 Hecun Zou
Hecun Zou Lan-Xiang Wu
Lan-Xiang Wu Lihong Tan2,3
Lihong Tan2,3 Fei-Fei Shang
Fei-Fei Shang Hong-Hao Zhou
Hong-Hao Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 25 May 2020
Sec. Molecular and Cellular Oncology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00347
This article is part of the Research TopicThe Role of Epigenetic Modifications in Cancer ProgressionView all 18 articles
Single-nucleotide variants (SNVs) are the most common genetic variants and universally present in the human genome. Genome-wide association studies (GWASs) have identified a great number of disease or trait-associated variants, many of which are located in non-coding regions. Long intergenic non-protein coding RNAs (lincRNAs) are the major subtype of long non-coding RNAs; lincRNAs play crucial roles in various disorders and cellular models via multiple mechanisms. With rapid growth in the number of the identified lincRNAs and genetic variants, there is great demand for an investigation of SNVs in lincRNAs. Hence, in this article, we mainly summarize the significant role of SNVs within human lincRNA regions. Some pivotal variants may serve as risk factors for the development of various disorders, especially cancer. They may also act as important regulatory signatures involved in the modulation of lincRNAs in a tissue- or disorder-specific manner. An increasing number of researches indicate that lincRNA variants would potentially provide additional options for genetic testing and disease risk assessment in the personalized medicine era.
Single-nucleotide variant (SNV), also known as single-nucleotide polymorphism (SNP), is the variant of a single nucleotide that occurs at a specific genomic position. It is the most common type of genetic variants, which has long been confirmed in various loci of the genome (Human Genome Structural Variation Working Group, Eichler et al., 2007). In the past few decades, genetic variants have been typically used to dissect complex human disorders through research on candidate genes, particularly genome-wide association study (GWAS), an observational study of the genome-wide set of genetic variants in different individuals, which is performed to identify whether any variant is associated with the phenotypes. As a representative of a large-scale variant analysis, it has provided an approach to identifying potential genetic variant loci associated with heterogeneous disorders, including cancer susceptibility (Freedman et al., 2011). With the development of emerging technologies, such as microarray-based genotyping and high-throughput next-generation sequencing, it offers a novel avenue for the clinical application of genetic variants (GTEx Consortium et al., 2017). As might be expected, the role of genetic variants in understanding the pathogenesis of diseases, therapeutic response, and even ultimately personalized medicine will be indispensable in the near future. Based on the implementation of the International HapMap Project and the 1000 Genomes Project, great breakthroughs have been achieved in the research field of genetic variants, particularly focusing on some variants of protein-coding genes (Human Genome Structural Variation Working Group, Eichler et al., 2007; Genomes Project et al., 2015). However, genetic variants, especially SNVs, not only occur to protein-coding sequences, but many of them also fall within non-coding regions or the intergenic regions between two genes. For instance, a considerable genetic component has been confirmed to be involved in the susceptibility of various cancers; the genomic contexts of cancer-associated SNVs (SNPs) have been analyzed within a comprehensive GWAS catalog. Of these risk variants, less than 10% are mapped in protein-coding regions, whereas most of them are located in the intronic or intergenic regions (Figure 1A), it brings forward the issue of these non-coding loci and their importance role in cancer research (Hindorff et al., 2009).
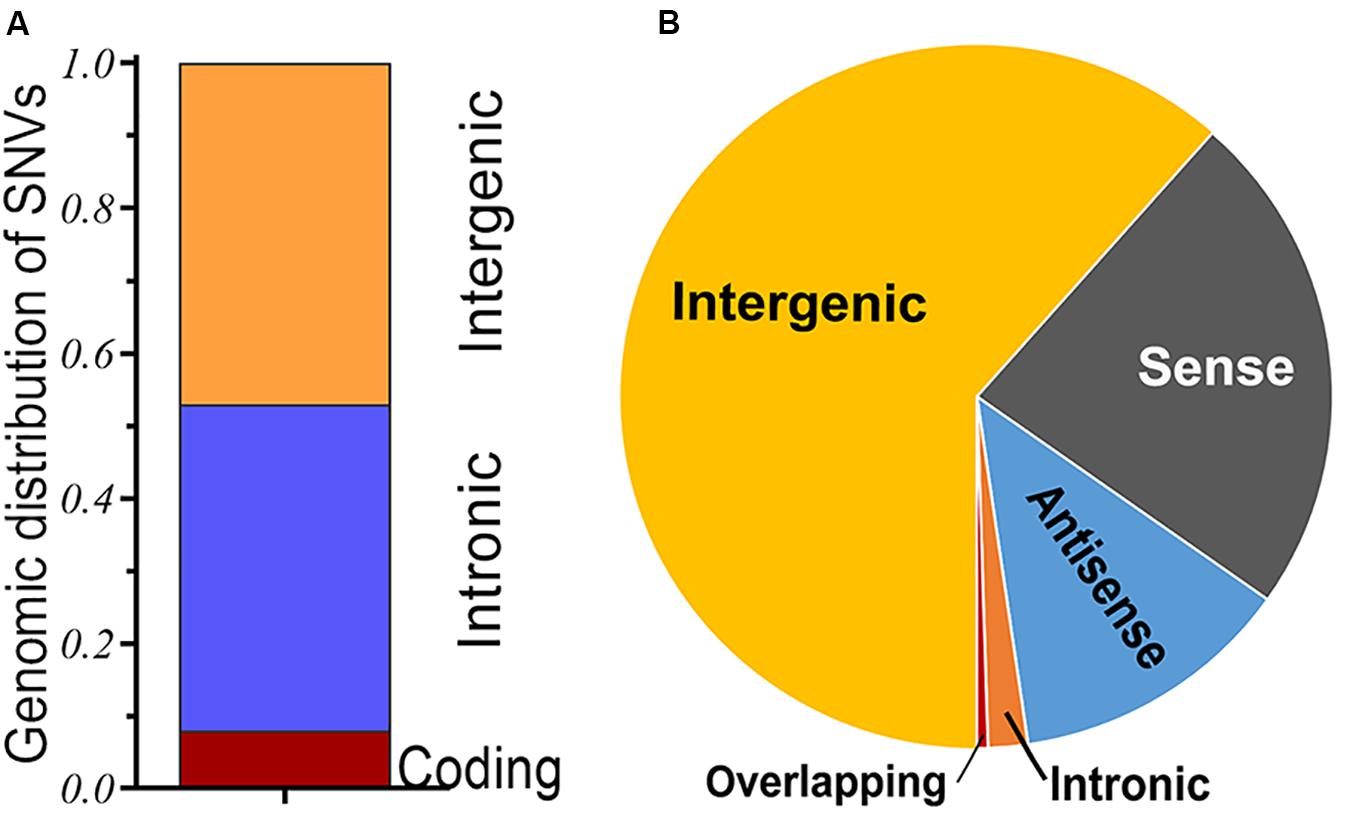
Figure 1. (A) Genomic distribution of single-nucleotide variants in cancers; a majority of cancer-associated single-nucleotide variants (SNVs) [single-nucleotide polymorphisms (SNPs)] are found in the intergenic or intronic regions, and only small numbers are located in protein-coding regions of the human genome. (B) Classification of long non-coding RNA (lncRNA) transcripts; long intergenic non-protein coding RNA (lincRNA) is a major subtype of lncRNA.
The Human Genome Project (HGP) has determined the whole sequence of nucleotide base pairs that compose the human genome and initially provided approximately 20,000 proteins that could serve as therapeutic targets (Venter et al., 2001). Subsequent large-scale annotation efforts, such as the Encyclopedia of DNA Elements (ENCODE) project, surprisingly, have identified hundreds of thousands of non-coding RNAs, which were previously regarded as “junk DNA” (The Encode Project Consortium, 2012). Among them, a great quantity of long non-coding RNAs (lncRNAs) are transcribed in mammalian genomes. Based on their locations and characteristics, lncRNAs can be placed into five broad categories: (1) intergenic, (2) antisense, (3) sense, (4) intronic, and (5) overlapping (Ponting et al., 2009; Derrien et al., 2012). Thereinto, long/large intergenic non-protein coding RNAs (lincRNAs), which are located within the genomic interval between two coding genes, are the major subtypes of lncRNAs accounting for approximately 63% (Figure 1B). Compared with other lncRNAs, molecules for which we know next to nothing about, lincRNAs are generally unexplored and have yet to be elucidated. About half of these lincRNAs are transcribed from the vicinity (<10 kb) of protein-coding loci and more likely to be involved in cis-regulatory of the expression level of adjacent genes; other transcripts that are well away from an adjacent gene seem to have little chance of cis-regulatory within the nearby region. Although they rarely form triplexes within double-stranded DNA owing to their poor complementarity to sequences elsewhere within the genome, these lincRNAs often act as trans-regulatory players within some ribonucleoprotein complexes (Ulitsky and Bartel, 2013). LincRNAs may have crucial roles in various disorders and cellular models via multiple mechanisms. Alterations in the levels of lincRNA expression have been linked to the occurrence of various disorders, such as cancers; they may act as tumor suppressors or proto-oncogenes (Huarte, 2015). Currently, advances in high-throughput RNA sequencing and computing approaches allow for an unparalleled analysis of transcriptomes. Of the diverse kinds of RNA transcripts, lincRNAs are attractive as they can be found out from the existing RNA-seq datasets through available bioinformatics methods (Cabili et al., 2011).
According to recent reports from the ENCODE project, thousands and thousands of variant loci are present in the non-coding regions of the human genome, and total number continues to increase (Schaub et al., 2012). Generally, genetic variants, such as SNVs, which occur to the non-coding loci, are more frequently than in conservative protein-coding genes regions. A large number of GWAS-identified SNVs loci reside in the regions that encode lincRNAs, indicating that these variants of lincRNAs may play a crucial role in the susceptibility of diseases. More than three quarters of disease-associated genetic variants are remarkably overlapped in promoter or enhancer regions, suggesting that SNVs may serve as an important player in the regulation of transcript levels (Hindorff et al., 2009). Therefore, identification of such variant loci and elucidation of their biological functions would be of profound significance in understanding the etiology of disorders and in promoting novel approaches for the diagnosis, prevention, and treatment of disorder.
As a matter of fact, the occurrence of complex diseases (e.g., cancer) is related to multiple factors, including genetic, environmental, and lifestyle. Among them, genetic factors are of particular interest, just as GWASs and next-generation sequencing studies have greatly broadened the understanding of genetic variants that confer risk of diseases. Numerous genetic variants in lincRNA regions have been determined to be associated with the susceptibility of heterogeneous diseases, especially multiple types of cancer. Herein, we reviewed some lincRNAs that encompass disease or trait-associated variants (Tables 1, 2).
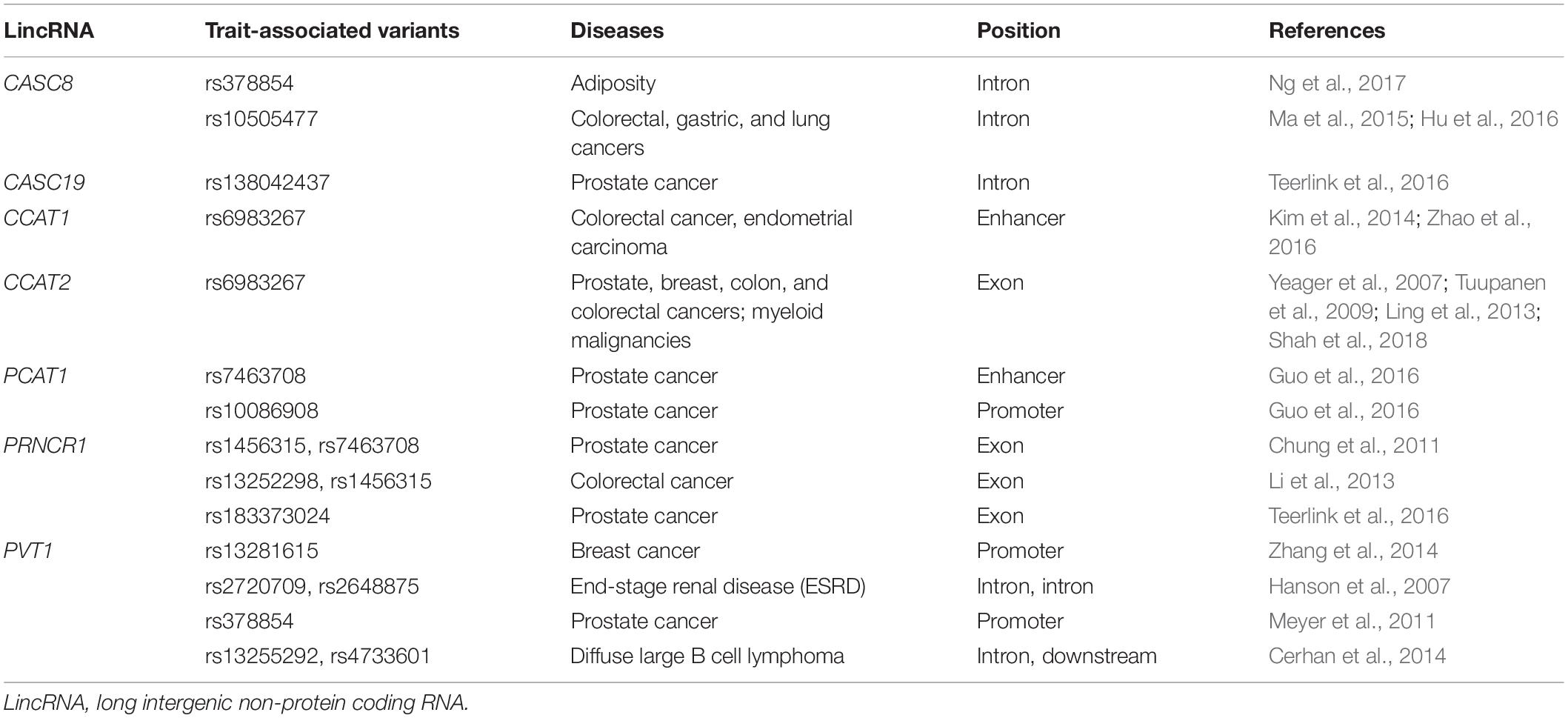
Table 1. Overviews of trait-associated variants on the chr8q24 locus.
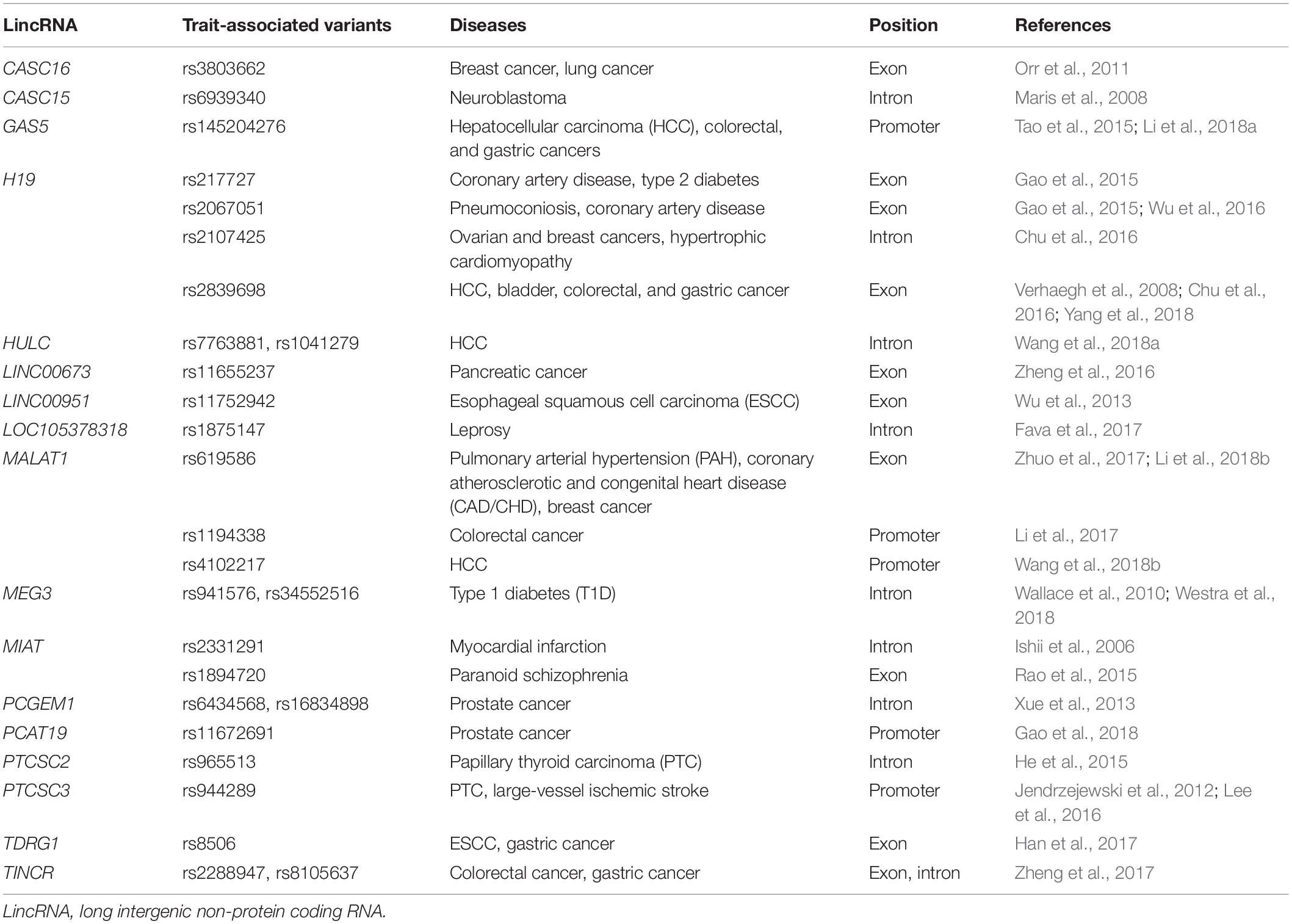
Table 2. Overviews of other lincRNAs encompassing trait-associated variants.
Genome-wide association studies have pointed to the chr8q24 genomic locus as a hotspot for cancer-associated variants owing to the large density, more strength, and high allele frequency of these variants (Yeager et al., 2007; Tuupanen et al., 2009). Even though chromosome 8q24 has been considered as a “gene desert” region owing to the absence of functionally annotated genes, with the only notable exception of the frequently amplified MYC (a proto-oncogene involved in tumorigenesis) (Chung et al., 2011). Surprisingly, large-scale studies have revealed that several lincRNAs are transcribed from the chr8q24 locus, such as CCAT1 (Kim et al., 2014), CCAT2 (Ling et al., 2013), PVT1 (Hanson et al., 2007), PCAT1 (Guo et al., 2016), and PRNCR1 (Li et al., 2013); all of these encompass multiple cancer-associated variants. For instance, lincRNA CCAT2 (Colon Cancer-Associated Transcript 2, also termed LINC00873), a transcript spanning SNV rs6983267, is associated with an increased risk for prostate, breast, colon, and colorectal cancers (Yeager et al., 2007; Tuupanen et al., 2009; Ling et al., 2013). CCAT2 is overexpressed in various types of cancers and may contribute to tumor growth, metastasis, and chromosomal instability by increasing MYC expression (Ling et al., 2013). LincRNA PRNCR1 has been reported to be involved in prostate carcinogenesis and may play an oncogene role via modulating the androgen receptor (Chung et al., 2011), PRNCR1 variants, especially rs1456315, are associated with the susceptibility of prostate and colorectal cancers (Li et al., 2013; Teerlink et al., 2016). Through an integrative analysis of the lncRNA transcriptome and GWAS data, Guo et al. (2016) have identified a prostate cancer-associated transcript PCAT1 and 10 risk loci on the chr8q24.21, including PCAT1 variants rs10086908 and rs7463708, which are significantly associated with prostate cancer susceptibility. As for PVT1 (also termed LINC00079), a GWAS analysis has identified that its variants rs13255292 and rs4733601 are associated with the susceptibility of diffuse large B cell lymphoma (Cerhan et al., 2014). Other independent SNVs (e.g., rs2720709 and rs2648875), which are mapped on PVT1, especially contributes to the development of end-stage renal disease (ESRD) in patients with type 2 diabetes (Hanson et al., 2007). A recent meta-analysis has summarized the relationship between two common variants (rs10505477 and rs7837328) in the intronic region of CASC8 (LINC00860) at 8q24 locus with the risk of cancers (Cui et al., 2018), including colorectal, gastric, and lung cancers (Ma et al., 2015; Hu et al., 2016). Another intronic loci rs378854 is related to adiposity in the individuals of African ancestry (Ng et al., 2017).
The H19 (also termed LINC00008) is located in chromosome 11p15.5, a paternally imprinted onco-fetal gene, which is typically down-regulated in adult tissues but can be overexpressed in multiple types of solid cancer. LincRNA H19 expression is closely related to tumor growth, metastasis, recurrence, and clinical prognosis (Ge et al., 2018). H19 variants are involved in the susceptibility of multiple diseases. A meta-analysis study has indicated that variant T allele of rs2107425 is correlated with a decreased risk of developing cancers (e.g., breast, ovarian, lung, and bladder cancers) (Chu et al., 2016; Wu et al., 2017), whereas variant rs2839698 is associated with an increased risk of digestive cancers (colorectal and gastric cancers) via up-regulating H19 expression; of note, there is no significant association observed between rs217727 variant and cancers susceptibility (Chu et al., 2016). However, in other reports, H19 rs217727 has been linked to the risk of hepatocellular carcinoma (HCC) (Ge et al., 2018), oral squamous cell carcinoma (OSCC), and bladder cancer in the Chinese population (Guo Q. Y. et al., 2017). For coronary artery disease (CAD), the T variant of rs217727 is associated with an increased risk, whereas rs2067051 A variant is linked to a decreased risk (Gao et al., 2015). H19 rs217727, but not rs2107425 variant, is associated with susceptibility of women with preeclampsia (PE) (Harati-Sadegh et al., 2018). Additionally, maternally transmitted fetal H19 variants (e.g., rs217727, rs2071094, and rs10732516), along with paternal IGF2 variants, are independently correlated with the placental DNA methylation levels (Marjonen et al., 2018) and birth weight of newborns (Petry et al., 2011).
LincRNA MALAT1 (metastasis-associated lung adenocarcinoma transcript 1, also termed LINC00047) has rs619586 A > G variant, which is significantly associated with the susceptibility of pulmonary arterial hypertension (PAH), and the carriers with variant G genotypes have a decreased PAH risk (Zhuo et al., 2017). Recent study has suggested that rs619586 AG/GG genotypes could reduce the risks of coronary atherosclerotic heart disease and congenital heart disease (CHD) by regulating MALAT1 expression (Li et al., 2018b). Another report has showed that MALAT1 is overexpressed in colorectal cancers and that SNV rs1194338 mapping to its promoter region is significantly associated with a decreased risk of colorectal cancer (Li et al., 2017). Moreover, the large-scale case–control association studies have identified a novel myocardial infarction-associated transcript, MIAT (also termed LINC00066), which encompasses rs2331291, and other variants confer the susceptibility of myocardial infarction (Ishii et al., 2006). As a component of the nuclear matrix, MIAT is mainly expressed in neurons, Rao et al. (2015) have reported that SNV rs1894720 is correlated with paranoid schizophrenia susceptibility, and MIAT may contribute to the pathogenesis of schizophrenia.
In addition to the above lincRNA molecules, recent studies have identified many other cancer-associated variants within lincRNA regions. For example, the tissue differentiation-inducing non-protein coding RNA (TINCR), also termed LINC00036, is essential for somatic tissue differentiation and tumor progression (Kretz et al., 2013). It has been demonstrated that two variants of TINCR (rs2288947 and rs8105637) are significantly correlated with the susceptibility and lymph node metastasis of colorectal cancer (Zheng et al., 2017); the lincRNA TINCR rs2288947 G allele and rs8113645 A allele genotypes could reduce the risk of gastric cancer. HULC, an HCC up-regulated lncRNA, also termed LINC00078, and its variants (rs7763881 and rs1041279) are linked to the susceptibility of HCC (Wang et al., 2018a). In thyroid carcinoma, several papillary thyroid carcinoma susceptibility candidates, such as PTCSC2, contain a risk-variant rs965513, and PTCSC3 encompasses rs944289; two lincRNA expression levels are strongly down-regulated in thyroid carcinoma tissues (Jendrzejewski et al., 2012; He et al., 2015). Additionally, GWAS analyses have identified five tag-SNVs, including rs944289 located in PTCSC3, are associated with large-vessel ischemic stroke (Lee et al., 2016). Xue et al. (2013) have reported that a prostate cancer gene expression marker, PCGEM1 (LINC00071), containing two risk-SNVs (rs6434568 C and rs16834898 A alleles) that are associated with a decreased risk of prostate cancer. Another prostate cancer risk-associated allele rs75823044 mapping to promoter of LINC00676 is almost exclusively found in African ancestry populations (Conti et al., 2017). In a GWAS analysis, five common variants including rs3803662 on the exon of CASC16 (LINC00918) have been identified to contribute to the susceptibility of lung and breast cancers (Orr et al., 2011). Furthermore, the colorectal cancer risk-SNV rs11776042 is located in the promoter of LNC00964, in which lincRNA is significantly decreased in colorectal cancer tissues (Chu et al., 2015). For tumor suppressor lncRNA GAS5, an insertion/deletion variant of rs145204276 is associated with the susceptibility of HCC (Tao et al., 2015) and colorectal and gastric cancers (Li et al., 2018a).
Except for cancer susceptibility, some lincRNA variants are found to be associated with the risk of other heterogeneous diseases. GWAS and expression quantitative trait locus (eQTL) analyses have identified a risk factor for pathological inflammatory responses of leprosy, SNV rs1875147, which is an eQTL variant for lincRNA LOC105378318 located in chromosome 10p21.2 (Fava et al., 2017). Rautanen et al. have found a variant rs140817150 in the intron of LOC107986770, which may be correlated with bacteremia susceptibility in African children (Kenyan Bacteraemia Study Group et al., 2016). A systematic analysis highlights some variant loci in lncRNA regions linked to cardiometabolic disorders; one of them, lincRNA LOC157273 harboring rs4841132, is linked to the regulation of serum lipid cholesterol (Ghanbari et al., 2018). Shyn et al.’s (2011) GWAS analysis has identified a major depressive disorder (MDD) risk-associated variant rs12526133, which resides in exon of LINC01108, in which lincRNA is overexpressed in patients with MDD. Moreover, the maternally expressed imprinted gene, MEG3 (also termed LINC00023), containing variants rs941576 (Wallace et al., 2010) and rs34552516 (Westra et al., 2018), which is found to be associated with susceptibility of type 1 diabetes. Nikpay et al.’s (2015) comprehensive GWAS meta-analyses have reported an association of CAD susceptibility with several SNVs, such as rs1870634, which is located in the downstream of LINC00841, and its GG genotype is strongly linked to CAD risk and has a higher frequency in CAD patients.
In addition to disease susceptibility, trait-associated SNVs are widely used for the indication of clinicopathological characteristics, prognosis, and treatment response (Gong et al., 2017). For example, with regard to a neuroblastoma-associated variant rs6939340, which is mapped on the intronic locus of lincRNA CASC15 and NBAT1, neuroblastoma individuals with the risk alleles are more likely to have clinical aggressive presentation, including metastatic disease, tumor with MYCN amplification, and disease relapse (Maris et al., 2008). Two independent cohort studies have observed that risk-SNV rs2608053 of PVT1 is correlated to the survival outcome of patients with classical Hodgkin lymphoma (Ghesquieres et al., 2018). For multiple sclerosis, several risk loci of PVT1 may contribute to the prediction of an optimal response to treatment with glatiramer acetate (Kulakova et al., 2017). LincRNA H19 variants have been found to increase the risk of ischemic strokes, and the up-regulated H19 may induce cerebral ischemia reperfusion injury by activating autophagy (Wang et al., 2017). Recent studies have reported that H19 rs2839698 variant may serve as an indicator for the increased risk and poor prognosis of HCC (Yang et al., 2018). Among individuals with coal workers’ pneumoconiosis (CWP), carriers of H19 rs2067051 CT/TT genotypes are associated with a decreased risk; H19 rs2067051 may be a possible biomarker for CWP prevention (Wu et al., 2016). A case–control study has shown that lincRNA MALAT1 variant rs4102217 is related to increased HCC risks; this SNV may be a potential predictor for the risk and prognosis of patients with HCC (Wang et al., 2018b). Another MALAT1 rs3200401 T allele has been found to confer better survival for patients with advanced lung adenocarcinoma (Wang et al., 2017). Furthermore, TDRG1 (testis development related 1, also termed LINC00532) is overexpressed in esophageal squamous cell carcinoma (ESCC) tissues; the AA genotype of variant rs8506 is linked to an increased risk of ESCC; this risk allele may regulate TDRG1 expression by disrupting the sponge binding of miR-526b; high TDRG1 expression and rs8506 A allele variant may contribute to the advanced tumor–node–metastasis stage and poor survival for ESCC patients (Han et al., 2017). Recent GWAS analyses have demonstrated that variant rs11672691 of PCAT19 (LINC01190) on 19q13 is positively related to aggressive prostate cancer. Further cohort studies have confirmed the association of rs11672691 with clinical characteristics of aggressive disease, including high tumor stage, prostate-specific antigen (PSA) progression, and development of castration-resistant prostate cancer (CRPC). The risk GG genotype of rs11672691 is also associated with a poor prognosis for patients with prostate cancer (Gao et al., 2018). These results highlight the clinical potential of trait-associated SNV, which may serve as risk stratification markers for the management of cancer patients.
Recent GWAS analyses have identified two common SNVs (rs4476990 and rs3802201), in which mapping to MIR2052HG may affect the recurrence risk of breast cancer patients treated with aromatase inhibitors. Expressions of MIR2052HG and estrogen receptor α (ERα, encoded by ESR1 gene) are induced by aromatase inhibitors and estrogen in a variant-dependent manner. MIR2052HG could sustain the levels of ERα via promoting AKT/FOXO3-mediated ESR1 transcription and limiting the ubiquitin-mediated ERα degradation. Its risk variant genotypes could enhance ERα binding to estrogen response elements and result in an alteration of response to aromatase inhibitors treatment for cancer patients (Ingle et al., 2016). In the evaluation of adverse reaction for lung cancer patients receiving platinum-based chemotherapy, the variants CASC8 rs10505477 (Hu et al., 2016) and ANRIL rs1333049 are correlated with overall toxicity, especially severe hematologic and gastrointestinal toxicity; lincRNA MEG3 rs116907618 is correlated with severe gastrointestinal toxicity; these variants may be considered as biomarkers for the evaluation of platinum-based treatment (Gong et al., 2017). Moreover, the rs10505477 GG genotype of CASC8 is also associated with tumor size, lymph node metastasis, and tumor–node–metastasis stage and may contribute to the survival for gastric cancers patients (Ma et al., 2015). In nasopharyngeal carcinoma (NPC), lncRNA GAS5 variant rs2067079 is associated with an increased risk of severe myelosuppression and neutropenia, whereas rs6790 may decrease the incidence rate of toxic reactions induced by chemo-radiotherapy in NPC patients (Guo Z. et al., 2017). Functional genomic studies have revealed that GAS5 promoter encompassing SNV rs55829688 (T > C), which up-regulates GAS5 expression via interacting with transcription factor TP63, may aggravate myelosuppression and result in a poor prognosis for patients with acute myeloid leukemia (AML) (Yan et al., 2017). Additionally, GWAS analyses have identified that some genetic variants are correlated with the pharmacokinetics of psychotropic drugs, such as variant rs16935279 located in an intron of LINC01592; its C allele carriers have a lower metabolism rate for anti-epileptic drugs (Athanasiu et al., 2015).
Genome-wide association studies have identified a lot of trait-associated variants, most of which reside in non-coding regions of the human genome. However, the specific functional mechanism of genetic variants still remains confused, which is one of the major challenges for post-GWAS research (Schaub et al., 2012). The regulatory elements are mainly located within regions of non-coding DNA and play critical roles in the transcription of target genes. Emerging studies have showed that these regulatory elements can affect the expression of lincRNAs and other related genes via long-range chromatin interactions in a cell-type- or tissue-specific manner. Many genetic variants reside in the regulatory element regions of lincRNAs and may disrupt the interaction of transcription factors with a region containing SNVs (Figures 2A,B). The mapping of SNVs to lincRNA regulatory regions (especially promoters and enhancers) may indicate a potential impact of these variants on the transcription of target genes (GTEx Consortium et al., 2017).
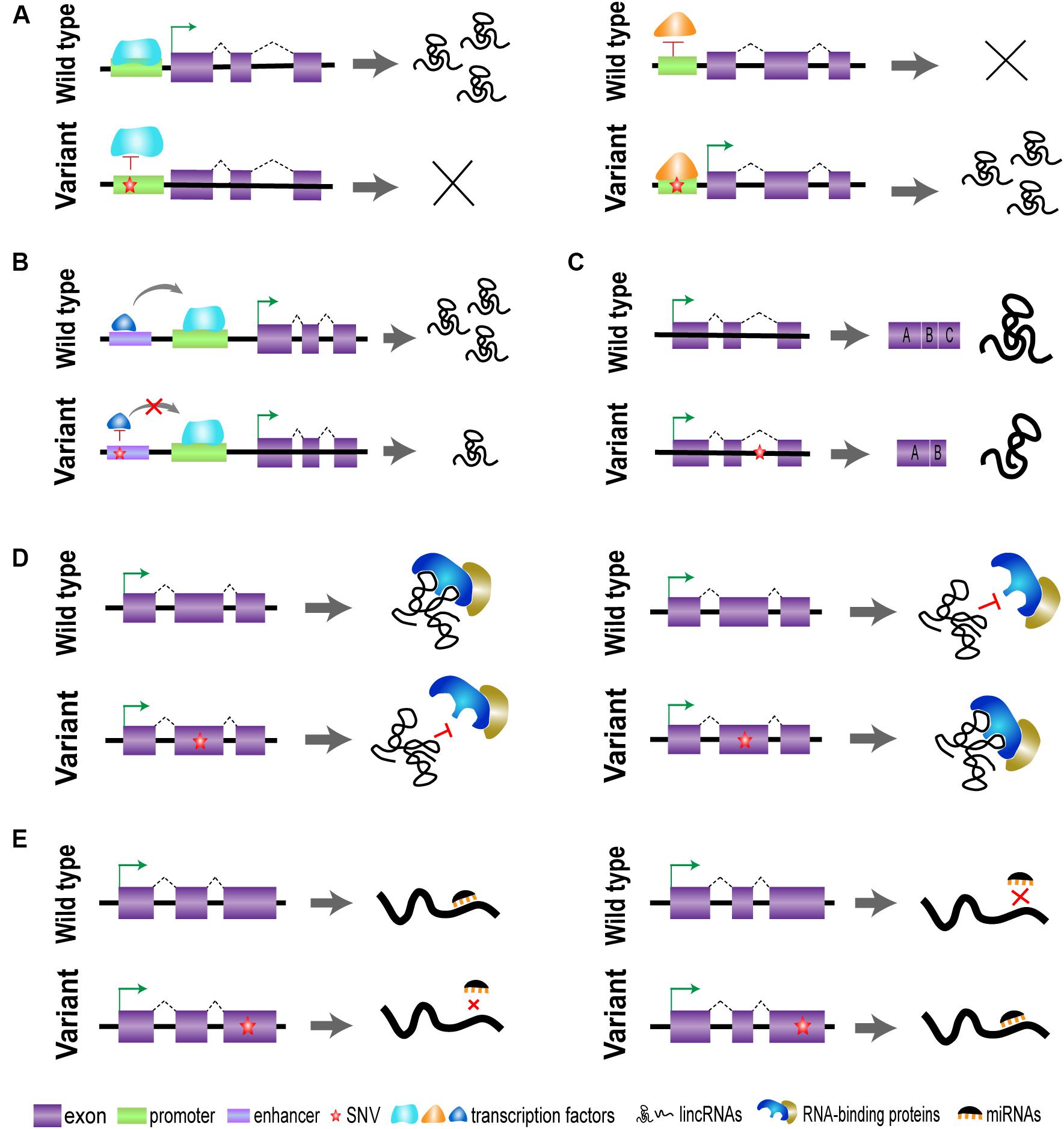
Figure 2. Graphical representations of the driving effect of variants [single-nucleotide variants (SNVs)] on long intergenic non-protein coding RNA (lincRNA) regions. (A) Genetic variants located in promoter may affect the binding of transcription factors and regulate the transcription of lincRNAs. (B) Genetic variants on enhancer affect the binding of transcription co-regulators and regulate lincRNA expression. (C) Genetic variants on intron may affect the process of splicing and stability of lincRNA conformation. (D) Genetic variants located in exons affect the lincRNA secondary structure, lincRNA stability, and interactive function. (E) Genetic variants on exons may affect the sponging of microRNAs (miRNAs).
Many genetic variants are located in the upstream of MYC, a gene desert on 8q24, which is related to the susceptibility of multiple cancers. Some observations, such as chromosome conformation capture (3C) assays, histone acetylation, and methylation marks analyses, have demonstrated that these regulatory regions containing SNVs may serve as enhancers for MYC gene in a tissue-specific manner. Functional investigations suggest that lincRNA CCAT2 augments the binding of transcription factor (TCF7L2 or TCF4) to MYC promoter region, activates WNT signaling, and increases the expression of target genes, especially the MYC proto-oncogene (Pomerantz et al., 2009; Ling et al., 2013). Although there is a disputable association between variant rs6983267 and MYC expression (Tuupanen et al., 2009), its risk G alleles produce more CCAT2 transcripts, which are exclusively retained in the nucleus. Interestingly, a risk-SNV rs6983267 also contributes to increased expression of CCAT1 (Zhao et al., 2016); an adjacent lincRNA of CCAT2, through affecting the long-range chromosomal interaction of MYC enhancer or CCAT1 promoter, then results in a cell-cycle regulation and tumor development (Kim et al., 2014). Guo et al. (2016) have reported that a prostate cancer risk-associated T allele of rs7463708 at lincRNA PCAT1 exhibited enhancer activity, through modulating the binding of novel transcription factor ONECUT2 with a distal enhancer that loops to the PCAT1 promoter; this process increases PCAT1 expression upon prolonged androgen treatment and promotes prostate transformation. Moreover, another prostate cancer risk-SNV rs378854 G alleles are also found to increase the expression of PVT1 oncogene by regulating an interaction of transcription factor YY1 with the promoters of PVT1 or MYC genes (Meyer et al., 2011). Similarly, the GG genotypes of rs13281615 increase PVT1 transcription and promote cell proliferation in breast cancer (Zhang et al., 2014). Overexpression of PVT1 may contribute to high levels of MYC mRNA and protein, along with an increased copy number, eventually leading to tumorigenesis (Zou et al., 2017). These results demonstrate the association of genetic variants with lincRNA transcription, although further studies are needed to reveal the relationship of these SNVs and lincRNAs on chromosome 8q24 locus.
Some SNVs reside in gene promoter regions and may influence the transcriptional expression of their target genes. Through an eQTL analysis of candidate genes and genetic variants in different tissues, an endometriosis risk-SNV rs3820282 is found to down-regulate LINC00339 expression by affecting the activity of LINC00339 promoter (Powell et al., 2016). Tao et al. (2015) have reported that an indel variant rs145204276 in the promoter region of GAS5 contributes to the up-regulation of GAS5 via affecting the methylation status of GAS5 promoter and regulating its transcriptional activity, thereby bringing its proto-oncogene role into play. Furthermore, the variant rs944289 of PTCSC3 is reported to reside in a binding site for CCAAT/enhancer binding proteins (C/EBPα/β); this variant may affect the activity of PTCSC3 promoter and down-regulate its transcript, then resulting in an abnormal expression of downstream genes and the progression of papillary thyroid carcinoma (Jendrzejewski et al., 2012).
Notably, a gene promoter region is likely to overlap with another super-enhancer locus, suggesting it that may have enhancer-like roles. In these interactions, lincRNA loci may serve as both target genes of its SNVs and the distal regulatory elements of other related genes. Integrative functional genomic and epigenomic analyses have identified that osteoporosis risk-associated SNV rs6426749 may act as a distal variant-specific enhancer and play a pivotal role in bone metabolism. Risk rs6426749 G alleles can affect the enhancer activity by binding to transcription factor TFAP2A; a thin process may increase transcription of LINC00339 and modulate the expression of downstream gene via long-range chromatin loop formation in osteoblast cells (Chen et al., 2018). Recent studies have reported that prostate cancer risk-associated G allele of rs11672691 is associated with an increased expression of lincRNA PCAT19 and oncogene CEACAM21; SNV rs11672691 is located in an enhancer element and may alter the binding site of its oncogenic transcription factor HOXA2. CRISPR/Cas9-mediated interference and activation assays have demonstrated that rs11672691 variant is involved in the regulation of its eQTL genes PCAT19 and CEACAM21 expression and affects the cells’ aggressive property in prostate cancers (Gao et al., 2018). In another alternative mechanism, risk variant rs11672691 is associated with the decreased levels of a short isoform of PCAT19 (PCAT19-short) and increased levels of a long isoform (PCAT19-long). This risk SNV locus is bifunctional with both promoter and enhancer activity, which maps to a promoter of PCAT19-short and the third intron of PCAT19-long. Risk allele rs11672691 and its linkage disequilibrium SNV rs887391 may alter the binding profiles of transcription factors NKX3.1 and YY1, thereby elevating the abundance of PCAT19-long through promoter-enhancer switching. Ultimately, it gives rise to an increased formation of the HNRNPAB-PCAT19-long complex to activate a subset of cell-cycle genes and promote prostate cancer aggression (Hua et al., 2018).
Another causative cis-regulatory mechanism has been constructed via integrative genomic analyses; the breast cancer-associated variant rs4415084 is located in a GATA3-binding motif of LINC02224, which refers to the differential GATA3 binding and chromatin accessibility, thereby promoting the transcription of LINC02224 and MRPS30 genes (Zhang et al., 2018). It is reasonable to postulate that the interactions of lincRNA, trait-associated variants, and regulatory factor may contribute to the development of specific disorders.
Currently, genetic variants in potential lincRNA regions have attracted increasing interest; it has been established that many SNVs are associated with susceptibility of multiple diseases. It is evident that the expression and function of lincRNAs may be influenced by its SNVs in a cell-type- or tissue-specific manner. A comprehensive analysis has suggested that genetic variants in lincRNA regions also possibly affect the process of splicing and stability of lincRNA conformation, thereby leading to a modification of their interacting partners, as shown in Figures 2C–E (Hon et al., 2017).
Several observations, such as eQTL and DNAase peak assays, indicate that genetic variants that occurred in exons of lincRNAs may change the lincRNA secondary structure, thereby affecting its stability, interactive properties, and regulatory functions (Khurana et al., 2016). For example, lincRNA CCAT2 could act as a scaffold or assembly platform and modulate the alternative splicing of glutaminase (GLS) pre-mRNA via directly binding to a Cleavage Factor I (CFIm) complex. However, SNV rs6983267 (G/T) may affect the interaction of CCAT2 with CFIm complex by changing lincRNA secondary structure and initiating a domino effect mechanism; this process leads to allele-specific reprogramming of cellular energy metabolism in colon cancers (Redis et al., 2016). Moreover, by using allele-specific CCAT2 transgenic mice, recently, Shah et al. (2018) have revealed that overexpression of CCAT2 may lead to genomic instability and myeloid malignancies; the SNV rs6983267-specific RNA-editing induces the dysregulation of a genome-wide gene expression by down-regulating EZH2, a histone-lysine N-methyltransferase, which then results in the impairment of immune processes and development of myelodysplastic neoplasms in vivo. In another study, Sur IK and his colleagues have generated mice lacking a myc enhancer region spanning risk-SNV rs6983267; the mutant mice have not showed an overt phenotype but confer resistance to intestinal tumorigenesis induced by APCmin mutation (Sur et al., 2012). These studies indicate that cancer risk-associated variants identified from the human genome may also exert a functional effect for animals in vivo.
It is worth noting that lincRNAs have a long average length and that their exon regions contain numerous trait-associated variants; significant alterations of lincRNA secondary structure may be caused by its SNVs on exon loci. Many variants such as PRNCR1 (prostate cancer-associated non-coding RNA) are located in exon regions, for example, rs1456315 G/A; it has been predicted to affect the lincRNA secondary structure of PRNCR1 (Chung et al., 2011) and then alter lincRNA stability and conformation, even giving rise to the modification of its interacting partners. Xue et al. (2015) have also reported that SNV rs7958904 G/C in an exon region does not affect transcription activity of HOTAIR; however, in in silico analyses, it is shown to alter the RNA secondary structure of HOTAIR. These findings indicate that genetic variants, especially SNVs in exon loci, may play a different role via affecting the lincRNA structure.
Not surprisingly, it has been documented that some microRNAs (miRNAs) can function in a non-canonical manner to regulate lincRNA expression levels or directly interact with lincRNA molecules. The competing endogenous RNA (ceRNA) is a mechanism that lncRNA could competitively bind or sponge miRNAs, such as ceRNA MALAT1; its exon locus contains a variant rs619586 A > G, which can significantly up-regulate the expression of XBP1 (X box-binding protein 1) by sponging miR-214 and then suppressing the proliferation and migration of vascular endothelial cells in vitro (Zhuo et al., 2017). In another case, variant rs11752942 of LINC00951 exon is linked to the susceptibility of ESCC; risk G alleles of rs11752942 may decrease the expression levels of LINC00951 via affecting the binding of miR-149-3p, thereby regulating cell proliferation and tumor growth (Wu et al., 2013). Intriguingly, recent studies have demonstrated that pancreatic cancer risk-SNV rs11655237 G > A in the LINC00673 exon region is likely to create a target site for miR-1231 binding and reduces the function of LINC00673 in an allele-specific manner. Down-regulation of LINC00673 may attenuate the interaction of PTPN11 with an E3 ubiquitin ligase PRPF19 and suppress the ubiquitin-mediated PTPN11 degradation; these processes enhance an oncogenic signaling whereas diminish STAT1-dependent anti-oncogenic signaling in cancer cells (Zheng et al., 2016). These findings highlight the regulatory relationships of miRNAs with lincRNAs in a variant-specific manner and may offer a wider field for future research on lincRNA.
As summarized above, genetic variants play a very significant role in the transcription and biological function of lincRNAs, contributing to various disease susceptibility, progression, prognosis, and treatment response. Genetic variants may act as a driving factor to affect the role of lincRNAs; just like a driver who drives a vehicle, analogously, lincRNA variants may vividly serve as a putative driver to regulate the lincRNA molecules.
Driver identification is a challenging task, owing to their complex and diverse modes of action and the inadequate understanding of non-coding regions; the computational prediction of non-coding drivers is even more challenging than that of protein-coding drivers. In addition, non-coding variants are more abundant than protein-coding genes; hence, the key variants with functional significance have to be distinguished from a larger set of passenger events (Khurana et al., 2016). Currently, several online databases have been constructed to describe genomic variants in lncRNA regions, such as lincSNP, lncRNASNP2, and LncVar. More specifically, lincSNP 2.0 is an integrated database to identify and annotate disease-associated SNVs on human lincRNAs and their transcription factor binding sites (Ning et al., 2017). LncRNASNP2 is an updated database of comprehensive information about SNVs or mutations in human and mouse lncRNAs, as well as their impacts on lncRNA structure and potential function on miRNA binding (Miao et al., 2018). LncVar provides genetic variants associated with lncRNAs in multiple species and their effects on biological function of lncRNAs (Chen et al., 2017). Furthermore, a large number of GWAS analyses have successfully identified an array of genetic variants that are associated with various types of human disorders (MacArthur et al., 2017). Numerous public databases have been set out to provide a comprehensive description of genetic variants and GWAS data in the human genome with high impact (Genomes Project et al., 2015). A brief overview of these databases with their key features and corresponding references is presented in Table 3.

Table 3. Some databases that may be used for driver identification.
Functional annotations and linkage disequilibrium analyses of genetic variants can be performed based on public databases and bioinformatic methods. Among tag-SNVs with strong linkage disequilibrium, significant genotype-specific effects on lincRNA expression can be observed by eQTL analysis (GTEx Consortium et al., 2017). Subsequently, according to ChIP-Seq data from the ENCODE database1, some trait-associated SNVs can be picked out; those variants mapping to cis-regulatory motifs may affect the binding activities of many interrelated transcription factors, including EZH2, CHD1, TCF7L2, and CTCF. These transcription factors may be closely related to the occurrence and progression of various human disorders, such as enhancer of zeste homologue 2 (EZH2), which is overexpressed in several human tumors and accounts for the aggressiveness and unfavorable prognoses of various cancers.
Many functional verification studies of genetic variants have focused on protein-coding regions of the human genome. With an expanding appreciation that non-coding variants play a crucial role in the development of disorders, several recent studies have set out to explore approaches to evaluate the function of non-coding variants (Khurana et al., 2016). For example, experimental methods used to understand the effects of cis-regulatory variants within a promoter or enhancer region on cellular biological functions is summarized as follows. A main strategy is required to introduce the sequence variants, the mutated DNA fragment can be constructed via site-directed mutagenesis, CRISPR–Cas system (Konermann et al., 2015) or oligonucleotide synthesis. Subsequently, the functional output of non-coding variants should be detected through several methods, either luciferase reporter assays or high-throughput sequencing-based assays, such as cis-regulatory element analysis by sequencing (CRE-seq) (Kwasnieski et al., 2014) and self-transcribing active regulatory region sequencing (STARR-seq) (Arnold et al., 2013). Furthermore, functional verification is required to determine the direct biological significances, such as the oncogenic properties, which can be manifested though cancer cellular experiments (e.g., cell proliferation, cell cycle, cell death, migration, and invasion tests) along with in vivo model assays. In addition, other approaches are needed to be explored to demonstrate the effects of genetic variants within introns, exons, or intergenic regions. For instance, genetic variants mapping to exons of a lincRNA may alter the lincRNA secondary structure, which can be partly predicted using RNAfold web server (Hofacker and Stadler, 2006). The 5′ UTR (un-translated region) variants may affect the process of splicing and stability of RNA conformation, a functional splicing reporter minigene assay should be used to assess the effect of genetic variants on RNA splicing (Giorgi et al., 2015). Through the aforementioned knowable strategies, comprehensive functional verification of non-coding variants is very important to understand their biological consequence; there is an urgent need to explore more practical methods and strategies for functional verification research.
Single-nucleotide variants are the most common genetic variants and universally present in the human genome, including non-coding regions. One current belief is that heterogeneous disease (e.g., cancers susceptibility) may be caused by the accumulation of multiple driving genetic variations. GWASs have identified a large number of disease or trait-associated SNVs, and many of those are located in non-coding regions of the human genome. The functions of genetic variants are generally unknown and remain to be elucidated. One critical common viewpoint is that the significance of lincRNA variants depends on their genomic position. Certain SNVs are located in regulatory regions of lincRNA genes; it is found to affect the binding efficiency of transcription factor; it is known to possibly regulate the transcription of lincRNAs and other related genes in a cell-type- or tissue-specific manner. The mapping of SNVs to lincRNA transcript itself potentially affects the process of splicing and stability of lincRNA conformation or modulates the lincRNA secondary structure; these effects may lead to an alteration of the interactive properties and regulatory functions of lincRNA (Khurana et al., 2016). Collectively, these findings indicate that genetic variants in lincRNA regions may serve as a regulatory signature for early events, which illustrate the genomic background of lincRNA differential expressions in a tissue- or disorder-specific manner.
Considering their important regulatory role, lincRNAs may serve as the promising biomarkers for the diagnosis, prognosis, and treatment response of various diseases (Zou et al., 2018). With their characteristic of tissue and disease specificity, lincRNAs may be explored as target molecules for personalized medicine in the future (Huarte, 2015). Currently, molecular targets drug approved by the US Food and Drug Administration (FDA) are mainly derived from proteins. However, owing to the finiteness of druggable protein genes in the human genome, the expansion of potentially druggable targets may need to include lncRNA molecules. One lincRNA PCA3-based test (the PROGENSA PCA3 assay approved by the FDA) has been used as a marker for the detection of prostate cancers (Evaluation of Genomic Applications in Practice and Prevention [EGAPP] Working Group, 2014). Moreover, a novel treatment strategy differs from the classical small molecules and antibodies that mainly target proteins. RNA-targeting therapeutics refer to the use of oligonucleotides to target primarily RNA involved in various diseases for therapeutic efforts. Two major approaches are employed to target RNA: double-stranded RNA-mediated interference (RNAi) and antisense oligonucleotides (ASOs) (Kole et al., 2012). Currently, both methods are in clinical trials. Among them, nusinersen (Spinraza), an ASO-targeting drug for spinal muscular atrophy (SMA), has been approved by the FDA (Finkel et al., 2017); patisiran, an RNAi therapeutic strategy for hereditary transthyretin amyloidosis (hATTR), has also shown promising results (Adams et al., 2018). Hence, we can expect that lincRNA molecules will provide additional options for RNA therapeutics. Importantly, disease-associated variants are found to exhibit a higher frequency in non-coding regions, which encompass enhancers, promoters, and other regulatory elements. It is likely that the role of genetic variants in lincRNA regions should be characterized at the regulatory network level. Genetic variants may offer the possibility to make use of the information from adjacent protein-coding or non-coding regions to link with heterogeneous diseases. Therefore, a combination of SNVs, lincRNAs, and proteins may bring personalized medicine closer to clinical applications in the foreseeable future (Li et al., 2019).
Previous studies may appear to be a slightly biased against the genetic variants that are located in non-coding regions, as their significant roles have not yet been explored to the same extent as those of protein-coding genes. In particular, for the disease-associated variants in lincRNA regions, whether functionally affected or altered in lincRNA expression by risk variants, it may be responsible for the disease development and its pathogenesis. Verification of the mechanisms requires a detailed understanding of the lincRNA structure and function, and a suitable experimental system to distinguish the subtle differences caused by genetic variants. Although it is difficult to describe the consequences of genetic variants in non-coding regions, more emerging technologies and approaches are urgently needed to explore the driving effects of genetic variants on lincRNA regions.
HZ consulted relevant literatures, finished the manuscript, and completed English revision. LT completed the figures and tables. F-FS provided constructive feedback and guidance. L-XW and H-HZ completed critical revisions and proofread the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81603201 and 81473284). This work was also supported by funds from the Joint International Research Laboratory of Reproduction and Development, Institute of life Sciences, Chongqing Medical University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adams, D., Gonzalez-Duarte, A., O’riordan, W. D., Yang, C. C., Ueda, M., Kristen, A. V., et al. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21. doi: 10.1056/NEJMoa1716153
Arnold, C. D., Gerlach, D., Stelzer, C., Boryn, L. M., Rath, M., and Stark, A. (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. doi: 10.1126/science.1232542
Athanasiu, L., Smorr, L. L., Tesli, M., Rossberg, J. I., Sonderby, I. E., Spigset, O., et al. (2015). Genome-wide association study identifies common variants associated with pharmacokinetics of psychotropic drugs. J. Psychopharmacol. 29, 884–891. doi: 10.1177/0269881115584469
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi: 10.1101/gad.17446611
Cerhan, J. R., Berndt, S. I., Vijai, J., Ghesquieres, H., Mckay, J., Wang, S. S., et al. (2014). Genome-wide association study identifies multiple susceptibility loci for diffuse large B cell lymphoma. Nat. Genet. 46, 1233–1238. doi: 10.1038/ng.3105
Chen, X., Hao, Y., Cui, Y., Fan, Z., He, S., Luo, J., et al. (2017). LncVar: a database of genetic variation associated with long non-coding genes. Bioinformatics 33, 112–118. doi: 10.1093/bioinformatics/btw581
Chen, X. F., Zhu, D. L., Yang, M., Hu, W. X., Duan, Y. Y., Lu, B. J., et al. (2018). An Osteoporosis Risk SNP at 1p36.12 Acts as an Allele-Specific Enhancer to Modulate LINC00339 Expression via Long-Range Loop Formation. Am. J. Hum. Genet. 102, 776–793. doi: 10.1016/j.ajhg.2018.03.001
Chu, H., Xia, L., Qiu, X., Gu, D., Zhu, L., Jin, J., et al. (2015). Genetic variants in noncoding PIWI-interacting RNA and colorectal cancer risk. Cancer 121, 2044–2052. doi: 10.1002/cncr.29314
Chu, M., Yuan, W., Wu, S., Wang, Z., Mao, L., Tian, T., et al. (2016). Quantitative assessment of polymorphisms in H19 lncRNA and cancer risk: a meta-analysis of 13,392 cases and 18,893 controls. Oncotarget 7, 78631–78639. doi: 10.18632/oncotarget.12530
Chung, S., Nakagawa, H., Uemura, M., Piao, L., Ashikawa, K., Hosono, N., et al. (2011). Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 102, 245–252. doi: 10.1111/j.1349-7006.2010.01737.x
Conti, D. V., Wang, K., Sheng, X., Bensen, J. T., Hazelett, D. J., Cook, M. B., et al. (2017). Two novel susceptibility loci for prostate cancer in men of african ancestry. J. Natl. Cancer Inst. 109:djx084. doi: 10.1093/jnci/djx084
Cui, Z., Gao, M., Yin, Z., Yan, L., and Cui, L. (2018). Association between lncRNA CASC8 polymorphisms and the risk of cancer: a meta-analysis. Cancer Manag. Res. 10, 3141–3148. doi: 10.2147/CMAR.S170783
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Evaluation of Genomic Applications in Practice and Prevention [EGAPP] Working Group (2014). Recommendations from the EGAPP Working Group: does PCA3 testing for the diagnosis and management of prostate cancer improve patient health outcomes? Genet. Med. 16, 338–346. doi: 10.1038/gim.2013.141
Fava, V. M., Manry, J., Cobat, A., Orlova, M., Van Thuc, N., Moraes, M. O., et al. (2017). A genome wide association study identifies a lncRna as risk factor for pathological inflammatory responses in leprosy. PLoS Genet. 13:e1006637. doi: 10.1371/journal.pgen.1006637
Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732. doi: 10.1056/NEJMoa1702752
Freedman, M. L., Monteiro, A. N., Gayther, S. A., Coetzee, G. A., Risch, A., Plass, C., et al. (2011). Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 43, 513–518. doi: 10.1038/ng.840
Gao, P., Xia, J. H., Sipeky, C., Dong, X. M., Zhang, Q., Yang, Y., et al. (2018). Biology AND CLINICAL IMPLICATIONS OF the 19q13 aggressive prostate cancer susceptibility locus. Cell 174, 576.e18–589.e18. doi: 10.1016/j.cell.2018.06.003
Gao, W., Zhu, M., Wang, H., Zhao, S., Zhao, D., Yang, Y., et al. (2015). Association of polymorphisms in long non-coding RNA H19 with coronary artery disease risk in a Chinese population. Mutat. Res. 772, 15–22. doi: 10.1016/j.mrfmmm.2014.12.009
Ge, L., Wang, Q., Hu, S., and Yang, X. (2018). Rs217727 polymorphism in H19 promotes cell apoptosis by regulating the expressions of H19 and the activation of its downstream signaling pathway. J. Cell Physiol. 234, 7279–7291. doi: 10.1002/jcp.27485
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Ghanbari, M., Peters, M. J., De Vries, P. S., Boer, C. G., Van Rooij, J. G. J., Lee, Y. C., et al. (2018). A systematic analysis highlights multiple long non-coding RNAs associated with cardiometabolic disorders. J. Hum. Genet. 63, 431–446. doi: 10.1038/s10038-017-0403-x
Ghesquieres, H., Larrabee, B. R., Casasnovas, O., Maurer, M. J., Mckay, J. D., Ansell, S. M., et al. (2018). A susceptibility locus for classical Hodgkin lymphoma at 8q24 near MYC/PVT1 predicts patient outcome in two independent cohorts. Br. J. Haematol. 180, 286–290. doi: 10.1111/bjh.14306
Giorgi, G., Casarin, A., Trevisson, E., Dona, M., Cassina, M., Graziano, C., et al. (2015). Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin. Chem. Lab. Med 53, 1719–1723. doi: 10.1515/cclm-2014-1047
Gong, W. J., Peng, J. B., Yin, J. Y., Li, X. P., Zheng, W., Xiao, L., et al. (2017). Association between well-characterized lung cancer lncRNA polymorphisms and platinum-based chemotherapy toxicity in Chinese patients with lung cancer. Acta Pharmacol. Sin. 38, 581–590. doi: 10.1038/aps.2016.164
GTEx Consortium, Data Analysis &Coordinating Center [LDACC], Analysis Working Group, Statistical Methods groups, Analysis Working Group, Enhancing GTEx groups et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. doi: 10.1038/nature25160
Guo, H., Ahmed, M., Zhang, F., Yao, C. Q., Li, S., Liang, Y., et al. (2016). Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 48, 1142–1150. doi: 10.1038/ng.3637
Guo, Q. Y., Wang, H., and Wang, Y. (2017). LncRNA H19 polymorphisms associated with the risk of OSCC in Chinese population. Eur. Rev. Med. Pharmacol. Sci. 21, 3770–3774.
Guo, Z., Wang, Y., Zhao, Y., Jin, Y., An, L., Wu, B., et al. (2017). Genetic polymorphisms of long non-coding RNA GAS5 predict platinum-based concurrent chemoradiotherapy response in nasopharyngeal carcinoma patients. Oncotarget 8, 62286–62297. doi: 10.18632/oncotarget.19725
Han, L., Liu, S., Liang, J., Guo, Y., Shen, S., Guo, X., et al. (2017). A genetic polymorphism at miR-526b binding-site in the lincRNA-NR_024015 exon confers risk of esophageal squamous cell carcinoma in a population of North China. Mol. Carcinog. 56, 960–971. doi: 10.1002/mc.22549
Hanson, R. L., Craig, D. W., Millis, M. P., Yeatts, K. A., Kobes, S., Pearson, J. V., et al. (2007). Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes Metab. Res. Rev. 56, 975–983. doi: 10.2337/db06-1072
Harati-Sadegh, M., Kohan, L., Teimoori, B., and Salimi, S. (2018). The long non-coding RNA H19 rs217727 polymorphism is associated with PE susceptibility. J. Cell. Biochem. 119, 5473–5480. doi: 10.1002/jcb.26708
He, H., Li, W., Liyanarachchi, S., Srinivas, M., Wang, Y., Akagi, K., et al. (2015). Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc. Natl. Acad. Sci. U.S.A. 112, 6128–6133. doi: 10.1073/pnas.1506255112
Hindorff, L. A., Sethupathy, P., Junkins, H. A., Ramos, E. M., Mehta, J. P., Collins, F. S., et al. (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367. doi: 10.1073/pnas.0903103106
Hofacker, I. L., and Stadler, P. F. (2006). Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 22, 1172–1176. doi: 10.1093/bioinformatics/btl023
Hon, C. C., Ramilowski, J. A., Harshbarger, J., Bertin, N., Rackham, O. J., Gough, J., et al. (2017). An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204. doi: 10.1038/nature21374
Hu, L., Chen, S. H., Lv, Q. L., Sun, B., Qu, Q., Qin, C. Z., et al. (2016). Clinical Significance of Long Non-Coding RNA CASC8 rs10505477 polymorphism in lung cancer susceptibility, platinum-based chemotherapy response, and toxicity. Int. J. Environ. Res. Public Health 13:545. doi: 10.3390/ijerph13060545
Hua, J. T., Ahmed, M., Guo, H., Zhang, Y., Chen, S., Soares, F., et al. (2018). Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 174, 564.e18–575.e18. doi: 10.1016/j.cell.2018.06.014
Huang, D., Yi, X., Zhang, S., Zheng, Z., Wang, P., Xuan, C., et al. (2018). GWAS4D: multidimensional analysis of context-specific regulatory variant for human complex diseases and traits. Nucleic Acids Res. 46, W114–W120. doi: 10.1093/nar/gky407
Huarte, M. (2015). The emerging role of lncRNAs in cancer. Nat. Med. 21, 1253–1261. doi: 10.1038/nm.3981
Human Genome Structural Variation Working Group, Eichler, E. E., Nickerson, D. A., Altshuler, D., Bowcock, A. M., Brooks, L. D., et al. (2007). Completing the map of human genetic variation. Nature 447, 161–165. doi: 10.1038/447161a
Ingle, J. N., Xie, F., Ellis, M. J., Goss, P. E., Shepherd, L. E., Chapman, J. W., et al. (2016). Genetic polymorphisms in the long noncoding RNA MIR2052HG offer a pharmacogenomic basis for the response of breast cancer patients to aromatase inhibitor therapy. Cancer Res. 76, 7012–7023. doi: 10.1158/0008-5472.CAN-16-1371
Ishii, N., Ozaki, K., Sato, H., Mizuno, H., Saito, S., Takahashi, A., et al. (2006). Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 51, 1087–1099. doi: 10.1007/s10038-006-0070-9
Jendrzejewski, J., He, H., Radomska, H. S., Li, W., Tomsic, J., Liyanarachchi, S., et al. (2012). The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. U.S.A. 109, 8646–8651. doi: 10.1073/pnas.1205654109
Kenyan Bacteraemia Study Group, Wellcome Trust, Case Control, C., Rautanen, A., Pirinen, M., Mills, T. C., et al. (2016). Polymorphism in a lincRNA associates with a doubled risk of pneumococcal bacteremia in kenyan children. Am. J. Hum. Genet. 98, 1092–1100. doi: 10.1016/j.ajhg.2016.03.025
Khurana, E., Fu, Y., Chakravarty, D., Demichelis, F., Rubin, M. A., and Gerstein, M. (2016). Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 17, 93–108. doi: 10.1038/nrg.2015.17
Kim, T., Cui, R., Jeon, Y. J., Lee, J. H., Lee, J. H., Sim, H., et al. (2014). Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc. Natl. Acad. Sci. U.S.A. 111, 4173–4178. doi: 10.1073/pnas.1400350111
Kole, R., Krainer, A. R., and Altman, S. (2012). RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 11, 125–140. doi: 10.1038/nrd3625
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. doi: 10.1038/nature14136
Kretz, M., Siprashvili, Z., Chu, C., Webster, D. E., Zehnder, A., Qu, K., et al. (2013). Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493, 231–235. doi: 10.1038/nature11661
Kulakova, O., Bashinskaya, V., Kiselev, I., Baulina, N., Tsareva, E., Nikolaev, R., et al. (2017). Pharmacogenetics of glatiramer acetate therapy for multiple sclerosis: the impact of genome-wide association studies identified disease risk loci. Pharmacogenomics 18, 1563–1574. doi: 10.2217/pgs-2017-0058
Kwasnieski, J. C., Fiore, C., Chaudhari, H. G., and Cohen, B. A. (2014). High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 24, 1595–1602. doi: 10.1101/gr.173518.114
Lee, T. H., Ko, T. M., Chen, C. H., Lee, M. T., Chang, Y. J., Chang, C. H., et al. (2016). Identification of PTCSC3 as a novel locus for large-vessel ischemic stroke: a genome-wide association Study. J. Am. Heart Assoc. 5:e003003. doi: 10.1161/JAHA.115.003003
Li, C., Rao, T., Chen, X., Zou, Z., Wei, A., Tang, J., et al. (2019). HLA-B∗35:01 allele is a potential biomarker for predicting polygonum multiflorum-induced liver injury in humans. Hepatology 70, 346–357. doi: 10.1002/hep.30660
Li, L., Sun, R., Liang, Y., Pan, X., Li, Z., Bai, P., et al. (2013). Association between polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of colorectal cancer. J. Exp. Clin. Cancer Res. 32:104. doi: 10.1186/1756-9966-32-104
Li, Q., Ma, G., Sun, S., Xu, Y., and Wang, B. (2018a). Polymorphism in the promoter region of lncRNA GAS5 is functionally associated with the risk of gastric cancer. Clin. Res. Hepatol. Gastroenterol. 42, 478–482. doi: 10.1016/j.clinre.2018.01.006
Li, Q., Zhu, W., Zhang, B., Wu, Y., Yan, S., Yuan, Y., et al. (2018b). The MALAT1 gene polymorphism and its relationship with the onset of congenital heart disease in Chinese. Biosci. Rep. 38:BSR20171381. doi: 10.1042/BSR20171381
Li, Y., Bao, C., Gu, S., Ye, D., Jing, F., Fan, C., et al. (2017). Associations between novel genetic variants in the promoter region of MALAT1 and risk of colorectal cancer. Oncotarget 8, 92604–92614. doi: 10.18632/oncotarget.21507
Ling, H., Spizzo, R., Atlasi, Y., Nicoloso, M., Shimizu, M., Redis, R. S., et al. (2013). CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 23, 1446–1461. doi: 10.1101/gr.152942.112
Ma, G., Gu, D., Lv, C., Chu, H., Xu, Z., Tong, N., et al. (2015). Genetic variant in 8q24 is associated with prognosis for gastric cancer in a Chinese population. J. Gastroenterol. Hepatol. 30, 689–695. doi: 10.1111/jgh.12801
MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901. doi: 10.1093/nar/gkw1133
Maris, J. M., Mosse, Y. P., Bradfield, J. P., Hou, C., Monni, S., Scott, R. H., et al. (2008). Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 358, 2585–2593. doi: 10.1056/NEJMoa0708698
Marjonen, H., Auvinen, P., Kahila, H., Tsuiko, O., Koks, S., Tiirats, A., et al. (2018). rs10732516 polymorphism at the IGF2/H19 locus associates with genotype-specific effects on placental DNA methylation and birth weight of newborns conceived by assisted reproductive technology. Clin. Epigenetics 10:80. doi: 10.1186/s13148-018-0511-2
Meyer, K. B., Maia, A. T., O’reilly, M., Ghoussaini, M., Prathalingam, R., Porter-Gill, P., et al. (2011). A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet. 7:e1002165. doi: 10.1371/journal.pgen.1002165
Miao, Y. R., Liu, W., Zhang, Q., and Guo, A. Y. (2018). lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 46, D276–D280. doi: 10.1093/nar/gkx1004
Ng, M. C. Y., Graff, M., Lu, Y., Justice, A. E., Mudgal, P., Liu, C. T., et al. (2017). Discovery and fine-mapping of adiposity loci using high density imputation of genome-wide association studies in individuals of African ancestry: African ancestry anthropometry genetics Consortium. PLoS Genet. 13:e1006719. doi: 10.1371/journal.pgen.1006719
Nikpay, M., Goel, A., Won, H. H., Hall, L. M., Willenborg, C., Kanoni, S., et al. (2015). A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. doi: 10.1038/ng.3396
Ning, S., Yue, M., Wang, P., Liu, Y., Zhi, H., Zhang, Y., et al. (2017). LincSNP 2.0: an updated database for linking disease-associated SNPs to human long non-coding RNAs and their TFBSs. Nucleic Acids Res. 45, D74–D78. doi: 10.1093/nar/gkw945
Orr, N., Cooke, R., Jones, M., Fletcher, O., Dudbridge, F., Chilcott-Burns, S., et al. (2011). Genetic variants at chromosomes 2q35, 5p12, 6q25.1, 10q26.13, and 16q12.1 influence the risk of breast cancer in men. PLoS Genet. 7:e1002290. doi: 10.1371/journal.pgen.1002290
Petry, C. J., Seear, R. V., Wingate, D. L., Acerini, C. L., Ong, K. K., Hughes, I. A., et al. (2011). Maternally transmitted foetal H19 variants and associations with birth weight. Hum. Genet. 130, 663–670. doi: 10.1007/s00439-011-1005-x
Pomerantz, M. M., Ahmadiyeh, N., Jia, L., Herman, P., Verzi, M. P., Doddapaneni, H., et al. (2009). The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 41, 882–884. doi: 10.1038/ng.403
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Powell, J. E., Fung, J. N., Shakhbazov, K., Sapkota, Y., Cloonan, N., Hemani, G., et al. (2016). Endometriosis risk alleles at 1p36.12 act through inverse regulation of CDC42 and LINC00339. Hum. Mol. Genet. 25, 5046–5058. doi: 10.1093/hmg/ddw320
Rao, S. Q., Hu, H. L., Ye, N., Shen, Y., and Xu, Q. (2015). Genetic variants in long non-coding RNA MIAT contribute to risk of paranoid schizophrenia in a Chinese Han population. Schizophr. Res. 166, 125–130. doi: 10.1016/j.schres.2015.04.032
Redis, R. S., Vela, L. E., Lu, W., Ferreira De Oliveira, J., Ivan, C., Rodriguez-Aguayo, C., et al. (2016). Allele-specific reprogramming of cancer metabolism by the long non-coding RNA CCAT2. Mol. Cell. 61:640. doi: 10.1016/j.molcel.2016.02.006
Schaub, M. A., Boyle, A. P., Kundaje, A., Batzoglou, S., and Snyder, M. (2012). Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759. doi: 10.1101/gr.136127.111
Shah, M. Y., Ferracin, M., Pileczki, V., Chen, B., Redis, R., Fabris, L., et al. (2018). Cancer-associated rs6983267 SNP and its accompanying long noncoding RNA CCAT2 induce myeloid malignancies via unique SNP-specific RNA mutations. Genome Res. 28, 432–447. doi: 10.1101/gr.225128.117
Shyn, S. I., Shi, J., Kraft, J. B., Potash, J. B., Knowles, J. A., Weissman, M. M., et al. (2011). Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol. Psychiatry 16, 202–215. doi: 10.1038/mp.2009.125
Sur, I. K., Hallikas, O., Vaharautio, A., Yan, J., Turunen, M., Enge, M., et al. (2012). Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 338, 1360–1363. doi: 10.1126/science.1228606
Tao, R., Hu, S., Wang, S., Zhou, X., Zhang, Q., Wang, C., et al. (2015). Association between indel polymorphism in the promoter region of lncRNA GAS5 and the risk of hepatocellular carcinoma. Carcinogenesis 36, 1136–1143. doi: 10.1093/carcin/bgv099
Teerlink, C. C., Leongamornlert, D., Dadaev, T., Thomas, A., Farnham, J., Stephenson, R. A., et al. (2016). Genome-wide association of familial prostate cancer cases identifies evidence for a rare segregating haplotype at 8q24.21. Hum. Genet. 135, 923–938. doi: 10.1007/s00439-016-1690-6
The Encode Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Tuupanen, S., Turunen, M., Lehtonen, R., Hallikas, O., Vanharanta, S., Kivioja, T., et al. (2009). The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 41, 885–890. doi: 10.1038/ng.406
Ulitsky, I., and Bartel, D. P. (2013). lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46.
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The sequence of the human genome. Science 291, 1304–1351.
Verhaegh, G. W., Verkleij, L., Vermeulen, S. H., Den Heijer, M., Witjes, J. A., and Kiemeney, L. A. (2008). Polymorphisms in the H19 gene and the risk of bladder cancer. Eur. Urol. 54, 1118–1126. doi: 10.1016/j.eururo.2008.01.060
Wallace, C., Smyth, D. J., Maisuria-Armer, M., Walker, N. M., Todd, J. A., and Clayton, D. G. (2010). The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 42, 68–71. doi: 10.1038/ng.493
Wang, B. G., Lv, Z., Ding, H. X., Fang, X. X., Wen, J., Xu, Q., et al. (2018a). The association of lncRNA-HULC polymorphisms with hepatocellular cancer risk and prognosis. Gene 670, 148–154. doi: 10.1016/j.gene.2018.05.096
Wang, B. G., Xu, Q., Lv, Z., Fang, X. X., Ding, H. X., Wen, J., et al. (2018b). Association of twelve polymorphisms in three onco-lncRNA genes with hepatocellular cancer risk and prognosis: a case-control study. World J. Gastroenterol. 24, 2482–2490. doi: 10.3748/wjg.v24.i23.2482
Wang, J. Z., Xiang, J. J., Wu, L. G., Bai, Y. S., Chen, Z. W., Yin, X. Q., et al. (2017). A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: a survival cohort analysis. BMC Cancer 17:167. doi: 10.1186/s12885-017-3151-6
Westra, H. J., Martinez-Bonet, M., Onengut-Gumuscu, S., Lee, A., Luo, Y., Teslovich, N., et al. (2018). Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 50, 1366–1374. doi: 10.1038/s41588-018-0216-7
Wu, H., Zheng, J., Deng, J., Hu, M., You, Y., Li, N., et al. (2013). A genetic polymorphism in lincRNA-uc003opf.1 is associated with susceptibility to esophageal squamous cell carcinoma in Chinese populations. Carcinogenesis 34, 2908–2917. doi: 10.1093/carcin/bgt252
Wu, N. Y., Huang, H. S., Chao, T. H., Chou, H. M., Fang, C., Qin, C. Z., et al. (2017). Progesterone prevents high-grade serous ovarian cancer by inducing necroptosis of p53-defective fallopian tube epithelial cells. Cell Rep. 18, 2557–2565. doi: 10.1016/j.celrep.2017.02.049
Wu, Q., Yan, W., Han, R., Yang, J., Yuan, J., Ji, X., et al. (2016). Polymorphisms in long noncoding RNA H19 contribute to the protective effects of coal workers’ pneumoconiosis in a chinese population. Int. J. Environ. Res. Public Health 13:903. doi: 10.3390/ijerph13090903
Xue, Y., Gu, D., Ma, G., Zhu, L., Hua, Q., Chu, H., et al. (2015). Genetic variants in lncRNA HOTAIR are associated with risk of colorectal cancer. Mutagenesis 30, 303–310. doi: 10.1093/mutage/geu076
Xue, Y., Wang, M., Kang, M., Wang, Q., Wu, B., Chu, H., et al. (2013). Association between lncrna PCGEM1 polymorphisms and prostate cancer risk. Prostate Cancer Prostatic Dis. 16, 139–144. doi: 10.1038/pcan.2013.6
Yan, H., Zhang, D. Y., Li, X., Yuan, X. Q., Yang, Y. L., Zhu, K. W., et al. (2017). Long non-coding RNA GAS5 polymorphism predicts a poor prognosis of acute myeloid leukemia in Chinese patients via affecting hematopoietic reconstitution. Leuk. Lymphoma 58, 1948–1957. doi: 10.1080/10428194.2016.1266626
Yang, M. L., Huang, Z., Wang, Q., Chen, H. H., Ma, S. N., Wu, R., et al. (2018). The association of polymorphisms in lncRNA-H19 with hepatocellular cancer risk and prognosis. Biosci. Rep. 38:BSR20171652. doi: 10.1042/BSR20171652
Yeager, M., Orr, N., Hayes, R. B., Jacobs, K. B., Kraft, P., Wacholder, S., et al. (2007). Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 39, 645–649. doi: 10.1016/j.eururo.2007.06.013
Zhang, Y., Manjunath, M., Zhang, S., Chasman, D., Roy, S., and Song, J. S. (2018). Integrative genomic analysis predicts causative Cis-regulatory mechanisms of the breast cancer-associated genetic variant rs4415084. Cancer Res. 78, 1579–1591. doi: 10.1158/0008-5472.CAN-17-3486
Zhang, Z., Zhu, Z., Zhang, B., Li, W., Li, X., Wu, X., et al. (2014). Frequent mutation of rs13281615 and its association with PVT1 expression and cell proliferation in breast cancer. J. Genet. Genomics 41, 187–195. doi: 10.1016/j.jgg.2014.03.006
Zhao, X., Wei, X., Zhao, L., Shi, L., Cheng, J., Kang, S., et al. (2016). The rs6983267 SNP and long non-coding RNA CARLo-5 are associated with endometrial carcinoma. Environ. Mol. Mutagen. 57, 508–515. doi: 10.1002/em.22031
Zheng, J., Huang, X., Tan, W., Yu, D., Du, Z., Chang, J., et al. (2016). Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 48, 747–757. doi: 10.1038/ng.3568
Zheng, Y., Yang, C., Tong, S., Ding, Y., Deng, W., Song, D., et al. (2017). Genetic variation of long non-coding RNA TINCR contribute to the susceptibility and progression of colorectal cancer. Oncotarget 8, 33536–33543. doi: 10.18632/oncotarget.16538
Zhuo, Y., Zeng, Q., Zhang, P., Li, G., Xie, Q., and Cheng, Y. (2017). Functional polymorphism of lncRNA MALAT1 contributes to pulmonary arterial hypertension susceptibility in Chinese people. Clin. Chem. Lab. Med 55, 38–46. doi: 10.1515/cclm-2016-0056
Zou, H., Wen, C., Peng, Z., Shao, Y., Hu, L., Li, S., et al. (2018). P4HB and PDIA3 are associated with tumor progression and therapeutic outcome of diffuse gliomas. Oncol. Rep. 39, 501–510. doi: 10.3892/or.2017.6134
Keywords: single-nucleotide variant, long/large intergenic non-protein coding RNA, disease susceptibility, transcription, biological function
Citation: Zou H, Wu L-X, Tan L, Shang F-F and Zhou H-H (2020) Significance of Single-Nucleotide Variants in Long Intergenic Non-protein Coding RNAs. Front. Cell Dev. Biol. 8:347. doi: 10.3389/fcell.2020.00347
Received: 18 February 2020; Accepted: 20 April 2020;
Published: 25 May 2020.
Edited by:
Hongquan Zhang, Peking University Health Science Center, ChinaCopyright © 2020 Zou, Wu, Tan, Shang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei-Fei Shang, c2hhbmdmZmVpQHNpbmEuY29t; Hong-Hao Zhou, aGh6aG91MjAwNUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.