 116, 3611–3622. doi: 10.1242/jcs.00666
Yoshida, N. (2006). Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An. Acad. Bras. Ciênc. 78, 87–111. doi: 10.1590/S0001-37652006000100010
Keywords: Trypanosoma cruzi, Leishmania, vacuole, parasitophorous vacuole, intracellular pathogen
Citation: Batista MF, Nájera CA, Meneghelli I and Bahia D (2020) The Parasitic Intracellular Lifestyle of Trypanosomatids: Parasitophorous Vacuole Development and Survival. Front. Cell Dev. Biol. 8:396. doi: 10.3389/fcell.2020.00396
Received: 13 February 2020; Accepted: 29 April 2020;
Published: 10 June 2020.
Edited by:
Si-Yang Huang, Yangzhou University, ChinaReviewed by:
Wanderley De Souza, Federal University of Rio de Janeiro, BrazilCopyright © 2020 Batista, Nájera, Meneghelli and Bahia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Bahia, ZGlhbmFiYWhpYUBob3RtYWlsLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.