 Tae-Un Han
Tae-Un Han Richard Sam
Richard Sam Ellen Sidransky
Ellen Sidransky- Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
Parkinson disease, the second most common movement disorder, is a complex neurodegenerative disorder hallmarked by the accumulation of alpha-synuclein, a neural-specific small protein associated with neuronal synapses. Mutations in the glucocerebrosidase gene (GBA1), implicated in the rare, autosomal recessive lysosomal disorder Gaucher disease, are the most common known genetic risk factor for Parkinson disease. Insights into the inverse relationship between glucocerebrosidase and alpha-synuclein have led to new therapeutic approaches for the treatment of Gaucher disease and GBA1-associated Parkinson disease. Unlike the current drugs used to treat Gaucher disease, which are highly expensive and do not cross the blood-brain-barrier, new small molecules therapies, including competitive and non-competitive chaperones that enhance glucocerebrosidase levels are being developed to overcome these limitations. Some of these include iminosugars, ambroxol, other competitive glucocerebrosidase inhibitors, and non-inhibitory chaperones or activators that do not compete for the active site. These drugs, which have been shown in different disease models to increase glucocerebrosidase activity, could have potential as a therapy for Gaucher disease and GBA1- associated Parkinson disease. Some have been demonstrated to reduce α-synuclein levels in pre-clinical studies using cell-based or animal models of GBA1-associated Parkinson disease, and may also have utility for idiopathic Parkinson disease.
Introduction
Gaucher disease (GD), first described in 1882 by Dr. Phillipe Gaucher, is an inborn error of metabolism due to mutations in the gene GBA1, encoding the lysosomal enzyme glucocerebrosidase (GCase) (Sidransky, 2012). GCase, a lysosomal hydrolase, cleaves both glucosylceramide (GlcCer) to ceramide and glucose, and glucosylsphingosine (GlcSph) into sphingosine and glucose. This rare autosomal recessive inherited disorder occurs in approximately 1:40,000–60,000 live births in the general population, whereas among Ashkenazi Jews the frequency increases to around 1:850 (Horowitz et al., 1998). In patients with GD, “Gaucher cells,” macrophages engorged with lysosomal GlcCer, present in the spleen, liver, and bone marrow, lead to organomegaly and inflammation (Pastores et al., 2000; Beutler and Grabowski, 2001; Sidransky, 2012). Clinically, there are three types of GD. Type 1 or non-neuronopathic GD presents with organomegaly, bone manifestations, anemia, and thrombocytopenia (Pastores et al., 2000; Beutler and Grabowski, 2001). Acute neuronopathic GD2, the rarest form with devastating and progressive neurodegeneration including bulbar findings, seizures, and opisthotonos, results in death during infancy or early childhood (Beutler and Grabowski, 2001; Sidransky, 2004; Weiss et al., 2015). Chronic neuronopathic GD3 is characterized by a slower progression of neurological symptoms, with a later onset. These patients have extensive visceral and skeletal involvement, but also may exhibit myoclonic epilepsy, ataxia, impaired eye movements, developmental delay, or intellectual deterioration (Sidransky, 2004, 2012; Gupta et al., 2011; Sidransky and Lopez, 2012). There is considerable phenotypic variation within each type of GD, including rare patients who develop Parkinson disease (PD).
Parkinson disease, first described in 1817 by James Parkinson, is found in 2–3% of individuals over age 65 (Poewe et al., 2017). This disease is characterized by the loss of dopaminergic (DA) neurons in the substantia nigra, as well as by Lewy bodies containing alpha-synuclein (α-Syn) (Spillantini et al., 1997; Surmeier et al., 2017), which ultimately result in the classically associated symptoms, tremor, postural instability, bradykinesia, and rigidity (Poewe et al., 2017). Currently, mutations in GBA1 are one of the most common genetic risk factors for PD, as well as dementia with Lewy bodies (DLB). Mutations in GBA1 were found in 5–20% of sporadic PD cases with an estimated odds ratio of 5.4. In DLB the odds ratio was 8.3 (Sidransky et al., 2009; Nalls et al., 2013; Mullin et al., 2019). Patients with GBA1-associated PD (GBA1-PD) resemble those with sporadic PD but exhibit an earlier disease onset. The lysosome, the central player uniting GD and PD, is the main organelle responsible for α-Syn degradation (Sala et al., 2016). While the mechanism underlying the association between GD and PD still remains controversial, one hypothesis proposed posits that elevated substrate levels resulting from diminished GCase could stabilize soluble oligomeric α-Syn intermediates, enhancing oligomerization and accumulation of α-Syn within lysosomes (Yap et al., 2011; Taguchi et al., 2017). Increased levels of α-Syn in DA neurons could also inhibit the translocation of GCase from the endoplasmic reticulum (ER) to lysosomes which could lead to a gradual increase of the lipid substrates within the lysosome (Mazzulli et al., 2016a). This positive feedback loop with the combination of inhibited GCase trafficking and α-Syn accumulation may result in neurodegeneration (Balestrino and Schapira, 2018; Stojkovska et al., 2018). Furthermore, even patients with PD without GBA1 mutations have lower levels of GCase, suggesting that in idiopathic PD, reduced GCase activity may also contribute to disease progression (Gegg et al., 2012; Murphy et al., 2014).
Due to this apparent reciprocal association between GCase and α-Syn, attention is being directed toward designing therapeutics for GD with implications for PD. Currently, patients with type 1 GD are commonly treated with enzyme replacement therapy (ERT) (Jung et al., 2016) which alleviates the hematological, visceral, and sometimes skeletal manifestations, but does not cross the blood-brain-barrier, and thus does not impact neurological involvement or PD manifestations (Valayannopoulos, 2013). Substrate reduction therapy (SRT), a second approach for treating GD, focuses on upstream targets, ultimately inhibiting the accumulation of GlcCer (Lukina et al., 2019). Two SRT drugs approved by FDA have shown efficacy in type 1 GD (Schiffmann et al., 2008; Bennett and Turcotte, 2015) and some cross the blood-brain-barrier. However, the current SRTs have not been efficacious for neuronopathic GD (nGD) (Bennett and Turcotte, 2015; Mistry et al., 2018; Zimran et al., 2018) and there are several pharmacokinetic limitations (Henley et al., 2014).
Pharmacological chaperones, small molecules designed to bind to a specific target protein and assist in the folding of the protein, are also being developed as an alternative treatment approach for GD or GD-associated PD. They further function by stabilizing the enzyme to prevent misfolding and to enhance accurate translocation of the protein from the ER to the Golgi and lysosome (Compain et al., 2006; Lieberman et al., 2007). Since most mutations encountered in GD or GBA1-PD are missense mutations leading to misfolded enzyme, pharmacological chaperones which cross the blood-brain-barrier provide an attractive therapeutic strategy. Here we review studies of chaperones as a treatment for GD and GBA1-PD, focusing on preclinical developments.
Iminosugars
The first chaperone considered for GCase was an iminosugar, N-(n-nonyl) deoxynojirimycin (NN-DNJ). Sawkar et al. (2002) reported that the addition of NN-DNJ to fibroblast culture medium increased the activity of GCase in N370S mutant and wild-type but not L444P mutant cells. However, NN-DNJ also inhibits α-glucosidase and GlcCer synthase, rendering it difficult to use as a target-specific drug. Another iminosugar, α-1-C-Nonyl-DIX that specifically inhibits GCase, but not α-glucosidase, doubled the residual cellular activity of GCase in N370S/N370S fibroblasts (Compain et al., 2006). Bicyclic nojirimycin (NJ) analogs with a sp2-iminosugar structure were found to be stronger chaperones as well as selective and competitive inhibitors of lysosomal GCase (Luan et al., 2009, 2010; Tiscornia et al., 2013). Some of the new sp2-iminosugar derivatives inhibited enzyme activity by 10-fold at pH 5 (Mena-Barragan et al., 2016).
Another type of iminosugar, isofagomine (IFG), was reported to bind to the GCase active site, increase GCase activity in cell lysates and restore lysosomal trafficking of GCase in N370S cells (Lieberman et al., 2007). Incubation of GD patient-derived lymphoblastoid or fibroblast lines with IFG increased GCase activity 3.5- and 1.3-fold and reduced endogenous GlcCer levels (Khanna et al., 2010). Testing the effects of IFG on GCase activity in a complex nGD mouse model, 4L;C∗ (V394L/V394L + saposin C−/−) showed that IFG administration extended lifespan and increased GCase activity and protein levels in the brain and visceral tissue, with attenuation of proinflammatory responses (Sun et al., 2011). Further, in vivo tests of IFG in homozygous V394L, D409H, and D409V mice, which are nGD models demonstrated increased GCase activity in visceral tissues and brain extracts (Sun et al., 2012). Whether the effect of IFG on wild-type GCase could be beneficial in synucleinopathies was evaluated using mice overexpressing human wild-type α-Syn (Thy1- α-Syn). Treating with IFG (AT2101) orally for 4 months improved motor function, diminished microglial inflammatory response in the substantia nigra, reduced α-Syn immunoreactivity in nigral DA neurons, and reduced small α-Syn aggregates (Richter et al., 2014). Another study in 4L;C∗ mice reported that IFG did not alter the GlcCer and GlcSph accumulation, but attenuated disease progression and altered global expression profiles of brain mRNA and miRNAs (Dasgupta et al., 2015). Treating Drosophila manipulated to express human wild-type, N370S and L444P GBA1, with IFG resulted in decreased ER stress and preserved motor function, suggesting that IFG might have potential as a PD therapy (Sanchez-Martinez et al., 2016).
De La Mata et al. reported that a distinct iminosugar, NAdBT-AIJ restored mitochondrial dysfunction and GCase activity in L444P mice in combination with coenzyme Q10 (De La Mata et al., 2015). Another pyrrolidine-based iminosugar, α-1-C-tridecyl-DAB (5j) was shown to function as a chaperone inhibitor of GCase enhancing enzyme activity at concentrations 10 times lower than IFG (Kato et al., 2016).
Ambroxol
Ambroxol (ABX), a drug used to treat airway mucus hypersecretion and hyaline membrane disease in newborns, was found to be a pH-dependent, mixed-type inhibitor of GCase, and thus a potential therapy for GD. With its inhibitory activity maximal at neutral pH, ABX is found in the ER, and is undetectable at the acidic pH of lysosomes (Maegawa et al., 2009). GCase activity was enhanced in ABX-treated GD fibroblasts and lymphoblasts. Furthermore, modeling studies indicated that ABX interacts with both active site and non-active site residues of GCase (Maegawa et al., 2009). The tolerability and efficacy of ABX was evaluated in 12 patients with type 1 GD not receiving ERT. While only three patients who continued on ABX for a year had improved platelet counts and decreased organ volumes, the others remained stable, supporting the need for a larger clinical trial (Zimran et al., 2013). The chaperone activity and cytotoxicity of ABX was tested in vitro, demonstrating low cytotoxicity and significantly increased GCase activity in GD and GD+PD fibroblasts with different GD mutations, without any serious adverse effects (Bendikov Bar et al., 2013; Luan et al., 2013; McNeill et al., 2014). Another study in GBA1-mutant fibroblasts showed that ABX enhanced GCase activity by increasing Sap C and LIMP-2 protein levels (Ambrosi et al., 2015). Testing in vivo efficacy of 12 days of ABX in wild-type, L444P carrier and transgenic mice overexpressing human α-Syn demonstrated increased brain GCase activity and decreased total and phosphorylated α-Syn levels (Migdalska-Richards et al., 2016). In nonhuman primates, daily administration of ABX increased brain GCase activity, supporting clinical testing in humans (Migdalska-Richards et al., 2017a). In Drosophila melanogaster with a mutated GBA1b ortholog, treatment with ABX did not rescue GCase activity, but did ameliorate the unfolded protein response, inflammation and neuroinflammation, and enhance the life span (Cabasso et al., 2019). Several pilot clinical studies of ABX were performed in patients with nGD (Narita et al., 2016; Pawlinski et al., 2018; Kim et al., 2019). One study in five patients with GD3 showed that high-dose oral ABX had good safety and tolerability, significantly increased lymphocyte GCase activity, permeated the blood–brain barrier, and decreased GlcSph levels in the cerebrospinal fluid. The investigators reported that myoclonus, seizures, and pupillary light reflex dysfunction improved, leading to the recovery of gross motor function in two patients (Narita et al., 2016). Another study of the long-term safety and efficacy of combined high-dose ABX up to 21 mg/kg/day) and ERT in GD3 showed that during the first 2 years seizure frequency and neurocognitive function worsened, but after the ABX dosage was increased to 27 mg/kg/day, seizure frequency markedly decreased from the baseline, neurocognitive function improved and the drug was tolerated without severe adverse events (Kim et al., 2019). However, these are primarily antidotal reports, and a double-blind placebo-control study is needed. ABX is currently being tested under a single-center phase II clinical trial in 75 subjects with mild to moderate PD by randomizing participants into ABX high-dose (1,050 mg/day), low-dose (525 mg/day), or placebo group (Silveira et al., 2019), and reasonable cerebrospinal fluid levels were attained (Mullin et al., 2020).
Non-Inhibitory Chaperones
A major limitation of inhibitory chaperones is that the chaperone activity must be balanced against the functional inhibition of GCase. In contrast, non-inhibitory chaperones can assist in the folding of mutant GCase in the ER and the translocation to lysosomes without interfering with the active site of the enzyme, and thus can restore enzyme activity in the lysosome. Several non-inhibitory compounds were identified by a high throughput screening (HTS) assay using an extract of spleen from a patient with GD as the source of mutant enzyme (Goldin et al., 2012). In this screen, saposin C and other potential cofactors were present in the extract, likely enhancing the detection of non-inhibitory chaperones. The activities of the compounds were confirmed in subsequent cell-based assays using patient-derived fibroblasts. The screen yielded novel pyrazolopyrimidine-based non-inhibitory pharmacological chaperones (Patnaik et al., 2012). Aflaki et al. (2014) assessed the efficacy of one non-inhibitory chaperone identified by HTS as a drug candidate for GD or GBA1-PD. Since fibroblasts do not store GlcCer, monocyte-derived macrophages from 20 patients with GD and GD iPSC-differentiated macrophages were examined. The non-inhibitory chaperone, NCGC758, enhanced GCase activity, reduced glycolipid storage, and normalized chemotaxis and the production of reactive oxygen species in the macrophages. NCGC758 also reversed inflammatory defects in GD macrophages by inducing autophagy and reducing IL-1b secretion (Aflaki et al., 2016b). Another lead non-inhibitory small molecule, NCGC607, was tested in dopaminergic (DA) neurons differentiated from type 1 and 2 GD and GD+PD iPSCs (Aflaki et al., 2016a). NCGC607 restored GCase activity and protein levels, and reduced glycolipid storage in GD DA neurons, indicating its therapeutic potential for GD. In addition, NCGC607 reduced α-Syn levels in DA neurons from patients with parkinsonism, suggesting that NCGC607 or a derivative of this lead compound may have efficacy as a treatment for PD (Aflaki et al., 2016a). NCGC758 also reduced α-Syn levels and improved lysosomal function in iPSC-derived DA neurons differentiated from patients with GD and PD (Mazzulli et al., 2016b).
Other Small Molecules
Histone deacetylase inhibitors (HDACi) have also been explored for the treatment of GD by modulating a GCase-associated ubiquitin–proteasome pathway (Lu et al., 2011). It was shown that a known HDACi (SAHA) and a unique small-molecule HDACi (LB-205) rescued GCase levels and increased enzymatic activity in fibroblasts derived from patients with GD1 and GD2. It was further shown that HDACi inhibits the deacetylation of heat shock protein (HSP90), resulting in impaired recognition of the mutant peptide by HSP90, thus protecting GCase from degradation (Yang et al., 2013).
Recently, S-181, a new small-molecule capable of stabilizing wild-type GCase was developed. S-181 increased GCase activity in iPSC-derived DA neurons from patients with idiopathic PD, as well as in patients with PD carrying GBA1 mutation c.84dupG and mutations in other PD genes (Burbulla et al., 2019). S-181 treatment in these DA neurons reversed pathogenic phenotypes including the reduced accumulation of oxidized dopamine. It was also shown in Table 1 that treating wild-type and heterozygous D409V mice with S-181 increased GCase activity in both, resulting in reduction of the lipid substrates and α-Syn in brain (Burbulla et al., 2019).
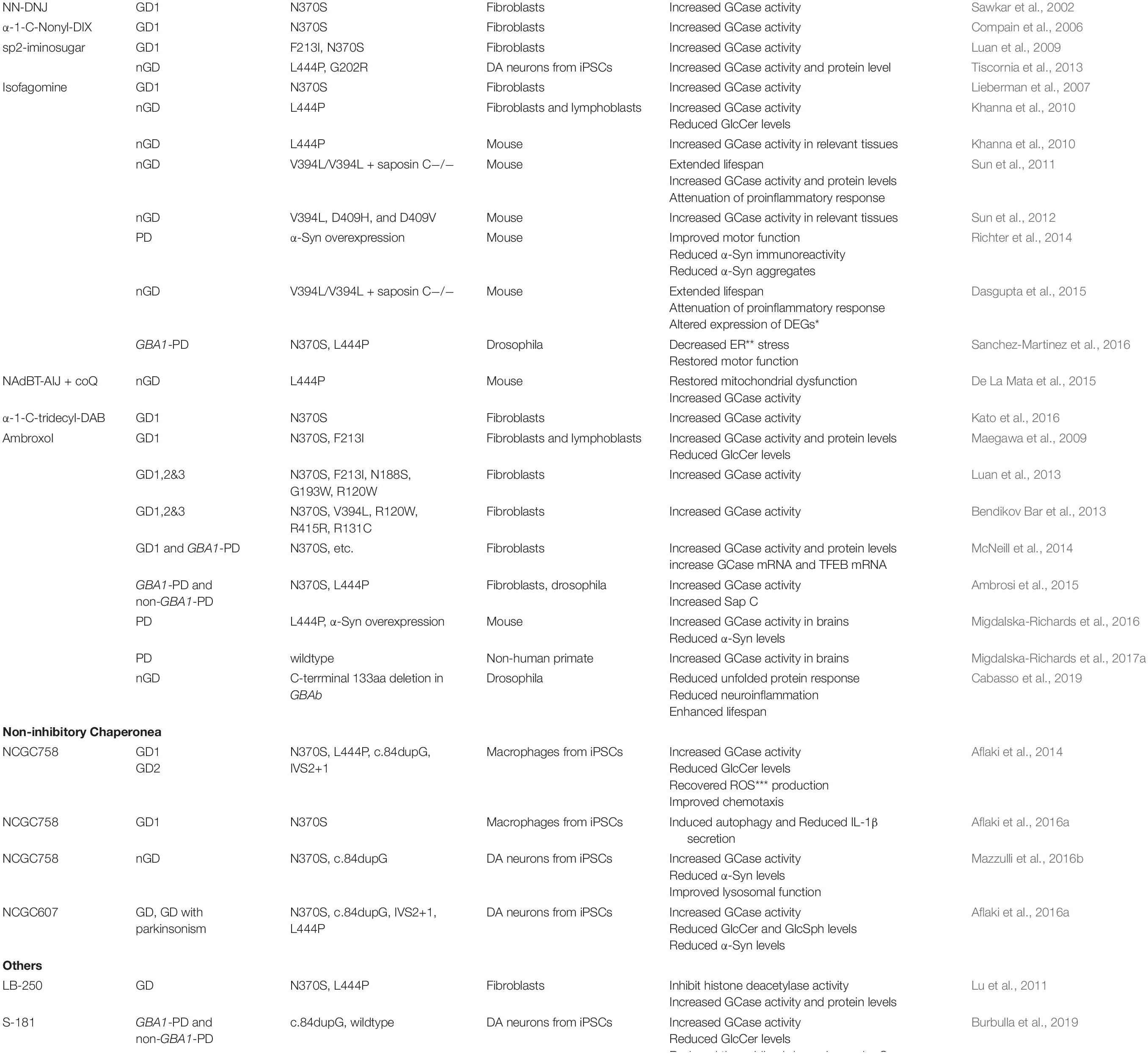
Table 1. Small molecules considered as therapeutics for GD and GBA1-associated PD.
Discussion
There are several FDA-approved ERT (Cerezyme, etc.) and SRT (Zaversca, etc.) drugs to treat GD. However, ERT does not have utility for GBA1-PD since it does not cross the blood-brain- barrier and since substrate accumulation in GBA1-PD is minimal, it is unlikely that SRT will be efficacious (Sidransky et al., 2019). Small molecule pharmacological chaperones have been developed to overcome these limitations. The first iminosugar developed, NN-DNJ, increased GCase activity in cells from patients with GD (Sawkar et al., 2002) by strong binding to GCase (Thirumal Kumar et al., 2019). However, NN-DNJ only increases enzymatic activity in lines with specific GBA1 mutations (Sawkar et al., 2002; Thirumal Kumar et al., 2019) and also inhibits GlcCer synthase activity in a dose-dependent manner (Compain et al., 2006). Isofagomine (IFG) appears to have more target-specific inhibition and broader pre-clinical efficacy in both in vitro and in vivo models of different types of GD and PD. However, like other inhibitory small molecule chaperones, IFG also causes dosage-dependent inhibition of GCase, limiting its clinical utility. A phase II clinical trial of IFG failed to improve clinical symptoms in patients with GD, likely due to dosing challenges. Ambroxol (ABX) has been evaluated as an inhibitory chaperone with fewer side effects and better efficacy, because its inhibitory effect does not occur at the low pH present in lysosomes. A recent comparative molecular docking analysis showed that ABX has more broad binding affinity toward GCase than NN-DNJ and other SRT drugs (Thirumal Kumar et al., 2019). A phase II clinical study of ABX for PD is currently underway (Silveira et al., 2019).
Non-inhibitory chaperones are highly attractive candidate drugs for the treatment of GD or GBA1-PD because they avoid the primary problem associated with inhibitory chaperones, the inhibitory competition with substrates of GCase in the lysosome. A few non-inhibitory chaperones identified by HTS were shown to have disease-reversing effects in cellular models of GD or GBA1-PD (Aflaki et al., 2014, 2016a). However, there are also difficulties in developing non-inhibitory chaperones. The binding of non-inhibitory chaperones to sites other than the active site makes it difficult to perform structure-guided optimization of drug efficacy and challenging to evaluate the potential activity of candidate molecules from HTS. To resolve this problem, visualizing the activity of endogenous levels of GCase in live cells is required. This might be achieved through the development of a fluorescence-based substrate probe representing GCase activity in lysosomes of live cells (Jung et al., 2016).
To increase the likelihood of success in clinical trials, a properly designed pre-clinical drug testing strategy is essential. Thus, appropriate in vitro and in vivo disease models which mimic key features of the disease are cessary. DA neurons differentiated from patient iPSCs accumulate α-Syn and oxidized dopamine, and thus can be used to test the efficacy of small molecules for PD (Aflaki et al., 2016a; Burbulla et al., 2019). However, mouse models accurately replicating PD phenotypes are still needed. A previous study showed that mice carrying the L444P mutation had increased stability of over-expressed human wild-type and A53T mutant α-Syn (Fishbein et al., 2014). However, the L444P mice did not demonstrate accumulation of α-Syn and there was no PD phenotypes in the L444P carrier mice (Migdalska-Richards et al., 2017b). While heterozygous L444P mice overexpressing mutant α-Syn had exacerbated motor deficits (Fishbein et al., 2014) and enhanced dopaminergic neurodegeneration (Migdalska-Richards et al., 2017b), it is not clear whether this phenotype can be used to establish drug efficacy, especially as in patients, the α-Syn levels are not as high, and the protein is not mutated. To identify and optimize candidate non-inhibitory chaperones, new methods for evaluating GCase activity in live cells and more appropriate GBA1-PD animal models need to be developed. Three dimensional in-vitro cell culture systems can be also considered for drug screening and preclinical validation of candidate drugs, because they more closely resembles the in-vivo cell environment than routine in-vitro cell culture (Langhans, 2018).
Author Contributions
T-UH contributed to the conceptual idea, reviewed, analyzed the literature, and wrote the manuscript and table. RS assisted in compiling background material and in writing the manuscript. ES provided the conceptual idea, reviewed, and edited the manuscript.
Funding
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aflaki, E., Borger, D. K., Moaven, N., Stubblefield, B. K., Rogers, S. A., Patnaik, S., et al. (2016a). A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016
Aflaki, E., Moaven, N., Borger, D. K., Lopez, G., Westbroek, W., Chae, J. J., et al. (2016b). Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15, 77–88. doi: 10.1111/acel.12409
Aflaki, E., Stubblefield, B. K., Maniwang, E., Lopez, G., Moaven, N., Goldin, E., et al. (2014). Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med. 6:240ra273. doi: 10.1126/scitranslmed.3008659
Ambrosi, G., Ghezzi, C., Zangaglia, R., Levandis, G., Pacchetti, C., and Blandini, F. (2015). Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 82, 235–242. doi: 10.1016/j.nbd.2015.06.008
Balestrino, R., and Schapira, A. H. V. (2018). Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist 24, 540–559. doi: 10.1177/1073858417748875
Bendikov Bar, I., Maor, G., Filocamo, M., and Horowitz, M. (2013). Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. doi: 10.1016/j.bcmd.2012.10.007
Bennett, L. L., and Turcotte, K. (2015). Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des. Devel. Ther. 9, 4639–4647. doi: 10.2147/DDDT.S77760
Beutler, E., and Grabowski, G. A. (2001). “Gaucher disease,” in The Metabolic & Molecular Bases of Inherited Disease, ed. C. R. Scriver (New York: McGraw-Hill), 3635–3668.
Burbulla, L. F., Jeon, S., Zheng, J., Song, P., Silverman, R. B., and Krainc, D. (2019). A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 11:eaau6870. doi: 10.1126/scitranslmed.aau6870
Cabasso, O., Paul, S., Dorot, O., Maor, G., Krivoruk, O., Pasmanik-Chor, M., et al. (2019). Drosophila melanogaster mutated in its GBA1b ortholog recapitulates neuronopathic gaucher disease. J. Clin. Med. 8:1420. doi: 10.3390/jcm8091420
Compain, P., Martin, O. R., Boucheron, C., Godin, G., Yu, L., Ikeda, K., et al. (2006). Design and synthesis of highly potent and selective pharmacological chaperones for the treatment of Gaucher’s disease. Chembiochem 7, 1356–1359. doi: 10.1002/cbic.200600217
Dasgupta, N., Xu, Y. H., Li, R., Peng, Y., Pandey, M. K., Tinch, S. L., et al. (2015). Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 24, 7031–7048. doi: 10.1093/hmg/ddv404
De La Mata, M., Cotan, D., Oropesa-Avila, M., Garrido-Maraver, J., Cordero, M. D., Villanueva Paz, M., et al. (2015). Pharmacological chaperones and coenzyme Q10 treatment improves mutant β-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci. Rep. 5:10903. doi: 10.1038/srep10903
Fishbein, I., Kuo, Y. M., Giasson, B. I., and Nussbaum, R. L. (2014). Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 137, 3235–3247. doi: 10.1093/brain/awu291
Gegg, M. E., Burke, D., Heales, S. J., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Goldin, E., Zheng, W., Motabar, O., Southall, N., Choi, J. H., Marugan, J., et al. (2012). High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS One 7:e29861. doi: 10.1371/journal.pone.0029861
Gupta, N., Oppenheim, I. M., Kauvar, E. F., Tayebi, N., and Sidransky, E. (2011). Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol. Dis. 46, 75–84. doi: 10.1016/j.bcmd.2010.08.012
Henley, W. E., Anderson, L. J., Wyatt, K. M., Nikolaou, V., Anderson, R., and Logan, S. (2014). The NCS-LSD cohort study: a description of the methods and analyses used to assess the long-term effectiveness of enzyme replacement therapy and substrate reduction therapy in patients with lysosomal storage disorders. J. Inherit. Metab. Dis. 37, 939–944. doi: 10.1007/s10545-014-9679-6
Horowitz, M., Pasmanik-Chor, M., Borochowitz, Z., Falik-Zaccai, T., Heldmann, K., Carmi, R., et al. (1998). Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum. Mutat. 12, 240–244. doi: 10.1002/(sici)1098-1004(1998)12:4<240::aid-humu4>3.0.co;2-j
Jung, O., Patnaik, S., Marugan, J., Sidransky, E., and Westbroek, W. (2016). Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteomics 13, 471–479. doi: 10.1080/14789450.2016.1174583
Kato, A., Nakagome, I., Sato, K., Yamamoto, A., Adachi, I., Nash, R. J., et al. (2016). Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 14, 1039–1048. doi: 10.1039/c5ob02223a
Khanna, R., Benjamin, E. R., Pellegrino, L., Schilling, A., Rigat, B. A., Soska, R., et al. (2010). The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of β-glucosidase. FEBS J. 277, 1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x
Kim, Y. M., Yum, M. S., Heo, S. H., Kim, T., Jin, H. K., Bae, J. S., et al. (2019). Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J. Med. Genet. 57, 124–131. doi: 10.1136/jmedgenet-2019-106132
Langhans, S. A. (2018). Three-Dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 9:6. doi: 10.3389/fphar.2018.00006
Lieberman, R. L., Wustman, B. A., Huertas, P., Powe, A. C., Pine, C. W., Khanna, R., et al. (2007). Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 3, 101–107. doi: 10.1038/nchembio850
Lu, J., Yang, C., Chen, M., Ye, D. Y., Lonser, R. R., Brady, R. O., et al. (2011). Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 108, 21200–21205. doi: 10.1073/pnas.1119181109
Luan, Z., Higaki, K., Aguilar-Moncayo, M., Li, L., Ninomiya, H., Nanba, E., et al. (2010). A fluorescent sp2-iminosugar with pharmacological chaperone activity for gaucher disease: synthesis and intracellular distribution studies. Chembiochem 11, 2453–2464. doi: 10.1002/cbic.201000323
Luan, Z., Higaki, K., Aguilar-Moncayo, M., Ninomiya, H., Ohno, K., Garcia-Moreno, M. I., et al. (2009). Chaperone activity of bicyclic nojirimycin analogues for Gaucher mutations in comparison with N-(n-nonyl)deoxynojirimycin. Chembiochem 10, 2780–2792. doi: 10.1002/cbic.200900442
Luan, Z., Li, L., Higaki, K., Nanba, E., Suzuki, Y., and Ohno, K. (2013). The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 35, 317–322. doi: 10.1016/j.braindev.2012.05.008
Lukina, E., Watman, N., Dragosky, M., Lau, H., Avila Arreguin, E., Rosenbaum, H., et al. (2019). Outcomes after 8 years of eliglustat therapy for Gaucher disease type 1: Final results from the Phase 2 trial. Am. J. Hematol. 94, 29–38. doi: 10.1002/ajh.25300
Maegawa, G. H., Tropak, M. B., Buttner, J. D., Rigat, B. A., Fuller, M., Pandit, D., et al. (2009). Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. doi: 10.1074/jbc.M109.012393
Mazzulli, J. R., Zunke, F., Isacson, O., Studer, L., and Krainc, D. (2016a). α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. U.S.A. 113, 1931–1936. doi: 10.1073/pnas.1520335113
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016b). Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
McNeill, A., Magalhaes, J., Shen, C., Chau, K. Y., Hughes, D., Mehta, A., et al. (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495. doi: 10.1093/brain/awu020
Mena-Barragan, T., Garcia-Moreno, M. I., Nanba, E., Higaki, K., Concia, A. L., Clapes, P., et al. (2016). Inhibitor versus chaperone behaviour of D-fagomine, DAB and LAB sp(2)-iminosugar conjugates against glycosidases: a structure-activity relationship study in Gaucher fibroblasts. Eur. J. Med. chem. 121, 880–891. doi: 10.1016/j.ejmech.2015.08.038
Migdalska-Richards, A., Daly, L., Bezard, E., and Schapira, A. H. (2016). Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 80, 766–775. doi: 10.1002/ana.24790
Migdalska-Richards, A., Ko, W. K. D., Li, Q., Bezard, E., and Schapira, A. H. V. (2017a). Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 71:e21967. doi: 10.1002/syn.21967
Migdalska-Richards, A., Wegrzynowicz, M., Rusconi, R., Deangeli, G., Di Monte, D. A., Spillantini, M. G., et al. (2017b). The L444P Gba1 mutation enhances α-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 140, 2706–2721. doi: 10.1093/brain/awx221
Mistry, P. K., Balwani, M., Baris, H. N., Turkia, H. B., Burrow, T. A., Charrow, J., et al. (2018). Safety, efficacy, and authorization of eliglustat as a first-line therapy in Gaucher disease type 1. Blood Cells Mol. Dis. 71, 71–74. doi: 10.1016/j.bcmd.2018.04.001
Mullin, S., Hughes, D., Mehta, A., and Schapira, A. H. V. (2019). Neurological effects of glucocerebrosidase gene mutations. Eur. J. Neurol. 26:e0388-29. doi: 10.1111/ene.13837
Mullin, S., Smith, L., Lee, K., D’souza, G., Woodgate, P., Elflein, J., et al. (2020). Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 13:e194611. doi: 10.1001/jamaneurol.2019.4611
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848. doi: 10.1093/brain/awt367
Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., Mckeith, I. G., Chinnery, P. F., et al. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735. doi: 10.1001/jamaneurol.2013.1925
Narita, A., Shirai, K., Itamura, S., Matsuda, A., Ishihara, A., Matsushita, K., et al. (2016). Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann. Clin. Transl. Neurol. 3, 200–215. doi: 10.1002/acn3.292
Pastores, G. M., Patel, M. J., and Firooznia, H. (2000). Bone and joint complications related to Gaucher disease. Curr. Rheumatol. Rep. 2, 175–180. doi: 10.1007/s11926-000-0059-x
Patnaik, S., Zheng, W., Choi, J. H., Motabar, O., Southall, N., Westbroek, W., et al. (2012). Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J. Med. Chem. 55, 5734–5748. doi: 10.1021/jm300063b
Pawlinski, L., Malecki, M. T., and Kiec-Wilk, B. (2018). The additive effect on the antiepileptic treatment of ambroxol in type 3 Gaucher patient. The early observation. Blood Cells Mol. Dis. 68, 192–193. doi: 10.1016/j.bcmd.2016.12.001
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Richter, F., Fleming, S. M., Watson, M., Lemesre, V., Pellegrino, L., Ranes, B., et al. (2014). A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 11, 840–856. doi: 10.1007/s13311-014-0294-x
Sala, G., Marinig, D., Arosio, A., and Ferrarese, C. (2016). Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front. Mol. Neurosci. 9:157. doi: 10.3389/fnmol.2016.00157
Sanchez-Martinez, A., Beavan, M., Gegg, M. E., Chau, K. Y., Whitworth, A. J., and Schapira, A. H. (2016). Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 6:31380. doi: 10.1038/srep31380
Sawkar, A. R., Cheng, W. C., Beutler, E., Wong, C. H., Balch, W. E., and Kelly, J. W. (2002). Chemical chaperones increase the cellular activity of N370S β -glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 99, 15428–15433. doi: 10.1073/pnas.192582899
Schiffmann, R., Fitzgibbon, E. J., Harris, C., Devile, C., Davies, E. H., Abel, L., et al. (2008). Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann. Neurol. 64, 514–522. doi: 10.1002/ana.21491
Sidransky, E. (2004). Gaucher disease: complexity in a “simple” disorder. Mol. Genet. Metab. 83, 6–15. doi: 10.1016/j.ymgme.2004.08.015
Sidransky, E. (2012). Gaucher disease: insights from a rare Mendelian disorder. Discov. Med. 14, 273–281.
Sidransky, E., Arkadir, D., Bauer, P., Dinur, T., Lopez, G., Rolfs, A., et al. (2019). Substrate reduction therapy for GBA1-associated Parkinsonism: are we betting on the wrong mouse? Mov. Disord. 35, 228–230. doi: 10.1002/mds.27903
Sidransky, E., and Lopez, G. (2012). The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. doi: 10.1016/S1474-4422(12)70190-4
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Silveira, C. R. A., Mackinley, J., Coleman, K., Li, Z., Finger, E., Bartha, R., et al. (2019). Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 19:20. doi: 10.1186/s12883-019-1252-3
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stojkovska, I., Krainc, D., and Mazzulli, J. R. (2018). Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 373, 51–60. doi: 10.1007/s00441-017-2704-y
Sun, Y., Liou, B., Xu, Y. H., Quinn, B., Zhang, W., Hamler, R., et al. (2012). Ex vivo and in vivo effects of isofagomine on acid β-glucosidase variants and substrate levels in Gaucher disease. J. Biol. Chem. 287, 4275–4287. doi: 10.1074/jbc.M111.280016
Sun, Y., Ran, H., Liou, B., Quinn, B., Zamzow, M., Zhang, W., et al. (2011). Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS One 6:e19037. doi: 10.1371/journal.pone.0019037
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Parkinson’s disease is not simply a prion disorder. J. Neurosci. 37, 9799–9807. doi: 10.1523/JNEUROSCI.1787-16.2017
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Thirumal Kumar, D., Iyer, S., Christy, J. P., Siva, R., Tayubi, I. A., George, P., et al. (2019). A comparative computational approach toward pharmacological chaperones (NN-DNJ and ambroxol) on N370S and L444P mutations causing Gaucher’s disease. Adv. Protein Chem. Struct. Biol. 114, 315–339. doi: 10.1016/bs.apcsb.2018.10.002
Tiscornia, G., Vivas, E. L., Matalonga, L., Berniakovich, I., Barragan Monasterio, M., Eguizabal, C., et al. (2013). Neuronopathic Gaucher’s disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 22, 633–645. doi: 10.1093/hmg/dds471
Valayannopoulos, V. (2013). Enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. Handb. Clin. Neurol. 113, 1851–1857. doi: 10.1016/B978-0-444-59565-2.00055-1
Weiss, K., Gonzalez, A., Lopez, G., Pedoeim, L., Groden, C., and Sidransky, E. (2015). The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 114, 110–122. doi: 10.1016/j.ymgme.2014.11.008
Yang, C., Rahimpour, S., Lu, J., Pacak, K., Ikejiri, B., Brady, R. O., et al. (2013). Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. U.S.A. 110, 966–971. doi: 10.1073/pnas.1221046110
Yap, T. L., Gruschus, J. M., Velayati, A., Westbroek, W., Goldin, E., Moaven, N., et al. (2011). α-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 286, 28080–28088. doi: 10.1074/jbc.M111.237859
Zimran, A., Altarescu, G., and Elstein, D. (2013). Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 50, 134–137. doi: 10.1016/j.bcmd.2012.09.006
Keywords: gaucher disease, Parkinson disease, lysosome, glucocerebrosidase, GBA1, small molecule chaperones
Citation: Han T-U, Sam R and Sidransky E (2020) Small Molecule Chaperones for the Treatment of Gaucher Disease and GBA1-Associated Parkinson Disease. Front. Cell Dev. Biol. 8:271. doi: 10.3389/fcell.2020.00271
Received: 25 January 2020; Accepted: 30 March 2020;
Published: 19 May 2020.
Edited by:
Miguel Weil, Tel Aviv University, IsraelReviewed by:
Pervin Rukiye Dincer, Hacettepe University, TurkeyMia Horowitz, Tel Aviv University, Israel
Copyright © 2020 Han, Sam and Sidransky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen Sidransky, sidranse@mail.nih.gov