 Masum M. Mia
Masum M. Mia Manvendra K. Singh
Manvendra K. Singh- 1Program in Cardiovascular and Metabolic Disorders, Duke-NUS Medical School, Singapore, Singapore
- 2National Heart Research Institute Singapore, National Heart Center, Singapore, Singapore
Heart disease continues to be the leading cause of morbidity and mortality worldwide. Cardiac malformation during development could lead to embryonic or postnatal death. However, matured heart tissue has a very limited regenerative capacity. Thus, loss of cardiomyocytes from injury or diseases in adults could lead to heart failure. The Hippo signaling pathway is a newly identified signaling cascade that modulates regenerative response by regulating cardiomyocyte proliferation in the embryonic heart, as well as in postnatal hearts after injury. In this review, we summarize recent findings highlighting the function and regulation of the Hippo signaling pathway in cardiac development and diseases.
Introduction
The Hippo signaling pathway components were first identified and characterized in Drosophila. However, the core components of the Hippo signaling pathway are highly conserved in mammals. The Hippo signaling pathway is a vital pathway that controls the development and homeostasis of many mammalian organs including heart by regulating cell proliferation, apoptosis, cell fate decisions, and stem cell self-renewal (Halder and Johnson, 2011; Zhao et al., 2011; Zhou et al., 2015). The mammalian core components of the Hippo signaling pathway are composed of a serine/threonine kinase cascade, transcriptional coactivators and transcription factors (Figure 1). Upon activation of a kinase cascade, the sterile 20-like protein kinase (Mst1/2) interacts with an adaptor protein Salvador (Salv) and phosphorylates many proteins including Salv, large tumor suppressor (Lats1/2) and Lats1/2-interacting proteins MOB1. This leads to phosphorylation, cytoplasmic retention, and degradation of the transcriptional coactivators, Yes-associated protein (Yap) and transcriptional coactivator with PDZ-binding motif (Taz, also known as Wwtr1). In contrast, inactivation of upstream kinases leads to nuclear translocation of both Yap and Taz, where they further interact with various transcription factors including TEA domain family member (Tead1-4). This results in regulation of transcriptional activity for downstream target genes involved in cell proliferation, survival and differentiation (Halder and Johnson, 2011; Heallen et al., 2011; Zhao et al., 2011; Zhou et al., 2015).
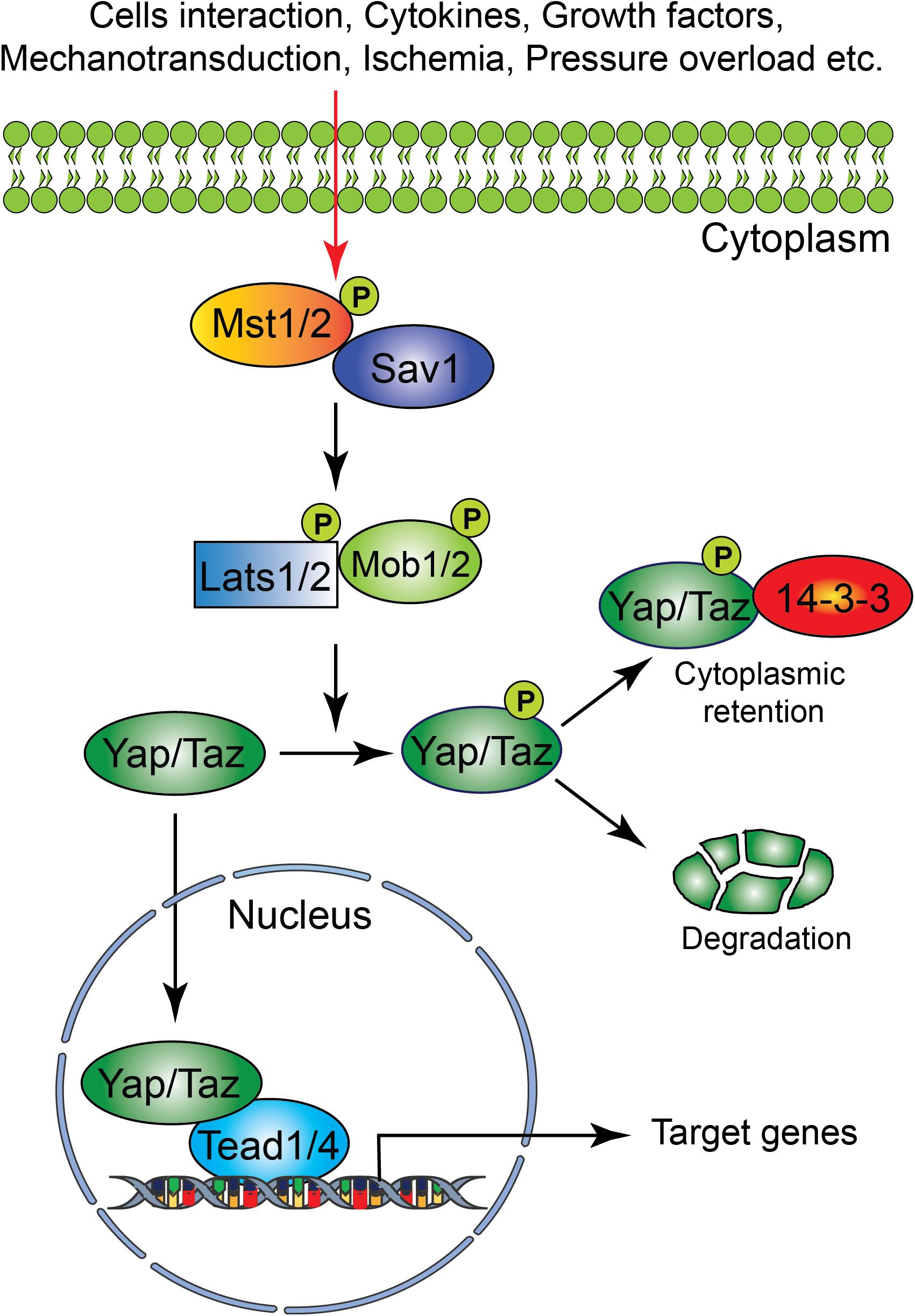
Figure 1. Schematic representation of the core components of the Hippo signaling pathway. Several physiological and pathological signals can activate the Hippo signaling pathway in the heart. The physiological signal includes cell–cell interaction, cytokines, and growth factors, whereas the pathological signal includes oxidative stress (ischemia-reperfusion injury), mechanical stress (pressure overload) and injury (myocardial infarction). The core Hippo signaling pathway consists of serine/threonine kinases, transcriptional coactivators, and transcription factors. Upon activation, the upstream kinases (Mst1/2, Lats1/2, Sav1, and MOB1) promote phosphorylation of downstream mediators Yap and Taz, resulting in their cytoplasmic retention or degradation. In contrast, inactivation of upstream kinases leads to nuclear translocation of Yap and Taz, where they bind to various transcription factors including Tead factors (Tead1-4) and regulate target gene expression.
The Hippo Signaling Pathway in Cardiac Development
The heart is the first functional organ formed during embryonic development. It pumps blood filled with oxygen and nutrients through the blood vessels to the tissues, required for further organogenesis. To maintain its function and carry out its physiologic cardiac output, the heart needs to develop properly. A smaller size heart is not able to eject sufficient blood into the circulation system, resulting in cardiac outflow disruption that leads to cardiomyopathies (diseases of the myocardium, or heart muscle affecting the pumping ability of the heart) (Heallen et al., 2011; Zhao et al., 2011). Deregulation of the Hippo signaling pathway leads to various congenital cardiac abnormalities. Hippo signaling components are expressed in the cells of all three cardiac layers (myocardium, epicardium, and endocardium), and play an important role during cardiac development (Figures 2, 3).
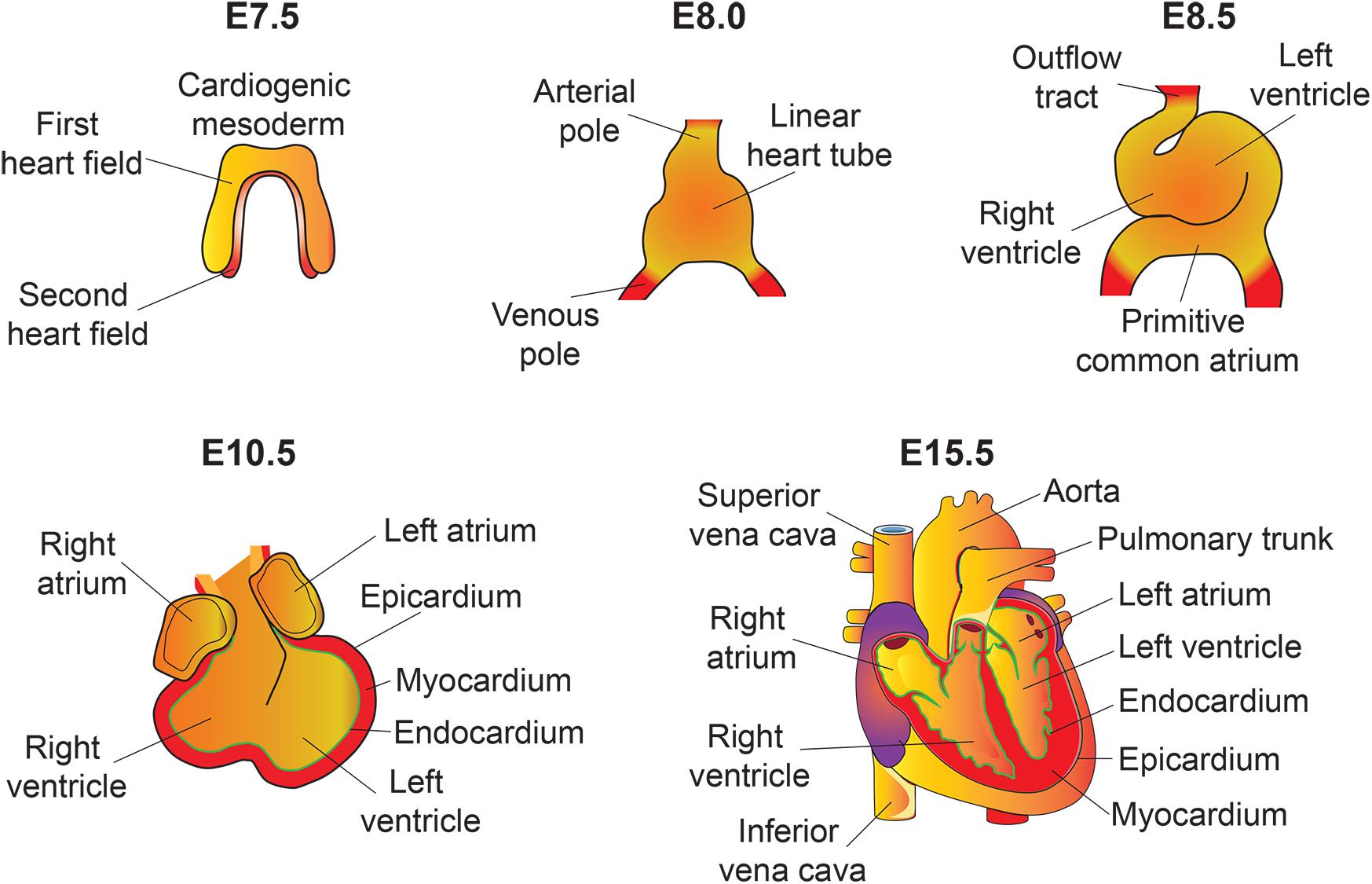
Figure 2. An overview of mouse heart development. Heart development begins with the specification of cardiogenic mesoderm cells in the primitive streak at E6.5. At E7.5, these mesodermal precursors migrate away from the primitive streak to form a bow-shaped structure called the cardiac crescent. Cardiac crescent can be divided into two major cardiac progenitor pools: the first and second heart field. The cardiac progenitor cells from the first heart field contribute to the linear heart tube, whereas the second heart field contribute to portions of the atria, the outflow tract, and the right ventricle. As embryonic development proceeds, the progenitor cells fuse at midline and form a primitive linear heart tube. At E8.5, the linear heart tube undergoes looping leading to formation of the outflow tract, primitive ventricles, and atria. At early stages, the heart consists of two layers: an inner endocardium and an outer myocardium. Between E9 and10.5, progenitor cells from different sources (including neural crest and proepicardial organ) migrate and contribute to the outflow tract and ventricular chambers. Myocardial layer expands and forms compact and trabecular myocardium. The proepicardial progenitor cells migrate over the heart surface and form epicardium. Epicardial-derived cells contribute to the formation of the coronary vasculature. Heart maturation involves a series of septation events and valve formation that results in a fully functional four-chambered heart integrated with the circulatory system by E15.5.
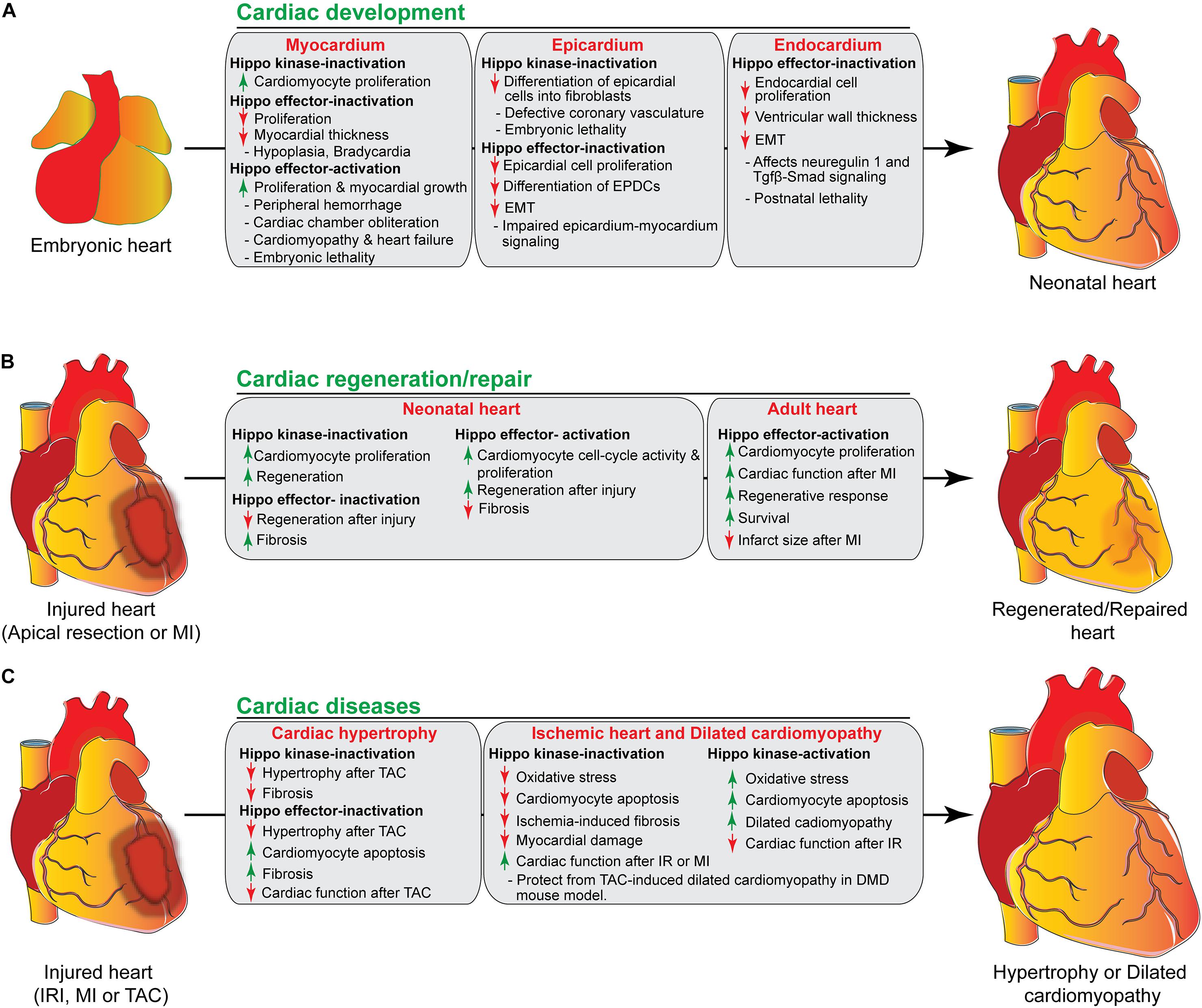
Figure 3. The role of Hippo signaling components in cardiac development, regeneration/repair, and diseases. (A) During cardiac development, activation/inactivation of the Hippo signaling components modulates proliferation and differentiation of cardiomyocytes, epicardial and endocardial cells, resulting in defective cardiogenesis and embryonic lethality as shown in the figure. (B) Activation/inactivation of the Hippo signaling components differently regulates the regenerative response of neonatal and adult hearts after injury. (C) Activation/inactivation of the Hippo signaling components leads to many cardiac diseases as described in the figure. EPDCs, epicardium-derived cells; EMT, endothelial mesenchymal transition; MI, myocardial infarction; IRI, ischemia/reperfusion injury, TAC, transverse aortic constriction; DMD, Duchenne Muscular Dystrophy.
Hippo Signaling Pathway in the Myocardium
Cardiomyocyte proliferation is vital for proper myocardial growth during heart development. A number of studies using either loss or gain of function genetic experiments have demonstrated that Hippo signaling pathway plays a central role in regulating cardiomyocyte proliferation during heart development to maintain a normal mammalian heart size (Tables 1, 2) (Heallen et al., 2011; Xin et al., 2011; von Gise et al., 2012). To investigate the role of Hippo pathway in cardiac development, Heallen et al. (2011) used gene inactivation approach to delete Sav1, Mst1/2, and Lats2 in the embryonic mouse heart by crossing their conditional-flox alleles with Nkx2.5Cre line. They found that cardiac-specific deletion of Sav1, Mst1/2 or Lats2 results in elevated cardiomyocyte proliferation, trabecular expansion, and thickening of compact ventricular myocardium leading to cardiomegaly (abnormal enlargement of the heart) without altering the cell size. However, the overall patterning of the heart was preserved in these mutants (Heallen et al., 2011). Further studies were also conducted to examine the effect of Yap’s gain and loss of function on cardiac development (Xin et al., 2011; von Gise et al., 2012). Cardiomyocyte specific deletion of Yap using Nkx2.5Cre or Tnnt2Cre driver resulted in impaired cardiomyocyte proliferation that led to lethal cardiac hypoplasia and severely thinned myocardial layers (Xin et al., 2011; von Gise et al., 2012). Conversely, cardiomyocyte-specific overexpression of activated Yap (YapS112A) resulted in enhanced cardiomyocyte proliferation leading to cardiac overgrowth in neonatal mice (Xin et al., 2011). Similarly, inducible expression of activated Yap (YapS127A) using Tnnt2Cre line showed peripheral hemorrhage, cardiomegaly and dramatic myocardial overgrowth causing cardiac chamber obliteration, all of which resulted in embryonic death by E15.5 (von Gise et al., 2012). Consistent with these findings, Monroe et al. demonstrated that overexpression of activated Yap in adult cardiomyocytes (αMHCCre–ERT2;Yap5SA) leads to increased proliferation, resulting in thickened ventricular walls and improved cardiac function (Monroe et al., 2019). Despite the improved cardiac function, these mice die within 4 days after the last dose of tamoxifen, given to induce the cre activity in cardiomyocytes. Furthermore, Monroe et al. demonstrated that Yap5SA overexpression induced adult cardiomyocytes into a fetal cell state by increasing the accessibility of embryonic cardiac enhancers in the adult heart (Monroe et al., 2019). In contrast to the well-established role of Yap in the heart, the role of Taz has not been studied extensively. Recently, Xin et al. (2013) demonstrated that cardiac-specific Taz knockout mice are viable and do not show any obvious cardiac defects. However, various combinations of Taz and Yap deletions in cardiomyocytes resulted in gene dosage-dependent cardiac defects. The cardiac defects in Yap/Taz mutants hearts were also associated with an increase in cardiomyocyte proliferation and a decrease in apoptosis (Xin et al., 2013). Furthermore, Murakami et al. (2005) revealed that the Hippo effector Taz physically interacts and modulates TBX5 transcriptional activity required for cardiomyogenesis. The Tead family transcription factors interact with both Yap and Taz via the N-terminal Tead binding domains in Yap/Taz. Disruption of Tead1 locus by a retroviral gene trapping resulted in formation of the thinned ventricular wall, enlarged pericardial cavity, bradycardia and embryonic lethality (Chen et al., 1994). Importantly, overexpression of Tead1 in the mouse heart induced cardiomyopathy and heart failure, suggesting that Tead1 is vital for proper heart development (Tsika et al., 2010).
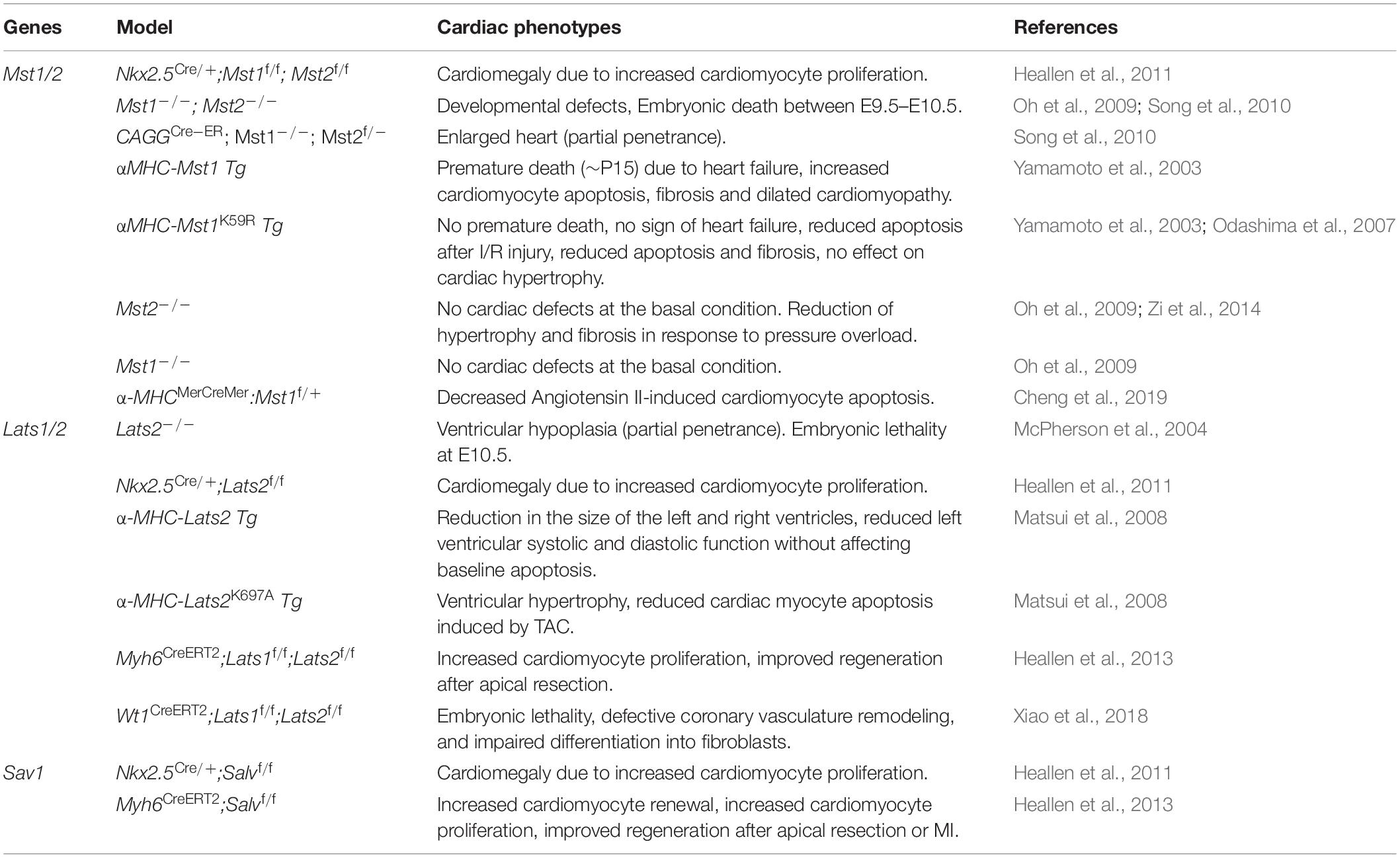
Table 1. Hippo signaling kinases in cardiac development and disease.
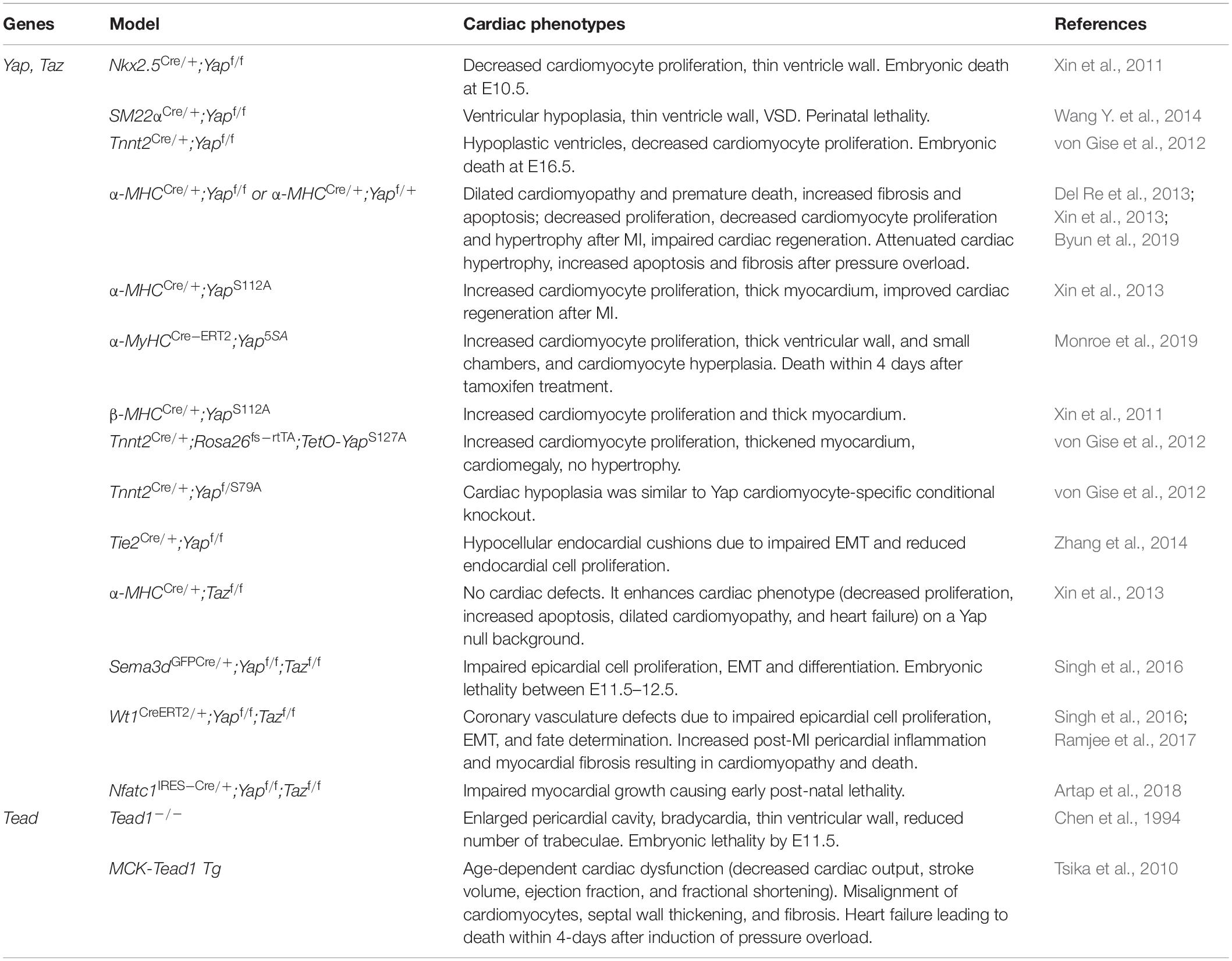
Table 2. Hippo signaling mediators in cardiac development and disease.
Multiple signaling pathways including Wnt-β-catenin, insulin-like growth factor (IGF), phosphoinositide 3-kinase (PI3K)–RACα, serine/threonine-protein kinase (AKT) and mammalian Target of Rapamycin (mTOR) have been reported to interact with the Hippo pathway to control cell growth and proliferation during cardiac development (Heallen et al., 2011; Xin et al., 2011). Wnt-β-catenin plays a crucial role in Hippo-Yap-mediated cardiac overgrowth. A recent study using cardiac-specific Salv knockout mice demonstrated an inhibitory role of Hippo signaling in the Wnt-β-catenin signaling pathway-dependent cardiac development. Transcriptional profiling of embryonic hearts from Salv knockout mice displayed elevated expression of canonical Wnt-β-catenin target genes required for heart development and those with anti-apoptosis function, including Sox2, Snai2, Birc2 and Birc5 (Heallen et al., 2011). Similarly, β-catenin nuclear localization was increased in the cardiomyocytes isolated from Salv knockout embryos (Heallen et al., 2011). Furthermore, biochemical studies showed that Yap-Tead forms complexes with β-catenin on the promoter region of Sox2 and Snai2 genes (Heallen et al., 2011). Collectively, these results suggest that Hippo signaling inhibit the interaction between Yap and β-catenin in the developing hearts to promote transcription of growth-related genes.
Another signaling pathway that has been demonstrated to cross-talk with Hippo is IGF pathway (Xin et al., 2011). Xin et al. (2011) generated mice overexpressing a constitutively active form of Yap (YapS112A) in the heart. Gene expression profiling of hearts expressing YapS112A revealed that Yap induces IGF signaling pathway genes such as Igf1, Igfbp2, and Igfbp3 (Xin et al., 2011). In addition to IGF pathway genes, Yap also induced β-catenin (Ctnnb1) and its downstream target genes including Kcne3, Ndrl, and Ier3, suggesting that β-catenin expression is stabilized in cardiomyocytes (Xin et al., 2011). Furthermore, increased PI3K and phosphorylated AKT levels were observed in YapS112A expressing cardiomyocytes, indicating increased IGF pathway activity (Xin et al., 2011). Enhanced IGF-AKT results in phosphorylation and inactivation of the β-catenin destruction complex component glycogen synthase kinase 3β (GSK3β), leading to β-catenin stabilization. Moreover, IGF inhibition decreases both Yap activity and β-catenin expression in cardiomyocyte. These suggest that activated Yap increases IGF signaling and stabilizes β-catenin by decreasing GSK3β phosphorylation (Xin et al., 2011). Collectively, they suggest that the Hippo-Yap pathway acts through cardiomyocytes to regulate myocardial growth.
Hippo Signaling Pathway in the Epicardium
The epicardium is the outermost mesothelial cell layer of the heart. It is essential for proper formation of the coronary vasculature. A number of studies have demonstrated that epicardium-derived cells (EPDCs) can differentiate into vascular smooth muscle cells, coronary endothelial cells and interstitial fibroblasts in the developing heart (Mikawa and Fischman, 1992; Gittenberger-de Groot et al., 1998; Lie-Venema et al., 2007; Red-Horse et al., 2010; Singh et al., 2011; Katz et al., 2012; Singh and Epstein, 2012; von Gise and Pu, 2012; Degenhardt et al., 2013; Epstein et al., 2015; Ramesh et al., 2016). The epicardium is also an important source of growth signals required for proliferation and differentiation of the underlying myocardial cells (Chen et al., 2002; Stuckmann et al., 2003; Lavine et al., 2005; Merki et al., 2005; Pennisi and Mikawa, 2009; Brade et al., 2011; Li et al., 2011; Zhou et al., 2011; Tian and Morrisey, 2012). Similarly, signals from the myocardium can reciprocally modulate epicardial cell behavior (Morabito et al., 2001; Chen et al., 2002; Smart et al., 2007; Mellgren et al., 2008; Bock-Marquette et al., 2009; Smith et al., 2011; Vega-Hernandez et al., 2011). To determine the role of the Hippo signaling pathway in coronary vessel development, Singh et al. (2016) generated an epicardial-specific Yap/Taz double mutant embryos using Sema3dGFPCre/+ and Wt1CreERT2/+ line. Consistent with the observed thin and fragmented myocardium and hemorrhage, the Yap/Taz double mutants die around E11.5-E12.5 from cardiovascular insufficiency. Epicardium-myocardium signaling was impaired due to Yap/Taz deletion in the epicardium. Inducible deletion of Yap/Taz demonstrated that epicardial cell proliferation, epithelial-to-mesenchymal transition, and EPDCs differentiation into cardiac lineages were affected in the double mutant embryos (Singh et al., 2016). In line with our findings, Xiao et al. (2018) demonstrated that the Hippo pathway kinases Lats1 and 2 play an essential role in epicardium development. Epicardial-specific deletion of Lats1 and 2 resulted in embryonic lethality by E15.5, and defected coronary vasculature remodeling in the double mutant embryos (Xiao et al., 2018). Using single-cell RNA sequencing approach, they showed that Lats1/2 deficiency in epicardial cells inhibited their differentiation into fibroblasts and remained at intermediate cell state with both epicardial and fibroblast-like features. These results suggest that Lats1/2 activity is required for EPDC progression from an epicardium-derived progenitor to fully differentiated cardiac fibroblasts (Xiao et al., 2018). Both genetic and pharmacological studies further revealed that fibroblasts differentiation is limited by Yap inhibition. Subsequently, this promotes matricellular factor dipeptidyl peptidase-4 expression that modulates the characteristics of extracellular matrix composition required for vascular remodeling during cardiac development (Xiao et al., 2018). Altogether, these findings suggest that the Hippo signaling pathway in the epicardium is necessary for cardiac fibroblast differentiation and coronary vessel development (Singh et al., 2016; Xiao et al., 2018).
Hippo Signaling Pathway in the Endocardium
Recently, Artap et al. (2018) reported that endocardial Hippo signaling is essential for embryonic heart myocardial growth (Artap et al., 2018). The myocardium’s growth and maturation are partly orchestrated by signals from the neighboring endocardium (Rentschler et al., 2002; Brutsaert, 2003; Sandireddy et al., 2019). Endothelial cells have been shown to express growth factors and cell surface ligands required for cardiac chamber development and maturation. For example, endocardial cells secrete neuregulin 1 (Nrg1) and express the cell surface ligand ephrin B2 (Efnb2), both of which signal to the adjacent myocardial cells to promote myocardium growth and maturation (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995). Artap et al. (2018) demonstrated that both Yap and Taz are expressed in both myocardial and endocardial cells of the developing heart. Endocardial-specific deletion of Yap and Taz using Nfatc1IRES–Cre/+ allele resulted in postnatal lethality (Artap et al., 2018). Ventricular wall thickness was also reduced in the double mutant embryos. They further showed that Yap/Taz deficiency in the endocardium led to reduced expression of Nrg1, an essential endocardial secreted factor that orchestrates the myocardium differentiation/phenotype. Interestingly, exogenous addition of recombinant Nrg1 partly rescued the myocardial phenotype of the mutant hearts. Moreover, they showed that Yap can bind to the promoter regions of Nrg1, indicating Yap-mediated regulation of Nrg1 in the endocardium is necessary for proper myocardial development (Artap et al., 2018). Similarly, conditional deletion of Yap in endothelial cells resulted in impaired endothelial mesenchymal transition (EMT) and reduced endocardial cell proliferation, leading to hypocellular endocardial cushions and embryonic lethality (Zhang et al., 2014). Zhang et al. (2014) further demonstrated that the interaction between Hippo signaling mediator Yap and TGFβ-smad (smad2/3/4) pathway is essential for proper atrioventricular cushion development. Loss of Yap function in the endothelial cells also disrupted the Tgfb-smad signaling, resulting in impaired activation of EMT genes such as Snail1, Twist1, and Slug (Zhang et al., 2014). Taken together, these results suggest that Hippo signaling pathway is required for the endocardial/endothelial cells to regulate normal heart development.
The Hippo Signaling Pathway in Cardiac Regeneration
Since Hippo signaling is reported as an important regulator of cardiomyocyte proliferation during embryonic development (Heallen et al., 2011; Xin et al., 2011; von Gise et al., 2012), it could potentially play a significant role in heart regeneration/repair. In contrast to the adult heart’s limited regenerative capacity, the neonatal mouse heart is capable to regenerate from myocardial injury. However, this capacity is lost by day seven postnatally (P7) (Porrello et al., 2011). Several studies have demonstrated that Hippo signaling pathway is also essential for cardiomyocyte proliferation in postnatal hearts and Yap activation can prolong the 1-week regenerative window. For example, Yap overexpression extends neonatal cardiomyocyte cell cycle activity and promotes regeneration (von Gise et al., 2012). Xin et al. (2013) demonstrated that cardiomyocyte-specific deletion of Yap impedes neonatal heart regeneration. Hearts from P2 neonatal wildtype mice were able to fully regenerate after myocardial injury, as evident by minimal fibrosis and healthy myocardial tissue distal to the ligature. In contrast, Yap mutant P2 hearts displayed extensive cardiac fibrosis after injury, suggesting impaired regenerative responses (Xin et al., 2013). To demonstrate that Yap activation is sufficient for postnatal heart regeneration, Xin et al. (2013) generated αMHC-YapS112A transgenic mice overexpressing activated Yap in the heart under the control of αMHC promoter. Both wildtype and αMHC-YapS112A transgenic mice were subjected to myocardial injuries at P7 and analyzed at P21. Wildtype mice showed extensive scar formation, loss of myocardial tissue, and ventricular dilation in their hearts after injury. In contrast, αMHC-YapS112A hearts were fully regenerated with minimal or no fibrosis (Xin et al., 2013). Consistent with these results, Heallen et al. (2013) showed that cardiomyocyte-specific inducible inactivation of Sav1 and Lats1/2 also increased cardiomyocyte proliferation in the neonatal heart and extended the neonatal regenerative window (Tables 1, 2).
Stimulating cellular proliferation is more challenging in adult cardiomyocyte than that in fetal or neonatal counterpart. To demonstrate whether Yap activation can promote mature heart regeneration by stimulating adult cardiomyocyte proliferation, Xin et al. (2013) performed myocardial injury in wildtype and αMHC-YapS112A transgenic mice at 4 weeks old and analyzed their hearts after 21 days. Similar to neonatal pups, adult αMHC-YapS112A transgenic mice had improved regenerative response after myocardial injury when compared with their wildtype littermate controls (Xin et al., 2013). Similarly, Lin et al. (2014) described that cardiac-specific Yap activation in adult mouse heart stimulated cardiomyocyte proliferation, improved cardiac function, survival and reduced infarction size after myocardial injury.
Recently, Tao et al. (2016) observed that Pitx2 expression was induced in injured Salv knockout hearts. Conditional deletion of Pitx2 in neonatal mouse heart resulted in impaired repair/regenerative response after apex resection, as evident by increased fibrosis and reduced cardiac function. However, Pitx2 gain of function in adult cardiomyocytes facilitated cardiac repair and function (Tao et al., 2016). Genetic studies further revealed that Pitx2-Yap interaction is essential for maintaining the reparative capacity of adult cardiomyocytes after cardiac tissue injury (Tao et al., 2016).
Extracellular matrix composition also plays an important role in regulating the proliferation and differentiation of neonatal cardiomyocytes (Bassat et al., 2017). Genetic deletion causing changes in extracellular matrix composition can limit the neonatal mouse hearts’ regenerative capacity. For example, conditional deletion of Agrin (Agrn) in the heart leads to impaired cardiac regeneration in neonatal mice after apex resection, evident by increased fibrosis and reduced cardiomyocyte proliferation and cardiac function (Bassat et al., 2017). The study further demonstrated that Agrin interacts with its receptor α-Dystroglycan (Dag1) and promotes disassembly of the dystrophin-glycoprotein complex (DGC), leading to Yap translocation to the nucleus, where it promotes cardiomyocyte proliferation (Bassat et al., 2017). The DGC complex components such as dystrophin and Dag1 play an important role in mediating interactions among the cytoskeleton, membrane, and extracellular matrix. Disruption of the DGC complex leads to destabilization of the plasma membrane, making the cardiomyocytes more susceptible to stretch- or contraction-induced injury and cell death (Weller et al., 1990; Petrof et al., 1993; Dellorusso et al., 2001). Consistent with these findings, Morikawa et al. (2017) also reported that Yap interaction with Dag1 prevents its nuclear translocation and inhibits cardiomyocyte proliferation. This interaction was further characterized (Morikawa et al., 2017) in a Mdx knockout mouse model (dystrophin loss of function). Using the neonatal cardiac apex resections injury model at P8, the authors showed that cardiomyocyte-specific Salv mutant hearts regenerated efficiently with little or no tissue fibrosis, while the control and Mdx (dystrophin mutant) hearts were unable to regenerate (Morikawa et al., 2017). Interestingly, Salv;Mdx double-knockout hearts regenerated efficiently with excessive myocardial growth and less fibrosis, suggesting an improvement in cardiac regeneration (Morikawa et al., 2017). These findings suggest that either inhibition of Hippo-kinases or Yap activation could be a potential therapeutic strategy to improve survival and cardiac function after myocardial injury.
The Hippo Signaling Pathway in Cardiac Diseases
Deregulation of the Hippo signaling pathway has been associated with various cardiac abnormalities including cardiac hypertrophy, heart failures, arrhythmogenic cardiomyopathy, dilated cardiomyopathy and ischemic heart disease (Tables 1, 2).
Cardiac Hypertrophy
In the developing hearts, cardiomyocyte proliferation is mainly initiated by pre-existing cardiomyocytes. However, since adult cardiomyocytes have restricted proliferative capacity, postnatal heart growth is governed by the physiological hypertrophy of the cardiomyocytes (Ahuja et al., 2007). Cardiac hypertrophy is also induced by pathological conditions such as myocardial ischemia/reperfusion (I/R) injury and myocardial infarction (MI). These conditions result in oxidative stress and irreversible loss of cardiomyocytes leading to heart failure. Adaptation to these adverse conditions causes insufficient contractile force where the ventricular cardiomyocytes undergo hypertrophy to increase the wall thickness and reduce wall stress, as well as cell death (Frey et al., 2004; Tardiff, 2006; Samak et al., 2016). Several studies have demonstrated that Yap activation under physiological and pathological conditions increases cardiomyocyte proliferation in both fetal and postnatal hearts without affecting its size (Xin et al., 2011, 2013; von Gise et al., 2012). Similarly, Lin et al. (2014) also observed that Yap activation increases cardiomyocyte proliferation without affecting hypertrophy and protects the heart from myocardial infarction. In contrast, Sadoshima and colleagues demonstrated that Yap prevents myocardial infarction by promoting not only cardiomyocyte survival and proliferation but also hypertrophy (Del Re et al., 2013). They further showed that cardiomyocyte survival resulting from Yap activation is mediated by either AKT/miR-206 or both, while its effect on hypertrophy is solely mediated by miR-206 (Yang et al., 2015). A recent clinical study demonstrated that Yap expression was increased whereas Mst1 expression was significantly reduced in heart samples from patients with hypertrophic cardiomyopathy (Wang P. et al., 2014). Similarly, Yap expression was also increased in mice with transverse aortic constriction (TAC), suggesting its important role in hypertrophic heart disease. Importantly, inhibition of Yap phosphorylation at serine 127 was reduced in both human and mouse tissues (Wang P. et al., 2014). Moreover, cardiomyocyte-specific overexpression of human Yap induced both in vitro and in vivo hypertrophy (Wang P. et al., 2014). Consistent with these findings, both Yap’s pro-hypertrophic and pro-survival functions were activated during TAC-induced adoptive hypertrophic response (Byun et al., 2019).
Several upstream Hippo signaling kinases are also implicated in cardiac hypertrophy. Inhibition or deletion of upstream Hippo kinases induces nuclear accumulation of Yap. Mst1 is also activated in response to I/R injury, myocardial infarction, and pressure overload. Surprisingly, neither cardiac-specific overexpression of wildtype nor the dominant-negative form of Mst1 (suppresses endogenous Mst1) caused any significant changes in cardiac function or cardiac hypertrophy in mice at baseline conditions (Yamamoto et al., 2003; Odashima et al., 2007). To further explain the role of Mst1, Del re et al. reported that elevation of Ras-association domain family 1 isoform A (Rassf1A), a physiological activator of Mst1, is associated with cardiomyocyte apoptosis and cardiac hypertrophy (Oceandy et al., 2009; Del Re et al., 2010). Rassf1A expression in the mouse heart is initially increased at 1 week but decreased after TAC-induced hypertrophy at 12 weeks. Rassf1A expression is also decreased in failing human hearts (Oceandy et al., 2009; Del Re et al., 2010). Subsequently, cardiomyocyte-specific ablation of Rassf1a or overexpression of Mst1-binding-mutant form of Rassf1A reduced TAC-induced hypertrophy and fibrosis in mice (Del Re et al., 2010). In contrast to Mst1, Mst2 knockout mice showed attenuated hypertrophy, while Mst2 overexpression increased hypertrophy in response to pressure overload (Zi et al., 2014). However, Mst2’s impact on hypertrophy was exerted through activation of the prohypertrophic Raf1-ERK1/2 pathway and not Yap (Zi et al., 2014). Similar to Mst kinases, Lats2 is also upregulated in response to pressure overload in mice (Matsui et al., 2008). Lats2 is also a negative regulator of cardiac hypertrophy (Matsui et al., 2008), where its overexpression did not affect cardiomyocyte apoptosis but rather reduced cardiomyocyte size. Cardiomyocyte-specific overexpression of a dominant-negative form of Lats2 notably increased ventricular hypertrophy and suppressed TAC-induced cardiomyocyte apoptosis (Matsui et al., 2008).
Sciarretta et al. (2015) recently demonstrated the cross-talk between mTOR and Hippo signaling pathway is essential for determining the cardiac response to pressure overload. The authors demonstrated that mTORC2, one of the multiprotein complexes mTOR forms with adaptor proteins, is a negative regulator of the Mst1 kinase, a key component of the Hippo signaling pathway (Sciarretta et al., 2015). mTORC2 reduces Mst1 homodimerization and its activity by phosphorylating specific serine residues in the SARAH domain. Rictor is the main components of the mTORC2 complex, where its deletion disrupts the whole complex (Wullschleger et al., 2006; Laplante and Sabatini, 2012). Sciarretta et al. (2015) showed that cardiac-specific mTORC2 disruption through rictor deletion in 6-month-old mice resulted in Mst1 activation, leading to dilation and impaired cardiac function. Increased cardiac fibrosis and apoptosis were also observed in rictor knockout mice, where they developed severe heart failure in response to pressure overload. In addition, compensatory hypertrophic response was also attenuated, suggesting that Rictor/mTORC2 regulates pressure overload-induced cardiac hypertrophy (Sciarretta et al., 2015). Collectively, Hippo-pathway plays an essential role in modulating the cardiac response to stress.
Ischemic Heart and Dilated Cardiomyopathy
Ischemia-reperfusion injury is one of the most common causes of cardiac dysfunction in human. It is widely accepted that cardiomyocyte death after IR injury is largely due to excessive production of reactive oxygen species (ROS) and activation of cell death pathways. The role of Hippo signaling pathway in regulating cardiomyocyte apoptosis has been extensively studied. Several studies have reported that activation of Hippo-kinase Mst1 is associated with cardiomyocyte apoptosis and dilated cardiomyopathy (Yamamoto et al., 2003; Odashima et al., 2007; Del Re et al., 2010). Mst1 is activated in vitro by pro-apoptotic stimuli in cardiomyocytes causing apoptosis in a kinase activity-dependent manner. Furthermore, Mst1 is activated in cardiomyocytes due to oxidative stress induced by I/R injury in the mouse heart (Yamamoto et al., 2003). Cardiac-specific overexpression of Mst1 in mice resulted in activated caspases, increased cardiomyocytes apoptosis and deterioration of cardiac function from heart dilation, all of which result in some premature death (Yamamoto et al., 2003). However, suppression of endogenous Mst1 with cardiac-specific overexpression of a dominant-negative form of Mst1 (Mst1K59R) inhibited cardiomyocytes apoptosis, reduced the extent of myocardial damage and improved cardiac function after I/R injury or myocardial infarction (Yamamoto et al., 2003; Odashima et al., 2007).
A recent study reported elevated phosphorylated Yap and Lats protein levels in the heart samples from patients with ischemic or non-ischemic heart failures (non-ischemic idiopathic end-stage cardiomyopathy) (Hou et al., 2017; Leach et al., 2017). However, no change in Salv levels was observed. Compared with control littermates, cardiomyocyte-specific inducible deletion of Salv in mice reduced ischemia-induced fibrosis and improved cardiac function (Leach et al., 2017). Consistent with these results, direct myocardial or systemic injection of adeno-associated virus 9 (AAV9) encoding short hairpin RNA (shRNA) against Salv improved cardiomyocyte cell cycle re-entry and cardiac function after myocardial infarction (Leach et al., 2017).
Duchenne Muscular Dystrophy (DMD) is a disorder where progressive muscle weakness causes dilated cardiomyopathy resulting in heart failure and myocardial fibrosis. DMD is caused by loss-of-function mutations in the dystrophin gene. To demonstrate the role of Hippo signaling pathway in dilated cardiomyopathy, Morikawa et al. (2017) generated Salv;Mdx double knockout mice and analyzed them after TAC, an overload model of dilated cardiomyopathy (Kassiri et al., 2005). Mdx hearts developed TAC-induced dilated cardiomyopathy with loss of cardiomyocytes, increased fibrosis and impaired cardiac function. In contrast, inactivation of the Hippo pathway in Mdx genetic background (Salv;Mdx double knockout) protected the heart from TAC-induced dilated cardiomyopathy by increasing Yap activity, thereby increasing cardiomyocyte proliferation and decreasing apoptosis (Morikawa et al., 2017).
Neurofibromin 2 (NF2) is a GTPase-activating protein that regulates the RAS signaling pathway (Matsuda et al., 2016). It has been described to modulate Mst1 and Lats2-mediated cardiomyocyte apoptosis after I/R injury (Matsuda et al., 2016). In response to oxidative stress triggered by I/R injury, NF2 is activated in the cardiomyocytes/myocardium. Knockdown of NF2 using siRNA inhibited Mst1-mediated apoptosis in cultured cardiomyocytes (Matsuda et al., 2016). Following I/R injury, cardiomyocyte-specific deletion of NF2 also demonstrated cardioprotective effect with reduced infarction and improved cardiac function (Matsuda et al., 2016; Mia et al., 2016). They also observed Yap activation in NF2 deficient heart after I/R injury and genetic ablation of Yap in NF2-deficient heart abolished its cardioprotective effects, as evident by enhanced apoptosis and increased infarction. Collectively, the Hippo-pathway plays a crucial role in controlling apoptosis and growth of cardiomyocytes, thereby regulating the size of ventricles and overall cardiac functions in the adult mouse heart.
Arrhythmogenic Cardiomyopathy
The Hippo-Yap pathway was also reported in the pathophysiology of arrhythmogenic cardiomyopathy (Chen et al., 2014). Arrhythmogenic cardiomyopathy is an inherited heart muscle disorder characterized by a progressive fibro-fatty replacement of the ventricular myocardium, ventricular arrhythmias and impaired ventricular systolic function leading to sudden cardiac death. Chen et al. (2014) demonstrated that the Hippo pathway is activated in the human heart and mouse models of arrhythmogenic cardiomyopathy. They showed that activation of the hippo pathway is due to changes at the intercalated disks, as shown by markedly reduced expression of several intercalated disks proteins such as JUP, PKP2, DSC2, DSG2, DSP, and GJA1 in human patients with advanced arrhythmogenic cardiomyopathy. These changes resulted in the reduced localization of activated protein kinase C-alpha (PKCα), which normally phosphorylates and inactivates NF2. Consequently, elevated expression of NF2 was observed in the heart samples from patients and preclinical mouse model with arrhythmogenic cardiomyopathy (Chen et al., 2014). Activation of NF2 leads to phosphorylation of Hippo components Mst1/2, Lats1/2, and Yap, suppressing the Yap-Tead target gene expression. Based on the reported crosstalk between Wnt/β-catenin and Hippo/Yap signaling pathways described above, the authors determined the status of the Wnt/β-catenin signaling in arrhythmogenic cardiomyopathy. Although the total β-catenin levels were unchanged, its localization to the intercalated disks was affected, leading to reduced Wnt/β-catenin transcriptional activity (Chen et al., 2014). Together, these results suggest that reduced Wnt/β-catenin and Hippo/Yap signaling are associated with enhanced adipogenesis in arrhythmogenic cardiomyopathy. Furthermore, enhanced adipogenesis was normalized in mice with Lats1/2 knockdown and those with active Yap expression. This suggests that activation of hippo kinases promote adipogenesis in arrhythmogenic cardiomyopathy (Chen et al., 2014). Collectively, these results demonstrate that activation of the Hippo pathway is mediated through the intercalated disks and thus provide a novel mechanism for the pathogenesis of arrhythmogenic cardiomyopathy.
Post-Myocardial Infarction (MI) Inflammation and Remodeling
Many preclinical studies have established the role of Hippo pathway members in cardiac development, regeneration, and homeostasis. Recent advances suggest that the Hippo signaling components are also critically involved in regulating inflammatory processes, including the immune response. For example, Yap mediates recruitment of tumor-associated macrophages required for immune evasion and tumor progression (Guo et al., 2017; Huang et al., 2017). Likewise, Hagenbeek et al. (2018) reported that both Yap and Taz play a diverse role in liver tumor development where Taz stimulated proinflammatory cytokine production and macrophage infiltration, while Yap promoted tumor formation. It is also evident that Yap plays a vital role in regulatory T cells (Treg) mediated suppression of antitumor immunity (Ni et al., 2018). Despite the well-established role of Hippo signaling components in regulating the inflammatory response in different pathophysiological conditions, their role in modulating post-MI inflammation and remodeling has not been fully investigated.
A growing number of studies demonstrated that epicardium is reactivated after myocardial infarction. Reactivated epicardial cells express genes that are normally expressed in the embryonic epicardium. These cells also proliferate and cover the damaged tissue. A subset of activated epicardial cells undergo EMT and differentiate into fibroblasts and smooth muscle cells, which further contribute to the repair process. Epicardium also secretes angiogenic and inflammatory factors in response to cardiac injury, which subsequently increases inflammatory cell influx to the infarcted area. Recently, Ramjee at al. (2017) reported that Hippo signaling in the epicardium plays a crucial role in the myocardial recovery after injury by recruiting regulatory T (Treg) cells, a subset of CD4+ T cells, to the injured myocardium. Treg cells have been shown to decrease the immune response and infarct scar size in the myocardium after injury (Hofmann et al., 2012; Nahrendorf and Swirski, 2014; Weirather et al., 2014). Ramjee et al. (2017) demonstrated that epicardial-specific deletion of Yap and Taz leads to profound post-injury pericardial inflammation and ventricular fibrosis due to decreased number of Treg cells in the injured myocardium, resulting in cardiomyopathy and increasing mortality. Yap/Taz also regulates IFN-γ expression in the activated epicardium. Loss of Yap/Taz in the epicardium resulted in downregulation of IFN-γ. Furthermore, exogenous administration of IFN-γ using hydrogel improved Treg cells infiltration into the injured myocardium and decreased ventricular fibrosis (Ramjee et al., 2017). Similarly, Yap activation through TLR3 signaling is essential for neonatal heart regeneration after MI (Wang et al., 2018). Although the role of Hippo signaling in non-immune cells associated inflammatory response is extensively studied, its functions in immune cells are not well established (Perez-Lopez et al., 2013; Ni et al., 2018). A recent study demonstrated that Yap deletion in the myeloid lineage protected the mice from inflammatory bowel disease (Zhou et al., 2019). These suggest that Hippo pathway components may play a significant role in inflammatory cells by modulating the post-MI inflammatory response, controlling adverse remodeling and facilitating cardiac function recovery after MI.
Conclusion
The Hippo signaling pathway has emerged as an essential regulator of cardiac development, growth and regeneration/repair after injury. In an animal model, deletion of Hippo-component Salv or activation of Yap improved cardiac function and survival after myocardial infarction (Lin et al., 2014; Leach et al., 2017). This suggests that Hippo signaling pathway can potentially be used as a target to improve cardiac regeneration/repair after injury. Although Yap activation appears to have short-term beneficial effects, its long-term consequences on the Hippo signaling pathway and cardiac injury remain to be elucidated. Some findings suggest that long-term Yap activation may have adverse effects on the heart (Wang P. et al., 2014; Monroe et al., 2019). This suggests that Yap activation in cardiomyocytes need to be tightly controlled to get the beneficial effects that improve the cardiac function after injury without major adverse effects. Recently, Hara et al. identified a compound that increased Yap-Tead activity and protected the heart against ischemic injury in mice (Hara et al., 2018). Although the Hippo signaling pathway has been extensively investigated in cardiomyocytes, its role in cardiac fibroblast and immune cells such as macrophages and T cells has not been well described. Since these cells play an important role in the repair process, further research is necessary to better understand how the Hippo signaling pathway regulates them in developing therapeutic interventions without potential adverse effects.
Author Contributions
MM wrote the initial draft of the manuscript. MS wrote and edited the final manuscript.
Funding
This work was supported by funds from the Duke-NUS Medical School Singapore, Goh Foundation, and Singapore NRF Fellowship (NRF-NRFF2016-01) to MS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to our colleagues whose work could not be cited due to space limitation.
References
Ahuja, P., Sdek, P., and MacLellan, W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544. doi: 10.1152/physrev.00032.2006
Artap, S., Manderfield, L. J., Smith, C. L., Poleshko, A., Aghajanian, H., See, K., et al. (2018). Endocardial hippo signaling regulates myocardial growth and cardiogenesis. Dev. Biol. 440, 22–30. doi: 10.1016/j.ydbio.2018.04.026
Bassat, E., Mutlak, Y. E., Genzelinakh, A., Shadrin, I. Y., Baruch Umansky, K., Yifa, O., et al. (2017). The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 547, 179–184. doi: 10.1038/nature22978
Bock-Marquette, I., Shrivastava, S., Pipes, G. C., Thatcher, J. E., Blystone, A., Shelton, J. M., et al. (2009). Thymosin beta4 mediated PKC activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J. Mol. Cell Cardiol. 46, 728–738. doi: 10.1016/j.yjmcc.2009.01.017
Brade, T., Kumar, S., Cunningham, T. J., Chatzi, C., Zhao, X., Cavallero, S., et al. (2011). Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 138, 139–148. doi: 10.1242/dev.054239
Brutsaert, D. L. (2003). Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 83, 59–115. doi: 10.1152/physrev.00017.2002
Byun, J., Del Re, D. P., Zhai, P., Ikeda, S., Shirakabe, A., Mizushima, W., et al. (2019). Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J. Biol. Chem. 294, 3603–3617. doi: 10.1074/jbc.RA118.006123
Chen, S. N., Gurha, P., Lombardi, R., Ruggiero, A., Willerson, J. T., and Marian, A. J. (2014). The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 114, 454–468. doi: 10.1161/CIRCRESAHA.114.302810
Chen, T., Chang, T. C., Kang, J. O., Choudhary, B., Makita, T., Tran, C. M., et al. (2002). Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev. Biol. 250, 198–207. doi: 10.1006/dbio.2002.0796
Chen, Z., Friedrich, G. A., and Soriano, P. (1994). Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes. Dev. 8, 2293–2301. doi: 10.1101/gad.8.19.2293
Cheng, Z., Zhang, M., Hu, J., Lin, J., Feng, X., Wang, S., et al. (2019). Cardiac-specific Mst1 deficiency inhibits ROS-mediated JNK signalling to alleviate Ang II-induced cardiomyocyte apoptosis. J. Cell Mol. Med. 23, 543–555. doi: 10.1111/jcmm.13958
Degenhardt, K., Singh, M. K., and Epstein, J. A. (2013). New approaches under development: cardiovascular embryology applied to heart disease. J. Clin. Invest. 123, 71–74. doi: 10.1172/JCI62884
Del Re, D. P., Matsuda, T., Zhai, P., Gao, S., Clark, G. J., Van Der Weyden, L., et al. (2010). Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Invest. 120, 3555–3567. doi: 10.1172/JCI43569
Del Re, D. P., Yang, Y., Nakano, N., Cho, J., Zhai, P., Yamamoto, T., et al. (2013). Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 288, 3977–3988. doi: 10.1074/jbc.M112.436311
Dellorusso, C., Crawford, R. W., Chamberlain, J. S., and Brooks, S. V. (2001). Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 22, 467–475.
Epstein, J. A., Aghajanian, H., and Singh, M. K. (2015). Semaphorin signaling in cardiovascular development. Cell Metab. 21, 163–173. doi: 10.1016/j.cmet.2014.12.015
Frey, N., Katus, H. A., Olson, E. N., and Hill, J. A. (2004). Hypertrophy of the heart: a new therapeutic target? Circulation 109, 1580–1589. doi: 10.1161/01.cir.0000120390.68287.bb
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394. doi: 10.1038/378390a0
Gittenberger-de Groot, A. C., Vrancken Peeters, M. P., Mentink, M. M., Gourdie, R. G., and Poelmann, R. E. (1998). Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 82, 1043–1052. doi: 10.1161/01.res.82.10.1043
Guo, X., Zhao, Y., Yan, H., Yang, Y., Shen, S., Dai, X., et al. (2017). Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes. Dev. 31, 247–259. doi: 10.1101/gad.294348.116
Hagenbeek, T. J., Webster, J. D., Kljavin, N. M., Chang, M. T., Pham, T., Lee, H. J., et al. (2018). The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Sci. Signal 11:eaaj1757. doi: 10.1126/scisignal.aaj1757
Halder, G., and Johnson, R. L. (2011). Hippo signaling: growth control and beyond. Development 138, 9–22. doi: 10.1242/dev.045500
Hara, H., Takeda, N., Kondo, M., Kubota, M., Saito, T., Maruyama, J., et al. (2018). Discovery of a small molecule to increase cardiomyocytes and protect the heart after ischemic injury. JACC Basic Transl. Sci. 3, 639–653. doi: 10.1016/j.jacbts.2018.07.005
Heallen, T., Morikawa, Y., Leach, J., Tao, G., Willerson, J. T., Johnson, R. L., et al. (2013). Hippo signaling impedes adult heart regeneration. Development 140, 4683–4690. doi: 10.1242/dev.102798
Heallen, T., Zhang, M., Wang, J., Bonilla-Claudio, M., Klysik, E., Johnson, R. L., et al. (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461. doi: 10.1126/science.1199010
Hofmann, U., Beyersdorf, N., Weirather, J., Podolskaya, A., Bauersachs, J., Ertl, G., et al. (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164
Hou, N., Wen, Y., Yuan, X., Xu, H., Wang, X., Li, F., et al. (2017). Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Exp. Mol. Pathol. 103, 267–275. doi: 10.1016/j.yexmp.2017.11.006
Huang, Y. J., Yang, C. K., Wei, P. L., Huynh, T. T., Whang-Peng, J., Meng, T. C., et al. (2017). Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. J. Hematol. Oncol. 10:60. doi: 10.1186/s13045-017-0421-3
Kassiri, Z., Oudit, G. Y., Sanchez, O., Dawood, F., Mohammed, F. F., Nuttall, R. K., et al. (2005). Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ. Res. 97, 380–390. doi: 10.1161/01.res.0000178789.16929.cf
Katz, T. C., Singh, M. K., Degenhardt, K., Rivera-Feliciano, J., Johnson, R. L., Epstein, J. A., et al. (2012). Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 22, 639–650. doi: 10.1016/j.devcel.2012.01.012
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lavine, K. J., Yu, K., White, A. C., Zhang, X., Smith, C., Partanen, J., et al. (2005). Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 8, 85–95. doi: 10.1016/j.devcel.2004.12.002
Leach, J. P., Heallen, T., Zhang, M., Rahmani, M., Morikawa, Y., Hill, M. C., et al. (2017). Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 550, 260–264. doi: 10.1038/nature24045
Lee, K. F., Simon, H., Chen, H., Bates, B., Hung, M. C., and Hauser, C. (1995). Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394–398. doi: 10.1038/378394a0
Li, P., Cavallero, S., Gu, Y., Chen, T. H., Hughes, J., Hassan, A. B., et al. (2011). IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 138, 1795–1805. doi: 10.1242/dev.054338
Lie-Venema, H., van den Akker, N. M., Bax, N. A., Winter, E. M., Maas, S., Kekarainen, T., et al. (2007). Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal 7, 1777–1798. doi: 10.1100/tsw.2007.294
Lin, Z., von Gise, A., Zhou, P., Gu, F., Ma, Q., Jiang, J., et al. (2014). Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 115, 354–363. doi: 10.1161/CIRCRESAHA.115.303632
Matsuda, T., Zhai, P., Sciarretta, S., Zhang, Y., Jeong, J. I., Ikeda, S., et al. (2016). NF2 activates hippo signaling and promotes ischemia/reperfusion injury in the heart. Circ. Res. 119, 596–606. doi: 10.1161/CIRCRESAHA.116.308586
Matsui, Y., Nakano, N., Shao, D., Gao, S., Luo, W., Hong, C., et al. (2008). Lats2 is a negative regulator of myocyte size in the heart. Circ. Res. 103, 1309–1318. doi: 10.1161/CIRCRESAHA.108.180042
McPherson, J. P., Tamblyn, L., Elia, A., Migon, E., Shehabeldin, A., Matysiak-Zablocki, E., et al. (2004). Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J. 23, 3677–3688. doi: 10.1038/sj.emboj.7600371
Mellgren, A. M., Smith, C. L., Olsen, G. S., Eskiocak, B., Zhou, B., Kazi, M. N., et al. (2008). Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ. Res. 103, 1393–1401. doi: 10.1161/CIRCRESAHA.108.176768
Merki, E., Zamora, M., Raya, A., Kawakami, Y., Wang, J., Zhang, X., et al. (2005). Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. U.S.A. 102, 18455–18460. doi: 10.1073/pnas.0504343102
Meyer, D., and Birchmeier, C. (1995). Multiple essential functions of neuregulin in development. Nature 378, 386–390. doi: 10.1038/378386a0
Mia, M. M., Chelakkot-Govindalayathil, A. L., and Singh, M. K. (2016). Targeting NF2-Hippo/Yap signaling pathway for cardioprotection after ischemia/reperfusion injury. Ann. Transl. Med. 4:545. doi: 10.21037/atm.2016.11.85
Mikawa, T., and Fischman, D. A. (1992). Retroviral analysis of cardiac morphogenesis: discontinuous formation of coronary vessels. Proc. Natl. Acad. Sci. U.S.A. 89, 9504–9508. doi: 10.1073/pnas.89.20.9504
Monroe, T. O., Hill, M. C., Morikawa, Y., Leach, J. P., Heallen, T., Cao, S., et al. (2019). YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev. Cell 48:e767. doi: 10.1016/j.devcel.2019.01.017
Morabito, C. J., Dettman, R. W., Kattan, J., Collier, J. M., and Bristow, J. (2001). Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 234, 204–215. doi: 10.1006/dbio.2001.0254
Morikawa, Y., Heallen, T., Leach, J., Xiao, Y., and Martin, J. F. (2017). Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 547, 227–231. doi: 10.1038/nature22979
Murakami, M., Nakagawa, M., Olson, E. N., and Nakagawa, O. (2005). A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in holt-oram syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 18034–18039. doi: 10.1073/pnas.0509109102
Nahrendorf, M., and Swirski, F. K. (2014). Regulating repair: regulatory T cells in myocardial infarction. Circ. Res. 115, 7–9. doi: 10.1161/circresaha.114.304295
Ni, X., Tao, J., Barbi, J., Chen, Q., Park, B. V., Li, Z., et al. (2018). YAP Is essential for treg-mediated suppression of antitumor immunity. Cancer Discov. 8, 1026–1043. doi: 10.1158/2159-8290.CD-17-1124
Oceandy, D., Pickard, A., Prehar, S., Zi, M., Mohamed, T. M., Stanley, P. J., et al. (2009). Tumor suppressor Ras-association domain family 1 isoform A is a novel regulator of cardiac hypertrophy. Circulation 120, 607–616. doi: 10.1161/CIRCULATIONAHA.109.868554
Odashima, M., Usui, S., Takagi, H., Hong, C., Liu, J., Yokota, M., et al. (2007). Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ. Res. 100, 1344–1352. doi: 10.1161/01.res.0000265846.23485.7a
Oh, S., Lee, D., Kim, T., Kim, T. S., Oh, H. J., Hwang, C. Y., et al. (2009). Crucial role for Mst1 and Mst2 kinases in early embryonic development of the mouse. Mol. Cell. Biol. 29, 6309–6320. doi: 10.1128/MCB.00551-09
Pennisi, D. J., and Mikawa, T. (2009). FGFR-1 is required by epicardium-derived cells for myocardial invasion and correct coronary vascular lineage differentiation. Dev. Biol. 328, 148–159. doi: 10.1016/j.ydbio.2009.01.023
Perez-Lopez, A., Rosales-Reyes, R., Alpuche-Aranda, C. M., and Ortiz-Navarrete, V. (2013). downregulates Nod-like receptor family CARD domain containing protein 4 expression to promote its survival in B cells by preventing inflammasome activation and cell death. J. Immunol. Salmonella 1201–1209 doi: 10.4049/jimmunol.1200415
Petrof, B. J., Shrager, J. B., Stedman, H. H., Kelly, A. M., and Sweeney, H. L. (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 90, 3710–3714.
Porrello, E. R., Mahmoud, A. I., Simpson, E., Hill, J. A., Richardson, J. A., Olson, E. N., et al. (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. doi: 10.1126/science.1200708
Ramesh, S., Singh, A., Cibi, D. M., Hausenloy, D. J., and Singh, M. K. (2016). in vitro culture of epicardial cells from mouse embryonic heart. J. Vis. Exp. 110:e53993. doi: 10.3791/53993
Ramjee, V., Li, D., Manderfield, L. J., Liu, F., Engleka, K. A., Aghajanian, H., et al. (2017). Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Invest. 127, 899–911. doi: 10.1172/JCI88759
Red-Horse, K., Ueno, H., Weissman, I. L., and Krasnow, M. A. (2010). Coronary arteries form by developmental reprogramming of venous cells. Nature 464, 549–553. doi: 10.1038/nature08873
Rentschler, S., Zander, J., Meyers, K., France, D., Levine, R., Porter, G., et al. (2002). Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc. Natl. Acad. Sci. U.S.A. 99, 10464–10469. doi: 10.1073/pnas.162301699
Samak, M., Fatullayev, J., Sabashnikov, A., Zeriouh, M., Schmack, B., Farag, M., et al. (2016). Cardiac hypertrophy: an introduction to molecular and cellular basis. Med. Sci. Monit. Basic Res. 22, 75–79. doi: 10.12659/MSMBR.900437
Sandireddy, R., Cibi, D. M., Gupta, P., Singh, A., Tee, N., Uemura, A., et al. (2019). Semaphorin 3E/PlexinD1 signaling is required for cardiac ventricular compaction. JCI Insight 4:125908. doi: 10.1172/jci.insight.125908
Sciarretta, S., Zhai, P., Maejima, Y., Del Re, D. P., Nagarajan, N., Yee, D., et al. (2015). mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 11, 125–136. doi: 10.1016/j.celrep.2015.03.010
Singh, A., Ramesh, S., Cibi, D. M., Yun, L. S., Li, J., Li, L., et al. (2016). Hippo signaling mediators yap and taz are required in the epicardium for coronary vasculature development. Cell Rep. 15, 1384–1393. doi: 10.1016/j.celrep.2016.04.027
Singh, M. K., and Epstein, J. A. (2012). Epicardium-derived cardiac mesenchymal stem cells: expanding the outer limit of heart repair. Circ. Res. 110, 904–906. doi: 10.1161/res.0b013e31825332a3
Singh, M. K., Lu, M. M., Massera, D., and Epstein, J. A. (2011). MicroRNA-processing enzyme Dicer is required in epicardium for coronary vasculature development. J. Biol. Chem. 286, 41036–41045. doi: 10.1074/jbc.M111.268573
Smart, N., Risebro, C. A., Melville, A. A., Moses, K., Schwartz, R. J., Chien, K. R., et al. (2007). Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445, 177–182. doi: 10.1038/nature05383
Smith, C. L., Baek, S. T., Sung, C. Y., and Tallquist, M. D. (2011). Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 108, e15–e26. doi: 10.1161/CIRCRESAHA.110.235531
Song, H., Mak, K. K., Topol, L., Yun, K., Hu, J., Garrett, L., et al. (2010). Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 107, 1431–1436. doi: 10.1073/pnas.0911409107
Stuckmann, I., Evans, S., and Lassar, A. B. (2003). Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 255, 334–349. doi: 10.1016/s0012-1606(02)00078-7
Tao, G., Kahr, P. C., Morikawa, Y., Zhang, M., Rahmani, M., Heallen, T. R., et al. (2016). Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123. doi: 10.1038/nature17959
Tardiff, J. C. (2006). Cardiac hypertrophy: stressing out the heart. J. Clin. Invest. 116, 1467–1470. doi: 10.1172/jci28884
Tian, Y., and Morrisey, E. E. (2012). Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ. Res. 110, 1023–1034. doi: 10.1161/CIRCRESAHA.111.243899
Tsika, R. W., Ma, L., Kehat, I., Schramm, C., Simmer, G., Morgan, B., et al. (2010). TEAD-1 overexpression in the mouse heart promotes an age-dependent heart dysfunction. J. Biol. Chem. 285, 13721–13735. doi: 10.1074/jbc.M109.063057
Vega-Hernandez, M., Kovacs, A., De Langhe, S., and Ornitz, D. M. (2011). FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 138, 3331–3340. doi: 10.1242/dev.064410
von Gise, A., Lin, Z., Schlegelmilch, K., Honor, L. B., Pan, G. M., Buck, J. N., et al. (2012). YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 109, 2394–2399. doi: 10.1073/pnas.1116136109
von Gise, A., and Pu, W. T. (2012). Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 110, 1628–1645. doi: 10.1161/CIRCRESAHA.111.259960
Wang, P., Mao, B., Luo, W., Wei, B., Jiang, W., Liu, D., et al. (2014). The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic Res. Cardiol. 109:435. doi: 10.1007/s00395-014-0435-8
Wang, Y., Hu, G., Liu, F., Wang, X., Wu, M., Schwarz, J. J., et al. (2014). Deletion of yes-associated protein (YAP) specifically in cardiac and vascular smooth muscle cells reveals a crucial role for YAP in mouse cardiovascular development. Circ. Res. 114, 957–965. doi: 10.1161/CIRCRESAHA.114.303411
Wang, X., Ha, T., Liu, L., Hu, Y., Kao, R., Kalbfleisch, J., et al. (2018). TLR3 mediates repair and regeneration of damaged neonatal heart through glycolysis dependent YAP1 regulated miR-152 expression. Cell Death. Differ. 25, 966–982. doi: 10.1038/s41418-017-0036-9
Weirather, J., Hofmann, U. D., Beyersdorf, N., Ramos, G. C., Vogel, B., Frey, A., et al. (2014). Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67. doi: 10.1161/CIRCRESAHA.115.303895
Weller, B., Karpati, G., and Carpenter, S. (1990). Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 100, 9–13. doi: 10.1016/0022-510x(90)90005-8
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi: 10.1016/j.cell.2006.01.016
Xiao, Y., Hill, M. C., Zhang, M., Martin, T. J., Morikawa, Y., Wang, S., et al. (2018). Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev. Cell 45:e156. doi: 10.1016/j.devcel.2018.03.019
Xin, M., Kim, Y., Sutherland, L. B., Murakami, M., Qi, X., McAnally, J., et al. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 13839–13844. doi: 10.1073/pnas.1313192110
Xin, M., Kim, Y., Sutherland, L. B., Qi, X., McAnally, J., Schwartz, R. J., et al. (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal 4:ra70. doi: 10.1126/scisignal.2002278
Yamamoto, S., Yang, G., Zablocki, D., Liu, J., Hong, C., Kim, S. J., et al. (2003). Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Invest. 111, 1463–1474. doi: 10.1172/jci17459
Yang, Y., Del Re, D. P., Nakano, N., Sciarretta, S., Zhai, P., Park, J., et al. (2015). miR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ. Res. 117, 891–904. doi: 10.1161/CIRCRESAHA.115.306624
Zhang, H., von Gise, A., Liu, Q., Hu, T., Tian, X., He, L., et al. (2014). Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 289, 18681–18692. doi: 10.1074/jbc.M114.554584
Zhao, B., Tumaneng, K., and Guan, K. L. (2011). The hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883. doi: 10.1038/ncb2303
Zhou, B., Honor, L. B., He, H., Ma, Q., Oh, J. H., Butterfield, C., et al. (2011). Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest. 121, 1894–1904. doi: 10.1172/JCI45529
Zhou, Q., Li, L., Zhao, B., and Guan, K. L. (2015). The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 116, 1431–1447. doi: 10.1161/circresaha.116.303311
Zhou, X., Li, W., Wang, S., Zhang, P., Wang, Q., Xiao, J., et al. (2019). YAP aggravates inflammatory bowel disease by regulating M1/M2 macrophage polarization and gut microbial homeostasis. Cell Rep. 27:e1175. doi: 10.1016/j.celrep.2019.03.028
Keywords: hippo signaling, cardiac development, hypertrophy, ischemia – reperfusion, cardiomyoapthies
Citation: Mia MM and Singh MK (2019) The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 7:211. doi: 10.3389/fcell.2019.00211
Received: 30 January 2019; Accepted: 12 September 2019;
Published: 01 October 2019.
Edited by:
Stephano Spano Mello, University of Rochester, United StatesReviewed by:
Lee Bardwell, University of California, Irvine, United StatesWenqi Wang, University of California, Irvine, United States
Todd R. Heallen, Texas Heart Institute, United States
Copyright © 2019 Mia and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manvendra K. Singh, manvendra.singh@duke-nus.edu.sg