 Cristina Garcia-Barcena1
Cristina Garcia-Barcena1 Juanma Ramirez
Juanma Ramirez Ugo Mayor
Ugo Mayor
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 04 February 2020
Sec. Cellular Biochemistry
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00039
This article is part of the Research Topic Probing the Ubiquitin Landscape View all 19 articles
E3 ubiquitin ligases are the ultimate enzymes involved in the transfer of ubiquitin to substrate proteins, a process that determines the fate of the modified protein. Numerous diseases are caused by defects in the ubiquitin-proteasome machinery, including when the activity of a given E3 ligase is hampered. Thus, inactivation of E3 ligases and the resulting effects at molecular or cellular level have been the focus of many studies during the last few years. For this purpose, site-specific mutation of key residues involved in either protein interaction, substrate recognition or ubiquitin transfer have been reported to successfully inactivate E3 ligases. Nevertheless, it is not always trivial to predict which mutation(s) will block the catalytic activity of a ligase. Here we review over 250 site-specific inactivating mutations that have been carried out in 120 human E3 ubiquitin ligases. We foresee that the information gathered here will be helpful for the design of future experimental strategies.
Ubiquitin is a 76-amino-acid protein, highly conserved among organisms (Zuin et al., 2014), used–through the ubiquitin-proteasome system- to regulate many cellular processes. Proteins are covalently modified on their Lys residues with ubiquitin via amide isopeptide linkages (Laney and Hochstrasser, 1999). Frequently, ubiquitinated proteins are targeted for degradation through the proteasomal system on an ATP hydrolysis-dependent manner (Hershko and Ciechanover, 1998; Komander and Rape, 2012). But protein ubiquitination participates in a plethora of additional cellular responses including regulation of gene expression, cell signalling, cell cycle, DNA repair and apoptosis (Pickart, 2001; Gilberto and Peter, 2017).
The ubiquitination reaction requires the coordinated action of three types of enzymes termed E1, E2, and E3. First, ubiquitin is activated with ATP in a process carried out by an activating E1 enzyme. Once ubiquitin is activated, it is transferred to the Cys on the active site of a conjugating E2 enzyme. Finally, ubiquitin is generally linked to a Lys of the target protein through an isopeptide bond, formed between the C-terminal carboxyl group of ubiquitin and the ε-amino group of the Lys. Substrate specificity in ubiquitination is attributed to E3 ligases, who are able to interact with both the ubiquitin-charged E2 and the substrates to be modified (Metzger et al., 2014). Like most post-translational modifications (PTMs), ubiquitination is reversible and deubiquitinating enzymes (DUBs) are responsible for hydrolysing the isopeptide bond between ubiquitin and substrate proteins or between ubiquitin molecules.
Proteins can be modified by ubiquitin in a wide range of manners. For instance, in addition to Lys, ubiquitin can be conjugated via a peptide bond to the N-terminal amino group of the substrates (Ciechanover and Ben-Saadon, 2004), as well as to Cys or Ser/Thr residues by thio- or oxy-ester bonds, respectively (Wang et al., 2012). Substrates can be mono-ubiquitinated, meaning modified in a single residue by only one ubiquitin. Multi-mono-ubiquitination occurs when several residues of a given protein are simultaneously modified with one ubiquitin each. Poly-ubiquitination occurs when the C-terminus of another ubiquitin associates to one of the seven Lys (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) or the N-terminal Met (Met1) on the previously added ubiquitin molecules. Consequently, a ubiquitin chain is formed on the target protein. Depending on how ubiquitin residues are bound together, different ubiquitin chain architectures can be formed: (i) homogenous, if the Lys used throughout the chain is the same (e.g., Lys48-linked chains), (ii) heterogeneous, if they alternate (e.g., Lys48-Lys11-linked chains) and (iii) branched, if multiple Lys of the same ubiquitin are modified at the same time. Altogether, ubiquitin can generate a huge amount of different types of modifications on any given protein (Komander and Rape, 2012). Consequently, ubiquitin-mediated cellular responses will depend not only on the specific residues of the substrate that are modified but also on the topology of the ubiquitin chains that are formed.
Eukaryotic cells express hundreds of ubiquitin E3 ligases, which can operate in different cellular contexts, respond to numerous cellular signals, and process diverse protein substrates (Zheng and Shabek, 2017). Ubiquitin E3 ligases have been classically classified in two different groups, based on conserved structural domains and the mechanism by which ubiquitin is transferred: RING (really interesting new gene)-type E3s and HECT (homologous to the E6AP carboxyl terminus)-type E3s. Whereas RING E3 ligases directly transfer the ubiquitin from the E2-ubiquitin complex to the substrate (Figure 1A), HECT-type E3s transfer ubiquitin to their own catalytic Cys before linking it to the substrate (Figure 1B; Deshaies and Joazeiro, 2009). Additionally, a third group of E3s, that combines features from both RING- and HECT-type E3 families, has been established: the RING between RING (RBR) family (Figure 1C). RBR and RING E3s share RING binding domains, but RBR family members have the ability to generate a thioester intermediate with ubiquitin, as HECT-type E3s do (Morreale and Walden, 2016).
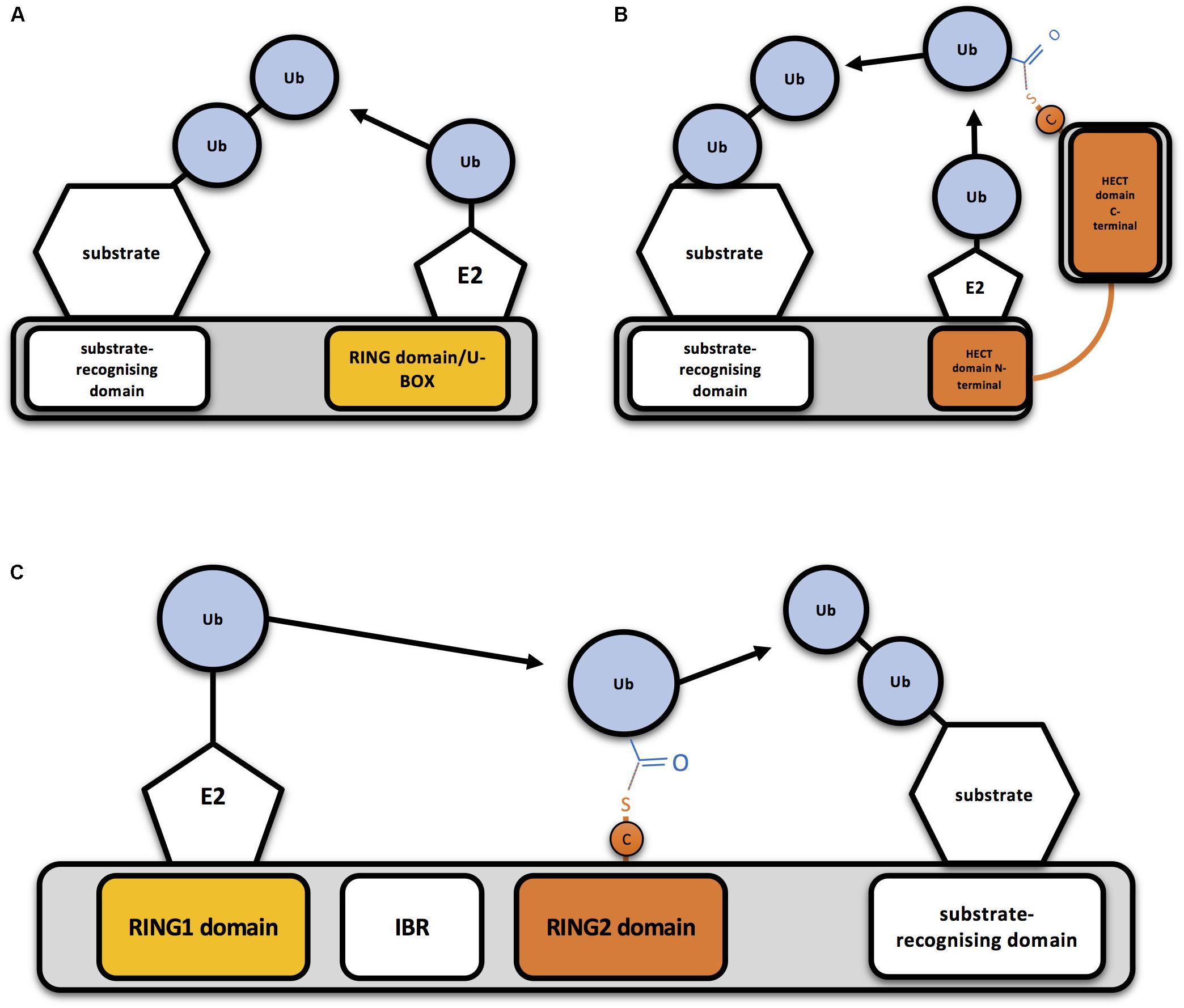
Figure 1. Mechanism of action of RING-, HECT- and RBR-type E3 ubiquitin ligases (A) Schematic representation of a RING-type ubiquitin E3 ligase. RING E3s bind both the E2-ubiquitin and the substrate to be ubiquitinated, so bringing them together allows direct conjugation of ubiquitin (Ub) on the substrate by the E2. A monomeric RING E3 ligase is shown for illustrative purposes. (B) Schematic representation of a HECT-type ubiquitin E3 ligase. Ubiquitin is transferred first to a cysteine (C) of the HECT domain through a thioester bond and then to the substrate. (C) Schematic representation of an RBR-type ubiquitin E3 ligase. Two RING domains are separated by an in-between-RING (IBR) domain. Ubiquitin is first transferred to a cysteine (C) of the second RING domain through a thioester bond and then to the substrate.
Typically, one E3 ligase is able to modify several substrates, as well as to bind different E2s. The same protein can, therefore, be ubiquitinated by different E2/E3 combinations, which will lead to different ubiquitination patterns (Metzger et al., 2014). Substrate recognition by HECT-type E3 ligases depends on protein-protein interactions that are mediated by specific motifs typically located in the N-terminal of the HECT domain (Scheffner and Kumar, 2014). Substrate recognition by RING-type E3s is achieved either through regions of the E3 other than the RING domain, in the case of monomeric E3s, or through substrate recognition elements in other domains, in the case of multi-subunit RING E3s (Metzger et al., 2014). On the other hand, some studies have reported that substrate proteins have a short linear sequence, known as degron, important in the regulation of protein degradation rates. Not all degron are ubiquitin-dependent, but if they are, it appears that they facilitate the recognition of the substrate protein by the E3 ligase. Degrons can be modified by kinases and other enzymes. These modifications appear to be crucial for timing the interaction between E3 and substrate, even though they are not always necessary and many substrates of HECT-type E3s and CRLs are able to recognise their substrates in their native forms (Kanelis et al., 2001; Kamadurai et al., 2009; Rotin and Kumar, 2009; Fukutomi et al., 2014; Muńoz-Escobar et al., 2015). In order to increase the specificity toward their substrates, many E3 ligases, such as TRIMs, are able to form homo- and heterodimers and recognise multiple degrons located in the same substrate (Li et al., 2014). Moreover, the effect is summatory and a robust degron may have the same effect as two weak degrons (Welcker et al., 2013).
The role mediated by E3 ligases is so crucial, that their activity must be tightly controlled in order to ensure they solely act when necessary. Oligomerisation is one of the mechanisms that modulate the activity of HECT- and RING-type E3s. For instance, structural studies suggest that the trimeric arrangement of E6AP activates the ligase (Ronchi et al., 2014), whereas homodimerisation of the HECT domain of HUWE1 results in enzyme inactivity (Sander et al., 2017). RING-type E3s can act as independent enzymes, but most of them tend to form homo- or heterodimers, and even more complex multi-subunit assemblies in order to mediate ubiquitination (Metzger et al., 2014). For instance, RING E3 ligases cIAP, RNF4, BIRC7, IDOL, CHIP, and Prp19 homodimerize, and RING domains of both units interact with E2 proteins. By contrast, RING-type E3 ligases BRCA1-BARD1, Mdm2-MdmX, and RING1B-Bmi1 form heterodimers. While BRCA1 and Mdm2 have the ability to interact with E2 proteins, their partners do not. But they function as enhancers of ligase activity and interact with substrates (Brzovic et al., 2001; Joukov et al., 2001; Wang et al., 2004; Cao et al., 2005).
In this review we aim to provide a detailed description of mutations in ubiquitin E3 ligases, with the outlook that such detailed and structured catalog of mutants will provide a pattern to be considered by future researchers when designing new mutations on their E3 ligases.
RING-type E3s are conserved from human to yeast. It is estimated that the human genome encodes above 600 different RING-type E3s. The RING domain was first characterised by Freemont et al. (1991). The canonical sequence for this 40–60 amino acid long domain is Cys-X2- Cys-X(9–39)-Cys-X(1–3) - His- X(2–3) -Cys-X2 -Cys- X(4–48)- Cys- X2-Cys. The conserved Cys residues (seven in total) and the single His are disposed in a “cross-brace” topology to coordinate two zinc ions and stabilise its structure (Figure 2; Deshaies and Joazeiro, 2009).
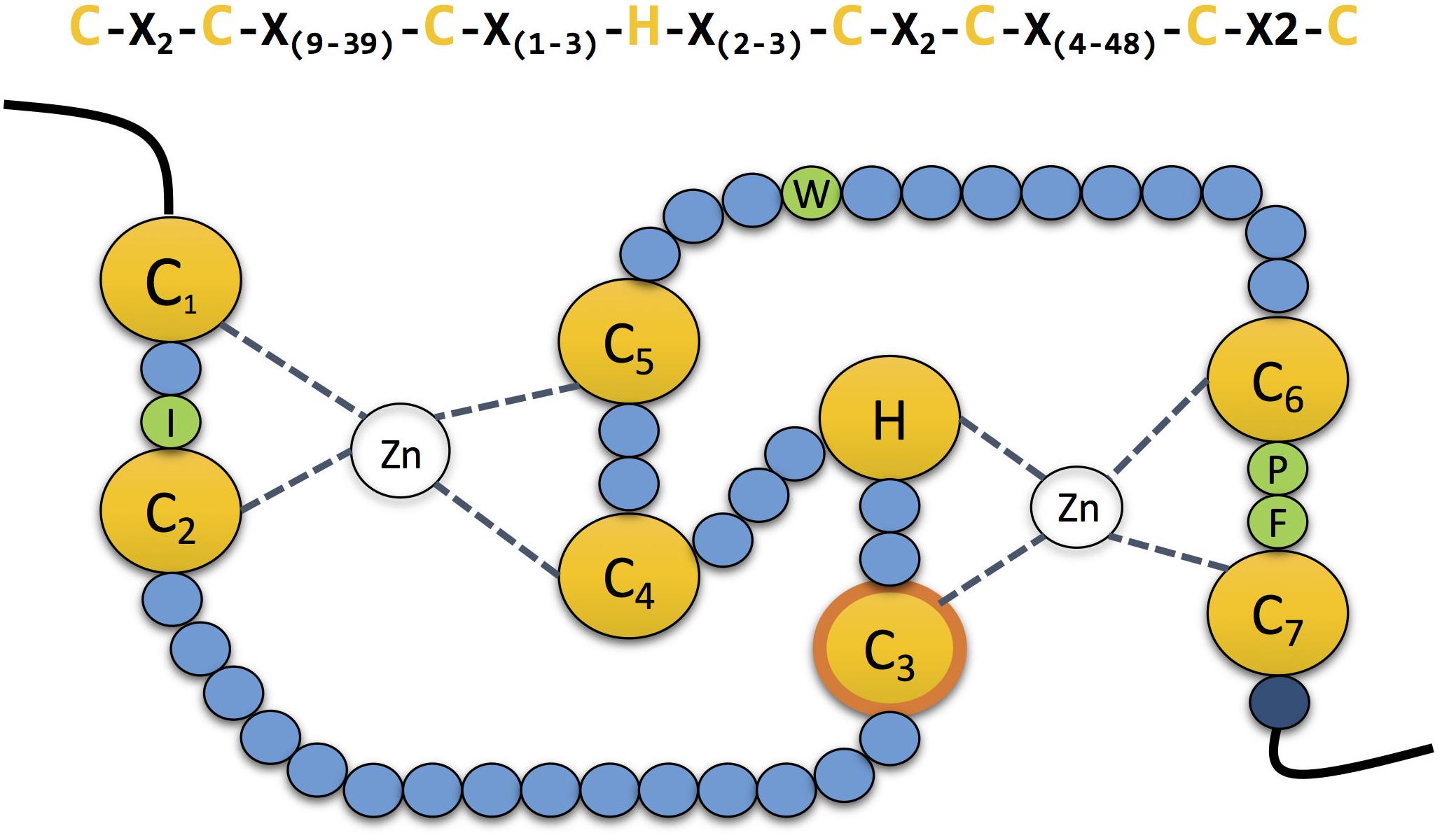
Figure 2. The zinc coordinating residues in RING domains. Schematic representation of the cross-brace” topology of RING domains. The RING domain contains seven conserved cysteines and one histidine (yellow) which are involved in the coordination of two atoms of zinc. The third cysteine mediates the ubiquitin transfer in the second RING domain in RBR E3 ubiquitin ligases (contour labelled in orange). Four conserved residues (green) guide the interaction with the E2 conjugating enzyme. Mutation of the last residue of the domain (dark blue), which is normally a positively charged arginine or lysine, compromises the stability of the adjacent cysteine, affecting the coordination of the zinc atom.
Initially, the role of RING domains was uncertain, although it was known they were involved in protein-protein interactions as well as in a wide range of cellular processes (Deshaies and Joazeiro, 2009). However, it was not until 1997 that the function of RING domains was elucidated by Bailly and co-workers (Bailly et al., 1997). Moreover, in 1999, Joazeiro and co-workers observed that the adapter protein c-Cbl bears two domains that act coordinately to mediate ubiquitination and subsequent degradation of substrates. Whereas the SH2 domain of c-Cbl served to recognize specific substrates, the RING domain was necessary to recruit and activate an ubiquitin-conjugating E2 (Joazeiro et al., 1999). After that, a similar role was conferred to a number of RING domain-containing proteins (Lorick et al., 1999). At present, it is accepted that the RING domain present in all RING E3s associates and activates E2-Ub conjugates promoting the direct transfer of ubiquitin from the E2 to the target protein (Figure 1A).
The interaction between the RING domain of E3 ligases and E2s was first elucidated with the crystal structure of Cbl’s RING domain bound to UbcH7 E2 (Zheng et al., 2000). The combination of many structural studies allowed the characterization of the four residues of each protein that play a crucial role in the interaction, those are shown in green in Figure 2. Located between Cys residues C1 and C2 of the RING domain, a hydrophobic residue (Ile, Leu or Val) interacts with two Pro residues from the E2. Those two prolines are localised in one of the two loops that compose the accessible surface of the E2 enzyme. Additionally, another hydrophobic residue (typically Trp, His or Leu) from the E3 interacts with a Phe and a Pro present on the second loop of the E2. Simultaneously, this Pro interacts with a Pro of the E3 located between Cys residues C6 and C7. Which in turn, is also connected to an Ala localised in the same loop of the E2. Finally, this same Ala of the E2 also interacts with a hydrophobic amino acid (typically Val, Phe or Ile) located straight after the Pro between C6 and C7 of the E3 (Deshaies and Joazeiro, 2009).
More recently, structural studies focused on RING-type E3:E2-Ub complexes have revealed the mechanism by which this class of ubiquitin ligases facilitates Ub transfer to substrate proteins. The E2-Ub complex has a flexible topology with multiple inter-domain configurations that are altered upon E3 binding (Pruneda et al., 2011). More precisely, binding of RING E3 reduces the dynamics of E2-Ub and stabilizes in an ensemble of closed conformations. This modification facilitates the reactivity for substrate Lys that can perform the corresponding nucleophilic attack (Pruneda et al., 2012; Soss et al., 2013). Studies carried out on dimeric E3s such as RNF4 or BIRC7 also support the same mechanism by showing that a positively charged residue (Arg or Lys) conserved in many RING E3s just straight after the last zinc-coordinating Cys supports the non-covalent interaction with the E2-Ub complex (Dou et al., 2012; Plechanovov et al., 2012).
As mentioned above, although some RING-type E3s act independently, they have the tendency to form homo- and heterodimers. Most RING-type E3s dimerise through their RING domain, such as RNF4 homodimers or MDM2/MDMX and BRAC1/BARD1 heterodimers (Brzovic et al., 2001; Linke et al., 2008; Liew et al., 2010). Nevertheless, there are exceptions. For instance, MARCH9 E3 ligase can form active dimers with RING-less variants (Hoer et al., 2007), whereas viral RING-type E3s MIR1 and MIR2 are believed to homodimerise via their transmembrane domain (Lehner et al., 2005). The tripartite motif (TRIM) family members in metazoans contain an additional domain termed B-box. Like the above mentioned RING domain, the B-box domain is a zinc-binding domain. However, whereas the RING domain is essential for E2 binding and E3 ligase activity, it has recently been shown that the B-box domain is involved in chain assembly rate modulation (Lazzari et al., 2019). Similarly, the U-box domain is also related to the RING domain, but unlike the B-box, it can interact with E2s. Additionally, the U-box domain has no coordinating zinc, so in order to ensure the stability of the structure, zinc-binding residues present in RING are replaced by charged and polar residues (Aravind and Koonin, 2000; Vander Kooi et al., 2006).
Since the coordination of the two atoms of zinc by the RING domain is crucial for E3 ligase activity, mutants that abolish such coordination have often been used to create ligase-dead versions of those E3 enzymes. Mutation of any of the conserved Cys and His involved in zinc binding should compromise the E3 activity, and so have all been, individually or jointly, mutated for that purpose (Figure 3). The mutated residue of choice to prevent E3 ligase activity appears the first conserved Cys (C1) of the RING domain, followed by the His (H), C2, C3, and C4. To our knowledge, C7 is the only key residue on the domain that has not been individually mutated for this purpose. However, it has been shown that simultaneous mutations on either C1+C7 or C6+C7 abolish the ligase activity of AMFR and some TRIM family members, respectively (Wang et al., 2014; Liu et al., 2017a; Lee et al., 2018a). As shown in Figure 3, many E3 ligases have been inactivated by simultaneous mutations on C1+C2. Less frequently, additional double mutations and even the triple C1+C2+C3 mutant have been efficiently applied to block the activity of distinct RING-type E3 ligases (Figure 3).
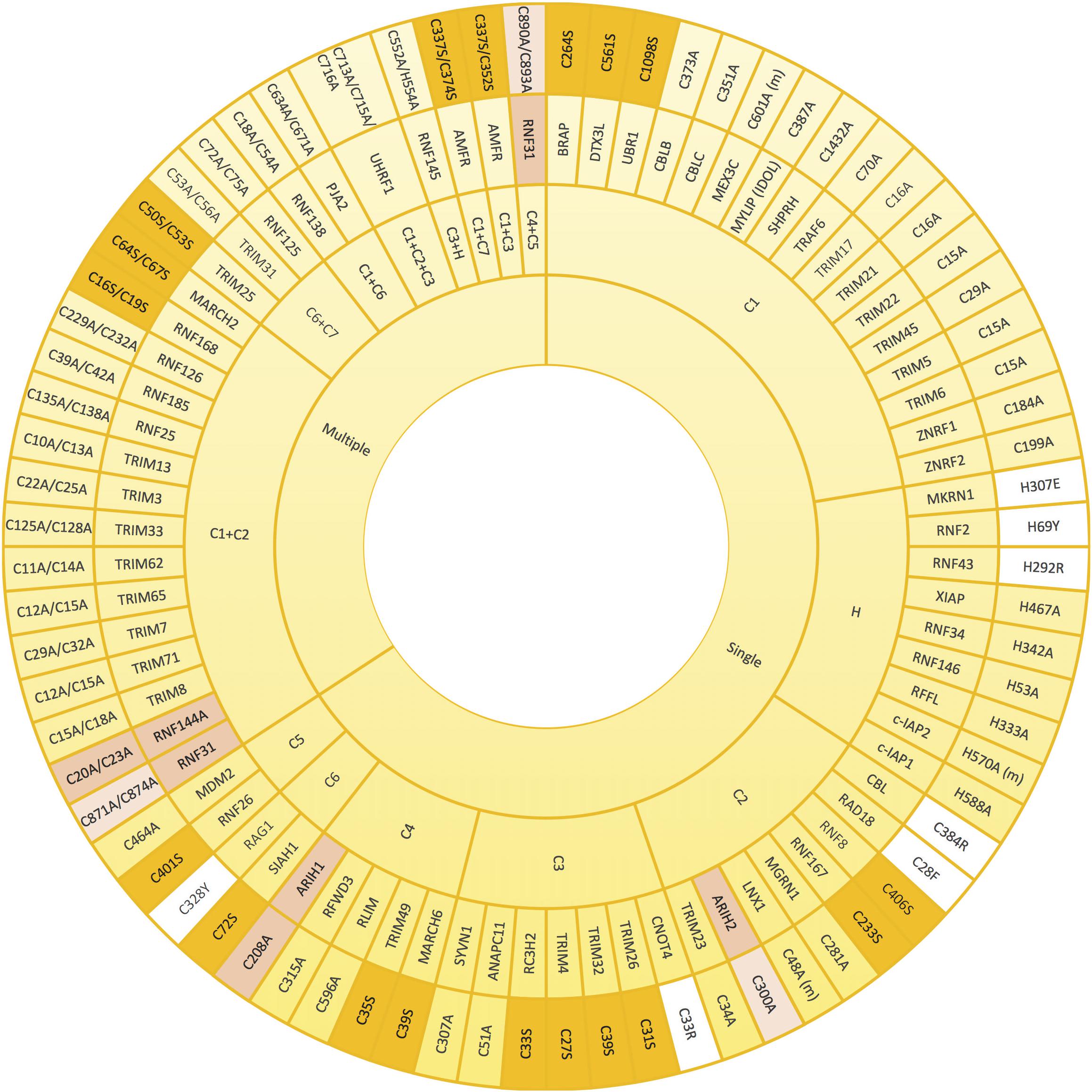
Figure 3. Mutations that alter zinc coordinating residues in RING domains. Wheel diagram showing the reported mutations in RING domains, classified first whether a unique (single) or various (multiple) residues were mutated simultaneously. Most of the inactive E3 enzymes have been obtained by mutating key residues into alanine (yellow). Lack of activity can also be acquired by mutations into serine (orange). Mutations into other residues have been also employed (white). Zinc coordinating residues of the first (pink) and the second (light pink) RING domains in RBR-type E3 ubiquitin ligases (pink) have also be modified in order to achieve inactivation. References to all the mutations shown in this figure are provided in Supplementary Table S1.
Zinc-coordinating Cys and His residues have been preferentially mutated into Ala in order to abolish the ubiquitin ligase activity of E3s (Figure 3). Nevertheless, in some cases, this type of substitution might be insufficient. In a recent research focused on studying TRIM27-dependent ubiquitination of UPS7, it was shown that a quadruple TRIM27 mutant, in which four zinc-coordinating residues of the RING domain (Cys16, Cys19, Cys31 and Cys33) were mutated into alanine, was still capable of ubiquitinating USP7. By contrast, the TRIM27 mutant, in which four zinc-binding residues of the B-box (Cys96, Cys99, His107, and Asp110) were simultaneously substituted by Ala, was incapable of ubiquitinating USP7 (not illustrated in Figure 3; Zaman et al., 2013). Moreover, it should be taken into account that in some cases a dominant negative effect may be acquired by the mutated E3 ligase. For example, CBL Cys381Ala mutant is not capable of ubiquitinating EGFR and thus, the subsequent desensitization of the receptor is abolished. However, CBL Cys381Ala mutant is still capable of interacting with EGFR, and consequently, competes with wild type CBL compromising CBL-mediated EGFR ubiquitination (Waterman et al., 1999). Similarly, the plant E3 ubiquitin ligase SINA1 mutant on the C2 of the RING domain Cys47Ser mutant retains dimerisation and substrate binding ability but lacks ubiquitination activity (den Herder et al., 2012).
Despite less frequently, in a number of investigations, the Cys involved in zinc coordination have also been efficiently mutated into serine. Indeed, this type of point mutation that results on E3 ligase inactivation has served to uncover, among others, the role of MDM2, RNF8, and SIAH1 RING E3s in cell cycle regulation, DNA damage response and Wnt signalling, respectively (Ji et al., 2017; Tian et al., 2017; Tripathi and Smith, 2017). Additionally, although there are fewer examples, it has been demonstrated that mutating the His into Glu, Tyr or Arg is sufficient to inactivate the ligase activity of MKRN1, RNF2, and RNF43 E3s, respectively (Xia et al., 2014; Loregger et al., 2015; Lee et al., 2018b; Figure 3). Similarly, it has been shown that mutating C2 of RAD18 and CBL into Phe and Arg, respectively, as well as substituting C3 of CNOT4 into Arg or C6 of RAG1 into Tyr has an inhibitory effect (Albert et al., 2002; Jones and Gellert, 2003; Williams et al., 2011; Javadi et al., 2013). It should be noted, however, that in search of structure-function relationships, the safest approach is to mutate into the smaller Ala residue (Fersht et al., 1999). Introducing larger residues might -in addition to preventing the coordination of the zinc- result in further distortions on the overall fold of the protein.
Especially in the absence of the molecular structure, deciding the residues that should be mutated might not always be straightforward, but appropriate sequence alignments can provide sufficient insight. For instance, TRIM37 has two adjacent Cys residues (Cys36 and Cys37) that could correspond to the C4 involved in zinc coordination (Supplementary Figure S1). Therefore, to ensure the inactivation of the enzyme, both Cys were simultaneously mutated (Kallijärvi et al., 2005; Wang et al., 2017). Similarly, ZNRF4 has two His nearby (His329 and His332) and in principle, either of them could be involved in coordinating zinc atoms. Once again, both His were mutated in order to obtain a catalytically inactive form of the E3 (Bist et al., 2017). Based on metal-binding studies, MDM2 His457 was initially confirmed to be the conserved His involved in zinc-coordination (Lai et al., 1998). Nevertheless, His452 is also essential, as demonstrated in auto-ubiquitination assays of this E3 ligase, with both His residues being necessary (Fang et al., 2000). It was later elucidated that His452 actually takes the place of the conserved Cys C3 in the zinc coordination, as illustrated in the sequence alignment in Figure 4B.
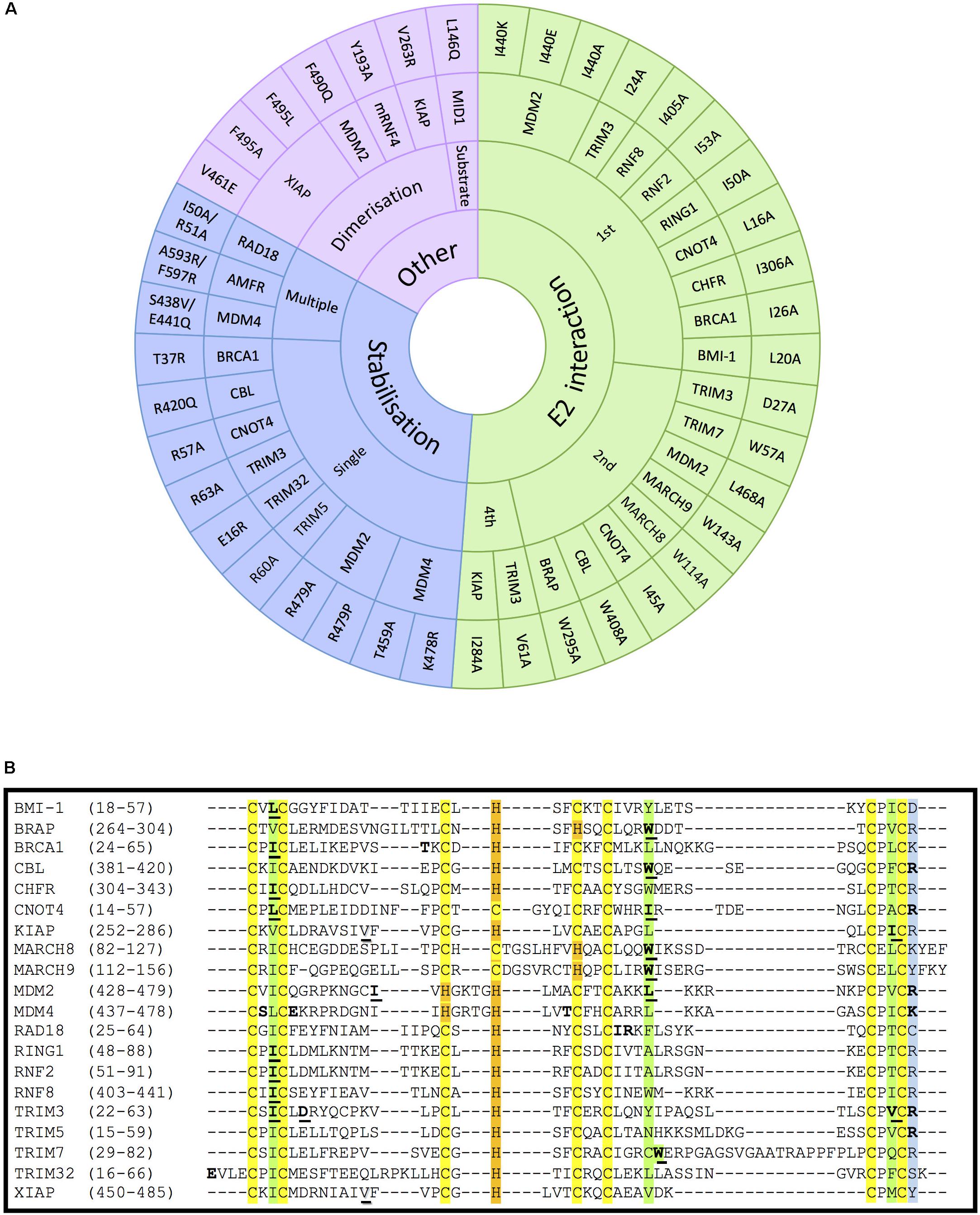
Figure 4. Mutations on RING- and RBR-type E3s that affect E2-interaction, domain stabilisation, protein dimerization or substrate recognition. (A) In RING-type E3 ubiquitin ligases, inactivation can be obtained by abolishing the interaction with E2 ubiquitin-conjugating enzymes (green). This has mostly been achieved by mutating the conserved 1st (I/L) and 2nd (W/I/L) hydrophobic residues indicated in Figure 2. Other mutations affecting the stabilisation of key residues of the domain (blue), dimerization or the interaction with a specific substrate also abolish the ligase activity (purple). For the stabilisation affecting mutations, those have been classified whether a unique (single) or various (multiple) residues were mutated simultaneously. References to all the mutations shown in this figure are provided in Supplementary Table S1. (B) Alignment of the RING domains of the RING-type E3 ligases involved in E2-interacting and stabilisation mutations within the RING domain. Conserved amino acids are highlighted in yellow and orange, respectively, for the Zn-coordinating Cys and His residues, and in green for the E2-interacting residues. The conserved positively charged residues at the end of the RING domain are highlighted in blue. Mutated E2-interacting residues are shown in bold and underlined. Mutated residues involved in stabilisation are shown in bold. Mutated residues involved in dimerisation are underlined and shadowed.
Additionally, there are few E3s bearing RING domains in which a non-conserved amino acid plays an indirect but pivotal role in the coordination of the zinc atom, and therefore, can be mutated in order to disrupt the activity of the ligase. For example, Thr455, which was originally believed to be directly involved in the zinc-coordination based on an incorrect primary sequence alignment, has been reported to abolish -upon its mutation- MDM2-dependent p53 ubiquitination (Boddy et al., 1994; Fang et al., 2000).
It has previously been described that RING E3s interact with E2-Ub conjugates via their RING domain to directly transfer the ubiquitin to the substrate protein. Therefore, disrupting the interaction between E2s and RING-type E3s has also been extensively used to block, or at least reduce ubiquitination mediated by RING E3s. All three key hydrophobic residues on E3s that mediate the interaction with E2s (shown in green in Figure 2) have been recurrently mutated to compromise the activity of the E3s. As shown in Figure 4A, numerous RING-type E3 ligases have been successfully inactivated by mutating the first Ile/Leu, the second Trp/Leu or the last Ile/Val into Ala. The first Ile/Leu has been mutated in BRCA1, BMI-1, CHFR, CNOT4, RING1, RNF2, RNF8 and TRIM3 (Albert et al., 2002; Eakin et al., 2007; Alchanati et al., 2009; Kim et al., 2010; Mallette et al., 2012; Raheja et al., 2014; Liu et al., 2018; Shen et al., 2018). The second Trp/Leu was mutated abolishing ligase activity in BRAP, CBL, MARCH8, MARCH9, MDM2, and TRIM7 (Joazeiro et al., 1999; Chen et al., 2012; Hayes et al., 2012; Chakraborty et al., 2015; Fan and Wang, 2017; Tan et al., 2019). Finally, the last Ile/Val was successfully mutated in KIAP and TRIM3 (Dou et al., 2012; Raheja et al., 2014). All these hydrophobic residues are conserved as seen in Figure 4B. However, to our knowledge, no one has mutated the E2-interacting Pro (located between C6 and C7) with the aim to disrupt the association with the E2 enzyme. Given the special properties of this cyclic amino acid, one certainly would have to be weary of additional conformational effects that could be caused by its mutation to Ala. Additionally, MDM2 mutant variants Ile440Glu and Ile440Lys prevent MDM2-dependent ubiquitination of p53, by disrupting the E2–ubiquitin binding by the E3 ligase without altering its RING domain structure (Nomura et al., 2017). This residue, however, is barely conserved across the different RING domains.
However, other types of mutations have also been efficiently applied to disrupt the interaction between E2s and E3s. For instance, one of the few U-box-type E3s that has been mutated is CHIP, also known as STUB1, which was inactivated by substituting His260 into Glu (Seo et al., 2018). Likewise, the U-box domain-containing UBE4B E3 can be inactivated by mutating a Pro (Pro1140) that is conserved among U-box-type E3 ligases (Pro269 in CHIP) into Ala (not included in Figure 4; Okumura et al., 2004; Li et al., 2018).
Many RING-type E3 ligases possess a conserved positively charged residue (Arg or Lys) in the last position of the RING domain, which appears to be essential for the ubiquitination activity of the E3. Nevertheless, it is still controversial whether the effect of mutating this residue results from the impaired interaction with E2s or from destabilization of the RING domain (Figure 4A, included in stabilization) (Albert et al., 2002; Linke et al., 2008; Lienlaf et al., 2011; Dou et al., 2012; Raheja et al., 2014; Nomura et al., 2017). But this uncertainty is not surprising given that mutations have been generated to substitute the positively charged residue by a very diverse choice of residues (mostly to Ala, but also to Glu, Pro and even Arg, as can be seen in Figure 4A. Future studies should preferably limit the mutations to substituting the positively charged residue by Ala.
As shown in Figure 4A, a number of other single point mutations, as well as multiple point mutations, have been generated along different positions of the RING domain to compromise protein stability and hence, E3 ligase activity, but no clear pattern can be predicted based on the studies reported so far. For example, the Tyr37Ala mutant in BRCA1 lack ligase activity, being therefore incapable of reversing γ-radiation hypersensitivity of BRCA1-null human breast cancer cells (Ruffner et al., 2001). In the case of the RAD18 ligase, the Ile50Ala/Arg51Ala inactive mutant allowed to study the formation of ternary complexes with RAD6A (Masuda et al., 2012); these two residues were selected due to being highly conserved among species.
RING-type E3s that act as dimers can also be inactivated by preventing their dimerization process. For instance, mutation of Val461Glu and Val263Arg within the RING domain diminishes oligomerisation and activity of XIAP and KIAP ligases, respectively (Poyurovsky et al., 2007; Dou et al., 2012; Nakatani et al., 2013). In other cases, however, the dimerization affecting residues are immediately after the RING domain (Supplementary Figure S2), as revealed for example by the mutation Phe490Gln in MDM2 (Poyurovsky et al., 2007). Another approach consists of inactivating oligomeric E3 ligases without affecting the oligomerisation process itself. For example, RNF4 Val134Ala and Ile153Ala mutants can form dimers but are catalytically incapacitated (Liew et al., 2010; Dou et al., 2012). Similarly, other E3 ligase mutants have been shown to act in a dominant negative due to their homo-dimeric nature. For example, mutant Fbw7 has a dominant-negative effect when dimerising with wild-type Fbw7, being able to effectively bind their substrate MYC but not to ubiquitinate and degrade it (Welcker et al., 2013).
Several experiments have also been carried out mutating specific residues on E3 ligases that are critical for the interaction with a given substrate, such as Leu146Gln mutation on the B-box containing E3 MID1 that cannot associate, nor ubiquitinate its substrate PP2A alpha-2 (Du et al., 2013; Figure 4).
The human HECT-type E3 family consists of 28 members that are divided into three different groups depending on their N-terminal domain architecture: (i) the NEDD4 subfamily, characterized by containing a C2 domain, a HECT domain and two to four WW domains, which bind to the PY motifs of target proteins (Staub et al., 1996; Kanelis et al., 2001); (ii) the HERC subfamily, which integrates at least one regulator chromosome condensation 1 (RCC1)-like domain (RLDs) and a reduced HECT domain; and (iii) the other HECT subfamily, that embrace HECT-type E3s not fitting the above mentioned two subfamilies.
Despite those differences, all HECT-type E3s share a ∼350 amino acid long HECT domain, that was first described in human papilloma virus E6 associated protein (E6AP) (Huibregtse et al., 1995). In the HECT domain, a conserved Cys forms thioester-linked-intermediate complexes with ubiquitin (Figure 1B), before being transferred and attached to the substrate through a transthiolation reaction. This conserved Cys is located in the C-terminal region of the HECT domain, while the E2 interacting site is localised in the N-terminal site (Figure 1B; Rotin and Kumar, 2009).
Given that an active site Cys is required for the formation of a thioester intermediate with ubiquitin, a typical approach is to mutate this specific Cys to generate ligase dead versions of HECT E3 ligases. As shown in Figure 5A, the majority of HECT-type E3 ligases have been inactivated by replacing this catalytic Cys by Ala. This approach has served to unveil, among others, the involvement of HERC3 in immune response (Hochrainer et al., 2015), the role of NEDD4L in EnaC receptor recycling (Zhou et al., 2007), and the contribution of SMURF1 to Axin degradation (Fei et al., 2013).
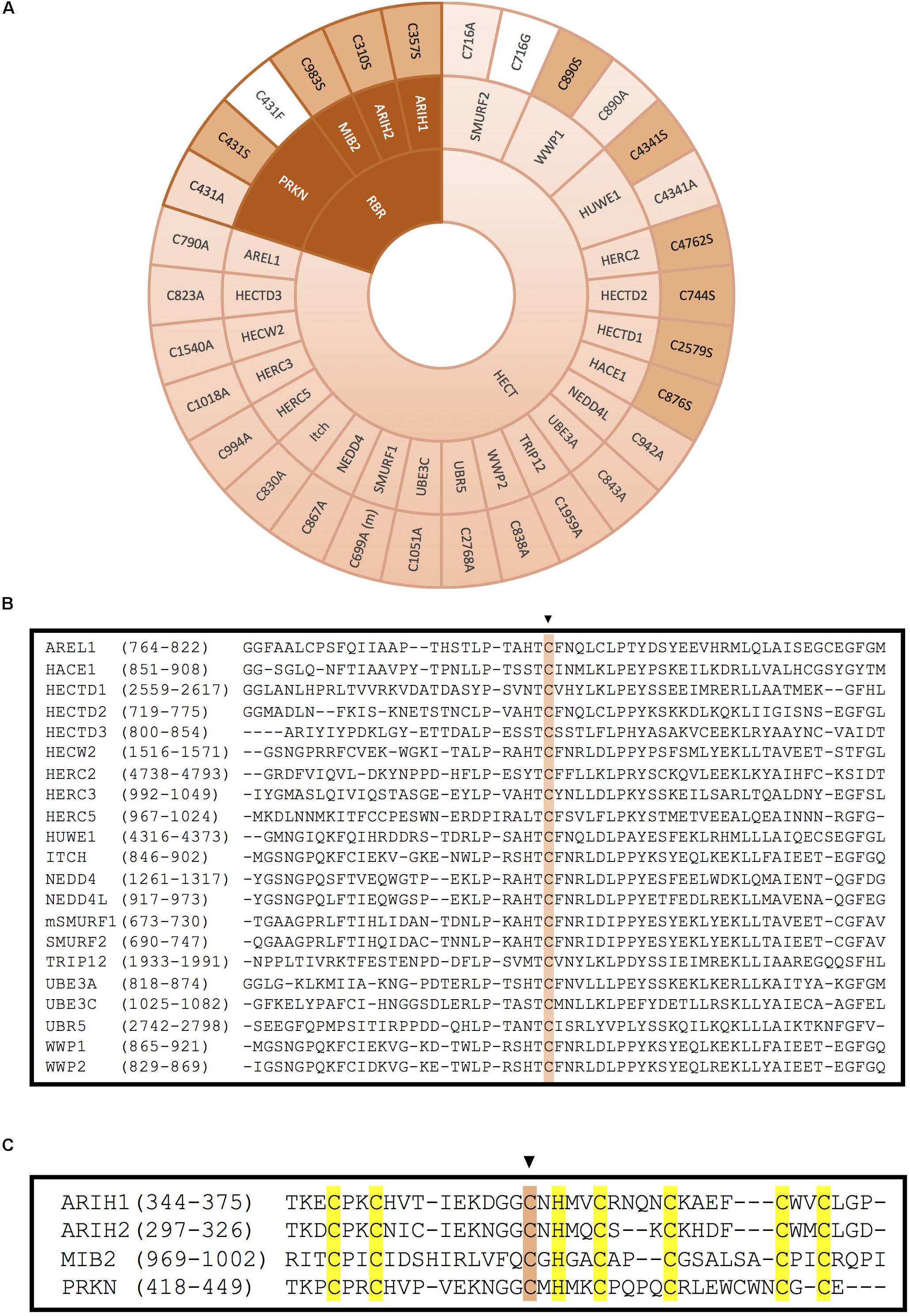
Figure 5. Inactivation of HECT- and RBR-type E3 ligases by mutation of the catalytic cysteine. (A) Mutation of the catalytic cysteine into an alanine (light pink), a serine (pink) or another residue (white) abolishes transference of ubiquitin onto the substrate. Inactivation of the catalytic cysteine of RBR-type E3 ubiquitin ligases is obtained by mutation of the third conserved cysteine in the second RING domain. (m) indicates that this mutation has been done in the mice homolog of the protein. References to all the mutations shown in this figure are provided in Supplementary Table S1. (B) Alignment of the HECT domains of the HECT-type E3 ligases. The conserved catalytic cysteine is highlighted in pink. (C) Alignment of the RING2 domains of the RBR-type E3 ligases. Conserved zinc-coordinating cysteines and histidine on the second RING domain of mutated RBR E3 ligases are highlighted in yellow and the mutated catalytic cysteine is highlighted in pink.
Less frequently, some ligase dead HECT-type E3s have been generated by substitution of the active Cys into Ser (Figure 5A). It has been reported that when the catalytic Cys of an E3 is mutated into Ser, the residue is still capable of binding through an oxyester bond with ubiquitin, but incapable to transfer it to substrates, which might result in a dominant-negative effect. In ubiquitination assays employing this type of ligase dead E3s, a stable monoubiquitinated version of the E3 has been detected (Lee et al., 2014). This approach has allowed, among other things to discover many substrates of distinct HECT-type E3 ligases. For instance, it was found that wild type version of HACE1 could ubiquitinate and target for degradation the small GTPase Rac1, but the Cys876Ser ligase dead version of the E3 ligase could not (Torrino et al., 2011). Similarly, HERC2 C4762S and HUWE C4341S mutants failed to ubiquitinate their substrates BRCA1 and N-Myc, respectively (Zhao et al., 2008; Wu et al., 2010). The sequence alignment for all the HECT domain E3 ligases illustrated in Figure 5A is shown around the catalytically active Cys in Figure 5B.
RING between RING family members contain two RING domains (RING1 and RING2) that are separated by an in-between-RING (IBR) zinc-binding domain. Morett and Bork first characterised these domains in 1999 in a sequence profile-based characterisation (Morett and Bork, 1999). In the process of confirming reports that UbcH7 could also interact with RBR E3s, they discovered that these RBR E3s act as RING/HECT hybrids. The first RING domain serves as the E2 binding platform, while the C3 of the second RING serves as the active site that mediates ubiquitination similarly to HECT E3 ligases (Wenzel et al., 2011; Figures 1C, 5C).
As it happens with HECT-type E3, the mutation of the catalytic Cys in the RING2 of RBR E3s results in the inactivation of these enzymes. However, unlike in HECT-type E3 ligases, in RBR E3s the active Cys has been mostly substituted by Ser, and less by Ala (Figure 5A). For example, C983S substitution in MIB2 resulted in ligase inactivation, and therefore, prevented ubiquitination of its substrate TANK-binding kinase 1 (Ye et al., 2014). Similarly, mutating the active Cys of ARIH2 (also called TRIAD1) into Ser or Ala completely abolished autoubiquitination of the RBR-type E3 ligase. Parkison disease has been shown to develop in patients carrying a Cys431Phe mutation at the catalytic Cys of the RBR-type E3 ligase PRKN; those mutants have also been characterized in the lab (Sarraf et al., 2013), in addition to the more common substitutions to Ser and Ala (Liu et al., 2017b; Xin et al., 2018).
In order to generate ligase dead versions of RBR-type E3s, it has been also shown to be plausible to preserve the active Cys, and instead mutate the zinc-coordinating residues in either of the two RING domains, substituting by Ala one or several of those key residues. For instance, ARIH1 and RNF144A have been successfully inactivated by modifying their RING1 domain (Figure 3, dark pink). Whereas mutating C4 of ARIH1 (Cys208) was sufficient to inhibit the ligase, Cys20 and Cys23 (C1+C2) were simultaneously modified to block the catalytic activity of the RBR-type E3 RNF144A (Ho et al., 2014; von Stechow et al., 2015). On the contrary, ARIH2 and RNF31 have been inactivated by mutating their RING2 domain zinc-coordinating Cys residues (Figure 3, light pink). Cells expressing an ARIH1 mutant in which the C2 of the RING2 domain was mutated into Ala (ARIH2 Cys300Ala mutant) was no longer able to ubiquitinate NLRPL3 (Kawashima et al., 2017). Similarly, Smit and co-workers generated various ligase dead versions of RNF31 by mutating simultaneously Cys871 and C874 (C1+C2) or Cys890 and Cys892 (C4+C5) of the RING2 domain (Smit et al., 2012).
Mutations on E3 ligases have been associated with a number of diseases, including neurological disorders (George et al., 2018; Osinalde et al., 2019). Thus, understanding their mechanism of action, as well as identifying which substrates are regulated by each E3 at different developmental stages and cell types, will provide invaluable knowledge that might contribute to develop therapeutic strategies to treat these diseases. Generation of E3 ligase dead mutants can certainly provide crucial information for this purpose. While the use of gene silencing techniques might be more appropriate to study the phenotypes derived from the loss of function of E3 ligases, the overexpression of ligase death versions can provide information about (i) the E2 enzymes they work with, (ii) substrate recognition domains and (iii) existing mechanism that regulate their activity. Additionally, a number of biochemical experiments do benefit from comparing the ectopic expression of wild type active E3 ligases with their mutated inactive variants.
As evident from all the examples shown in this review, there are multiple options to disrupt the activity of an E3 ligase. As illustrated by the sequence alignment in Figure 4B, the first necessary step is to identify which are the key residues in our ligase of interest. This is an essential step to ensure that any mutagenesis performed has a higher chance of success in disrupting the E3 ligase activity. For example, not all cysteine residues within a RING domain are involved in zinc coordination, as can be seen in the sequence alignment of Mdm2 in Figure 4B. When this cysteine of the Mdm2 RING domain was mutated (Kostic et al., 2006) the zinc coordination was maintained and no disruption to the ubiquitination activity of Mdm2 was detectable.
It is worth mentioning that mutating key residues involved either in the coordination of the zinc ions, dimerisation, proteins stabilization or E2 interaction might not always be sufficient to abolish the catalytic activity of the E3 ligase. The resulting mutation replacing the original residue that is substituted can actually be determinant in order to have a functional effect. For instance, mutating Phe495 of XIAP into either Ala, Tyr or Trp completely prevents E3 ligase autoubiquitination. However, XIAP Phe495Leu mutants appear to be functionally wild-type like (Nakatani et al., 2013); but might not be that surprising given the partial hydrophobic similarity between those two amino acids.
As illustrated within this review, so far one of the most frequent approaches for RING E3 ligases has been to mutate the residues involved in the zinc coordination (Cys and His residues, shaded in yellow and orange, respectively, in Figure 4B). Eliminating the zinc coordination on the RING domain is well known to severely disrupt the ubiquitination activity of those E3 ligases. However, this breakdown of the global structural integrity of the RING construct might lead to a severe effect in the folding and expression levels of the E3 ligase (Chasapis et al., 2010). Therefore, for certain experiments might be more effective to generate less disruptive point mutations. For example, the mutation of the hydrophobic residues (Ile, Leu, Trp, Val, shaded in green in Figure 4B) that mediate the interaction with the E2 conjugating enzyme, as demonstrated for a number of RING E3 ligases. To our knowledge this approach has not yet been employed for the E2-interacting RING domain of RBR E3 ligases, but it should indeed be an interesting experiment to perform.
Another approach that has been used as well is to eliminate by mutagenesis the positive charge of the Lys or Arg residue located straight after the last zinc-coordinating Cys of the RING domain. It is yet unclear, however, whether the effect caused by this mutation is on the interaction with E2s or from destabilization of the RING domain.
Mutations on the active Cys of HECT- and RBR-type E3 ligases are very straight forward, as they generate, without further effect to the structure and stability of the E3, ligase-dead versions of these enzymes. Those are of good value to be used as the best control in experiments overexpressing the wild type ligase, for example, to identify substrates in an unbiased manner. Additionally, if mutating the active site Cys to Ser, the formation of an oxyester to ubiquitin can be used with the aim to obtain a dominant-negative version of the ligase; the E3 will recruit the E2 and the substrate but the ubiquitination reaction cannot proceed since the ubiquitin cannot be released once it has conjugated to the E3.
To investigate the regulation of a specific protein by a particular HECT or RBR E3, however, it might be more suitable to mutate the ligase at the substrate recognition motif. Moreover, in some cases, as is the case of some RING E3s, the inactivation of E3 enzymes is not achieved by a single point mutation, even though such residue is defined as a key amino acid involved in substrate recognition. Hence, in such situations, several residues must be simultaneously mutated in order to disrupt the E3 ligase function. The generation and usage of E3 mutants have revealed unexpected and important lessons about the complexity of this family of enzymes. Nevertheless, a complete understanding of E3 ligases still requires more research, in which the generation of novel E3 ligase mutants will undoubtedly be decisive.
CG-B designed and wrote the first draft. All authors contributed to this manuscript.
This work was supported by Spanish MINECO (grant SAF2016-76898-P) cofinanced with FEDER funds. JR was funded with a postdoctoral fellowship from the University of the Basque Country (UPV/EHU).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Jesus Mari Arizmendi for sharing the laboratory with our group. We would also like to thank all the people in the lab.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00039/full#supplementary-material
Albert, T. K., Hanzawa, H., Legtenberg, Y. I. A., de Ruwe, M. J., van den Heuvel, F. A. J., Collart, M. A., et al. (2002). Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 21, 355–364. doi: 10.1093/emboj/21.3.355
Alchanati, I., Teicher, C., Cohen, G., Shemesh, V., Barr, H. M., Nakache, P., et al. (2009). The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2α cleavage complex - A potentially new drug target. PLoS One 4:e8104. doi: 10.1371/journal.pone.0008104
Aravind, L., and Koonin, E. V. (2000). The U box is a modified RING finger - a common domain in ubiquitination. Curr. Biol. 10, R132–R134. doi: 10.1016/S0960-9822(00)00398-395
Bailly, V., Lauder, S., Prakash, S., and Prakash, L. (1997). Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 272, 23360–23365. doi: 10.1074/jbc.272.37.23360
Bist, P., Cheong, W. S., Ng, A., Dikshit, N., Kim, B.-H., Pulloor, N. K., et al. (2017). E3 Ubiquitin ligase ZNRF4 negatively regulates NOD2 signalling and induces tolerance to MDP. Nat. Commun. 8:15865. doi: 10.1038/ncomms15865
Boddy, M. N., Freemont, P. S., and Borden, K. L. B. (1994). The p53-associated protein MDM2 contains a newly characterized zinc-binding domain called the RING finger. Trends Biochem. Sci. 19, 198–199. doi: 10.1016/0968-0004(94)90020-90025
Brzovic, P. S., Rajagopal, P., Hoyt, D. W., King, M. C., and Klevit, R. E. (2001). Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 8, 833–837. doi: 10.1038/nsb1001-833
Cao, R., Tsukada, Y. I., and Zhang, Y. (2005). Role of Bmi-1 and Ring1A in H2A ubiquitylation and hox gene silencing. Mol. Cell 20, 845–854. doi: 10.1016/j.molcel.2005.12.002
Chakraborty, A., Diefenbacher, M. E., Mylona, A., Kassel, O., and Behrens, A. (2015). The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nat. Commun. 6:6782. doi: 10.1038/ncomms7782
Chasapis, C. T., Loutsidou, A. K., Orkoula, M. G., and Spyroulias, G. A. (2010). Zinc binding properties of engineered ring finger domain of arkadia e3 ubiquitin ligase. Bioinorg. Chem. Appl. 2010, 1–7. doi: 10.1155/2010/323152
Chen, R., Li, M., Zhang, Y., Zhou, Q., and Shu, H. (2012). The E3 ubiquitin ligase MARCH8 negatively regulates IL-1 β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 109, 14128–14133. doi: 10.1073/pnas.1205246109
Ciechanover, A., and Ben-Saadon, R. (2004). N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 14, 103–106. doi: 10.1016/j.tcb.2004.01.004
den Herder, G., Yoshida, S., Antolín-Llovera, M., Ried, M. K., and Parniskea, M. (2012). Lotus japonicus E3 ligase SEVEN IN ABSENTIA4 destabilizes the symbiosis receptor-like Kinase SYMRK and negatively regulates rhizobial infection. Plant Cell 24, 1691–1707. doi: 10.1105/tpc.110.082248
Deshaies, R. J., and Joazeiro, C. A. P. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. doi: 10.1146/annurev.biochem.78.101807.093809
Dou, H., Buetow, L., Sibbet, G. J., Cameron, K., and Huang, D. T. (2012). BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 19, 876–883. doi: 10.1038/nsmb.2379
Du, H., Huang, Y., Zaghlula, M., Walters, E., Cox, T. C., and Massiah, M. A. (2013). The MID1 E3 ligase catalyzes the polyubiquitination of Alpha4 (α4), a regulatory subunit of protein phosphatase 2A (PP2A): novel insights into MID1-mediated regulation of PP2A. J. Biol. Chem. 288, 21341–21350. doi: 10.1074/jbc.M113.481093
Eakin, C. M., Maccoss, M. J., Finney, G. L., and Klevit, R. E. (2007). Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 104, 5794–5799. doi: 10.1073/pnas.0610887104
Fan, C., and Wang, X. (2017). Mdm2 Splice isoforms regulate the p53 / Mdm2 / Mdm4 regulatory circuit via RING domain-mediated ubiquitination of p53 and Mdm4. Cell Cycle 16, 660–664. doi: 10.1080/15384101.2017.1288327
Fang, S., Jensen, J. P., Ludwig, R. L., Vousden, K. H., and Weissman, A. M. (2000). Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 275, 8945–8951. doi: 10.1074/jbc.275.12.8945
Fei, C., Li, Z., Li, C., Chen, Y., Chen, Z., He, X., et al. (2013). Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/β-catenin signaling. Mol. Cell. Biol. 33, 4095–4105. doi: 10.1128/MCB.00418-413
Fersht, A., Julet, M. R., and Britch, J. (1999). Structure and Mechanicsm in Protein Science - A. Fersht (W H Freeman, 1999). Singapore: World Scientific.
Freemont, P. S., Hanson, I. M., and Trowsdale, J. (1991). A novel cysteine-rich sequence motif. Cell 64, 483–484. doi: 10.1016/0092-8674(91)90229-r
Fukutomi, T., Takagi, K., Mizushima, T., Ohuchi, N., and Yamamoto, M. (2014). Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 34, 832–846. doi: 10.1128/MCB.01191-1113
George, A. J., Hoffiz, Y. C., Charles, A. J., Zhu, Y., and Mabb, A. M. (2018). A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front. Genet. 9:29. doi: 10.3389/fgene.2018.00029
Gilberto, S., and Peter, M. (2017). Dynamic ubiquitin signaling in cell cycle regulation. J. Cell Biol. 216, 2259–2271. doi: 10.1083/jcb.201703170
Hayes, S. D., Liu, H., MacDonald, E., Sanderson, C. M., Coulson, J. M., Clague, M. J., et al. (2012). Direct and indirect control of mitogen-activated protein kinase pathway-associated components, BRAP/IMP E3 ubiquitin ligase and CRAF/RAF1 kinase, by the deubiquitylating enzyme USP15. J. Biol. Chem. 287, 43007–43018. doi: 10.1074/jbc.M112.386938
Hershko, A., and Ciechanover, A. (1998). the ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Ho, S.-R., Mahanic, C. S., Lee, Y.-J., and Lin, W.-C. (2014). RNF144A, an E3 ubiquitin ligase for DNA-PKcs, promotes apoptosis during DNA damage. Proc. Natl. Acad. Sci. U.S.A. 111, E2646–E2655. doi: 10.1073/pnas.1323107111
Hochrainer, K., Pejanovic, N., Olaseun, V. A., Zhang, S., Iadecola, C., and Anrather, J. (2015). The ubiquitin ligase HERC3 attenuates NF-κB-dependent transcription independently of its enzymatic activity by delivering the RelA subunit for degradation. Nucleic Acids Res. 43, 9889–9904. doi: 10.1093/nar/gkv1064
Hoer, S., Smith, L., and Lehner, P. J. (2007). MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett. 581, 45–51. doi: 10.1016/j.febslet.2006.11.075
Huibregtse, J. M., Scheffner, M., Beaudenon, S., and Howley, P. M. (1995). A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 5249. doi: 10.1073/pnas.92.11.5249-a
Javadi, M., Richmond, T. D., Huang, K., and Barber, D. L. (2013). CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. J. Biol. Chem. 288, 19459–19470. doi: 10.1074/jbc.M113.475087
Ji, L., Jiang, B., Jiang, X., Charlat, O., Chen, A., Mickanin, C., et al. (2017). The SIAH E3 ubiquitin ligases promote Wnt/β-catenin signaling through mediating Wnt-induced Axin degradation. Genes Dev. 31, 904–915. doi: 10.1101/gad.300053.117
Joazeiro, C. A., Wing, S. S., Huang, H., Leverson, J. D., Hunter, T., and Liu, Y. C. (1999). The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312. doi: 10.1126/science.286.5438.309
Jones, J. M., and Gellert, M. (2003). Autoubiquitylation of the V(D)J recombinase protein RAG1. Proc. Natl. Acad. Sci. U.S.A. 100, 15446–15451. doi: 10.1073/pnas.2637012100
Joukov, V., Chen, J., Fox, E. A., Green, J. B. A., and Livingston, D. M. (2001). Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. U.S.A. 98, 12078–12083. doi: 10.1073/pnas.211427098
Kallijärvi, J., Lahtinen, U., Hämäläinen, R., Lipsanen-Nyman, M., Palvimo, J. J., and Lehesjoki, A.-E. (2005). TRIM37 defective in mulibrey nanism is a novel RING finger ubiquitin E3 ligase. Exp. Cell Res. 308, 146–155. doi: 10.1016/j.yexcr.2005.04.001
Kamadurai, H. B., Souphron, J., Scott, D. C., Duda, D. M., Miller, D. J., Stringer, D., et al. (2009). Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol. Cell 36, 1095–1102. doi: 10.1016/j.molcel.2009.11.010
Kanelis, V., Rotin, D., and Forman-Kay, J. D. (2001). Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat. Struct. Biol. 8, 407–412. doi: 10.1038/87562
Kawashima, A., Karasawa, T., Tago, K., Kimura, H., Kamata, R., Usui-Kawanishi, F., et al. (2017). ARIH2 Ubiquitinates NLRP3 and negatively regulates nlrp3 inflammasome activation in macrophages. J. Immunol. 199, 3614–3622. doi: 10.4049/jimmunol.1700184
Kim, J. M., Cho, E. N., Kwon, Y. E., Bae, S. J., Kim, M., and Seol, J. H. (2010). CHFR functions as a ubiquitin ligase for HLTF to regulate its stability and functions. Biochem. Biophys. Res. Commun. 395, 515–520. doi: 10.1016/j.bbrc.2010.04.052
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi: 10.1146/annurev-biochem-060310-170328
Kostic, M., Matt, T., Martinez-Yamout, M. A., Dyson, H. J., and Wright, P. E. (2006). Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J. Mol. Biol. 363, 433–450. doi: 10.1016/j.jmb.2006.08.027
Lai, Z., Freedman, D. A., Levine, A. J., and McLendon, G. L. (1998). Metal and RNA binding properties of the hdm2 RING finger domain. Biochemistry 37, 17005–17015. doi: 10.1021/bi980596r
Laney, J. D., and Hochstrasser, M. (1999). Substrate targeting in the ubiquitin system. Cell 97, 427–430. doi: 10.1016/S0092-8674(00)80752-80757
Lazzari, E., El-Halawany, M., De March, M., Valentino, F., Cantatore, F., Migliore, C., et al. (2019). Analysis of the zn-binding domains of TRIM32, the E3 ubiquitin ligase mutated in limb girdle muscular dystrophy 2H. Cells 8:254. doi: 10.3390/cells8030254
Lee, J. M., Choi, S. S., Lee, Y. H., Khim, K. W., Yoon, S., Kim, B.-G., et al. (2018a). The E3 ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Exp. Mol. Med. 50:135. doi: 10.1038/s12276-018-0162-6
Lee, M. S., Han, H. J., Han, S. Y., Kim, I. Y., Chae, S., Lee, C. S., et al. (2018b). Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nat. Commun. 9:3404. doi: 10.1038/s41467-018-05721-5724
Lee, S. Y., Ramirez, J., Franco, M., Lectez, B., Gonzalez, M., Barrio, R., et al. (2014). Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci. 71, 2747–2758. doi: 10.1007/s00018-013-1526-1527
Lehner, P. J., Hoer, S., Dodd, R., and Duncan, L. M. (2005). Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol. Rev. 207, 112–125. doi: 10.1111/j.0105-2896.2005.00314.x
Li, X., Zhong, L., Wang, Z., Chen, H., Liao, D., Zhang, R., et al. (2018). Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 8, 3643–3653. doi: 10.7150/thno.26021
Li, Y., Wu, H., Wu, W., Zhuo, W., Liu, W., Zhang, Y., et al. (2014). Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 24, 762–765. doi: 10.1038/cr.2014.46
Lienlaf, M., Hayashi, F., Di Nunzio, F., Tochio, N., Kigawa, T., Yokoyama, S., et al. (2011). Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5αrh: structure of the RING domain of TRIM5. J. Virol. 85, 8725–8737. doi: 10.1128/jvi.00497-411
Liew, C. W., Sun, H., Hunter, T., and Day, C. L. (2010). RING domain dimerization is essential for RNF4 function. Biochem. J. 431, 23–29. doi: 10.1042/BJ20100957
Linke, K., Mace, P. D., Smith, C. A., Vaux, D. L., Silke, J., and Day, C. L. (2008). Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 15, 841–848. doi: 10.1038/sj.cdd.4402309
Liu, B., Zhang, M., Chu, H., Zhang, H., Wu, H., Song, G., et al. (2017a). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224. doi: 10.1038/ni.3641
Liu, J., Zhang, C., Zhao, Y., Yue, X., Wu, H., Huang, S., et al. (2017b). Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 8:1823. doi: 10.1038/s41467-017-01947-w
Liu, S., Jiang, M., Wang, W., Liu, W., Song, X., Ma, Z., et al. (2018). Nuclear RNF2 inhibits interferon function by promoting K33-linked STAT1 disassociation from DNA. Nat. Immunol. 19, 41–52. doi: 10.1038/s41590-017-0003-0
Loregger, A., Grandl, M., Mejías-Luque, R., Allgäuer, M., Degenhart, K., Haselmann, V., et al. (2015). The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated β-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 8, 1–13. doi: 10.1126/scisignal.aac6757
Lorick, K. L., Jensen, J. P., Fang, S., Ong, A. M., Hatakeyama, S., and Weissman, A. M. (1999). RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 96, 11364–11369. doi: 10.1073/pnas.96.20.11364
Mallette, F. A., Mattiroli, F., Cui, G., Young, L. C., Hendzel, M. J., Mer, G., et al. (2012). RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878. doi: 10.1038/emboj.2012.47
Masuda, Y., Suzuki, M., Kawai, H., Suzuki, F., and Kamiya, K. (2012). Asymmetric nature of two subunits of RAD18, a RING-type ubiquitin ligase E3, in the human RAD6A-RAD18 ternary complex. Nucleic Acids Res. 40, 1065–1076. doi: 10.1093/nar/gkr805
Metzger, M. B., Pruneda, J. N., Klevit, R. E., and Weissman, A. M. (2014). RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60. doi: 10.1016/j.bbamcr.2013.05.026
Morett, E., and Bork, P. (1999). A novel transactivation domain in parkin. Trends Biochem. Sci. 24, 229–231. doi: 10.1016/S0968-0004(99)01381-X
Morreale, F. E., and Walden, H. (2016). Types of ubiquitin ligases. Cell 165, 248–248.e1. doi: 10.1016/j.cell.2016.03.003
Muńoz-Escobar, J., Matta-Camacho, E., Kozlov, G., and Gehring, K. (2015). The MLLE domain of the ubiquitin ligase UBR5 binds to its catalytic domain to regulate substrate binding. J. Biol. Chem. 290, 22841–22850. doi: 10.1074/jbc.M115.672246
Nakatani, Y., Kleffmann, T., Linke, K., Condon, S. M., Hinds, M. G., and Day, C. L. (2013). Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem. J. 450, 629–638. doi: 10.1042/BJ20121702
Nomura, K., Klejnot, M., Kowalczyk, D., Hock, A. K., Sibbet, G. J., Vousden, K. H., et al. (2017). Structural analysis of MDM2 RING separates degradation from regulation of p53 transcription activity. Nat. Struct. Mol. Biol. 24, 578–587. doi: 10.1038/nsmb.3414
Okumura, F., Hatakeyama, S., Matsumoto, M., Kamura, T., and Nakayama, K. I. (2004). Functional regulation of FEZ1 by the U-box-type ubiquitin ligase E4B contributes to neuritogenesis. J. Biol. Chem. 279, 53533–53543. doi: 10.1074/jbc.M402916200
Osinalde, N., Duarri, A., Ramirez, J., Barrio, R., Perez de Nanclares, G., and Mayor, U. (2019). Impaired proteostasis in rare neurological diseases. Semin. Cell Dev. Biol. 93, 164–177. doi: 10.1016/j.semcdb.2018.10.007
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Plechanovov, A., Jaffray, E. G., Tatham, M. H., Naismith, J. H., and Hay, R. T. (2012). Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120. doi: 10.1038/nature11376
Poyurovsky, M. V., Priest, C., Kentsis, A., Borden, K. L. B., Pan, Z. Q., Pavletich, N., et al. (2007). The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 26, 90–101. doi: 10.1038/sj.emboj.7601465
Pruneda, J. N., Littlefield, P. J., Soss, S. E., Nordquist, K. A., Chazin, W. J., Brzovic, P. S., et al. (2012). Structure of an E3:E2 Ub Complex Reveals an Allosteric Mechanism Shared among RING/U-box Ligases. Mol. Cell 47, 933–942. doi: 10.1016/j.molcel.2012.07.001
Pruneda, J. N., Stoll, K. E., Bolton, L. J., Brzovic, P. S., and Klevit, R. E. (2011). Ubiquitin in motion: structural studies of the ubiquitin-conjugating enzyme ubiquitin conjugate. Biochemistry 50, 1624–1633. doi: 10.1021/bi101913m
Raheja, R., Liu, Y., Hukkelhoven, E., Yeh, N., and Koff, A. (2014). The ability of TRIM3 to induce growth arrest depends on RING-dependent E3 ligase activity. Biochem. J. 458, 537–545. doi: 10.1042/BJ20131288
Ronchi, V. P., Klein, J. M., Edwards, D. J., and Haas, A. L. (2014). The active form of E6-associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J. Biol. Chem. 289, 1033–1048. doi: 10.1074/jbc.M113.517805
Rotin, D., and Kumar, S. (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409. doi: 10.1038/nrm2690
Ruffner, H., Joazeiro, C. A., Hemmati, D., Hunter, T., and Verma, I. M. (2001). Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. U.S.A. 98, 5134–5139. doi: 10.1073/pnas.081068398
Sander, B., Xu, W., Eilers, M., Popov, N., and Lorenz, S. (2017). A conformational switch regulates the ubiquitin ligase HUWE1. eLife 6:e21036. doi: 10.7554/eLife.21036
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Scheffner, M., and Kumar, S. (2014). Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta 1843, 61–74. doi: 10.1016/j.bbamcr.2013.03.024
Seo, J. H., Agarwal, E., Bryant, K. G., Caino, M. C., Kim, E. T., Kossenkov, A. V., et al. (2018). Syntaphilin ubiquitination regulates mitochondrial dynamics and tumor cell movements. Cancer Res. 78, 4215–4228. doi: 10.1158/0008-5472.CAN-18-0595
Shen, J., Li, P., Shao, X., Yang, Y., Liu, X., Feng, M., et al. (2018). The E3 ligase ring1 targets p53 for degradation and promotes cancer cell proliferation and survival. Cancer Res. 78, 359–371. doi: 10.1158/0008-5472.CAN-17-1805
Smit, J. J., Monteferrario, D., Noordermeer, S. M., Van, W. J., Reijden, B. A., and Van Der Sixma, T. K. (2012). The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 31, 3833–3844. doi: 10.1038/emboj.2012.217
Soss, S. E., Klevit, R. E., and Chazin, W. J. (2013). Activation of UbcH5c Ub is the result of a shift in interdomain motions of the conjugate bound to U-box E3 ligase E4B. Biochemistry 52, 2991–2999. doi: 10.1021/bi3015949
Staub, O., Dho, S., Henry, P., Correa, J., Ishikawa, T., McGlade, J., et al. (1996). WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 15, 2371–2380. doi: 10.1002/j.1460-2075.1996.tb00593.x
Tan, C., Byrne, E. F. X., Ah-Cann, C., Call, M. J., and Call, M. E. (2019). A serine in the first transmembrane domain of the human E3 ubiquitin ligase MARCH9 is critical for down-regulation of its protein substrates. J. Biol. Chem. 294, 2470–2485. doi: 10.1074/jbc.RA118.004836
Tian, H., Tackmann, N. R., Jin, A., Zheng, J., and Zhang, Y. (2017). Inactivation of the MDM2 RING domain enhances p53 transcriptional activity in mice. J. Biol. Chem. 292, 21614–21622. doi: 10.1074/jbc.RA117.000122
Torrino, S., Visvikis, O., Doye, A., Boyer, L., Stefani, C., Munro, P., et al. (2011). The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 21, 959–965. doi: 10.1016/j.devcel.2011.08.015
Tripathi, E., and Smith, S. (2017). Cell cycle-regulated ubiquitination of tankyrase 1 by RNF8 and ABRO1/BRCC36 controls the timing of sister telomere resolution. EMBO J. 36, 503–519. doi: 10.15252/embj.201695135
Vander Kooi, C. W., Ohi, M. D., Rosenberg, J. A., Oldham, M. L., Newcomer, M. E., Gould, K. L., et al. (2006). The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry 45, 121–130. doi: 10.1021/bi051787e
von Stechow, L., Typas, D., Carreras Puigvert, J., Oort, L., Siddappa, R., Pines, A., et al. (2015). The E3 ubiquitin ligase ARIH1 protects against genotoxic stress by initiating a 4EHP-Mediated mRNA translation arrest. Mol. Cell. Biol. 35, doi: 10.1128/mcb.01152-1114
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004). Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878. doi: 10.1038/nature02985
Wang, Q., Liu, X., Cui, Y., Tang, Y., Chen, W., Li, S., et al. (2014). The E3 Ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933. doi: 10.1016/j.immuni.2014.11.011
Wang, W., Xia, Z.-J., Farré, J.-C., and Subramani, S. (2017). TRIM37, a novel E3 ligase for PEX5-mediated peroxisomal matrix protein import. J. Cell Biol. 216, 2843–2858. doi: 10.1083/jcb.201611170
Wang, X., Herr, R. A., and Hansen, T. H. (2012). Ubiquitination of substrates by esterification. Traffic 13, 19–24. doi: 10.1111/j.1600-0854.2011.01269.x
Waterman, H., Levkowitz, G., Alroy, I., and Yarden, Y. (1999). The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J. Biol. Chem. 274, 22151–22154. doi: 10.1074/jbc.274.32.22151
Welcker, M., Larimore, E. A., Swanger, J., Bengoechea-Alonso, M. T., Grim, J. E., Ericsson, J., et al. (2013). Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev. 27, 2531–2536. doi: 10.1101/gad.229195.113
Wenzel, D. M., Lissounov, A., Brzovic, P. S., and Klevit, R. E. (2011). UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108. doi: 10.1038/nature09966
Williams, S. A., Longerich, S., Sung, P., Vaziri, C., and Kupfer, G. M. (2011). The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117, 5078–5087. doi: 10.1182/blood-2010-10-311761
Wu, W., Sato, K., Koike, A., Nishikawa, H., Koizumi, H., Venkitaraman, A. R., et al. (2010). HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 70, 6384–6392. doi: 10.1158/0008-5472.CAN-10-1304
Xia, P., Wang, S., Huang, G., Du, Y., Zhu, P., Li, M., et al. (2014). RNF2 is recruited by WASH to ubiquitinate AMBRA1 leading to downregulation of autophagy. Cell Res. 24, 943–958. doi: 10.1038/cr.2014.85
Xin, D., Gu, H., Liu, E., and Sun, Q. (2018). Parkin negatively regulates the antiviral signaling pathway by targeting TRAF3 for degradation. J. Biol. Chem. 293, 11996–12010. doi: 10.1074/jbc.RA117.001201
Ye, J. S., Kim, N., Lee, K. J., Nam, Y. R., Lee, U., and Joo, C. H. (2014). Lysine 63-linked TANK-binding kinase 1 ubiquitination by mindbomb E3 ubiquitin protein ligase 2 is mediated by the mitochondrial antiviral signaling protein. J. Virol. 88, 12765–12776. doi: 10.1128/JVI.02037-14
Zaman, M. M.-U., Nomura, T., Takagi, T., Okamura, T., Jin, W., Shinagawa, T., et al. (2013). Ubiquitination-deubiquitination by the TRIM27-USP7 complex regulates TNF-α-induced apoptosis. Mol. Cell. Biol. 33, 4971–4984. doi: 10.1128/MCB.00465-413
Zhao, X., Heng, J. I.-T., Guardavaccaro, D., Jiang, R., Pagano, M., Guillemot, F., et al. (2008). The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 10, 643–653. doi: 10.1038/ncb1727
Zheng, N., and Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157. doi: 10.1146/annurev-biochem-060815-14922
Zheng, N., Wang, P., Jeffrey, P. D., and Pavletich, N. P. (2000). Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 102, 533–539. doi: 10.1016/s0092-8674(00)00057-x
Zhou, R., Patel, S. V., and Snyder, P. M. (2007). Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 282, 20207–20212. doi: 10.1074/jbc.M611329200
Keywords: ubiquitin, E3, mutation, ligase, inactivation
Citation: Garcia-Barcena C, Osinalde N, Ramirez J and Mayor U (2020) How to Inactivate Human Ubiquitin E3 Ligases by Mutation. Front. Cell Dev. Biol. 8:39. doi: 10.3389/fcell.2020.00039
Received: 17 October 2019; Accepted: 16 January 2020;
Published: 04 February 2020.
Edited by:
Benedikt M. Kessler, University of Oxford, United KingdomReviewed by:
Richard Chahwan, University of Zurich, SwitzerlandCopyright © 2020 Garcia-Barcena, Osinalde, Ramirez and Mayor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ugo Mayor, dWdvLm1heW9yQGVodS5ldXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.