 Joshua D. Webster
Joshua D. Webster Domagoj Vucic
Domagoj Vucic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 21 May 2020
Sec. Signaling
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00365
This article is part of the Research TopicTNFR Superfamily Oligomerization and SignalingView all 10 articles
Tumor necrosis factor alpha (TNF; TNFα) is a critical regulator of immune responses in healthy organisms and in disease. TNF is involved in the development and proper functioning of the immune system by mediating cell survival and cell death inducing signaling. TNF stimulated signaling pathways are tightly regulated by a series of phosphorylation and ubiquitination events, which enable timely association of TNF receptors-associated intracellular signaling complexes. Disruption of these signaling events can disturb the balance and the composition of signaling complexes, potentially resulting in severe inflammatory diseases.
TNF is a type II transmembrane protein that is expressed at the plasma membrane as a trimer (Vassalli, 1992). Cleavage by tumor necrosis factor converting enzyme (TACE) can generate a soluble ligand that propagates signaling by binding to two receptors – TNFR1 (CD120a) and TNFR2 (CD120b) (Black et al., 1997; Moss et al., 1997). TNFR1 associates strongly with both membrane-bound and soluble TNF, while TNFR2 has much higher binding affinity for membrane-bound TNF (Grell et al., 1995). The extracellular region of both receptors has four homologous cysteine-rich domains (CRDs) but their intracellular regions are structurally different. The intracellular portion of TNFR1 possesses a protein-binding region called a death domain (DD), which allows homo- and hetero-typic interactions with other DD-containing proteins. TNFR2, on the other hand, has a TNF Receptor Associated Factor (TRAF) binding site that interacts with the TRAF family of signaling adaptors (Grell et al., 1995; Reddy et al., 2000). The distinct expression profiles and stark difference in the intracellular regions of the TNF receptors greatly influence their physiological roles and cellular activity. Through engaging DD adaptors, broadly expressed TNFR1 can activate proliferative nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling as well as cell death (Wallach et al., 1999; Sessler et al., 2013). On the other hand, TNFR2 is mostly expressed in immune and endothelial tissues. In addition, since it lacks a DD, TNFR2 cannot stimulate cell death, but uses TRAF recruitment to trigger NF-κB and MAPK activation (Wallach et al., 1999; Sessler et al., 2013). Due to its wide spectrum of cellular activities and ubiquitous expression, TNFR1 plays a prevailing role in TNF signaling and will be more extensively covered in this article.
Binding of TNF to TNFR1 triggers receptor trimerization and leads to the assembly of the TNFR1-associated signaling complex (complex I) (Figure 1). Within complex I, the adaptor proteins receptor interacting protein 1 (RIP1; RIPK1) and TNF receptor associated death domain (TRADD) are recruited to TNFR1 through their respective death domains (Micheau and Tschopp, 2003). TRADD then recruits adaptor proteins TRAF2 and TRAF5, which enables the engagement of the E3 ligases cellular inhibitors of apoptosis 1 and 2 (c-IAP1, c-IAP2) and subsequent ubiquitination of various components of complex I (Bertrand et al., 2008; Mahoney et al., 2008; Varfolomeev et al., 2008; Dynek et al., 2010). c-IAP1/2 promote self-ubiquitination and ubiquitination of RIP1 with K63-, K48-, and K11-linked chains, which are critical for TNFR1 complex I signaling (Bertrand et al., 2008; Mahoney et al., 2008; Varfolomeev et al., 2008; Dynek et al., 2010). K63-linked polyubiquitin chains conjugated onto c-IAP1/2 allow the recruitment of the linear ubiquitin chain assembly complex (LUBAC), which generates linear ubiquitin chains on several molecules including RIP1, TNFR1, LUBAC itself, and NF-κB essential modulator (NEMO) (Haas et al., 2009; Tokunaga et al., 2009; Ikeda et al., 2011; Tokunaga et al., 2011; Varfolomeev and Vucic, 2016). The LUBAC complex consists of adaptor proteins SHANK-associated RH-domain interactor (SHARPIN) and heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1L), and the E3 enzyme HOIL-1L-interacting protein (HOIP) (Tokunaga and Iwai, 2012). LUBAC produces linear or M1-linked ubiquitin chains by catalyzing a head-to-tail ubiquitination where a peptide bond between the N-terminal methionine of ubiquitin and the C-terminal glycine of the next ubiquitin is generated (Kirisako et al., 2006; Tokunaga et al., 2009). The diverse ensemble of polyubiquitin chains assembled during TNF-induced activation of NF-κB and MAPK includes, but is not limited to, K11, K48, K63, and linear chains (Dynek et al., 2010; Gerlach et al., 2011). This set of polyubiquitin chains provides a docking platform for the recruitment and retention of the signaling kinase complexes consisting of kinases IKKα and IKKβ (inhibitor of kappa B kinase 1 and 2) and the adaptor NEMO (IKKγ; IKK complex), and transforming growth factor beta-activated kinase 1 (TAK1) along with its partners, the K63-linked ubiquitin binding proteins TAK1-binding proteins 2 and 3 (TAB2/3) (Figure 1; Shim et al., 2005; Haas et al., 2009). The recruitment of kinase complexes leads to the activation of NF-κB and MAPK signaling and subsequent gene activation and expression of pro-inflammatory cytokines, such as interleukin 6 and 8 (IL-6, IL-8), and pro-survival proteins like c-IAP2 and the caspase-8 inhibitor cellular FLICE inhibitory protein (cFLIP) (Scheidereit, 2006).
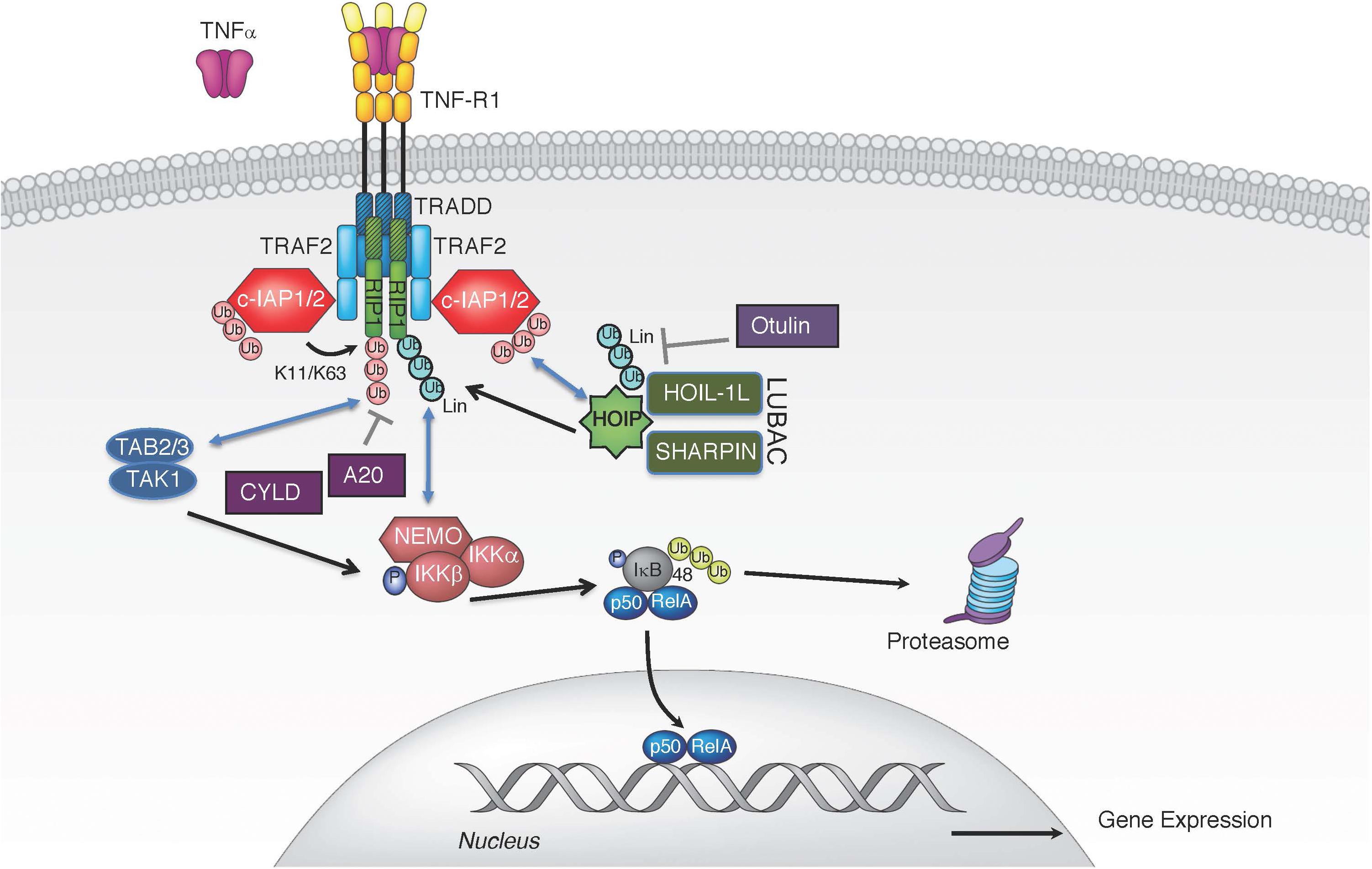
Figure 1. TNF induced canonical NF-κB pathway. TNF stimulation triggers the recruitment of TRADD, TRAF2, RIP1, and c-IAP1/2 to TNFR1. E3 ligases c-IAP1/2 polyubiquitinate themselves and RIP1 with K11 and K63 ubiquitin linkages, creating a platform for further recruitment of LUBAC. LUBAC mediates linear polyubiquitin, resulting in gene expression via the IKK complex. Several DUBs have been implicated in the regulation of TNFR1-associated complex I by removing linear (CYLD and OTULIN) or K63-linked polyubiquitin chains (A20 and CYLD).
The specific polyubiquitination pattern on RIP1 that keeps it in complex I for proper downstream activation of NF-κB and MAPKs is fine-tuned by the activation of E3 ligases, such as c-IAP1/2 (Bertrand et al., 2008; Mahoney et al., 2008; Varfolomeev et al., 2008; Silke and Vucic, 2014). The combined deletion of c-IAP1 and c-IAP2 in mice results in embryonic lethality and severe liver and intestinal damage in the adulthood, which can be rescued by TNFR1 knock-out or by TNF blockade, further emphasizing the functional and genetic relationship between these E3 ligases and TNF signaling (Moulin et al., 2012; Zhang J. et al., 2019). However, these complexes are also governed through negative regulation by deubiquitinases (DUBs). TNF stimulation also leads to transcriptional upregulation of deubiquitinases tumor necrosis factor alpha-induced protein 3 (TNFAIP3 or A20) and OTU domain DUB 7B (also known as Cezanne) whose DUB activity can dampen NF-κB signaling (Wertz et al., 2004; Enesa et al., 2008). A20 is an ubiquitin chain-binding enzyme that removes K63-linked ubiquitin chains from RIP1 to reduce NF-κB activation. Binding of linear ubiquitin chains via its zinc finger 7 motif is critical for A20’s recruitment to TNFR1 complex and suppression of inflammatory signaling (Tokunaga et al., 2012; Martens A. et al., 2020; Razani et al., 2020). Consequently, deletion of A20 results in enhanced RIP1 ubiquitination and inflammation (Wertz et al., 2004; Zhou et al., 2016a). Cylindromatosis (CYLD) is another DUB whose recruitment to complex I can dampen NF-κB activation by hydrolyzing the K63-linked and linear polyubiquitin chains from the complex I components (Brummelkamp et al., 2003; Kovalenko et al., 2003; Trompouki et al., 2003). CYLD is recruited to the TNFR1 complex via the adaptor protein SPATA2, which binds the PUB (peptide:N-glycanase/UBA/X-containing protein) domain of HOIP through its PIM (PUB-interaction motif) (Elliott et al., 2016; Kupka et al., 2016; Schlicher et al., 2016; Wagner et al., 2016; Wei et al., 2017). Consequently, the absence of SPATA2, just like CYLD loss, enhances TNF stimulated NF-κB activation and dampens cell death (Elliott et al., 2016; Kupka et al., 2016; Schlicher et al., 2016; Wagner et al., 2016; Wei et al., 2017). Unlike these DUBs, which do not have a strict ubiquitin chain specificity, OTULIN (OTU deubiquitinase with linear specificity, also known as FAM105B or Gumby) binds the PUB domain of HOIP and selectively removes linear ubiquitin chains on LUBAC components thereby keeping uncontrolled TNF-associated inflammation in check (Fiil et al., 2013; Keusekotten et al., 2013; Elliott et al., 2014; Damgaard et al., 2016; Zhou et al., 2016b). Thus, a tightly controlled balance of E3 ligases and DUBs in the assembly and disassembly of diverse polyubiquitin chains on RIP1 and other signaling components is clearly needed for the appropriate level of signaling by complex I and corresponding gene activation.
Dynamic changes of post-translational modifications of RIP1 and other components of TNFR1-associated signaling complexes can trigger a switch from inflammatory gene signaling to cell death via apoptosis or necroptosis (Figure 2). RIP1-dependent and RIP1-independent apoptotic signaling complexes can form in response to inhibited or altered NF-κB signaling (e.g., IKKβ or TAK1 inhibitors, genetic deletion of NF-κB) or the presence of transcriptional or translational inhibitors like actinomycin D or cycloheximide, respectively (Rubin et al., 1988; Micheau and Tschopp, 2003; Shan et al., 2018). A cytosolic complex II centered on TRADD recruits Fas-associated death domain (FADD) to activate caspase-8 and cause apoptotic cell death (Van Antwerp et al., 1996; Micheau and Tschopp, 2003; Wang et al., 2008). For RIP1-dependent apoptotic complex II, distinct ubiquitin modifications play a critical regulatory role in dictating the fate of cells. When E3 ligases c-IAP1/2 and LUBAC are degraded or absent, unmodified RIP1 dissociates from receptor-associated signaling complex I and associates with FADD through binding of their DDs (Micheau and Tschopp, 2003; Bertrand et al., 2008; Wang et al., 2008). FADD recruits pro-caspase 8 and/or its catalytically inactive homolog FLIP to form the death platform complex II using death effector domain (DED) interactions (Majkut et al., 2014). RIP1 dependent apoptosis can be further modulated by additional signaling proteins and E3 ligases (NEK1, APC11, LRKK2, and Cbl) that regulate the transition of RIP1 from complex I to complex II (Amin et al., 2018). Thus, the ubiquitination status of RIP1 determines the switch of RIP1 between pro-survival gene activation and cell death.
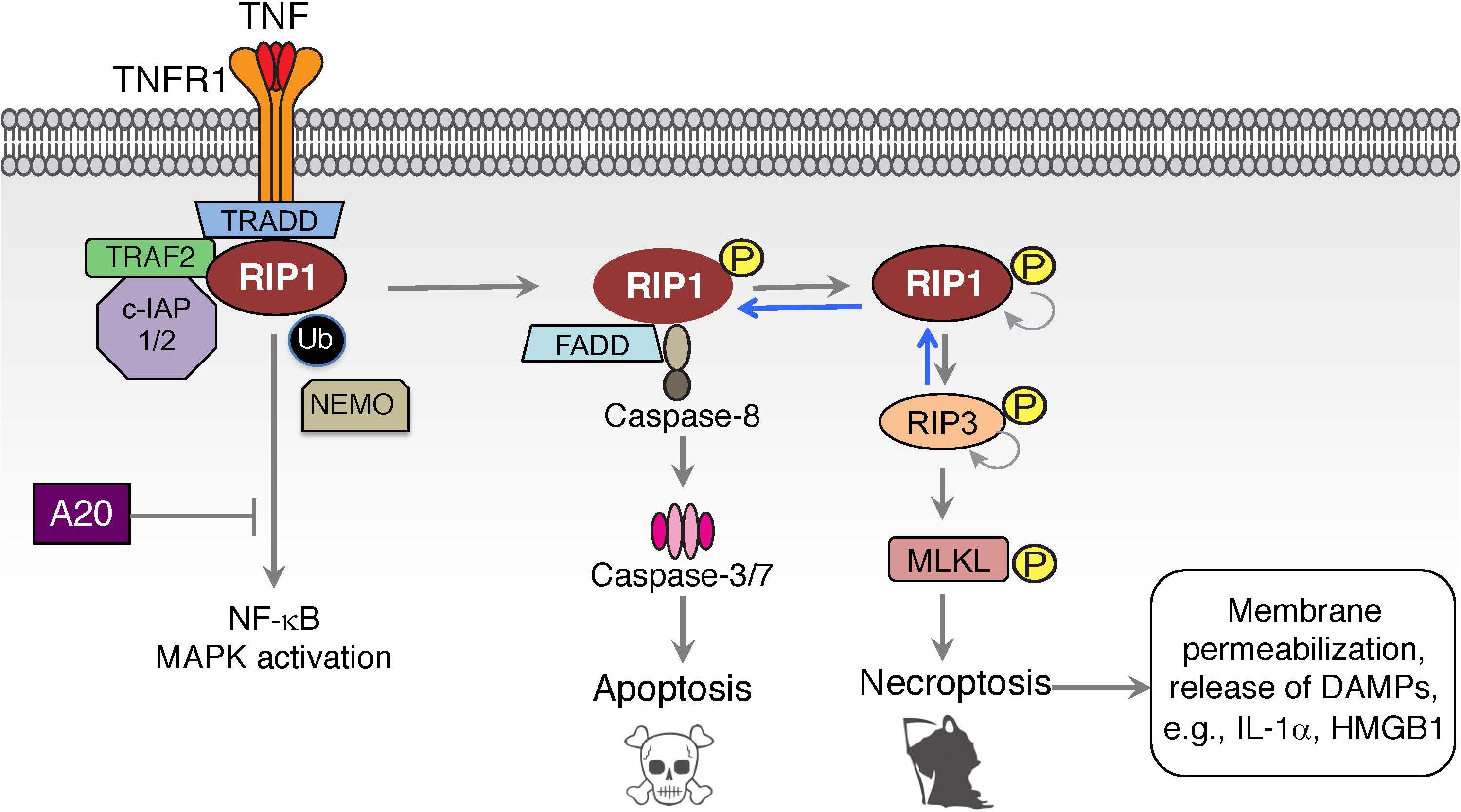
Figure 2. TNF induced cell death signaling. Inhibition of NF-κB and MAPK signaling can divert TNF-mediated signaling to the formation of an intracellular complex II centered on FADD and caspase-8 in a RIP1-dependent apoptotic cell death. This cell death pathway can be augmented by A20 or by the absence of the E3 ligases c-IAP1/2 or LUBAC, thereby eliminating the ubiquitination of complex I components and promoting the switch to complex II. Activation of RIP1-dependent cell death under caspase-8 inhibited or deficient conditions can lead to a necroptotic form of cell death that is mediated by kinase activity of RIP1 and RIP3, and results in the activation of MLKL and membrane permeabilization.
If caspase-8 is insufficiently activated or inhibited in complex II, RIP1 can autophosphorylate at S166 and bind RIP3 using their RIP homology interaction motifs (RHIM) leading to the formation of the necrosome (Sun et al., 2002; He et al., 2009; Wu et al., 2014; Laurien et al., 2020). Unlike complex I, where RIP1 kinase activity is dispensable, TNF stimulated necrosome formation is dependent on RIP1 kinase activity (Cho et al., 2009; He et al., 2009; Laurien et al., 2020). Within the necrosome, RIP3 undergoes auto-phosphorylation at S227 in human, and T231 and S232 in mouse RIP3 that is crucial for the execution of necroptotic cell death (Cho et al., 2009; He et al., 2009; Chen et al., 2013). Accordingly, genetic inactivation or chemical inhibition of their kinase functions blocks RIP1/3 dependent necroptotic cell death (Degterev et al., 2008; He et al., 2009; Newton et al., 2014). RIP3 phosphorylates necroptosis mediator mixed lineage kinase domain-like (MLKL) at residues T357 and S358 in human, and S345, S347, and T349 in mouse MLKL within its carboxy-terminal pseudokinase domain to execute necroptosis (Sun et al., 2012; Chen et al., 2013; Murphy et al., 2013; Khan et al., 2014; Wang et al., 2014; Rodriguez et al., 2016). How MLKL facilitates cell death is not entirely clear, but it does involve the disruption of cell membrane integrity. RIP3-phosphorylated MLKL undergoes a conformational change that exposes the N-terminal domain of MLKL, promoting its oligomerization and translocation to the membranes (Murphy et al., 2013; Dondelinger et al., 2014; Wang et al., 2014). Membrane associated MLKL may interfere directly with cell integrity by oligomeric insertion into the membrane thus causing membrane disruption/permeabilization/perturbation (Cai et al., 2014; Dondelinger et al., 2014; Hildebrand et al., 2014; Su et al., 2014; Wang et al., 2014). The pore-forming capacity of necroptosis results in a strong pro-inflammatory signal, a feature that places this cell death pathway at the core of many inflammatory and tissue-damage related diseases. However, RIP1 autophosphorylation can result in RIP1-dependent apoptosis as well, and a number of in vivo inflammatory animal models involve a mixture of RIP1-dependent apoptosis and necroptosis as we will describe later in this article (Patel et al., 2020; Webster et al., 2020).
While TNF’s importance in driving inflammatory diseases is well-established, the recent identification of patients with defects in TNF signaling components have shown the importance of tightly regulating this pathway and the potential consequences of its dysregulation (Manthiram et al., 2017; Oda and Kastner, 2017; Figure 3). In vitro and in vivo studies have suggested the critical role ubiquitin plays in TNF signaling, both in enabling signal complex formation and in protein degradation. For example, chronic proliferative dermatitis (cpdm) mice were originally characterized as a strain of C57BL/KaLawRij mice that developed eosinophilic dermatitis with epidermal hyperplasia, multi-systemic inflammation, and defects in lymphoid development (HogenEsch et al., 1993, 1999; Gijbels et al., 1996). Subsequent studies demonstrated that this phenotype was due to a spontaneous mutation in the Sharpin gene that resulted in diminished expression of SHARPIN and the other LUBAC components HOIP and HOIL-1L (Seymour et al., 2007; Gerlach et al., 2011; Tokunaga et al., 2011). Studies in these mice suggested that LUBAC mediated linear ubiquitination plays an important role in modulating inflammatory and cell death signaling downstream of TNFR1. However, the clinical validation of these observations has been more recently evident through the identification of patients with mutations in the genes encoding LUBAC components HOIP and HOIL-1L, and in mutations in the genes encoding the deubiquitinases OTULIN and A20.
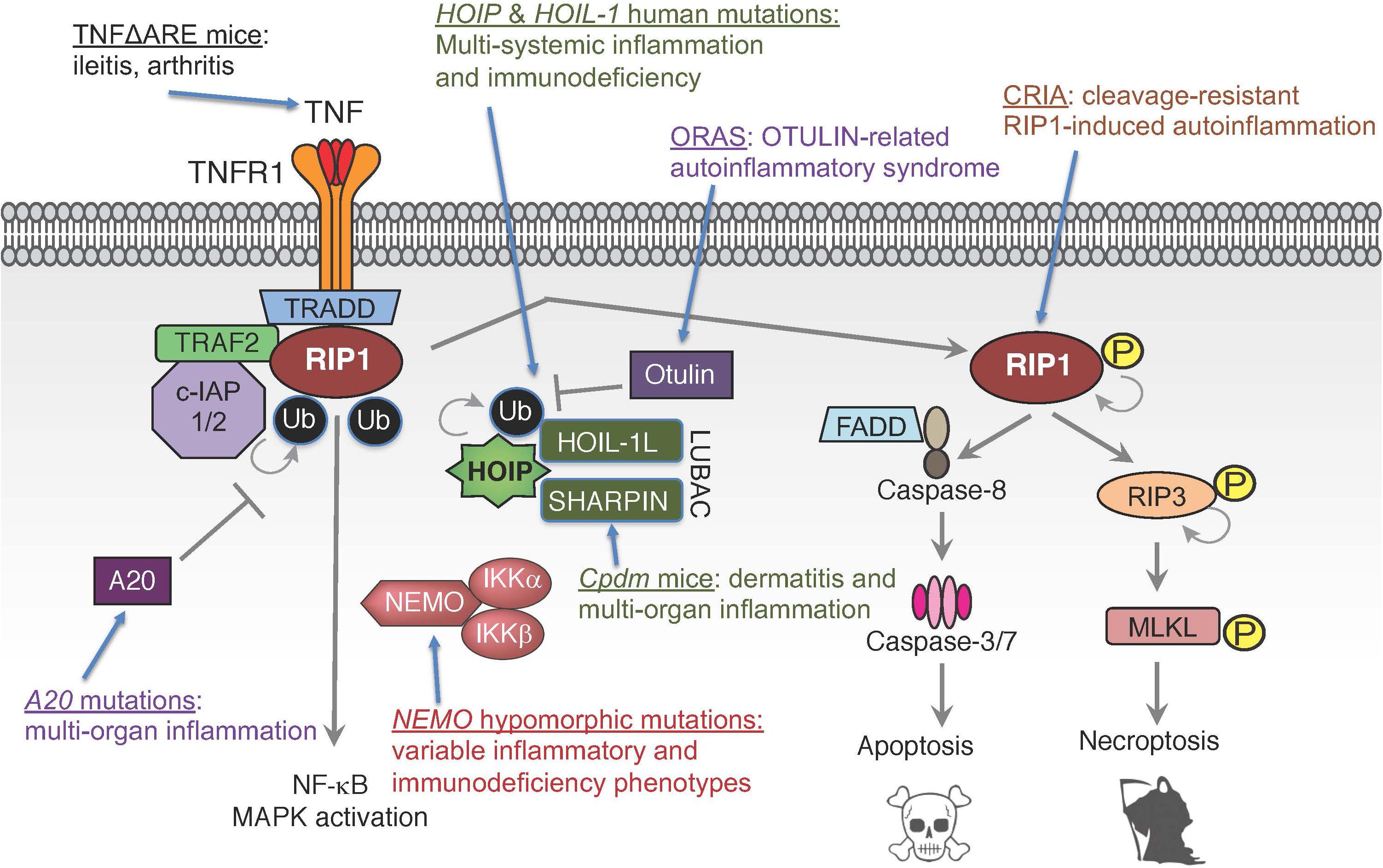
Figure 3. Human mutations and genetic models in TNF signaling pathways. Mutations in multiple components of TNF signaling leading to functional dysregulation of E3 ligase complex LUBAC, deubiquitinases A20 and OTULIN, ubiquitin-binding protein NEMO, ligand TNF or pro-death kinase RIP1 can cause inflammatory diseases and/or immunodeficiency.
Mutations in HOIL-1L were originally reported in 2012, in two families with immunodeficiency, as characterized by recurrent pyogenic infections, multi-systemic inflammation, and amylopectinosis (Boisson et al., 2012). The single described patient in the first family had a homozygous deletion of 2 nucleotides resulting in a premature stop codon. Patients in a second family had partial deletions of one allele and a nonsense point mutation in the second allele; suggesting an autosomal recessive mode of inheritance in both families. These HOIL-1L mutations caused an approximately 50% decrease in SHARPIN expression and near complete loss of HOIP expression. Loss of LUBAC expression resulted in impaired NF-κB activity in response to interleukin-1 beta (IL-1β) and, to a lesser extent, TNF in the patients’ fibroblasts. Similarly, the patients’ B cell responses to Toll-like receptor (TLR) 7 and 8 agonists, IL-1β, and CD40-ligand (CD40L) were also impaired. Interestingly, LUBAC deficiency had an opposing effect in monocytes. Specifically, monocytes had approximately four-fold increased IL-6 production following IL-1β stimulation and enhanced responses to TLR1 and 2 agonists (Boisson et al., 2012). While the patients described above presented with immune dysregulation, other HOIL-1L mutant patients present with primary myopathy and less frequent to no evidence of immune dysfunction (Nilsson et al., 2013; Wang et al., 2013). These patients presented with muscle weakness in adolescence that progresses over time, and a subset of patients develop dilated cardiomyopathy (Nilsson et al., 2013; Wang et al., 2013). Histologically, myofibers contain periodic acid-Schiff positive, amylase-resistant inclusions characteristic of polyglucosan (Nilsson et al., 2013). The reason that some patients with HOIL-1L mutations present for myopathy while others present for immunodeficiency is not fully understood, but the phenotype might be influenced by the location of the mutation in the gene (Nilsson et al., 2013).
An autosomal recessive missense mutation in HOIP has also been identified in one patient. This mutation resulted in a loss of HOIP protein and reduced levels of SHARPIN and HOIL-1L, resulting in LUBAC deficiency (Boisson et al., 2015). A second patient was identified with compound heterozygous HOIP polymorphisms. These polymorphisms caused alternative RNA splicing that resulted in truncated HOIP protein and LUBAC destabilization (Oda et al., 2019). Clinical and biochemical phenotypes in HOIP mutant patients mirrored patients with HOIL-1L mutations. Specifically, patients with mutations or polymorphisms in HOIP presented with multi-systemic inflammation and immunodeficiency characterized by recurrent infections, chronic diarrhea, and antibody deficiency (Boisson et al., 2015; Oda et al., 2019). Patients’ fibroblasts had blunted NF-κB responses to IL-1β and TNF, and their B cells had reduced response to CD40L (Boisson et al., 2015). Additionally, similar to HOIL-1L deficient patients, monocytes derived from these patients had increased response to IL-1β, resulting in elevated IL-6 and IL-1β production (Boisson et al., 2015).
Characterization of these patients confirms the critical role of LUBAC-mediated linear ubiquitination in NF-κB driven immune responses of fibroblasts and lymphocytes, and demonstrates that loss of this signaling has significant consequences including immunodeficiency and subsequent recurrent infections. However, the paradoxical increase in proinflammatory signaling in monocytes, which likely accounts for the concurrent multi-systemic inflammation, suggests that the role of LUBAC is dependent on cellular context and tight regulation of this pathway is critical to modulate inflammatory responses.
Deubiquitinases are critical to counter-regulate ubiquitin ligase activities. Just as c-IAP1/2 and LUBAC play fundamental roles in establishing signaling complexes downstream of the TNF receptor, deubiquitinases like OTULIN, CYLD, and A20 play equally important roles in modulating these complexes. Patients with reduced OTULIN expression due to autosomal recessive mutations develop fevers, dermatitis, and panniculitis (Damgaard et al., 2016, 2019; Zhou et al., 2016b; Nabavi et al., 2019). Comparable phenotypes are observed in patients with autosomal dominant mutations in the gene encoding A20, TNFAIP3. Specifically, these patients present with early onset systemic inflammation including arthritis, ophthalmitis, and oral and genital ulcers (Zhou et al., 2016a). Initial characterization of cells from affected patients revealed that TNF stimulated peripheral blood mononuclear cells (PBMCs) and fibroblasts from OTULIN and TNFAIP3 mutant patients have increased NF-κB activity compared to controls and increased p38 phosphorylation in fibroblasts, which is associated with increased ubiquitination (Zhou et al., 2016a,b). These changes were correlated with increased serum cytokines in TNFAIP3 mutant patients (Zhou et al., 2016a), and increased LPS-induced production of interferon-gamma, IL-1β, IL-6, IL-12, and IL-18 in whole blood samples of OTULIN deficient patients (Zhou et al., 2016b). Fibroblasts from a subsequently identified patient with a unique homozygous OTULIN mutation had reduced NFκB and p38 activity in response to TNF (Damgaard et al., 2019). The differences between these patients and their responses to TNF is unclear, since mutations characterized in both studies reportedly resulted in decreased OTULIN activity (Zhou et al., 2016b; Damgaard et al., 2019; Nabavi et al., 2019). Interestingly, although the later study found that OTULIN deficient fibroblasts were hypo-responsive to TNF and shOTULIN THP-1 cells were hyper-responsive to TNF, both cell types had increased susceptibility to cell death induced by the combination of TNF and cyclohexamide (Damgaard et al., 2019), suggesting that cell death is a common end product of dysregulation of this pathway. The clinical data and cellular characterization of HOIP, HOIL-1L, OTULIN, and A20 deficient patients highlight the essential role of ubiquitination in modulating TNF signaling. On the surface, the data suggest that too little ubiquitination (e.g., HOIP or HOIL-1L deficiency) results in dampening of the immune response, and persistent ubiquitination (e.g., OTULIN or A20 deficiency) causes autoinflammation. However, there are added complexities to this perspective, as noted in the increased IL-1β response in HOIP and HOIL-1L deficient monocytes.
Aside from autoinflammation, A20 mutations also occur in approximately 12% of B cell lymphomas, with the highest incidence in mucosa-associated lymphoid tissue (MALT) lymphoma (Kato et al., 2009). Reconstitution of an A20 deficient lymphoma cell line with wild-type A20 resulted in decreased proliferation, increased apoptosis, and decreased NF-κB signaling. Similarly, A20 expressing cells transplanted into immunodeficient mice failed to develop tumors, as opposed to mock transfected cells, which developed tumors (Kato et al., 2009). Therefore, A20 does not only regulate NF-κB signaling in the context of normal immune responses, but it also appears to act as a tumor suppressor, regulating NF-κB signaling in the context of tumorigenesis.
Disease-associated mutations have also been identified in genes that encode target proteins of ubiquitination including RIP1 and NEMO. Patients with autosomal recessive RIP1 deficiency are immunodeficient, as characterized by lymphopenia and recurrent infections, and develop inflammatory enterocolitis that resembles inflammatory bowel disease (IBD) (Cuchet-Lourenco et al., 2018; Abed et al., 2019; Uchiyama et al., 2019). Similar to patients with HOIP and HOIL-1L deficiencies, their fibroblasts had reduced MAPK and NF-κB signaling in response to TNF and polyinosinic:polycytidylic [poly(I:C)] (Cuchet-Lourenco et al., 2018). This was coupled with increased fibroblast death that appeared to be driven by necroptosis, as indicated by RIP3 and MLKL phosphorylation (Cuchet-Lourenco et al., 2018). However, while ex vivo stimulation of patients’ monocytes with LPS produced less IL-6, TNF, and IL-12 in response to LPS, they had increased IL-1β production (Cuchet-Lourenco et al., 2018), suggesting increased inflammasome activation. In addition to mutations that result in RIP1 deficiency, mutations in the caspase-8 cleavage site of RIP1 (D324) have also been identified in patients with periodic fevers and lymphadenopathy (Lalaoui et al., 2020; Tao et al., 2020). Peripheral blood mononuclear cells from these patients had enhanced susceptibility to both apoptotic and necroptotic stimuli, and increased pro-inflammatory cytokine production including IL-6, TNF, interferon-gamma, and IL-10 (Lalaoui et al., 2020; Tao et al., 2020). Together, these results highlight and validate RIP1’s unique physiological role in TNF signaling, as a mediator of pro-inflammatory signaling and as a regulator of cell death.
Mutations in IKBKG, the gene that encodes NEMO, are associated with both incontinentia pigmenti (Smahi et al., 2000) and X-linked recessive ectodermal dysplasia with immunodeficiency (Zonana et al., 2000; Doffinger et al., 2001). X-linked recessive ectodermal dysplasia with immunodeficiency is associated with hypomorphic mutations and the clinical phenotype is highly variable and may include recurrent infections, hyper-IgM levels, ectodermal dysplasia including coning teeth and hypodontia, inability to sweat, lymphedema, and osteopetrosis. This diverse presentation is due to both the variety of mutations that occur in these patients, but also the diversity of receptors associated with NF-κB signaling including ectodysplasin-A receptor, TNFR1, CD40, and receptor activator of NF-κB (RANK) (Zonana et al., 2000; Doffinger et al., 2001; Miot et al., 2017). While immunodeficiency due to both inadequate NF-κB mediated innate responses and CD40 signal in B cells is a primary medical concern in these patients, hematopoietic stem cell transplantation does not alleviate all of the associated disease. For instance, many patients have persistent colitis, even post-transplantation, which suggests epithelial specific defects are also important in the clinical phenotype (Miot et al., 2017).
Identification and characterization of patients with monogenic defects in TNF signaling components has provided critical insights into the significance of these proteins in regulating inflammatory signaling, and provides clinical context as to how dysfunction in this pathway can manifest in disease. While these clinical data are invaluable, there are experimental limits to what can be studied in patients and patient-derived samples. Therefore, spontaneous and genetically engineered mouse models have proven valuable tools to further interrogate TNF signaling pathways, to model diseases where these pathways likely play a role, and to identify how these pathways can be modulated when they go awry. A clear example of the value of these mouse models is the coincidental reporting of cleavage resistant RIP1 mutations in patients with periodic fevers and the description of knock-in mice with complementary mutations (Newton et al., 2019; Zhang X. et al., 2019; Lalaoui et al., 2020; Tao et al., 2020). Genetic experiments in these mouse models demonstrated that observed clinical phenotypes were likely driven, at least in part, by TNFR1 and RIP1 kinase dependent apoptosis, but also highlight the complex role RIP1 plays in control both inflammatory and cell death pathways (Newton et al., 2019; Zhang X. et al., 2019; Lalaoui et al., 2020).
SHARPIN-deficient cpdm mice were the first LUBAC deficient mice characterized. SHARPIN deficiency results in reduced, but not eliminated, LUBAC activity and therefore is best characterized as a hypomorphic mouse (Seymour et al., 2007; Gerlach et al., 2011; Tokunaga et al., 2011). The most prominent phenotype in cpdm mice is eosinophilic dermatitis (HogenEsch et al., 1993) that begins around 1 week of age and progresses to severe disease by 6 weeks of age (Gijbels et al., 1996). Inflammatory infiltrates are also present in the joints, liver, and lung (Zhang et al., 2009) of these mice. Additionally, these mice develop eosinophilic esophagitis (Chien et al., 2015) and have hypoplastic lymphoid tissues (HogenEsch et al., 1999). While systemic immune infiltrates are partially dependent on lymphocytes, dermatitis in these mice is lymphocyte independent, indicating that this is an auto-inflammatory rather than autoimmune process (Potter et al., 2014). Loss of TNF or TNFR1 protects cpdm mice from both dermatitis and systemic inflammation, suggesting TNF signaling is the primary driver of inflammation (Gerlach et al., 2011; Kumari et al., 2014; Rickard et al., 2014). RIP1 kinase inhibition is also protective against dermatitis and reduces systemic inflammation in cpdm mice (Berger et al., 2014; Patel et al., 2020; Webster et al., 2020). Cpdm mice that express catalytically inactive RIP1K45A do not develop dermatitis or systemic inflammation (Berger et al., 2014), and treatment with a RIP1 inhibitor, even starting at 6 weeks of age when there is disease induction, provides significant amelioration of the dermatitis and reduces immune infiltrates in the liver (Webster et al., 2020). Interestingly, while RIP3 loss delays the development of dermatitis in SHARPIN deficient mice (Kumari et al., 2014; Rickard et al., 2014), MLKL loss does not affect the development of dermatitis (Rickard et al., 2014). Consistent with this data, caspase-3 is robustly activated in the epidermis of SHARPIN deficient mice (Liang et al., 2011; Kumari et al., 2014; Rickard et al., 2014; Webster et al., 2020), while phosphorylated RIP3 positive cells were rarely detected in the dermis (Webster et al., 2020). Furthermore, the loss of 1 caspase-8 allele in addition to RIP3 deficiency prevented the development of inflammatory lesions in most cpdm mice (Rickard et al., 2014). Together, this suggests that while RIP1 kinase activity drives the inflammation in cpdm mice, RIP1 is only partially signaling through RIP3 and the inflammation is primarily driven by apoptosis rather than necroptosis. Therefore, in some contexts, especially when epithelial barriers are disrupted, excessive apoptosis can be pro-inflammatory. Loss of caspase-1 also prevents the development of inflammatory lesions in cpdm mice. This protection is thought to be due to SHARPIN’s role in regulating caspase-1 activity in a LUBAC independent manner (Nastase et al., 2016).
Aside from the inflammatory lesions in the skin, joints and visceral organs, cpdm mice also have defective lymphoid development that includes altered splenic architecture and absence of Peyer’s patches (HogenEsch et al., 1999). Loss of TNF or TNFR1 does not restore the splenic architecture in cpdm mice (Gerlach et al., 2011; Kumari et al., 2014), but this is suspected to be partially due to the intrinsic defects in lymphoid development in the absence of TNFR1 signaling (Kumari et al., 2014). Similarly, caspase-1 deficient cpdm mice do not develop normal lymphoid architecture (Nastase et al., 2016). However, Peyer’s patches were restored in Rip3–/–Casp8± cpdm mice (Rickard et al., 2014). The role of RIP1 kinase activity in the lymphoid phenotype of cpdm mice is not well characterized as evaluations of lymphoid tissues in cpdm with catalytically inactive RIP1K45A have not been reported (Berger et al., 2014). While treatment with a RIP1 inhibitor after the onset of dermatitis did not restore the lymphoid architecture, this might be due to the late timing of the intervention and it is possible that germline loss of RIP1 kinase activity may restore the lymphoid architecture (Webster et al., 2020).
In contrast to the hypomorphic phenotype of cpdm mice, Hoip and Hoil-1l knock-out mice die around embryonic day 10.5 due to increased endothelial cell death and vascular collapse, most notably in the yolk sac (Peltzer et al., 2014; Peltzer et al., 2018). The timing of this is notable because this is also the stage when Caspase-8 knock-out mice die due to RIP3 dependent necroptosis (Varfolomeev et al., 1998; Kaiser et al., 2011). While loss of caspase-8 and RIP3, loss of RIP1 catalytic activity due to the expression of catalytically inactive RIP1K45A, or loss of TNF signaling can extend survival to later embryonic stages in HOIL-1L deficient mice, only the combined loss of RIP1, RIP3, and caspase-8 is protective, which suggests cell death is a primary driver of embryonic lethality in these mice (Peltzer et al., 2018). Epidermal specific deletion of Hoip and Hoil-1l results in dermatitis in the perinatal period and death by post-partum day 6. Similar lesions are observed following inducible deletion of Hoip in adult mice (Taraborrelli et al., 2018). Dermatitis in epithelial-specific knock-out mice indicates that the inflammation is driven by an epithelial autonomous process, rather than being initiated by aberrant immune cell signaling. Similar to SHARPIN deficient mice, increased cell death, as evidenced by increased cleaved caspase-3 immunolabeling and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), was observed in the epidermis of these mice. In germline epidermal specific Hoip and Hoil-1l knock-out mice, cell death is apparent at embryonic day 18.5 and precedes inflammatory cell infiltration into the dermis, suggesting that cell death is a cause rather than a consequence of inflammation. The significance of cell death in driving dermatitis is further supported by the fact that loss of caspase-8 and either MLKL or RIP3 is protective in these mice (Taraborrelli et al., 2018). Interestingly, while dermatitis appears to be solely driven by TNFR1 signaling in SHARPIN deficient mice, loss of TNFR1 only delays the onset of dermatitis to approximately 70 days, which appears to be due to concurrent signaling through other death receptors including TNF-related apoptosis inducing ligand (TRAIL) receptor and CD95. Additionally, in contrast to SHARPIN deficient mice, loss of RIP1 kinase activity through the expression of catalytically inactive RIP1D138N does not show dramatic protection in these mice. However, the combination of small molecule RIP1 kinase inhibition and loss of TNFR1 expression provides more efficient protection in Hoil-1l knock-out mice, suggesting that RIP1 inhibition can provide a benefit independent of TNFR1 in some circumstances (Taraborrelli et al., 2018).
Both the similarities and differences between SHARPIN deficient mice that have hypomorphic LUBAC function and epidermal specific Hoip and Hoil-1L knock-out mice provide key insights into this pathway and its role in disease. Firstly, disruption in TNF stimulated linear ubiquitination can result in severe dermatitis. Cell death, predominantly apoptosis, is a key driver of inflammation in the skin of these mice, and inhibition of cell death can rescue the inflammation. This suggests that modulation of cell death pathways should be further considered for inflammatory skin diseases. Secondly, RIP1 inhibition was more effective when LUBAC activity was reduced rather than when it was eliminated (Berger et al., 2014; Taraborrelli et al., 2018). However, there was a benefit to RIP1 inhibition in addition to TNFR1 loss in the HOIP and HOIL-1L deficient mice (Taraborrelli et al., 2018), suggesting that the efficacy of RIP1 inhibition as a single agent might be context and disease specific. Additionally, the synergistic role of TNFR1 loss and RIP1 inhibition suggests that RIP1 inhibition is not just another means to disrupt the TNF signaling pathway, but scenarios where anti-TNFs and RIP1 inhibitors could be used in combination should be explored further.
Inducible inactivation of OTULIN’s DUB function in adult mice results in extensive hepatocyte and intestinal crypt cell death, and inflammation, primarily driven by myeloid cells, in the heart and liver (Heger et al., 2018). Similarly, co-deletion of Birc2 and Birc3, which encode c-IAP1 and c-IAP2, respectively, in adult mice results in extensive hepatocyte death and crypt degeneration with intestinal villous atrophy and secondary inflammation (Zhang J. et al., 2019). In both OTULIN and c-IAP1/2 deficient mice, cell death in the liver and intestines was associated with extensive cleaved caspase-3 immunolabeling, suggesting a predominance of apoptosis (Heger et al., 2018; Zhang J. et al., 2019). While loss of RIP3 alone does not prevent lesions in either mouse, loss of caspase-8 and RIP3 rescued both the cell death and, to a significant degree, the associated inflammation. Since caspase-8 loss is embryonic lethal due to RIP3 mediated necroptosis, it is impossible to determine the independent contribution of apoptosis. However, the lack of protection with RIP3 loss alone and the extensive cleaved caspase-3 labeling suggests that apoptosis is the primary driver of the pro-inflammatory phenotype in these mice. In line with these observations in systemic, inducible Otulin or Birc2/3 knock-out mice, hepatocyte specific deletion of Otulin results in hepatocyte apoptosis with resultant compensatory hyperplasia and inflammation that can progress to hepatocellular carcinoma (Damgaard et al., 2020; Verboom et al., 2020). Increased cell death and steatosis is evident in these mice by postnatal day 9. Interestingly, steatosis and increased liver enzymes were also identified in an OTULIN deficient patient (Damgaard et al., 2020). This hepatic injury can be alleviated by the loss of RIP1 kinase activity due to expression of the RIP1D138N kinase dead protein, and more completely rescued by hepatocyte-specific Fadd deletion, indicating that the injury is driven by apoptosis signaling (Verboom et al., 2020), although loss of TNFR1 is not sufficient to protect against liver pathology in these mice (Damgaard et al., 2020). mTOR signaling is also increased in livers with hepatocyte-specific OTULIN deficiency. While treatment with the mTOR inhibitor rapamycin reduced the proliferative lesions and fibrosis in these mice, it did not reduce serum alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels, which suggests that while mTOR may be important for the proliferative response, it might not be the driver of the initial hepatocyte injury (Damgaard et al., 2020).
In contrast to germline loss of other components of the ubiquitin machinery, including LUBAC, OTULIN, and c-IAP1/2, that result in embryonic lethality, A20 deficient mice survive to birth, but subsequently develop multi-systemic inflammation that includes dermatitis, hepatitis, nephritis, enteritis, and arthritis (Lee et al., 2000). Inflammation in these mice appears to be lymphocyte independent because there was no protection when A20 deficient mice were crossed to Rag1 knock-out mice (Lee et al., 2000). Consistent with their development of multi-systemic inflammation, A20 knock-out mice have increased susceptibility to LPS and TNF, and this is associated with persistent NF-κB signaling in mouse embryonic fibroblasts (MEFs) (Lee et al., 2000). Loss of RIP3 or RIP1 kinase activity due the D138A kinase dead mutation significantly prolongs the survival of A20 deficient mice; however, a similar benefit is not observed in Mlkl knock-out mice (Onizawa et al., 2015; Newton et al., 2016). The difference in protection between RIP3 and MLKL deficient mice highlights the potential for necroptotic-independent functions of RIP3, which are not fully characterized.
The role of TNF in inflammatory bowel disease and rheumatoid arthritis has been well established both in mouse models and in clinical practice (Williams et al., 1992; Elliott et al., 1993; Targan et al., 1997; Kontoyiannis et al., 1999). Constitutive TNF over-expression in the TNFΔARE mice, which have increased Tnf mRNA production and stability, develop Crohn’s-like ileitis that can progress to transmural and granulomatous inflammation and arthritis (Kontoyiannis et al., 1999). Ileitis even develops when TNF over-expression is restricted to intestinal enterocytes, although the disease onset and progression is delayed compared to mice with systemic TNF over-expression (Roulis et al., 2011; Bamias et al., 2013a). However, while TNF signaling in enterocytes causes apoptosis, it is not sufficient to cause ileitis, indicating the importance of paracrine signaling in other stromal and immune cells, rather than just enterocyte-restricted autocrine signaling (Roulis et al., 2011). Considering the permissive effects of TNF and the complexity of inflammatory bowel disease, it should not be surprising that disease progression requires an interplay of the epithelial, stromal, and hematopoietic compartments. Arthritis in TNFΔARE mice is characterized by synovial hyperplasia and myeloid infiltrates that progresses to cartilage and bone erosion and fibrosis, resulting in pannus (Kontoyiannis et al., 1999). Similarly, bone phenotype spontaneous mutation 1 (BPSM1) mice that have increased TNF expression due to a spontaneous insertion of a small interspersed element (SINE) in the 3’ untranslated region of Tnf, develop severe, progressive arthritis and valvular endocarditis with aortic aneurysm (Lacey et al., 2015). Development of arthritis requires local TNF production, as evidenced by the lack of joint changes in TNFΔARE mice with intestinal-specific TNF hyper-secretion (Bamias et al., 2013b). In BPSM1 mice, bone marrow transplantation of wild-type or BPSM1 cells and genetic crosses with Tnfr1–/– mice suggest that while myeloid cells are necessary for TNF production in this model, TNFR1 signaling on non-hematopoietic cells, presumable synoviocytes, is required for the development of arthritis (Lacey et al., 2015). TNF blockade is also protective in collagen-induced and anti-collagen antibody-induced arthritis models (Williams et al., 1992; Patel et al., 2020), and this is consistent with the clinical benefit of TNF blockade in rheumatoid arthritis patients. While RIP1 inhibition provided a similar benefit in anti-collagen antibody-induced arthritis model, there was no synergistic benefit in combining TNF and RIP1 inhibition (Patel et al., 2020). This is in contrast to the added protective benefit of combinatorial blockade in the development of dermatitis in LUBAC deficient mice (Taraborrelli et al., 2018). Therefore, the benefit of combination therapies is likely to be context specific and requires further exploration.
Patients with hypomorphic NEMO mutations frequently develop colitis. The fact that this colitis is not responsive to hematopoietic stem cell transplantation suggests that NEMO deficiency has cell autonomous effects in intestinal enterocytes (Miot et al., 2017). This has been studied in mice by using a Cre recombinase driven by the villin promoter to specifically delete Nemo from intestinal epithelial cells. Enterocyte-specific NEMO loss results in severe colitis, particularly in the proximal colon, and small intestinal crypt cell death with Paneth cell loss (Nenci et al., 2007; Vlantis et al., 2016). While TNFR1 loss and germ-free conditions protect against colitis, increased cell death in the small intestine remains (Vlantis et al., 2016). Similar to SHARPIN-mutant mice, RIP3 loss affords inconsistent and incomplete protection in NEMO deficient mice, while inactivation of RIP1 kinase activity via RIP1D138N or RIP3 and FADD combined ablation provide complete protection (Vlantis et al., 2016). Pharmacologic RIP1 inhibition is similarly fully protective in these mice (Patel et al., 2020). Again, this suggests that RIP1 mediated apoptosis can drive both extensive tissue damage and inflammation in the context of dysfunctional TNF signaling.
While TNF signaling is biased toward cell death pathways in NEMO deficient mice, presumably in part due to a lack of NF-κB signaling, enterocytes with overactive NF-κB signaling are also sensitive to TNF-induced cell death. IKKβ (EE)IEC mice have constitutive NF-κB signaling in intestinal epithelial cells (Guma et al., 2011). These mice have increased sensitivity to LPS due to MAPK mediated TNF production (Guma et al., 2011). TNF stimulation in enteroids from these mice causes intestinal epithelial cell apoptosis, as noted by increased cleaved caspase-3 and caspase-8. Genetic loss of RIP1 catalytic activity through expression of RIP1D138N protected enterocytes from TNF induced apoptosis, while RIP3 loss was not protective. Similarly, both genetic and pharmacologic RIP1 inactivation protected these mice from LPS induced intestinal cell death in vivo (Wong et al., 2020). Together, the increased susceptibility of both the IKKβ (EE)IEC mice and NEMO deficient mice to TNF-induced apoptosis suggests that NF-κB signaling needs to be tightly controlled and dysregulation in either direction may shift TNF signaling from a pro-survival to a pro-death pathway. Interestingly, RIP1 kinase activity is a potent driver of cell death in both scenarios. This further strengthens the hypothesis that RIP1 inhibition may provide a therapeutic benefit to IBD patients.
The ATG16L1T300A polymorphism is associated with Crohn’s disease, and ATG16L1 has an important role in Paneth cell survival and function (Cadwell et al., 2008). Norovirus infected mice with reduced ATG16L1 expression have decreased and disorganized Paneth cell granules and decreased lysozyme, and similar defects have been identified in Crohn’s disease patients (Cadwell et al., 2008, 2010). ATG16L1 deficient mice also have increased susceptibility to dextran sodium sulfate (DSS)-induced colitis and, in the presence of norovirus infection, develop small intestinal villous atrophy and have loss of Paneth cells. This small intestinal pathology is driven by increased epithelial TNF production and subsequent cell death, and is protected by RIP1 kinase inhibition (Matsuzawa-Ishimoto et al., 2017). Increased Paneth cell death has been identified in the ileum of Crohn’s disease patients, and treatment of control patient biopsies with TNF has been shown to reduce Paneth cell-associated lysozyme mRNA, which can be rescued by Nec-1, a RIP1 inhibitor (Gunther et al., 2011). Considering the importance of Paneth cells in producing anti-microbial peptides and innate immune responses in the intestine, TNF mediated Paneth cell death may play an important role in the pathogenesis of Crohn’s disease. Given the protection observed with RIP1 inhibitors in the survival of mouse and human Paneth cells, and the intrinsic role of RIP1 kinase activity in intestinal pathology secondary to NFκB dysregulation, RIP1 inhibitors should be further evaluated in the treatment of inflammatory bowel disease.
While TNF inhibition is efficacious in the treatment of many inflammatory disease, it is also associated with immunosuppression and increased risk of infections, and many patients are refractory to TNF inhibitors (Taylor and Feldmann, 2009; Adegbola et al., 2018). RIP1 inhibition may provide an alternative mechanism to treat inflammatory diseases with no known risk of immunosuppression (Shan et al., 2018; Yuan et al., 2019). While the Rip1 knock-out mouse dies in the perinatal period due to RIP3 mediated inflammation and caspase 8 mediated intestinal apoptosis (Kelliher et al., 1998; Dillon et al., 2014; Kaiser et al., 2014), catalytically dead Rip1 knock-in (RIP1 KD) mice are viable and healthy, even when aged to 18 months (Berger et al., 2014; Kaiser et al., 2014; Newton et al., 2014; Polykratis et al., 2014; Webster et al., 2020). RIP1D138N KD mice were able to clear both vaccinia virus and mouse gammaherpesvirus, MHV68, at a similar rate compared to wild-type mice and these mice showed no immunologic dysfunction following MHV68 infection (Webster et al., 2020). This suggests that while RIP1 scaffolding functions are essential for survival, RIP1 kinase activity can be inhibited without detrimental effects.
As described above, RIP1 kinase inhibition is protective against inflammation in the skin, intestines, and joints secondary to dysfunctions in TNF and NF-κB signaling (Berger et al., 2014; Vlantis et al., 2016; Patel et al., 2020; Webster et al., 2020; Wong et al., 2020). Patients with mutations in these pathways have variably responded to different biologics including anti-IL-1, anti-TNF, and anti-IL-6 molecules (Damgaard et al., 2016; Zhou et al., 2016b; Lalaoui et al., 2020; Tao et al., 2020). It still is to be seen whether these patients would benefit from RIP1 inhibitors. The role of RIP1 kinase activity in inflammation is also evident in the TNF-induced systemic inflammatory response syndrome (SIRS) model, in which genetic or pharmacologic RIP1 inhibition is protective (Newton et al., 2014; Polykratis et al., 2014; Newton et al., 2016; Patel et al., 2020). Interestingly, in some disease models, such as anti-collagen antibody-induced arthritis, combined RIP1 and TNF inhibition does not show a synergistic effect suggesting these proteins are working on a linear pathway (Patel et al., 2020). However, in other models, such as HOIP and HOIL-1L deficient mice, TNF and RIP1 inhibition plays a synergistic role (Taraborrelli et al., 2018), indicating that combination therapies might be efficacious in some diseases. Although RIP1 has been implicated in numerous disease models, the results have not always been reproducible (Newton et al., 2016; Patel et al., 2020; Webster et al., 2020). Therefore, more studies are needed to define the context and potential combination therapies that will provide the maximal benefit for RIP1 inhibition. However, the true test of RIP1 inhibition in inflammatory diseases will be in clinical trials.
To date, GlaxoSmithKline (GSK) and Denali have tested their RIP1 inhibitors in clinical settings and reported that GSK2982772 and DNL104 were generally well tolerated in human subjects (Harris et al., 2017; Weisel et al., 2017; Grievink et al., 2020; Jensen et al., 2020; Martens S. et al., 2020). Denali’s brain-penetrant RIP1 inhibitor DNL104 did not cause any central nervous system toxicities but 37% percent of subjects receiving multiple doses of DNL104 had post-dose liver toxicity (Grievink et al., 2020). Denali has, in the meantime, terminated clinical examination of DNL104 and in collaboration with Sanofi entered another RIP1 inhibitor, DNL747, in clinical trials for Alzheimer’s disease, amylotrophic lateral sclerosis, and multiple sclerosis (Jensen et al., 2020; Martens S. et al., 2020). GSK2982772 is a systemic, non-brain penetrant RIP1 inhibitor was well-tolerated with no serious adverse events (AEs) and no suggestion of a safety concern (Weisel et al., 2017). Encouraged by favorable safety data, GSK has entered GSK2982772 into small phase 2 clinical trials for psoriasis, rheumatoid arthritis, and ulcerative colitis. So far, GSK2982772 has not shown significant therapeutic benefit in psoriasis or rheumatoid arthritis (clinicatrials.gov), while the data from the ulcerative colitis trial are still pending. GSK has also ventured into cancer trials with a different RIP1 inhibitor, GSK3145095 (Harris et al., 2019). However, that particular trial, designed to test the ability of RIP1 inhibitor to provide benefit in pancreatic and other solid tumors, was relatively quickly terminated (Martens S. et al., 2020). This may not come as a complete surprise given that protective role of RIP1 inhibition in pancreatic cancer was never fully validated (Patel et al., 2020). Thus, although RIP1 inhibition presents an attractive opportunity to target TNF mediated inflammatory diseases, further efforts are needed to fully explore this therapeutic strategy.
JW and DV wrote and contributed to this manuscript and approved this submission.
JW and DV were employees and shareholders at Genentech-Roche.
Abed, M., Verschueren, E., Budayeva, H., Liu, P., Kirkpatrick, D. S., Reja, R., et al. (2019). The Gag protein PEG10 binds to RNA and regulates trophoblast stem cell lineage specification. PLoS One 14:e0214110. doi: 10.1371/journal.pone.0214110
Adegbola, S. O., Sahnan, K., Warusavitarne, J., Hart, A., and Tozer, P. (2018). Anti-TNF Therapy in Crohn’s Disease. Int. J. Mol. Sci. 19:2244.
Amin, P., Florez, M., Najafov, A., Pan, H., Geng, J., Ofengeim, D., et al. (2018). Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 115, E5944–E5953. doi: 10.1073/pnas.1806973115
Bamias, G., Dahman, M. I., Arseneau, K. O., Guanzon, M., Gruska, D., Pizarro, T. T., et al. (2013a). Intestinal-specific TNFalpha overexpression induces Crohn’s-like ileitis in mice. PLoS One 8:e72594. doi: 10.1371/journal.pone.0072594
Bamias, G., Stamatelopoulos, K., Zampeli, E., Protogerou, A., Sigala, F., Papamichael, C., et al. (2013b). Circulating levels of TNF-like cytokine 1A correlate with the progression of atheromatous lesions in patients with rheumatoid arthritis. Clin. Immunol. 147, 144–150. doi: 10.1016/j.clim.2013.03.002
Berger, S. B., Kasparcova, V., Hoffman, S., Swift, B., Dare, L., Schaeffer, M., et al. (2014). Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480. doi: 10.4049/jimmunol.1400499
Bertrand, M. J., Milutinovic, S., Dickson, K. M., Ho, W. C., Boudreault, A., Durkin, J., et al. (2008). cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700. doi: 10.1016/j.molcel.2008.05.014
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733. doi: 10.1038/385729a0
Boisson, B., Laplantine, E., Dobbs, K., Cobat, A., Tarantino, N., Hazen, M., et al. (2015). Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939–951. doi: 10.1084/jem.20141130
Boisson, B., Laplantine, E., Prando, C., Giliani, S., Israelsson, E., Xu, Z., et al. (2012). Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13, 1178–1186. doi: 10.1038/ni.2457
Brummelkamp, T. R., Nijman, S. M., Dirac, A. M., and Bernards, R. (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB-. Nature 424, 797–801. doi: 10.1038/nature01811
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. doi: 10.1038/nature07416
Cadwell, K., Patel, K. K., Maloney, N. S., Liu, T. C., Ng, A. C., Storer, C. E., et al. (2010). Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145. doi: 10.1016/j.cell.2010.05.009
Cai, Z., Jitkaew, S., Zhao, J., Chiang, H. C., Choksi, S., Liu, J., et al. (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65. doi: 10.1038/ncb2883
Chen, W., Zhou, Z., Li, L., Zhong, C. Q., Zheng, X., Wu, X., et al. (2013). Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261. doi: 10.1074/jbc.M112.435545
Chien, S. J., Silva, K. A., Kennedy, V. E., HogenEsch, H., and Sundberg, J. P. (2015). The pathogenesis of chronic eosinophilic esophagitis in SHARPIN-deficient mice. Exp. Mol. Pathol. 99, 460–467. doi: 10.1016/j.yexmp.2015.08.012
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi: 10.1016/j.cell.2009.05.037
Cuchet-Lourenco, D., Eletto, D., Wu, C., Plagnol, V., Papapietro, O., Curtis, J., et al. (2018). Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813. doi: 10.1126/science.aar2641
Damgaard, R. B., Elliott, P. R., Swatek, K. N., Maher, E. R., Stepensky, P., Elpeleg, O., et al. (2019). OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11:e9324. doi: 10.15252/emmm.201809324
Damgaard, R. B., Jolin, H. E., Allison, M. E. D., Davies, S. E., Titheradge, H. L., McKenzie, A. N. J., et al. (2020). OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 27, 1457–1474. doi: 10.1038/s41418-020-0532-1
Damgaard, R. B., Walker, J. A., Marco-Casanova, P., Morgan, N. V., Titheradge, H. L., Elliott, P. R., et al. (2016). The deubiquitinase OTULIN Is an essential negative regulator of inflammation and autoimmunity. Cell 166,, 1215.e20–1230.e20. doi: 10.1016/j.cell.2016.07.019
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Dillon, C. P., Weinlich, R., Rodriguez, D. A., Cripps, J. G., Quarato, G., Gurung, P., et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. doi: 10.1016/j.cell.2014.04.018
Doffinger, R., Smahi, A., Bessia, C., Geissmann, F., Feinberg, J., Durandy, A., et al. (2001). X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB- signaling. Nat. Genet. 27, 277–285. doi: 10.1038/85837
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. doi: 10.1016/j.celrep.2014.04.026
Dynek, J. N., Goncharov, T., Dueber, E. C., Fedorova, A. V., Izrael-Tomasevic, A., Phu, L., et al. (2010). c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. Embo J. 29, 4198–4209. doi: 10.1038/emboj.2010.300
Elliott, M. J., Maini, R. N., Feldmann, M., Long-Fox, A., Charles, P., Katsikis, P., et al. (1993). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheumatism 36, 1681–1690. doi: 10.1002/art.23362
Elliott, P. R., Leske, D., Hrdinka, M., Bagola, K., Fiil, B. K., McLaughlin, S. H., et al. (2016). SPATA2 links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol. Cell 63, 990–1005. doi: 10.1016/j.molcel.2016.08.001
Elliott, P. R., Nielsen, S. V., Marco-Casanova, P., Fiil, B. K., Keusekotten, K., Mailand, N., et al. (2014). Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 54, 335–348. doi: 10.1016/j.molcel.2014.03.018
Enesa, K., Zakkar, M., Chaudhury, H., Luong le, A., Rawlinson, L., Mason, J. C., et al. (2008). NF-κB- suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in Pro-inflammatory signaling. J. Biol, Chem. 283, 7036–7045. doi: 10.1074/jbc.M708690200
Fiil, B. K., Damgaard, R. B., Wagner, S. A., Keusekotten, K., Fritsch, M., Bekker-Jensen, S., et al. (2013). OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 50, 818–830. doi: 10.1016/j.molcel.2013.06.004
Gerlach, B., Cordier, S. M., Schmukle, A. C., Emmerich, C. H., Rieser, E., Haas, T. L., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596. doi: 10.1038/nature09816
Gijbels, M. J., Zurcher, C., Kraal, G., Elliott, G. R., HogenEsch, H., Schijff, G., et al. (1996). Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am. J. Pathol. 148, 941–950.
Grell, M., Douni, E., Wajant, H., Lohden, M., Clauss, M., Maxeiner, B., et al. (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83, 793–802. doi: 10.1016/0092-8674(95)90192-2
Grievink, H. W., Heuberger, J., Huang, F., Chaudhary, R., Birkhoff, W. A. J., Tonn, G. R., et al. (2020). DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Cli. Pharmacol. Ther. 107, 406–414. doi: 10.1002/cpt.1615
Guma, M., Stepniak, D., Shaked, H., Spehlmann, M. E., Shenouda, S., Cheroutre, H., et al. (2011). Constitutive intestinal NF-κB- does not trigger destructive inflammation unless accompanied by MAPK activation. J. Exp. Med. 208, 1889–1900. doi: 10.1084/jem.20110242
Gunther, C., Martini, E., Wittkopf, N., Amann, K., Weigmann, B., Neumann, H., et al. (2011). Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339. doi: 10.1038/nature10400
Haas, T. L., Emmerich, C. H., Gerlach, B., Schmukle, A. C., Cordier, S. M., Rieser, E., et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844. doi: 10.1016/j.molcel.2009.10.013
Harris, P. A., Berger, S. B., Jeong, J. U., Nagilla, R., Bandyopadhyay, D., Campobasso, N., et al. (2017). Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261. doi: 10.1021/acs.jmedchem.6b01751
Harris, P. A., Marinis, J. M., Lich, J. D., Berger, S. B., Chirala, A., Cox, J. A., et al. (2019). Identification of a RIP1 kinase inhibitor clinical Candidate (GSK3145095) for the treatment of pancreatic cancer. Med. Chem. Lett. 10, 857–862. doi: 10.1021/acsmedchemlett.9b00108
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. doi: 10.1016/j.cell.2009.05.021
Heger, K., Wickliffe, K. E., Ndoja, A., Zhang, J., Murthy, A., Dugger, D. L., et al. (2018). OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124. doi: 10.1038/s41586-018-0256-2
Hildebrand, J. M., Tanzer, M. C., Lucet, I. S., Young, S. N., Spall, S. K., Sharma, P., et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 15072–15077. doi: 10.1073/pnas.1408987111
HogenEsch, H., Gijbels, M. J., Offerman, E., van Hooft, J., van Bekkum, D. W., and Zurcher, C. (1993). A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 143, 972–982.
HogenEsch, H., Janke, S., Boggess, D., and Sundberg, J. P. (1999). Absence of Peyer’s patches and abnormal lymphoid architecture in chronic proliferative dermatitis (cpdm/cpdm) mice. J. Immunol. 162, 3890–3896.
Ikeda, F., Deribe, Y. L., Skanland, S. S., Stieglitz, B., Grabbe, C., and Franz-Wachtel, M. (2011). SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB- activity and apoptosis. Nature 471, 637–641. doi: 10.1038/nature09814
Jensen, S., Seidelin, J. B., LaCasse, E. C., and Nielsen, O. H. (2020). SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci. Signal. 13:aax8295. doi: 10.1126/scisignal.aax8295
Kaiser, W. J., Daley-Bauer, L. P., Thapa, R. J., Mandal, P., Berger, S. B., Huang, C., et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U.S.A. 111, 7753–7758. doi: 10.1073/pnas.1401857111
Kaiser, W. J., Upton, J. W., Long, A. B., Livingston-Rosanoff, D., Daley-Bauer, L. P., Hakem, R., et al. (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. doi: 10.1038/nature09857
Kato, M., Sanada, M., Kato, I., Sato, Y., Takita, J., Takeuchi, K., et al. (2009). Frequent inactivation of A20 in B-cell lymphomas. Nature 459, 712–716.
Kelliher, M. A., Grimm, S., Ishida, Y., Kuo, F., Stanger, B. Z., and Leder, P. (1998). The death domain kinase RIP mediates the TNF-induced NF-κB- signal. Immunity 8, 297–303. doi: 10.1016/s1074-7613(00)80535-x
Keusekotten, K., Elliott, P. R., Glockner, L., Fiil, B. K., Damgaard, R. B., Kulathu, Y., et al. (2013). OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312–1326. doi: 10.1016/j.cell.2013.05.014
Khan, N., Lawlor, K. E., Murphy, J. M., and Vince, J. E. (2014). More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr. Opin. Immunol. 26C, 76–89. doi: 10.1016/j.coi.2013.10.017
Kirisako, T., Kamei, K., Murata, S., Kato, M., Fukumoto, H., Kanie, M., et al. (2006). A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo J. 25, 4877–4887. doi: 10.1038/sj.emboj.7601360
Kontoyiannis, D., Pasparakis, M., Pizarro, T. T., Cominelli, F., and Kollias, G. (1999). Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398. doi: 10.1016/s1074-7613(00)80038-2
Kovalenko, A., Chable-Bessia, C., Cantarella, G., Israel, A., Wallach, D., and Courtois, G. (2003). The tumour suppressor CYLD negatively regulates NF-κB- signalling by deubiquitination. Nature 424, 801–805. doi: 10.1038/nature01802
Kumari, S., Redouane, Y., Lopez-Mosqueda, J., Shiraishi, R., Romanowska, M., and Lutzmayer, S. (2014). Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 3:e03422. doi: 10.7554/eLife.03422
Kupka, S., De Miguel, D., Draber, P., Martino, L., Surinova, S., Rittinger, K., et al. (2016). SPATA2-Mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep. 16, 2271–2280. doi: 10.1016/j.celrep.2016.07.086
Lacey, D., Hickey, P., Arhatari, B. D., O’Reilly, L. A., Rohrbeck, L., Kiriazis, H., et al. (2015). Spontaneous retrotransposon insertion into TNF 3′UTR causes heart valve disease and chronic polyarthritis. Proc. Natl. Acad. Sci. U.S.A. 112, 9698–9703. doi: 10.1073/pnas.1508399112
Lalaoui, N., Boyden, S. E., Oda, H., Wood, G. M., Stone, D. L., Chau, D., et al. (2020). Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108. doi: 10.1038/s41586-019-1828-5
Laurien, L., Nagata, M., Schunke, H., Delanghe, T., Wiederstein, J. L., Kumari, S., et al. (2020). Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11:1747. doi: 10.1038/s41467-020-15466-8
Lee, E. G., Boone, D. L., Chai, S., Libby, S. L., Chien, M., Lodolce, J. P., et al. (2000). Failure to regulate TNF-induced NF-κB- and cell death responses in A20-deficient mice. Science 289, 2350–2354. doi: 10.1126/science.289.5488.2350
Liang, Y., Seymour, R. E., and Sundberg, J. P. (2011). Inhibition of NF-κB- signaling retards eosinophilic dermatitis in SHARPIN-deficient mice. J. Invest Dermatol. 131, 141–149. doi: 10.1038/jid.2010.259
Mahoney, D. J., Cheung, H. H., Mrad, R. L., Plenchette, S., Simard, C., Enwere, E., et al. (2008). Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-κB- activation. Proc. Natl. Acad. Sci. U.S.A. 105, 11778–11783. doi: 10.1073/pnas.0711122105
Majkut, J., Sgobba, M., Holohan, C., Crawford, N., Logan, A. E., Kerr, E., et al. (2014). Longley, differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat. Commun. 5:3350. doi: 10.1038/ncomms4350
Manthiram, K., Zhou, Q., Aksentijevich, I., and Kastner, D. L. (2017). The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 18, 832–842. doi: 10.1038/ni1117-1271a
Martens, A., Priem, D., Hoste, E., Vetters, J., Rennen, S., Catrysse, L., et al. (2020). Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat. Immunol. 21, 381–387. doi: 10.1038/s41590-020-0621-9
Martens, S., Hofmans, S., Declercq, W., Augustyns, K., and Vandenabeele, P. (2020). Inhibitors targeting RIPK1/RIPK3: Old and new drugs. Trends Pharmacol. Scie. 41, 209–224. doi: 10.1016/j.tips.2020.01.002
Matsuzawa-Ishimoto, Y., Shono, Y., Gomez, L. E., Hubbard-Lucey, V. M., Cammer, M., Neil, J., et al. (2017). Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 214, 3687–3705. doi: 10.1084/jem.20170558
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. doi: 10.1016/s0092-8674(03)00521-x
Miot, C., Imai, K., Imai, C., Mancini, A. J., Kucuk, Z. Y., Kawai, T., et al. (2017). Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood 130, 1456–1467. doi: 10.1182/blood-2017-03-771600
Moss, M. L., Jin, S. L., Milla, M. E., Bickett, D. M., Burkhart, W., and Carter, H. L. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385, 733–736. doi: 10.1038/385733a0
Moulin, M., Anderton, H., Voss, A. K., Thomas, T., Wong, W. W., Bankovacki, A., et al. (2012). IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 31, 1679–1691. doi: 10.1038/emboj.2012.18
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. doi: 10.1016/j.immuni.2013.06.018
Nabavi, M., Shahrooei, M., Rokni-Zadeh, H., Vrancken, J., Changi-Ashtiani, M., and Darabi, K. (2019). Auto-inflammation in a patient with a novel homozygous OTULIN mutation. J. Clin. Immunol. 39, 138–141. doi: 10.1007/s10875-019-00599-3
Nastase, M. V., Zeng-Brouwers, J., Frey, H., Hsieh, L. T., Poluzzi, C., and Beckmann, J. (2016). An Essential role for SHARPIN in the regulation of caspase 1 activity in sepsis. Am. J. Pathol. 186, 1206–1220. doi: 10.1016/j.ajpath.2015.12.026
Nenci, A., Becker, C., Wullaert, A., Gareus, R., van Loo, G., Danese, S., et al. (2007). Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561. doi: 10.1038/nature05698
Newton, K., Dugger, D. L., Maltzman, A., Greve, J. M., Hedehus, M., and Martin-McNulty, B. (2016). RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576. doi: 10.1038/cdd.2016.46
Newton, K., Dugger, D. L., Wickliffe, K. E., Kapoor, N., de Almagro, M. C., Vucic, D., et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. doi: 10.1126/science.1249361
Newton, K., Wickliffe, K. E., Dugger, D. L., Maltzman, A., Roose-Girma, M., Dohse, M., et al. (2019). Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431. doi: 10.1038/s41586-019-1548-x
Nilsson, J., Schoser, B., Laforet, P., Kalev, O., Lindberg, C., Romero, N. B., et al. (2013). Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann. Neurol 74, 914–919. doi: 10.1002/ana.23963
Oda, H., Beck, D. B., Kuehn, H. S., Sampaio Moura, N., Hoffmann, P., Ibarra, M., et al. (2019). Second Case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front. Immunol. 10:479. doi: 10.3389/fimmu.2019.00479
Oda, H., and Kastner, D. L. (2017). Genomics, biology, and human illness: advances in the monogenic autoinflammatory diseases. Rheum. Dis. Clin. North Am. 43, 327–345. doi: 10.1016/j.rdc.2017.04.011
Onizawa, M., Oshima, S., Schulze-Topphoff, U., Oses-Prieto, J. A., Lu, T., Tavares, R., et al. (2015). The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627. doi: 10.1038/ni0715-785c
Patel, S., Webster, J. D., Varfolomeev, E., Kwon, Y. C., Cheng, J. H., Zhang, J., et al. (2020). RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 27, 161–175. doi: 10.1038/s41418-019-0347-0
Peltzer, N., Darding, M., Montinaro, A., Draber, P., Draberova, H., Kupka, S., et al. (2018). LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117. doi: 10.1038/s41586-018-0064-8
Peltzer, N., Rieser, E., Taraborrelli, L., Draber, P., Darding, M., Pernaute, B., et al. (2014). AHOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165. doi: 10.1016/j.celrep.2014.08.066
Polykratis, A., Hermance, N., Zelic, M., Roderick, J., Kim, C., Van, T. M., et al. (2014). Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543. doi: 10.4049/jimmunol.1400590
Potter, C. S., Wang, Z., Silva, K. A., Kennedy, V. E., Stearns, T. M., Burzenski, L., et al. (2014). Chronic proliferative dermatitis in Sharpin null mice: development of an autoinflammatory disease in the absence of B and T lymphocytes and IL4/IL13 signaling. PLoS One 9:e85666. doi: 10.1371/journal.pone.0085666
Razani, B., Whang, M. I., Kim, F. S., Nakamura, M. C., Sun, X., and dvincula, R. A. (2020). Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis-like disease and inflammation. Nat. Immunol. 21, 422–433. doi: 10.1038/s41590-020-0634-4
Reddy, P., Slack, J. L., Davis, R., Cerretti, D. P., Kozlosky, C. J., Blanton, R. A., et al. (2000). Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 275, 14608–14614. doi: 10.1074/jbc.275.19.14608
Rickard, J. A., Anderton, H., Etemadi, N., Nachbur, U., Darding, M., Peltzer, N., et al. (2014). TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 3:e03464. doi: 10.7554/eLife.03464
Rodriguez, D. A., Weinlich, R., Brown, S., Guy, C., Fitzgerald, P., Dillon, C. P., et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88. doi: 10.1038/cdd.2015.70
Roulis, M., Armaka, M., Manoloukos, M., Apostolaki, M., and Kollias, G. (2011). Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl. Acad. Sci. US.A. 108, 5396–5401. doi: 10.1073/pnas.1007811108
Rubin, B. Y., Smith, L. J., Hellermann, G. R., Lunn, R. M., Richardson, N. K., and Anderson, S. L. (1988). Correlation between the anticellular and DNA fragmenting activities of tumor necrosis factor. Cancer Res. 48, 6006–6010.
Scheidereit, C. (2006). IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene 25, 6685–6705. doi: 10.1038/sj.onc.1209934
Schlicher, L., Wissler, M., Preiss, F., Brauns-Schubert, P., Jakob, C., Dumit, V., et al. (2016). SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB- signaling and cell death. EMBO Rep. 17, 1485–1497. doi: 10.15252/embr.201642592
Sessler, T., Healy, S., Samali, A., and Szegezdi, E. (2013). Structural determinants of DISC function: new insights into death receptor-mediated apoptosis signalling. Pharmacol. Ther. 140, 186–199. doi: 10.1016/j.pharmthera.2013.06.009
Seymour, R. E., Hasham, M. G., Cox, G. A., Shultz, L. D., Hogenesch, H., Roopenian, D. C., et al. (2007). Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 8, 416–421. doi: 10.1038/sj.gene.6364403
Shan, B., Pan, H., Najafov, A., and Yuan, J. (2018). Necroptosis in development and diseases. Genes Dev. 32, 327–340.
Shim, J. H., Xiao, C., Paschal, A. E., Bailey, S. T., Rao, P., Hayden, M. S., et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681. doi: 10.1101/gad.1360605
Silke, J., and Vucic, D. (2014). IAP family of cell death and signaling regulators. Methods Enzymol. 545, 35–65. doi: 10.1016/B978-0-12-801430-1.00002-0
Smahi, A., Courtois, G., Vabres, P., Yamaoka, S., Heuertz, S., Munnich, A., et al. (2000). Genomic rearrangement in NEMO impairs NF-κB- activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 405, 466–472. doi: 10.1038/35013114
Su, L., Quade, B., Wang, H., Sun, L., Wang, X., and Rizo, J. (2014). A plug release mechanism for membrane permeation by MLKL. Structure 22, 1489–1500. doi: 10.1016/j.str.2014.07.014
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sun, X., Yin, J., Starovasnik, M. A., Fairbrother, W. J., and Dixit, V. M. (2002). Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511. doi: 10.1074/jbc.M109488200
Tao, P., Sun, J., Wu, Z., Wang, S., Wang, J., Li, W., et al. (2020). A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114. doi: 10.1038/s41586-019-1830-y
Taraborrelli, L., Peltzer, N., Montinaro, A., Kupka, S., Rieser, E., Hartwig, T., et al. (2018). LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 9:3910. doi: 10.1038/s41467-018-06155-8
Targan, S. R., Hanauer, S. B., van Deventer, S. J., Mayer, L., Present, D. H., Braakman, T., et al. (1997). A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s disease cA2 study group. N. Engl. J. Med. 337, 1029–1035. doi: 10.1056/NEJM199710093371502
Taylor, P. C., and Feldmann, M. (2009). Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 578–582. doi: 10.1038/nrrheum.2009.181
Tokunaga, F., and Iwai, K. (2012). LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 14, 563–572. doi: 10.1016/j.micinf.2012.01.011
Tokunaga, F., Nakagawa, T., Nakahara, M., Saeki, Y., Taniguchi, M., Sakata, S., et al. (2011). SHARPIN is a component of the NF-κB–activating linear ubiquitin chain assembly complex. Nature 471, 633–636. doi: 10.1038/nature09815
Tokunaga, F., Nishimasu, H., Ishitani, R., Goto, E., Noguchi, T., Mio, K., et al. (2012). Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB- regulation. EMBO J. 31, 3856–3870. doi: 10.1038/emboj.2012.241
Tokunaga, F., Sakata, S., Saeki, Y., Satomi, Y., Kirisako, T., Kamei, K., et al. (2009). Involvement of linear polyubiquitylation of NEMO in NF-κB- activation. Nat. Cell Biol. 11, 123–132. doi: 10.1038/ncb1821
Trompouki, E., Hatzivassiliou, E., Tsichritzis, T., Farmer, H., Ashworth, A., and Mosialos, G. (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-κB- activation by TNFR family members. Nature 424, 793–796. doi: 10.1038/nature01803
Uchiyama, Y., Kim, C. A., Pastorino, A. C., Ceroni, J., Lima, P. P., de Barros Dorna, M., et al. (2019). Primary immunodeficiency with chronic enteropathy and developmental delay in a boy arising from a novel homozygous RIPK1 variant. J. Hum. Genet. 64, 955–960. doi: 10.1038/s10038-019-0631-3
Van Antwerp, D. J., Martin, S. J., Kafri, T., Green, D. R., and Verma, I. M. (1996). Suppression of TNF-alpha-induced apoptosis by NF-κB-. Science 274, 787–789. doi: 10.1126/science.274.5288.787
Varfolomeev, E., Goncharov, T., Fedorova, A. V., Dynek, J. N., Zobel, K., Deshayes, K., et al. (2008). c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299. doi: 10.1074/jbc.C800128200
Varfolomeev, E., and Vucic, D. (2016). Intracellular regulation of TNF activity in health and disease. Cytokine 101, 26–32. doi: 10.1016/j.cyto.2016.08.035
Varfolomeev, E. E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J. S., and Mett, I. L. (1998). Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276. doi: 10.1016/s1074-7613(00)80609-3
Vassalli, P. (1992). The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10, 411–452.
Verboom, L., Martens, A., Priem, D., Hoste, E., Sze, M., Vikkula, H., et al. (2020). OTULIN prevents liver inflammation and hepatocellular carcinoma by inhibiting FADD- and RIPK1 kinase-mediated hepatocyte apoptosis. Cell Rep. 30, 2237.e6–2247.e6. doi: 10.1016/j.celrep.2020.01.028
Vlantis, K., Wullaert, A., Polykratis, A., Kondylis, V., Dannappel, M., Schwarzer, R., et al. (2016). RIP kinase 1-Mediated epithelial cell death and chronic intestinal inflammation by NF-κB–dependent and -independent functions. Immunity 44, 553–567. doi: 10.1016/j.immuni.2016.02.020
Wagner, S. A., Satpathy, S., Beli, P., and Choudhary, C. (2016). SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 35, 1868–1884. doi: 10.15252/embj.201694300
Wallach, D., Varfolomeev, E. E., Malinin, N. L., Goltsev, Y. V., Kovalenko, A. V., and Boldin, M. P. (1999). Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev, Immunol. 17, 331–367. doi: 10.1146/annurev.immunol.17.1.331
Wang, H., Sun, L., Su, L., Rizo, J., Liu, L., Wang, L. F., et al. (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. doi: 10.1016/j.molcel.2014.03.003
Wang, K., Kim, C., Bradfield, J., Guo, Y., Toskala, E., Otieno, F. G., et al. (2013). Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med., 5:67. doi: 10.1186/gm471
Wang, L., Du, F., and Wang, X. (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703. doi: 10.1016/j.cell.2008.03.036
Webster, J. D., Kwon, Y. C., Park, S., Zhang, H., Corr, N., Ljumanovic, N., et al. (2020). RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J. Leukoc Biol. doi: 10.1002/JLB.3MA1219-398R [Epub ahead of print].
Wei, R., Xu, L. W., Liu, J., Li, Y., Zhang, P., Shan, B., et al. (2017). SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev. 31, 1162–1176. doi: 10.1101/gad.321075.118
Weisel, K., Scott, N. E., Tompson, D. J., Votta, B. J., Madhavan, S., Povey, K., et al. (2017). Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 5:e00365. doi: 10.1002/prp2.365
Wertz, I. E., O’Rourke, K. M., Zhou, H., Eby, M., Aravind, L., Seshagiri, S., et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694–699. doi: 10.1038/nature02794
Williams, R. O., Feldmann, M., and Maini, R. N. (1992). Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. U.S.A. 89, 9784–9788. doi: 10.1073/pnas.89.20.9784
Wong, J., Garcia-Carbonell, R., Zelic, M., Ho, S. B., Boland, B. S., Yao, S. J., et al. (2020). RIPK1 mediates TNF-induced intestinal crypt apoptosis during chronic NF-κB- activation. Cell Mol. Gastroenterol. Hepatol. 9, 295–312. doi: 10.1016/j.jcmgh.2019.10.002
Wu, X. N., Yang, Z. H., Wang, X. K., Zhang, Y., Wan, H., Song, Y., et al. (2014). Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720. doi: 10.1038/cdd.2014.77
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33. doi: 10.1038/s41583-018-0093-1
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi: 10.1126/science.1172308
Zhang, J., Webster, J. D., Dugger, D. L., Goncharov, T., Roose-Girma, M., Hung, J., et al. (2019). Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep. 27, 2679.e3–2689.e3. doi: 10.1016/j.celrep.2019.04.111
Zhang, X., Dowling, J. P., and Zhang, J. (2019). RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 10:245. doi: 10.1038/s41419-019-1490-8
Zhou, Q., Wang, H., Schwartz, D. M., Stoffels, M., Park, Y. H., Zhang, Y., et al. (2016a). Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 48, 67–73. doi: 10.1038/ng.3459
Zhou, Q., Yu, X., Demirkaya, E., Deuitch, N., Stone, D., Tsai, W. L., et al. (2016b). Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. US.A. 113, 10127–10132. doi: 10.1073/pnas.1612594113
Zonana, J., Elder, M. E., Schneider, L. C., Orlow, S. J., Moss, C., Golabi, M., et al. (2000). A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am. J. Hum. Genet. 67, 1555–1562. doi: 10.1086/316914
Keywords: TNF, RIP1 (RIPK1), RIP3 kinase, NEMO, necroptois, apoptosis, RIPK1 inhibitors
Citation: Webster JD and Vucic D (2020) The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 8:365. doi: 10.3389/fcell.2020.00365
Received: 18 March 2020; Accepted: 23 April 2020;
Published: 21 May 2020.
Edited by:
Cristian Roberto Smulski, Bariloche Atomic Centre (CNEA), ArgentinaReviewed by:
Fuminori Tokunaga, Osaka City University, JapanCopyright © 2020 Webster and Vucic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua D. Webster, d2Vic3Rlci5qb3NodWFAZ2VuZS5jb20=; Domagoj Vucic, ZG9tYWdvakBnZW5lLmNvbQ==; dnVjaWMuZG9tYWdvakBnZW5lLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.