
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 06 December 2019
Sec. Cell Growth and Division
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00322
This article is part of the Research Topic Phosphorylation-Dependent Peptidyl-Prolyl Cis/Trans Isomerase PIN1 View all 13 articles
 Songyin Huang1†
Songyin Huang1† Yang Chen1†Zhi-Mei Liang2,3Na-Na Li2,3Yujie Liu2,4Yinghua Zhu2,3Dingzhun Liao5Xiao Zhen Zhou6
Yang Chen1†Zhi-Mei Liang2,3Na-Na Li2,3Yujie Liu2,4Yinghua Zhu2,3Dingzhun Liao5Xiao Zhen Zhou6 Kun Ping Lu6
Kun Ping Lu6 Yandan Yao2,4*
Yandan Yao2,4* Man-Li Luo2,3*
Man-Li Luo2,3*Breast cancer is the most prevalent tumor in women worldwide and about 70% patients are estrogen receptor positive. In these cancer patients, resistance to the anticancer estrogen receptor antagonist tamoxifen emerges to be a major clinical obstacle. Peptidyl-prolyl isomerase Pin1 is prominently overexpressed in breast cancer and involves in tamoxifen-resistance. Here, we explore the mechanism and effect of targeting Pin1 using its chemical inhibitor all-trans retinoic acid (ATRA) in the treatment of tamoxifen-resistant breast cancer. We found that Pin1 was up-regulated in tamoxifen-resistant human breast cancer cell lines and tumor tissues from relapsed patients. Pin1 overexpression increased the phosphorylation of ERα on S118 and stabilized ERα protein. ATRA treatment, resembling the effect of Pin1 knockdown, promoted ERα degradation in tamoxifen-resistant cells. Moreover, ATRA or Pin1 knockdown decreased the activation of ERK1/2 and AKT pathways. ATRA also reduced the nuclear expression and transcriptional activity of ERα. Importantly, ATRA inhibited cell viability and proliferation of tamoxifen-resistant human breast cancer cells in vitro. Slow-releasing ATRA tablets reduced the growth of tamoxifen-resistant human breast cancer xenografts in vivo. In conclusion, ATRA-induced Pin1 ablation inhibits tamoxifen-resistant breast cancer growth by suppressing multifactorial mechanisms of tamoxifen resistance simultaneously, which demonstrates an attractive strategy for treating aggressive and endocrine-resistant tumors.
Breast cancer is a leading cause of cancer-related death in female (Chen et al., 2016). In all breast cancer patients, approximately 70% patients are estrogen receptor positive (Deroo and Korach, 2006; Yager and Davidson, 2006; Nilsson et al., 2011). Although selective estrogen receptor modulator such as tamoxifen are effective for ER positive patient, approximately 30% of patients are not sensitive to tamoxifen treatment at the beginning, and over 50% of initial effective patients finally suffer from tamoxifen-resistance (TAMR) (Osborne and Schiff, 2011). The mechanism of TAMR is still not completely known. The possible molecular mechanisms include, but not limited to, the alteration of estrogen receptor transcriptional co-regulatory proteins (Shao et al., 2004; Girault et al., 2006), cross-talk between receptor tyrosine kinase signaling and estrogen receptor (Stenoien et al., 2001), non-canonical transcriptional activation of estrogen receptor (Anbalagan and Rowan, 2015), the expression of specific microRNAs (Miller et al., 2008), etc. Given that many studies have demonstrated that estrogen receptors play a central role in TAMR (Wijayaratne and McDonnell, 2001; Marsh et al., 2017; Zhang et al., 2017), blocking estrogen receptor related pathways is an attractive strategy to treat TAMR breast cancer.
Pin1 is a peptidyl-prolyl cis/trans isomerase (PPIase), which specifically recognizes pSer/Thr-Pro motifs of proteins and catalyzes their trans-cis conformational change (Lu and Zhou, 2007). Pin1 plays a vital role in cancer development by regulating more than 40 oncoproteins and over 20 tumor suppressors, therefore promoting cancer growth and cancer stem cell tumorigenesis (Zhou and Lu, 2016). Pin1 has been found to be up-regulated in tamoxifen-resistant breast cancer (Stanya et al., 2008; Namgoong et al., 2010; Khanal et al., 2012). Overexpression of Pin1 reduces the protein stability of estrogen receptor transcriptional co-regulatory protein SMRT (Stanya et al., 2008), as well as regulates the transcription function of ERα (Rajbhandari et al., 2012, 2015). Knockdown of Pin1 by siRNA inhibits the viability of TAMR breast cancer cells (Namgoong et al., 2010), indicating that Pin1 might be a promising therapeutic target for tamoxifen-resistant breast cancer. However, due to the lack of appropriate Pin1 inhibitors, it is challenging to evaluate the effect of targeting Pin1 on overcoming TAMR. Recently, Wei et al. has discovered all-trans retinoic acid (ATRA) as a specific Pin1 chemical inhibitor (Wei et al., 2015). ATRA has been used to induce differentiation and treat acute promyelocytic leukemia (APL). In APL, ATRA facilitates PML–RAR-α degradation, thereby suppresses APL stem cells (Huang et al., 1988; de The and Chen, 2010; Sanz and Lo-Coco, 2011). Wei et al. (2015) has found that besides RAR, Pin1 is a key target of ATRA in APL and breast cancer. ATRA directly and selectively binds to and degrades active Pin1, thereby inhibiting multiple Pin1-regulated cancer driving pathways.
In the current study, we explored the effects of ATRA in inhibiting Pin1 and treating tamoxifen-resistant breast cancer in vitro and in vivo. Our experiments showed that Pin1 was up-regulated in tamoxifen-resistant cells and increased ERα protein stability. ATRA treatment accelerated ERα protein turnover, reduced ERα transcriptional activity, and decreased the phosphorylation of AKT and ERK1/2 simultaneously, which further inhibited ERα activation. Thus, ATRA induced the degradation of Pin1 and suppressed cell viability and proliferation of tamoxifen-resistant breast cancer cells. More importantly, slow-releasing ATRA tablets showed remarkable anti-tumor effects in the tamoxifen-resistant xenograft model. Therefore, targeting Pin1 by ATRA promised a new potential approach to treat tamoxifen-resistant breast cancer.
The human breast cancer cell lines MCF7 and T47D were purchased from American Type Culture Collection (Manassas, VA, United States) and cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, United States) supplemented with 10% fetal bovine serum (FBS, GBICO). Tamoxifen resistant cell lines (MCF-7R and T47DR) were kindly provided by Dr. Qiang Liu as gift, and were cultured in no-phenol red 1640 medium (Life Technologies, United States) supplemented with 10% charcoal-stripped FBS (cFBS) (HyClone, United States) and 1 μM 4-hydroxytamoxifen (Sigma-Aldrich, St. Louis, MO, United States). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Antibodies of ERα was from cell signaling technology (8644) and Abcam (16660). Antibodies of Pin1 was from R&D (MAB2294) and Abnova (MAB12340). Phospho-ERα S118 antibody was from Abcam (ab32396). Phospho-ERα S167 antibody was from cell signaling technology (64508). ATRA was from Sigma (R2625). Tamoxifen was from Sigma (H6278). Pin1 shRNA targeting sequence: CCACCGTCACACAGTATTTAT; Pin1 siRNA-1 targeting sequence: TCAGGCCGAGTGTACTACT; Pin1 siRNA-2 targeting sequence: GCTCAGGCCGAGTGTACTA; RARα siRNA-1 targeting sequence: CCAGCTCACAGAACTGCTT; RARα siRNA-2 targeting sequence: TTCCGCACGTAGACCTT TAGC; ERα siRNA-1 targeting sequence: CAGGCCAAATTCA GATAAT; ERα siRNA-2 targeting sequence: GGTCCAC CTTCTAGAATGT.
Six-well plates were seeded 2000 cells per well. Cells were treated with vehicle (DMSO), 10 μM ATRA, 10 μM tamoxifen (TAM) or ATRA plus TAM, and medium were changed every 3 days. 14 days later cells were fixed with methanol, stained with 0.5% crystal violet.
Cell viability was measured using the Cell Titer Glo reagent (Promega). The cells were plated in 96-well plates at 1 × 103 cells per well and maintained at 37°C. At the indicated time points, cell viability was measured according to the manufacturer’s instructions.
Total RNA was extracted from cells using Trizol (life, United States). MMLV kit (life, United States) was used to generate cDNA. Real time PCR were performed using Toyobo SYBR GREEN. The primers used were as follows: Pin1 (forward, 5′- AGCTCAGGCCGAGTGTACTA-3′; reverse, 5′-CCTTGGTCCGGGTGATCTTC -3′); growth regulation by estrogen in breast cancer 1 (GREB1) (forward, 5′-GTGGT AGCCGAGTGGACAAT-3′; reverse, 5′-ATTTG TTTCCAGCC CTCCTT-3′); progesterone receptor (PGR) (forward, 5′-GG CATGGTCCTTGGAGGT -3′; reverse, 5′-CCACTGGCTGTGG GAGAG-3′); c-Myc (forward, 5′- TACAACACCC GAGC AAGGAC-3′; reverse, 5′-GAGGCTGCTGGTTTTCCACT-3′); β-actin (forward, 5′-GGAAGGGGGACGGGGACAGC-3′; reverse, 5′- GGAGGAGCAAG GAGCGGGAG-3′).
Cells were lysed with RIPA buffer containing 0.1% protease inhibitors or phosphatase inhibitors (Life, United States). The supernatant of lysate was separated by electrophoresis and blotted onto a PVDF membrane, then blocked with 5% skim milk at room temperature for 1 h. The blots were incubated with the following antibody at 4°C overnight: ERα (1:1000, CST, #8644); Pin1 (1:1000, R&D, #MAB2294); phospho-ERα S118 (1:1000, CST, #ab32396); AKT (1:1000, CST); Flag-tag (1:5000, Sigma, United States); phospho-AKT (1:1000, CST); phospho-c-Raf (1:1000, CST); phospho-MEK1/2 (1:1000, CST); ERK1/2 (1:1000, CST); phospho-ERK1/2 (1:1000, CST); phospho-ERαS167 (1:1000, CST, #64508); β-Actin (1:2000, CST); GAPDH (1:2000, proteintech). After incubation with HRP-conjugated secondary antibodies at room temperature for 1 h, all blots were detected by an enhanced chemiluminescence (ECL) and were scanned using ChemiDocTM XRS + imaging system (Bio-Rad, Hercules, CA, United States).
MCF-7R and T47DR cells were fixed in 4% polyoxymethylene at 4°C for 20 min, washed with PBS and permeabilized in 0.1% Triton X-100 at room temperature for 10 min. Cells were then blocked in 10% goat serum at room temperature for 30 min, and incubated with ERα antibody (1:100, Abcam, #16660) in 10% goat serum at 4°C overnight. Cells were washed, incubated with secondary antibodies at room temperature for 1 h, washed, incubated with DAPI at room temperature for 15 min. Slides were then covered with fluorescently quencher 30 μl, sealed and photographed with an Olympus confocal microscope.
Nude mice were purchased from Laboratory Animal Service Center, Sun Yat-sen University. The experiment protocol was approved by the Animal Care and Use Committee of Sun Yat-sen University. 2 × 106 MCF-7R cells were mixed with an equal volume of matrigel (Corning) and injected into the mammary fat pads of 4 week-old female BALB/c nude mice. One week later, when tumor size reached ∼100 mm3, the tumor-bearing mice were randomized into treatment groups. 21-days ATRA tablets were implanted under neck skin. Tamoxifen was injected at 4 mg/kg per day. Tumor volume was measured every 3 days.
Tumor samples were obtained from patients with ER positive breast cancer who underwent tamoxifen therapy in Sun Yat-sen Memorial Hospital. All samples were collected from patients with informed consent, and all related procedures were performed with the approval of the internal review and ethics boards of Sun Yat-sen Memorial Hospital. Immunohistochemistry staining for Pin1 and ERα was performed as described previously (Luo et al., 2015; Zhang et al., 2016). Briefly, sodium citrate was used to repair tissue antigen. Incubation of primary antibodies (Pin1, 1:50, Abnova, #MAB12340; ERα, 1:50, CST, #16660) was carried out at 4°C overnight. The slides were incubated with HRP-conjugated secondary antibodies at room temperature for 1 h, washed, visualized with DAB solution, followed by staining with hematoxylin. Immunostaining results was analyzed by ImageJ software.
All data are presented as the means ± SD. Student’s t-test was used to analysis the significance between two experimental groups, and ANOVA test was used to analysis among three or more groups. P < 0.05 was considered significant. All the statistical analyses were performed using SPSS20.
We established tamoxifen-resistant human breast cancer cell lines MCF-7 and T47D by long-term exposure to tamoxifen (Herman and Katzenellenbogen, 1996; Knowlden et al., 2003; Chu et al., 2015). We confirmed the resistance of these cells by showing that the viability of resistance cells was significantly higher than parental cells and apoptosis were remarkable lower in the presence of 1 μM tamoxifen (Chu et al., 2015). We found that both Pin1 protein and mRNA were up-regulated in tamoxifen-resistant MCF-7 (MCF-7R) and T47D (T47DR) cells, comparing to parental cells (Figures 1A–E and Supplementary Figure S5), which was consistent with previous reports that Pin1 was overexpressed in TAMR human breast cancer tissues (Namgoong et al., 2010; Khanal et al., 2012).
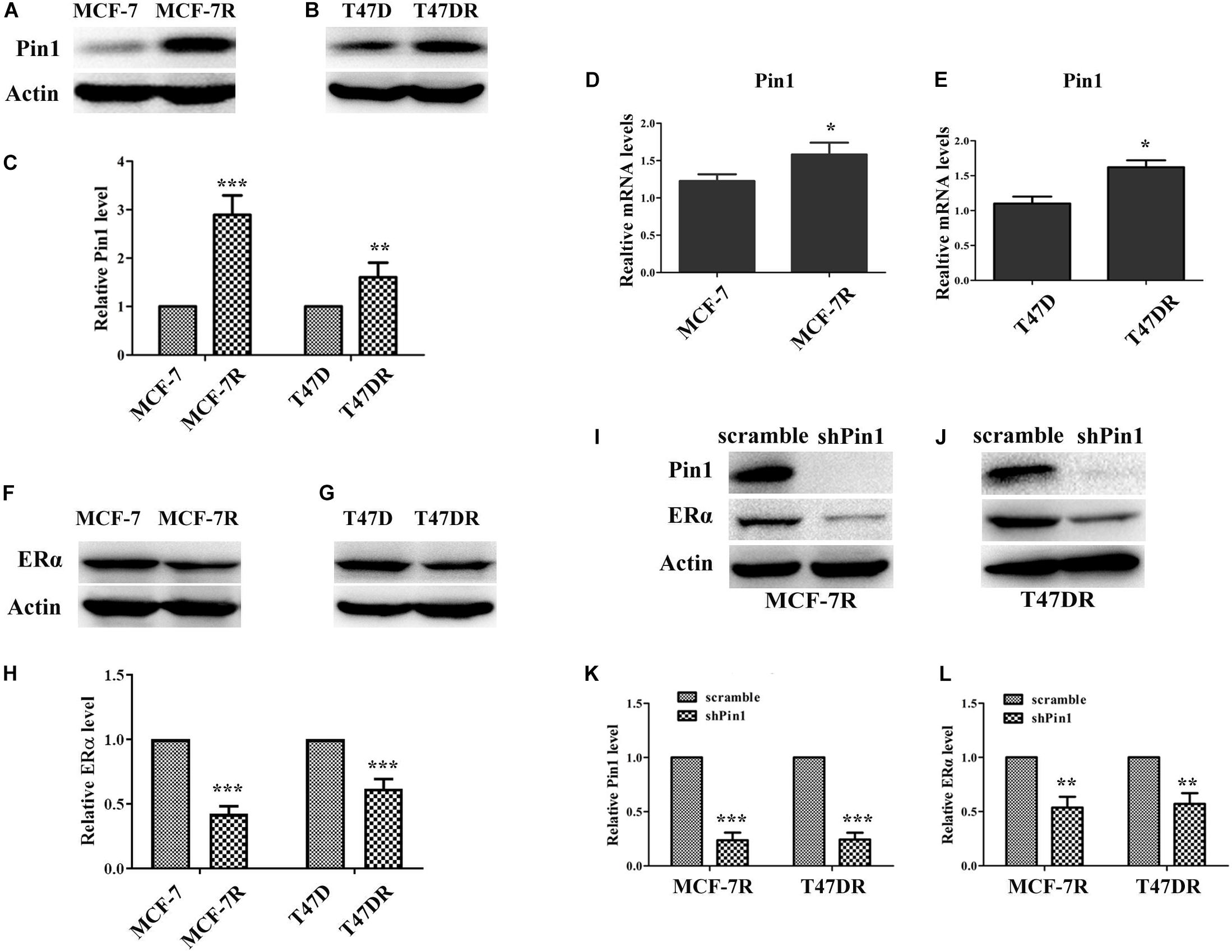
Figure 1. Pin1 is overexpressed in tamoxifen resistant breast cancer cells. (A,B) Pin1 is up-regulated in tamoxifen resistant breast cancer cells. Pin1 protein was detected by western blot in parental (MCF-7 and T47D) and tamoxifen resistant (MCF-7R and T47DR) cells. (C) Quantification of Pin1 levels in parental and tamoxifen resistant cells. Western blot bands in panels (A,B) were quantified by densitometric scan and represented as a relative ratio to control samples. Data are represented as means ± SD for three independent experiments. (D,E) Pin1 mRNA is up-regulated in tamoxifen resistant breast cancer cells, as detected by qRT-PCR. (F–H) The ERα protein level in parental and resistant breast cancer cells. Western blot bands were quantified in panel (H). (I–L) Pin1 knockdown decreases the level of ERα in MCF-7R and T47DR cells. Western blot bands were quantified in panels (K,I). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Although ERα was not so indispensable for TAMR cells as for parental cells, depleting ERα still further limited the growth of TAMR cells (Xiong et al., 2017). Indeed, through a variety of mechanisms, TMAR breast cancer cells made full use of remaining ERα to escape from the impact of tamoxifen (Osborne and Schiff, 2005; Johnston, 2010; Marsh et al., 2017). Here we examined the ERα level in TAMR cells, and found that ERα protein was down-regulated in TAMR cells (Figures 1F–H and Supplementary Figure S5), as shown previously (Stone et al., 2013; Lu et al., 2016). Given that ERα was a known Pin1 substrate which was positively regulated by Pin1 (Rajbhandari et al., 2012, 2015). We asked why Pin1 level was high while ERα level was low in TAMR cells. We found that Pin1 knockdown further decreased ERα level in TAMR cell lines (Figures 1I–L and Supplementary Figure S5). These results suggest that Pin1 is up-regulated and helps maintain ERα levels in TAMR cells even although ERα levels in these cells are low.
Next, we detected Pin1 and ERα protein levels in tumor tissues of recurrent ER positive breast cancer patients, who have received tamoxifen treatment. Pin1 protein level was significantly higher in recurrent tumors comparing with primary tumors (P = 0.004) (Figures 2A,B). More importantly, the expression level of Pin1 was associated with ERα in these tissues (Figures 2C,D). Together, Pin1 was up-regulated in both TAMR human breast cancer cell lines and relapsed tumor tissues, which positively correlated with ERα expression.
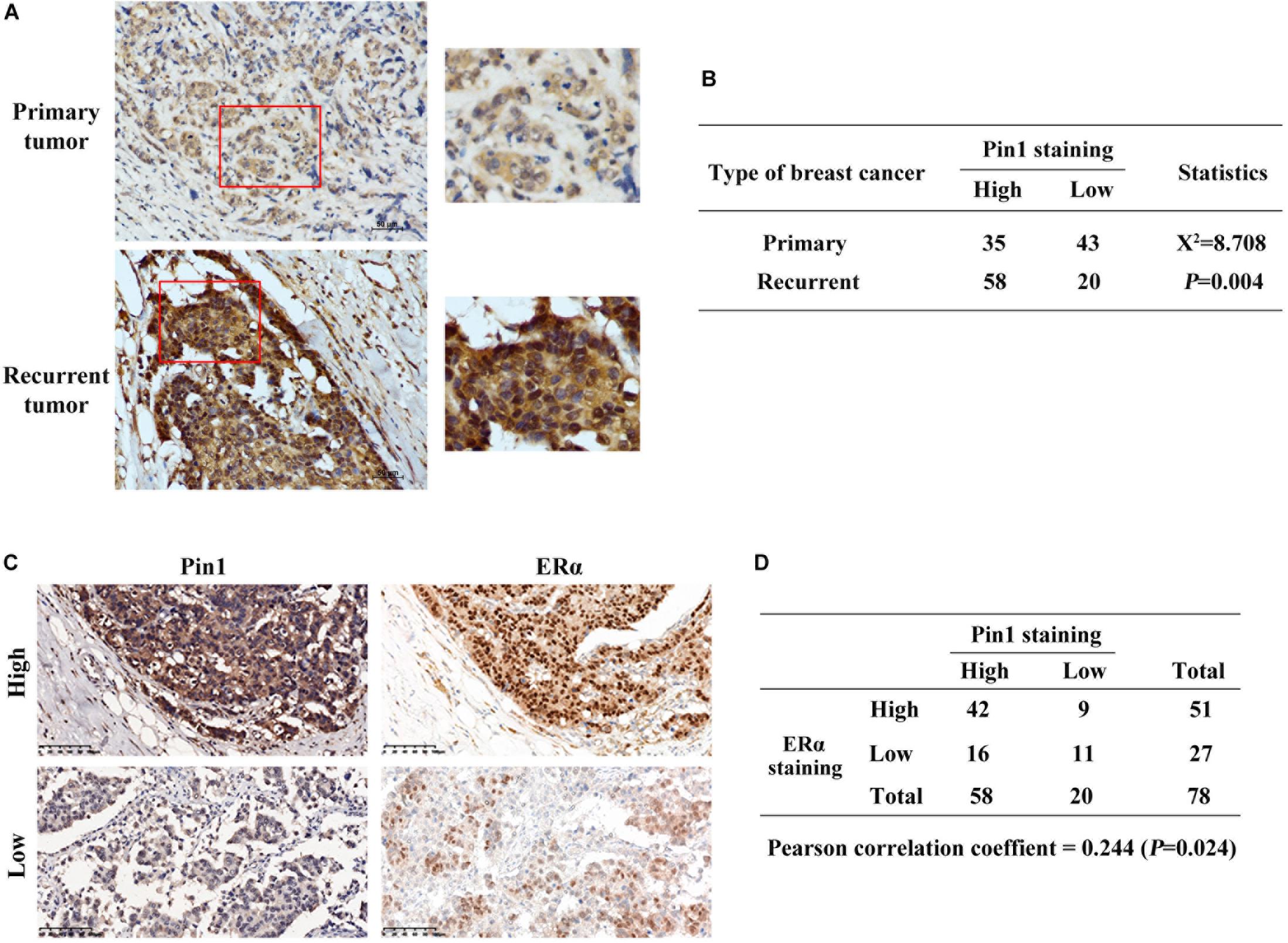
Figure 2. Pin1 is up-regulated in recurrent tumors and positively correlates with ERα in breast cancer tissues. (A) Representative immunohistochemistry image shows that Pin1 expression is higher in recurrent tamoxifen treated tumors than primary breast cancer tissues. Tissues in the red frames in left panels were magnified in the right panels. Scale bars, 50 μm. (B) Statistic analysis shows that Pin1 expression is significantly different in primary and recurrent tumors. P = 0.004. (C,D) Pin1 expression correlates with ERα expression in recurrent breast cancer tissues. Representative image (A) showed high and low levels of Pin1 and ERα. Pearson correlation of Pin1 and ERα was analyzed in panel (B). Scale bars, 100 μm.
To explore the effect of Pin1 on regulating ERα, and more importantly to test whether ATRA was effective in inhibiting Pin1’s function on ERα, we first examined whether overexpressing Pin1 affected ERα protein level. As expected, estradiol (E2) could induce down-regulation of ERα protein (Figures 3A,B and Supplementary Figure S6), which was due to ligand-dependent degradation (Wijayaratne and McDonnell, 2001; Nonclercq et al., 2004). We found that not only enforced Pin1 expression (Flag-Pin1) rescued the ERα expression, but the Pin1 inhibitor ATRA reversed this effect in both MCF-7 and MCF-7R cells (Figures 3A,B and Supplementary Figures S1A,B), suggesting that ATRA specifically inhibiting Pin1 from protecting the degradation of ERα.
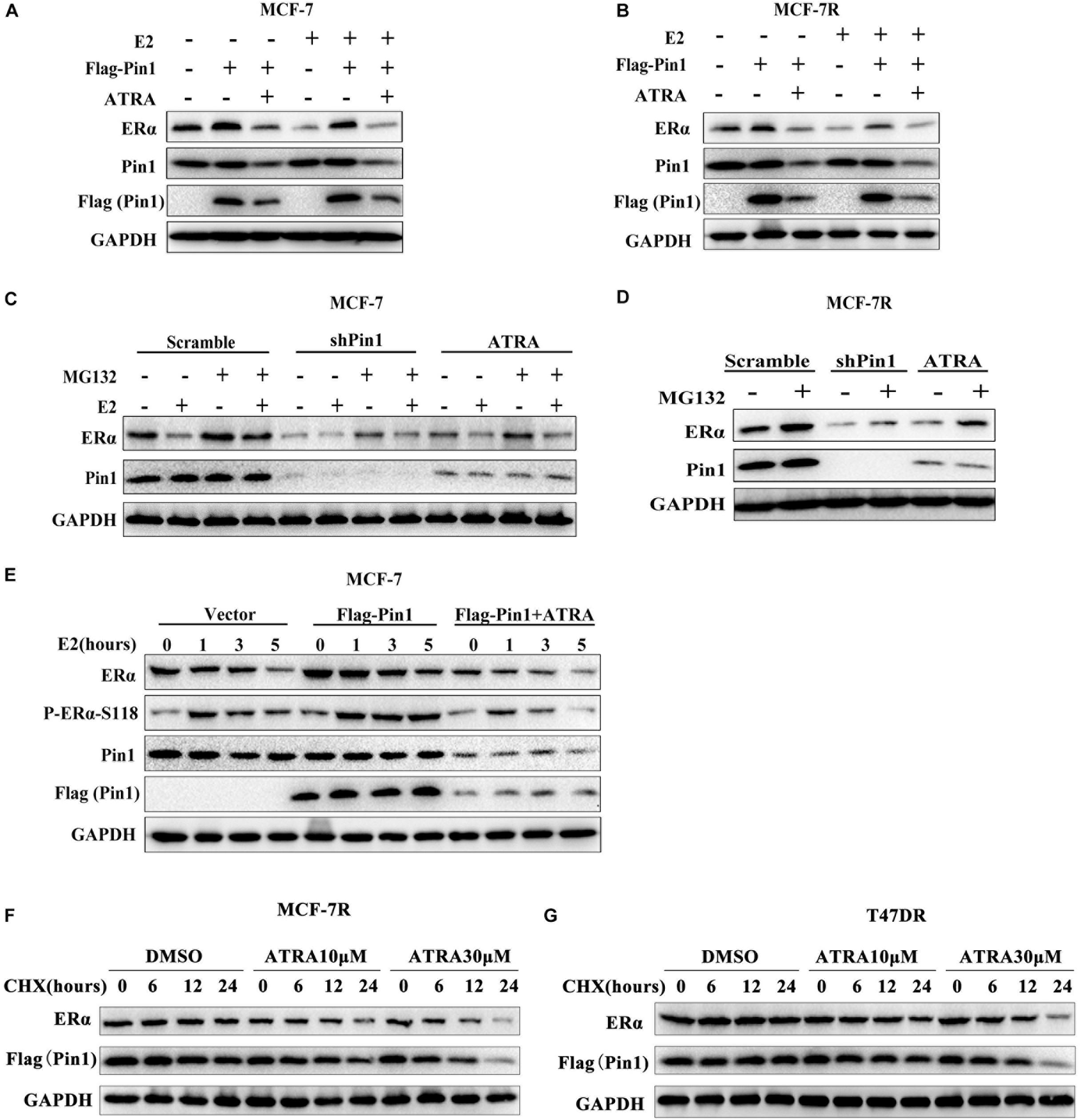
Figure 3. ATRA promotes proteasome-mediated degradation of ERα by blocking Pin1. (A,B) Ectopic expression of Pin1 up-regulates ERα, but ATRA abrogates the effect. MCF-7 (A) and MCF-7R (B) cells transfected with Flag-Pin1 or empty vector were pretreated with 10 μM ATRA for 72 h, followed by 10nM E2 or EtOH treatment for 5 h. (C,D) Pin1 knockdown or ATRA treatment promotes ERα degradation. MCF-7 (C) or MCF-7R (D) were treated with 10 μM MG132 for 4 h before harvesting. (E) Overexpression of Pin1 stabilizes pS118-ERα, but ATRA abrogates the effect. MCF-7 cells transfected with Flag-Pin1 or empty vector were pretreated with 10 μM ATRA for 72 h, followed by 10nM E2 treatment for 0, 1, 3, and 5 h. (F,G) ATRA promotes Pin1 and ERα degradation in MCF-7R and T47DR cells. Cells were pretreated with ATRA and treated with CHX for indicated time course.
Next, to confirm the effect of ATRA on ERα protein degradation, MCF-7 and MCF-7R cells stably knocking down Pin1 with shRNA were treated with or without proteasome inhibitor MG132. Contrary to overexpression experiments, Pin1 knockdown promoted E2-induced ERα degradation (Figures 3C,D and Supplementary Figures S1C,D). Notably, ATRA had the same effects as shPin1 both in MCF-7 and MCF-7R cells (Figures 3C,D and Supplementary Figures S1C,D). One of the vital regulatory element governing ERα protein turnover is Ser118 phosphorylation of the N-terminus, which is phosphorylated by ERK1/2 as well as other kinases, and regulated by Pin1 (Rajbhandari et al., 2012, 2014, 2015). Therefore we speculated that ATRA promoted ERα protein turnover through ERα-pS118. Indeed, overexpressing Pin1 increased the phosphorylation of S118 as well as total ERα level, suggesting that Pin1 prevented the turnover of pERα, whereas ATRA reversed Pin1’s effect (Figure 3E and Supplementary Figures S1E,F).
To directly examine whether ATRA could promote the degradation of Pin1 and ERα in tamoxifen-resistant cells, we treated MCF-7R and T47DR cells with ATRA, followed by cycloheximide (CHX) and detected the protein levels at different time points. Our data showed that ATRA promoted the degradation of both Pin1 and ERα in TAMR breast cancer cells in a dose dependent manner (Figures 3F,G and Supplementary Figures S1G–J). In addition, we treated MCF-7R and T47DR cells with increasing doses of ATRA for different length of time, and found that both Pin1 and ERα protein levels indeed reduced (Supplementary Figures S2A,B, S9). Together, these data demonstrate that overexpressing Pin1 in breast cancer cells protects the ERα protein from degradation. ATRA blocks the up-regulated Pin1 in tamoxifen-resistant cells, thereby promoting the degradation of remaining ERα in tamoxifen-resistant cells, which suggests that ATRA may be able to overcome TAMR by eradicate ERα.
Several kinase pathways have been reported to involve in the growth of TAMR breast cancer cells, including AKT and ERK1/2 (Svensson et al., 2005; Garcia-Becerra et al., 2012). AKT phosphorylates ERα on S167 (Sun et al., 2001), and ERK1/2 phosphorylates ERα on S104/S106, S167, and S118 (Ali et al., 1993; Arnold et al., 1995; Endoh et al., 1999; Sun et al., 2001; Sheeler et al., 2003). Notably, phosphorylation of S118 and S167 induces estrogen-independent activation of ERα (Garcia-Becerra et al., 2012). Moreover, AKT and ERK1/2 activity are also regulated by Pin1 (Liao et al., 2009; Luo et al., 2015). Thus, we explored the effects of ATRA in inhibiting these pathways in MCF-7R and T47DR cells. ATRA treatment didn’t alter the total expression of AKT or ERK1/2, but reduced the level of phosphorylated AKT, MEK1/2, ERK1/2 and Raf (Figures 4A,B). In consistence with decreased activity of these pathways, phosphorylation of ERα on S167 and S118 were also inhibited, resembling the effect of Pin1 knockdown (Figures 4A,B and Supplementary Figure S7).
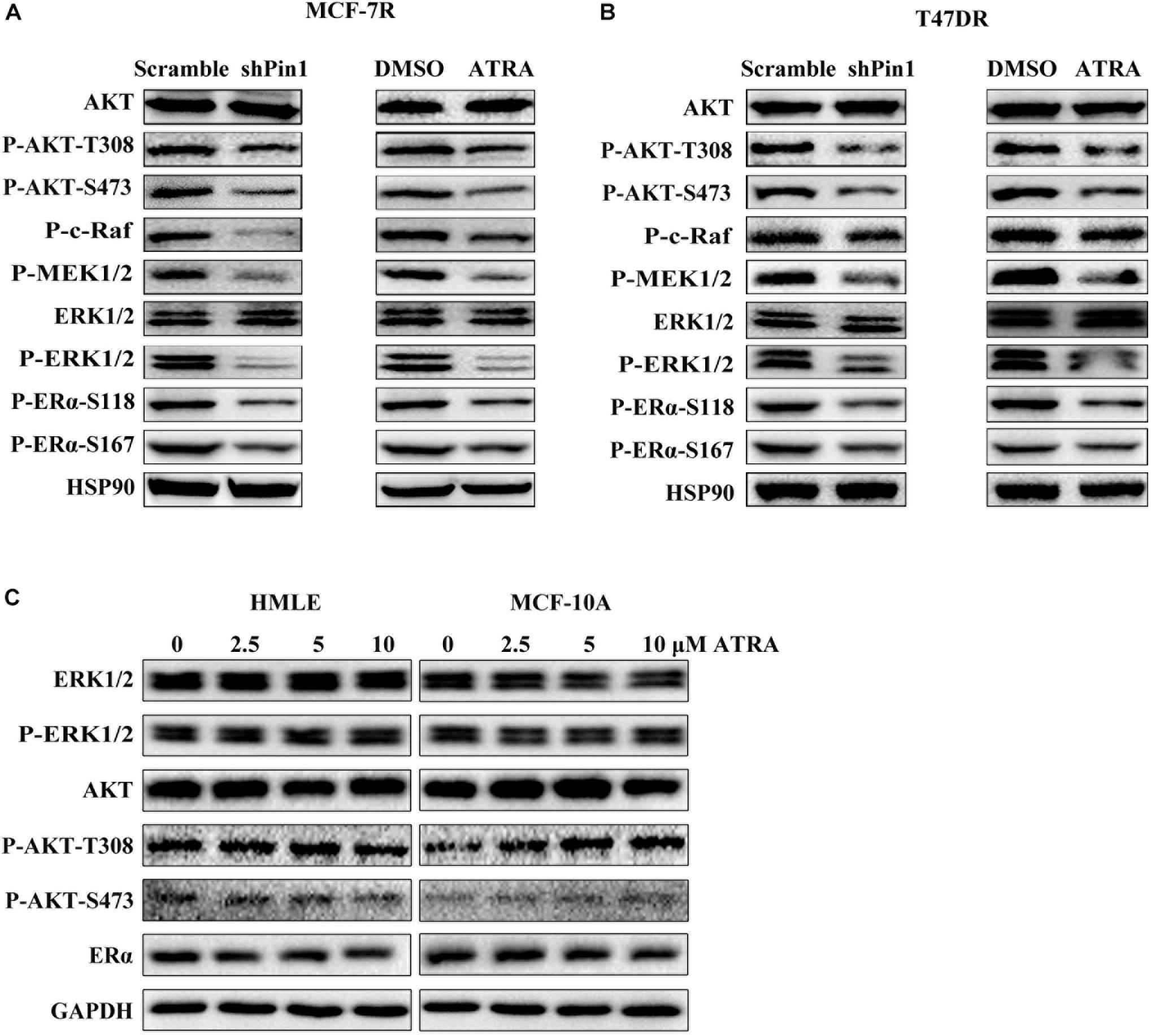
Figure 4. ATRA and Pin1 knockdown inhibits the activation of AKT and ERK1/2 signal pathways. (A) Pin1 knockdown or ATRA treatment inhibits the activation of AKT and ERK1/2 signal pathways simultaneously in MCF-7R cells. Cells were infected with lentivirus expressing scramble or Pin1 shRNA, or treated with 10 μM ATRA for 72 h. (B) Pin1 knockdown or ATRA treatment decreases the activation of AKT and ERK1/2 signal pathways simultaneously in T47DR cells. (C) ATRA treatment doesn’t affect ERα, AKT, and ERK1/2 in HMLE and MCF-10A cells.
To assess the effect of ATRA on ER signaling in normal cells, we treated immortalized mammary epithelial cells MCF-10A and HMLE with different doses of ATRA. These two cell lines expressed very low level of Pin1, comparing to breast cancer cell lines (Wei et al., 2015). We found that ATRA almost had no effect on the protein level of ERα, or P-ERK1/2 and P-AKT (Figure 4C). Hence, ATRA had the unique potential to simultaneously block multiple signal pathways in TAMR breast cancer cells.
In addition, RARα, another ATRA target, has been indicated to play a role in tamoxifen resistance of breast cancer (Johansson et al., 2013). Using siRNAs, we knocked down either Pin1 or RARα in MCF-7R cells with or without ATRA treatment (Supplementary Figures S3A,B). The total and phosphorylated levels of ERα only decreased in Pin1-silencing, but not RARα-silencing cells (Supplementary Figures S3C, S10). As ATRA can still target other proteins to regulate ERα, our data indicate that ATRA may mainly act on Pin1 to regulate ERα.
To determine whether ATRA affected the transcriptional function of ERα in tamoxifen-resistant cells, we first examined ERα subcellular expression by immunofluorescence. Parental or resistant MCF-7 and T47D cells were treated with 10 μM ATRA for 72 h. The nuclear staining of ERα was dramatically reduced by ATRA treatment in all cell lines (Figures 5A–D), indicating a decreased transcriptional activity of ERα. Next, we detected the transcription of three known ERα regulatory genes, including PGR, GREB1, and c-Myc (Lee and Gorski, 1996; Bosch et al., 2015; Wu et al., 2018). The mRNA levels of these three genes were decreased after ATRA treatment (Figures 5E–H). These data suggest that ATRA suppresses ERα transcriptional function in vitro.
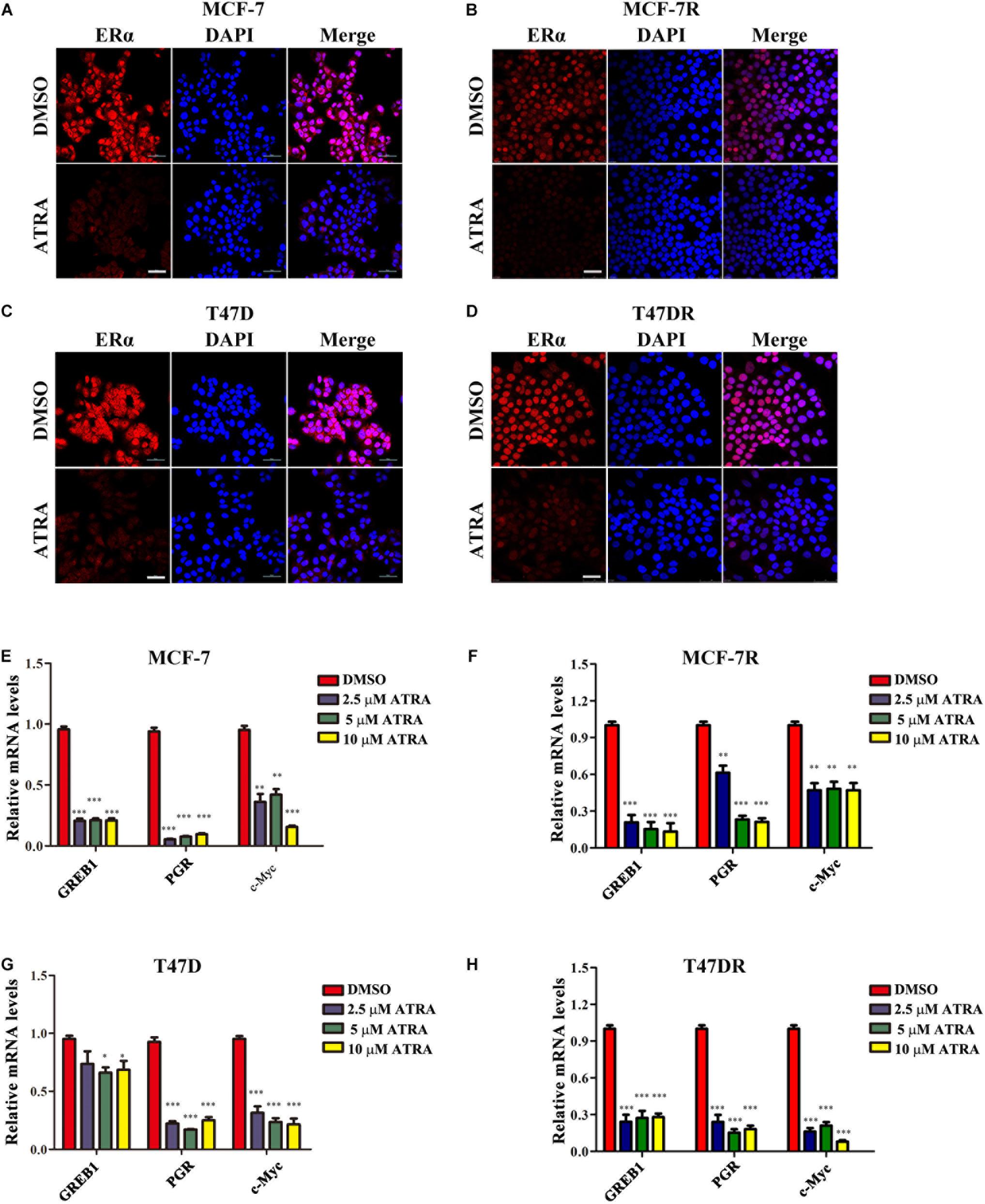
Figure 5. ATRA suppresses nuclear expression and transactivation of ERα. (A,B) ATRA decreases the level of nuclear ERα in MCF-7 and MCF-7R. Cells were treated with 10 μM ATRA for 72 h before immunofluorescence staining. Scale bars, 50 μm. (C,D) ATRA decreases the level of nuclear ERα in T47D and T47DR. Cells were treated with10 μM ATRA for 72 h before immunofluorescence staining. Scale bars, 50 μm. (E–H) ATRA suppresses the transcription of ERα target genes GREB1, PGR, and c-Myc. Cells were treated with ATRA (2.5, 5.0, and10 μM) for 48 h. Expression of ERα downstream genes were detected by qRT-PCR, and normalized to β-ACTIN expression in DMSO treated cells. Error bars denote the SD of three biological replicates, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Although our data demonstrated that ATRA targeted Pin1 to promote ERα protein degradation, decrease ERα transcriptional activity, and inhibit AKT and ERK1/2 pathway, the therapeutic potential of ATRA in treating tamoxifen-resistant breast cancer was still not clear. We thus evaluated the effects of ATRA on cell viability and foci formation of parental and TAMR cells. As expected, tamoxifen treatment reduced the growth of parental cells, but not the TAMR cells, whereas ATRA suppressed the proliferation of both parental and TAMR cells (Figures 6A–D). Moreover, ATRA potentiated tamoxifen therapeutic effect in both parental and TAMR cells (Figures 6A–D). In the colony formation experiments, ATRA showed similar effects as in the proliferation assay (Figures 6E–G).
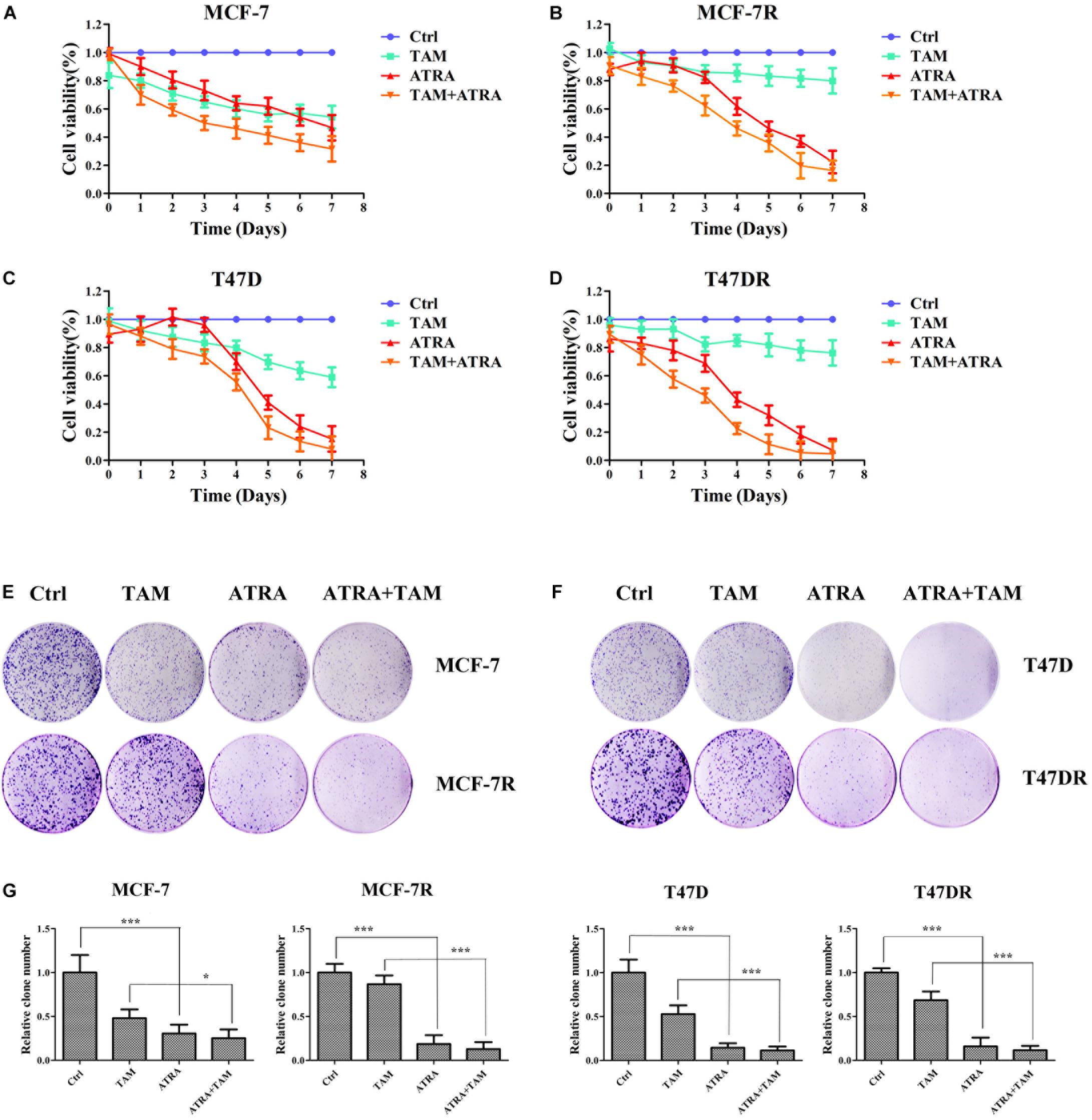
Figure 6. ATRA inhibits the cell growth of tamoxifen resistant breast cancer cells. (A–D) The inhibitory effect of ATRA and tamoxifen on cell viability of parental and tamoxifen resistant breast cancer cells. Viability of drug treated cells was normalized to control untreated cells (Ctrl). (E,F) The inhibitory effect of ATRA and tamoxifen on foci formation of parental and tamoxifen resistant breast cancer cells. (G) Quantification of foci formation by ImageJ software. ∗P < 0.05 and ∗∗∗P < 0.001, as determined by Student’s t-test. Bar graphs are means ± SD by three independent experiments.
We have shown that ATRA-induced Pin1 degradation reduces the protein expression of ERα in tamoxifen-resistant breast cancer cells. To confirm that ERα contributes to tamoxifen resistance in our TAMR cell model, we used siRNAs to knock down ERα in MCF-7R and T47DR. ERα siRNAs dramatically suppressed the proliferation of these cells upon tamoxifen treatment (Supplementary Figures S4A,B, S10), suggesting that ERα indeed contributed to TAMR in these cells.
To investigate the effects of ATRA on Pin1-low cells, we treated MCF-10A and HMLE with different doses of ATRA. ATRA exhibited very limited inhibitory effects on cell viability of these epithelial cells (Figures 7A,B), likely because ATRA selectively targets active Pin1 in cancer cells, but not in normal cells with low Pin1 levels (Wei et al., 2015). Thus these results demonstrated that ATRA inhibits cell growth of TAMR breast cancer, with little effects on normal cells.
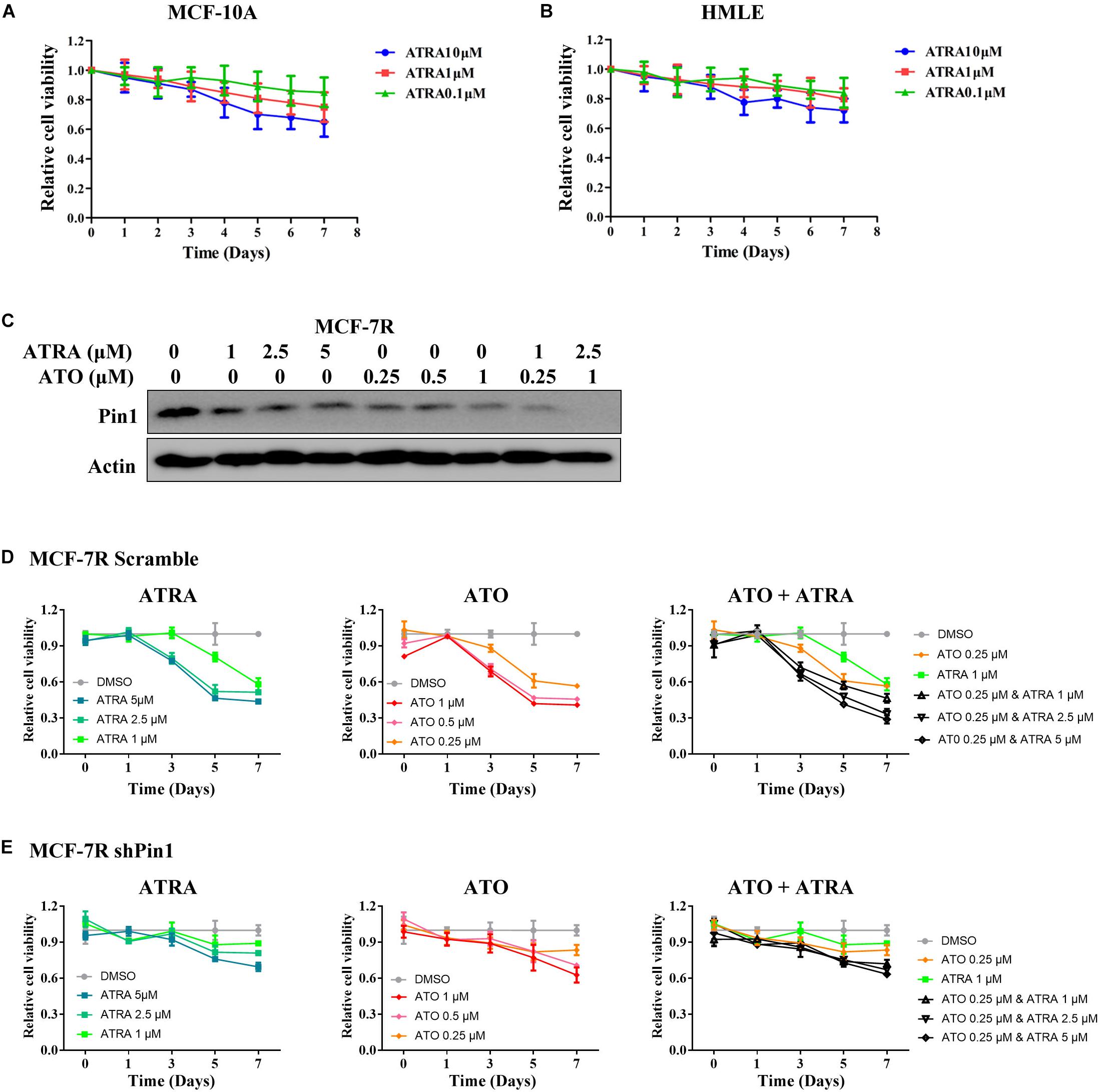
Figure 7. ATRA and other Pin1 inhibitor has limited inhibitory effect on Pin1-low expressing cells. (A,B) ATRA shows little inhibitory effect on the viability of MCF-10A and HMLE cells. Cells were treated with increasing doses of ATRA for 7 days. (C) The effect of ATRA and ATO on reducing the level of Pin1 protein in MCF-7R cells. The concentration of ATRA and ATO were added to the culture medium as indicated for 48 h. (D,E) ATRA and ATO show much less inhibitory effect on the proliferation of shPin1 cells than that of control MCF-7R cells. MCF-7R cells stably expressing scramble or Pin1 shRNA were treated with ATRA and ATO for 7 days.
We also treated MCF-7R and T47DR cells that had knocked down Pin1 with ATRA and ATO, a newly identified Pin1 inhibitor (Kozono et al., 2018). Both ATRA and ATO showed much less inhibitory effect on the proliferation of shPin1 cells than that of control MCF-7R cells (Figures 7C–E and Supplementary Figure S8). Notably, although either ATRA or ATO could inhibit the proliferation of TAMR cells, the combination of ATRA + ATO effectively suppressed the cell viability in low dose (Figures 7D,E).
Given the remarkable effects of ATRA on inhibiting ERα, AKT, and ERK1/2, as well as cell proliferation in tamoxifen-resistant breast cancer in vitro, we next asked whether ATRA had therapeutic effect against TAMR breast tumors in vivo. We established MCF-7R xenografts and implanted 21-day slow-releasing ATRA tablets in nude mice. Tamoxifen showed no therapeutic effect on TAMR xenografts, whereas ATRA remarkably inhibited the growth of TAMR breast cancer cells in vivo (Figures 8A–C). In addition, ATRA significantly suppressed Pin1, ERα, as well as the phosphorylation of AKT and ERK1/2 in the xenografts (Figure 8D and Supplementary Figure S8). Therefore, ATRA is effective in overcoming tamoxifen resistance in vivo.
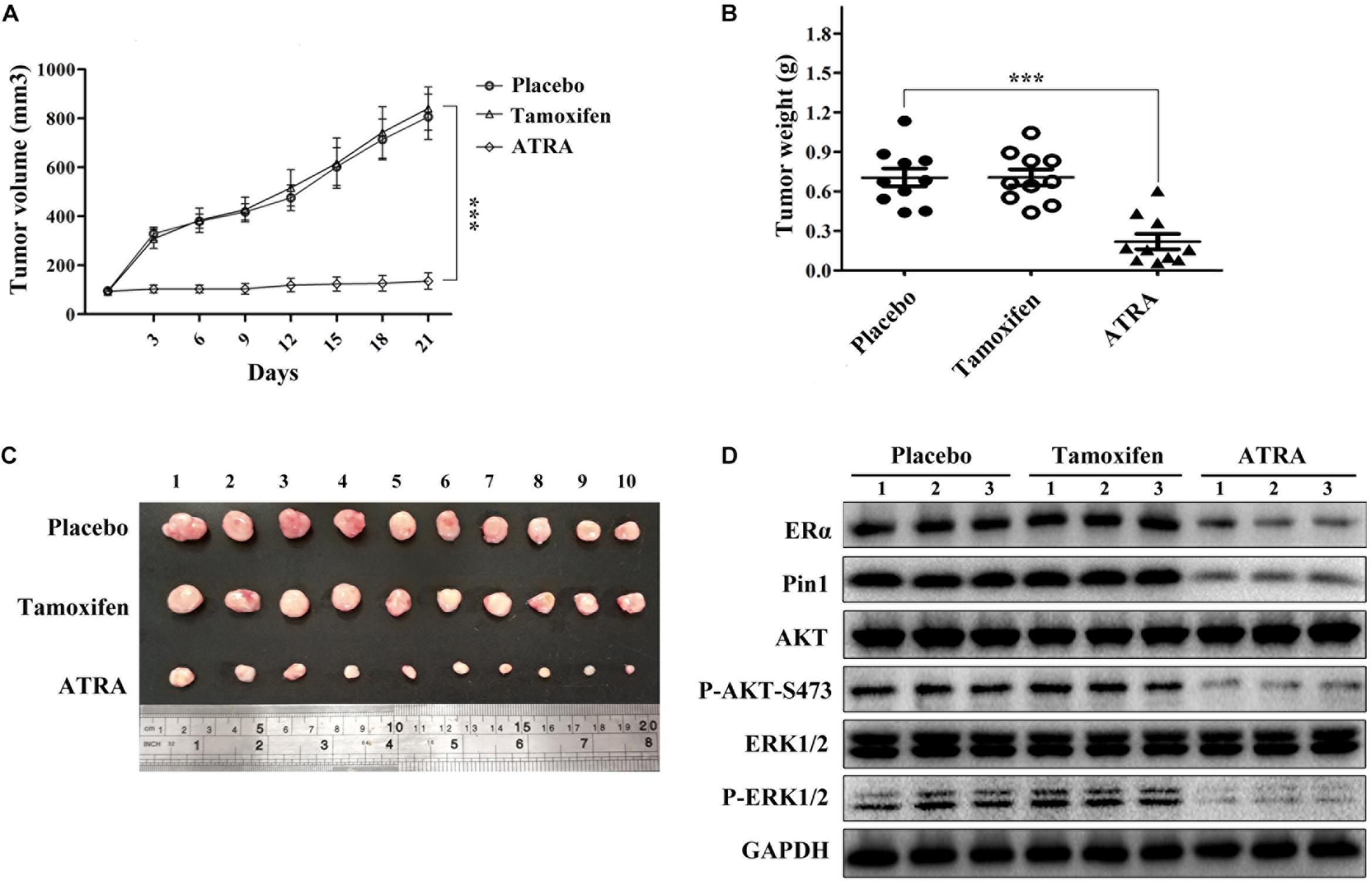
Figure 8. ATRA suppresses the growth of TAMR breast cancer in vivo. (A) Growth curve of MCF-7R xenografts in nude mice. Tumor volumes were measured every 3 days. Error bars represent SD of ten mice, ∗∗∗P < 0.001. (B) Slow-releasing ATRA pellet inhibits the tumor growth of MCF-7R xenografts in nude mice. The tumor weight represented the mean ± SD of each group, ∗∗∗P < 0.001. (C) MCF-7R xenografts were collected 3 weeks after treatment. (D) Representative immunoblots show Pin1, ERα, P-AKT, and P-ERK1/2 levels in MCF-7R xenografts.
Tamoxifen resistance is one of the major hurdles in treating breast cancer. A large body of evidence suggests that modulation of ERα pathway and activation of pro-survival pathways are important factors of tamoxifen resistance (Cui et al., 2015; Ferraiuolo et al., 2017; Marsh et al., 2017; Zhang et al., 2017). Our study demonstrated that Pin1 was up-regulated in tamoxifen-resistant breast cancer cells and relapsed breast cancer tissues. ATRA-induced Pin1 degradation decreased the protein stability and transcription activity of ERα, as well as reduced the phosphorylation of pro-survival kinases AKT and ERK1/2 in tamoxifen-resistant breast cancer cells. Moreover, targeting Pin1 by ATRA inhibited cell growth in vitro, and exhibited anti-tumor effects in vivo against tamoxifen-resistant breast cancer. Our data suggest that ATRA is a potent drug in treating tamoxifen-resistant breast cancer via suppressing multifactorial mechanisms of tamoxifen resistance.
Compelling evidence has demonstrated that decreased ERα expression and function contributes to intrinsic and acquired tamoxifen resistance (Cui et al., 2015; Ferraiuolo et al., 2017; Marsh et al., 2017; Zhang et al., 2017). Various clinical and experimental models suggest that tumor cells with acquired resistance to tamoxifen express low level of ERα (Stone et al., 2013; Lu et al., 2016). In the presence of tamoxifen, the resistant cells can still activate ERα, but through a ligand-independent way (He et al., 2018), or rely on non-ERα growth-promoting pathways for survival (Hur et al., 2004; Cannings et al., 2007; Mohseni et al., 2014). Thus the low level of ERα is one of the key resources of growth signal that are available for the resistant cells to utilize. Previous study showed that Pin1 inhibited phosphorylation-dependent ubiquitination and degradation of ERα in breast cancer cells (Rajbhandari et al., 2014). Here we found that Pin1 was up-regulated in tamoxifen-resistant breast cancer cells. This up-regulated Pin1 prevented ERα from degradation, which substantially enhanced the ERα level in tamoxifen-resistant cells. Although ERα level was low in these resistant cells, it would be even lower if Pin1 was not up-regulated. In our clinical samples, ERα expression was high in more than 60% of recurrent breast cancer tissues. This may be because Pin1 is frequently highly expressed in recurrent tumors, therefore preventing ERα from degradation, which substantially enhances the ERα level in relapsed tumors. Notably, this increased ERα, just as the low level of ERα in the resistant cells, is very likely activated via ligand independent way. This is supported by the evidence that phosphorylation of key serine residues of ERα, in particular serine 118 and 167, promotes re-activation of ERα in a ligand-independent manner (Garcia-Becerra et al., 2012). Pin1 has been reported to bind specifically to pS118 ERα to isomerize the serine118-proline119 bond (Rajbhandari et al., 2012). Therefore, Pin1 overexpression promotes the growth of tamoxifen-resistant breast cancer cells by up-regulating the ligand-independent ERα activity.
In addition to the effects on ERα stabilization, isomerization of phosphorylated ERα by Pin1 directly increases endogenous ERα DNA binding activity (Rajbhandari et al., 2015). Our study showed that inhibiting Pin1 by ATRA suppressed nuclear ERα expression and the transcription of ERα target genes. Moreover, previous data suggest that besides affecting ERα, Pin1 may promote tamoxifen resistance of breast cancer by activating growth-promoting pathways (Khanal et al., 2012), down-regulating SMRT (Stanya et al., 2008) and cyclin dependent kinase (Khanal et al., 2012), facilitating tumor angiogenesis (Kim et al., 2009b, 2012) and epithelial–mesenchymal transition (Kim et al., 2009a). However, few studies assess the potential of Pin1 inhibitor in treating TAMR breast cancer in vitro and in vivo. Our study showed that ATRA decreased ERα level both in vitro and in vivo. Moreover, ATRA down-regulated phosphorylation of ERα at S118 and two important pro-survival kinases ERK1/2 and AKT. Thus, ATRA inhibits Pin1 to overcome tamoxifen resistance in breast cancer cells at least at three levels: (1) promotes the degradation of ligand independent ERα, (2) suppresses the transactivation of ERα, (3) inhibits alternative growth pathways. Indeed, our results showed that ATRA exhibited potent anti-tumor activity against tamoxifen-resistant breast cancer in vitro and in vivo.
All-Trans Retinoic Acid has been used to treat APL for a long period of time. Recently Wei et al. (2015) has discovered that ATRA is a Pin1 inhibitor which binds to Pin1’s active site and accelerated its degradation. These findings make it possible to expand the application of ATRA to treat more types of cancer, especially solid tumors, because Pin1 is overexpressed in a wide range of human cancers and regulates multiple cancer-driving pathways (Lu and Hunter, 2014; Zhou and Lu, 2016). Besides Wei et al. demonstrated that ATRA-induced Pin1 ablation inhibits triple-negative breast cancer cell growth (Wei et al., 2015), Liao et al. also reported the anti-tumor effect of ATRA in hepatocellular carcinoma (HCC) in vitro and in vivo (Liao et al., 2017; Yang et al., 2018). New Pin1 inhibitors have also been discovered to suppress the growth of cancer cells (Campaner et al., 2017; Kozono et al., 2018), even the tumor-initiating cells, as Pin1 promotes the self-renewal of these stem-like cancer cells (Luo et al., 2014; Rustighi et al., 2014, 2017). Our data showed that ATRA suppressed the cell proliferation of tamoxifen-resistant breast cancer cells, and effectively reduced the tumor growth of tamoxifen-resistant xenografts by promoting ERα degradation, decreasing ERα transactivation, and inhibits the activation of ERK1/2 and AKT. Given that multiple survival pathways and factors contribute to tamoxifen resistance, blocking a single pathway may be ineffective to overcome the resistance. Therefore, ATRA may have the advantage of suppressing multifactorial mechanisms of tamoxifen resistance simultaneously.
Currently there are still obstacles of using ATRA to treat solid tumors in human. Regular ATRA has a half-life of only 45 min in humans. Although slow-releasing pellets can be implanted subcutaneously in mice, the formulation of ATRA pellet is different from that used for oral administration or intravenous injection in human therapies, and can’t be applied to human yet. Novel controlled releasing formulation of ATRA for effective cancer therapy are being developed actively (Westervelt et al., 2002; Tsimberidou et al., 2006; Yang et al., 2018). In addition, ATRA concentration is high in treating solid tumors. Similar to the previous report that the combination of ATRA + ATO inhibited tumor-initiating cells (Kozono et al., 2018), we found that this combination could reduce ATRA concentration and effectively inhibited the growth of TAMR cells. Thus, studies are ongoing to increase the efficacy of ATRA by improving its formulation, or using ATRA as part of combination therapies.
In summary, our data have shown for the first time that targeting Pin1 by ATRA effectively inhibits the growth of tamoxifen resistant breast cancer. This new approach represents a potential therapeutic strategy for intrinsic tamoxifen-resistant patients and relapsed ERα-positive breast cancer patients. Our findings shed new light on the molecular mechanism of ATRA in overcoming tamoxifen resistance and warrant future preclinical and clinical studies of ATRA in treating the tamoxifen resistant breast cancers.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Internal review and ethics boards of Sun Yat-sen Memorial Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Animal Care and Use Committee of Sun Yat-sen University.
M-LL and SH: conception and design. YL and YZ: development of methodology. YC, Z-ML, N-NL, and DL: acquisition of data. YC and SH: analysis and interpretation of data. M-LL and YY: writing, review, and/or revision of the manuscript. XZ and KL: technical and material support. M-LL and YY: study supervision.
This work was supported by grants from the National Natural Science Foundation of China (81572890, 81772837, and 81872370), Guangdong Science and Technology Department (2017B030314026), Guangdong Natural Science Foundation (2018A0303130322), the Science and Technology Foundation of the Guangdong Province (2014A050503029 and 2019A050510016), Sun Yat-sen Initiative Program for Scientific Research (YXQH201701), and Elite Young Scholar Program of Sun Yat-sen Memorial Hospital (Y201701).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2019.00322/full#supplementary-material
Ali, S., Metzger, D., Bornert, J. M., and Chambon, P. (1993). Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 12, 1153–1160. doi: 10.1002/j.1460-2075.1993.tb05756.x
Anbalagan, M., and Rowan, B. G. (2015). Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell Endocrinol. 418(Pt 3), 264–272. doi: 10.1016/j.mce.2015.01.016
Arnold, S. F., Obourn, J. D., Jaffe, H., and Notides, A. C. (1995). Phosphorylation of the human estrogen receptor by mitogen-activated protein kinase and casein kinase II: consequence on DNA binding. J. Steroid Biochem. Mol. Biol. 55, 163–172. doi: 10.1016/0960-0760(95)00177-2
Bosch, A., Li, Z., Bergamaschi, A., Ellis, H., Toska, E., Prat, A., et al. (2015). PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 7:283ra51. doi: 10.1126/scitranslmed.aaa4442
Campaner, E., Rustighi, A., Zannini, A., Cristiani, A., Piazza, S., Ciani, Y., et al. (2017). A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 8:15772. doi: 10.1038/ncomms15772
Cannings, E., Kirkegaard, T., Tovey, S. M., Dunne, B., Cooke, T. G., and Bartlett, J. M. (2007). Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res. Treat. 102, 173–179. doi: 10.1007/s10549-006-9323-8
Chen, W., Zheng, R., Baade, P. D., Zhang, S., Zeng, H., Bray, F., et al. (2016). Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132. doi: 10.3322/caac.21338
Chu, J., Zhu, Y., Liu, Y., Sun, L., Lv, X., Wu, Y., et al. (2015). E2F7 overexpression leads to tamoxifen resistance in breast cancer cells by competing with E2F1 at miR-15a/16 promoter. Oncotarget 6, 31944–31957. doi: 10.18632/oncotarget.5128
Cui, J., Yang, Y., Li, H., Leng, Y., Qian, K., Huang, Q., et al. (2015). MiR-873 regulates ERalpha transcriptional activity and tamoxifen resistance via targeting CDK3 in breast cancer cells. Oncogene 34, 3895–3907. doi: 10.1038/onc.2014.430
de The, H., and Chen, Z. (2010). Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783. doi: 10.1038/nrc2943
Deroo, B. J., and Korach, K. S. (2006). Estrogen receptors and human disease. J. Clin. Invest. 116, 561–570. doi: 10.1172/jci27987
Endoh, H., Maruyama, K., Masuhiro, Y., Kobayashi, Y., Goto, M., Tai, H., et al. (1999). Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Mol. Cell Biol. 19, 5363–5372. doi: 10.1128/mcb.19.8.5363
Ferraiuolo, R. M., Tubman, J., Sinha, I., Hamm, C., and Porter, L. A. (2017). The cyclin-like protein, SPY1, regulates the ERalpha and ERK1/2 pathways promoting tamoxifen resistance. Oncotarget 8, 23337–23352. doi: 10.18632/oncotarget.15578
Garcia-Becerra, R., Santos, N., Diaz, L., and Camacho, J. (2012). Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol. Sci. 14, 108–145. doi: 10.3390/ijms14010108
Girault, I., Bieche, I., and Lidereau, R. (2006). Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 54, 342–351. doi: 10.1016/j.maturitas.2006.06.003
He, H., Sinha, I., Fan, R., Haldosen, L. A., Yan, F., Zhao, C., et al. (2018). c-Jun/AP-1 overexpression reprograms ERalpha signaling related to tamoxifen response in ERalpha-positive breast cancer. Oncogene 37, 2586–2600. doi: 10.1038/s41388-018-0165-8
Herman, M. E., and Katzenellenbogen, B. S. (1996). Response-specific antiestrogen resistance in a newly characterized MCF-7 human breast cancer cell line resulting from long-term exposure to trans-hydroxytamoxifen. J. Steroid Biochem. Mol. Biol. 59, 121–134. doi: 10.1016/s0960-0760(96)00114-8
Huang, M. E., Ye, Y. C., Chen, S. R., Chai, J. R., Lu, J. X., Zhoa, L., et al. (1988). Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72, 567–572. doi: 10.1182/blood.v72.2.567.567
Hur, J., Chesnes, J., Coser, K. R., Lee, R. S., Geck, P., Isselbacher, K. J., et al. (2004). The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc. Natl. Acad. Sci. U.S.A. 101, 2351–2356. doi: 10.1073/pnas.0307337101
Johansson, H. J., Sanchez, B. C., Mundt, F., Forshed, J., Kovacs, A., Panizza, E., et al. (2013). Retinoic acid receptor alpha is associated with tamoxifen resistance in breast cancer. Nat. Commun. 4:2175. doi: 10.1038/ncomms3175
Johnston, S. R. (2010). New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res. 16, 1979–1987. doi: 10.1158/1078-0432.CCR-09-1823
Khanal, P., Yun, H. J., Lim, S. C., Ahn, S. G., Yoon, H. E., Kang, K. W., et al. (2012). Proyl isomerase Pin1 facilitates ubiquitin-mediated degradation of cyclin-dependent kinase 10 to induce tamoxifen resistance in breast cancer cells. Oncogene 31, 3845–3856. doi: 10.1038/onc.2011.548
Kim, J. A., Kim, M. R., Kim, O., Phuong, N. T., Yun, J., Oh, W. K., et al. (2012). Amurensin G inhibits angiogenesis and tumor growth of tamoxifen-resistant breast cancer via Pin1 inhibition. Food Chem. Toxicol. 50, 3625–3634. doi: 10.1016/j.fct.2012.07.027
Kim, M. R., Choi, H. K., Cho, K. B., Kim, H. S., and Kang, K. W. (2009a). Involvement of Pin1 induction in epithelial-mesenchymal transition of tamoxifen-resistant breast cancer cells. Cancer Sci. 100, 1834–1841. doi: 10.1111/j.1349-7006.2009.01260.x
Kim, M. R., Choi, H. S., Yang, J. W., Park, B. C., Kim, J. A., and Kang, K. W. (2009b). Enhancement of vascular endothelial growth factor-mediated angiogenesis in tamoxifen-resistant breast cancer cells: role of Pin1 overexpression. Mol. Cancer Ther. 8, 2163–2171. doi: 10.1158/1535-7163.MCT-08-1061
Knowlden, J. M., Hutcheson, I. R., Jones, H. E., Madden, T., Gee, J. M., Harper, M. E., et al. (2003). Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044. doi: 10.1210/en.2002-220620
Kozono, S., Lin, Y. M., Seo, H. S., Pinch, B., Lian, X., Qiu, C., et al. (2018). Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun. 9:3069. doi: 10.1038/s41467-018-05402-2
Lee, Y. J., and Gorski, J. (1996). Estrogen-induced transcription of the progesterone receptor gene does not parallel estrogen receptor occupancy. Proc. Natl. Acad. Sci. U.S.A. 93, 15180–15184. doi: 10.1073/pnas.93.26.15180
Liao, X. H., Zhang, A. L., Zheng, M., Li, M. Q., Chen, C. P., Xu, H., et al. (2017). Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways. Sci. Rep. 7:43639. doi: 10.1038/srep43639
Liao, Y., Wei, Y., Zhou, X., Yang, J. Y., Dai, C., Chen, Y. J., et al. (2009). Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 28, 2436–2445. doi: 10.1038/onc.2009.98
Lu, K. P., and Zhou, X. Z. (2007). The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916. doi: 10.1038/nrm2261
Lu, R., Hu, X., Zhou, J., Sun, J., Zhu, A. Z., Xu, X., et al. (2016). COPS5 amplification and overexpression confers tamoxifen-resistance in ERalpha-positive breast cancer by degradation of NCoR. Nat. Commun. 7:12044. doi: 10.1038/ncomms12044
Lu, Z., and Hunter, T. (2014). Prolyl isomerase Pin1 in cancer. Cell Res. 24, 1033–1049. doi: 10.1038/cr.2014.109
Luo, M. L., Gong, C., Chen, C. H., Hu, H., Huang, P., Zheng, M., et al. (2015). The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via Erk signaling activation. Cell Rep. 11, 111–124. doi: 10.1016/j.celrep.2015.03.002
Luo, M. L., Gong, C., Chen, C. H., Lee, D. Y., Hu, H., Huang, P., et al. (2014). Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 74, 3603–3616. doi: 10.1158/0008-5472.CAN-13-2785
Marsh, L. A., Carrera, S., Shandilya, J., Heesom, K. J., Davidson, A. D., Medler, K. F., et al. (2017). BASP1 interacts with oestrogen receptor alpha and modifies the tamoxifen response. Cell Death Dis. 8:e2771. doi: 10.1038/cddis.2017.179
Miller, T. E., Ghoshal, K., Ramaswamy, B., Roy, S., Datta, J., Shapiro, C. L., et al. (2008). MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 283, 29897–29903. doi: 10.1074/jbc.M804612200
Mohseni, M., Cidado, J., Croessmann, S., Cravero, K., Cimino-Mathews, A., Wong, H. Y., et al. (2014). MACROD2 overexpression mediates estrogen independent growth and tamoxifen resistance in breast cancers. Proc. Natl. Acad. Sci. U.S.A. 111, 17606–17611. doi: 10.1073/pnas.1408650111
Namgoong, G. M., Khanal, P., Cho, H. G., Lim, S. C., Oh, Y. K., Kang, B. S., et al. (2010). The prolyl isomerase Pin1 induces LC-3 expression and mediates tamoxifen resistance in breast cancer. J. Biol. Chem. 285, 23829–23841. doi: 10.1074/jbc.M109.092874
Nilsson, S., Koehler, K. F., and Gustafsson, J. A. (2011). Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 10, 778–792. doi: 10.1038/nrd3551
Nonclercq, D., Journe, F., Body, J. J., Leclercq, G., and Laurent, G. (2004). Ligand-independent and agonist-mediated degradation of estrogen receptor-alpha in breast carcinoma cells: evidence for distinct degradative pathways. Mol. Cell Endocrinol. 227, 53–65. doi: 10.1016/j.mce.2004.07.003
Osborne, C. K., and Schiff, R. (2005). Estrogen-receptor biology: continuing progress and therapeutic implications. J. Clin. Oncol. 23, 1616–1622. doi: 10.1200/jco.2005.10.036
Osborne, C. K., and Schiff, R. (2011). Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62, 233–247.
Rajbhandari, P., Finn, G., Solodin, N. M., Singarapu, K. K., Sahu, S. C., Markley, J. L., et al. (2012). Regulation of estrogen receptor alpha N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol. Cell Biol. 32, 445–457. doi: 10.1128/MCB.06073-11
Rajbhandari, P., Ozers, M. S., Solodin, N. M., Warren, C. L., and Alarid, E. T. (2015). Peptidylprolyl isomerase Pin1 directly enhances the DNA binding functions of estrogen receptor alpha. J. Biol. Chem. 290, 13749–13762. doi: 10.1074/jbc.M114.621698
Rajbhandari, P., Schalper, K. A., Solodin, N. M., Ellison-Zelski, S. J., Ping Lu, K., Rimm, D. L., et al. (2014). Pin1 modulates ERalpha levels in breast cancer through inhibition of phosphorylation-dependent ubiquitination and degradation. Oncogene 33, 1438–1447. doi: 10.1038/onc.2013.78
Rustighi, A., Zannini, A., Campaner, E., Ciani, Y., Piazza, S., and Del Sal, G. (2017). PIN1 in breast development and cancer: a clinical perspective. Cell Death Dif. 24, 200–211. doi: 10.1038/cdd.2016.122
Rustighi, A., Zannini, A., Tiberi, L., Sommaggio, R., Piazza, S., Sorrentino, G., et al. (2014). Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med. 6, 99–119. doi: 10.1002/emmm.201302909
Sanz, M. A., and Lo-Coco, F. (2011). Modern approaches to treating acute promyelocytic leukemia. J. Clin. Oncol. 29, 495–503. doi: 10.1200/jco.2010.32.1067
Shao, W., Keeton, E. K., McDonnell, D. P., and Brown, M. (2004). Coactivator AIB1 links estrogen receptor transcriptional activity and stability. Proc. Natl. Acad. Sci. U.S.A. 101, 11599–11604. doi: 10.1073/pnas.0402997101
Sheeler, C. Q., Singleton, D. W., and Khan, S. A. (2003). Mutation of serines 104, 106, and 118 inhibits dimerization of the human estrogen receptor in yeast. Endocr. Res. 29, 237–255. doi: 10.1081/erc-120022321
Stanya, K. J., Liu, Y., Means, A. R., and Kao, H. Y. (2008). Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J. Cell Biol. 183, 49–61. doi: 10.1083/jcb.200806172
Stenoien, D. L., Patel, K., Mancini, M. G., Dutertre, M., Smith, C. L., O’Malley, B. W., et al. (2001). FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat. Cell Biol. 3, 15–23. doi: 10.1038/35050515
Stone, A., Cowley, M. J., Valdes-Mora, F., McCloy, R. A., Sergio, C. M., Gallego-Ortega, D., et al. (2013). BCL-2 hypermethylation is a potential biomarker of sensitivity to antimitotic chemotherapy in endocrine-resistant breast cancer. Mol. Cancer Ther. 12, 1874–1885. doi: 10.1158/1535-7163.MCT-13-0012
Sun, M., Paciga, J. E., Feldman, R. I., Yuan, Z., Coppola, D., Lu, Y. Y., et al. (2001). Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 61, 5985–5991.
Svensson, S., Jirstrom, K., Ryden, L., Roos, G., Emdin, S., Ostrowski, M. C., et al. (2005). ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene 24, 4370–4379. doi: 10.1038/sj.onc.1208626
Tsimberidou, A. M., Tirado-Gomez, M., Andreeff, M., O’Brien, S., Kantarjian, H., Keating, M., et al. (2006). Single-agent liposomal all-trans retinoic acid can cure some patients with untreated acute promyelocytic leukemia: an update of The University of Texas M. D. Anderson Cancer Center series. Leuk. Lymphoma 47, 1062–1068. doi: 10.1080/10428190500463932
Wei, S., Kozono, S., Kats, L., Nechama, M., Li, W., Guarnerio, J., et al. (2015). Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 21, 457–466. doi: 10.1038/nm.3839
Westervelt, P., Pollock, J. L., Oldfather, K. M., Walter, M. J., Ma, M. K., Williams, A., et al. (2002). Adaptive immunity cooperates with liposomal all-trans-retinoic acid (ATRA) to facilitate long-term molecular remissions in mice with acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 99, 9468–9473. doi: 10.1073/pnas.132657799
Wijayaratne, A. L., and McDonnell, D. P. (2001). The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 276, 35684–35692. doi: 10.1074/jbc.m101097200
Wu, Y., Zhang, Z., Cenciarini, M. E., Proietti, C. J., Amasino, M., Hong, T., et al. (2018). Tamoxifen resistance in breast cancer is regulated by the EZH2-ERalpha-GREB1 transcriptional axis. Cancer Res. 78, 671–684. doi: 10.1158/0008-5472.CAN-17-1327
Xiong, R., Zhao, J., Gutgesell, L. M., Wang, Y., Lee, S., Karumudi, B., et al. (2017). Novel Selective Estrogen Receptor Downregulators (SERDs) developed against treatment-resistant breast cancer. J. Med. Chem. 60, 1325–1342. doi: 10.1021/acs.jmedchem.6b01355
Yager, J. D., and Davidson, N. E. (2006). Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 354, 270–282.
Yang, D., Luo, W., Wang, J., Zheng, M., Liao, X. H., Zhang, N., et al. (2018). A novel controlled release formulation of the Pin1 inhibitor ATRA to improve liver cancer therapy by simultaneously blocking multiple cancer pathways. J. Control Release 269, 405–422. doi: 10.1016/j.jconrel.2017.11.031
Zhang, J., Zhou, C., Jiang, H., Liang, L., Shi, W., Zhang, Q., et al. (2017). ZEB1 induces ER-alpha promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis. 8, e2732. doi: 10.1038/cddis.2017.154
Zhang, X., Peng, D., Xi, Y., Yuan, C., Sagum, C. A., Klein, B. J., et al. (2016). G9a-mediated methylation of ERalpha links the PHF20/MOF histone acetyltransferase complex to hormonal gene expression. Nat. Commun. 7:10810. doi: 10.1038/ncomms10810
Keywords: ATRA, Pin1, breast cancer, tamoxifen, ERα
Citation: Huang S, Chen Y, Liang Z-M, Li N-N, Liu Y, Zhu Y, Liao D, Zhou XZ, Lu KP, Yao Y and Luo M-L (2019) Targeting Pin1 by All-Trans Retinoic Acid (ATRA) Overcomes Tamoxifen Resistance in Breast Cancer via Multifactorial Mechanisms. Front. Cell Dev. Biol. 7:322. doi: 10.3389/fcell.2019.00322
Received: 20 September 2019; Accepted: 22 November 2019;
Published: 06 December 2019.
Edited by:
Tae Ho Lee, Fujian Medical University, ChinaReviewed by:
Giannino Del Sal, Consorzio Interuniversitario per le Biotecnologie, ItalyCopyright © 2019 Huang, Chen, Liang, Li, Liu, Zhu, Liao, Zhou, Lu, Yao and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yandan Yao, eWFveWFuZEBtYWlsLnN5c3UuZWR1LmNu; Man-Li Luo, bHVvbWxpQG1haWwuc3lzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.