 Alvaro Gonzalez-Rajal
Alvaro Gonzalez-Rajal Jordan F. Hastings
Jordan F. Hastings D. Neil Watkins
D. Neil Watkins David R. Croucher
David R. Croucher Andrew Burgess
Andrew Burgess- 1ANZAC Research Institute, Concord, NSW, Australia
- 2The Kinghorn Cancer Centre, Garvan Institute of Medical Research, Sydney, NSW, Australia
- 3Research Institute in Oncology and Hematology, Cancer Care Manitoba, Winnipeg, MB, Canada
- 4Department of Internal Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
- 5St Vincent's Hospital Clinical School, University of New South Wales, Sydney, NSW, Australia
- 6The University of Sydney Concord Clinical School, Faculty of Medicine and Health, Sydney, NSW, Australia
Introduction
Lung cancer accounts for approximately 11% of all cancer cases, however the 5-year survival rate is often below 20%. Consequently, lung cancer is the leading cause of cancer related mortality worldwide (Bray et al., 2018). There are two major types of lung cancer; small cell lung cancer (SCLC), which accounts for ~15% of cases and non-small cell lung cancer (NSCLC), which accounts for ~85% (Herbst et al., 2018). NSCLC is further separated into lung adenocarcinoma (LUAD, ~50%), squamous cell carcinoma (~30%) and multiple smaller subtypes (~20%). Notably, up to 75% of NSCLC patients are diagnosed with advanced stage III/IV lung cancer (Walters et al., 2013), limiting surgical intervention.
While smoking is strongly associated with all lung cancer types, at least 20% of LUAD cases are from non–or never smokers (Herbst et al., 2018). Furthermore, while LUAD is characterized by a high somatic mutation rate, with deletion or mutation of TP53 occurring in up to 46% of cases, <20% of patients carry targetable mutations such as those within EGFR, ALK, or BRAF or NTRK (Arbour and Riely, 2019). Consequently, the overwhelming majority of LUAD patients receive platinum-based chemotherapy as standard of care.
Unfortunately, response rates to platinum in LUAD are below 30%, due to innate/acquired resistance and rate-limiting side-effects such as nephrotoxicity (Marini et al., 2018). Importantly, potential synergy between platinum chemotherapy and immunotherapy has emerged as a therapeutic opportunity in LUAD (Mathew et al., 2018). Therefore, improving platinum efficacy and identifying mechanism of resistance could significantly improve patient outcomes. In this opinion article, we cover several of the latest landmark publications that shed new light on the mechanisms of platinum resistance in LUAD.
Overview of Platinum Chemotherapy
The anti-tumor abilities of cisplatin were identified over 50 years ago (Rosenberg et al., 1969). Since then platinum has become one of the most successful chemotherapeutics developed. It is essentially curative in testicular cancer, with survival rates >90% (Koster et al., 2013). It is also used with varying degrees of success to treat ovarian, head and neck, bladder and cervical cancer. Second and third generation cisplatin analogs have now been developed with the aim of lessening nephrotoxicity, neurotoxicity, ototoxicity, or providing better bioavailability and overcoming tumor resistance. Of these, carboplatin and oxiplatin are the most well-known, however nedaplatin, heptaplatin, lobaplatin and satraplatin are also used clinically (Wang and Lippard, 2005).
Cisplatin and its derivates rely on their platinum group to exert killing. Platinum compounds can bind to many biological targets including DNA, RNA, and proteins (Stordal and Davey, 2007). The binding of cisplatin to DNA forms platinum-DNA adducts (Figure 1), which must be repaired by the cell. Approximately 90% of cisplatin-induced adducts are intra-strand crosslinks that are rapidly repaired mostly by the base-excision and nucleotide excision repair (BER, NER) pathways during G1 phase (Slyskova et al., 2018). In contrast, inter-strand crosslinks (ICL) represent <5% of cisplatin-induced adducts but are far more difficult for cells to remove as they are “hidden” within the DNA helix. ICLs prevent the unzipping of the double helix, creating a physical barrier to efficient DNA replication. The removal, largely by the Fanconi anemia (FA) pathway (Michl et al., 2016; Niraj et al., 2019; Smogorzewska, 2019), results in the formation of single and double strand breaks (SSBs and DSBs). The damaged DNA is then repaired by either the high-fidelity homologous recombination (HR) pathway during S/G2-phase (Karanam et al., 2012) or by the error-prone non-homologous end joining (NHEJ) pathway in G1 phase (Enoiu et al., 2012). The extent of, or failure to repair the DNA damage caused by cisplatin can result in cell death, accounting for the cytotoxic mode of action for most platinum agents. The exception is oxiplatin, which kill cells through increasing ribosome biogenesis stress (Bruno et al., 2017). For simplicity, here we will only focus on the mechanisms of cisplatin resistance in LUAD.
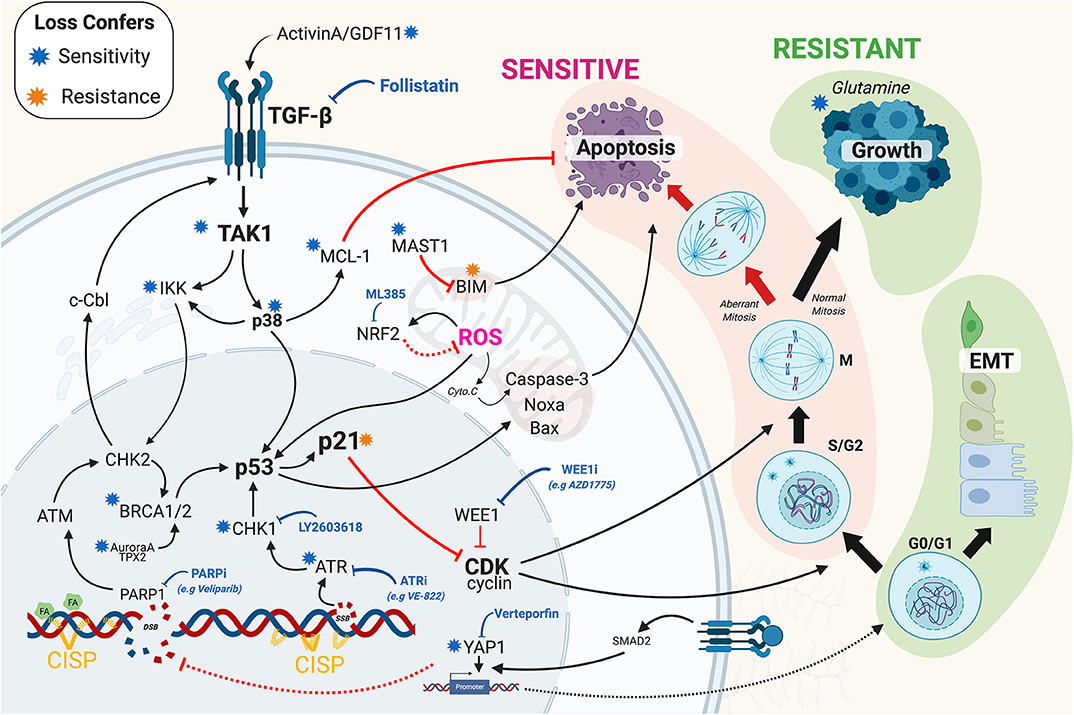
Figure 1. Overview of the recent mechanisms of resistance identified in lung adenocarcinoma. Briefly, mechanism of resistance work to increase repair of DNA damage and or prevent the triggering of cell death through inhibition of apoptotic signaling. Black solid arrows indicated activation. Red arrows indicate inhibition. Solid and dashed lines indicate direct and indirect mechanisms. Blue and orange stars indicate that loss (e.g., by RNAi, CRISPR, or chemical inhibition) confers sensitivity and resistance to cisplatin, respectively. Blue block arrow indicates potential therapeutic cisplatin synergistic treatment options. CISP, cisplatin; EMT, epithelial-mesenchymal transition; SSB/DSB, single/double strand breaks; FA, Fanconi Anemia pathway; ROS, reactive oxygen species; Cyto.C, Cytochrome C. The Figure created with BioRender.com.
Screening for Platinum Sensitisers
To date over 147 mechanisms of platinum resistance have been proposed (Stewart, 2007), yet there remains a lack of viable clinical options to improve response rates. To overcome this, several recent publications (Cheng et al., 2016; Jhuraney et al., 2016; Jin et al., 2018; Marini et al., 2018; Ding et al., 2019; Hsu C.-H et al., 2019), have looked at potential mechanisms of resistance in LUAD using unbiased screens, and detailed preclinical models. Based on these new data, it is clear that the main points of resistance arise from alterations to DNA repair, TGF-β signaling, cell cycle and apoptosis (Figure 1). Put simply, the ability of cisplatin to kill cells requires actively cycling cells that generate sufficient DNA damage and a functional apoptotic pathway to induce death. Consequently, disruption at any point along these pathways can prevent cell death, thereby reducing sensitivity to platinum mediated killing. Conversely, synergising therapies in general either block inhibitory cell death pathways, thereby lowering the threshold required to trigger death or increase the amount of damage induced by platinum.
DNA Repair and Resistance to Platinum
The DNA Damage response (DDR), has been extensively reviewed (Jackson and Bartek, 2009; O'Connor, 2015; Pearl et al., 2015; Matt and Hofmann, 2016), as has its role in LUAD (O'Grady et al., 2014). Briefly, the DDR comprises of several functional layers including sensors (e.g., MRN complex, RPA, ATRIP), signaling kinases (e.g., ATM, ATR), damage mediators (e.g., 53BP1, BRCA1/2, H2AX), downstream kinases (e.g., CHK1/2), and cell cycle checkpoint effectors (e.g., p53, p21, WEE1). Unsurprisingly, defects at each level have been reported to regulate sensitivity to cisplatin in a variety of cancers including LUAD. Perhaps the best example of this is the well-reported link between BRCA1/2 mutations and sensitivity to cisplatin in breast cancer (Tutt et al., 2018). Similarly, the BRACness phenotype, which is defined as any defect that impacts HR repair and phenocopies the mutation or loss BRCA1/2 (Byrum et al., 2019b), is also strongly linked with sensitivity to platinum and PARP inhibitors (Ding et al., 2019), especially in ovarian (Pillay et al., 2019) and breast cancer (Tung and Garber, 2018). The links with BRCA1/2 mutations, BRACness and cisplatin sensitivity are less clear in LUAD. Although recent reports indicate that DNA methyltransferase inhibitors can induce a BRACness phenotype in NSCLC cells, sensitizing them to PARP inhibitors (Abbotts et al., 2019), and hence may extend to other DNA damage chemotherapies such as cisplatin (Figure 1). Early preclinical studies showed significant promise for directly inhibiting ATR kinase activity (Hall et al., 2014; Vendetti et al., 2015) to enhance cisplatin killing of LUAD cells. Interestingly, inhibition of ATM does not appear to synergise with cisplatin (Schmitt et al., 2017), although it may reduce the metastatic potential of cisplatin resistant LUAD cells (Shen et al., 2019). Furthermore, co-depletion of ATM and MCL-1 can re-sensitize cells to cisplatin (Zhang et al., 2017). While phase 1/2 trials of the CHK1 inhibitor LY2603618 in combination with cisplatin showed promising anti-tumor activity, but also caused significant thromboembolic side-effects (Wehler et al., 2017), indicating that despite promising results in SCLC (Sen et al., 2017; Hsu W.-H. et al., 2019; Nagel et al., 2019), Chk1 inhibitors may not translate to LUAD. Indirectly targeting the DDR has also shown some promise, with inhibition of the JMJD2 histone demethylase family re-sensitizing resistant LUAD to cisplatin by preventing ATR association to sites of DNA damage, thereby weakening the DDR (Duan et al., 2019). Similarly, targeting specific forms of the PP2A phosphatase complex (PPP2R2A2), which are responsible for dephosphorylating and inactivating ATM and ATR, enhanced sensitivity to PARP inhibition in LUAD by maintaining the DDR response (Kalev et al., 2012). What is becoming clear is that there are a number of non-canonical DDR pathways, many of which become upregulated during oncogenesis and can increase replication fork stability and counterbalance BRACness and BRCA mutations (Chaudhuri et al., 2016). A surprising recent example is the discovery that the mitotic kinase Aurora A and its targeting factor TPX2 can regulate 53BP1 and HR repair in a pathway parallel to BRCA1 (Byrum et al., 2019a), possibly explaining why shRNA knockdown of Aurora A sensitized LUAD cells to cisplatin (Cheng et al., 2016). These results highlight the need for additional research that maps all of the pathways regulating the DDR in LUAD.
TGF-β Signaling, EMT and Resistance
The sensing and repair of cisplatin adducts does not happen in isolation from the rest of the cell or its local environment. The DDR signaling pathway is intimately integrated into multiple signaling networks, with a prime example being the transforming growth factor β (TGF-β) pathway. TGF-β regulates a multitude of cellular pathways including the DDR, cellular proliferation and the epithelial-mesenchymal transition (EMT). It plays both positive and negative roles in cancer development and progression. In established tumors, high TGF-β expression can drive metastasis, tumor heterogeneity and chemoresistance (Li J. et al., 2019). We recently demonstrated that members of the TGF-β pathway, including ACVR1B, TGFBR1, TAK1 and GDF11, mediated innate cisplatin resistance in LUAD (Figure 1), a possible consequence of epithelial airway cell lineage (Kretser et al., 2011). Critically, inhibition of activin receptor signaling reversed the resistance, as did blockage of activin A and GDF11 by the endogenous protein Follistatin (Marini et al., 2018). The mechanisms for TGF-β resistance are multifaceted, likely acting to suppress cell proliferation, apoptosis, and the DDR. In support, the antiapoptotic protein MCL-1 decreased upon chemical inhibition of the TGF-β pathway in cisplatin treated cells (Marini et al., 2018). TAK1 has also recently been shown to phosphorylate p38 MAPK and IKKα after DNA damage (Colomer et al., 2019), promoting ATM phosphorylation and increasing DNA repair, leading to chemoresistance. In turn, ATM can feedback into the TGF-β pathway, phosphorylating c-Cbl, stabilizing TβRII receptor and activating TFG-β signaling (Li Y. et al., 2019), creating a positive feedback loop (Figure 1). TGF-β can also drive EMT (Hao et al., 2019) and chemoresistance (Fischer et al., 2015), in part due increased YAP1 mediated transcription of TGF-β target genes (Pefani et al., 2016). Consequently, TGFBR1 and YAP1 inhibitors have been shown to be synergistic in GATA4 deficient (Hao et al., 2019) and EGFR-mutant (Cheng et al., 2016) lung cancers, respectively (Gao et al., 2019), offering another potential therapeutic approach to enhancing cisplatin selectivity.
Cell Cycle and Apoptosis
In general, non-cycling cells are more resistant to cytotoxic chemotherapies such as cisplatin, however, proliferating cells that increase repair or reduce death signaling are more resistant, and often more deadly. Once a proliferating cell encounters DNA damage it must halt cell cycle progression so that repair can occur. If the damage is deemed too great, then apoptosis will be initiated, thereby preventing the damage being passed on to subsequent generations. The key central regulator of this decision pathway is p21waf1/kip, which inhibits G1 and G2 cell cycle progression (Burgess et al., 2019) and blocks caspase 3 dependent apoptosis (Suzuki et al., 2000). Interestingly, intermediate “goldilocks” levels of p21 strongly correlate with continued cell proliferation post cisplatin exposure, while low or high levels result in damaged cells undergoing senescence (Hsu C.-H et al., 2019). Similarly, over-riding the protective cell cycle checkpoints in S and G2 phase through WEE1 inhibition has also shown promise, especially in p53 null and mutant cell lines (Jhuraney et al., 2016; Richer et al., 2017). Interestingly, some resistant cycling cells become highly dependent on glutamine for a multitude of metabolic reactions. Consequently, removal of glutamine makes resistant cells highly sensitive to cisplatin, and lowers the threshold required to trigger apoptosis (Guidi and Longo, 2018). Similarly, metformin, which blocks glucose uptake and ATP production, has also been linked with increasing sensitivity to cisplatin (Liu et al., 2017; Riaz et al., 2019). While, inhibition of NRF2, which protects against hypoxia and reactive oxygen species (ROS), synergises with cisplatin by enhancing DNA damage (Singh et al., 2016; Shi et al., 2019). Notably, NRF2 is commonly upregulated in LUAD by KRAS (Tao et al., 2014) and mutant p53 (Tung et al., 2015). Disrupting apoptosis is another common mechanism, with upregulation of MAST1 in LUAD cells resulting in a rewiring of downstream MEK signaling and a reduction in pro-apoptotic protein Bim (Figure 1), thereby increasing the threshold required to trigger apoptosis (Jin et al., 2018). Likewise, mutations in SET containing 2 (SETD2), a histone methyltransferase, confers cisplatin resistance in LUAD by altering ERK signaling and inhibiting apoptosis (Kim et al., 2019). While, as mention, TAK1-p38 signaling results in an increase in anti-apoptotic MCL1 levels, raising the threshold required to trigger apoptosis (Marini et al., 2018).
Discussion and Conclusion
A more complete understanding of the signaling, repair and apoptotic networks that are re-wired in LUAD will be key to improving platinum efficacy in LUAD. In addition, better temporal information on the dynamic nature of the signaling responses will greatly aid in the identification and prediction of resistance mechanisms. Any models will need to take into account cell cycle status, repair pathway and apoptotic thresholds in order to identify suitable synergising treatments. Finally, better preclinical models that more accurately model the dosing of platinum will be essential. Currently, the majority of studies rely on prolonged exposure, often >10-fold higher than what is achievable in patients (Urien and Lokiec, 2004; Jacobs et al., 2005). Screening of synergistic treatments using this extreme exposure may have increased the rate of false positives and failure of some preclinical studies to translate clinically. This is further cofounded by the disparate effect that platinum has on various organs (Yimit et al., 2019), especially the kidneys. Consequently, treatments such as Follistatin (Marini et al., 2018), which not only protect these vital organs but also enhance tumor selective killing, may have significant clinical potential. In summary, the advent of large-scale screens combined with detailed preclinical studies has given a greater understanding of the mechanisms of cisplatin resistance in LUAD, breathing new life into this stalwart of chemotherapy.
Author Contributions
AG-R and JH co-wrote the initial draft. DW and DC co-wrote and edited the manuscript. AB conceived and wrote the article.
Funding
AB was supported by a NBCF Investigator Initiated Research Scheme (IIRS-18-103). DC is also supported by NBCF Investigator Initiated Research Schemes (IIRS-18-121 and IIRS-20-032).
Conflict of Interest
DW is a coinventor on a patent application relating to components of this work (U.S. 20180125936-A1).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbotts, R., Topper, M. J., Biondi, C., Fontaine, D., Goswami, R., Stojanovic, L., et al. (2019). DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. U.S.A. 116, 22609–22618. doi: 10.1073/pnas.1903765116
Arbour, K. C., and Riely, G. J. (2019). Systemic therapy for locally advanced and metastatic non-small cell lung cancer. JAMA 322, 764–774. doi: 10.1001/jama.2019.11058
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Bruno, P. M., Liu, Y., Park, G. Y., Murai, J., Koch, C. E., Eisen, T. J., et al. (2017). A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 23, 4614471. doi: 10.1038/nm.4291
Burgess, A., Vuong, J., Marzec, K. A., Lichtenberg, U. N., de O'Donoghue, S. I., and Jensen, L. J. (2019). SnapShot: S-phase entry and exit. Cell 179, 802–802.e1. doi: 10.1016/j.cell.2019.09.031
Byrum, A. K., Carvajal-Maldonado, D., Mudge, M. C., Valle-Garcia, D., Majid, M. C., Patel, R., et al. (2019a). Mitotic regulators TPX2 and aurora A protect DNA forks during replication stress by counteracting 53BP1 function. J. Cell Biol. 218, 422–432. doi: 10.1083/jcb.201803003
Byrum, A. K., Vindigni, A., and Mosammaparast, N. (2019b). Defining and modulating ‘BRCAness.' Trends. Cell. Biol 29, 740–751. doi: 10.1016/j.tcb.2019.06.005
Chaudhuri, A. R., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J.-E., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535:3823387. doi: 10.1038/nature19826
Cheng, H., Zhang, Z., Rodriguez-Barrueco, R., Borczuk, A., Liu, H., Yu, J., et al. (2016). Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 7, 28976–28988. doi: 10.18632/oncotarget.6721
Colomer, C., Margalef, P., Villanueva, A., Vert, A., Pecharroman, I., Solé, L., et al. (2019). IKKα kinase regulates the DNA damage response and drives chemo-resistance in cancer. Mol. Cell 75, 669-682.e5. doi: 10.1016/j.molcel.2019.05.036
Ding, H., Zhao, J., Zhang, Y., Yu, J., Liu, M., Li, X., et al. (2019). Systematic analysis of drug vulnerabilities conferred by tumor suppressor loss. Cell Reports 27, 3331–3344.e6. doi: 10.1016/j.celrep.2019.05.043
Duan, L., Perez, R. E., Chastain, P. D., Mathew, M. T., Bijukumar, D. R., and Maki, C. G. (2019). JMJD2 promotes acquired cisplatin resistance in non-small cell lung carcinoma cells. Oncogene 38, 5643–5657. doi: 10.1038/s41388-019-0814-6
Enoiu, M., Jiricny, J., and Schärer, O. D. (2012). Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 40, 8953–8964. doi: 10.1093/nar/gks670
Fischer, K. R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S. T. C., et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527:472. doi: 10.1038/nature15748
Gao, L., Hu, Y., Tian, Y., Fan, Z., Wang, K., Li, H., et al. (2019). Lung cancer deficient in the tumor suppressor GATA4 is sensitive to TGFBR1 inhibition. Nat. Commun. 10:1665. doi: 10.1038/s41467-019-09295-7
Guidi, N., and Longo, V. D. (2018). Periodic fasting starves cisplatin-resistant cancers to death. EMBO J. 37:e99815. doi: 10.15252/embj.201899815
Hall, A. B., Newsome, D., Wang, Y., Boucher, D. M., Eustace, B., Gu, Y., et al. (2014). Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 5, 5674–5685. doi: 10.18632/oncotarget.2158
Hao, Y., Baker, D., Dijke, P., and ten (2019). TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 20:2767. doi: 10.3390/ijms20112767
Herbst, R. S., Morgensztern, D., and Boshoff, C. (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. doi: 10.1038/nature25183
Hsu, C.-H., Altschuler, S. J., and Wu, L. F. (2019). Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 178, 361–373. doi: 10.1016/j.cell.2019.05.041
Hsu, W.-H., Zhao, X., Zhu, J., Kim, I.-K., Rao, G., McCutcheon, J., et al. (2019). Chk1 inhibition enhances cisplatin cytotoxicity and overcomes cisplatin resistance in small cell lung cancer by promoting mitotic cell death. J. Thorac. Oncol. 14, 1032–1045. doi: 10.1016/j.jtho.2019.01.028
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jacobs, S. S., Fox, E., Dennie, C., Morgan, L. B., McCully, C. L., and Balis, F. M. (2005). Plasma and cerebrospinal fluid pharmacokinetics of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin. Cancer Res. 11:166911674. doi: 10.1158/1078-0432.CCR-04-1807
Jhuraney, A., Woods, N. T., Wright, G., Rix, L., Kinose, F., Kroeger, J. L., et al. (2016). PAXIP1 potentiates the combination of WEE1 Inhibitor AZD1775 and platinum agents in lung cancer. Mol. Cancer Ther. 15, 1669–1681. doi: 10.1158/1535-7163.MCT-15-0182
Jin, L., Chun, J., Pan, C., Li, D., Lin, R., Alesi, G. N., et al. (2018). MAST1 drives cisplatin resistance in human cancers by rewiring cRaf-independent MEK activation. Cancer Cell 34, 315–330.e7. doi: 10.1016/j.ccell.2018.06.012
Kalev, P., Simicek, M., Vazquez, I., Munck, S., Chen, L., Soin, T., et al. (2012). Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res. 72, 6414–6424. doi: 10.1158/0008-5472.CAN-12-1667
Karanam, K., Kafri, R., Loewer, A., and Lahav, G. (2012). Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 47, 320–329. doi: 10.1016/j.molcel.2012.05.052
Kim, I.-K., McCutcheon, J. N., Rao, G., Liu, S. V., Pommier, Y., Skrzypski, M., et al. (2019). Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene 38, 180–193. doi: 10.1038/s41388-018-0429-3
Koster, R., van Vugt, M. A., Timmer-Bosscha, H., Gietema, J. A., and de Jong, S. (2013). Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 15:e12. doi: 10.1017/erm.2013.13
Kretser, D. M., de O'Hehir, R. E., Hardy, C. L., and Hedger, M. P. (2011). The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell Endocrinol. 359, 101–106. doi: 10.1016/j.mce.2011.10.009
Li, J., Shen, C., Wang, X., Lai, Y., Zhou, K., Li, P., et al. (2019). Prognostic value of TGF-β in lung cancer: systematic review and meta-analysis. Bmc Cancer 19:691. doi: 10.1186/s12885-019-5917-5
Li, Y., Liu, Y., Chiang, Y. J., Huang, F., Li, Y., Li, X., et al. (2019). DNA daimage activates TGF-β signaling via ATM-c-Cbl-mediated stabilization of the Type II receptor TβRII. Cell Reports 28, 735–745.e4. doi: 10.1016/j.celrep.2019.06.045
Liu, Y., He, C., and Huang, X. (2017). Metformin partially reverses the carboplatin-resistance in NSCLC by inhibiting glucose metabolism. Oncotarget 8:75206775216. doi: 10.18632/oncotarget.20663
Marini, K. D., Croucher, D. R., McCloy, R. A., Vaghjiani, V., Gonzalez-Rajal, A., Hastings, J. F., et al. (2018). Inhibition of activin signaling in lung adenocarcinoma increases the therapeutic index of platinum chemotherapy. Sci. Transl. Med. 10:eaat3504. doi: 10.1126/scitranslmed.aat3504
Mathew, M., Enzler, T., Shu, C. A., and Rizvi, N. A. (2018). Combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol. Therapeut. 186, 130–137. doi: 10.1016/j.pharmthera, 2018.01.003
Matt, S., and Hofmann, T. G. (2016). The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol. Life Sci. 73, 2829–2850. doi: 10.1007/s00018-016-2130-4
Michl, J., Zimmer, J., and Tarsounas, M. (2016). Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. Embo J. 35, 909–923. doi: 10.15252/embj.201693860
Nagel, R., Avelar, A. T., Aben, N., Proost, N., van de Ven, M., van der Vliet, J., et al. (2019). Inhibition of the replication stress response is a synthetic vulnerability in SCLC that acts synergistically in combination with cisplatin. Mol. Cancer Ther. 18, 762–770. doi: 10.1158/1535-7163.MCT-18-0972
Niraj, J., Färkkilä, A., and D'Andrea, A. D. (2019). The fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 3, 457–478. doi: 10.1146/annurev-cancerbio-030617-050422
O'Connor, M. J. (2015). Targeting the DNA damage response in cancer. Mol. Cell 60, 547–560. doi: 10.1016/j.molcel.2015.10.040
O'Grady, S., Finn, S. P., Cuffe, S., Richard, D. J., O'Byrne, K. J., and Barr, M. P. (2014). The role of DNA repair pathways in cisplatin resistant lung cancer. Cancer Treatment Rev. 40, 116111170. doi: 10.1016/j.ctrv.2014.10.003
Pearl, L. H., Schierz, A. C., Ward, S. E., Al-Lazikani, B., and Pearl, F. M. G. (2015). Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180. doi: 10.1038/nrc3891
Pefani, D.-E., Pankova, D., Abraham, A. G., Grawenda, A. M., Vlahov, N., Scrace, S., et al. (2016). TGF-β targets the hippo pathway scaffold RASSF1A to facilitate YAP/SMAD2 nuclear translocation. Mol. Cell 63, 156–166. doi: 10.1016/j.molcel.2016.05.012
Pillay, N., Tighe, A., Nelson, L., Littler, S., Coulson-Gilmer, C., Bah, N., et al. (2019). DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-Ribose) glycohydrolase inhibitors. Cancer Cell 35, 519–533.e8. doi: 10.1016/j.ccell.2019.02.004
Riaz, M. A., Sak, A., Erol, Y. B., Groneberg, M., Thomale, J., and Stuschke, M. (2019). Metformin enhances the radiosensitizing effect of cisplatin in non-small cell lung cancer cell lines with different cisplatin sensitivities. Sci Rep-uk 9:1282. doi: 10.1038/s41598-018-38004-5
Richer, A. L., Cala, J. M., O'Brien, K., Carson, V. M., Inge, L. J., and Whitsett, T. G. (2017). WEE1 kinase inhibitor AZD1775 has preclinical efficacy in LKB1-deficient non-small cell lung cancer. Cancer Res 77, 4663–4672. doi: 10.1158/0008-5472.CAN-16-3565
Rosenberg, B., Vancamp, L., Trosko, J. E., and and, Mansour, V. H. (1969). Platinum compounds: a new class of potent antitumour agents. Nature 222, 385–386. doi: 10.1038/222385a0
Schmitt, A., Knittel, G., Welcker, D., Yang, T.-P., George, J., Nowak, M., et al. (2017). ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res 77, 3040–3056. doi: 10.1158/0008-5472.CAN-16-3398
Sen, T., Tong, P., Stewart, C. A., Cristea, S., Valliani, A., Shames, D. S., et al. (2017). CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res. 77, 3870–3884. doi: 10.1158/0008-5472.CAN-16-3409
Shen, M., Xu, Z., Xu, W., Jiang, K., Zhang, F., Ding, Q., et al. (2019). Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Canc. Res. 38:149. doi: 10.1186/s13046-019-1161-8
Shi, Y., Fan, S., Wu, M., Zuo, Z., Li, X., Jiang, L., et al. (2019). YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat. Commun. 10:4892. doi: 10.1038/s41467-019-12801-6
Singh, A., Venkannagari, S., Oh, K. H., Zhang, Y.-Q., Rohde, J. M., Liu, L., et al. (2016). Small molecule inhibitor of NRF2 Selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. Acs Chem. Biol. 11, 3214–3225. doi: 10.1021/acschembio.6b00651
Slyskova, J., Sabatella, M., Ribeiro-Silva, C., Stok, C., Theil, A. F., Vermeulen, W., et al. (2018). Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 46, 9537–9549. doi: 10.1093/nar/gky764
Smogorzewska, A. (2019). Fanconi anemia: a paradigm for understanding DNA repair during replication. Blood 134, SCI-32-SCI-32. doi: 10.1182/blood-2019-121229
Stewart, D. J. (2007). Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hemat. 63, 12–31. doi: 10.1016/j.critrevonc.2007.02.001
Stordal, B., and Davey, M. (2007). Understanding cisplatin resistance using cellular models. IUBMB Life 59:6966699. doi: 10.1080/15216540701636287
Suzuki, A., Kawano, H., Hayashida, M., Hayasaki, Y., Tsutomi, Y., and Akahane, K. (2000). Procaspase 3/p21 complex formation to resist Fas-mediated cell death is initiated as a result of the phosphorylation of p21 by protein kinase A. Cell Death Differ. 7, 721–728. doi: 10.1038/sj.cdd.4400706
Tao, S., Wang, S., Moghaddam, S. J., Ooi, A., Chapman, E., Wong, P. K., et al. (2014). Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 74, 7430–7441. doi: 10.1158/0008-5472.CAN-14-1439
Tung, M.-C., Lin, P.-L., Wang, Y.-C., He, T.-Y., Lee, M.-C., Yeh, S. D., et al. (2015). Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 6, 41692–41705. doi: 10.18632/oncotarget.6150
Tung, N. M., and Garber, J. E. (2018). BRCA1/2 testing: therapeutic implications for breast cancer management. Brit. J. Cancer 119, 141–152. doi: 10.1038/s41416-018-0127-5
Tutt, A., Tovey, H., Cheang, M. C. U., Kernaghan, S., Kilburn, L., Gazinska, P., et al. (2018). Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat. Med. 24, 628–637. doi: 10.1038/s41591-018-0009-7
Urien, S., and Lokiec, F. (2004). Population pharmacokinetics of total and unbound plasma cisplatin in adult patients. British J. Clin. Pharmacol. 57:7567763. doi: 10.1111/j.1365-2125.2004.02082.x
Vendetti, F. P., Lau, A., Schamus, S., Conrads, T. P., O'Connor, M. J., and Bakkenist, C. J. (2015). The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 6, 44289–44305. doi: 10.18632/oncotarget.6247
Walters, S., Maringe, C., Coleman, M. P., Peake, M. D., Butler, J., Young, N., et al. (2013). Lung cancer survival and stage at diagnosis in Australia, Canada, Denmark, Norway, Sweden and the UK: a population-based study, 2004-2007. Thorax 68, 551–564. doi: 10.1136/thoraxjnl-2012-202297
Wang, D., and Lippard, S. J. (2005). Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4, 307–320. doi: 10.1038/nrd1691
Wehler, T., Thomas, M., Schumann, C., Bosch-Barrera, J., Segarra, N. V., Dickgreber, N. J., et al. (2017). A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 108, 212–216. doi: 10.1016/j.lungcan.2017.03.001
Yimit, A., Adebali, O., Sancar, A., and Jiang, Y. (2019). Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 10:309. doi: 10.1038/s41467-019-08290-2
Keywords: cisplatin, TGF-β, DNA damage, DNA repair, follistatin, cell cycle, p21, p53
Citation: Gonzalez-Rajal A, Hastings JF, Watkins DN, Croucher DR and Burgess A (2020) Breathing New Life into the Mechanisms of Platinum Resistance in Lung Adenocarcinoma. Front. Cell Dev. Biol. 8:305. doi: 10.3389/fcell.2020.00305
Received: 17 December 2019; Accepted: 07 April 2020;
Published: 08 May 2020.
Edited by:
Weimin Li, Washington State University, United StatesReviewed by:
Joseph William Landry, Virginia Commonwealth University, United StatesCopyright © 2020 Gonzalez-Rajal, Hastings, Watkins, Croucher and Burgess. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew Burgess, YW5kcmV3LmJ1cmdlc3NAc3lkbmV5LmVkdS5hdQ==