 Ninaad Lasrado
Ninaad Lasrado Bharathi Yalaka
Bharathi Yalaka Jay Reddy
Jay Reddy- 1School of Veterinary Medicine and Biomedical Sciences, University of Nebraska–Lincoln, Lincoln, NE, United States
- 2Bristol-Myers Squibb – Hopewell, Pennington, NJ, United States
Inflammatory heart disease (IHD) is a group of diseases that includes pericarditis, myocarditis, and endocarditis. Although males appear to be more commonly affected than females, IHD can be seen in any age group. While the disease can be self-limiting leading to full recovery, affected individuals can develop chronic disease, suggesting that identification of primary triggers is critical for successful therapies. Adding to this complexity, however, is the fact that IHD can be triggered by a variety of infectious and non-infectious causes that can also occur as secondary events to primary insults. In this review, we discuss the immunological insights into the development of IHD as well as a mechanistic understanding of the disease process in animal models.
Introduction
The immune system has evolved to fight infections. Upon exposure to microbial infections, the innate immune cells swiftly come to the body’s defense, and, as infections get established, cells of the adaptive immune system respond to prevent future recurrences. Nevertheless, the immune system is not expected to recognize self-tissues as foreign. Should such misdirected responses occur, the autoimmunity that ensues needs to be distinguished from auto inflammatory diseases. However, both are marked by dysregulated and chronic activation of the immune system against self-antigens in the genetically predisposed individuals.
Autoimmunity can occur in any healthy individuals as evidenced by the detection of low levels of autoantibodies (Waldner, 2009; Lleo et al., 2010). Thus, autoimmunity and autoimmune disease should be distinguished clinically in that autoimmune disease occurs only when autoimmunity leads to tissue damage disrupting the functions of affected organs. The National Institutes of Health estimates that approximately 23 million people (up to 8% of the total population) could be affected by some form of approximately 80 known autoimmune diseases (Selgrade et al., 1999). In fact, autoimmune disease represents the fourth largest cause of disability among women in the United States, and is the eighth leading cause of death in women between the ages of 15 and 64 (Fairweather and Rose, 2004; Benros et al., 2014). Additionally, diagnosis of autoimmune diseases may take up to 5 years, and as many as five doctors may see a patient before proper diagnosis is made (Cassell and Rose, 2003). Economically, annual direct health care costs resulting from autoimmune diseases are estimated to be $100 billion (2005). Mechanistically, autoimmune diseases are considered to be disorders of the adaptive immune system. While autoantibodies mediate tissue damage via activation of complement proteins and leukocytes, autoreactive T cells induce damage via delayed-type hypersensitivity (DTH) reaction by secreting primarily T helper (Th)1, (interferon-γ) and Th17 (interleukin-17 family) cytokines. Two major factors have been implicated in the development of autoimmune responses: genetic susceptibility and environmental triggers such as exposure to microbes. Hypothetically, if ∼8% of the U.S. population are affected with some form of autoimmune disease, then it is hard to envision a scenario in which genetic defects exist in all of those affected. Thus, both genetic and environmental factors appear to be inseparable for occurrence of organ-specific autoimmune diseases that also include diseases of the cardiovascular system.
Conversely, auto inflammatory diseases involve direct participation of cells of the innate immune system, importantly neutrophils and macrophages. Cells of the adaptive immune system including major histocompatibility complex (MHC) alleles as well as infectious agents are excluded as the causative factors of auto inflammatory diseases (McGonagle and McDermott, 2006). These disorders could be monogenetic (involvement of a single gene) (Drenth et al., 1999) or polygenetic (involvement of more than one gene) (Doria et al., 2012). For example, mutations in inflammasome-related proteins, particularly NOD-like receptor (NLR) genes, are commonly implicated in auto inflammatory disorders with a phenotype of recurrent inflammation and periodic fever attacks (Zhong et al., 2016). Nevertheless, auto inflammatory diseases can affect the cardiovascular system with a phenotype of myocarditis, pericarditis/inflammatory recurrent acute pericarditis (IRAP), as might occur in sarcoidosis and eosinophilic granulomatosis associated polyangiitis (Dennert et al., 2010a; Bocoum et al., 2011).
In this review, we discuss the mechanistic insights into the development of inflammatory heart disease (IHD), a group of disorders that affect different layers of the heart (Figure 1). These include pericarditis (inflammation of the outer membranous sac), myocarditis (inflammation of the heart muscle), and endocarditis (inflammation of the inner lining of the heart and heart valves and/or coronary arteries) (Fairweather and Rose, 2005; Lu et al., 2015).
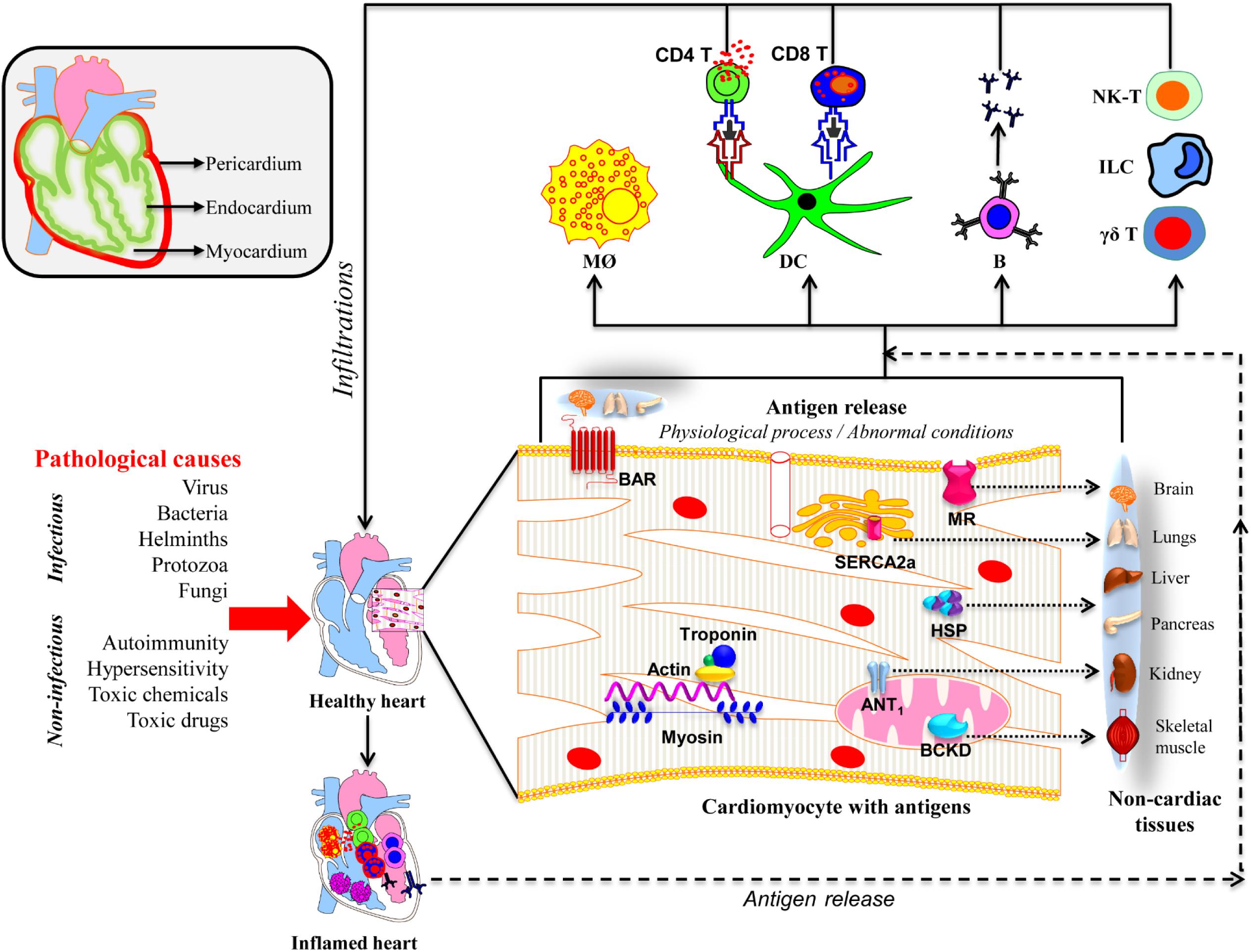
Figure 1. Proposed mechanisms of inflammatory heart disease. All three layers (pericardium, myocardium, and endocardium) individually or together can be affected by various infectious and non-infectious causes. A variety of innate immune cells like neutrophils, macrophages, dendritic cells including γδ T cells, ILCs, and NK-T cells can infiltrate and take part in tissue destruction. The inflammatory damage can also lead to the release of cardiac antigens such as cardiac myosin, actin, cTnI, ANT, BAR, BCKD, SERCA2a, laminin, muscarinic receptor (MR), and heat shock proteins (HSP). The newly released antigens can trigger autoimmune responses by activating T cells and B cells in the draining lymph nodes, which can in turn infiltrate the heart and aggravate inflammation. It is also possible that the dying cardiac myocytes can be engulfed by macrophages as a part of the cleaning process by phagocytosis, and possibly, autophagy, and trigger pathogenic autoimmune responses in genetically susceptible individuals. Alternatively, misfolded or mutated or modified self-proteins as might occur with mitochondrial proteins can be seen by the immune system as foreign, resulting in pathological autoimmune responses. It also is possible that some of the intracellular proteins like HSPs may bear sequences similar to microbial HSPs leading to the generation of cross-reactive immune responses. In all these scenarios, self-antigens can be presented to both CD4 and CD8 T cells, and B cells. They mediate inflammation through, respectively, DTH, cytolysis and complement activation. However, it is to be noted that several putative cardiac antigens can be promiscuously expressed in non-cardiac tissues such as brain, lungs, liver, pancreas, kidney, and skeletal muscle, raising the question whether autoreactive responses generated in response to cardiac damage can also inflict damage in other organs.
Pericarditis
The pericardium is made up of an outer single-layered fibrous membranous sac, and an inner double-layered serous sac that covers the heart and great blood vessels. Pericarditis, although self-limiting, can be complicated by pericardial effusion or pericardial constriction (Troughton et al., 2004). While, 40–85% of patients affected with pericarditis may have unknown etiologies, recurrences can occur in up to 20–50% of those affected (Brucato et al., 2008; Fancello et al., 2014). Acute pericarditis can be seen in all age groups, but males are more commonly affected than females in the age group of 20–50 years (Ariyarajah and Spodick, 2007). Estimates indicate that 0.1% of people admitted to hospitals with chest pain can have pericarditis (Imazio and Trinchero, 2007). Similarly, 5% of those admitted with chest pain in emergencies unrelated to myocardial infarction might have pericarditis (Imazio et al., 2010; LeWinter, 2014). The disease is self-limiting in most patients (70–90%) and respond well to anti-inflammatory medications like aspirin or non-steroidal anti-inflammatory drugs with colchicine, corticosteroids, and anakinra (IL-1 receptor antagonist) (Imazio et al., 2010). A variety of infectious and non-infectious triggers have been implicated in the causation of pericarditis (Table 1). Microbes frequently associated with pericarditis include bacteria [Staphylococcus aureus, Streptococcus, Haemophilus, Coxiella burnetti, and Mycobacterium tuberculosis (M. tb)] (Pankuweit et al., 2005; Brucato et al., 2008), viruses [Echovirus, Coxsackievirus B (CVB), parvovirus B19, human herpes virus 6 (HHV6), Epstein-Barr Virus (EBV), human immunodeficiency virus (HIV), and influenza B virus (IBV)] (Brucato et al., 2008). The disease can also arise from co-infections caused by multiple organisms such as S. aureus and M. tb in immune-suppressive individuals as might occur in HIV infection imposing a clinical challenge to target specific pathogens for therapy particularly in patients with recurrences (Lamas et al., 2019).

Table 1. Potential major causes of inflammatory heart disease.
The most alarming complication of acute pericarditis is IRAP, which occurs in 15–30% of patients due to auto inflammatory or autoimmune events (Lazaros et al., 2017). Auto inflammatory reactions in IRAP can result from virus infections such as Echovirus, CVB, and bacteria like M. tb, but the affected individuals should be negative for microbe-specific RNA and antibodies (IgM) to qualify for IRAP-diagnosis (Imazio et al., 2010; Bogdanos et al., 2013; LeWinter, 2014). However, the IRAP patients can carry anti-heart (AHA: myosin heavy chain β and β and myosin light chain-1v isoform) and anti-intercalated-disk autoantibodies, in addition to non-cardiac autoantibodies (Maisch et al., 1991; Latif et al., 1993). Impaired function of the innate immune system resulting in auto inflammatory pericarditis can occur due to mutations in immune response genes. Two such examples include mutations in MEFV and TNFRSF1A that cause Mediterranean fever and TNF receptor-associated periodic syndrome, respectively (Cantarini et al., 2010; Maggiolini et al., 2011).
As for autoimmune pericarditis, involvement of the pericardium has been reported in systemic autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), progressive systemic sclerosis, Sjögrens syndrome, and polyarthritis, but the affected patients can remain asymptomatic (Cantarini et al., 2015). Pericardial fluid, and not plasma samples, may contain inflammatory mediators like IL-6, IL-8, and IFN-γ with a preferential detection of anti-myolemma over anti-sarcolemma antibodies, implying that local autoimmune events can occur specific to the heart (Pankuweit et al., 2000). Recent reports indicate that serum carcinoembryonic antigen cell adhesion molecule 1 and MHC class I chain-related protein A can be used as biomarkers and prognostic markers in pericarditis patients, respectively, whereas the appearance of cardiac troponin-T (cTnT) signifies occurrence of acute and recurrent pericarditis (Hamm et al., 1997; Gamaza-Chulián et al., 2014).
Myocarditis
Myocarditis may involve cardiac myocytes, interstitial, or vascular elements of the heart that can be manifested as perimyocarditis involving the pericardium. Affected patients may show clinical manifestations of disease or may remain asymptomatic, but histopathologic changes can be detected in those affected (Fabre and Sheppard, 2006). Myocarditic hearts can contain variable numbers of lymphocytes and macrophages, but antibody-mediated injury also can be expected (Cooper, 2009; Schultz et al., 2009). The disease is generally regarded as an underdiagnosed cause of acute heart failure, and sudden death or dilated cardiomyopathy (DCM) can be expected in young adults (Drory et al., 1991). The annual global prevalence of myocarditis has been estimated to be ∼22 cases per 100,000 patients (Drory et al., 1991; Roth et al., 2015), and 1–5% of acute viral infections may have myocardial involvement (Fairweather and Rose, 2005). Furthermore, myocarditis is relatively more common in young men than their female counterparts, indicating that sex hormones can influence the disease outcome (Kytö et al., 2013). While virus-induced myocarditis in neonates and children may lead to fulminant myocarditis, lymphocytic or giant cell myocarditis is generally noted in the median age group of 42–43 years (Rose, 2016). Approximately 10–20% of those affected with acute myocarditis as young adults develop chronic myocarditis, DCM, and congestive heart failure. Approximately half of these patients undergo heart transplantation due to the lack of effective treatment options (Caforio et al., 2010; Rose, 2016).
Myocarditis can occur in association with a wide spectrum of infectious agents, systemic diseases, and hypersensitivity to drugs and toxins (Table 1). While viral infections caused by enteroviruses like CVB, adenoviruses, parvovirus B19, CMV, EBV, HIV, hepatitis C virus, and influenza virus are commonly suspected as causes of myocarditis in the developed world (Pollack et al., 2015), rheumatic carditis/diphtheria caused by Streptococcus aureus and Chagas disease caused by Trypanosoma cruzi are implicated in developing countries (Anez et al., 1999; Grumbach et al., 1999; Arbustini et al., 2000; Nolte et al., 2000; Boruah et al., 2010). More recently, it has been noted that patients receiving checkpoint inhibitors for tumors can develop autoimmune myocarditis, raising the question whether anti-tumor T cells may recognize cardiac antigens by cross-reactivity with microbial antigens (Brumbaugh et al., 2019; Martin Huertas et al., 2019). In support of this proposition, it was recently shown translationally that commensal bacteria can promote inflammatory cardiomyopathy by elevating Bacteroides-specific CD4 T cell and B cell responses in human myocarditis patients (Gil-Cruz et al., 2019). This observation was also complemented in the mouse model, where cardiac myosin-specific Th17 cells could be activated by cross-reactive mimic peptides from Bacteroides thetaiotaomicron. These findings suggest that the microbiome could predispose genetically susceptible individuals undergoing checkpoint inhibitors to develop myocarditis, leading to the suggestion that they can potentially be treated with antibiotics (Gil-Cruz et al., 2019).
In North America, myocarditis/DCM has been frequently associated with enterovirus infections like CVB, and approximately 50% of DCM patients can carry CVB-reactive antibodies, and viral RNA can be detected in 70% of them (Baboonian and McKenna, 2003; Fujinami et al., 2006). However, other viruses namely, parvovirus B19 and HHV-6 are now being increasingly reported in association with myocarditis/DCM (Andreoletti et al., 2009; Pollack et al., 2015). The CVB infection is generally used to understand how viral infections can trigger autoimmune responses. CVB pathogenesis involves both virus-induced (direct) and host-induced (indirect) mechanisms (Cihakova and Rose, 2008). Because it is a cardiotropic and lytic virus, CVB can directly lyse cardiomyocytes during viral replication. Experimentally, CVB infection in mice can have two phases in continuum: acute myocarditis (viral) up to day 14–18 and chronic myocarditis (non-viral) beyond 18 days (Rose et al., 1986). During the initial stages of CVB infection, activation of innate immune cells results in the production of the pro-inflammatory cytokines IL-1, IL-6, TNF-β, soluble IL-1R-like 1 also called, sST2 and IFNs (Fairweather et al., 2003; Nishimura et al., 2018). Such proteins can potentially be used as markers of myocarditis. For example, it has been recently shown that men with myocarditis had higher levels of sST2 than women. This finding was later confirmed experimentally, where gonadectomy with testosterone, but not estradiol replacement, led to increased levels of sST2 in male mice with myocarditis (Coronado et al., 2019). These data also add credence to the relevance of experimental models to human diseases. As to adaptive immune response, Th cells appear to play a decisive role in the induction of chronic myocarditis/DCM, but involve complex mechanisms. While, the Th1 cells can protect acute myocarditis and prevent viral replication (Fairweather et al., 2004; Fairweather et al., 2005), Th2 cells, although they have the ability to attenuate acute myocarditis via anti-inflammatory cytokines and Treg cells (Frisancho-Kiss et al., 2007; Liu and Huber, 2011), can induce acute cardiac remodeling leading to chronic myocarditis/DCM (Abston et al., 2012). Conversely, Th17 cells contribute to the development of both acute myocarditis (but without affecting viral replication), as well as cardiac remodeling/DCM events and heart failure (Fairweather et al., 2012) suggesting that the disease course of viral myocarditis may be dictated by nature and the amount of Th cytokines being produced. Nonetheless, these findings raise a question as to the mechanisms of generation of pathogenic cardiac-reactive T cells, should they be antigen-specific.
One mechanism is molecular mimicry, which involves the generation of cross-reactive immune responses against self-antigens. In fact, sequence similarities of up to 40% have been detected between CVB viral protein1 (VP1), streptococcal M protein, and cardiac myosin; synthetic peptides containing these sequences block monoclonal antibody (mAb) reactivity to streptococcal M protein (Cunningham et al., 1992). Furthermore, murine anti-streptococcal mAbs that cross-react with streptococcal M protein and human cardiac myosin were found to neutralize CVB viruses (CVB3 and CVB4) and poliovirus type 1 (Cunningham et al., 1992). Additionally, T lymphocytes from CVB-infected animals can respond to peptides from streptococcal M protein (Huber et al., 1994). Similarly, the N-terminal region four peptide from streptococcus could induce IHD in MRL/++ mice, whereas induction of anergy to this peptide protects mice from developing CVB-induced myocarditis (Huber and Cunningham, 1996).
Another mechanism is epitope spreading which may occur as a result of release of self-antigens primarily by viral damage, leading to induction of autoimmunity secondarily (Fujinami et al., 2006). In addressing this phenomenon, we had demonstrated that A/J mice infected with CVB produce Myhc-α 334-352-specific CD4 T cell responses that are pathogenic in nature (Gangaplara et al., 2012). Using MHC class II dextramers specific to Myhc 334–352, we showed that Myhc-specific, CD4 T cells can infiltrate into the hearts of animals infected with CVB during the post infectious phase of CVB infection (Gangaplara et al., 2012). By expanding this observation, we may be able to determine whether autoreactive T cells specific to multiple cardiac antigens could contribute to CVB pathogenesis. This effort, however, requires identification of myocarditogenic epitopes for other putative cardiac antigens implicated in myocarditis/DCM patients (Figure 1).
Endocarditis
Endocarditis can also be caused by infective or non-infective agents (Table 1), that have a male preponderance (Aksoy et al., 2007). Incidence of infective endocarditis in developed countries is 3–10 cases per 100,000 people, and the disease prevalence is expected to be high in developing countries (Duval et al., 2012; Selton-Suty et al., 2012). Endocarditic lesions, primarily called vegetation, consist of microorganisms, inflammatory cells, platelets, and fibrin. Microbes such as S. aureus, Streptococcus species, Enterococcus species, and HACEK Gram-negative bacteria (Haemophilus species, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella corrodens and Kingella species) account for more than 90% of endocarditis cases that may have a nosocomial or odontogenic origin (Table 1; Murdoch et al., 2009; Raza et al., 2010; Ashrafi et al., 2012; Selton-Suty et al., 2012). However, highest mortalities occur with S. aureus and the non-HACEK group pathogens such as Candida albicans (Ashrafi et al., 2012). Additionally, infective endocarditis also can occur in intravenous drug abusers or in association with HIV infection, diabetes mellitus, rheumatoid arthritis, Sjögrens syndrome and cancer (Miro et al., 2002; Filizcan et al., 2004; Sughimoto et al., 2006; Yoshioka et al., 2018; Sanaiha et al., 2019; Table 1). These observations suggest that multiple pathomechanisms may contribute to the development of endocarditis.
Damage to the endocardium can result from valve sclerosis, rheumatic valvulitis, or direct colonization/vegetation by bacteria like S. aureus and Streptococcus species (Cahill and Prendergast, 2016). The main sequelae of endocarditis include damage to the heart valve and endothelial scarring, leading to congestive heart failure as might occur in autoimmune rheumatic diseases. Endocarditis resulting from Streptococcus species., has been used as a prototypic disease model to address how cross-reactive autoimmune responses can lead to heart dysfunction (Krisher and Cunningham, 1985; Cunningham et al., 1986). It has been shown that mAbs specific to Streptococcus M protein or the group A streptococcal epitope N-acetyl-βD-glucosamine can cross-react with cardiac myosin and laminin (Krisher and Cunningham, 1985; Galvin et al., 2000). The finding that anti-streptococcal antibodies cross-react with human and canine cardiac sarcolemma proteins but not kidney or skeletal muscle, suggests that sarcoplasmic antigens can be targeted in autoimmune endocarditis (Dale and Beachey, 1982; Cunningham et al., 1986).
Animal models have been developed to study the immune pathology of endocarditis and valvular inflammation. Intraperitoneal injection of cell lysate from group A streptococci into Swiss Webster mice results in aortic and mitral valve stenosis, chronic inflammation, and scarring that mimic human chronic rheumatic carditis (Cromartile and Craddock, 1966; Cunningham et al., 1984). In the Lewis rat model, immunization with streptococcal M protein leads to the induction of cross-reactive T cells that are pathogenic in nature (Quinn et al., 2001). Similarly, Lewis rats immunized with M protein can develop valvulitis in mitral valves, but with minimal myocardial involvement (Quinn et al., 2001). Expectedly, lymph node cells obtained from immunized animals respond to human cardiac myosin, but not human and rabbit skeletal myosin and actin, although both human cardiac and skeletal myosin share extensive sequence identity (Quinn et al., 2001). However, it is interesting that, in spite of protein sequence identity between streptococcal M proteins and cardiac myosin, myocardial involvement was noted to be minimal relative to valvulitis in the immunized Lewis rats. Likewise, laminin present in the valves has been shown to cross-react with both anti-streptococcal and myosin antibodies, and cross-reactive T cells generated by immunization with laminin can induce valvulitis without significant involvement of the myocardium (Quinn et al., 2001).
Endocarditis can be seen in patients with RA, SLE, and rheumatic fever, in which both joint and cardiovascular systems could be affected. RA patients can also carry cardiac-specific antibodies (Breed and Binstadt, 2015). To study the immune-pathogenic mechanism of inflammatory arthritis, the K/BxN TCR transgenic mouse model has been developed (Monach et al., 2008). The transgenic mice express TCR specific to glucose-6-phosphate isomerase (GPI), which plays a key role in catalyzing the interconversion of glucose-6-phosphate to fructose-6-phosphate (Chang and Wei, 2011). GPI-primed B cells produce autoantibodies that can induce arthritis (Monach et al., 2007). Recent studies suggest that transgenic mice can develop both arthritis and endocarditis, but through two independent pathways. While complement C5 protein is critical for development of arthritis, but not endocarditis, occurrence of endocarditis, but not arthritis, requires the participation of FcRγ (Binstadt et al., 2009). Adoptive transfer of serum from transgenic mice into naïve animals led to the development of arthritis that was devoid of endocarditis, implying that arthritis is an antibody-mediated disease (Maccioni et al., 2002). Conversely, animals depleted of CD4+ T cells fail to develop endocarditis, but arthritis remains unaffected, suggesting that T cells play a critical role in the development of endocarditis (Binstadt et al., 2009). Such disease models may be helpful in studying the immune mechanisms of rheumatic diseases that involve both synovial joints and/or the endocardium.
Discussion
As described above, etiologically, IHD can be triggered by a wide range of microbes, therefore posing a clinical challenge, that necessitates identification of triggers to initiate pathogen-specific therapies. Such avenues are clinically complicated, since affected patients can have co-infections caused by microbes belonging to different classes. For example, HIV patients can develop pericarditis in association with S. aureus and M. tb infections (Lamas et al., 2019). Similarly, myocarditis can result from co-infection with HHV-6 and parvovirus B19 (Rohayem et al., 2001). Likewise, some infections like EBV and parvovirus B19, may occur as a result of their reactivation, respectively, in CVB3 and hepatitis virus, and HHV-6 infections (Bock et al., 2014; Atti et al., 2017). Such scenarios are likely because up to 95% of the United States population is affected with EBV infection (Atti et al., 2017). Furthermore, tissue destruction may not necessarily result directly from cardiotropic pathogens like CVB3 and adenovirus. For example, parvovirus B19 targeting endothelial cells can induce myocarditis via endothelial dysfunction and initiate inflammation (Bock et al., 2010).
Nevetheless, following infection-tiggered IHD, the immune system in immune-competent individuals is expected to repair the damaged tissue with or without successful clearance of primary offenders. But such an ability of the immune system to self-cure is relatively limited for cardiac cells, especially adult cardiac myocytes because of their limited regenerative capacity (Kikuchi and Poss, 2012). This scenario can be complicated by the potential occurrence of secondary damage resulting from autoimmune responses in genetically susceptible individuals. In addressing this phenomenon, we have noted that CVB3 infection can lead to the generation of cardiac myosin-specific T cells that can contribute to post-infectious myocarditis in the absence of detectable virions in infected animals (Gangaplara et al., 2012). Based on this observation, we envision that cardiac-reactive T cells specific for multiple autoantigens could be generated in infections caused by cardiotropic pathogens that may have implications in diagnosis and therapy of IHD.
Diagnostically, it is extremely challenging to identify the causative agents for IHD with certainty, especially if polymicrobes (co-infections) are involved. Additionally, one or more manifestions of IHD (pericarditis, myocarditis, or endocarditis) can potentially occur concurrently in affected patients. Nonetheless, it may be possible to develop diagnostic panels for commonly found pathogens like viruses and bacteria. These may include PCR-based molecular markers or antibody assays with the challenge that clinical specimens, especially endomycardial biopsies, need to be collected in a timely fashion to make a reliable diagnosis. This is especially important in chronically affected individuals with no overt clinical signs suggestive of known microbial infections. Such an approach, if supported by routinely used serum biomarkers namely, cardiac troponin I and T, creatinine kinase-MB, C-reactive protein, myoglobin, natriuetic peptides, and sST2 (Biomarkers, 2014; Nishimura et al., 2018; Coronado et al., 2019; Kottwitz et al., 2020) may have diagnostic value. A limitation of this approach is that the offending agents would be gone by that time, except being suspects, which may not alter treatment approaches to be followed clinically.
Thus, the complex nature of IHD is therapeutically challenging in the medical management of affected individuals. Although, various clinical trials reported thus far have yielded mixed successes, two major approaches seem to be encouraging. These include immune therapies (administration of intravenous immunoglobulins or immunoglobulin adsorption), and anti-viral agents. The former approach appeared to be effective in patients with giant cell myocarditis and sarcoidosis who are negative for viruses (Pollack et al., 2015). Conversely, this approach offered no advantage in those positive for viruses (Pollack et al., 2015). The latter group of patients, however, can potentially be treated with anti-viral agents as reported in parvovirus B19 (peramivir) and influenza (acyclovir) infections, with or without immune suppressive therapy, respectively (Dennert et al., 2010b; Baik et al., 2015). Although such outcomes are rarely reported, the therapeutic value of anti-viral compounds can be validated only in large clinical trials that involve careful recruitment of patient populations and appropriate controls. Finally, as to autoimmunity, use of general immune suppressants potentially elimimate most naïve lymphocytes that can seriously impair a host’s defense mechanisms against microbes. Thus, immune strategies are expected to target disease-inducing, autoreactive T cells or B cells by sparing naïve repertoires. To this end, various selective immune suppressants have been investigated that may hold promise in the future.
Author Contributions
All authors contributed to the synthesis of literature and writing the manuscript.
Funding
This work was partially supported by the Transformational grant from the American Heart Association [18TPA34170206].
Conflict of Interest
BY was employed by Bristol-Myers Squibb.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AHA, anti-heart antibodies; ANT, adenine nucleotide translocator; BAR, beta adrenergic receptor; BCKD, branched chain ketoacid dehydrogenase; CMV, cytomegalovirus; cTnI, cardiac troponin I; cTnT, cardiac troponin T; CVB, Coxsackievirus B; DCM, dilated cardiomyopathy; DTH, delayed-type hypersensitivity; EBV, Epstein-Barr virus; GPI, glycosyl phosphatidyl inositol; HHV, human herpesvirus; HIV, human immunodeficiency virus; HSPs, heat shock proteins; IBV, influenza B virus; IFN, interferon; IHD, inflammatory heart disease; IL, interleukin; ILCs, innate lymphoid cells; IRAP, inflammatory recurrent acute pericarditis; mAb, monoclonal antibody; MHC, major histocompatibility complex; M. tb, Mycobacterium tuberculosis; NK-Ts, natural killer-T cells; NLR, NOD-like receptor; PCR, polymerase chain reaction; RA, rheumatoid arthritis; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a; SLE, systemic lupus erythematosus; TCR, T cell receptor; Th, T helper; TNF, tumor necrosis factor.
References
Abston, E. D., Coronado, M. J., Bucek, A., Bedja, D., Shin, J., Kim, J. B., et al. (2012). Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin. Dev. Immunol. 2012:129486. doi: 10.1155/2012/129486
Aksoy, O., Meyer, L. T., Cabell, C. H., Kourany, W. M., Pappas, P. A., and Sexton, D. J. (2007). Gender differences in infective endocarditis: pre- and co-morbid conditions lead to different management and outcomes in female patients. Scand. J. Infect. Dis. 39, 101–107. doi: 10.1080/00365540600993285
Andreoletti, L., Leveque, N., Boulagnon, C., Brasselet, C., and Fornes, P. (2009). Viral causes of human myocarditis. Arch. Cardiovasc. Dis. 102, 559–568. doi: 10.1016/j.acvd.2009.04.010
Anez, N., Carrasco, H., Parada, H., Crisante, G., Rojas, A., Fuenmayor, C., et al. (1999). Myocardial parasite persistence in chronic chagasic patients. Am. J. Trop. Med. Hyg. 60, 726–732. doi: 10.4269/ajtmh.1999.60.726
Arbustini, E., Porcu, E., Bellini, O., Grasso, M., Pilotto, A., Dal Bello, B., et al. (2000). Enteroviral infection causing fatal myocarditis and subclinical myopathy. Heart 83, 86–90. doi: 10.1136/heart.83.1.86
Ariyarajah, V., and Spodick, D. H. (2007). Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol. Rev. 15, 24–30. doi: 10.1097/01.crd.0000210645.89717.34
Ashrafi, R., McKay, E., Ebden, L., Jones, J., Davis, G. K., and Burgess, M. I. (2012). Endocarditis in a large district general hospital: A study of the microbiological spectrum between 2000 and 2011. Exp. Clin. Cardiol. 17, 175–178.
Atti, V., Anderson, N. M., and Day, M. B. (2017). Coxsackie myocarditis and hepatitis with reactivated epstein-barr virus (EBV): a case report. Am. J. Case Rep. 18, 166–169. doi: 10.12659/ajcr.900096
Baboonian, C., and McKenna, W. (2003). Eradication of viral myocarditis. J. Am. Coll. Cardiol. 42, 466–472.
Baik, S. H., Jeong, H. S., Kim, S. J., Yoon, Y. K., Sohn, J. W., and Kim, M. J. (2015). A case of influenza associated fulminant myocarditis successfully treated with intravenous peramivir. Infect. Chemother. 47, 272–277. doi: 10.3947/ic.2015.47.4.272
Benros, M. E., Eaton, W. W., and Mortensen, P. B. (2014). The epidemiologic evidence linking autoimmune diseases and psychosis. Biol. Psychiatry 75, 300–306. doi: 10.1016/j.biopsych.2013.09.023
Binstadt, B. A., Hebert, J. L., Ortiz-Lopez, A., Bronson, R., Benoist, C., and Mathis, D. (2009). The same systemic autoimmune disease provokes arthritis and endocarditis via distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 106, 16758–16763. doi: 10.1073/pnas.0909132106
Biomarkers, E. X. G. O. (2014). Biomarkers in cardiology–part 1–in heart failure and specific cardiomyopathies. Arq. Bras. Cardiol. 103, 451–459. doi: 10.5935/abc.20140184
Bock, C. T., Duchting, A., Utta, F., Brunner, E., Sy, B. T., Klingel, K., et al. (2014). Molecular phenotypes of human parvovirus B19 in patients with myocarditis. World J. Cardiol. 6, 183–195. doi: 10.4330/wjc.v6.i4.183
Bock, C. T., Klingel, K., and Kandolf, R. (2010). Human parvovirus B19-associated myocarditis. N. Engl. J. Med. 362, 1248–1249.
Bocoum, A., Daumas, A., Cammilleri, S., Bernard, F., Rossi, P., Bagneres, D., et al. (2011). Recurrent pericarditis revealing a systemic sarcoidosis. La Revue de medecine interne/fondee. Par Soc. Nat. Francaise Med. Int. 32, 575–579. doi: 10.1016/j.revmed.2011.03.007
Bogdanos, D. P., Smyk, D. S., Invernizzi, P., Rigopoulou, E. I., Blank, M., Pouria, S., et al. (2013). Infectome: a platform to trace infectious triggers of autoimmunity. Autoimmun. Rev. 12, 726–740. doi: 10.1016/j.autrev.2012.12.005
Boruah, P., Shetty, S., and Kumar, S. S. (2010). Acute streptococcal myocarditis presenting as acute ST-elevation myocardial infarction. J. Invasive Cardiol. 22, E189–E191.
Breed, E. R., and Binstadt, B. A. (2015). Autoimmune valvular carditis. Curr. Allergy Asthma Rep. 15:491. doi: 10.1007/s11882-014-0491-z
Brucato, A., Maestroni, S., Cumetti, D., Thiella, G., Alari, G., Brambilla, G., et al. (2008). Recurrent pericarditis: infectious or autoimmune? Autoimmun. Rev. 8, 44–47. doi: 10.1016/j.autrev.2008.07.024
Brumbaugh, A. D., Narurkar, R., Parikh, K., Fanucchi, M., and Frishman, W. H. (2019). Cardiac immune-related adverse events in immune checkpoint inhibition therapy. Cardiol. Rev. 27, 97–107. doi: 10.1097/CRD.0000000000000217
Caforio, A. L., Brucato, A., Doria, A., Brambilla, G., Angelini, A., Ghirardello, A., et al. (2010). Anti-heart and anti-intercalated disk autoantibodies: evidence for autoimmunity in idiopathic recurrent acute pericarditis. Heart 96, 779–784. doi: 10.1136/hrt.2009.187138
Cahill, T. J., and Prendergast, B. D. (2016). Infective endocarditis. Lancet 387, 882–893. doi: 10.1016/S0140-6736(15)00067-7
Cantarini, L., Lopalco, G., Selmi, C., Napodano, S., De Rosa, G., Caso, F., et al. (2015). Autoimmunity and autoinflammation as the yin and yang of idiopathic recurrent acute pericarditis. Autoimmun. Rev. 14, 90–97. doi: 10.1016/j.autrev.2014.10.005
Cantarini, L., Lucherini, O., Baldari, C., Laghi Pasini, F., and Galeazzi, M. (2010). Familial clustering of recurrent pericarditis may disclose tumour necrosis factor receptor-associated periodic syndrome. Clin. Exp. Rheumatol. 28, 405–407.
Cassell, D. K., and Rose, N. R. (2003). The Encyclopedia of Autoimmune Diseases. New York, N.Y: Facts On File.
Chang, X., and Wei, C. (2011). Glycolysis and rheumatoid arthritis. Int. J. Rheumatic Dis. 14, 217–222. doi: 10.1111/j.1756-185X.2011.01598.x
Cihakova, D., and Rose, N. R. (2008). Pathogenesis of myocarditis and dilated cardiomyopathy. Adv. Immunol. 99, 95–114. doi: 10.1016/S0065-2776(08)00604-4
Coronado, M. J., Bruno, K. A., Blauwet, L. A., Tschope, C., Cunningham, M. W., Pankuweit, S., et al. (2019). Elevated Sera sST2 is associated with heart failure in men < / = 50 years old with myocarditis. J. Am. Heart Assoc. 8:e008968. doi: 10.1161/JAHA.118.008968
Cromartile, W. J., and Craddock, J. G. (1966). Rheumatic-like cardiac lesions in mice. Science 154, 285–287. doi: 10.1126/science.154.3746.285
Cunningham, M., Hall, N., Krisher, K., and Spanier, A. (1986). A study of anti-group A streptococcal monoclonal antibodies cross-reactive with myosin. J. Immunol. 136, 293–298.
Cunningham, M. W., Antone, S. M., Gulizia, J. M., McManus, B. M., Fischetti, V. A., and Gauntt, C. J. (1992). Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc. Natl. Acad. Sci. U.S.A. 89, 1320–1324. doi: 10.1073/pnas.89.4.1320
Cunningham, M. W., Krisher, K., and Graves, D. (1984). Murine monoclonal antibodies reactive with human heart and group A streptococcal membrane antigens. Infect. Immun. 46, 34–41. doi: 10.1128/iai.46.1.34-41.1984
Dale, J. B., and Beachey, E. H. (1982). Protective antigenic determinant of streptococcal M protein shared with sarcolemmal membrane protein of human heart. J. Exp. Med. 156, 1165–1176. doi: 10.1084/jem.156.4.1165
Dennert, R. M., van Paassen, P., Schalla, S., Kuznetsova, T., Alzand, B. S., Staessen, J. A., et al. (2010a). Cardiac involvement in Churg-Strauss syndrome. Arthr. Rheumatol. 62, 627–634.
Dennert, R., Velthuis, S., Westermann, D., Donker, D., Schalla, S., van Suylen, R. J., et al. (2010b). Parvovirus-B19-associated fulminant myocarditis successfully treated with immunosuppressive and antiviral therapy. Antivir. Ther. 15, 681–685. doi: 10.3851/IMP1563
Doria, A., Dayer, J.-M., and Punzi, L. (2012). Autoinflammatory diseases: how to put the fire inside the body out? Autoimmun. Rev. 12, 1–4. doi: 10.1016/j.autrev.2012.07.013
Drenth, J. P., Cuisset, L., Grateau, G., Vasseur, C., van de Velde-Visser, S. D., de Jong, J. G., et al. (1999). Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat. Genet. 22, 178–181. doi: 10.1038/9696
Drory, Y., Turetz, Y., Hiss, Y., Lev, B., Fisman, E. Z., Pines, A., et al. (1991). Sudden unexpected death in persons < 40 years of age. Am. J. Cardiol. 68, 1388–1392. doi: 10.1016/0002-9149(91)90251-f
Duval, X., Delahaye, F., Alla, F., Tattevin, P., Obadia, J.-F., Le Moing, V., et al. (2012). Temporal trends in infective endocarditis in the context of prophylaxis guideline modifications: three successive population-based surveys. J. Am. Coll. Cardiol. 59, 1968–1976. doi: 10.1016/j.jacc.2012.02.029
Fabre, A., and Sheppard, M. N. (2006). Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart 92, 316–320. doi: 10.1136/hrt.2004.045518
Fairweather, D., Frisancho-Kiss, S., Yusung, S. A., Barrett, M. A., Davis, S. E., Gatewood, S. J., et al. (2004). Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am. J. Pathol. 165, 1883–1894. doi: 10.1016/s0002-9440(10)63241-5
Fairweather, D., Frisancho-Kiss, S., Yusung, S. A., Barrett, M. A., Davis, S. E., Steele, R. A., et al. (2005). IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J. Immunol. 174, 261–269. doi: 10.4049/jimmunol.174.1.261
Fairweather, D., and Rose, N. R. (2004). Women and autoimmune diseases. Emerg. Infect. Dis. 10:2005.
Fairweather, D., and Rose, N. R. (2005). Inflammatory heart disease: a role for cytokines. Lupus 14, 646–651. doi: 10.1191/0961203305lu2192oa
Fairweather, D., Stafford, K. A., and Sung, Y. K. (2012). Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 24, 401–407. doi: 10.1097/BOR.0b013e328353372d
Fairweather, D., Yusung, S., Frisancho, S., Barrett, M., Gatewood, S., Steele, R., et al. (2003). IL-12 receptor β1 and Toll-like receptor 4 increase IL-1β-and IL-18-associated myocarditis and coxsackievirus replication. J. Immunol. 170, 4731–4737. doi: 10.4049/jimmunol.170.9.4731
Fancello, L., Monteil, S., Popgeorgiev, N., Rivet, R., Gouriet, F., Fournier, P. E., et al. (2014). Viral communities associated with human pericardial fluids in idiopathic pericarditis. PLoS One 9:e93367. doi: 10.1371/journal.pone.0093367
Filizcan, U., Cetemen, S., Enc, Y., Cakmak, M., Goksel, O., and Eren, E. (2004). Candida albicans endocarditis and a review of fungal endocarditis: case report. Heart Surg. Forum 7, E312–E314.
Frisancho-Kiss, S., Davis, S. E., Nyland, J. F., Frisancho, J. A., Cihakova, D., Barrett, M. A., et al. (2007). Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J. Immunol. 178, 6710–6714. doi: 10.4049/jimmunol.178.11.6710
Fujinami, R. S., Von Herrath, M. G., Christen, U., and Whitton, J. L. (2006). Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin. Microbiol. Rev. 19, 80–94. doi: 10.1128/cmr.19.1.80-94.2006
Galvin, J. E., Hemric, M. E., Ward, K., and Cunningham, M. W. (2000). Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J. Clin. Invest. 106, 217–224. doi: 10.1172/jci7132
Gamaza-Chulián, S., León-Jiménez, J., Recuerda-Núñez, M., Camacho-Freire, S., Gutiérrez-Barrios, A., and Vargas-Machuca, J. C. (2014). Cardiac troponin-T in acute pericarditis. J. Cardiovasc. Med. 15, 68–72.
Gangaplara, A., Massilamany, C., Brown, D. M., Delhon, G., Pattnaik, A. K., Chapman, N., et al. (2012). Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-α-reactive CD4 T cells in A/J mice. Clin. Immunol. 144, 237–249. doi: 10.1016/j.clim.2012.07.003
Gil-Cruz, C., Perez-Shibayama, C., De Martin, A., Ronchi, F., van der Borght, K., Niederer, R., et al. (2019). Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 366, 881–886. doi: 10.1126/science.aav3487
Grumbach, I. M., Heim, A., Pring-Akerblom, P., Vonhof, S., Hein, W., Müller, G., et al. (1999). Adenoviruses and enteroviruses as pathogens in myocarditis and dilated cardiomyopathy. Acta Cardiol. 54, 83–88.
Hamm, C. W., Goldmann, B. U., Heeschen, C., Kreymann, G., Berger, J., and Meinertz, T. (1997). Emergency room triage of patients with acute chest pain by means of rapid testing for cardiac troponin T or troponin I. N. Eng. J. Med. 337, 1648–1653. doi: 10.1056/nejm199712043372302
Huber, S., Moraska, A., and Cunningham, M. (1994). Alterations in major histocompatibility complex association of myocarditis induced by coxsackievirus B3 mutants selected with monoclonal antibodies to group A streptococci. Proc. Natl. Acad. Sci. U.S.A. 91, 5543–5547. doi: 10.1073/pnas.91.12.5543
Huber, S. A., and Cunningham, M. W. (1996). Streptococcal M protein peptide with similarity to myosin induces CD4 + T cell-dependent myocarditis in MRL/ + + mice and induces partial tolerance against Coxsakieviral myocarditis. J. Immunol. 156, 3528–3534.
Imazio, M., Spodick, D. H., Brucato, A., Trinchero, R., and Adler, Y. (2010). Controversial issues in the management of pericardial diseases. Circulation 121, 916–928. doi: 10.1161/circulationaha.108.844753
Imazio, M., and Trinchero, R. (2007). Current and future treatment for pericarditis. Future Cardiol. 3, 623–634. doi: 10.2217/14796678.3.6.623
Kikuchi, K., and Poss, K. D. (2012). Cardiac regenerative capacity and mechanisms. Annu. Rev. Cell Dev. Biol. 28, 719–741. doi: 10.1146/annurev-cellbio-101011-155739
Kottwitz, J., Bruno, K. A., Berg, J., Salomon, G. R., Fairweather, D., Elhassan, M., et al. (2020). Myoglobin for detection of high-risk patients with acute myocarditis. J. Cardiovasc. Transl. Res. doi: 10.1007/s12265-020-09957-8 [Epub ahead of print].
Krisher, K., and Cunningham, M. W. (1985). Myosin: a link between streptococci and heart. Science 227, 413–416.
Kytö, V., Sipilä, J., and Rautava, P. (2013). The effects of gender and age on occurrence of clinically suspected myocarditis in adulthood. Heart 99, 1681–1684. doi: 10.1136/heartjnl-2013-304449
Lamas, E. S., Bononi, R. J. R., Bernardes, M., Pasin, J. L., Soriano, H. A. D., Martucci, H. T., et al. (2019). Acute purulent pericarditis due co-infection with Staphylococcus aureus and Mycobacterium tuberculosis as first manifestation of HIV infection. Oxf. Med. Case Rep. 2019:omy127. doi: 10.1093/omcr/omy127
Latif, N., Baker, C. S., Dunn, M. J., Rose, M. L., Brady, P., and Yacoub, M. H. (1993). Frequency and specificity of antiheart antibodies in patients with dilated cardiomyopathy detected using SDS-PAGE and western blotting. J. Am. Coll. Cardiol. 22, 1378–1384. doi: 10.1016/0735-1097(93)90546-d
Lazaros, G., Antonatou, K., and Vassilopoulos, D. (2017). The therapeutic role of Interleukin-1 inhibition in idiopathic recurrent pericarditis: current evidence and future challenges. Front. Med. 4:78. doi: 10.3389/fmed.2017.00078
Liu, W., and Huber, S. A. (2011). Cross-talk between cd1d-restricted nkt cells and gammadelta cells in t regulatory cell response. Virol. J. 8:32. doi: 10.1186/1743-422X-8-32
Lleo, A., Invernizzi, P., Gao, B., Podda, M., and Gershwin, M. E. (2010). Definition of human autoimmunity—autoantibodies versus autoimmune disease. Autoimmun. Rev. 9, A259–A266. doi: 10.1016/j.autrev.2009.12.002
Lu, L., Sun, R., Liu, M., Zheng, Y., and Zhang, P. (2015). The inflammatory heart diseases: causes, symptoms, and treatments. Cell Biochem. Biophys. 72, 851–855. doi: 10.1007/s12013-015-0550-7
Maccioni, M., Zeder-Lutz, G., Huang, H., Ebel, C., Gerber, P., Hergueux, J., et al. (2002). Arthritogenic monoclonal antibodies from K/BxN mice. J. Exp. Med. 195, 1071–1077. doi: 10.1084/jem.20011941
Maggiolini, S., Tiberti, G., Cantarini, L., Carbone, C., Mariani, S., Achilli, F., et al. (2011). Large pericardial effusion in a family with recurrent pericarditis: a report of probable X-linked transmission. Exp. Clin. Cardiol. 16:54.
Maisch, B., Drude, L., Hengstenberg, C., Hufnagel, G., Schönian, U., and Schwab, D. (1991). Cytolytic anticardiac membrane antibodies in the pathogenesis of myopericarditis. Postgrad.Med. J. 68, S11–S16.
Martin Huertas, R., Saavedra Serrano, C., Perna, C., Ferrer Gomez, A., and Alonso Gordoa, T. (2019). Cardiac toxicity of immune-checkpoint inhibitors: a clinical case of nivolumab-induced myocarditis and review of the evidence and new challenges. Cancer Manag. Res. 11, 4541–4548. doi: 10.2147/cmar.s185202
McGonagle, D., and McDermott, M. F. (2006). A proposed classification of the immunological diseases. PLoS Med. 3:e297. doi: 10.1371/journal.pmed.0030297
Miro, J. M., del Rio, A., and Mestres, C. A. (2002). Infective endocarditis in intravenous drug abusers and HIV-1 infected patients. Infect. Dis. Clin. North Am. 16, 273–295. doi: 10.1016/s0891-5520(01)00008-3
Monach, P., Hattori, K., Huang, H., Hyatt, E., Morse, J., Nguyen, L., et al. (2007). The K/BxN mouse model of inflammatory arthritis: theory and practice. Arthr. Res. 2, 269–282. doi: 10.1007/978-1-59745-402-5_20
Monach, P. A., Mathis, D., and Benoist, C. (2008). The K/BxN arthritis model. Curr. Protoc. Immunol. Chapter 15:Unit 15.22. doi: 10.1002/0471142735.im1522s81
Murdoch, D. R., Corey, G. R., Hoen, B., Miró, J. M., Fowler, V. G., Bayer, A. S., et al. (2009). Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the international collaboration on endocarditis–prospective cohort study. Arch. Int. Med. 169, 463–473. doi: 10.1001/archinternmed.2008.603
National Institutes of Health (2005). The Autoimmune Diseases Coordinating Committee. Bethesda, MD: The Institutes.
Nishimura, M., Sharim, J., Horiuchi, Y., Barnett, O., Wettersten, N., and Maisel, A. (2018). Soluble ST2: a biomarker to monitor heart failure progression and treatment. J. Clin. Prevent. Cardiol. 7, 148–153.
Nolte, K. B., Alakija, P., Oty, G., Shaw, M. W., Subbarao, K., Guarner, J., et al. (2000). Influenza A virus infection complicated by fatal myocarditis. Am. J. Forensic Med. Pathol. 21, 375–379. doi: 10.1097/00000433-200012000-00016
Pankuweit, S., Ristic, A. D., Seferovic, P. M., and Maisch, B. (2005). Bacterial pericarditis: diagnosis and management. Am. J. Cardiovasc. Drugs 5, 103–112. doi: 10.2165/00129784-200505020-00004
Pankuweit, S., Wädlich, A., Meyer, E., Portig, I., Hufnagel, G., and Maisch, B. (2000). Cytokine activation in pericardial fluids in different forms of pericarditis. Herz 25, 748–754. doi: 10.1007/pl00001993
Pollack, A., Kontorovich, A. R., Fuster, V., and Dec, G. W. (2015). Viral myocarditis–diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 12, 670–680. doi: 10.1038/nrcardio.2015.108
Quinn, A., Kosanke, S., Fischetti, V. A., Factor, S. M., and Cunningham, M. W. (2001). Induction of autoimmune valvular heart disease by recombinant streptococcal M protein. Infect. Immun. 69, 4072–4078. doi: 10.1128/iai.69.6.4072-4078.2001
Raza, S. S., Sultan, O. W., and Sohail, M. R. (2010). Gram-negative bacterial endocarditis in adults: state-of-the-heart. Expert. Rev. Anti Infect. Ther. 8, 879–885. doi: 10.1586/eri.10.76
Rohayem, J., Dinger, J., Fischer, R., Klingel, K., Kandolf, R., and Rethwilm, A. (2001). Fatal myocarditis associated with acute parvovirus B19 and human herpesvirus 6 coinfection. J. Clin. Microbiol. 39, 4585–4587. doi: 10.1128/jcm.39.12.4585-4587.2001
Rose, N. R. (2016). Viral myocarditis. Curr. Opin. Rheumatol. 28, 383–389. doi: 10.1097/BOR.0000000000000303
Rose, N. R., Wolfgram, L. J., Herskowitz, A., and Beisel, K. W. (1986). Postinfectious autoimmunity: two distinct phases of coxsackievirus B3-induced myocarditis. Ann. N. Y. Acad. Sci. 475, 146–156. doi: 10.1111/j.1749-6632.1986.tb20864.x
Roth, G. A., Huffman, M. D., Moran, A. E., Feigin, V., Mensah, G. A., Naghavi, M., et al. (2015). Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation 132, 1667–1678. doi: 10.1161/CIRCULATIONAHA.114.008720
Sanaiha, Y., Lyons, R., and Benharash, P. (2019). Infective endocarditis in intravenous drug users. Trends Cardiovasc. Med. 5, 307–316.
Schultz, J. C., Hilliard, A. A., Cooper, L. T., and Rihal, C. S. (2009). “Diagnosis and treatment of viral myocarditis,” in Proceedings of the Mayo Clinic, (Amsterdem: Elsevier).
Selgrade, M., Cooper, G. S., Germolec, D. R., and Heindel, J. J. (1999). Linking environmental agents and autoimmune disease: an agenda for future research. Environ. Health Perspect. 107(Suppl. 5), 811. doi: 10.2307/3434345
Selton-Suty, C., Célard, M., Le Moing, V., Doco-Lecompte, T., Chirouze, C., Iung, B., et al. (2012). Preeminence of Staphylococcus aureus in infective endocarditis: a 1-year population-based survey. Clin. Infect. Dis. 54, 1230–1239. doi: 10.1093/cid/cis199
Sughimoto, K., Nakano, K., Gomi, A., Nakatani, H., Nakamura, Y., and Sato, A. (2006). Infective endocarditis associated with Sjogren’s syndrome. Asian Cardiovasc. Thorac. Ann. 14, e115–e117.
Waldner, H. (2009). The role of innate immune responses in autoimmune disease development. Autoimmun. Rev. 8, 400–404. doi: 10.1016/j.autrev.2008.12.019
Yoshioka, D., Toda, K., Yokoyama, J. Y., Matsuura, R., Miyagawa, S., Kainuma, S., et al. (2018). Diabetes mellitus adversely affects mortality and recurrence after valve surgery for infective endocarditis. J. Thorac. Cardiovasc. Surg. 155, 1021–1029e1025.
Keywords: autoimmune diseases, myocarditis, pericarditis, endocarditis, microbes, inflammatory heart disease, bacteria
Citation: Lasrado N, Yalaka B and Reddy J (2020) Triggers of Inflammatory Heart Disease. Front. Cell Dev. Biol. 8:192. doi: 10.3389/fcell.2020.00192
Received: 25 January 2020; Accepted: 06 March 2020;
Published: 24 March 2020.
Edited by:
Francesco Ria, Catholic University of the Sacred Heart, ItalyReviewed by:
Sally A. Huber, The University of Vermont, United StatesDeLisa Fairweather, Mayo Clinic, United States
Copyright © 2020 Lasrado, Yalaka and Reddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jay Reddy, bnJlZGR5MkB1bmwuZWR1