 Huiling Xiang
Huiling Xiang Feng Zhu
Feng Zhu Zhifeng Xu
Zhifeng Xu Jing Xiong
Jing Xiong- 1Department of Nephrology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Inflammasomes, multiprotein complex induced by harmful factors in the body, play a crucial role in innate immunity. Activation of inflammasomes lead to the activation of casepase-1 and then the secretion of inflammatory cytokines, including IL-1β and IL-18, subsequently leading to a type of cell death called pyroptosis. There are two types of signaling pathways involved in the process of inflammasome activation: the canonical and the non-canonical signaling pathway. The canonical signaling pathway is mainly dependent on casepase-1; the non-canonical signal pathway, which was recently discovered, is mainly dependent on caspase-11, but is also meditated by caspase-4, caspase-5, and caspase-8. Kidney inflammation is basically associated with inflammatory factor exudation and inflammatory cell infiltration. Several studies have showed that inflammasomes are closely related to kidney diseases, especially the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome, which play a role in regulating kidney inflammation and fibrosis. In this review, we focus on the relationship between inflammasomes and kidney diseases, especially the role of the NLRP3 inflammasome in different kinds of kidney disease via both canonical and non-canonical signal pathways.
Introduction
Inflammasomes are sensors of innate immunity that are triggered in response to danger signals. They consist kinds of multiprotein complex that include Toll-like receptors (TLRs) and nucleotide-biding oligomerization-domain protein (NOD)-like receptors (NLRs) which trigger inflammation in various diseases especially in immune diseases (Schroder and Tschopp, 2010; Lamkanfi and Dixit, 2012). Inflammasomes have also been related with intestine, lung, neurological diseases and cardiovascular diseases, but how is the inflammasome assembled and even functions in these diseases remains unclear (Opipari and Franchi, 2015; Chen et al., 2016a; Wang et al., 2016; Pinkerton et al., 2017; Lang et al., 2018; Zhang et al., 2020).
The basic structure of an inflammasome consists of a receptor, adaptor, and effector. The receptors are classified based on their structural features and include TLRs, NLRs, Absent in Melanoma 2-Like receptors (ALRs), and pyrin domain (Pyrin) (Sharma and Kanneganti, 2016). The adaptor molecule, apoptosis-associate speck-like protein (ASC), enables the recruitment and activation of caspase-1, which is the effector (Broz and Dixit, 2016). The effector molecule pro-caspase-1 induces maturation of pro-IL-1β and pro-IL-18, causing the cell pyroptosis (Lamkanfi, 2011; Lamkanfi and Dixit, 2014). Innate immunity detects the inflammasome or dangers signals mostly via pattern-recognition receptors (PRRs). There are two kinds of PRRs in innate immune recognition-one recognizes pathogen-associated molecular patterns (PAMPs) and the other danger-associated molecular patterns (DAMPs) (Janeway and Medzhitov, 2002). PAMPs are mostly outer conserved molecules on microbes, while the DAMPs are basically derived from host cells and deliver the signal of injury or cell death (Kanneganti et al., 2007). In the presence of a signal, the PAMPs or DAMPs activate the PRRs on NLRs or TLRs, which triggers the ASC adaptor to activate the effector caspase-1 or caspase-11. The activation of these molecules can lead to the secretion of proinflammatory cytokine pro-IL-1β, pro-IL-18 as well as the cleavage of gasdermin D (GSDMD), thereby triggering cell pyroptosis (Conforti-Andreoni et al., 2011; Kayagaki et al., 2011; Kovacs and Miao, 2017). The canonical pathway involving caspase-1 has been highly researched; however, the role and underlying mechanism of the inflammasomes, particularly involving caspase-11 or other effectors like caspase-4, caspase-5, and caspase-8, which were recently identified to be part of a non-canonical signaling pathway, remains poorly understood (Gurung et al., 2014; Vigano et al., 2015).
Depending on their differences in structure and activation, the inflammasomes are divided into NLRP1, NOD-, LRR- and pyrin domain-containing 3 (NLRP3), NLRC4, IPAF, and AIM2 inflammasomes (de Zoete et al., 2014). Of these, the NLRP3 has been extensively researched and characterized. In this review, we focus on the NLRP3 inflammasome in kidney diseases both in canonical and non-canonical signaling pathway, and provide an update on its roles in kidney disease and discuss their potential therapeutic effects for the future application.
NLRP3 Inflammasomes and Kidney Diseases
The NLRP3 inflammasome is a member of the NLR families of proteins, which was firstly named in 2002 (Martinon et al., 2002). The NLR families include 23 human genes and 34 mouse genes that can form the inflammasome complex (Tian et al., 2009; Rathinam et al., 2012a). Although NLR genes are expressed in kidney, there is no evidence for a direct link between inflammasome and kidney disease (Vilaysane et al., 2010). NLR family members share the same central NACHT and C-terminal LRR domains, with the different subtypes based on their N-terminus. The common N-terminal domains include acidic transactivation, pyrin, caspase recruitment domain (CARD), and baculoviral inhibitory repeat-like domains (Ting et al., 2008). NLRP3 contains PYD at its D-terminal, which allows its contact with the adapter via PYD-PYD interactions (Sutterwala et al., 2014). Canonical NLRP3 inflammasomes often react with varieties of PRRs, including the danger signal reproduced by itself, bacterial as well as its ligands such as the bacterial DNA, muramyl dipeptide, ATP, and even some virus (Man and Kanneganti, 2015). Non-canonical NLRP3 inflammasomes respond mainly to gram-negative bacteria (Rathinam et al., 2012b).
Although the accurate mechanism of the activation of NLRP3 still remains unknown, some models have been proposed, including the ion fuel model, the reactive oxygen species (ROS) model, and the lysosome rupture model (de Zoete et al., 2014). In ion fuel model, the activation of NLRP3 is majorly meditated by the K+ efflux, but also by some other ions like H+ or Ca+ (Pétrilli et al., 2007; Ichinohe et al., 2010; Lee et al., 2012). The ROS model is based on the fact that ROS from the NADPH oxidase system can accelerate the activation of NLRP3 inflammasome, and inhibiting the ROS can inhibit the activation, although studies have also shown that the activation of NLRP3 in a model of lacking NADPH (Latz, 2010; van Bruggen et al., 2010). Therefore, the role of ROS in the activation of inflammasome requires further thorough investigation. In the lysosome rupture model, the NLRP3 inflammasome is thought to be activated upon sensing the large molecules such as uric acid and cholesterol leading to the lysosome rupture; this reflects the key role of inflammasome in kidney disease and atherosclerosis (Duewell et al., 2010; Gaidt et al., 2017).
Many factors can contribute to the development of kidney diseases, such as sepsis, chemotherapy, contrast agents, infection, and kidney injury that can turn into the end stage of renal disease, following which the only option a patient has is hemodialysis or kidney transplantation (D’Agati and Appel, 1997; Arany and Safirstein, 2003; McCullough, 2008; Chawla et al., 2014; Bellomo et al., 2017). At present, the kidney diseases are mainly diagnosed by renal pathology and most of these diseases have not been thoroughly studied and remain poorly understood. Almost all kidney diseases have the same trait of cytokines expression and infiltration of immune cells, which indicates that the kidney is a target in immune response (Kurts et al., 2013). The NLRP3 genes are found to express in renal dendritic cells and macrophages, while other non-immune cells seem do not release IL-1β (Martinon et al., 2009; Lichtnekert et al., 2011). Previous study has confirmed that kidney diseases are related to inflammasomes and this association might be direct or indirect (Mulay et al., 2013; Xiong et al., 2015). The activation of inflammasomes in kidney aggravates the damage, while inhibiting certain signaling pathways of inflammasome often alleviates kidney injury. Thus, inflammasome might be a potential target in the treatment of renal disease, suggesting development of novel effective therapeutics for kidney diseases.
Canonical Signal Pathway
Mechanism of Canonical Signal Pathway
The canonical pathway of the activation of inflammasomes is dependent on caspase-1 (Lamkanfi and Dixit, 2014). The adopter protein ASC in inflammasomes possesses a caspase binding site (Sharma and Kanneganti, 2016). Upon any stimuli harmful to the body, the PAMPs or DAMPs activated PRRs trigger the relevant signal pathway as the first protective barrier of the innate immunity (Akira et al., 2006). After detection of risk factors, the ASC recruits the caspase-1, which is cleaved into caspase-1 p10 and caspase-1 p20 subunits (Guo et al., 2015), which meditate the transformation of the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms IL-1β and IL-18 as well as the cleavage of GSDMD. These molecules are related to the NF-kB and MAPK signaling pathways as well as other signaling pathways, which subsequently lead to cell pyrotosis (Fann et al., 2018), and further lead to the damage of organs and dysfunction of the body.
Inflammasomes in Acute Kidney Injury
Acute kidney injury (AKI) is a kind of clinical syndrome characterized by a rapid decrease of the kidney function and involves increase of metabolites such as creatinine (Levey and James, 2017). Many factors can result in AKI, such as the sepsis, ischemia reperfusion, chemotherapy and contrast agents (Ozkok and Edelstein, 2014; Gómez and Kellum, 2016; Fähling et al., 2017; Yang et al., 2017). AKI can also occur upon exposure of the body to harmful factors such as the lipopolysaccharide and cisplatin, which activate the innate immunity and inflammasomes (Leemans et al., 2014).
Studies have indicated that the inflammasome plays a role in the AKI model of Ischemia/reperfusion (I/R), cisplatin, sepsis, rhabdomyolysis, and contrast agents (Zhang et al., 2014; Komada et al., 2015; Shen et al., 2016; Nazir et al., 2017). It has already been verified that blocking the NLRP3 inflammasome signal pathway often alleviates the kidney injury in clinical trials (Fan et al., 2019). I/R can directly cause the AKI and the activation of NLRP3 inflammasome (Wen et al., 2018). Recent study showed that mitochondrial reactive oxygen species (mROS)-medicated activation of NLRP3 inflammasomes aggravates the renal injury (Liu et al., 2014). The use of mitochondria target antioxidants Mito TEMPO or the thioredoxin-interacting protein (TXNIP) siRNA can inhibit the production of the NLRP3, which confirmed that the NLRP3 inflammasome could be activated through mROS-TXNIP-NLRP3 signal pathway in I/R injury (Wen et al., 2018).
SIRT family proteins also play a certain role in inflammatory response. SIRT1 reduces ROS production and exerts its anti-inflammatory effect, although whether it can directly regulate inflammation remains controversial (Yeung et al., 2004). In a sepsis AKI model, SIRT3 was shown to have a protective effect on renal mitochondrial injury by reducing the production of ROS, and decreasing the release of IL-1β and IL-18 (Zhao et al., 2016). Overexpressing of SIRT3 promoted the autophagy and reduced the inflammasomes which are assembling in kidney injury caused by sepsis (Zhao et al., 2018). The use of cisplatin is often limited by the presence of AKI during chemotherapy (Huang et al., 2019). Research shows that cisplatin may inhibit autophagy and activate NLRP3 leading to the kidney injury (Qu et al., 2018). However, SIRT3 can protect from the AKI caused by cisplatin by inducing autophagy (Zhao et al., 2018). Moreover, Astragaloside IV can protect from cisplatin-induced kidney injury by inducing the autophagy and suppressing the NF-kB signal pathway, thereby down-regulating the expression of NLRP3 inflammasome (Qu et al., 2019). Moreover, the caspase inhibition can reduce the production of caspase-1 and IL-1β, and thus protect from the cisplatin-induced kidney injury (Lee et al., 2015).
In a model of rhabdomyolysis-induced acute kidney injury (RIAKI), activation and assembly of NLRP3 inflammasomes preceded infiltration of renal immune cells such as macrophages (Komada et al., 2015). Anisodamine shows a protective role in RIAKI by inhibiting the endoplasmic reticulum stress related to thioredoxin-interaction protein (TXNIP)/NLRP3 inflammasome (Yuan et al., 2017). NLRP3 inflammasome could also meditate contrast agent-induced AKI by regulating the cell apoptosis; indeed, NLRP3 inhibition could reduce the rate of apoptosis and the production of ROS in contrast agent-induced AKI (Shen et al., 2016; Tan et al., 2017).
All of these new findings confirmed that the close relationship between acute kidney disease and inflammasome. Though the inflammasome as a therapeutic target has not been widely considered in clinical research, these findings indicate the potential of the inflammasome for developing novel treatment against acute kidney diseases.
Inflammasomes in Chronic Kidney Disease
Chronic kidney disease (CKD) is always the end step of many kidney diseases. CKD is divided into five stages based on glomerular filtration rate, and the different stages show different clinical features (Levey et al., 2003). The basic feature of CKD is persistent renal tubular injury and renal fibrosis. CKD has become a huge burden in many countries due to its high morbidity and lack of effective treatment. Recently, many researchers have proved that interfering with the activation of inflammasome could regulate CKD, indicating that the inflammasome is involved in the CKD pathogenesis (Vilaysane et al., 2010). IL-18 and caspase-1 were found to be expressed in renal tubular epithelium as well as in patients with CKD (Matsumoto and Kanmatsuse, 2001; Gauer et al., 2007). In a unilateral ureteral obstruction (UUO) model, caspase-1, IL-1β, and IL-18 showed increased expression, leading to NLRP3 activation, While in NLRP3 gene knock out mice, less tubular injury and fibrosis were observed after UUO (Vilaysane et al., 2010).
The evaluation of the association between CKD and inflammasome in recent study is mainly focused on the mechanism of mitochondria, aldosterone, proteinuria, and calcinosis (Nishi et al., 2013; Ding et al., 2016; Gong et al., 2016; Anders et al., 2018). Recently, mROS was confirmed related to the CKD, and silencing the NLRP3 gene could alleviate the associated mitochondrial dysfunction and renal fibrosis (Guo et al., 2017). Aldosterone can directly damage the renal tubule, which is a key point in CKD pathogenesis. Kadoya et al. (2015) showed that high level of aldosterone could activate the inflammasomes via mROS. However, Bi et al. (2018) showed that the Mn(III) tetrakis (4-benzoic acid) porphyrin chloride (MnTABP) can significantly improve the morphology and function of mitochondria and limit the activation of NLRP3 inflammasome, finally reducing the renal injury caused by aldosterone. Proteinuria is an independent risk factor of kidney injury and is considered as an important clinical symptom. A Study showed that proteinuria could function as a DAMP, leading to the renal tubular inflammation via mROS-meditated activation of the NLRP3 inflammasome (Liu et al., 2014).
Calcium carbonate crystal deposition is a crucial factor in the mechanism of tubular injury and fibrosis, which also involves the NLRP3 inflammasome. Anders et al. (2018) showed that calcium oxalate crystal deposition activates the NLRP3 inflammasome via the TGFR signaling pathway, and not through IL-1β, as is commonly thought. Furthermore, it has been shown that NLRP3 can directly promote the TGF-β signal and activation of R-Smad independent of inflammasome (Wang et al., 2013), which offers new insights into the function and potential treatments of NLRP3 inflammasome in kidney disease. In addition, there are other potential treatment or drugs can reduce or inhibit tubular damage by interfering with the signal pathway of NLRP3 inflammasome; these include mitochondria target antioxidants, compound K, Neferine, allopurinol, and ghrelin (Ding et al., 2017; Foresto-Neto et al., 2018; Hsu et al., 2019; Ling et al., 2019).
Though these potential treatments are still under investigation, it will be a promising therapy for CKD in the near future.
Inflammasomes in Diabetic Nephropathy
Diabetic nephropathy (DN) is one of the most common microvascular complications in diabetic patients (Flyvbjerg, 2017). It often eventually turns into end stage renal disease, after which the patients can only be sustained through dialysis. In addition, there are still other cardiovascular syndromes that also do not have effective treatments. Although there are many symptomatic treatment measures, the innate mechanism needs to be thoroughly studied and effective treatments need to be urgently developed. Diabetic nephropathy is characterized by typical aseptic inflammation, and the role of inflammasomes in diabetes has also been discovered, as NLRP3 activation was found in diabetic patients as well as podocytes and endothelial cell injury (Boini et al., 2014; Chen et al., 2015; Shahzad et al., 2015; Koka et al., 2019). Studies found that high glucose may be involved in the occurrence of DN by regulating the activation of inflammasome (Chen et al., 2016b; Fu et al., 2017). Another study focus on the mitochondrial ROS-TXNIP-NLRP3 signal pathway to examine the relationship between DN and NLRP3 inflammasome (Feng et al., 2016). The excessive production of mROS can activate the NLRP3 inflammasome through the TRX/TXNIP, but the function of this signal pathway in diabetes pathway remains unknown (Liu et al., 2018). ROS signal pathway has been shown to be closely related to the DN (Qiu and Tang, 2016). Zhou et al. (2018) proved that high glucose can induce the activation of ROS-meditated activation of the NLRP3 inflammasome. Xu et al. (2018) showed that TXNIP can induce the oxidative stress response in glomerular mesangial cells, while Tan et al. (2015) found that TXNIP inhibited tubule-interstitial compartment from acting as an important meditator in the process of tubule-interstitial fibrosis in DN. Moreover, Han et al. (2018) used an antioxidant targeting mtROS (MitoQ) in mice and confirmed that mtROS-TXNIP-NLRP3 inflammasome pathway activation was key for the DN. Apart from these, other potential mechanisms are also under investigation.
Interestingly, Yi et al. (2017) has found that lincRNA-Gm4419 can regulate the inflammasome and tubular fibrosis during DN through the NF-kB/NLRP3 signal pathway. They reported abnormal expression of a number of lncRNAs, including that of Gm4419, in diabetic kidney organization, and showed that the knockdown of lincRNA-Gm4419 could inhibit the inflammasome in kidney (Yi et al., 2017). In another study, it was found that acid ceramidase (AC) deficiency significantly promoted the activation of NLRP3 inflammasome and the secretion of exosomes; the latter further promoted the release of IL-1β in the diabetes model. This study not only confirmed the relationship between inflammasomes and diabetes, but also confirmed the role of exosomes in the development of diabetes (Yuan et al., 2019). This research offers a new perspective on the relationship between inflammasome and kidney disease.
As a potential therapeutic agents against inflammation and DN, Compound K can inhibit the TXNIP/NLRP3 signal pathway and ameliorate the insulin resistance to protect the tubular inflammation (Chen et al., 2016c). Dihydroquercetin also shows a protective role in DN by suppressing the ROS and the NLRP3 inflammasome (Ding et al., 2018). However, further research is required to find and validate effective drugs concerning the role of NLRP3 inflammasome.
Inflammasomes in IgA Nephropathy
IgA nephropathy (IGAN) is the most common primary glomerular disease, and is characterized by accumulation or deposition of IgA in kidney (Magistroni et al., 2015). Most such patients eventually show end stage of kidney disease, making it urgent to find effective treatment. As already indicated, the inflammasome has a close relationship with kidney; however, whether it plays a role in the IGAN remains unknown. Recently, Tsai et al. (2017) found that IgA immune complexes can activate the inflammasome through the ROS in macrophage and induce the secretion of IL-1β and caspase-1, which indicated that the NLRP3 might participate in the inflammation in IGAN. Moreover, other factors that regulate NLRP3 activation can relieve the inflammation in IgA to some degree. For example, Antroquinonol can reduce the ROS produced by the IgA-immune complexes-primed macrophages (Yang et al., 2013), while triptolide treatment significantly reduces IL-1β and IL-18 levels and may have anti-inflammatory effects by down-regulating the expression of NLRP3 and TLR4 (He L. et al., 2015). However, there is still a long way to uncover the detailed mechanism of inflammasome in IGAN such as the relationship between IgA-related immune response and inflammasome, offering potential therapeutic approaches.
Inflammasomes in Other Kidney Diseases
Hyperhomocysteinemia is an important risk factor for the development of glomerular injury and sclerosis (Zhang et al., 2012). Activation of NLRP3 inflammasome has been shown to be involved in podocyte injury and glomerular sclerosis in hyperhomocysteinemia, and inhibition of NADPH oxidase or knockdown of ASC or caspase-1 inhibition may play a protective effect (Zhang et al., 2012; Abais et al., 2013). These studies confirmed the role of NLRP3 inflammasome in glomerular injury caused by hyperhomocysteinemia. The NADPH oxidase participates in the activation of NLRP3 inflammasome and the downstream H2O2 is mainly involved (Abais et al., 2014).
Activation of NLRP3 inflammasome is also involved in the development of obesity-related kidney disease. In ASC (+/+) mice, high fat diet promoted podocyte damage and the copolymerization of ASC and NLRP3 complex, while in ASC (−/−) mice, it could not (Boini et al., 2014). In acid sphingomyelinase (ASM) knockout mice, less caspase-1 and IL-1β production are observed, which suggests that the ASM may participate in the activation of NLRP3 inflammasome during the development of obesity-related kidney disease (Boini et al., 2016).
In addition, NLRP3 inflammasome also plays an important role in crystalline nephropathy. Some crystals, including sodium urate and calcium oxalate, can activate NLRP3 inflammasome (Hutton et al., 2016). Uric acid crystals are engulfed by lysosomes, which leads to lysosome rupture and then activation of NLRP3 inflammasome (Isaka et al., 2016). Blocking the activation of NLRP3 inflammasome, such as blocking IL-1β, may reduce the progression of crystalline nephropathy (Mulay et al., 2013).
Non-canonical Signal Pathway
Mechanism of Non-canonical Pathway
The recently identified non-canonical pathway of the activation of the inflammasome is mainly dependent on caspase-11, and also by caspase-4, caspase-5, and caspase-8. Murine caspase-11, also called caspase-4, is a critical regulatory factor in the activation of caspase-1 in the bacterial infections. Human caspase-4/caspase-5, speculated to be homologous with murine caspase-11, can regulate the release of inflammatory cytokines by binding LPS directly via CARD domains in an LPS stimulated monocytes model (Vigano et al., 2015). Caspase-11, caspase-4, and caspase-5 can all directly bind the LPS, inducing the production of IL-1β (Yang et al., 2015). In the non-canonical pathway, the inflammasomes first assemble due to detection of PAMPs or DAMPs by PRRs, leading to the activation of caspase-11, which in turn has two kinds of effects. One is that the caspase-11 activation leads directly macrophage death, and the other effect is that the caspase-11 functions as a binding partner in activation of caspase-1, leading to the release of the pro-IL-1β and pro-IL-18 and subsequent pyroptosis; that indicates that the caspase-11 can regulate activation of caspase-1 (Kayagaki et al., 2011; Broz et al., 2012; Vigano and Mortellaro, 2013). In a model of septic shock, the caspase-11 was overexpressed in macrophage and it could directly regulate the caspase-1 and caspase-3 (Kang et al., 2002). Another study also found that the LPS-induced caspase-11 in mice or caspase-4 in human can trigger the cleavage of GSDMD and its subsequent insertion into the cell membrane, leading to pyroptosis (Kayagaki et al., 2015). It has also shown that the caspase-11-meditated response to cytoplasmic LPS activates GSDMD, which is essential for pyroptosis and the secretion of IL-1β (He W.T. et al., 2015, Shi et al., 2015). Though the mechanism of GSDMD and its relationship with IL-1β remains to be further investigated, these studies have provided new insights for both canonical and non-canonical signaling pathway in the activation of inflammasome.
Caspase-8 is a kind of initiator caspase that can activate the downstream effectors caspase-3 and caspase-7 (Lavrik and Krammer, 2012). Caspase-8 and its adaptor FAS-associated death domain protein (FADD) can trigger the activation of inflammasome by inducing the release of IL-1β in the absence of caspase-1/caspase-11, thereby indicating the presence of another non-canonical pathway (Bossaller et al., 2012; Gringhuis et al., 2012).
Non-canonical Function of NLRP3 Inflammasome in Kidney Disease
Caspase-1, IL-1β, and IL-18 cannot be activated after knockout of NLRP3 inflammasome, suggesting that NLRP3 is essential for the production of these inflammatory components (Shigeoka et al., 2010). Previous studies have confirmed the role of inflammasomes in kidney immunity, supporting the theory that knocking out the inflammasome genes may also alleviate the injury.
In AKI, deficiency of NLRP3 inflammasome provides a protective effect in I/R induced AKI, although blocking IL-1β and IL-18 does not show any protective effect (Iyer et al., 2009; Shigeoka et al., 2010). This implies that NLRP3 inflammasome not only induces the kidney injury by meditating the production of IL-1β and IL-18, but might also be involved in other non-canonical signaling pathway. Knockout of both NLRP3 and ASC showed a protective effect in kidney injury, suggesting that they may function together. Chun et al. confirmed that caspase-8 can form a protein complex with NLRP3 and ASC to regulate apoptosis in a non-canonical manner in epithelial cells (Chung et al., 2016). Interestingly, NLRP3 knockout showed no protective effects in cisplatin-induced AKI, which further confirms the non-canonical effect of NLRP3 (Kim et al., 2013); however, its role in cisplatin model remains to be further elucidated.
In CKD, increased expression of NLRP3 inflammasome promotes renal epithelial-mesenchymal transformation (EMT) which is related to the phosphorylation of Smad-2 and Smad-3, and this effect is independent of caspase-1, ASC, IL-1β, or IL-18 (Anders and Lech, 2013). The NLRP3 inflammasome has also been identified to affect the TGF-β signal pathway and activate R-Smads independently of the caspase-1, IL-1β, and IL-18 signaling pathway (Wang et al., 2013). In oxalate-induced nephrocalcinosis, NLRP3 and ASC deficient mice failed to develop nephrocalcinosis, suggesting the protective effects of NLRP3 and ASC deficiency (Anders et al., 2018). However, the use of IL-1β inhibitors could not achieve the protective effect, revealing that NLRP3 inflammasome might act on nephrocalcinosis through other non-canonical pathway, possibly involving macrophage polarization and fibrosis (Anders et al., 2018). The molecular mechanism underlying these non-canonical signaling pathways and whether they involve caspase-11, caspase-8, or other unknown pathways requires further investigation, and the existing research progress will also provide a new understanding of the pathogenesis of kidney diseases.
Conclusion and Future Perspectives
The NLRP3 inflammasome is an intermediate medium for the rapid response of the body to the external injury, including activation of inflammatory cytokine and pyroptotic cell death. The inflammasomes activation by the PRRs and then result in the activation of caspase-1 and then lead to the secretion of IL-1β and IL-18 also with pyroptosis. In the process of NLRP3 activation, both the canonical signal pathway and non-canonical signaling pathways have been shown to play a role in the immune response to the external or internal harmful signals (Figure 1). The relationship of inflammasomes with many diseases, such as lung disease, intestine disease, neurological disease and cardiovascular diseases, has already been tested. In kidney, inflammasomes play a different role in various diseases, and mainly induce the inflammation and dysfunction of kidney; interfering the process of NLRP3 inflammasome activation can regulate the kidney injury. Through the canonical pathway, molecules like SIRT3 can inhibit the kidney injury by decreasing the release of IL-1β and IL-18. In the non-canonical pathway, caspase-8 can form a protein complex with NLRP3 and ASC to regulate apoptosis (Figure 2). Although current studies have confirmed the relationship with NLRP3 inflammasome and kidney disease, the mechanism needs further elucidation. The canonical pathway has been studied extensively, but its clinical applicability to treatment of disease is still ongoing; the non-canonical pathway, which was relatively recently identified, is currently under investigation, and it is hoped that the research will provide insights into mechanisms of kidney disease and its prevention. The existing ways and other unknown mechanisms for non-canonical pathway might be hot topics for future research. In conclusion, a full understanding of the mechanism of inflammasome in kidney disease may help comprehending the pathogenesis of renal disease and solving some of the current clinical problems.
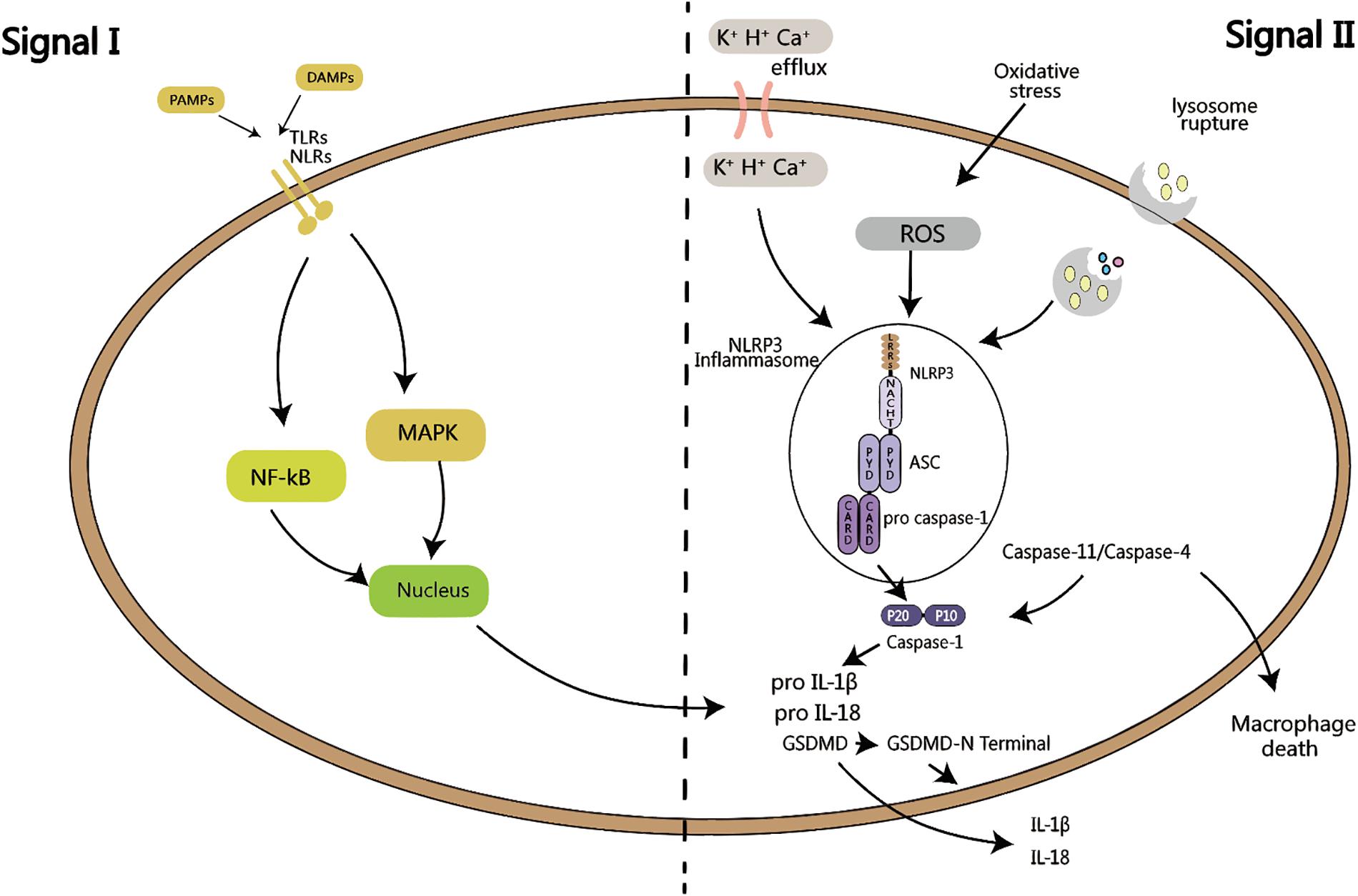
Figure 1. Activation of inflammasome requires two signals: signal I is PAMPs or DAMPs close to the cell, sensed by TLRs or NLRs, and activate MAPK or NF-kB signaling pathway, which in turns produces pro-IL-1β and pro-IL-18. Signal II is K+, H+ or Ca+ efflux, or oxidative stress activation by ROS, or lysosomal rupture, leading to the activation of NLRP3 inflammasome. ASC recruits pro caspase-1, which is then is cleaved into active p20 and p10 fragments, followed by the maturation of IL-1β and IL-18, as well as the cleavage of GSDMD, leading to cell pyroptosis. Both of these signals are called the canonical pathway of inflammation activation. When signal II is mediated by caspase-11/caspase-4, it is called the non-canonical activation pathway of inflammasome.
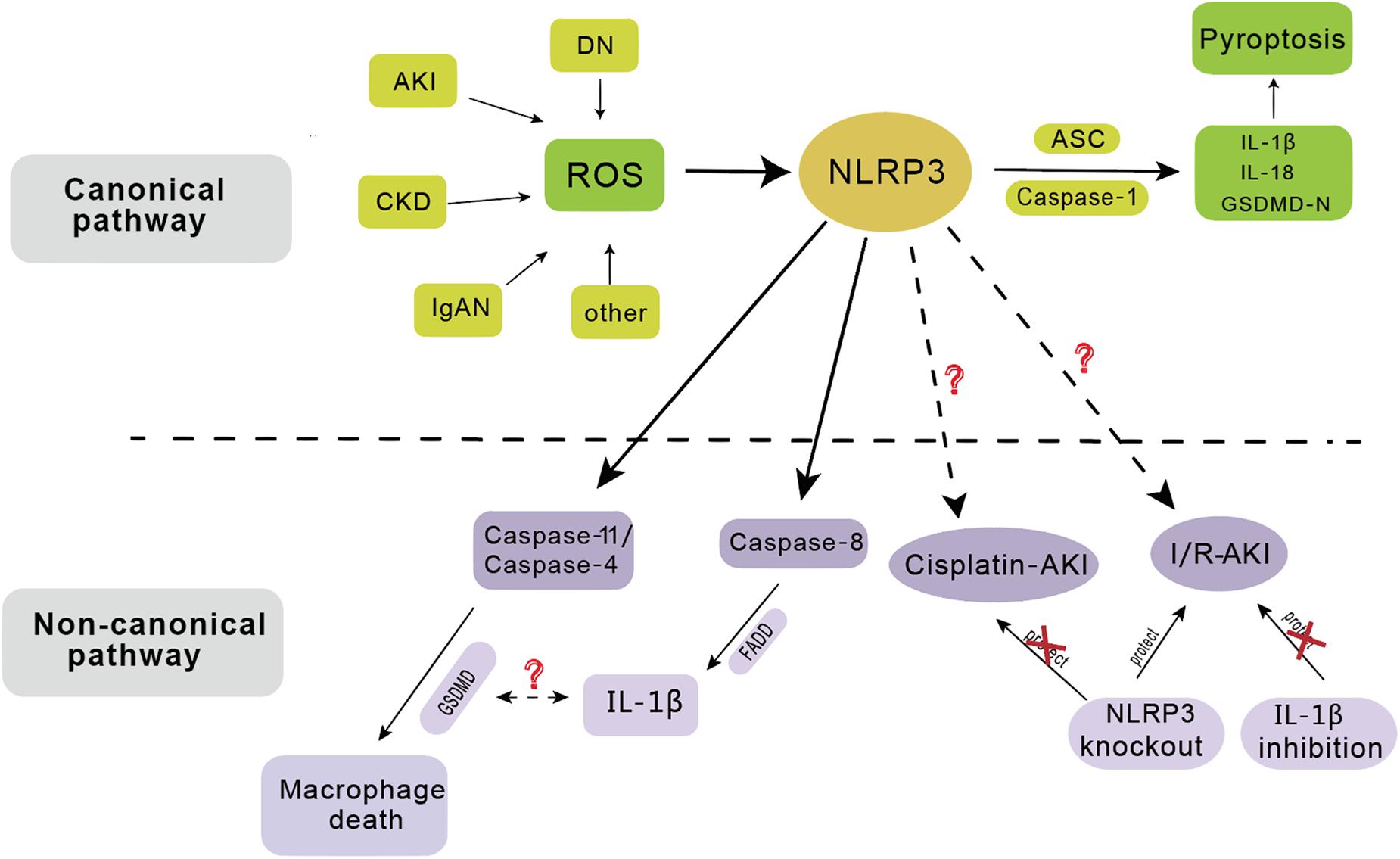
Figure 2. The activation of NLRP3 inflammasome has involved in the pathogenesis of several renal diseases including acute kidney injury (AKI), chronic kidney disease (CKD), diabetic nephropathy (DN), IgA nephropathy (IgAN), etc. In the canonical pathway, NLRP3 functions as a pattern recognition receptor for danger signal. Stimulation with danger or inflammatory signals triggers the formation of a large multimolecular complex, namely, NLRP3 inflammasome, where caspase-1 is activated to cleave its substrates including the precursors of inflammatory cytokine IL-1β to its bioactive form and leads to cell death called pyroptosis. In the non-canonical pathway, NLRP3 inflammasome mainly dependent on caspase-11/caspase-4 or caspase-8, leading to macrophage death. NLRP3 gene knockout has protective effect on ischemia/reperfusion induced AKI (I/R-AKI), but not on cisplatin induced AKI. IL-1β inhibition has no protective effect on I/R-AKI.
Author Contributions
HX and FZ wrote the manuscript. ZX contributed to the preparation of this manuscript. JX organized and reviewed the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81770736), the Doctoral Fund of the Ministry of Education of China (No. 200804871122), the Natural Science Foundation of Hubei Province (No. 2014CFB197), the Chinese Medical Association (No. 14050510588), the Huazhong University of Science and Technology (No. 2015LC033), and the Research-based Clinician Grants Program of Tongji Medical College, Huazhong University of Science and Technology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abais, J. M., Xia, M., Li, G., Gehr, T. W., Boini, K. M., and Li, P.-L. (2014). Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic. Biol. Med. 67, 211–220. doi: 10.1016/j.freeradbiomed.2013.10.009
Abais, J. M., Zhang, C., Xia, M., Liu, Q., Gehr, T. W., Boini, K. M., et al. (2013). NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid. Redox Signal. 18, 1537–1548. doi: 10.1089/ars.2012.4666
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Anders, H. J., and Lech, M. (2013). NOD-like and Toll-like receptors or inflammasomes contribute to kidney disease in a canonical and a non-canonical manner. Kidney Int. 84, 225–228. doi: 10.1038/ki.2013.122
Anders, H. J., Suarez-Alvarez, B., Grigorescu, M., Foresto-Neto, O., Steiger, S., and Desai, J. (2018). The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 93, 656–669. doi: 10.1016/j.kint.2017.09.022
Bellomo, R., Kellum, J. A., Ronco, C., Wald, R., Martensson, J., Maiden, M., et al. (2017). Acute kidney injury in sepsis. Intensive Care Med. 43, 816–828. doi: 10.1007/s00134-017-4755-7
Bi, X., Wang, J., Liu, Y., Wang, Y., and Ding, W. (2018). MnTBAP treatment ameliorates aldosterone-induced renal injury by regulating mitochondrial dysfunction and NLRP3 inflammasome signalling. Am. J. Transl. Res. 10, 3504–3513.
Boini, K. M., Xia, M., Abais, J. M., Li, G., Pitzer, A. L., Gehr, T. W., et al. (2014). Activation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencing. Biochim. Biophys. Acta 1843, 836–845. doi: 10.1016/j.bbamcr.2014.01.033
Boini, K. M., Xia, M., Koka, S., Gehr, T. W., and Li, P. L. (2016). Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene. Oncotarget 7, 19031–19044. doi: 10.18632/oncotarget.8023
Bossaller, L., Chiang, P. I, Schmidt-Lauber, C., Ganesan, S., Kaiser, W. J., and Rathinam, V. A. (2012). Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 189, 5508–5512. doi: 10.4049/jimmunol.1202121
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Broz, P., Ruby, T., Belhocine, K., Bouley, D. M., Kayagaki, N., and Dixit, V. M. (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. doi: 10.1038/nature11419
Chawla, L. S., Eggers, P. W., Star, R. A., and Kimmel, P. L. (2014). Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 371, 58–66.
Chen, W., Wang, J., Luo, Y., Wang, T., Li, X., Li, A., et al. (2016a). Ginsenoside Rb1 and compound K improve insulin signaling and inhibit ER stress-associated NLRP3 inflammasome activation in adipose tissue. J. Ginseng. Res. 40, 351–358. doi: 10.1016/j.jgr.2015.11.002
Chen, Y., Pitzer, A. L., Li, X., Li, P. L., Wang, L., and Zhang, Y. (2015). Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J. Cell. Mol. Med. 19, 2715–2727. doi: 10.1111/jcmm.12657
Chen, Y., Wang, L., Pitzer, A. L., Li, X., Li, P. L., and Zhang, Y. (2016b). Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 94, 1335–1347. doi: 10.1007/s00109-016-1481-5
Chen, Y., Yuan, M., Xia, M., Wang, L., Zhang, Y., and Li, P. L. (2016c). Instant membrane resealing in nlrp3 inflammmasome activation of endothelial cells. Front. Biosci. (Landmark Ed) 21, 635–650. doi: 10.2741/4411
Chung, H., Vilaysane, A., Lau, A., Stahl, M., Morampudi, V., and Bondzi-Simpson, A. (2016). NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 23, 1331–1346. doi: 10.1038/cdd.2016.14
Conforti-Andreoni, C., Ricciardi-Castagnoli, P., and Mortellaro, A. (2011). The inflammasomes in health and disease: from genetics to molecular mechanisms of autoinflammation and beyond. Cell. Mol. Immunol. 8, 135–145. doi: 10.1038/cmi.2010.81
de Zoete, M. R., Palm, N. W., Zhu, S., and Flavell, R. A. (2014). Inflammasomes. Cold Spring Harb. Perspect. Biol. 6:a016287. doi: 10.1101/cshperspect.a016287
Ding, T., Wang, S., Zhang, X., Zai, W., Fan, J., and Chen, W. (2018). Kidney protection effects of dihydroquercetin on diabetic nephropathy through suppressing ROS and NLRP3 inflammasome. Phytomedicine 41, 45–53. doi: 10.1016/j.phymed.2018.01.026
Ding, W., Guo, H., Xu, C., Wang, B., Zhang, M., and Ding, F. (2016). Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 7, 17479–17491. doi: 10.18632/oncotarget.8243
Ding, W., Liu, T., Bi, X., and Zhang, Z. (2017). Mitochondria-targeted antioxidant Mito-Tempo protects against aldosterone-induced renal injury In Vivo. Cell. Physiol. Biochem. 44, 741–750. doi: 10.1159/000485287
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., and Bauernfeind, F. G. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Fähling, M., Seeliger, E., Patzak, A., and Persson, P. B. (2017). Understanding and preventing contrast-induced acute kidney injury. Nat. Rev. Nephrol. 13, 169–180. doi: 10.1038/nrneph.2016.196
Fan, J., Xie, K., Wang, L., Zheng, N., and Yu, X. (2019). Roles of inflammasomes in inflammatory kidney diseases. Mediators Inflamm. 2019:2923072. doi: 10.1155/2019/2923072
Fann, D. Y., Lim, Y. A., Cheng, Y. L., Lok, K. Z., Chunduri, P., and Baik, S. H. (2018). Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 55, 1082–1096. doi: 10.1007/s12035-017-0394-9
Feng, H., Gu, J., Gou, F., Huang, W., Gao, C., and Chen, G. (2016). High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J. Diabetes Res. 2016:6973175. doi: 10.1155/2016/6973175
Flyvbjerg, A. (2017). The role of the complement system in diabetic nephropathy. Nat. Rev. Nephrol. 13, 311–318. doi: 10.1038/nrneph.2017.31
Foresto-Neto, O., Ávila, V. F., Arias, S. C. A., Zambom, F. F. F., Rempel, L. C. T., and Faustino, V. D. (2018). NLRP3 inflammasome inhibition ameliorates tubulointerstitial injury in the remnant kidney model. Lab. Invest. 98, 773–782. doi: 10.1038/s41374-018-0029-4
Fu, Y., Wu, N., and Zhao, D. (2017). Function of NLRP3 in the pathogenesis and development of diabetic nephropathy. Med. Sci. Monit. 23, 3878–3884. doi: 10.12659/msm.903269
Gaidt, M. M., Ebert, T. S., Chauhan, D., Ramshorn, K., Pinci, F., and Zuber, S. (2017). The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171, 1110.e–1124.e. doi: 10.1016/j.cell.2017.09.039
Gauer, S., Sichler, O., Obermüller, N., Holzmann, Y., Kiss, E., and Sobkowiak, E. (2007). IL-18 is expressed in the intercalated cell of human kidney. Kidney Int. 72, 1081–1087. doi: 10.1038/sj.ki.5002473
Gómez, H., and Kellum, J. A. (2016). Sepsis-induced acute kidney injury. Curr. Opin. Crit. Care 22, 546–553.
Gong, W., Mao, S., Yu, J., Song, J., Jia, Z., Huang, S., et al. (2016). NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am. J. Physiol. Renal Physiol. 310, F1081–F1088. doi: 10.1152/ajprenal.00534.2015
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Theelen, B., van der Vlist, M., Boekhout, T., et al. (2012). Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254. doi: 10.1038/ni.2222
Guo, H., Bi, X., Zhou, P., Zhu, S., and Ding, W. (2017). NLRP3 deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm. 2017:8316560. doi: 10.1155/2017/8316560
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. doi: 10.1038/nm.3893
Gurung, P., Anand, P. K., Malireddi, R. S., Walle, L. V., Van Opdenbosch, N., and Dillon, C. P. (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846. doi: 10.4049/jimmunol.1302839
Han, Y., Xu, X., Tang, C., Gao, P., Chen, X., and Xiong, X. (2018). Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 16, 32–46. doi: 10.1016/j.redox.2018.02.013
He, L., Peng, X., Liu, G., Tang, C., Liu, H., Liu, F., et al. (2015). Anti-inflammatory effects of triptolide on IgA nephropathy in rats. Immunopharmacol. Immunotoxicol. 37, 421–427. doi: 10.3109/08923973.2015.1080265
He, W. T., Wan, H., Hu, L., Chen, P., Wang, X., and Huang, Z. (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298. doi: 10.1038/cr.2015.139
Hsu, W. H., Hua, K. F., Tuan, L. H., Tsai, Y. L., Chu, L. J., and Lee, Y. C. (2019). Compound K inhibits priming and mitochondria-associated activating signals of NLRP3 inflammasome in renal tubulointerstitial lesions. Nephrol. Dial. Transplant. 35, 74–85. doi: 10.1093/ndt/gfz073
Huang, T. H., Wu, T. H., Guo, Y. H., Li, T. L., Chan, Y. L., and Wu, C. J. (2019). The concurrent treatment of Scutellaria baicalensis Georgi enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced cachexia and acute kidney injury. J. Ethnopharmacol. 243:112075. doi: 10.1016/j.jep.2019.112075
Hutton, H. L., Ooi, J. D., Holdsworth, S. R., and Kitching, A. R. (2016). The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 21, 736–744. doi: 10.1111/nep.12785
Ichinohe, T., Pang, I. K., and Iwasaki, A. (2010). Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410. doi: 10.1038/ni.1861
Isaka, Y., Takabatake, Y., Takahashi, A., Saitoh, T., and Yoshimori, T. (2016). Hyperuricemia-induced inflammasome and kidney diseases. Nephrol. Dial. Transplant. 31, 890–896. doi: 10.1093/ndt/gfv024
Iyer, S. S., Pulskens, W. P., Sadler, J. J., Butter, L. M., Teske, G. J., and Ulland, T. K. (2009). Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 106, 20388–20393. doi: 10.1073/pnas.0908698106
Janeway, C. A., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Kadoya, H., Satoh, M., Sasaki, T., Taniguchi, S., Takahashi, M., and Kashihara, N. (2015). Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 29, 3899–3910. doi: 10.1096/fj.15-271734
Kang, S. J., Wang, S., Kuida, K., and Yuan, J. (2002). Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 9, 1115–1125. doi: 10.1038/sj.cdd.4401087
Kanneganti, T. D., Lamkanfi, M., and Núñez, G. (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559. doi: 10.1016/j.immuni.2007.10.002
Kayagaki, N., Stowe, I. B., Lee, B. L., O’Rourke, K., Anderson, K., and Warming, S. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., and Dong, J. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kim, H. J., Lee, D. W., Ravichandran, K., Keys, D. O., Akcay, A., and Nguyen, Q. (2013). NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 346, 465–472. doi: 10.1124/jpet.113.205732
Koka, S., Xia, M., Zhang, C., Zhang, Y., Li, P. L., and Boini, K. M. (2019). Podocyte NLRP3 inflammasome activation and formation by adipokine visfatin. Cell. Physiol. Biochem. 53, 355–365. doi: 10.33594/000000143
Komada, T., Usui, F., Kawashima, A., Kimura, H., Karasawa, T., and Inoue, Y. (2015). Role of NLRP3 inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci. Rep. 5:10901. doi: 10.1038/srep10901
Kovacs, S. B., and Miao, E. A. (2017). Gasdermins: effectors of pyroptosis. Trends Cell Biol. 27, 673–684. doi: 10.1016/j.tcb.2017.05.005
Kurts, C., Panzer, U., Anders, H.-J., and Rees, A. J. (2013). The immune system and kidney disease: basic concepts and clinical implications. Nat. Rev. Immunol. 13, 738–753. doi: 10.1038/nri3523
Lamkanfi, M. (2011). Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11, 213–220. doi: 10.1038/nri2936
Lamkanfi, M., and Dixit, V. M. (2012). Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161. doi: 10.1146/annurev-cellbio-101011-155745
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Lang, Y., Chu, F., Shen, D., Zhang, W., Zheng, C., and Zhu, J. (2018). Role of inflammasomes in neuroimmune and neurodegenerative diseases: a systematic review. Mediators Inflamm. 2018:1549549. doi: 10.1155/2018/1549549
Latz, E. (2010). NOX-free inflammasome activation. Blood 116, 1393–1394. doi: 10.1182/blood-2010-06-287342
Lavrik, I. N., and Krammer, P. H. (2012). Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 19, 36–41. doi: 10.1038/cdd.2011.155
Lee, D. W., Faubel, S., and Edelstein, C. L. (2015). A pan caspase inhibitor decreases caspase-1, IL-1α and IL-1β, and protects against necrosis of cisplatin-treated freshly isolated proximal tubules. Ren. Fail. 37, 144–150. doi: 10.3109/0886022x.2014.970194
Lee, G. S., Subramanian, N., Kim, A. I, Aksentijevich, I., Goldbach-Mansky, R., and Sacks, D. B. (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. doi: 10.1038/nature11588
Leemans, J. C., Kors, L., Anders, H. J., and Florquin, S. (2014). Pattern recognition receptors and the inflammasome in kidney disease. Nat. Rev. Nephrol. 10, 398–414. doi: 10.1038/nrneph.2014.91
Levey, A. S., Coresh, J., Balk, E., Kausz, A. T., Levin, A., Steffes, M. W., et al. (2003). National kidney foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann. Intern. Med. 139, 137–147.
Levey, A. S., and James, M. T. (2017). Acute kidney injury. Ann. Intern. Med. 167, Itc66–Itc80. doi: 10.7326/AITC201711070
Lichtnekert, J., Kulkarni, O. P., Mulay, S. R., Rupanagudi, K. V., Ryu, M., and Allam, R. (2011). Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS One 6:e26778. doi: 10.1371/journal.pone.0026778
Ling, L., Yang, M., Ding, W., and Gu, Y. (2019). Ghrelin attenuates UUO-induced renal fibrosis via attenuation of Nlrp3 inflammasome and endoplasmic reticulum stress. Am. J. Transl. Res. 11, 131–141.
Liu, D., Xu, M., Ding, L. H., Lv, L. L., Liu, H., Ma, K. L., et al. (2014). Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: a novel mechanism of albumin-induced tubulointerstitial inflammation. Int. J. Biochem. Cell Biol. 57, 7–19. doi: 10.1016/j.biocel.2014.09.018
Liu, Q., Zhang, D., Hu, D., Zhou, X., and Zhou, Y. (2018). The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124. doi: 10.1016/j.molimm.2018.09.010
Magistroni, R., D’Agati, V. D., Appel, G. B., and Kiryluk, K. (2015). New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 88, 974–989. doi: 10.1038/ki.2015.252
Man, S. M., and Kanneganti, T. D. (2015). Regulation of inflammasome activation. Immunol. Rev. 265, 6–21. doi: 10.1111/imr.12296
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi: 10.1146/annurev.immunol.021908.132715
Matsumoto, K., and Kanmatsuse, K. (2001). Augmented interleukin-18 production by peripheral blood monocytes in patients with minimal-change nephrotic syndrome. Am. J. Nephrol. 21, 20–27. doi: 10.1159/000046214
McCullough, P. A. (2008). Contrast-induced acute kidney injury. J. Am. Coll. Cardiol. 51, 1419–1428.
Mulay, S. R., Kulkarni, O. P., Rupanagudi, K. V., Migliorini, A., Darisipudi, M. N., and Vilaysane, A. (2013). Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Invest. 123, 236–246. doi: 10.1172/jci63679
Nazir, S., Gadi, I., Al-Dabet, M. M., Elwakiel, A., Kohli, S., and Ghosh, S. (2017). Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130, 2664–2677. doi: 10.1182/blood-2017-05-782102
Nishi, Y., Satoh, M., Nagasu, H., Kadoya, H., Ihoriya, C., and Kidokoro, K. (2013). Selective estrogen receptor modulation attenuates proteinuria-induced renal tubular damage by modulating mitochondrial oxidative status. Kidney Int. 83, 662–673. doi: 10.1038/ki.2012.475
Opipari, A., and Franchi, L. (2015). Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm. Bowel Dis. 21, 173–181. doi: 10.1097/MIB.0000000000000230
Ozkok, A., and Edelstein, C. L. (2014). Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014:967826. doi: 10.1155/2014/967826
Pétrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. doi: 10.1038/sj.cdd.4402195
Pinkerton, J. W., Kim, R. Y., Robertson, A. A. B., Hirota, J. A., Wood, L. G., and Knight, D. A. (2017). Inflammasomes in the lung. Mol. Immunol. 86, 44–55.
Qiu, Y. Y., and Tang, L. Q. (2016). Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 114, 251–264. doi: 10.1016/j.phrs.2016.11.004
Qu, X., Gao, H., Tao, L., Zhang, Y., Zhai, J., and Song, Y. (2018). Autophagy inhibition-enhanced assembly of the NLRP3 inflammasome is associated with cisplatin-induced acute injury to the liver and kidneys in rats. J. Biochem. Mol. Toxicol. 33:e22208. doi: 10.1002/jbt.22228
Qu, X., Gao, H., Tao, L., Zhang, Y., Zhai, J., Sun, J., et al. (2019). Astragaloside IV protects against cisplatin-induced liver and kidney injury via autophagy-mediated inhibition of NLRP3 in rats. J. Toxicol. Sci. 44, 167–175. doi: 10.2131/jts.44.167
Rathinam, V. A., Vanaja, S. K., and Fitzgerald, K. A. (2012a). Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342. doi: 10.1038/ni.2237
Rathinam, V. A., Vanaja, S. K., Waggoner, L., Sokolovska, A., Becker, C., and Stuart, L. M. (2012b). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. doi: 10.1016/j.cell.2012.07.007
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Shahzad, K., Bock, F., Dong, W., Wang, H., Kopf, S., and Kohli, S. (2015). Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 87, 74–84. doi: 10.1038/ki.2014.271
Sharma, D., and Kanneganti, T. D. (2016). The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J. Cell Biol. 213, 617–629. doi: 10.1083/jcb.201602089
Shen, J., Wang, L., Jiang, N., Mou, S., Zhang, M., and Gu, L. (2016). NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci. Rep. 6:34682. doi: 10.1038/srep34682
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., and Huang, H. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shigeoka, A. A., Mueller, J. L., Kambo, A., Mathison, J. C., King, A. J., Hall, W. F., et al. (2010). An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 185, 6277–6285. doi: 10.4049/jimmunol.1002330
Sutterwala, F. S., Haasken, S., and Cassel, S. L. (2014). Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 1319, 82–95. doi: 10.1111/nyas.12458
Tan, C. Y., Weier, Q., Zhang, Y., Cox, A. J., Kelly, D. J., and Langham, R. G. (2015). Thioredoxin-interacting protein: a potential therapeutic target for treatment of progressive fibrosis in diabetic nephropathy. Nephron 129, 109–127. doi: 10.1159/000368238
Tan, X., Zheng, X., Huang, Z., Lin, J., Xie, C., and Lin, Y. (2017). Involvement of S100A8/A9-TLR4-NLRP3 inflammasome pathway in contrast-induced acute kidney injury. Cell. Physiol. Biochem. 43, 209–222. doi: 10.1159/000480340
Tian, X., Pascal, G., and Monget, P. (2009). Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol. Biol. 9:202. doi: 10.1186/1471-2148-9-202
Ting, J. P.-Y., Lovering, R. C., Alnemri, E. S., Bertin, J., Boss, J. M., and Davis, B. (2008). The NLR gene family: an official nomenclature. Immunity 28, 285–287.
Tsai, Y. L., Hua, K. F., Chen, A., Wei, C. W., Chen, W. S., and Wu, C. Y. (2017). NLRP3 inflammasome: pathogenic role and potential therapeutic target for IgA nephropathy. Sci. Rep. 7:41123. doi: 10.1038/srep41123
van Bruggen, R., Köker, M. Y., Jansen, M., van Houdt, M., Roos, D., and Kuijpers, T. W. (2010). Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115, 5398–5400. doi: 10.1182/blood-2009-10-250803
Vigano, E., Diamond, C. E., Spreafico, R., Balachander, A., Sobota, R. M., and Mortellaro, A. (2015). Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 6:8761. doi: 10.1038/ncomms9761
Vigano, E., and Mortellaro, A. (2013). Caspase-11: the driving factor for noncanonical inflammasomes. Eur. J. Immunol. 43, 2240–2245. doi: 10.1002/eji.201343800
Vilaysane, A., Chun, J., Seamone, M. E., Wang, W., Chin, R., and Hirota, S. (2010). The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 21, 1732–1744. doi: 10.1681/ASN.2010020143
Wang, L., Chen, Y., Li, X., Zhang, Y., Gulbins, E., and Zhang, Y. (2016). Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 7, 73229–73241. doi: 10.18632/oncotarget.12302
Wang, W., Wang, X., Chun, J., Vilaysane, A., Clark, S., and French, G. (2013). Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J. Immunol. 190, 1239–1249. doi: 10.4049/jimmunol.1201959
Wen, Y., Liu, Y. R., Tang, T. T., Pan, M. M., Xu, S. C., and Ma, K. L. (2018). mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int. J. Biochem. Cell Biol. 98, 43–53. doi: 10.1016/j.biocel.2018.02.015
Xiong, J., Wang, Y., Shao, N., Gao, P., Tang, H., Su, H., et al. (2015). The expression and significance of NLRP3 inflammasome in patients with primary glomerular diseases. Kidney Blood Press. Res. 40, 344–354. doi: 10.1159/000368511
Xu, W., Wang, L., Li, J., Cai, Y., and Xue, Y. (2018). TXNIP mediated the oxidative stress response in glomerular mesangial cells partially through AMPK pathway. Biomed. Pharmacother. 107, 785–792. doi: 10.1016/j.biopha.2018.08.067
Yang, J., Zhao, Y., and Shao, F. (2015). Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 32, 78–83. doi: 10.1016/j.coi.2015.01.007
Yang, K., Li, W. F., Yu, J. F., Yi, C., and Huang, W. F. (2017). Diosmetin protects against ischemia/reperfusion-induced acute kidney injury in mice. J. Surg. Res. 214, 69–78. doi: 10.1016/j.jss.2017.02.067
Yang, S. M., Ka, S. M., Hua, K. F., Wu, T. H., Chuang, Y. P., and Lin, Y. W. (2013). Antroquinonol mitigates an accelerated and progressive IgA nephropathy model in mice by activating the Nrf2 pathway and inhibiting T cells and NLRP3 inflammasome. Free Radic. Biol. Med. 61, 285–297. doi: 10.1016/j.freeradbiomed.2013.03.024
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., and Frye, R. A. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. doi: 10.1038/sj.emboj.7600244
Yi, H., Peng, R., Zhang, L. Y., Sun, Y., Peng, H. M., Liu, H. D., et al. (2017). LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 8:e2583. doi: 10.1038/cddis.2016.451
Yuan, X., Bhat, O. M., Lohner, H., Zhang, Y., and Li, P. L. (2019). Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: evidence from endothelium-specific deletion of Asah1 gene. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864:158532. doi: 10.1016/j.bbalip.2019.158532
Yuan, X., Zheng, Y., Chen, C., and Wang, C. (2017). Anisodamine inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in rhabdomyolysis-induced acute kidney injury. Apoptosis 22, 1524–1531. doi: 10.1007/s10495-017-1414-y
Zhang, C., Boini, K. M., Xia, M., Abais, J. M., Li, X., Liu, Q., et al. (2012). Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 60, 154–162. doi: 10.1161/HYPERTENSIONAHA.111.189688
Zhang, Y., Wang, Y. T., Koka, S., Zhang, Y., Hussain, T., and Li, X. (2020). Simvastatin improves lysosome function via enhancing lysosome biogenesis in endothelial cells. Front. Biosci. (Landmark Ed) 25:283–298. doi: 10.2741/4807
Zhang, Y., Yuan, F., Cao, X., Zhai, Z., GangHuang, Du, X., et al. (2014). P2X7 receptor blockade protects against cisplatin-induced nephrotoxicity in mice by decreasing the activities of inflammasome components, oxidative stress and caspase-3. Toxicol. Appl. Pharmacol. 281, 1–10. doi: 10.1016/j.taap.2014.09.016
Zhao, W., Zhang, L., Chen, R., Lu, H., Sui, M., Zhu, Y., et al. (2018). SIRT3 protects against acute kidney injury via AMPK/mTOR-regulated autophagy. Front. Physiol. 9:1526. doi: 10.3389/fphys.2018.01526
Zhao, W. Y., Zhang, L., Sui, M. X., Zhu, Y. H., and Zeng, L. (2016). Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci. Rep. 6:33201. doi: 10.1038/srep33201
Keywords: kidney, inflammasome, NLRP3, acute kidney injury, chronic kidney diseases
Citation: Xiang H, Zhu F, Xu Z and Xiong J (2020) Role of Inflammasomes in Kidney Diseases via Both Canonical and Non-canonical Pathways. Front. Cell Dev. Biol. 8:106. doi: 10.3389/fcell.2020.00106
Received: 14 September 2019; Accepted: 10 February 2020;
Published: 27 February 2020.
Edited by:
Zhengchao Wang, Fujian Normal University, ChinaReviewed by:
Fengyi Wan, Johns Hopkins University, United StatesGuangbi Li, Virginia Commonwealth University, United States
Krishna M. Boini, University of Houston, United States
Fan Yi, Shandong University, China
Copyright © 2020 Xiang, Zhu, Xu and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Xiong, amluZ3hpb25nQGh1c3QuZWR1LmNu
†These authors have contributed equally to this work