 Hyun-Ju Cho
Hyun-Ju Cho Yufei Shan
Yufei Shan Niteace C. Whittington
Niteace C. Whittington Susan Wray
Susan Wray
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Cell Dev. Biol. , 11 July 2019
Sec. Cell Adhesion and Migration
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00121
This article is part of the Research Topic Cell Migration in Neurological Disorders View all 6 articles
The development of Gonadotropin releasing hormone-1 (GnRH) neurons is important for a functional reproduction system in vertebrates. Disruption of GnRH results in hypogonadism and if accompanied by anosmia is termed Kallmann Syndrome (KS). From their origin in the nasal placode, GnRH neurons migrate along the olfactory-derived vomeronasal axons to the nasal forebrain junction and then turn caudally into the developing forebrain. Although research on the origin of GnRH neurons, their migration and genes associated with KS has identified multiple factors that influence development of this system, several aspects still remain unclear. This review discusses development of the olfactory system, factors that regulate GnRH neuron formation and development of the olfactory system, migration of the GnRH neurons from the nose into the brain, and mutations in humans with KS that result from disruption of normal GnRH/olfactory systems development.
Unraveling how specialized neurons arise from heterogenous cell populations and then migrate to their appropriate location has important implications for understanding the development and progression of neuronal disorders. Proper establishment of the Gonadotropin releasing hormone-1 (GnRH) system is crucial for function of the reproduction system in vertebrates. GnRH neurons originate within the nasal placode (Schwanzel-Fukuda and Pfaff, 1989; Wray et al., 1989a,b), a region that also gives rise to olfactory sensory neurons (both those that sense odors as well as those that sense pheromones) and olfactory ensheathing cells (OECs). As the nasal placode invaginates to form the main olfactory epithelium (OE) and the vomeronasal organ (VNO), GnRH neurons migrate out of the VNO to the brain along axons that are covered by OECs (Figure 1; Wray, 2002a). Once within the forebrain, GnRH neurons function in hormone signaling through the hypothalamic-pituitary-gonadal (HPG) axis. In humans, improper development of the nasal placode and/or migration of GnRH neurons results in various forms of hypogonadism, including Kallmann Syndrome (KS) which is characterized by anosmia and lack of sexual development. Ongoing studies continue to support the occurrence of oligogenism in KS patients, indicating that a combination of mutations or rare variants on two or more genes can underlie the disease (di/oligogenic disorder). As such, one needs to understand the cellular components that GnRH neurons are exposed to, how they interact, which are redundant and who may compensate. In addition, the question remains as to the diversity of GnRH cells themselves – Do subpopulations exist (expressing different receptors perhaps), responding to different cues, to ensure some GnRH cells reach the brain and the animal can reproduce? Since the development of the GnRH system is intimately entwined with the development of the olfactory system, factors that regulate development of the nasal placode and thus GnRH cells are discussed in Section “Development of the Olfactory Placode and GnRH Neurons”. Section “Migration of GnRH Neurons From the Nose to the Brain” focuses on factors that influence outgrowth of olfactory axons and migration of the GnRH neurons. Then, in the last section, we discuss mutations in humans with KS that result from disruption of normal development of the GnRH/olfactory systems and present new candidate genes that might contribute to KS based on bioinformatic analysis of known KS genes.
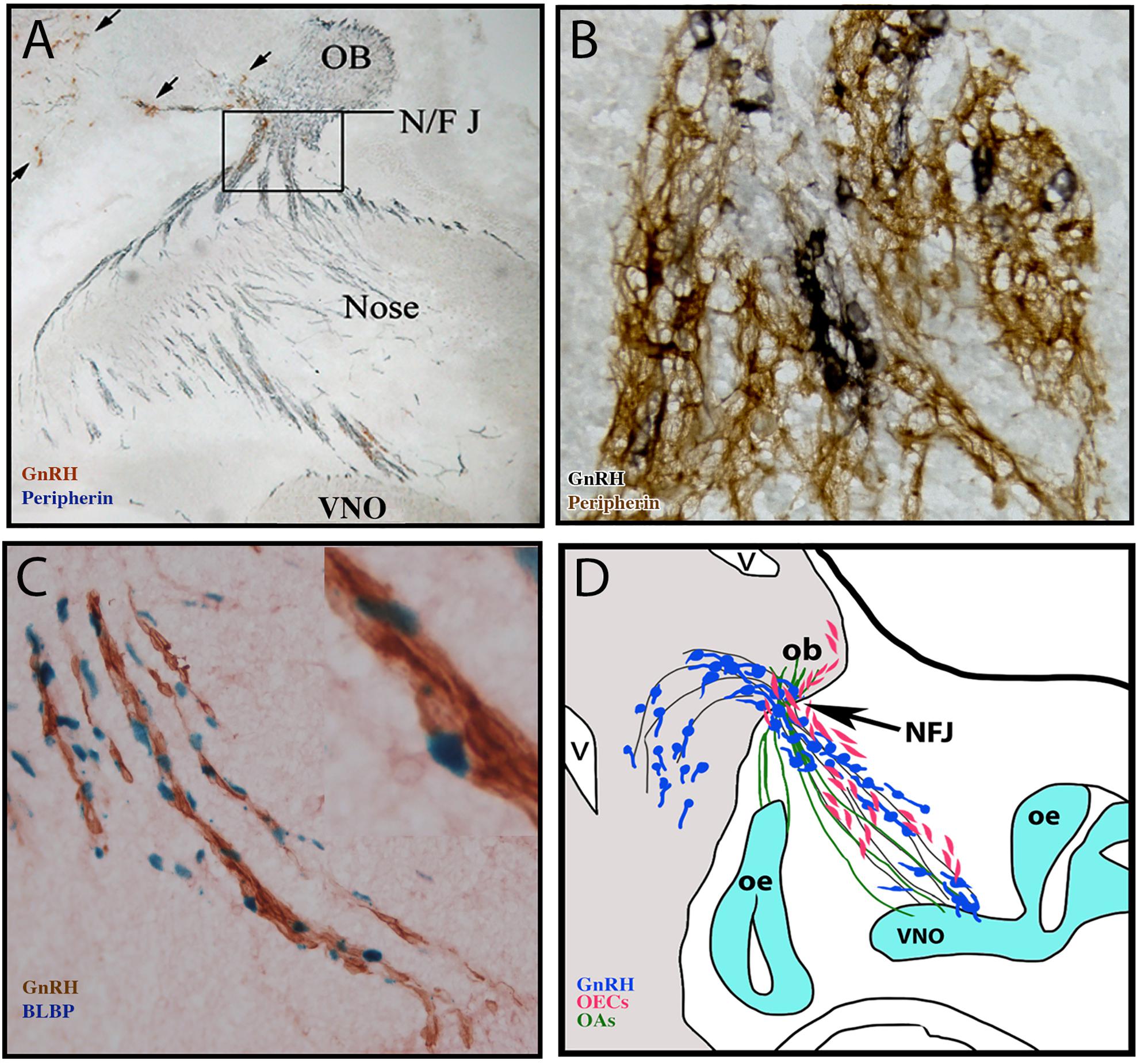
Figure 1. Multiple components form the GnRH migratory bundle during development. (A) Immunocytochemistry staining of E14.5 mouse section. Brown, GnRH (black arrows); Blue, peripherin (marking olfactory axons). Axons and cells must travel through the cribriform plate at the NFJ (boxed area) (B) Immunocytochemistry staining of GnRH (black) and peripherin (brown) at the nasal forebrain junction (NF/J). (C) Staining of GnRH neurons (brown) and BLBP (blue nuclei, labeling OECs) on the migratory route. (D) Schematic of multiple components forming the GnRH migratory bundle in mouse at E14.5. GnRH neurons (blue) migrate along VNO/terminal nerve axons (black lines) into the forebrain. Other VNO sensory axons bundle with olfactory sensory axons (green) and OECs (pink) enter the olfactory bulb.
Olfactory placodes (OP) are complex structures forming as bilateral transient thickenings of non-neural ectoderm at the ventrorostral region of the developing embryonic head (Figure 2A). They arise from pre-placodal ectoderm (reviewed in Saint-Jeannet and Moody, 2014) and intermixing of migratory cranial neural crest cells and ectodermal cells occur in the OP in vertebrates (Baker and Bronner-Fraser, 2001; Whitlock, 2005b; Forni et al., 2011). These neural crest cells give rise to the OECs (Forni et al., 2011; Katoh et al., 2011). However, the contribution of neural crest to other cell types in the OP is still being debated. Transgenic reporter mice were used to map the development of ectoderm and neural crest cells arising in the OE and VNO, and neural crest cells were found to give rise to a subpopulation of progenitors, sensory neurons, support cells and GnRH cells (Forni et al., 2011) (Figure 2A).
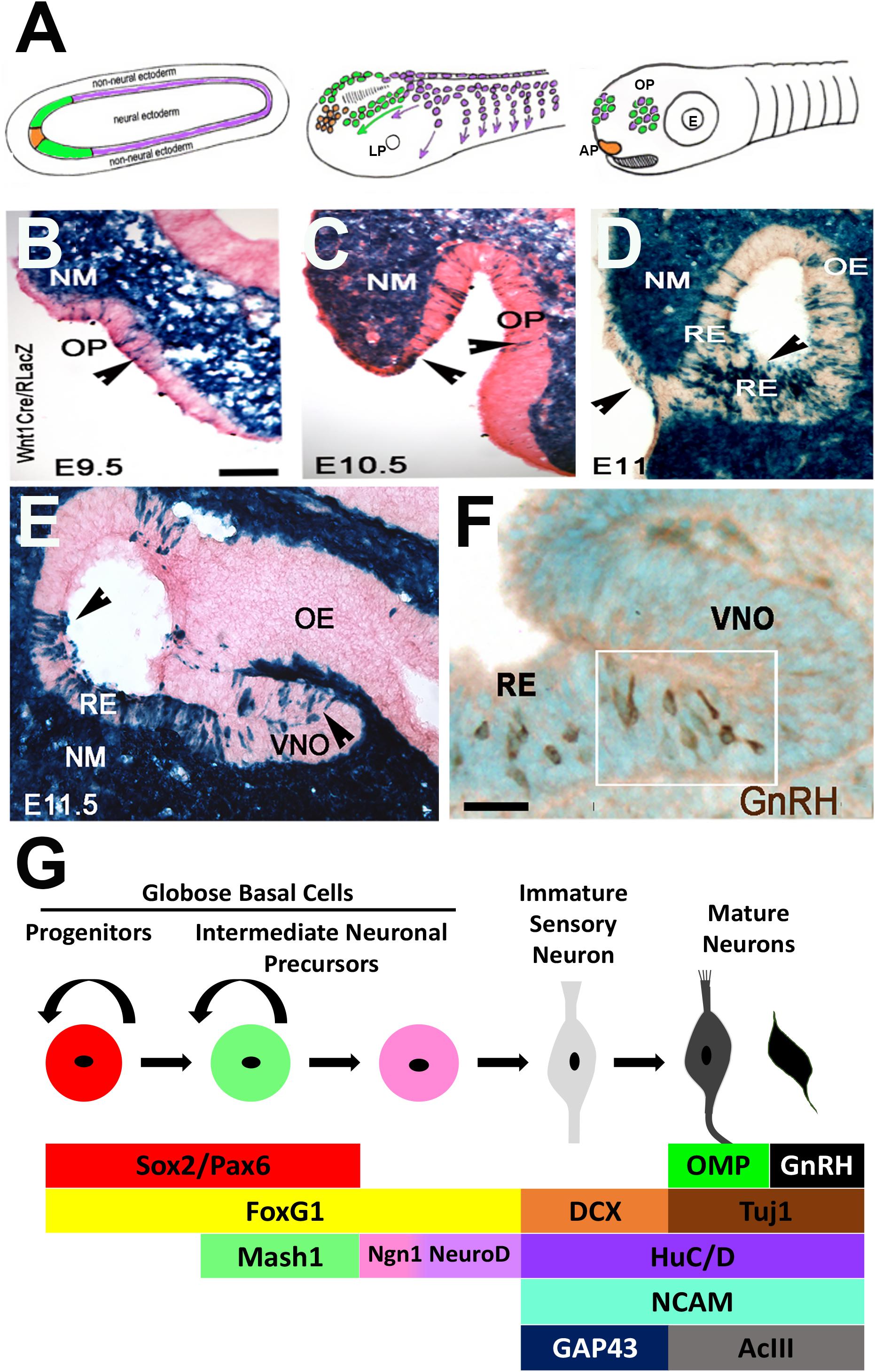
Figure 2. Development of the olfactory system. (A) Schematic of olfactory placode formation. Cell populations at the border of the neural plate: preplacodal ectoderm (green), anterior pituitary placode (orange), and pre-migratory neural crest cells (purple), migrate rostrally and intermix to form the olfactory placodes. (B–E) X-Gal staining on nasal sections of E9.5–E11.5 neural crest reporter mouse (Wnt1Cre/RLacZ) showing Wnt1 expression (blue) in cells within the invaginating olfactory pit (OP, arrowheads), olfactory epithelium (OE), vomeronasal organ (VNO) and throughout the nasal mesenchyme (NM) and respiratory epithelium (RE). (F) Immunocytochemistry staining of E11.5 OE showing GnRH neurons (brown) located at the border between the RE and developing VNO. (G) Schematic of olfactory neurogenesis. Cell types in the OE can be identified by expression of specific factors, which direct cells to remain as cycling progenitors or to undergo neuronal differentiation to form sensory and/or GnRH neurons (Adapted from Whitlock et al., 2006; Forni et al., 2011).
In mice, the OP forms as an epithelial sheet at embryonic day (E) 9.5 (Figures 2B–F). During the next 24 h, this sheet of cells invaginates to form the olfactory pit, which is the beginning of the nasal cavity and comprised of OE and surrounding nasal mesenchyme. At E11.5 the pit deepens to form the nostrils. The olfactory pit contains neural and non-neural structures and forms sensory and respiratory epithelium. The respiratory epithelium is located at the rostral portion and is the source of FGF8 and BMP4 expression that define the borders of the neurogenic sensory epithelium niche (Forni et al., 2013). The sensory epithelium, located within the invaginating OP, is layered and contains stem and progenitor cells, sensory neurons, GnRH neurons, sustentacular support cells and mucus-producing Bowman’s glands. Early on, OECs appear to move to the lamina propria beneath the basal membrane where they start to surround olfactory axons. Following invagination, GnRH neurons delaminate from the OE and migrate with immature neurons (Fornaro et al., 2003) and OECs to the brain along axonal projections. As development progresses, the OE forms into the VNO and turbinates of the main olfactory epithelium (mOE). The VNO is formed medially in the ventral region of the OE and gives rise to pheromone receptor cells that project to the accessory olfactory bulb, whereas the mOE generates odor receptors that project to the main olfactory bulb. Disruption of several of these sequences have consequences on GnRH neuronal specification and migration and thus reproductive capacity of the animal.
Cell stages can be identified by the expression of specific transcription factors and histological arrangement within the OE. Progenitor cells are first located on both the apical and basal sides of the OE, but later become restricted to the basal side and are identified as horizontal and globose basal cells. Globose basal cells are actively dividing progenitor cells that give rise to neurons (Figure 2G), while horizontal basal cells are stem cells that give rise neuronal and non-neuronal cells in the OE (Schwob et al., 2017). Multiple transcriptions factors have been identified as important for this process. PAX6 is required for initial establishment of placodal fate. Both PAX6 and SOX2 regulate the progression of olfactory neurogenesis (Donner et al., 2007; Tucker et al., 2010; Moody and LaMantia, 2015), holding olfactory progenitors in an undifferentiated state and preventing OSN formation (Packard et al., 2016). PAX6-null mice do not develop an eye or nasal placode, and thus lack olfactory cells and GnRH neurons (Dellovade et al., 1998). FOXG1 is only expressed in ventral progenitor cells, yet it plays a global role in OE formation. The loss of FOXG1 results in a diminished OE without neuronal differentiation (Duggan et al., 2008), and a loss of proliferation and/or differentiation (Kawauchi et al., 2009). Analysis of GnRH neurons and OECs was not examined. MASH1 (or ASCL1) is a basic helix-loop-helix (bHLH) transcription factor that is expressed in intermediate neuronal precursors (Figure 2G) that will give rise to OSNs, both in the OE and VNO. MASH1 is essential for proper olfactory neuron development (Cau et al., 1997; Castro et al., 2011), activating Notch signaling in the OE (Cau et al., 2002), which is required for differentiation. Sensory neurons of the VNO are also dependent on MASH1 for their proper development, with the size of the accessory olfactory bulb reduced when MASH1 is knocked out (Murray et al., 2003). While MASH1 regulates olfactory neurogenesis, it does not regulate OP formation or OE development, as a deletion of MASH1 results in an OE that still expresses SOX2 and has no defects in OE structure (Kawauchi et al., 2004). MASH1 activates a number of downstream targets, including Neurogenin-1, another bHLH proneural transcription factor. Neurogenin-1 marks basal cells (Ma et al., 1997) that give rise to daughter cells that exit the cell cycle to become immature OSNs and activates expression of NEUROD1, which is expressed in post-mitotic immature OSNs (Cau et al., 1997). Once intermediate progenitor cells commit to the olfactory neuronal lineage and differentiate, they become bipolar and start migrating away from the basal layer, and express markers such as NCAM (Calof and Chikaraishi, 1989), microtubule protein Doublecortin (DCX) (Francis et al., 1999; Gleeson et al., 1999), and HuC/D (Okano and Darnell, 1997; Fornaro et al., 2001) and GAP-43 (Pellier et al., 1994). As they mature, OSNs extend their cilia into the nasal cavity on the apical surface for odor detection, and project their OEC-wrapped axons to the olfactory bulb (Kawauchi et al., 2009). Molecules such as TUJ1 (De Carlos et al., 1996), adenylyl cyclase (ACIII) (Wong et al., 2000) and Olfactory Marker Protein (OMP) (Margolis, 1972) are expressed in mature OSNs (Figure 2G). In neurogenin-1 mutant OE, most OSNs fail to differentiate even though basal progenitors are still generated (Cau et al., 2002). Notably, GnRH cells do not appear to arise from MASH1 positive progenitors (Kramer and Wray, 2000a), suggesting differences among intermediate neuronal precursors in the VNO.
Thirty years after the identification of the nasal placode as the source of forebrain GnRH cells, the lineage and specification of GnRH neurons still remains unclear. Lineage tracing and ablation studies in mouse have shown that GnRH cells in the OE arise from two precursor populations having ectodermal and neural crest lineages (reviewed in Wray, 2002a; Forni and Wray, 2012). The formation of GnRH neurons has also been analyzed using an activating enhancer binding protein 2 (AP2) alpha mutant mouse. AP2alpha is expressed in respiratory epithelium. In these mutants, GnRH neurons were ectopically expressed although the morphology of the OE and VNO was conserved (Kramer et al., 2000). Unfortunately, embryonic stages earlier than E12.5 were not examined, but these data suggest alternative regulation of GnRH and OSN development and add to the argument that lineage and specification of GnRH neurons and sensory neurons diverge early. Identifying a marker that distinguishes GnRH cell lineage has been difficult. In mice, GnRH cells mature at approximately E11.5 and begin migrating out of the OE toward the forebrain. It was hypothesized that two bilateral groups of 50 progenitors (100 in total) give rise to a population of about 800 GnRH cells at E12.5 (Wray et al., 1989a). Nestin was found to be expressed in primary GnRH cells prior to onset of GnRH mRNA expression but was downregulated during GnRH differentiation (Kramer and Wray, 2000a). Studies using the mouse GnRH promoter from mice have identified several transcriptional promoter regions that could regulate the expression of GnRH (Iyer et al., 2010). Similar studies have been done in immoraltilized GnRH cell lines (Kim et al., 2011) and in tilapia (Kitahashi et al., 2005). Yet no transcription factor found is restricted to the GnRH cell population. Supporting the need for multiple factors is the work by Lund et al. (2016) on human pluripotent stem cells. In this work, the authors were able to differentiate stem cells to neural progenitor cells expressing FOXG1, EMX2, and PAX6 by dual SMAD inhibition, and then GnRH expressing cells by treating with FGF8, followed by a Notch inhibitor. Thus, a combination of transcription factors and molecular signaling appear to play pivotal roles in the spatiotemporal development of GnRH cells. However, recent work in zebrafish suggests Islet-1 as a potential early marker of GnRH cells (Aguillon et al., 2018). In addition to its expression in the brain and motor neurons of spinal cord (Ericson et al., 1992), Islet-1 was reported in the OP, exclusively in GnRH3 precursor cells (GnRH3 in zebrafish is the ortholog of mouse GnRH1, referred to as GnRH in this paper (reviewed in Forni and Wray, 2012). To determine the origin of these Islet-1/GnRH cells, transgenics and fate mapping experiments were done, and identified solely pre-placodal ectoderm origins and no neural crest derived lineage, consistent with some studies in zebrafish and chicken embryos (reviewed in Forni and Wray, 2012; Steventon et al., 2014; Suzuki and Osumi, 2015). However, the Islet-1 data directly opposes data which suggested that zebrafish GnRH3 derived from SOX10-dependent neural crest (Whitlock, 2005a) and work in mouse that reported up to 30% of GnRH cells are of neural crest origin (Forni et al., 2011). Thus, whether Islet-1 marks the entire GnRH population or only the ectodermally derived cells needs to be further examined in multiple species.
Fibroblast growth factor (FGF) signaling plays diverse roles during embryonic development, including embryonic patterning and neural development. In the developing OP, FGF8 is expressed in the respiratory epithelium and is required for craniofacial development (Kawauchi et al., 2005; Forni et al., 2013). In FGF8 mutants, OE structures failed to form (Kawauchi et al., 2005). FGF activity prevents prospective placodal cells from acquiring epidermal fate but it alone is not sufficient to specify sensory epithelium (Sjodal et al., 2007; Maier et al., 2010). Loss of FGF8 also affects GnRH development. FGF8 hypomorphic mice (with 50% retention of FGF8 expression), which only survive up to birth, lack GnRH neurons and fibers in the brain (Falardeau et al., 2008). Work in chicken indicates that FGF8 maintains the undifferentiated nature of progenitor cells (Sabado et al., 2012; Chung et al., 2016). This suggests that the timing of FGF8 signaling is important for GnRH neuron formation, and that expression in the OP region must be downregulated before differentiation of GnRH neurons occurs. As one would predict, FGFR1, the receptor for FGF8, is important in OE development. Acute pharmacological inhibition of FGFR1 between E9.5 and E10.5 in mouse resulted in smaller OE, less invagination, and a reduction in expression of critical OE transcriptional regulators PAX6 and NGN1. Additionally, there was reduction in cell proliferation, increase in cell death and reduction in cranial nerve axon growth (Karpinski et al., 2016). Studies in chick embryo explants revealed that FGFR1 uses PI3K pathway, particularly the p110-alpha isoform to regulate GnRH migration and olfactory sensory neuronal outgrowth (Hu et al., 2013). Loss of FGFR1 in mice results in disruption of OB development and a reduction in GnRH neuron numbers (Chung and Tsai, 2010). Mutations in components of the FGF signaling pathway (FGF8, FGF17 and FGFR1) have been identified in KS patients (Section “Spectrum of Genetic Disorders in GnRH Cell Deficiency and Kallmann Syndrome”).
Bone Morphogenetic Protein (BMP), a member of the transforming growth factor (TGF)-β superfamily, plays an important role in formation of the cranial placodes and neurogenesis in the OE. BMP signaling promotes formation of epidermis at the expense of neural induction and promotes respiratory epithelial cell fates (Maier et al., 2010). BMP is regulated by Noggin, a signaling protein involved in patterning and formation of dorsal structures. Our group has shown that BMP originates from nasal mesenchyme, and its expression, along with antagonist Noggin, is crucial in defining neuronal versus epidermal fates in the developing OP (Forni et al., 2013). Moreover, the combination of BMP inhibition with FGF signaling is required for inducing olfactory cell fates (Sjodal et al., 2007; Maier et al., 2010; Forni and Wray, 2015). GnRH neurons form in the VNO at the border between non-neural and neural epithelium, adjacent to mesenchymally expressed Noggin (Forni and Wray, 2015). Changes in BMP and Noggin (via loss of FGF8) also alter the neurogenic pattern of the OE and caused a reduction in the number of GnRH neurons in the developing embryo (Forni et al., 2013).
Wingless-Int (Wnt) signaling, patterns the anterior–posterior axis of the developing embryo and must be antagonized (along with BMP antagonism and FGF activation) for neural induction, cranial placode formation, and cell specification. Wnt is expressed in neural crest derived cells, including OECs and some progenitors, neurons and sustentacular cells of the OE (Forni et al., 2011; Katoh et al., 2011; Wang et al., 2011; Forni and Wray, 2012). In the canonical Wnt (beta-catenin dependent) pathway, Wnt binds to its transmembrane receptor Frizzled (Frz), which stabilizes beta-catenin and directs gene expression. Canonical Wnt is involved in OSN axon growth through various Wnt and Frz family members (Rodriguez-Gil and Greer, 2008). Activation of canonical Wnt signaling is also necessary and sufficient to drive the transition of horizontal basal olfactory cells from a resting to an activated neurogenic state to generate more OSNs. Additionally, loss of Wnt via beta-catenin knockout mice led to premature differentiation of progenitors and ultimately OSNs, while gain of function maintained a progenitor state (Fletcher et al., 2017).
Retinoic Acid (RA) signaling is crucial for craniofacial development. RA inhibits differentiation and maintains self-renewing progenitors before they commit to neurogenesis (Sabado et al., 2012). RA signaling is often analyzed via retinaldehyde dehydrogenases (RALDHs), which convert the retinaldehyde intermediate (retinal) to active RA. RALDH3 is the only member expressed within the neuroepithelium of the developing OE. RALDH3 mediates RA activity in the invaginating olfactory pit (Kawauchi et al., 2004). RA signaling also coordinates with FGF during OE development. RA and FGF pathways oppose each other to pattern the OP and to regulate the transition from self-renewing to neurogenic progenitor cells (Tucker et al., 2010). Inhibition of FGF signaling expands RALDH3 (Kawauchi et al., 2005). In chick, RA represses the formation of GnRH precursors at the time of specification but does not interfere with their subsequent development (Sabado et al., 2012).
Migration of GnRH cells in nasal regions is intimately entwined with both olfactory axon outgrowth and OEC migration. Many molecules directing this process have been identified and will be discussed below. Much less is known about the migration of GnRH cells once they cross into the forebrain and turn caudally toward the hypothalamus, as well as mechanisms that stop their migration and allow them, from multiple rostrocaudal locations, to then target their axons to the median eminence. Several model systems exist to study migration of the GnRH cells. These include in vivo analysis using different species, in situ slice preparations, immortalized cell lines, and explant models. The latter, described below, has been used by several groups and takes advantage of the embryonic origin of the GnRH cells, allowing one to study neuronal movement in primary GnRH cells devoid of brain tissue while maintaining many of the components surrounding the GnRH cells in vivo (Figure 3). In vivo, slice and explants models are feasible, because of the early expression of GnRH, allowing one to monitor their development.
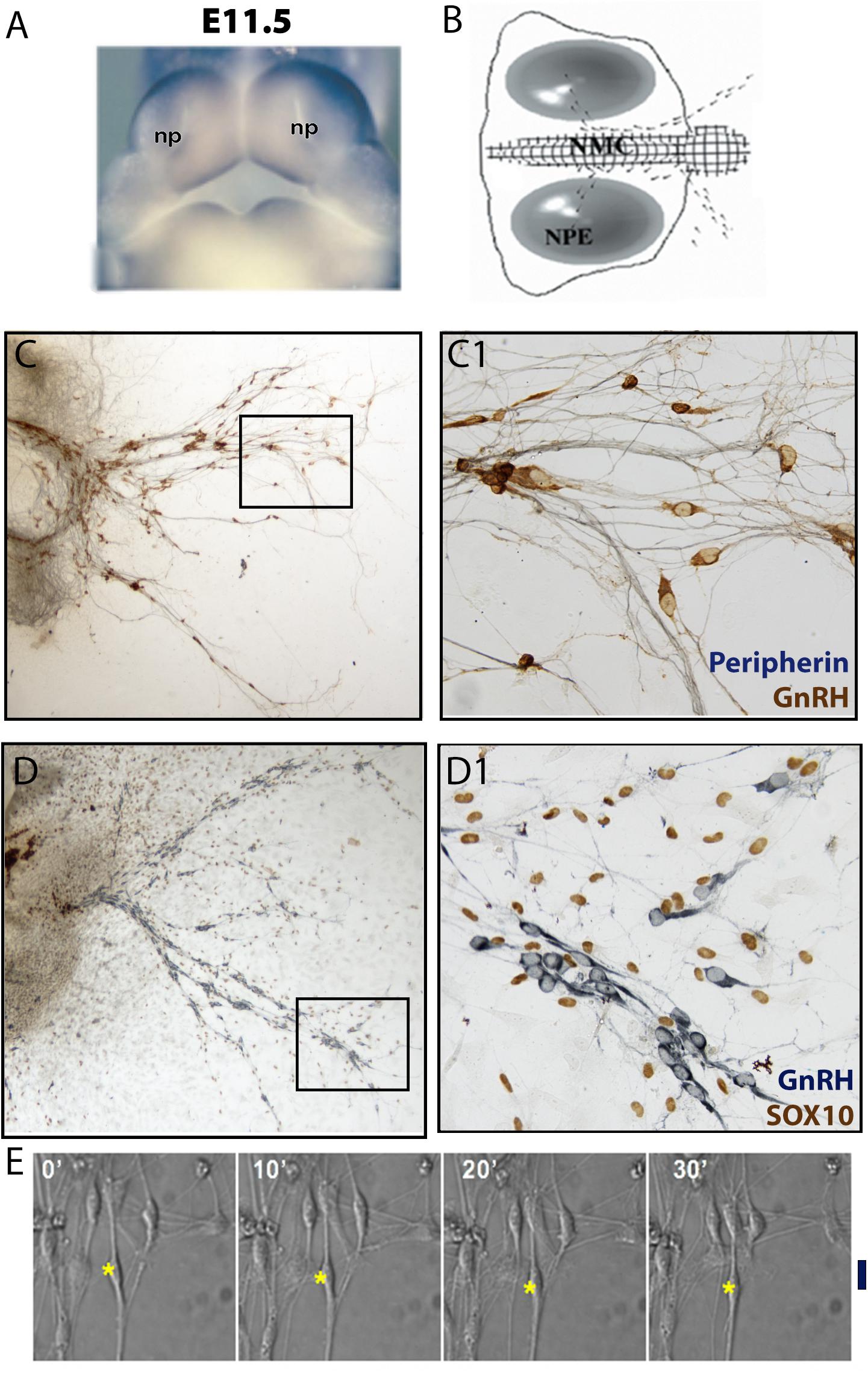
Figure 3. Nasal explant model and migration assay. (A) Picture of E11.5 mouse embryo showing the location of the nasal pits (NP). (B) Schematic of a nasal explant after 4 days in vitro (div); nasal midline cartilage (NMC) and olfactory/VNO epithelium (NPE). GnRH neurons (black dots) migrate out of the nasal pit, toward the NMC, and then into the explant periphery. (C) 4div explant stained for peripherin (blue, marking olfactory axons) and GnRH (brown) (D) 4div explant stained for GnRH (blue) and SOX10 (brown, marking olfactory ensheathing cells nuclei). Boxed areas are magnified in panels (C1,D1), respectively. (E) Example of migration assay using a 4div nasal explant. GnRH cell movement is recorded within a 30 min time frame. ∗, nucleus location of GnRH neuron; Black line, linear distance moved after 30 min.
GnRH neuronal migration was first categorized as axophilic/neurophilic since they used olfactory sensory axons/terminal nerve fibers to reach the forebrain (Wray et al., 1989a; Taroc et al., 2017). However, it is now clear that the GnRH cells in nasal regions migrate on tracks composed of (1) a subset of olfactory axon bundles originating from sensory cells in the VNO, (2) terminal nerve bundles/transient axons, (3) blood vessels, (4) other neurons and (5) OECs (Wray, 2002b, 2010; Forni and Wray, 2012). Although many diverse components are involved in the migration of GnRH cells, the overall process is maintained across vertebrates, as recent studies highlight the similarities between development of this system in mouse and human (Casoni et al., 2016). It should be noted however, that the location of the cells can vary depending on the species and may be a consequence of the species’ specific brain development as the cells migrate in. Yet, if one compares the development of GnRH cells in mouse and human, one finds that the GnRH cells become postmitotic early, rapidly increase in number and begin their migration quickly (Figure 4A; Wray et al., 1989b; Casoni et al., 2016). No sex differences are seen and although most GnRH cells integrate into the hypothalamic-pituitary axis and reside within the preoptic area (POA)/hypothlamus, scattered cells are seen in other regions including the hippocampus, cerebral cortex, and amygdala (Hoffman et al., 1992; Casoni et al., 2016). Although species differences in GnRH cell location exist, there are two unifying principles of GnRH cell distribution. First, they avoid defined cytoarchitectonically identified nuclei and second, they lie in a continuum extending from the olfactory bulbs, at varying distances, through the midline septum to the ventral hypothalmus. In mice, primates and humans, a relatively large population of GnRH cells in the ventral telencephalon has been identified (Quanbeck et al., 1997; Casoni et al., 2016), however, in mice and primates, these cells differ from the neuroendocrine GnRH expressing cells in their reactivity to anti-mammalian GnRH antisera (Quanbeck et al., 1997; Casoni et al., 2016; Wray personal observation). In primates, this population was shown to originate from the olfactory placode before the formation of the olfactory pit, and migrate into the brain along the olfactory nerve, rather than the terminal nerve, and eventually settle in striatal and limbic structures. In both mice and primates, these cells are smaller than neuroendocrine GnRH cells. In mice, these cells are not detected pre- or post-natally using in situ hybridization for GnRH mRNA (Wray personal observation) and their fate remains unknown. All data presented below refers to the neuroendocrine GnRH cell population.
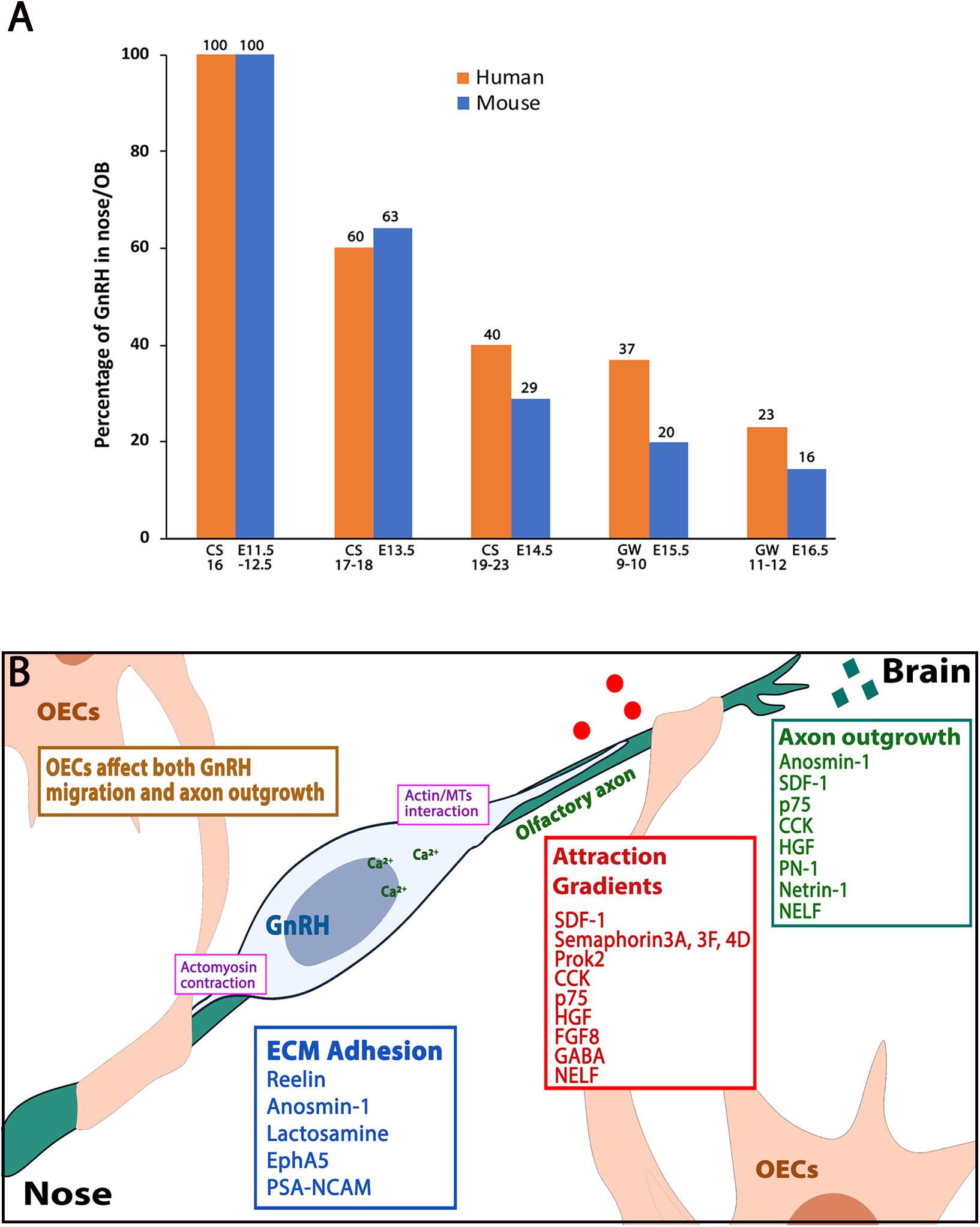
Figure 4. Human-mouse GnRH distribution plot and molecules affecting GnRH neuronal migration. (A) Plot showing percentage of GnRH neurons that reside in nose/OB area between human (orange bars) and mouse (blue bars) at five developmental windows. CS: carnegie stage, GW: gestation week, E: embryonic stage. Data adapted from Casoni et al. (2016) and Wray et al. (1989b). (B) Schematic showing a GnRH neuron (blue) along the olfactory axon bundles (green), which are ensheathed by OECs (beige). Categories of axon outgrowth cues (green box), migration attraction gradients cues (red box) and ECM adhesion molecules (blue box) are indicated. Actin/microtubule interactions and actomyosin contractions (pink boxes) occur at the leading process and the trailing process of the migrating GnRH neuron.
In mammals, GnRH neurons migrate along axons of cells that reside within the VNO (Figure 1). These axons, together with OECs and non-GnRH neurons, form a track/bundle – this is the migratory pathway on which GnRH cells move from the VNO to the cribriform plate. This bundle converges with axons and OECs from the mOE and targets the cribriform plate. Once through the cribriform plate and meninges, OECs and axons from sensory neurons in mOE and VNO turn rostrally to enter the olfactory bulb. GnRH cells on the other hand, follow a transient pathway/nervus terminalus (TN), caudally turning away from the olfactory bulb toward the hypothalamus (Wray et al., 1994; Tobet and Schwarting, 2006; Taroc et al., 2017). This nerve is transient in some species including humans (Fuller and Burger, 1990; Muller and O’Rahilly, 2004; Whitlock, 2004). VNO derived fibers in the nasal region are positive for TAG-1 (transiently expressed axonal surface glycoprotein, (Yoshida et al., 1995) and DCC (Deleted in Colorectal Carcinoma, netrin-1 receptor) (Deiner et al., 1997). The expression of these two markers is not detectable on the caudal trajectory. However, knocking out DCC results in the caudal trajectory turning toward the cortex (Schwarting et al., 2001) and misdirected GnRH cells (Deiner and Sretavan, 1999). In the olfactory sensory system of some vertebrates (zebrafish, xenopus, etc.) (Demski and Schwanzel-Fukuda, 1987; Muske and Moore, 1988; Mousley et al., 2006; Zhao et al., 2013), the TN remains after birth. Taroc et al. (2017) suggested that mouse GnRH neurons can properly migrate via TN fibers without the development of the OB and olfactory/VNO axonal connections. Further investigations are needed to distinguish what extracellular cues are guiding the TN outgrowth specifically. Some of the molecules guiding olfactory axons outgrowth are listed in Table 1.
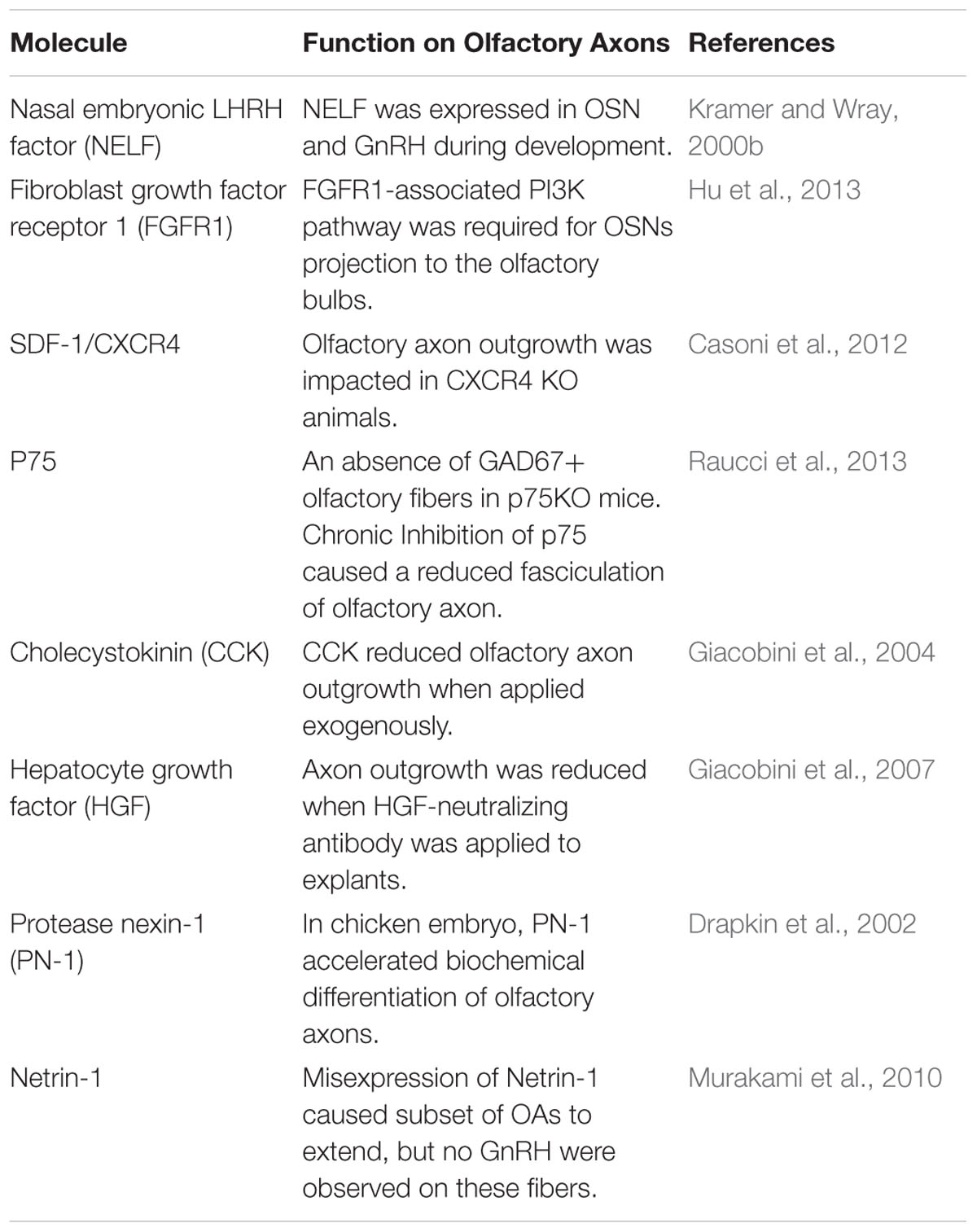
Table 1. Guidance molecules involved in embryonic olfactory axon outgrowth.
Olfactory ensheathing cells migrate along the olfactory axons through the nasal region, ensheathing the olfactory axon bundles going into the OB, and also reside in the external layer of the OB (Doucette, 1990; Cummings and Brunjes, 1997; Barnett, 2004; Miller et al., 2010). They are not detected on the caudal projection that GnRH neurons follow toward the hypothalamus. However, in nasal regions they are in close proximity to GnRH cells (Geller et al., 2013; Raucci et al., 2013; Dairaghi et al., 2018). OECs express p75NTR (nerve growth factor receptors) (Yan et al., 2001). Signaling via p75NTR has been shown to regulate OEC migration (Raucci et al., 2013). p75NTR KO mice showed reduced olfactory axons and OECs in the nasal region (Raucci et al., 2013). A subpopulation of GnRH cells also express p75NTR. Although ablation of p75NTR did not alter the total number of GnRH neurons, a small population of GnRH neurons migrated further in the hypothalamus in the p75NTR KO mice (Raucci et al., 2013). Whether this change is direct, or indirect via changes in OECs is unclear. Sox10 is expressed by migrating neural crest cells and required for the specification and differentiation of neural crest-derived Schwann cells and normal OEC differentiation (Barraud et al., 2013). Disruption of OECs by SOX10 mutations caused embryonic olfactory axons to accumulate in the ventromedial nerve layer in the OB, and slower migration of GnRH neurons into the forebrain was reported at early ages (Barraud et al., 2013). SOX10 mutations have been identified in KS patients (see below).
OSNs express distinct receptors and project to specific glomeruli in the main olfactory bulb or accessory olfactory bulb (Sakano, 2010). Independent of the location of the OSN, or their target, all sensory axons must traverse the cribriform plate to gain access to the central nervous system (CNS). This process is also critical for GnRH cells to enter the brain. Thus, outgrowth of fibers from the mOE can impact GnRH cell migration specifically at the cribriform plate. This was observed in Fez family zinc-finger protein 1 (Fezf1) KO mice. FEZF1 is a transcription factor that is essential for olfactory development and olfactory axon projection to the olfactory bulb (Watanabe et al., 2009). Experiments indicate that FEZF1 is required for the penetration of olfactory axons through the CNS basal lamina and thus for them to innervate the OB. When this does not occur, an axonal tangle results which halts GnRH neuronal migration as well. A homozygous loss of function in FEZF1 has been identified in KS patients, who showed attenuated OBs and absence of puberty (Kotan et al., 2014) (see Section “Spectrum of Genetic Disorders in GnRH Cell Deficiency and Kallmann Syndrome”).
While GnRH neurons have a unique migration route (from outside the brain), like other types of migrating neurons they require cell–cell adhesive mechanisms to remain on the olfactory axon bundles, as well as a variety of guidance molecule gradients (secreted peptides, growth factors, cytokines and chemokines etc.) to direct them. Nasal explant systems have been developed to study GnRH migration (Terasawa et al., 1993; Fueshko and Wray, 1994; Kanaho et al., 2009). These explant models: (1) contain a large number of primary GnRH neurons, olfactory sensory neurons and OECs; (2) allow one to perform single cell tracing and imaging; (3) are accessible to both molecular and pharmacological manipulations (Figure 3) (Wray et al., 1994; Klenke and Taylor-Burds, 2012). Although no brain is present, the GnRH cells mature in this model with respect to both processing of the peptide, extension of processes and cellular properties (Wray et al., 1994; Klenke and Taylor-Burds, 2012; Constantin, 2017), suggesting that the cells in explants are equivalent to their counterparts in vivo, aging appropriate. In explants generated from mice, olfactory axon outgrowth, OEC migration and GnRH cell migration occur between days 1 and 7 (Wray et al., 1996). However, after day 4, migration rates decrease (Casoni et al., 2016), similar to what would occur in vivo, as many of the cells have reached their final location by E15.5-E18.5. Certainly, when possible, parallel in vivo studies are run. Often the results are comparable, but not always, leaving one to search for redundancy in vivo that might not be present in explants or a maturation event that was not triggered in the explant. However, in the mouse nasal explant model, GnRH neurons migrate at 12–16 μm/h, and exhibit saltatory movement (Casoni et al., 2012), similar to GnRH neurons in studied in slices (Bless et al., 2005), and glial fiber mediated migration in vivo (Edmondson and Hatten, 1987). Thus, studying GnRH migration in primary nasal explant using techniques like live cell imaging, post hoc immunolabeling, calcium recording, and pharmacological/genetic manipulation has enhanced our understanding of how GnRH neurons develop and mature.
For a cell to migrate properly, the coordination of extracellular guidance cues and intracellular cytoskeletal elements is required. Actin filaments, microtubule bundles and centrosomes, plus cytoskeletal associated proteins are involved in migration (Coles and Bradke, 2015). In a migrating cell, cortical actin filaments are located underneath the plasma membrane and interact with microtubules, via multiple actin/tubulin binding proteins (Rodriguez et al., 2003). Intracellular signals control the extension and retraction of microtubule bundles, which creates a “pulling force.” Saltatory movement has been characterized in many types of migrating neurons (Schaar and McConnell, 2005), including GnRH neurons (Casoni et al., 2012). The sequence of events in migrating GnRH cells includes: (1) the leading process extends and contacts on the scaffolding that supports the migration (glial fiber, sensory axons, ECM, etc.), (2) the centrosome moves forward into a bulging area in the leading process, (3) the microtubule bundles form a cage around the nucleus, and the “pulling force” from actin/microtubule interaction pulls the nucleus toward the centrosome, and (4) myosin II, at the rear end of the moving cell, generates a “pushing force” by actinomyosin contraction (Hutchins et al., 2013). The saltatory movement pattern of GnRH neurons is similar to radial migrating cells in the developing cortices (Edmondson and Hatten, 1987; Schaar and McConnell, 2005) and tangential migrating neurons in medial ganglionic eminence (Bellion et al., 2005). Ca2+ signaling is known to be involved in the transduction of extracellular cues and conformational change of actin mesh (Komuro and Kumada, 2005; Fahrion et al., 2012). Hutchins and Wray (2014) showed that by blocking or enhancing Ca2+ signaling, GnRH neuronal migration was inhibited or increased accordingly. Furthermore, live actin tracing in migrating GnRH neurons revealed that leading process actin mesh translocation and rear actin contraction corresponded with Ca2+ signaling (as observed in glial mediated neuronal migration, (Solecki et al., 2009), and microtubule plus end capturing in the actin mesh was reduced when Ca2+ signaling was inhibited via the calcium channel (IP3 receptors) blocker, 2-APB (Hutchins and Wray, 2014). Cortactin is known to promote F-actin polymerization and cellular motility in fibroblast (Nakane et al., 2012). Sirtuin1 (a NAD-Dependent Protein Deacetylase Sirtuin-1) deacetylates cortactin and was in GN11 cells (an immortalized GnRH cell line). Sirtuin1 deficient mice have an IHH phenotype due to defective GnRH neuronal migration (Di Sante et al., 2015). Sirtuin1-Cortactin co-localization in the GN11 cells was FGF8/FGFR1 dependent, suggesting that FGF signaling maybe involved in GnRH neuronal migration as well as OP development.
The semaphorin (SEMA) proteins, acting as guidance cues, are necessary for appropriate development of the GnRH/olfactory system. Disruption of three class 3 SEMAs, SEMA3A, SEMA3F and SEMA3E perturbs GnRH development (Messina and Giacobini, 2013; Cariboni et al., 2018). SEMA3A and SEMA3F, are involved in proper GnRH migration. SEMA3A binds to neuropilin (NRP) 1 and 2 (Kolodkin et al., 1997; Reza et al., 1999). In mice, lack of SEMA3A altered VNO nerves and inhibited GnRH cell migration (Cariboni et al., 2011) while, lack of either SEMA3E or its receptor, PLXND1, increased apoptosis of GnRH neurons and reduced GnRH terminals in the median eminence (Cariboni et al., 2015). SEMA4D is expressed on the GnRH cell migratory pathway, with higher expression in the hypothalamic area (Giacobini et al., 2008). SEMA4D binds to PLEXINB1, which is robustly expressed in the OP during development (Perala et al., 2005) as well as on migrating GnRH cells (Giacobini et al., 2008). PlexinB1 KO mice show a delay in GnRH migration and reduced GnRH cells in adult brains. SEMA3A, SEMA3E and SEMA7A mutations have been reported in KS patients (Hanchate et al., 2012; Young et al., 2012; Kansakoski et al., 2014) (see below).
Reelin is an ECM glycoprotein. Reelin has a canonical pathway where it binds to low-density lipoprotein receptor (VLDLR) and apolipoprotein R receptor 2 [APOER2, (Hiesberger et al., 1999)], and eventually leads to the degradation of Disabled 1 (DAB1). Non-canonical pathways of reelin (independent of DAB1 mechanism) were reported in migrating interneurons in the olfactory bulb (Hellwig et al., 2012), lymphatic vascular development (Lutter et al., 2012) and hypothalamic GnRH neurons (Cariboni et al., 2005). However, a recent study (Dairaghi et al., 2018) showed that both VLDLR, APOER2 and DAB1 were, in fact, expressed in GnRH cells as well as in OECs. Although immunodepletion of Reelin using an explant model inhibited GnRH and OEC migration, no change in GnRH or OEC distribution was detected in homozygous reelin mutant mice.
Several G protein-coupled receptors and their ligands modulate GnRH cell movement. Stromal cell derived factor-1 (SDF-1) and its receptor chemokine receptor type 4 (CXCR4) regulate neuronal migration and axonal pathfinding in a number of brain regions (Stumm and Hollt, 2007). CXCR4 is detected on cells in the OE and on GnRH neurons during development. An SDF-1 signal is seen in GnRH cells as well as cells in the midline cartilage and the mesenchyme surrounding the OE (Schwarting et al., 2007; Toba et al., 2008). Both CXCR4 and SDF-1 are also expressed on the olfactory nerves. Cxcr4 homozygous KO animals die at E18.5, but younger embryos have disrupted GnRH cell migration and inhibited olfactory axon outgrowth (Schwarting et al., 2007; Toba et al., 2008). SDF-1/CXCR4 signaling accelerates the movement of GnRH neurons in explants, utilizing GIRK channels to promote GnRH movement (Casoni et al., 2012). Prokineticin 2 (PROK2) has been reported to be associated with olfactory bulb development and GnRH cell migration (Ng et al., 2005). KO mice of PROK2 or its receptor PROKR2 show a decrease in GnRH cells in the forebrain (Matsumoto et al., 2006; Zhao et al., 2019). However, ProkR2 KO mice have a reduced OB size, whereas Prok2 KO mice have normal sized OB (Martin et al., 2011). Mutations of PROK2 and PROKR2 are seen in patients with KS (Dode et al., 2006). Expression of the neuropeptide cholecystokinin (CCK) and its receptors CCK-1R and CCK-2R are seen in developing OE, olfactory axons, and the VNO (Giacobini et al., 2004). GnRH neurons express CCK-1R but not CCK-2R. Exogenous application of CCK to nasal explants reduced GnRH migration and olfactory axon outgrowth. In Cck1R KO mice, the total number of GnRH cells was identical in wild-type and mutant mice at E14.5. However, the number of GnRH neurons within the forebrain was significantly greater in Cck1R-/- embryos, consistent with an accelerated migratory process. These results indicate that CCK provides an inhibitory influence on GnRH neuronal migration, contributing to the appropriate entrance of these neuroendocrine cells into the brain. G protein-coupled receptor 37 (GPR37 or PAEL-r) and its ligand prosaposin (PSAP) have recently been shown to be expressed in developing GnRH neurons and OECs. Inhibition of GPR37 attenuates migrations of GnRH neurons and OECs, and acute migration assay shows that application of a GPR37 agonist accelerates GnRH migration. In GPR37 KO animals, GnRH development and migration were delayed. GPR37 KO animals also showed an altered OB development, with a thinner olfactory nerve layer present in adults (Saadi et al., 2019).
Hepatocyte growth factor (HGF), Gamma-amino butyric acid (GABA) and nasal embryonic LHRH factor (NELF) have also been shown to alter GnRH cell movement. HGF is a cytokine that binds to its receptor MET tyrosine kinase (Ebens et al., 1996; Giacobini et al., 2007). HGF is expressed in nasal mesenchyme during embryonic development with an increased concentration toward the nasal forebrain junction (Sonnenberg et al., 1993; Giacobini et al., 2007). MET is expressed in migratory GnRH neurons, but not in post-migratory GnRH cells (Giacobini et al., 2002). In mice, KO of tissue-type plasminogen activator (tPA, an HGF activator) reduced the total number of GnRH cells (Giacobini et al., 2007). In nasal explants, blocking HGF with a neutralizing antibody caused a reduction in GnRH cell migration and olfactory axon outgrowth. This effect was then rescued by exogenous application of HGF. During development GABA acts as an excitatory neurotransmitter. During GnRH cell migration, a subset of GnRH neurons (30%) express GABA (Tobet et al., 1996), as do the olfactory axons and migratory cells along the migratory route (Wray et al., 1996). Expression of GABAAR is seen on the majority of GnRH neurons, but with heterogeneous subunit compositions (Fueshko et al., 1998; Bless et al., 2000). The GABAAR agonist Muscimol inhibited GnRH cell migration and reduced GnRH fiber extension. The GABAA antagonist Bicucullin disorganized GnRH cells in the forebrain and dissociated GnRH cells from olfactory fibers (Bless et al., 2000). Activation of GABAAR in explant resulted in increased Ca2+ signaling and membrane depolarization in GnRH neurons and promoted GnRH migration (Moore and Wray, 2000), with GABA signaling tending to orient cell movement (Casoni et al., 2012). Kramer and Wray first identified nasal embryonic LHRH factor (NELF, now termed NSMF) in 2000, where NSMF expression was detected in mouse nasal epithelia, migrating GnRH neurons and olfactory axons (Kramer and Wray, 2000b). Both olfactory axon outgrowth and GnRH migration were attenuated after applying antisense oligonucleotides. Studies in Nsmf KO mouse are conflicting. One study reported that in females there was a decrease in GnRH number, as well as an impaired fertility (delayed vaginal opening, decreased uterine size, and reduced mean litter size). No delayed puberty was observed in males (Quaynor et al., 2015). However, in a second Nsmf KO model, no changes in GnRH neuronal number or reproductive function was observed (Spilker et al., 2016). To date, six variant transcripts of Nsmf have been identified (Quaynor et al., 2014). Further studies are needed to identify whether splice variant expression might account for these differences. A NSMF heterozygous missense (T480A) (Miura et al., 2004) and three cases of NSMF digenic mutations (Pitteloud et al., 2007; Xu et al., 2011) have been reported associated with KS patients.
Migrating neurons following a pathway must attach and subsequently detach to facilitate saltatory movement. A variety of adhesive molecules are implicated in this mechanism for GnRH neurons. NCAMs are surface associated adhesion molecules. PSA-NCAM is expressed in developing nervous system regions that are associated with migrating neurons and synaptogenesis (Kiss and Rougon, 1997). PSA-NCAM is seen on GnRH neurons and along the migratory pathway and is suggested to be involved in the pathway formation, as well as guiding GnRH neurons from the VNO to the brain (Schwanzel-Fukuda et al., 1992, 1996; Yoshida et al., 1995). PSA-NCAM is also seen on OECs (Franceschini and Barnett, 1996), though no functional studies have been done to date. The function of PSA-NCAM is believed to be highly dependent on the sialic acid polymer chain (Yang et al., 1994). Experiments in chick embryos examining a late migratory stage showed that removal of PSA (via Endo N, a PSA specific enzyme) from the PSA-NCAM molecule resulted in a significant reduction of caudal turning GnRH neurons in the forebrain, however, the morphology of the caudal turning fibers did not change (Murakami et al., 2000). Notably, PSA-NCAM expression is not detectable when GnRH neurons reach their destination (Murakami et al., 1991).
Glycoconjugates containing terminal lactosamine at the carbohydrate chains have been seen in neurons within the OE and the VNO. Beta1,3-N-acetylglucosaminyltransferase-1 (B3GNT1), an enzyme that regulates the synthesis of lactosamine, is expressed in cells in the OE and VNO, and migratory GnRH neurons. Lactosamine expression is high on migrating GnRH neurons but decreases dramatically in post-migratory GnRH cells. In B3gnt1 KO mice, more GnRH neurons were seen at E15 in olfactory region while fewer GnRH neurons were detected in the forebrain (Bless et al., 2006). Ephrins are a family of cell surface proteins that binds to Ephrin receptor tyrosine kinases. In a mouse model with overexpressed subtype 5 Ephrin type-A receptor (EPHA5), GnRH neuronal migration was disrupted. It is proposed that the overexpression of EPHA5 caused abnormal adhesion of GnRH/olfactory axons (Gamble et al., 2005).
Idiopathic hypogonadotropic hypogonadism (IHH) is a rare disorder that results from the failure of the normal episodic GnRH secretion, leading to delayed puberty and infertility. Since GnRH neurons arise in the olfactory placode and share developmental events with the olfactory system, approximately 60% of IHHs are accompanied by an impaired sense of smell (anosmia), which is categorized as KS. The remaining 40% of IHHs patients show a normal sense of smell (nIHH) (Sedlmeyer and Palmert, 2002; Lewkowitz-Shpuntoff et al., 2012). Notably, some mutations associated with IHH can cause either KS or nIHH, suggesting other mutations or underlying dysfunctions may be present. About 60% of patients with IHH are sporadic cases without family history, while ∼40% are familial cases who already have family member(s) with associated symptoms (Seminara et al., 1998; Oliveira et al., 2001). With the development of medical treatment for IHH, patient fertility rates have increased, increasing offspring and the proportion of familial cases among the IHH patient population. This phenomenon emphases the importance of understanding the underlying genetics of IHH to develop genetic diagnosis and genetic counseling for patients.
Idiopathic hypogonadotropic hypogonadism can be diagnosed at different developmental stages. Some patients with IHH show clinical features such as cryptorchidism and/or microphallus at birth, and may have biochemical evidence of low gonadotropins and sex steroids during the minipuberty (occurs at 3–6 months in humans, and is accompanied by a transient activation of the HPG axis and higher levels of gonadotropins/sex steroids) (Tapanainen et al., 1981; Kuiri-Hanninen et al., 2011a,b). However, most patients with IHH do not present with neonatal features and can only be diagnosed at the time of puberty. In either sex, the patients cannot develop secondary sex characteristics. In females this includes growth of body hair, start of menstruation (primary amenorrhea), and enlargement of breasts. In males this includes growth of facial and body hair, deepening of voice, enlargement of the scrotum and testes followed by growth of the penis (Aksglaede et al., 2009; Herman-Giddens et al., 2012; Boehm et al., 2015).
Variable phenotypes and penetrance of olfactory symptoms are observed in IHH patients. KS patients can exhibit an absence of smell (anosmia) to decreased sense of smell (hyposmia) (Pitteloud et al., 2006; Trarbach et al., 2006; Bianco and Kaiser, 2009). Olfactory function is evaluated by history and by formal diagnostic smell tests (Doty, 2007) which allow classification of the patient as anosmic, hyposmic, or normosmic. In families with KS affected members, anosmia has been reported to be present as an isolated symptom (Dode et al., 2003; Pitteloud et al., 2006). In addition to attenuated olfactory function, anatomical malformations of the olfactory bulbs are frequently observed in MRIs of KS patients, with partially developed (hypoplasia) or complete absence (aplasia) of the olfactory bulb reported (Truwit et al., 1993; Sato et al., 2004).
Kallmann syndrome patients can exhibit a wide spectrum of non-reproductive features resulting from aberrant developmental events including midline anomalies (a cleft lip/palate, high-arched palate, and dental agenesis) (Dode et al., 2003; Falardeau et al., 2008; Kim et al., 2008; Jongmans et al., 2009), ear impairment (hearing loss, external ear anomalies, and semicircular canal dysplasia) (Dode et al., 2003; Zenaty et al., 2006; Jongmans et al., 2009), eye problems (coloboma of the eye and abnormal eye movements) (Kim et al., 2008; Jongmans et al., 2009), unilateral renal agenesis (Wegenke et al., 1975; Georgopoulos et al., 2007), abnormalities of bones in limb and digit, and bimanual synkinesis (involuntary movement of one hand that are mirrored by the other hand) (Quinton et al., 2001; Costa-Barbosa et al., 2013).
The appearance of high throughput sequencing techniques and advances in neuroimaging technology have led to an impressive advance in the molecular genetics of KS in the past 30 years, from the first finding of a causative gene associated with KS, KAL1 (ANOS1), in 1991 (Franco et al., 1991; Legouis et al., 1991; Bick et al., 1992; Hardelin et al., 1992) to 25 genes today (see Table 2). It was believed that a mutation in a single gene was sufficient to cause KS (monogenic disorder) and the penetrance of the mutation in a patient resulted in phenotypic variability within and across families. However, recent studies show the existence and prevalence of oligogenism in KS patients, indicating that a combination of mutations or rare variants on two or more genes can underlie the disease (di/oligogenic disorder) (Sykiotis et al., 2010; Miraoui et al., 2013). In fact, oligogenism is now thought to explain the complex variability in some of the clinical phenotypes observed in IHH patients in general (Dode et al., 2006; Martin et al., 2011; Cox et al., 2018). The known genes involved in KS can be categorized by their inheritance pattern (X-linked, autosomal dominant, autosomal recessive, oligonism, and de novo) as shown in Table 2.
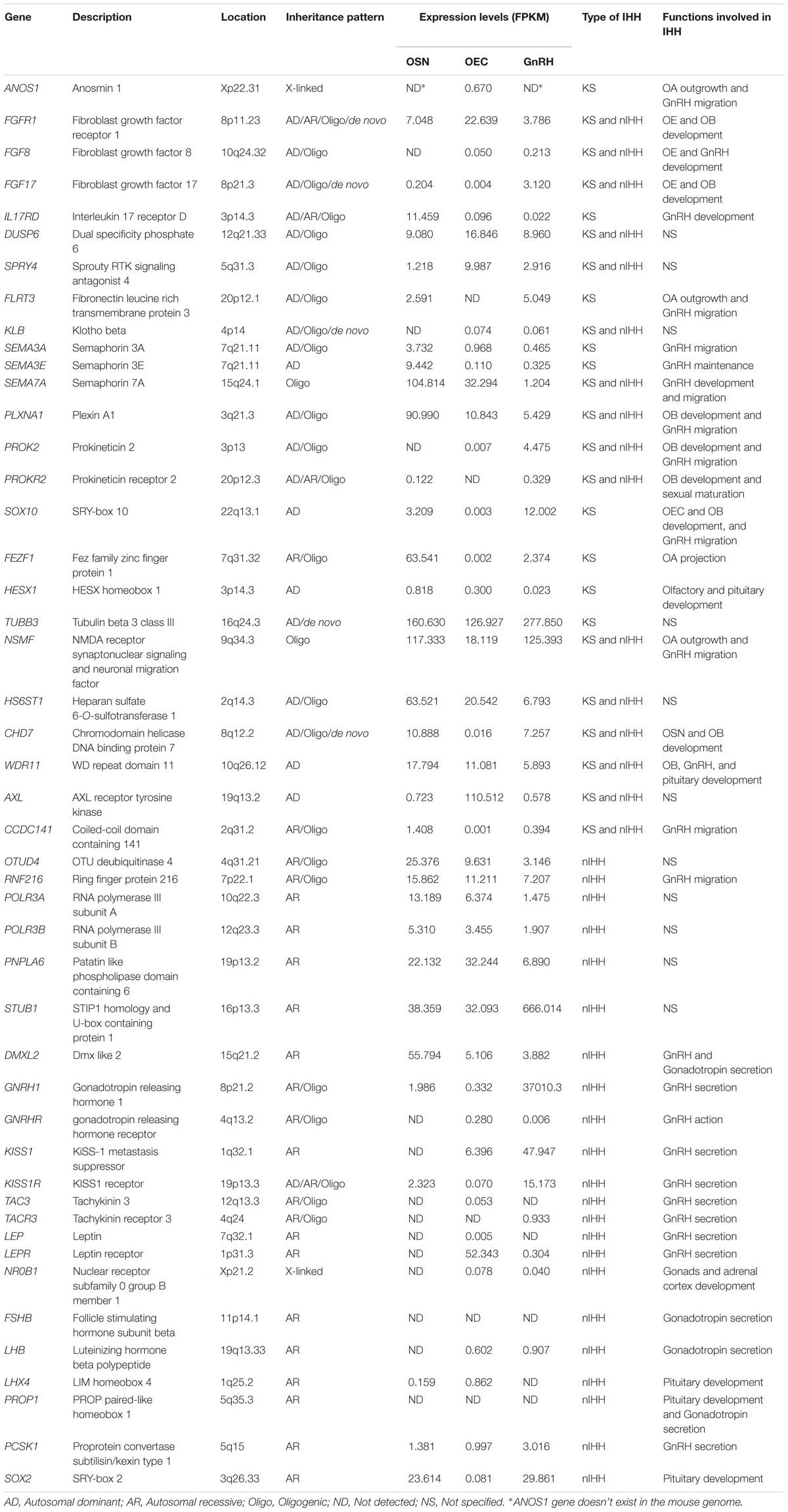
Table 2. The known causative genes related with idiopathic hypogonadotropic hypogonadism.
The genes mutated in KS patients include: ANOS1, FGFR1, FGF8, FGF17, IL17RD, DUSP6, SPRY4, FLRT3, KLB, SEMA3A, SEMA3E, SEMA7A, PLXNA1, PROK2, PROKR2, SOX10, FEZF1, HESX1, TUBB3, NSMF, HS6ST1, CHD7, WDR11, AXL, and CCDC141. Genes identified in only nIHH patients have a role in the development of the hypothalamic-pituitary axis (NR0B1), neuroendocrine physiology of the normal secretion of GnRH (GnRH1, KISS1, KISSIR, TAC3, TACR3, LEP, and LEPR) or its action on the pituitary (GNRHR) (Stamou and Georgopoulos, 2018), and will not be discussed here. The genes identified in patients with KS are primarily neurodevelopmental genes. Notably, if the GnRH cells fail to enter the forebrain, reproductive function will be lost, and the mutation removed from the population. In some of the mouse models examined, a subpopulation of GnRH cells is able to enter the CNS, and subfertility is observed, indicating that redundant systems are present to ensure GnRH cells enter the forebrain and maturation of the reproductive system occurs. The biological role of several KS associated genes have been discussed in the previous two sections. These include FGFR1, FGF8, SEMA3A, SEMA3E, SEMA7A, PLXNA1, PROK2, PROKR2, SOX10, CCDC141, FEZF1, ANOS1 and NSMF. Below the remaining KS genes are discussed and clinically relevant sections are added for some of the genes mentioned earlier.
ANOS1 was first identified as one of multiple genes lost in a large deletion on the X chromosome of a patient with that showed multiple clinical phenotypes such as short stature, chondrodysplasia punctata, intellectual disability, ichthyosis and KS (Franco et al., 1991; Legouis et al., 1991; Bick et al., 1992; Hardelin et al., 1992). Anosmin-1 is an ECM glycoprotein and a product of ANOS1 gene (de Castro et al., 2017). Anosmin-1 contains four consecutive fibronectin type III domains (FNIII), known to be present in the proteins involved in cell adhesion, tyrosine kinases and phosphatases (Franco et al., 1991; Hu and Bouloux, 2011). Unfortunately, the corresponding region of the X chromosome containing ANOS1 was lost in mouse. However, experiments in Medaka showed, consistent with the X-KS phenotype, antisense knockdown of the Medaka KAL1 ortholog resulted in the disruption of forebrain GnRH neuronal migration (Okubo et al., 2006). In addition, applying protein to rodents lacking the gene have suggested Anosmin-1 regulates cell adhesion (Soussi-Yanicostas et al., 1998; Bribian et al., 2008), migration (Cariboni et al., 2004), and neurite outgrowth and branching (Soussi-Yanicostas et al., 2002; Diaz-Balzac et al., 2015). Mutations in ANOS1, account for approximately 10–20% of KS cases. These mutations include frameshift, nonsense mutations, and a few missense mutations and splicing mutations have been identified (Albuisson et al., 2005; Pedersen-White et al., 2008; Goncalves et al., 2017; Maione et al., 2018). Almost all patients with an ANOS1 mutation suffer anosmia or hyposmia (Quinton et al., 2001; Goncalves et al., 2017; Nie et al., 2017). Synkinesis and renal agenesis are often observed in these patients but do not always co-segregated with the mutation in the pedigree. Hearing loss and vas deferens agenesis are also reported phenotypes occasionally seen in patients with ANOS1 mutation (Dode and Hardelin, 2010; Madhu et al., 2012; Costa-Barbosa et al., 2013; Xu et al., 2015).
FGF17 has strong sequence homology, similar splicing, and a similar FGFR1 binding specificity as FGF8 (Olsen et al., 2006; Valdes-Socin et al., 2014). During development, FGF17 expression overlaps with FGF8 in the olfactory placodes, where FGF17 is expressed specifically in the ectoderm at the medial side of the nasal pits from which GnRH cells eventually arise (Bachler and Neubuser, 2001). In addition, FGF17 is important for proper craniofacial development (Kasberg et al., 2013). Although its function is similar to FGF8, FGF17 has a milder phenotype when knocked out in mice. Fgf17 knockout mice survive into adulthood, display smaller olfactory bulbs but increased volume in the hypothalamus (Yu et al., 2011), and are fertile. In humans, FGF17 mutations have been identified in 2/199 individuals with KS (Miraoui et al., 2013). One of these mutations appeared with other mutations in FLRT3, HS6ST1, FGFR1. This same study also proposed that FGF17 served as an alternative ligand to FGF8b in GnRH neuron development, as both FGF8b and FGF17 bind FGFR1c, which is also mutated in some KS individuals.
Miraoui and colleagues (2013) used targeted Sanger sequencing to perform a functional candidate-gene approach on a large group of KS/nIHH cohorts (386 IHH probands, 199 KS and 187 nIHH), and identified mutations in genes from the FGF8 subfamily (FGF17 and FGF18) and their synexpression group (IL17RD, DUSP6, SPRY2, SPRY4, and FLRT3). They found mutations in five of seven genes screened: IL17RD, FGF17, DUSP6, SPRY4, and FLRT3. IL17RD was first identified as synexpression gene of Fgf genes during embryonic development (see Figure 5) and named as SEF (Similar Expression to Fgf genes). SEF expression is positively regulated by FGF and functions as an antagonist of FGF signaling in zebrafish or Xenopus laevis (Furthauer et al., 2002; Tsang et al., 2002). Seven different mutations on IL17RD have been identified from 8 patients, and 2 of them have additional mutations, one with FGFR1 and the other with KISS1R. All the patients with a mutation in IL17RD were anosmic. 75% of these patients exhibited hearing loss (Miraoui et al., 2013). Some of the patients also exhibited abnormal dentition, or low bone mass. A functional relation of IL17RD with the auditory system has been described in two models. Defected auditory development is observed in overexpressed chick and mutant mice (Abraira et al., 2007). These data suggest that mutations in genes encoding components of the FGF pathway are associated with complex modes of KS inheritance and act primarily as contributors to an oligogenic genetic architecture of this disease.
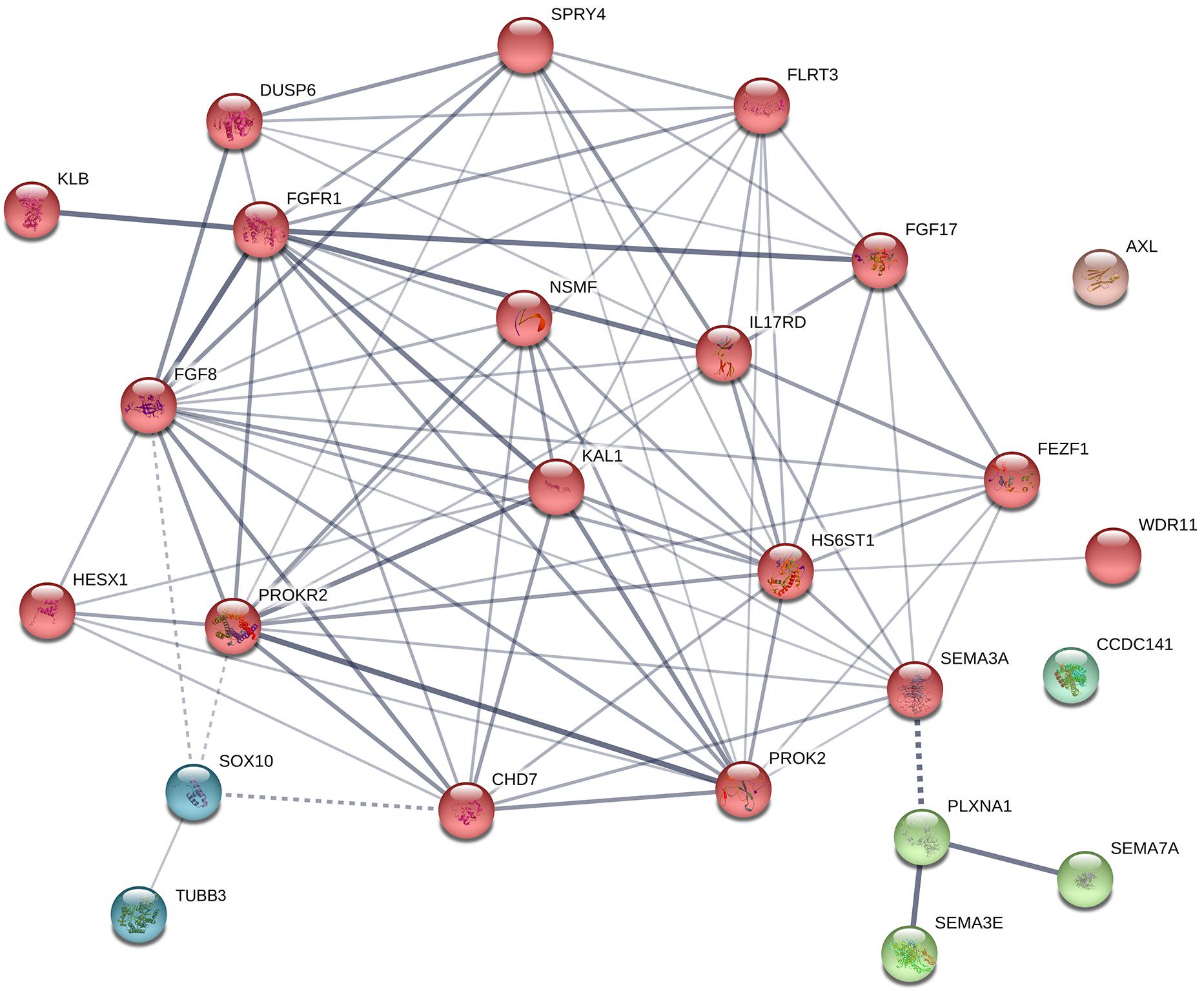
Figure 5. Protein–protein interaction network in known genes of KS. The diagram shows known and predicted interactions among KS causative genes listed in Table 2, created using String software version 11.0 with minimal confidence score of 0.4. Total 87 lines (interactions) appeared across 25 nodes. The colored nodes symbolize proteins which clustered by MCL algorithm. The thickness of the line represents confidence of protein–protein associations (with score 0.4, 0.7, and 0.9, relatively). Solid line, interactions within a cluster; Dotted line, interactions between clusters.
Dual specificity phosphatase 6 (DUSP6), encodes MKP3, a Mitogen-activated protein kinase (MAPK) phosphatase which inhibits the MAPK pathway, and therefore FGF signaling, via dephosphorylation of active extracellular-signal-regulated kinase (ERK), in negative feedback loop (Li et al., 2007). DUSP3/MKP3 is expressed during embryonic development in areas corresponding to FGF signaling including the forebrain and OE, and requires FGF signaling for its expression (Ishibashi et al., 2001; Kawakami et al., 2003; Li et al., 2007; Urness et al., 2008). The loss of MKP3 is postnatally lethal (Li et al., 2007). In humans, mutations in DUSP6 have been associated with KS (2.5%). Miraoui et al. (2013) identified 5 KS individuals with DUSP6 mutations, three of which occurred with other mutation in FGF8 network genes (SPRY4 and FGFR1).
Sprouty homolog 4 (SPRY4) is a cytoplasmic membrane-associated protein that inhibits receptor tyrosine kinase signaling and antagonizes FGF, EGF, and VEGF signaling via MAPK pathway inhibition (Hacochen et al., 1998; de Maximy et al., 1999). SPRY4 expression is regulated by FGF signaling (Minowada et al., 1999), and is found in the developing brain, nasal placode, inner ear, limb buds, branchial arches, tail bud, lungs, and areas of the developing gut (de Maximy et al., 1999; Zhang et al., 2001). In mice, the loss of SPRY4 resulted in dwarfism, polysyndactyly (a combination of polydactyly and syndactyly where extra digits are formed but contained in a single skin envelope), some abnormal tooth development, and mandible (jawbone) defects (Taniguchi et al., 2007). Mutations in SPRY4 were identified in 9 KS patients, and two showed loss of bone mass, three had hearing loss, and one had abnormal dentition. Three of the KS patients also had mutations in additional FGF8 network genes (DUSP6 and FGFR1, Miraoui et al., 2013).
FLRT3 is a putative type-I transmembrane protein (Lacy et al., 1999; Haines et al., 2006). FLRT3 expression overlaps with some FGF8 expression and is regulated by FGF signaling (Bottcher et al., 2004). FLRT3 interacts with FGFR1 and promotes FGF signaling via MAPK pathway during early development (Bottcher et al., 2004; Haines et al., 2006). FLRT3 is a cell surface adhesion protein that is critical for neurite outgrowth (Robinson et al., 2004; Tsuji et al., 2004), and interacts with the receptor ROBO1 for Netrin-1 dependent axon guidance (Leyva-Diaz et al., 2014). Flrt3 knockout mice die at early stages due multiple developmental defects (Egea et al., 2008; Maretto et al., 2008). Miraoui et al. (2013) identified 3 KS individuals (1.5%) with mutations in FLRT3. However, 2 of these patients also had mutations in other FGF-network-associated genes, including FGFR1.
Xu et al. (2017) examined the FGF21/FGFR1/Klotho Beta (KLB) signaling pathway that mediates the response to starvation and other metabolic stresses in 227 KS patients. KLB and FGFR1 are co-receptors for FGF21. KLB binds to the FGFR1, competing with FGF8 for the same binding pocket (Goetz et al., 2007, 2012; Kurosu et al., 2007; Ogawa et al., 2007; Owen et al., 2015; Misrahi, 2017). Large scale genetic screening revealed six different heterozygous KLB mutations in 11 KS patients. Some of these patients had a metabolic phenotype as well, such as obesity and insulin resistance. Mutations occurred as autosomal dominant, de novo, or oligogenic in a pedigree. 2/11 of these patients had no other identified mutation. The other patients were found to have mutations in FGF8 or PROKR2 or FGFR1 together with the KLB mutation. Double KO of two KLB homologs in C. elegance, klo-1 and klo-2, resulted in subfertility, with a 40% reduction of ∼40% egg laying compared to WT (Xu et al., 2017).
KO in Fezf1 was known to alter olfactory axon outgrowth and GnRH neuronal migration (Watanabe et al., 2009) (see Section “Migration of GnRH Neurons From the Nose to the Brain”). Two homozygous mutations in FEZF1 were identified from two different KS families through the exome sequencing and autozygosity mapping (Kotan et al., 2014). One missense mutation was detected together with a second homozygous mutation in the CCDC141 (Hutchins et al., 2016; Turan et al., 2017). These patients did not show any additional features associated with KS such as cleft palate, deafness, and synkinesis. The phenotype of the patients suggests that FEZF1 is spatially and temporally restricted in expression and functionally specific during embryogenesis. The FEZF1 missense mutation exhibited partial function in vitro as a transcriptional repressor suggesting the involvement of the CCDC141 mutation in the KS phenotype (Kotan et al., 2014).
A screen of KS patients identified three heterozygous missense HESX1 mutations (3.6%) (Newbern et al., 2013). HESX1 is a paired-like homeobox transcriptional repressor that is first expressed at the onset of gastrulation. The Hesx1 homozygous mutant mouse generated two classes of phenotypes. The most severe (class I) consisted of substantial reduction of the prospective forebrain, Rathke’s pouch, olfactory placodes and frontonasal mass along with loss of optic vesicles. Class II was milder with an overall reduction of the forebrain and less craniofacial dysplasia, thinner OE with reduced or absent VNO, abnormal Rathke’s pouch, and usually only one eye affected (Dattani et al., 1998). Because HESX1 is required for pituitary development, the KS patients with mutations in this gene may have low LH/FSH levels related to pituitary defects and/or defects in the development of the GnRH system. To date, a model to examine this issue has not been explored.
TUBB3 is originally identified as a genetic cause of congenital fibrosis of the extraocular muscles (CFEOM), a complex dysmotility disorder characterized by ptosis and strabismus, with or without additional neurological findings. However, detailed phenotypic analysis on patients with de novo heterozygous mutation c.1228G > A (p. E410K) showed additional phenotypes such as KS, intellectual disability, facial weakness, tracheomalacia, vocal cord paralysis and late onset cyclic vomiting and progressive peripheral neuropathy. TUBB3, primarily expressed in neurons, is a class III member of the beta-tubulin protein family, which heterodimerizes with alpha-tubulin to form microtubules. In vitro functional assays showed all the mutations identified in TUBB3 altered microtubule dynamics, and some of mutations (including c.1228G > A (p. E410K)) also perturbed interaction of microtubule with kinesin (Tischfield et al., 2010). Chew et al. (2013) suggested defining a distinctive “TUBB3 E410K syndrome.” All tested patients with E410K showed strong penetrance of the KS phenotype (hypoplasia to absent olfactory sulci, and olfactory bulbs, and hypogonadotropic hypogonadism) without any additional mutation in other candidate genes (Chew et al., 2013). However, in 2015, a female patient with a E410K mutation in TUBB3 was identified and exhibited normal fertility yet displayed all other features of the E410K syndrome, including anosmia. This heterozygous mutation was inherited by her three sons, as well as variable features of additional endocrine abnormalities (Balasubramanian et al., 2015). Tubb3 KO mice show no notable neurobehavioral or neuropathological deficits that overlap with above described patient (Latremoliere et al., 2018).
HS6ST1 is one of three vertebrate genes that encodes an enzyme with HS 6-O-sulfotransferase activity (Habuchi et al., 2000). HS6ST1 is strongly expressed in the olfactory nerves, and OE (Nishizuka and Arai, 1996; Sedita et al., 2004). Studies have suggested that anosmin-1 require heparan sulfate with specific 6-O-sulfate modifications to exert its function (Bulow et al., 2002; Loo and Salmivirta, 2002; Tornberg et al., 2011). Out of 338 KS/nIHH patients, 5 different heterozygous missense mutations in HS6ST1 were identified from 7 patients (5 KS patients and 2 nIHH patients) through targeted sanger sequencing (2% of IHH patients). Of these 5 mutations, 2 exhibited digenicity in KS patients, by co-existing with a second heterozygous mutation in FGFR1 or NSMF (Tornberg et al., 2011). Cleft palate occurred in some patients, but not always together with the second mutation, even in a same pedigree revealing variable expression of phenotype. Most Hs6st1-KO mice die during embryonic development, between E15.5 and the perinatal stage, but a few mice survive and as adults exhibited erroneous axon navigation in the optic chiasm (Pratt et al., 2006; Habuchi et al., 2007; Izvolsky et al., 2008). The fertility of Hs6st1 KO mice has not been reported.
CHD7 is expressed in undifferentiated neuroepithelium and neural crest originated mesenchyme (Sanlaville et al., 2006). Several other genes known to be regulated by CHD7 are essential for the neural crest transcriptional circuitry, such as SOX9, TWIST, and SLUG (Bajpai et al., 2010). Because of its early and wide expression during development, mutations in CHD7 are associated with problems throughout the entire body, and the disorder is categorized as CHARGE syndrome (Coloboma, Heart malformation, Atresia choanae, Retardation of growth and/or development, Genital anomalies, and Ear anomalies/deafness). Since IHH and olfactory defects are common phenotypes in both CHARGE syndrome and KS, KS is considered a part of the CHARGE syndrome spectrum (Pinto et al., 2005; Kim et al., 2008). Two recent studies showed that patients harboring missense mutations in CHD7 predominantly suffer KS, whereas truncating mutations (nonsense, frameshift, and splicing mutations) mostly result in traditional CHARGE syndrome (Balasubramanian et al., 2014; Marcos et al., 2014). CHD7 mutations are ∼1.5% of the KS patients (Balasubramanian and Crowley, 2017). Some patients with CHD7 mutations also possessed mutations in FGFR1 and SEMA3A, supporting oligogenism of KS. Several animal models of CHD7 has been generated, with the phenotype mimicking that seen in CHARGE patients. Nine ENU (N-ethyl-N-nitrosourea) induced Chd7 heterozygous mutant mice exhibited cleft palate, choanal atresia, septal defects of the heart, hemorrhages, prenatal death, vulva and clitoral defects, and keratoconjunctivitis sicca (Bosman et al., 2005). While a study of Chd7 null mice reported that the loss of a single allele of Chd7 results in smaller olfactory bulbs and reduced olfactory sensory neurons (Layman et al., 2009).
WD repeats are minimally conserved regions of approximately 40 amino acids that facilitate the formation of multiprotein complexes (Smith, 2008). They are widely expressed and poorly understood. In E10.5-E14.5 mice, WDR11 is expressed in the developing CNS except the spinal cord. Wdr11-null mice exhibit olfactory bulb dysgenesis, hypothalamic GnRH deficiency, pituitary dysgenesis, delayed puberty and reproductive dysfunctions, and obesity (Kim et al., 2018). Notably, intense expression is detected in the diencephalic neuroepithelium and the olfactory bulb neuroepithelium (Kim et al., 2010). Roles of WDR11 protein include regulating the expression of GnRH1 (Kim et al., 2010, 2018). Kim et al. (2010) found a heterozygout missense muation from KS patients which abolishes binding with its partner, EMX1. Only one mutation has been identified in KS while 6 other mutations are founded from nIHH and other HPG axis diseases (combined pituitary hormone deficiency, CPHD, and pituitary stalk interruption syndrome, PSIS) (Izumi et al., 2014; McCormack et al., 2017).
AXL encodes a receptor tyrosine kinase which is a member of the TYRO3, AXL, and MER (TAM) family and has been mutated in 1.9% of KS patients (Salian-Mehta et al., 2014). These receptor tyrosine kinases form homodimers or heterodimers with each other and have a multitude of functions in response to ligand binding, i.e., Growth arrest specific gene 6 (GAS6) and protein S (Allen et al., 2002; Pierce et al., 2008). Axl-/- and Tyro3-/- double KO mice showed a delay of first estrus, persistent abnormal estrus cyclicity, and selective loss of GnRH neurons (36%) in the ventral forebrain, and increased apoptosis along the GnRH migratory route (Pierce et al., 2008). Axl-/- only mice exhibited delayed first estrus and an increased interval between vaginal opening and first estrus. However, estrus cyclicity was normal in adult mice, suggesting compensation (Salian-Mehta et al., 2014).
Not a lot is known about CAMDI, a protein encoded by CCDC141. Experiments suggest that it is involved in radial migration, interacting with myosin II and DISC1, and regulates centrosome positioning during neuronal development (Fukuda et al., 2010). CAMDI can also bind to HDA C6 and inhibit the -tubulin deacetylase activity at the centrosome (Fukuda et al., 2016). Five different mutations (one nonsense and four missense) in the CCDC141 gene have been identified (Turan et al., 2017). Relevant to KS patients, Ccdc141 was found to be is expressed during development in migrating GnRH neurons, olfactory sensory neurons, nasal mesenchymal cells, (Hutchins et al., 2016). In vitro migration assays using siRNA showed knock down of Ccdc141 resulted in a 25% reduction in migration rates of GnRH neurons, but olfactory axon growth was not affected (Hutchins et al., 2016). Using Ccdc141 KO mice, Fukuda et al. (2016) reported delayed radial cell migration, abnormal neural circuit formation, and psychiatric behaviors, but that the mice were fertile. Based on the analysis of Ccdc141 splice variants in mice and humans (unpublished data obtained from open access databases) it is possible that the KO mouse used by Fukuda et al. (2016) still produces a Ccdc141 isoform(s) that may compensate in the GnRH system. To unveil the role of CCDC141 involvement with pathogenic mechanism of KS, a complete null model of Ccdc141 or mutation knock-in model will be required.
A protein–protein interaction network analysis of known genes of KS was performed to identify new candidates that may be involved in KS. The interactions across these 25 proteins were determined by STRING analysis1 (Figure 5). Most of proteins interact with each other, and can be clustered into three different groups, except CCDC141 and AXL. The main cluster (red colored) shows robust interactions among 18 proteins. The Semaphorins and their receptors (green colored) are linked to the main cluster through SEMA3A. TUBB3 and SOX10 (dark cyan colored) are linked to main cluster as well, through SOX10 and its interacting partners (FGF8, PROKR2, and CHD7). Based on the STRING database, 38 additional proteins that interact with known factors of KS were predicted (Figure 6). The interactions through these genes indicate that they might have a role in the development of the olfactory/GnRH systems. As such, these proteins are potential candidate genes for genetic screening of KS patients who don’t harbor mutations in any of the known genes associated with KS.
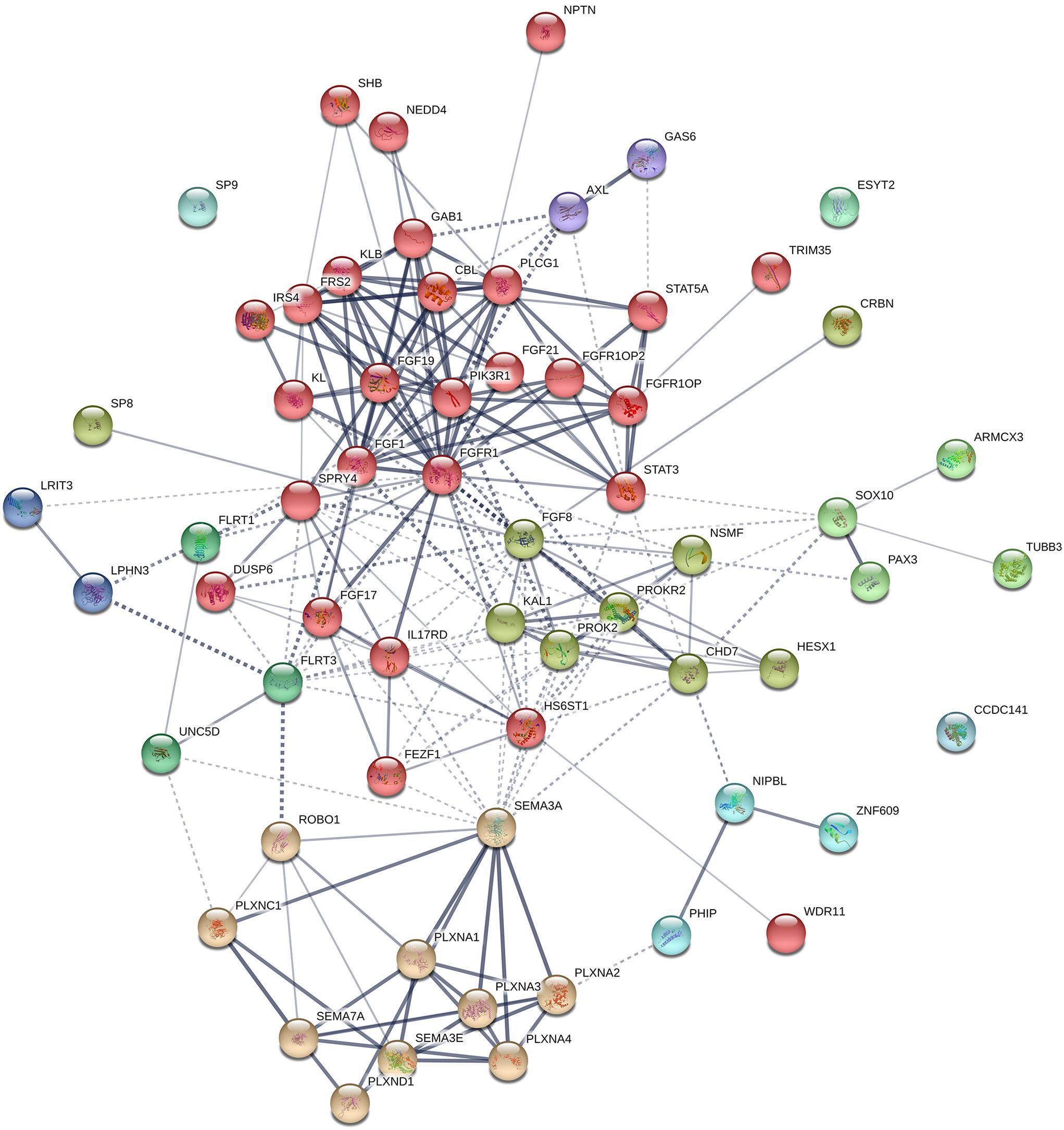
Figure 6. Protein–protein interaction network in known genes of KS and their interacting partners. The diagram shows known and predicted interactions among KS causative genes and their interacting proteins, created using String software version 11.0 with minimal confidence score of 0.4. Total 227 lines (interactions) appear across 63 nodes (25 known factors of KS and 38 additional interacting proteins). The colored nodes symbolized proteins which clustered by MCL algorithm. The thickness of the line represents confidence of protein–protein associations. Solid line, interactions within a cluster; Dotted line, interactions between clusters. Abbreviations for 38 proteins interacting with known KS factors: ARMCX3, Armadillo repeat-containing X-linked protein 3; CBL, E3 ubiquitin-protein ligase CBL; CRBN, Protein cereblon; ESYT2, Extended synaptotagmin-2; FGF1, Fibroblast growth factor 1; FGF19, Fibroblast growth factor 19; FGF21, Fibroblast growth factor 21; FGFR1OP, FGFR1 oncogene partner; FGFR1OP2, FGFR1 oncogene partner 2; FLRT1, Leucine-rich repeat transmembrane protein FLRT1; FRS2, Fibroblast growth factor receptor substrate 2; GAB1, GRB2-associated-binding protein 1; GAS6, Growth arrest-specific protein 6; IRS4, Insulin receptor substrate 4; KL, Klotho; LPHN3, Adhesion G protein-coupled receptor L3; LRIT3, Leucine-rich repeat; NEDD4, E3 ubiquitin-protein ligase NEDD4; NIPBL, Nipped-B-like protein; NPTN, Neuroplastin; PAX3, Paired box protein Pax-3; PHIP, PH-interacting protein; PIK3R1, Phosphatidylinositol 3-kinase regulatory subunit alpha; PLCG1, 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1; PLXNA2, Plexin-A2; PLXNA3, Plexin-A3; PLXNA4, Plexin-A4; PLXNC1, Plexin-C1; PLXND1, Plexin-D1; ROBO1, Roundabout homolog 1; SHB, SH2 domain-containing adapter protein B; SP8, Transcription factor Sp8; SP9, Transcription factor Sp9; STAT3, Signal transducer and activator of transcription 3; STAT5A, Signal transducer and activator of transcription 5A; TRIM35, Tripartite motif-containing protein 35; UNC5D, Netrin receptor UNC5D; ZNF609, Zinc finger protein 609.
Some of the genes involved in KS/nIHH have been studied to investigate their pathogenic mechanism in the disease and to get a better understating of their overall role in olfactory development and GnRH neuronal migration. However, many of the genes, at least those associated with KS, are broadly expressed during development rather than restricted to the nasal area. This makes it difficult to focus on the molecular function of these gene products at the cellular level, in the cell of interest. To begin to address this issue, we compared the expression of known causative genes of KS/nIHH in three different cell types involved in olfactory/GnRH development and function; OSNs, OECs and GnRH cells (Table 2). The expression levels are represented by FPKM (Fragments Per Kilobase Million) values were gathered from Gene Expression Omnibus (GEO) repository2 and other studies including (1) OSN data set (GSE53793) – collected from 4 weeks old OMP-GFP mice (6 homozygous and 8 heterozygous) by FACS of OMP-GFP positive cells and following RNA-Seq analysis (Kanageswaran et al., 2015), (2) OEC data set (GSE69312) collected from cultured human immortalized OEC cells and following RNA-Seq analysis (Dando et al., 2016), unfortunately such data from mice is currently not available) and (3) GnRH data set collected from adult GnRH-Cre/L10a-GFP mice by immunoprecipitation of GnRH specific GFP positive ribosome-mRNAs complex (polysomes) and following RNA-Seq analysis (Burger et al., 2018).
TUBB3, NSMF, and STUB1 are abundantly expressed in all of three cell types (18.119 ≤ FPKM ≤ 666.014). SOX2, SOX10, STUB1, GNRH1, KISS1, and KISS1R are highly expressed in GnRH cells (12.002 ≤ FPKM ≤ 37010.3). The role of SOX2 and SOX10 in GnRH cells is unclear. Certainly, the expression of GNRH1, KISS1, and KISS1R correlates with their role in GnRH neuronal function. IL17RD, FEZF1, HS6ST1, CHD7, WDR11, SEMA3E, SEMA7A, PLXNA1, OTUD4, RNF216, POLR3A, and DMXL2 showed the highest expression in OSNs among three cell types. The expression levels of FGFR1, DUSP6, SPRY4, AXL, PNPLA6, and LEPR are higher in OECs compare to other cell types. HS6ST1, WDR11, SEMA7A, RNF216, and PNPLA6 are relatively high in both OSNs and OECs. The differential expression of these genes may indicate the primary cell that is perturbed and underlies the KS phenotype. Many of genes related with nIHH (GNRHR, TAC3, TACR3, LEP, NR0B1, FSHB, LHB, LHX4, PROP1, and PCSK1) showed low expression (FPKM ≤ 3.016) in these 3 cell types suggesting an indirect role in either GnRH development or physiology.
The mean expression values of the genes related to olfactory symptoms (KS, magenta) are higher than the other genes (only identified in nIHH patients, blue) in OSN (KS, mean 28.36 ± SD 45.89, nIHH, mean 9.34 ± SD 15.15) and OECs (KS, mean 15.28 ± SD 32.54, nIHH, mean 7.37 ± SD 13.72) while the gene set identified in nIHH patients has a higher mean expression than KS genes in GnRH cells (KS, mean 19.79 ± SD 60.43, nIHH, mean 1718 ± SD 7884) (Figure 7). This is consistent with the KS phenotype and the role of OSNs and OECs in olfactory/GnRH development and GnRH cell migration. In addition, the genes showing high expression in the GnRH cells may be more important for GnRH cell maintanence and survival, and/or neuroendocrine function, which links these genes to the pathogenicity of IHH without olfactory problems. These results suggest that examining gene expression levels in each cell type may uncover the role of these genes in KS vs. nIHH.
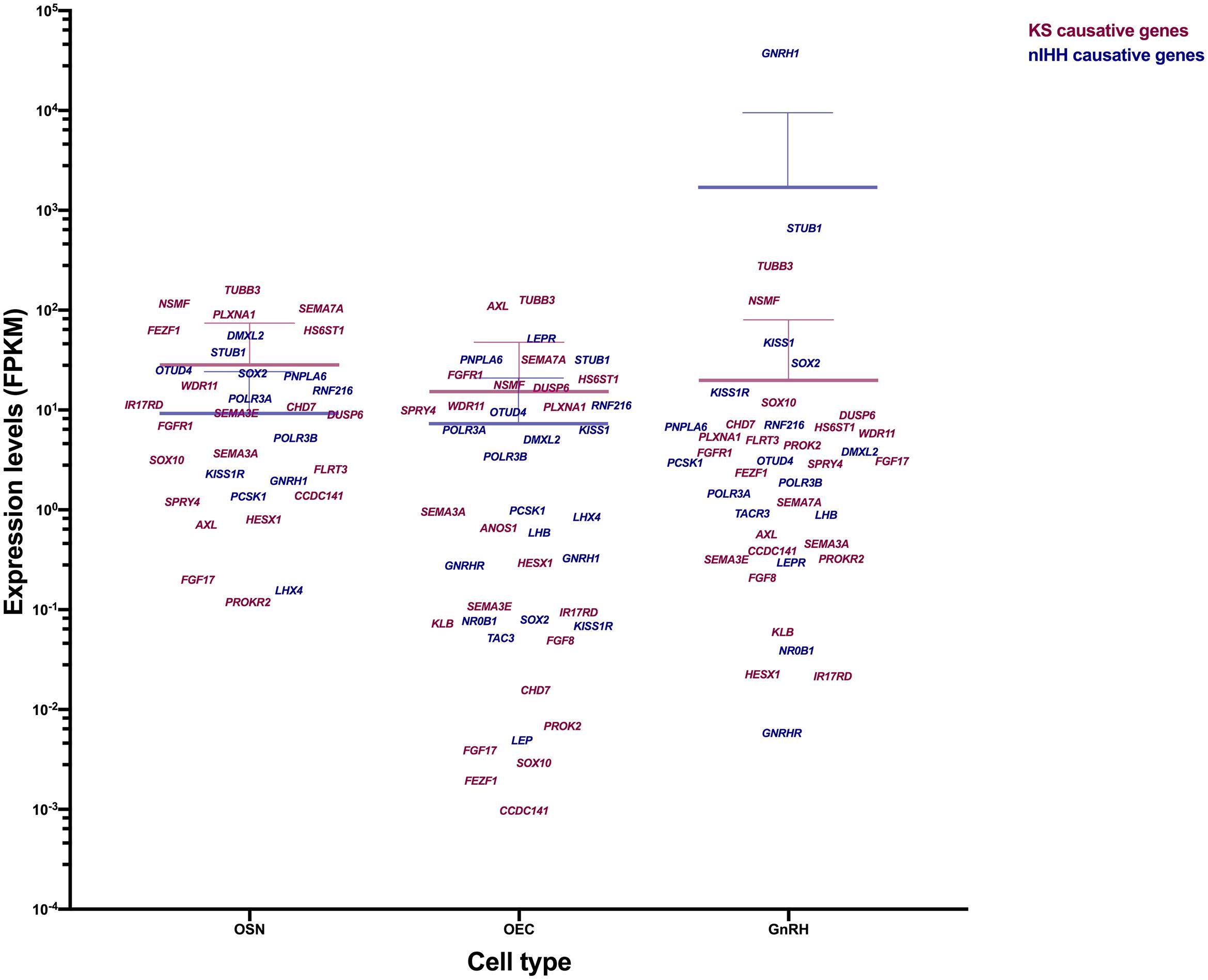
Figure 7. Expression level of genes causing KS vs. nIHH in OSN, OEC, and GnRH neurons. Genes related to KS (magenta) and genes only related to nIHH (blue) are plotted according to their expression level (FPKM) in the three different cell types. The gene expression values are gathered from previous studies and GEO database and compared in OSN (GSE53793), OEC (GSE69312) and GnRH neuron (Burger et al., 2018). The mean expression level and SD of gene group is represented as same color on the scattered plot.
Kallmann Syndrome is a rare disorder which shows high genetic heterogenicity and oligogenicity. We expect that the results from in silico analyses of known factors of KS, together with the updated knowledge on GnRH neuronal development/migration summarized in this review, provides fundamental data that (1) can be used to identify the remaining 60% of the genetic basis of KS, (2) provides new insight into the molecules/mechanisms by which neurons migrate to their appropriate location and our understanding of neuronal disorders associated with aberrant cell migration.
All datasets generated for this study are included in the manuscript and/or the supplementary files.
NW contributed to section “Development of the Olfactory Placode and GnRH Neurons.” YS contributed to section “Migration of GnRH Neurons From the Nose to the Brain.” H-JC contributed to sections “Spectrum of Genetic Disorders in GnRH Cell Deficiency and Kallmann Syndrome” and “Bioinformatic Analysis on KS Genes.” SW supervised the review. All authors wrote and reviewed the manuscript.
This work was supported by the Intramural Research Program of NINDS, NIH, Bethesda, MD, United States (NS002824-29).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all previous members of CDNS, who have contributed to the work discussed in this review.
Abraira, V. E., Hyun, N., Tucker, A. F., Coling, D. E., Brown, M. C., Lu, C., et al. (2007). Changes in Sef levels influence auditory brainstem development and function. J. Neurosci. 27, 4273–4282. doi: 10.1523/jneurosci.3477-06.2007
Aguillon, R., Batut, J., Subramanian, A., Madelaine, R., Dufourcq, P., Schilling, T. F., et al. (2018). Cell-type heterogeneity in the early zebrafish olfactory epithelium is generated from progenitors within preplacodal ectoderm. eLife 7:e32041. doi: 10.7554/eLife.32041
Aksglaede, L., Sorensen, K., Petersen, J. H., Skakkebaek, N. E., and Juul, A. (2009). Recent decline in age at breast development: the Copenhagen Puberty Study. Pediatrics 123, e932–e939. doi: 10.1542/peds.2008-2491
Albuisson, J., Pecheux, C., Carel, J. C., Lacombe, D., Leheup, B., Lapuzina, P., et al. (2005). Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2). Hum. Mutat. 25, 98–99. doi: 10.1002/humu.9298
Allen, M. P., Linseman, D. A., Udo, H., Xu, M., Schaack, J. B., Varnum, B., et al. (2002). Novel mechanism for gonadotropin-releasing hormone neuronal migration involving Gas6/Ark signaling to p38 mitogen-activated protein kinase. Mol. Cell. Biol. 22, 599–613. doi: 10.1128/mcb.22.2.599-613.2002
Bachler, M., and Neubuser, A. (2001). Expression of members of the Fgf family and their receptors during midfacial development. Mech. Dev. 100, 313–316. doi: 10.1016/s0925-4773(00)00518-9
Bajpai, R., Chen, D. A., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J., et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. doi: 10.1038/nature08733
Baker, C. V., and Bronner-Fraser, M. (2001). Vertebrate cranial placodes I. Embryonic induction. Dev. Biol. 232, 1–61. doi: 10.1006/dbio.2001.0156
Balasubramanian, R., Chew, S., MacKinnon, S. E., Kang, P. B., Andrews, C., Chan, W. M., et al. (2015). Expanding the phenotypic spectrum and variability of endocrine abnormalities associated with TUBB3 E410K syndrome. J. Clin. Endocrinol. Metab. 100, E473–E477. doi: 10.1210/jc.2014-4107
Balasubramanian, R., Choi, J. H., Francescatto, L., Willer, J., Horton, E. R., Asimacopoulos, E. P., et al. (2014). Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc. Natl. Acad. Sci. U.S.A. 111, 17953–17958. doi: 10.1073/pnas.1417438111
Balasubramanian, R., and Crowley, W. F. Jr. (2017). Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am. J. Med. Genet. C Semin. Med. Genet. 175, 507–515. doi: 10.1002/ajmg.c.31585
Barnett, S. C. (2004). Olfactory ensheathing cells: unique glial cell types? J. Neurotrauma 21, 375–382. doi: 10.1089/089771504323004520
Barraud, P., St John, J. A., Stolt, C. C., Wegner, M., and Baker, C. V. (2013). Olfactory ensheathing glia are required for embryonic olfactory axon targeting and the migration of gonadotropin-releasing hormone neurons. Biol. Open 2, 750–759. doi: 10.1242/bio.20135249
Bellion, A., Baudoin, J. P., Alvarez, C., Bornens, M., and Metin, C. (2005). Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 25, 5691–5699. doi: 10.1523/jneurosci.1030-05.2005
Bianco, S. D., and Kaiser, U. B. (2009). The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 5, 569–576. doi: 10.1038/nrendo.2009.177
Bick, D., Franco, B., Sherins, R. J., Heye, B., Pike, L., Crawford, J., et al. (1992). Brief report: intragenic deletion of the KALIG-1 gene in Kallmann’s syndrome. N. Engl. J. Med. 326, 1752–1755.
Bless, E., Raitcheva, D., Henion, T. R., Tobet, S., and Schwarting, G. A. (2006). Lactosamine modulates the rate of migration of GnRH neurons during mouse development. Eur. J. Neurosci. 24, 654–660. doi: 10.1111/j.1460-9568.2006.04955.x
Bless, E. P., Walker, H. J., Yu, K. W., Knoll, J. G., Moenter, S. M., Schwarting, G. A., et al. (2005). Live view of gonadotropin-releasing hormone containing neuron migration. Endocrinology 146, 463–468. doi: 10.1210/en.2004-0838
Bless, E. P., Westaway, W. A., Schwarting, G. A., and Tobet, S. A. (2000). Effects of gamma-aminobutyric acid(A) receptor manipulation on migrating gonadotropin-releasing hormone neurons through the entire migratory route in vivo and in vitro. Endocrinology 141, 1254–1262. doi: 10.1210/en.141.3.1254
Boehm, U., Bouloux, P. M., Dattani, M. T., de Roux, N., Dode, C., Dunkel, L., et al. (2015). Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 11, 547–564. doi: 10.1038/nrendo.2015.112
Bosman, E. A., Penn, A. C., Ambrose, J. C., Kettleborough, R., Stemple, D. L., and Steel, K. P. (2005). Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 14, 3463–3476. doi: 10.1093/hmg/ddi375
Bottcher, R. T., Pollet, N., Delius, H., and Niehrs, C. (2004). The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nat. Cell Biol. 6, 38–44. doi: 10.1038/ncb1082
Bribian, A., Esteban, P. F., Clemente, D., Soussi-Yanicostas, N., Thomas, J. L., Zalc, B., et al. (2008). A novel role for anosmin-1 in the adhesion and migration of oligodendrocyte precursors. Dev. Neurobiol. 68, 1503–1516. doi: 10.1002/dneu.20678
Bulow, H. E., Berry, K. L., Topper, L. H., Peles, E., and Hobert, O. (2002). Heparan sulfate proteoglycan-dependent induction of axon branching and axon misrouting by the Kallmann syndrome gene kal-1. Proc. Natl. Acad. Sci. U.S.A. 99, 6346–6351. doi: 10.1073/pnas.092128099
Burger, L. L., Vanacker, C., Phumsatitpong, C., Wagenmaker, E. R., Wang, L., Olson, D. P., et al. (2018). Identification of genes enriched in GnRH neurons by translating ribosome affinity purification and RNAseq in mice. Endocrinology 159, 1922–1940. doi: 10.1210/en.2018-00001
Calof, A. L. D., and Chikaraishi, M. (1989). Analysis of neurogenesis in a mammalian neuroepithelium: proliferation and differentiation of an olfactory neuron precursor in vitro. Neuron 3, 115–127. doi: 10.1016/0896-6273(89)90120-7
Cariboni, A., Andre, V., Chauvet, S., Cassatella, D., Davidson, K., Caramello, A., et al. (2015). Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J. Clin. Invest. 125, 2413–2428. doi: 10.1172/JCI78448
Cariboni, A., Davidson, K., Rakic, S., Maggi, R., Parnavelas, J. G., and Ruhrberg, C. (2011). Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum. Mol. Genet. 20, 336–344. doi: 10.1093/hmg/ddq468
Cariboni, A., Oleari, R., Lettieri, A., Paganoni, A., and Zanieri, L. (2018). Semaphorin signalling in GnRH neurons: from development to disease. Neuroendocrinology [Epub ahead of print].
Cariboni, A., Pimpinelli, F., Colamarino, S., Zaninetti, R., Piccolella, M., Rumio, C., et al. (2004). The product of X-linked Kallmann’s syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum. Mol. Genet. 13, 2781–2791. doi: 10.1093/hmg/ddh309
Cariboni, A., Rakic, S., Liapi, A., Maggi, R., Goffinet, A., and Parnavelas, J. G. (2005). Reelin provides an inhibitory signal in the migration of gonadotropin-releasing hormone neurons. Development 132, 4709–4718. doi: 10.1242/dev.02033
Casoni, F., Hutchins, B. I, Donohue, D., Fornaro, M., Condie, B. G., and Wray, S. (2012). SDF and GABA interact to regulate axophilic migration of GnRH neurons. J. Cell Sci. 125(Pt 21), 5015–5025. doi: 10.1242/jcs.101675
Casoni, F., Malone, S. A., Belle, M., Luzzati, F., Collier, F., Allet, C., et al. (2016). Development of the neurons controlling fertility in humans: new insights from 3D imaging and transparent fetal brains. Development 143, 3969–3981. doi: 10.1242/dev.139444
Castro, D. S., Martynoga, B., Parras, C., Ramesh, V., Pacary, E., Johnston, C., et al. (2011). A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev. 25, 930–945. doi: 10.1101/gad.627811
Cau, E., Casarosa, S., and Guillemot, F. (2002). Mash1 and Ngn1 control distinct steps of determination and differentiation in the olfactory sensory neuron lineage. Development 129, 1871–1880.
Cau, E., Gradwohl, G., Fode, C., and Guillemot, F. (1997). Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development 124, 1611–1621.
Chew, S., Balasubramanian, R., Chan, W. M., Kang, P. B., Andrews, C., Webb, B. D., et al. (2013). A novel syndrome caused by the E410K amino acid substitution in the neuronal beta-tubulin isotype 3. Brain 136(Pt 2), 522–535. doi: 10.1093/brain/aws345
Chung, W. C., Linscott, M. L., Rodriguez, K. M., and Stewart, C. E. (2016). The Regulation and function of fibroblast growth factor 8 and its function during gonadotropin-releasing hormone neuron development. Front. Endocrinol. 7:114. doi: 10.3389/fendo.2016.00114
Chung, W. C., and Tsai, P. S. (2010). Role of fibroblast growth factor signaling in gonadotropin-releasing hormone neuronal system development. Front. Horm. Res. 39:37–50. doi: 10.1159/000312692
Coles, C. H., and Bradke, F. (2015). Coordinating neuronal actin-microtubule dynamics. Curr. Biol. 25, R677–R691. doi: 10.1016/j.cub.2015.06.020
Constantin, S. (2017). Progress and challenges in the search for the mechanisms of pulsatile gonadotropin-releasing hormone secretion. Front. Endocrinol. 8:180. doi: 10.3389/fendo.2017.00180
Costa-Barbosa, F. A., Balasubramanian, R., Keefe, K. W., Shaw, N. D., Al-Tassan, N., Plummer, L., et al. (2013). Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J. Clin. Endocrinol. Metab. 98, E943–E953. doi: 10.1210/jc.2012-4116
Cox, K. H., Oliveira, L. M. B., Plummer, L., Corbin, B., Gardella, T., Balasubramanian, R., et al. (2018). Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum. Mol. Genet. 27, 338–350. doi: 10.1093/hmg/ddx404
Cummings, D. M., and Brunjes, P. C. (1997). The effects of variable periods of functional deprivation on olfactory bulb development in rats. Exp. Neurol. 148, 360–366. doi: 10.1006/exnr.1997.6660
Dairaghi, L., Flannery, E., Giacobini, P., Saglam, A., Saadi, H., Constantin, S., et al. (2018). Reelin can modulate migration of olfactory ensheathing cells and gonadotropin releasing hormone neurons via the canonical pathway. Front. Cell Neurosci. 12:228. doi: 10.3389/fncel.2018.00228
Dando, S. J., Ipe, D. S., Batzloff, M., Sullivan, M. J., Crossman, D. K., Crowley, M., et al. (2016). Burkholderia pseudomallei capsule exacerbates respiratory melioidosis but does not afford protection against antimicrobial signaling or bacterial killing in human olfactory ensheathing cells. Infect. Immun. 84, 1941–1956. doi: 10.1128/IAI.01546-15
Dattani, M. T., Martinez-Barbera, J. P., Thomas, P. Q., Brickman, J. M., Gupta, R., Martensson, I. L., et al. (1998). Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat. Genet. 19, 125–133. doi: 10.1038/477
De Carlos, J. A., Lopez-Mascaraque, L., and Valverde, F. (1996). Early olfactory fiber projections and cell migration into the rat telencephalon. Int. J. Dev. Neurosci. 14, 853–866.
de Castro, F., Seal, R., Maggi, R., and Group of Hgnc consultants for KAL1 nomenclature (2017). ANOS1: a unified nomenclature for Kallmann syndrome 1 gene (KAL1) and anosmin-1. Brief. Funct. Genomics 16, 205–210. doi: 10.1093/bfgp/elw037
de Maximy, A. A., Nakatake, Y., Moncada, S., Itoh, N., Thiery, J. P., and Bellusci, S. (1999). Cloning and expression pattern of a mouse homologue of Drosophila sprouty in the mouse embryo. Mech. Dev. 81, 213–216. doi: 10.1016/s0925-4773(98)00241-x
Deiner, M. S., Kennedy, T. E., Fazeli, A., Serafini, T., Tessier-Lavigne, M., and Sretavan, D. W. (1997). Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron 19, 575–589. doi: 10.1016/s0896-6273(00)80373-6
Deiner, M. S., and Sretavan, D. W. (1999). Altered midline axon pathways and ectopic neurons in the developing hypothalamus of netrin-1- and DCC-deficient mice. J. Neurosci. 19, 9900–9912. doi: 10.1523/jneurosci.19-22-09900.1999
Dellovade, T. L., Pfaff, D. W., and Schwanzel-Fukuda, M. (1998). The gonadotropin-releasing hormone system does not develop in small-eye (Sey) mouse phenotype. Dev. Brain Res. 107, 233–240. doi: 10.1016/s0165-3806(98)00007-8
Demski, L. S., and Schwanzel-Fukuda, M. (1987). The terminal nerve (nervus terminalis): structure, function, and evolution. Introduction. Ann. N. Y. Acad. Sci. 519, ix–xi.
Di Sante, G., Wang, L., Wang, C., Jiao, X., Casimiro, M. C., Chen, K., et al. (2015). Sirt1-deficient mice have hypogonadotropic hypogonadism due to defective GnRH neuronal migration. Mol. Endocrinol. 29, 200–212. doi: 10.1210/me.2014-1228
Diaz-Balzac, C. A., Lazaro-Pena, M. I., Ramos-Ortiz, G. A., and Bulow, H. E. (2015). The adhesion molecule KAL-1/anosmin-1 regulates neurite branching through a SAX-7/L1CAM-EGL-15/FGFR receptor complex. Cell Rep. 11, 1377–1384. doi: 10.1016/j.celrep.2015.04.057
Dode, C., and Hardelin, J. P. (2010). Clinical genetics of Kallmann syndrome. Ann. Endocrinol. 71, 149–157. doi: 10.1016/j.ando.2010.02.005
Dode, C., Levilliers, J., Dupont, J. M., De Paepe, A., Le Du, N., Soussi-Yanicostas, N., et al. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465. doi: 10.1038/ng1122
Dode, C., Teixeira, L., Levilliers, J., Fouveaut, C., Bouchard, P., Kottler, M. L., et al. (2006). Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2:e175. doi: 10.1371/journal.pgen.0020175
Donner, A. L., Episkopou, V., and Maas, R. L. (2007). Sox2 and Pou2f1 interact to control lens and olfactory placode development. Dev. Biol. 303, 784–799. doi: 10.1016/j.ydbio.2006.10.047
Doty, R. L. (2007). Office procedures for quantitative assessment of olfactory function. Am. J. Rhinol. 21, 460–473. doi: 10.2500/ajr.2007.21.3043
Doucette, R. (1990). Glial influences on axonal growth in the primary olfactory system. Glia 3, 433–449. doi: 10.1002/glia.440030602
Drapkin, P. T., Monard, D., and Silverman, A. J. (2002). The role of serine proteases and serine protease inhibitors in the migration of gonadotropin-releasing hormone neurons. BMC Dev. Biol. 2:1. doi: 10.1186/1471-213X-2-1
Duggan, C. D., DeMaria, S., Baudhuin, A., Stafford, D., and Ngai, J. (2008). Foxg1 is required for development of the vertebrate olfactory system. J. Neurosci. 28, 5229–5239. doi: 10.1523/JNEUROSCI.1134-08.2008
Ebens, A., Brose, K., Leonardo, E. D., Hanson, M. G. Jr., Bladt, F., Birchmeier, C., et al. (1996). Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 17, 1157–1172. doi: 10.1016/s0896-6273(00)80247-0
Edmondson, J. C., and Hatten, M. E. (1987). Glial-guided granule neuron migration in vitro: a high-resolution time-lapse video microscopic study. J. Neurosci. 7, 1928–1934. doi: 10.1523/jneurosci.07-06-01928.1987
Egea, J., Erlacher, C., Montanez, E., Burtscher, I., Yamagishi, S., Hess, M., et al. (2008). Genetic ablation of FLRT3 reveals a novel morphogenetic function for the anterior visceral endoderm in suppressing mesoderm differentiation. Genes Dev. 22, 3349–3362. doi: 10.1101/gad.486708
Ericson, J., Thor, S., Edlund, T., Jessell, T. M., and Yamada, T. (1992). Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet- 1. Science 256, 1555–1560. doi: 10.1126/science.1350865
Fahrion, J. K., Komuro, Y., Li, Y., Ohno, N., Littner, Y., Raoult, E., et al. (2012). Rescue of neuronal migration deficits in a mouse model of fetal Minamata disease by increasing neuronal Ca2+ spike frequency. Proc. Natl. Acad. Sci. U.S.A. 109, 5057–5062. doi: 10.1073/pnas.1120747109
Falardeau, J., Chung, W. C., Beenken, A., Raivio, T., Plummer, L., Sidis, Y., et al. (2008). Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 118, 2822–2831. doi: 10.1172/JCI34538
Fletcher, R. B., Das, D., Gadye, L., Street, K. N., Baudhuin, A., Wagner, A., et al. (2017). Deconstructing olfactory stem cell trajectories at single-cell resolution. Cell Stem Cell 20, 817–830.e8. doi: 10.1016/j.stem.2017.04.003
Fornaro, M., Geuna, S., Fasolo, A., and Giacobini-Robecchi, M. G. (2001). Evidence of very early neuronal migration from the olfactory placode of the chick embryo. Neuroscience 107, 191–197. doi: 10.1016/s0306-4522(01)00334-7
Fornaro, M., Geuna, S., Fasolo, A., and Giacobini-Robecchi, M. G. (2003). HuC/D confocal imaging points to olfactory migratory cells as the first cell population that expresses a post-mitotic neuronal phenotype in the chick embryo. Neuroscience 122, 123–128. doi: 10.1016/j.neuroscience.2003.07.004
Forni, P. E., Bharti, K., Flannery, E. M., Shimogori, T., and Wray, S. (2013). The indirect role of fibroblast growth factor-8 in defining neurogenic niches of the olfactory/GnRH systems. J. Neurosci. 33, 19620–19634. doi: 10.1523/JNEUROSCI.3238-13.2013
Forni, P. E., Taylor-Burds, C., Melvin, V. S., Williams, T., and Wray, S. (2011). Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J. Neurosci. 31, 6915–6927. doi: 10.1523/JNEUROSCI.6087-10.2011
Forni, P. E., and Wray, S. (2012). Neural crest and olfactory system: new prospective. Mol. Neurobiol. 46, 349–360. doi: 10.1007/s12035-012-8286-5
Forni, P. E., and Wray, S. (2015). GnRH, anosmia and hypogonadotropic hypogonadism–where are we? Front. Neuroendocrinol. 36:165–177. doi: 10.1016/j.yfrne.2014.09.004
Franceschini, I. A., and Barnett, S. C. (1996). Low-affinity NGF-receptor and E-N-CAM expression define two types of olfactory nerve ensheathing cells that share a common lineage. Dev. Biol. 173, 327–343. doi: 10.1006/dbio.1996.0027
Francis, F., Koulakoff, A., Boucher, D., Chafey, P., Schaar, B., Vinet, M.-C., et al. (1999). Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23, 247–256. doi: 10.1016/s0896-6273(00)80777-1
Franco, B., Guioli, S., Pragliola, A., Incerti, B., Bardoni, B., Tonlorenzi, R., et al. (1991). A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353, 529–536. doi: 10.1038/353529a0
Fueshko, S., and Wray, S. (1994). LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev. Biol. 166, 331–348. doi: 10.1006/dbio.1994.1319
Fueshko, S. M., Key, S., and Wray, S. (1998). GABA inhibits migration of luteinizing hormone-releasing hormone neurons in embryonic olfactory explants. J. Neurosci. 18, 2560–2569. doi: 10.1523/jneurosci.18-07-02560.1998
Fukuda, T., Nagashima, S., Abe, T., Kiyonari, H., Inatome, R., and Yanagi, S. (2016). Rescue of CAMDI deletion-induced delayed radial migration and psychiatric behaviors by HDAC6 inhibitor. EMBO Rep. 17, 1785–1798. doi: 10.15252/embr.201642416
Fukuda, T., Sugita, S., Inatome, R., and Yanagi, S. (2010). CAMDI, a novel disrupted in schizophrenia 1 (DISC1)-binding protein, is required for radial migration. J. Biol. Chem. 285, 40554–40561. doi: 10.1074/jbc.M110.179481
Fuller, G. N., and Burger, P. C. (1990). Nervus terminalis (cranial nerve zero) in the adult human. Clin. Neuropathol. 9, 279–283.
Furthauer, M., Lin, W., Ang, S. L., Thisse, B., and Thisse, C. (2002). Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 4, 170–174. doi: 10.1038/ncb750
Gamble, J. A., Karunadasa, D. K., Pape, J. R., Skynner, M. J., Todman, M. G., Bicknell, R. J., et al. (2005). Disruption of ephrin signaling associates with disordered axophilic migration of the gonadotropin-releasing hormone neurons. J. Neurosci. 25, 3142–3150. doi: 10.1523/jneurosci.4759-04.2005
Geller, S., Kolasa, E., Tillet, Y., Duittoz, A., and Vaudin, P. (2013). Olfactory ensheathing cells form the microenvironment of migrating GnRH-1 neurons during mouse development. Glia 61, 550–566. doi: 10.1002/glia.22455
Georgopoulos, N. A., Koika, V., Galli-Tsinopoulou, A., Spiliotis, B. E., Adonakis, G., Keramida, M. K., et al. (2007). Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil. Steril. 88, 1311–1317. doi: 10.1016/j.fertnstert.2006.12.044
Giacobini, P., Giampietro, C., Fioretto, M., Maggi, R., Cariboni, A., Perroteau, I., et al. (2002). Hepatocyte growth factor/scatter factor facilitates migration of GN-11 immortalized LHRH neurons. Endocrinology 143, 3306–3315. doi: 10.1210/en.2002-220146
Giacobini, P., Kopin, A. S., Beart, P. M., Mercer, L. D., Fasolo, A., and Wray, S. (2004). Cholecystokinin modulates migration of gonadotropin-releasing hormone-1 neurons. J. Neurosci. 24, 4737–4748. doi: 10.1523/jneurosci.0649-04.2004
Giacobini, P., Messina, A., Morello, F., Ferraris, N., Corso, S., Penachioni, J., et al. (2008). Semaphorin 4D regulates gonadotropin hormone-releasing hormone-1 neuronal migration through PlexinB1-Met complex. J. Cell Biol. 183, 555–566. doi: 10.1083/jcb.200806160
Giacobini, P., Messina, A., Wray, S., Giampietro, C., Crepaldi, T., Carmeliet, P., et al. (2007). Hepatocyte growth factor acts as a motogen and guidance signal for gonadotropin hormone-releasing hormone-1 neuronal migration. J. Neurosci. 27, 431–445. doi: 10.1523/jneurosci.4979-06.2007
Gleeson, J. G., Lin, P. T., Flanagan, L. A., and Walsh, C. A. (1999). Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23, 257–271. doi: 10.1016/s0896-6273(00)80778-3
Goetz, R., Beenken, A., Ibrahimi, O. A., Kalinina, J., Olsen, S. K., Eliseenkova, A. V., et al. (2007). Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 27, 3417–3428. doi: 10.1128/mcb.02249-06
Goetz, R., Ohnishi, M., Ding, X., Kurosu, H., Wang, L., Akiyoshi, J., et al. (2012). Klotho coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily ligands. Mol. Cell. Biol. 32, 1944–1954. doi: 10.1128/MCB.06603-11
Goncalves, C. I., Fonseca, F., Borges, T., Cunha, F., and Lemos, M. C. (2017). Expanding the genetic spectrum of ANOS1 mutations in patients with congenital hypogonadotropic hypogonadism. Hum. Reprod. 32, 704–711. doi: 10.1093/humrep/dew354
Habuchi, H., Nagai, N., Sugaya, N., Atsumi, F., Stevens, R. L., and Kimata, K. (2007). Mice deficient in heparan sulfate 6-O-sulfotransferase-1 exhibit defective heparan sulfate biosynthesis, abnormal placentation, and late embryonic lethality. J. Biol. Chem. 282, 15578–15588. doi: 10.1074/jbc.m607434200
Habuchi, H., Tanaka, M., Habuchi, O., Yoshida, K., Suzuki, H., Ban, K., et al. (2000). The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J. Biol. Chem. 275, 2859–2868. doi: 10.1074/jbc.275.4.2859
Hacochen, N., Kramer, S., Sutherland, D., Hiromi, Y., and Krasnow, M. A. (1998). sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the drosophila airways. Cell 92, 253–263. doi: 10.1016/s0092-8674(00)80919-8
Haines, B. P., Wheldon, L. M., Summerbell, D., Heath, J. K., and Rigby, P. W. (2006). Regulated expression of FLRT genes implies a functional role in the regulation of FGF signalling during mouse development. Dev. Biol. 297, 14–25. doi: 10.1016/j.ydbio.2006.04.004
Hanchate, N. K., Giacobini, P., Lhuillier, P., Parkash, J., Espy, C., Fouveaut, C., et al. (2012). SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 8:e1002896. doi: 10.1371/journal.pgen.1002896
Hardelin, J. P., Levilliers, J., del Castillo, I., Cohen-Salmon, M., Legouis, R., Blanchard, S., et al. (1992). X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc. Natl. Acad. Sci. U.S.A. 89, 8190–8194. doi: 10.1073/pnas.89.17.8190
Hellwig, S., Hack, I., Zucker, B., Brunne, B., and Junghans, D. (2012). Reelin together with ApoER2 regulates interneuron migration in the olfactory bulb. PLoS One 7:e50646. doi: 10.1371/journal.pone.0050646
Herman-Giddens, M. E., Steffes, J., Harris, D., Slora, E., Hussey, M., Dowshen, S. A., et al. (2012). Secondary sexual characteristics in boys: data from the pediatric research in office settings network. Pediatrics 130, e1058–e1068. doi: 10.1542/peds.2011-3291
Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J. A., et al. (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489. doi: 10.1016/s0896-6273(00)80861-2
Hoffman, G. E., Lee, W.-S., and Wray, S. (1992). “Gonadotropin releasing hormone (GnRH)”, in Neuroendocrinology, Chap. 8, ed. C. B. Nemeroff (Boca Raton, FL: CRC Press).
Hu, Y., and Bouloux, P. M. (2011). X-linked GnRH deficiency: role of KAL-1 mutations in GnRH deficiency. Mol. Cell. Endocrinol. 346, 13–20. doi: 10.1016/j.mce.2011.04.001
Hu, Y., Poopalasundaram, S., Graham, A., and Bouloux, P. M. (2013). GnRH neuronal migration and olfactory bulb neurite outgrowth are dependent on FGF receptor 1 signaling, specifically via the PI3K p110alpha isoform in chick embryo. Endocrinology 154, 388–399. doi: 10.1210/en.2012-1555
Hutchins, B. I., Klenke, U., and Wray, S. (2013). Calcium release-dependent actin flow in the leading process mediates axophilic migration. J. Neurosci. 33, 11361–11371. doi: 10.1523/JNEUROSCI.3758-12.2013
Hutchins, B. I., Kotan, L. D., Taylor-Burds, C., Ozkan, Y., Cheng, P. J., Gurbuz, F., et al. (2016). CCDC141 mutation identified in anosmic hypogonadotropic hypogonadism (Kallmann Syndrome) alters GnRH neuronal migration. Endocrinology 157, 1956–1966. doi: 10.1210/en.2015-1846
Hutchins, B. I., and Wray, S. (2014). Capture of microtubule plus-ends at the actin cortex promotes axophilic neuronal migration by enhancing microtubule tension in the leading process. Front. Cell Neurosci. 8:400. doi: 10.3389/fncel.2014.00400
Ishibashi, T., Shinogami, M., Ishimoto, S., Nibu, K., Suzuki, M., and Kaga, K. (2001). Identification of dual specificity phosphatases induced by olfactory bulbectomy in rat olfactory neuroepithelium. Brain Res. 902, 205–211. doi: 10.1016/s0006-8993(01)02386-1
Iyer, A. K., Miller, N. L., Yip, K., Tran, B. H., and Mellon, P. L. (2010). Enhancers of GnRH transcription embedded in an upstream gene use homeodomain proteins to specify hypothalamic expression. Mol. Endocrinol. 24, 1949–1964. doi: 10.1210/me.2010-0156
Izumi, Y., Suzuki, E., Kanzaki, S., Yatsuga, S., Kinjo, S., Igarashi, M., et al. (2014). Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil. Steril. 102, 1130–1136e1133. doi: 10.1016/j.fertnstert.2014.06.017
Izvolsky, K. I., Lu, J., Martin, G., Albrecht, K. H., and Cardoso, W. V. (2008). Systemic inactivation of Hs6st1 in mice is associated with late postnatal mortality without major defects in organogenesis. Genesis 46, 8–18. doi: 10.1002/dvg.20355
Jongmans, M. C., van Ravenswaaij-Arts, C. M., Pitteloud, N., Ogata, T., Sato, N., Claahsen-van der Grinten, H. L., et al. (2009). CHD7 mutations in patients initially diagnosed with Kallmann syndrome–the clinical overlap with CHARGE syndrome. Clin. Genet. 75, 65–71. doi: 10.1111/j.1399-0004.2008.01107.x
Kanageswaran, N., Demond, M., Nagel, M., Schreiner, B. S., Baumgart, S., Scholz, P., et al. (2015). Deep sequencing of the murine olfactory receptor neuron transcriptome. PLoS One 10:e0113170. doi: 10.1371/journal.pone.0113170
Kanaho, Y., Enomoto, M., Endo, D., Maehiro, S., Park, M. K., and Murakami, S. (2009). Neurotrophic effect of gonadotropin-releasing hormone on neurite extension and neuronal migration of embryonic gonadotropin-releasing hormone neurons in chick olfactory nerve bundle culture. J. Neurosci. Res. 87, 2237–2244. doi: 10.1002/jnr.22051
Kansakoski, J., Fagerholm, R., Laitinen, E. M., Vaaralahti, K., Hackman, P., Pitteloud, N., et al. (2014). Mutation screening of SEMA3A and SEMA7A in patients with congenital hypogonadotropic hypogonadism. Pediatr. Res. 75, 641–644. doi: 10.1038/pr.2014.23
Karpinski, B. A., Bryan, C. A., Paronett, E. M., Baker, J. L., Fernandez, A., Horvath, A., et al. (2016). A cellular and molecular mosaic establishes growth and differentiation states for cranial sensory neurons. Dev. Biol. 415, 228–241. doi: 10.1016/j.ydbio.2016.03.015
Kasberg, A. D., Brunskill, E. W., and Steven Potter, S. (2013). SP8 regulates signaling centers during craniofacial development. Dev. Biol. 381, 312–323. doi: 10.1016/j.ydbio.2013.07.007
Katoh, H., Shibata, S., Fukuda, K., Sato, M., Satoh, E., Nagoshi, N., et al. (2011). The dual origin of the peripheral olfactory system: placode and neural crest. Mol. Brain 4:34. doi: 10.1186/1756-6606-4-34
Kawakami, Y., Rodriguez-Leon, J., Koth, C. M., Buscher, D., Itoh, T., Raya, A., et al. (2003). MKP3 mediates the cellular response to FGF8 signaling in the vertebrate limb. Nat. Cell Biol. 5, 513–519. doi: 10.1038/ncb989
Kawauchi, S., Beites, C. L., Crocker, C. E., Wu, H. H., Bonnin, A., Murray, R., et al. (2004). Molecular signals regulating proliferation of stem and progenitor cells in mouse olfactory epithelium. Dev. Neurosci. 26, 166–180. doi: 10.1159/000082135
Kawauchi, S., Santos, R., Kim, J., Hollenbeck, P. L., Murray, R. C., and Calof, A. L. (2009). The role of foxg1 in the development of neural stem cells of the olfactory epithelium. Ann. N. Y. Acad. Sci. 1170, 21–27. doi: 10.1111/j.1749-6632.2009.04372.x
Kawauchi, S., Shou, J., Santos, R., Hebert, J. M., McConnell, S. K., Mason, I., et al. (2005). Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development 132, 5211–5223. doi: 10.1242/dev.02143
Kim, H. D., Choe, H. K., Chung, S., Kim, M., Seong, J. Y., Son, G. H., et al. (2011). Class-C SOX transcription factors control GnRH gene expression via the intronic transcriptional enhancer. Mol. Endocrinol. 25, 1184–1196. doi: 10.1210/me.2010-0332
Kim, H. G., Ahn, J. W., Kurth, I., Ullmann, R., Kim, H. T., Kulharya, A., et al. (2010). WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 87, 465–479. doi: 10.1016/j.ajhg.2010.08.018
Kim, H. G., Kurth, I., Lan, F., Meliciani, I., Wenzel, W., Eom, S. H., et al. (2008). Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 83, 511–519. doi: 10.1016/j.ajhg.2008.09.005
Kim, Y. J., Osborn, D. P., Lee, J. Y., Araki, M., Araki, K., Mohun, T., et al. (2018). WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. 19, 269–289. doi: 10.15252/embr.201744632
Kiss, J. Z., and Rougon, G. (1997). Cell biology of polysialic acid. Curr. Opin. Neurobiol. 7, 640–646. doi: 10.1016/s0959-4388(97)80083-9
Kitahashi, T., Sato, H., Sakuma, Y., and Parhar, I. S. (2005). Cloning and functional analysis of promoters of three GnRH genes in a cichlid. Biochem. Biophys. Res. Commun. 336, 536–543. doi: 10.1016/j.bbrc.2005.08.122
Klenke, U., and Taylor-Burds, C. (2012). Culturing embryonic nasal explants for developmental and physiological study. Curr. Protoc. Neurosci. Chapter 3: Unit 3.25.1-16. doi: 10.1002/0471142301.ns0325s59
Kolodkin, A. L., Levengood, D. V., Rowe, E. G., Tai, Y. T., Giger, R. J., and Ginty, D. D. (1997). Neuropilin is a semaphorin III receptor. Cell 90, 753–762. doi: 10.1016/s0092-8674(00)80535-8
Komuro, H., and Kumada, T. (2005). Ca2+ transients control CNS neuronal migration. Cell Calcium 37, 387–393. doi: 10.1016/j.ceca.2005.01.006
Kotan, L. D., Hutchins, B. I., Ozkan, Y., Demirel, F., Stoner, H., Cheng, P. J., et al. (2014). Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet. 95, 326–331. doi: 10.1016/j.ajhg.2014.08.006
Kramer, P. R., Guerrero, G., Krishnamurthy, R., Mitchell, P. J., and Wray, S. (2000). Ectopic expression of luteinizing hormone-releasing hormone and peripherin in the respiratory epithelium of mice lacking transcription factor AP-2alpha. Mech. Dev. 94, 79–94. doi: 10.1016/s0925-4773(00)00316-6
Kramer, P. R., and Wray, S. (2000a). Midline nasal tissue influences nestin expression in nasal-placode-derived luteinizing hormone-releasing hormone neurons during development. Dev. Biol. 227, 343–357. doi: 10.1006/dbio.2000.9896
Kramer, P. R., and Wray, S. (2000b). Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 14, 1824–1834.
Kuiri-Hanninen, T., Kallio, S., Seuri, R., Tyrvainen, E., Liakka, A., Tapanainen, J., et al. (2011a). Postnatal developmental changes in the pituitary-ovarian axis in preterm and term infant girls. J. Clin. Endocrinol. Metab. 96, 3432–3439. doi: 10.1210/jc.2011-1502
Kuiri-Hanninen, T., Seuri, R., Tyrvainen, E., Turpeinen, U., Hamalainen, E., Stenman, U. H., et al. (2011b). Increased activity of the hypothalamic-pituitary-testicular axis in infancy results in increased androgen action in premature boys. J. Clin. Endocrinol. Metab. 96, 98–105. doi: 10.1210/jc.2010-1359
Kurosu, H., Choi, M., Ogawa, Y., Dickson, A. S., Goetz, R., Eliseenkova, A. V., et al. (2007). Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282, 26687–26695. doi: 10.1074/jbc.m704165200
Lacy, S. E., Bonnemann, C. G., Buzney, E. A., and Kunkel, L. M. (1999). Identification of FLRT1, FLRT2, and FLRT3: a novel family of transmembrane leucine-rich repeat proteins. Genomics 62, 417–426. doi: 10.1006/geno.1999.6033
Latremoliere, A., Cheng, L., DeLisle, M., Wu, C., Chew, S., Hutchinson, E. B., et al. (2018). Neuronal-specific TUBB3 is not required for normal neuronal function but is essential for timely axon regeneration. Cell Rep. 24, 1865–1879.e9. doi: 10.1016/j.celrep.2018.07.029
Layman, W. S., McEwen, D. P., Beyer, L. A., Lalani, S. R., Fernbach, S. D., Oh, E., et al. (2009). Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum. Mol. Genet. 18, 1909–1923. doi: 10.1093/hmg/ddp112
Legouis, R., Hardelin, J. P., Levilliers, J., Claverie, J. M., Compain, S., Wunderle, V., et al. (1991). The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 67, 423–435. doi: 10.1016/0092-8674(91)90193-3
Lewkowitz-Shpuntoff, H. M., Hughes, V. A., Plummer, L., Au, M. G., Doty, R. L., Seminara, S. B., et al. (2012). Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J. Clin. Endocrinol. Metab. 97, E136–E144. doi: 10.1210/jc.2011-2041
Leyva-Diaz, E., del Toro, D., Menal, M. J., Cambray, S., Susin, R., Tessier-Lavigne, M., et al. (2014). FLRT3 is a Robo1-interacting protein that determines Netrin-1 attraction in developing axons. Curr. Biol. 24, 494–508. doi: 10.1016/j.cub.2014.01.042
Li, C., Scott, D. A., Hatch, E., Tian, X., and Mansour, S. L. (2007). Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 134, 167–176. doi: 10.1242/dev.02701
Loo, B. M., and Salmivirta, M. (2002). Heparin/Heparan sulfate domains in binding and signaling of fibroblast growth factor 8b. J. Biol. Chem. 277, 32616–32623. doi: 10.1074/jbc.m204961200
Lund, C., Pulli, K., Yellapragada, V., Giacobini, P., Lundin, K., Vuoristo, S., et al. (2016). Development of gonadotropin-releasing hormone-secreting neurons from human pluripotent stem cells. Stem Cell Rep. 7, 149–157. doi: 10.1016/j.stemcr.2016.06.007
Lutter, S., Xie, S., Tatin, F., and Makinen, T. (2012). Smooth muscle-endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J. Cell Biol. 197, 837–849. doi: 10.1083/jcb.201110132
Ma, Q., Sommer, L., Cserjesi, P., and Anderson, D. J. (1997). Mash1 and neurogenin1 expression patterns define complementary domains of neuroepithelium in the developing CNS and are correlated with regions expressing notch ligands. J. Neurosci. 17, 3644–3652. doi: 10.1523/jneurosci.17-10-03644.1997
Madhu, S. V., Kant, S., Holla, V. V., Arora, R., and Rathi, S. (2012). Unusual presentation of Kallmannn syndrome with contiguous gene deletion in three siblings of a family. Indian J. Endocrinol. Metab. 16(Suppl. 2), S326–S328. doi: 10.4103/2230-8210.104077
Maier, E., von Hofsten, J., Nord, H., Fernandes, M., Paek, H., Hebert, J. M., et al. (2010). Opposing Fgf and Bmp activities regulate the specification of olfactory sensory and respiratory epithelial cell fates. Development 137, 1601–1611. doi: 10.1242/dev.051219
Maione, L., Dwyer, A. A., Francou, B., Guiochon-Mantel, A., Binart, N., Bouligand, J., et al. (2018). GENETICS IN ENDOCRINOLOGY: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur. J. Endocrinol. 178, R55–R80. doi: 10.1530/EJE-17-0749
Marcos, S., Sarfati, J., Leroy, C., Fouveaut, C., Parent, P., Metz, C., et al. (2014). The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J. Clin. Endocrinol. Metab. 99, E2138–E2143. doi: 10.1210/jc.2014-2110
Maretto, S., Muller, P. S., Aricescu, A. R., Cho, K. W., Bikoff, E. K., and Robertson, E. J. (2008). Ventral closure, headfold fusion and definitive endoderm migration defects in mouse embryos lacking the fibronectin leucine-rich transmembrane protein FLRT3. Dev. Biol. 318, 184–193. doi: 10.1016/j.ydbio.2008.03.021
Margolis, F. (1972). A brain protein unique to the olfactory bulb. Proc. Natl. Acad. Sci. U.S.A. 69, 1221–1224. doi: 10.1073/pnas.69.5.1221
Martin, C., Balasubramanian, R., Dwyer, A. A., Au, M. G., Sidis, Y., Kaiser, U. B., et al. (2011). The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr. Rev. 32, 225–246. doi: 10.1210/er.2010-0007
Matsumoto, S., Yamazaki, C., Masumoto, K. H., Nagano, M., Naito, M., Soga, T., et al. (2006). Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. U.S.A. 103, 4140–4145. doi: 10.1073/pnas.0508881103
McCormack, S. E., Li, D., Kim, Y. J., Lee, J. Y., Kim, S. H., Rapaport, R., et al. (2017). Digenic inheritance of PROKR2 and WDR11 mutations in pituitary stalk interruption syndrome. J. Clin. Endocrinol. Metab. 102, 2501–2507. doi: 10.1210/jc.2017-00332
Messina, A., and Giacobini, P. (2013). Semaphorin signaling in the development and function of the gonadotropin hormone-releasing hormone system. Front. Endocrinol. 4:133. doi: 10.3389/fendo.2013.00133
Miller, A. M., Treloar, H. B., and Greer, C. A. (2010). Composition of the migratory mass during development of the olfactory nerve. J. Comp. Neurol. 518, 4825–4841. doi: 10.1002/cne.22497
Minowada, G., Jarvis, L. A., Chi, C. L., Neubuser, A., Sun, X., Hacochen, N., et al. (1999). Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126, 4465–4475.
Miraoui, H., Dwyer, A. A., Sykiotis, G. P., Plummer, L., Chung, W., Feng, B., et al. (2013). Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 92, 725–743. doi: 10.1016/j.ajhg.2013.04.008
Misrahi, M. (2017). beta-Klotho sustains postnatal GnRH biology and spins the thread of puberty. EMBO Mol. Med. 9, 1334–1337. doi: 10.15252/emmm.201708180
Miura, K., Acierno, J. S. Jr., and Seminara, S. B. (2004). Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J. Hum. Genet. 49, 265–268. doi: 10.1007/s10038-004-0137-4
Moody, S. A., and LaMantia, A. S. (2015). Transcriptional regulation of cranial sensory placode development. Curr. Top. Dev. Biol. 111, 301–350. doi: 10.1016/bs.ctdb.2014.11.009
Moore, J. P. Jr., and Wray, S. (2000). Luteinizing hormone-releasing hormone (LHRH) biosynthesis and secretion in embryonic LHRH. Endocrinology 141, 4486–4495. doi: 10.1210/endo.141.12.7814
Mousley, A., Polese, G., Marks, N. J., and Eisthen, H. L. (2006). Terminal nerve-derived neuropeptide y modulates physiological responses in the olfactory epithelium of hungry axolotls (Ambystoma mexicanum). J. Neurosci. 26, 7707–7717. doi: 10.1523/jneurosci.1977-06.2006
Muller, F., and O’Rahilly, R. (2004). Olfactory structures in staged human embryos. Cells Tissues Organs 178, 93–116. doi: 10.1159/000081720
Murakami, S., Ohki-Hamazaki, H., Watanabe, K., Ikenaka, K., and Ono, K. (2010). Netrin 1 provides a chemoattractive cue for the ventral migration of GnRH neurons in the chick forebrain. J. Comp. Neurol. 518, 2019–2034. doi: 10.1002/cne.22319
Murakami, S., Seki, T., Rutishauser, U., and Arai, Y. (2000). Enzymatic removal of polysialic acid from neural cell adhesion molecule perturbs the migration route of luteinizing hormone-releasing hormone neurons in the developing chick forebrain. J. Comp. Neurol. 420, 171–181. doi: 10.1002/(sici)1096-9861(20000501)420:2<171::aid-cne2>3.0.co;2-9
Murakami, S., Seki, T., Wakabayashi, K., and Arai, Y. (1991). The ontogeny of luteinizing hormone-releasing hormone (LHRH) producing neurons in the chick embryo: possible evidence for migrating LHRH neurons from the olfactory epithelium expressing a highly polysialylated neural cell adhesion molecule. Neurosci. Res. 12, 421–431. doi: 10.1016/0168-0102(91)90073-8
Murray, R., Navi, D., Fesenko, J., Lander, A., and Calof, A. L. (2003). Widespread defects in the primary olfactory pathway caused by loss of Mash1 function. J. Neurosci. 23, 1769–1780. doi: 10.1523/jneurosci.23-05-01769.2003
Muske, L. E., and Moore, F. L. (1988). The nervus terminalis in amphibians: anatomy, chemistry and relationship with the hypothalamic gonadotropin-releasing hormone system. Brain Behav. Evol. 32, 141–150. doi: 10.1159/000116541
Nakane, K., Fujita, Y., Terazawa, R., Atsumi, Y., Kato, T., Nozawa, Y., et al. (2012). Inhibition of cortactin and SIRT1 expression attenuates migration and invasion of prostate cancer DU145 cells. Int. J. Urol. 19, 71–79. doi: 10.1111/j.1442-2042.2011.02888.x
Newbern, K., Natrajan, N., Kim, H. G., Chorich, L. P., Halvorson, L. M., Cameron, R. S., et al. (2013). Identification of HESX1 mutations in Kallmann syndrome. Fertil. Steril. 99, 1831–1837. doi: 10.1016/j.fertnstert.2013.01.149
Ng, S. M., Kumar, Y., Cody, D., Smith, C., and Didi, M. (2005). The gonadotrophins response to GnRH test is not a predictor of neurological lesion in girls with central precocious puberty. J. Pediatr. Endocrinol. Metab. 18, 849–852.
Nie, M., Xu, H., Chen, R., Mao, J., Wang, X., Xiong, S., et al. (2017). Analysis of genetic and clinical characteristics of a Chinese Kallmann syndrome cohort with ANOS1 mutations. Eur. J. Endocrinol. 177, 389–398. doi: 10.1530/EJE-17-0335
Nishizuka, M., and Arai, Y. (1996). Glycosaminoglycans in the olfactory epithelium and nerve of chick embryos: an immunocytochemical study. Neurosci. Res. 24, 165–173. doi: 10.1016/0168-0102(95)00990-6
Ogawa, Y., Kurosu, H., Yamamoto, M., Nandi, A., Rosenblatt, K. P., Goetz, R., et al. (2007). BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. U.S.A. 104, 7432–7437. doi: 10.1073/pnas.0701600104
Okano, H., and Darnell, R. B. (1997). A hierarchy of Hu RNA binding proteins in developing and adult neurons. J. Neurosci. 17, 3024–3037. doi: 10.1523/jneurosci.17-09-03024.1997
Okubo, K., Sakai, F., Lau, E. L., Yoshizaki, G., Takeuchi, Y., Naruse, K., et al. (2006). Forebrain gonadotropin-releasing hormone neuronal development: insights from transgenic medaka and the relevance to X-linked Kallmann syndrome. Endocrinology 147, 1076–1084. doi: 10.1210/en.2005-0468
Oliveira, L. M., Seminara, S. B., Beranova, M., Hayes, F. J., Valkenburgh, S. B., Schipani, E., et al. (2001). The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J. Clin. Endocrinol. Metab. 86, 1532–1538. doi: 10.1210/jc.86.4.1532
Olsen, S. K., Li, J. Y., Bromleigh, C., Eliseenkova, A. V., Ibrahimi, O. A., Lao, Z., et al. (2006). Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 20, 185–198. doi: 10.1101/gad.1365406
Owen, B. M., Mangelsdorf, D. J., and Kliewer, S. A. (2015). Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 26, 22–29. doi: 10.1016/j.tem.2014.10.002
Packard, A. I., Lin, B., and Schwob, J. E. (2016). Sox2 and Pax6 play counteracting roles in regulating neurogenesis within the murine olfactory epithelium. PLoS One 11:e0155167. doi: 10.1371/journal.pone.0155167
Pedersen-White, J. R., Chorich, L. P., Bick, D. P., Sherins, R. J., and Layman, L. C. (2008). The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Hum. Reprod. 14, 367–370. doi: 10.1093/molehr/gan027
Pellier, V., Astic, L., Oestriecher, A. B., and Saucier, D. (1994). B-50 GAP-43 expression by the olfactory receptor cells and the neurons migrating from the olfactory placode in embryonic rats. Dev. Brain Res. 80, 63–72. doi: 10.1016/0165-3806(94)90090-6
Perala, N. M., Immonen, T., and Sariola, H. (2005). The expression of plexins during mouse embryogenesis. Gene Expr. Patterns 5, 355–362. doi: 10.1016/j.modgep.2004.10.001
Pierce, A., Bliesner, B., Xu, M., Nielsen-Preiss, S., Lemke, G., Tobet, S., et al. (2008). Axl and Tyro3 modulate female reproduction by influencing gonadotropin-releasing hormone neuron survival and migration. Mol. Endocrinol. 22, 2481–2495. doi: 10.1210/me.2008-0169
Pinto, G., Abadie, V., Mesnage, R., Blustajn, J., Cabrol, S., Amiel, J., et al. (2005). CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J. Clin. Endocrinol. Metab. 90, 5621–5626. doi: 10.1210/jc.2004-2474
Pitteloud, N., Meysing, A., Quinton, R., Acierno, J. S. Jr., Dwyer, A. A., Plummer, L., et al. (2006). Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol. Cell. Endocrinol. 254-255, 60–69. doi: 10.1016/j.mce.2006.04.021
Pitteloud, N., Quinton, R., Pearce, S., Raivio, T., Acierno, J., Dwyer, A., et al. (2007). Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 117, 457–463. doi: 10.1172/jci29884
Pratt, T., Conway, C. D., Tian, N. M., Price, D. J., and Mason, J. O. (2006). Heparan sulphation patterns generated by specific heparan sulfotransferase enzymes direct distinct aspects of retinal axon guidance at the optic chiasm. J. Neurosci. 26, 6911–6923. doi: 10.1523/jneurosci.0505-06.2006
Quanbeck, C., Sherwood, N. M., Millar, R. P., and Terasawa, E. (1997). Two populations of luteinizing hormone-releasing hormone neurons in the forebrain of the rhesus macaque during embryonic development. J. Comp. Neurol. 380, 293–309. doi: 10.1002/(sici)1096-9861(19970414)380:3<293::aid-cne1>3.3.co;2-y
Quaynor, S. D., Goldberg, L. Y., Ko, E. K., Stanley, R. K., Demir, D., Kim, H. G., et al. (2014). Differential expression of nasal embryonic LHRH factor (NELF) variants in immortalized GnRH neuronal cell lines. Mol. Cell. Endocrinol. 383, 32–37. doi: 10.1016/j.mce.2013.11.020
Quaynor, S. D., Ko, E. K., Chorich, L. P., Sullivan, M. E., Demir, D., Waller, J. L., et al. (2015). NELF knockout is associated with impaired pubertal development and subfertility. Mol. Cell. Endocrinol. 407, 26–36. doi: 10.1016/j.mce.2015.02.015
Quinton, R., Duke, V. M., Robertson, A., Kirk, J. M., Matfin, G., de Zoysa, P. A., et al. (2001). Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin. Endocrinol. 55, 163–174. doi: 10.1046/j.1365-2265.2001.01277.x
Raucci, F., Tiong, J. D., and Wray, S. (2013). P75 nerve growth factor receptors modulate development of GnRH neurons and olfactory ensheating cells. Front. Neurosci. 7:262. doi: 10.3389/fnins.2013.00262
Reza, J. N., Gavazzi, I., and Cohen, J. (1999). Neuropilin-1 is expressed on adult mammalian dorsal root ganglion neurons and mediates semaphorin3a/collapsin-1-induced growth cone collapse by small diameter sensory afferents. Mol. Cell. Neurosci. 14, 317–326. doi: 10.1006/mcne.1999.0786
Robinson, M., Parsons Perez, M. C., Tebar, L., Palmer, J., Patel, A., Marks, D., et al. (2004). FLRT3 is expressed in sensory neurons after peripheral nerve injury and regulates neurite outgrowth. Mol. Cell. Neurosci. 27, 202–214. doi: 10.1016/j.mcn.2004.06.008
Rodriguez, O. C., Schaefer, A. W., Mandato, C. A., Forscher, P., Bement, W. M., and Waterman-Storer, C. M. (2003). Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 5, 599–609. doi: 10.1038/ncb0703-599
Rodriguez-Gil, D. J., and Greer, C. A. (2008). Wnt/Frizzled family members mediate olfactory sensory neuron axon extension. J. Comp. Neurol. 511, 301–317. doi: 10.1002/cne.21834
Saadi, H., Shan, Y., Marazziti, D., and Wray, S. (2019). GPR37 signaling modulates migration of olfactory ensheathing cells and gonadotropin releasing hormone cells in mice. Front. Cell Neurosci. 13:200. doi: 10.3389/fncel.2019.00200
Sabado, V., Barraud, P., Baker, C. V., and Streit, A. (2012). Specification of GnRH-1 neurons by antagonistic FGF and retinoic acid signaling. Dev. Biol. 362, 254–262. doi: 10.1016/j.ydbio.2011.12.016
Saint-Jeannet, J. P., and Moody, S. A. (2014). Establishing the pre-placodal region and breaking it into placodes with distinct identities. Dev. Biol. 389, 13–27. doi: 10.1016/j.ydbio.2014.02.011
Sakano, H. (2010). Neural map formation in the mouse olfactory system. Neuron 67, 530–542. doi: 10.1016/j.neuron.2010.07.003
Salian-Mehta, S., Xu, M., Knox, A. J., Plummer, L., Slavov, D., Taylor, M., et al. (2014). Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 99, 1452–1460. doi: 10.1210/jc.2013-3426
Sanlaville, D., Etchevers, H. C., Gonzales, M., Martinovic, J., Clement-Ziza, M., Delezoide, A. L., et al. (2006). Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J. Med. Genet. 43, 211–217.
Sato, N., Katsumata, N., Kagami, M., Hasegawa, T., Hori, N., Kawakita, S., et al. (2004). Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J. Clin. Endocrinol. Metab. 89, 1079–1088. doi: 10.1210/jc.2003-030476
Schaar, B. T., and McConnell, S. K. (2005). Cytoskeletal coordination during neuronal migration. Proc. Natl. Acad. Sci. U.S.A. 102, 13652–13657. doi: 10.1073/pnas.0506008102
Schwanzel-Fukuda, M., Abraham, S., Crossin, K. L., Edelman, G. M., and Pfaff, D. W. (1992). Immunocytochemical demonstration of neural cell adhesion molecule (NCAM) along the migration route of luteinizing hormone-releasing hormone (LHRH) neurons in mice. J. Comp. Neurol. 321, 1–18. doi: 10.1002/cne.903210102
Schwanzel-Fukuda, M., Crossin, K. L., Pfaff, D. W., Bouloux, P. M., Hardelin, J. P., and Petit, C. (1996). Migration of luteinizing hormone-releasing hormone (LHRH) neurons in early human embryos. J. Comp. Neurol. 366, 547–557. doi: 10.1002/(sici)1096-9861(19960311)366:3<547::aid-cne12>3.3.co;2-c
Schwanzel-Fukuda, M., and Pfaff, D. W. (1989). Origin of luteinizing hormone-releasing hormone neurons. Nature 338, 161–164. doi: 10.1038/338161a0
Schwarting, G. A., Kostek, C., Bless, E. P., Ahmad, N., and Tobet, S. A. (2001). Deleted in colorectal cancer (DCC) regulates the migration of luteinizing hormone-releasing hormone neurons to the basal forebrain. J. Neurosci. 21, 911–919. doi: 10.1523/jneurosci.21-03-00911.2001
Schwarting, G. A., Wierman, M. E., and Tobet, S. A. (2007). Gonadotropin-releasing hormone neuronal migration. Semin. Reprod. Med. 25, 305–312. doi: 10.1055/s-2007-984736
Schwob, J. E., Jang, W., Holbrook, E. H., Lin, B., Herrick, D. B., Peterson, J. N., et al. (2017). Stem and progenitor cells of the mammalian olfactory epithelium: taking poietic license. J. Comp. Neurol. 525, 1034–1054. doi: 10.1002/cne.24105
Sedita, J., Izvolsky, K., and Cardoso, W. V. (2004). Differential expression of heparan sulfate 6-O-sulfotransferase isoforms in the mouse embryo suggests distinctive roles during organogenesis. Dev. Dyn. 231, 782–794. doi: 10.1002/dvdy.20173
Sedlmeyer, I. L., and Palmert, M. R. (2002). Delayed puberty: analysis of a large case series from an academic center. J. Clin. Endocrinol. Metab. 87, 1613–1620. doi: 10.1210/jc.87.4.1613
Seminara, S. B., Hayes, F. J., and Crowley, W. F. Jr. (1998). Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. Endocr. Rev. 19, 521–539. doi: 10.1210/edrv.19.5.0344
Sjodal, M., Edlund, T., and Gunhaga, L. (2007). Time of exposure to BMP signals plays a key role in the specification of the olfactory and lens placodes ex vivo. Dev. Cell 13, 141–149. doi: 10.1016/j.devcel.2007.04.020
Smith, T. F. (2008). Diversity of WD-repeat proteins. Subcell Biochem. 48, 20–30. doi: 10.1007/978-0-387-09595-0_3
Solecki, D. J., Trivedi, N., Govek, E. E., Kerekes, R. A., Gleason, S. S., and Hatten, M. E. (2009). Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron 63, 63–80. doi: 10.1016/j.neuron.2009.05.028
Sonnenberg, E., Weidner, K. M., and Birchmeier, C. (1993). Expression of the met-receptor and its ligand, HGF-SF during mouse embryogenesis. EXS 65, 381–394.
Soussi-Yanicostas, N., de Castro, F., Julliard, A. K., Perfettini, I., Chedotal, A., and Petit, C. (2002). Anosmin-1, defective in the X-linked form of Kallmann syndrome, promotes axonal branch formation from olfactory bulb output neurons. Cell 109, 217–228. doi: 10.1016/s0092-8674(02)00713-4
Soussi-Yanicostas, N., Faivre-Sarrailh, C., Hardelin, J. P., Levilliers, J., Rougon, G., and Petit, C. (1998). Anosmin-1 underlying the X chromosome-linked Kallmann syndrome is an adhesion molecule that can modulate neurite growth in a cell-type specific manner. J. Cell Sci. 111(Pt 19), 2953–2965.
Spilker, C., Grochowska, K. M., and Kreutz, M. R. (2016). What do we learn from the murine Jacob/Nsmf gene knockout for human disease? Rare Dis. 4, e1241361. doi: 10.1080/21675511.2016.1241361
Stamou, M. I., and Georgopoulos, N. A. (2018). Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 86, 124–134. doi: 10.1016/j.metabol.2017.10.012
Steventon, B., Mayor, R., and Streit, A. (2014). Neural crest and placode interaction during the development of the cranial sensory system. Dev. Biol. 389, 28–38. doi: 10.1016/j.ydbio.2014.01.021
Stumm, R., and Hollt, V. (2007). CXC chemokine receptor 4 regulates neuronal migration and axonal pathfinding in the developing nervous system: implications for neuronal regeneration in the adult brain. J. Mol. Endocrinol. 38, 377–382. doi: 10.1677/jme-06-0032
Suzuki, J., and Osumi, N. (2015). Neural crest and placode contributions to olfactory development. Curr. Top. Dev. Biol. 111, 351–374. doi: 10.1016/bs.ctdb.2014.11.010
Sykiotis, G. P., Plummer, L., Hughes, V. A., Au, M., Durrani, S., Nayak-Young, S., et al. (2010). Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. U.S.A. 107, 15140–15144. doi: 10.1073/pnas.1009622107
Taniguchi, K., Ayada, T., Ichiyama, K., Kohno, R., Yonemitsu, Y., Minami, Y., et al. (2007). Sprouty2 and Sprouty4 are essential for embryonic morphogenesis and regulation of FGF signaling. Biochem. Biophys. Res. Commun. 352, 896–902. doi: 10.1016/j.bbrc.2006.11.107
Tapanainen, J., Koivisto, M., Vihko, R., and Huhtaniemi, I. (1981). Enhanced activity of the pituitary-gonadal axis in premature human infants. J. Clin. Endocrinol. Metab. 52, 235–238. doi: 10.1210/jcem-52-2-235
Taroc, E. Z. M., Prasad, A., Lin, J. M., and Forni, P. E. (2017). The terminal nerve plays a prominent role in GnRH-1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol. Open 6, 1552–1568. doi: 10.1242/bio.029074
Terasawa, E., Quanbeck, C. D., Schulz, C. A., Burich, A. J., Luchansky, L. L., and Claude, P. (1993). A primary cell culture system of luteinizing hormone releasing hormone neurons derived from embryonic olfactory placode in the rhesus monkey. Endocrinology 133, 2379–2390. doi: 10.1210/en.133.5.2379
Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87. doi: 10.1016/j.cell.2009.12.011
Toba, Y., Tiong, J. D., Ma, Q., and Wray, S. (2008). CXCR4/SDF-1 system modulates development of GnRH-1 neurons and the olfactory system. Dev. Neurobiol. 68, 487–503. doi: 10.1002/dneu.20594
Tobet, S. A., Chickering, T. W., King, J. C., Stopa, E. G., Kim, K., Kuo-Leblank, V., et al. (1996). Expression of gamma-aminobutyric acid and gonadotropin-releasing hormone during neuronal migration through the olfactory system. Endocrinology 137, 5415–5420. doi: 10.1210/en.137.12.5415
Tobet, S. A., and Schwarting, G. A. (2006). Minireview: recent progress in gonadotropin-releasing hormone neuronal migration. Endocrinology 147, 1159–1165. doi: 10.1210/en.2005-1275
Tornberg, J., Sykiotis, G. P., Keefe, K., Plummer, L., Hoang, X., Hall, J. E., et al. (2011). Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. U.S.A. 108, 11524–11529. doi: 10.1073/pnas.1102284108
Trarbach, E. B., Costa, E. M., Versiani, B., de Castro, M., Baptista, M. T., Garmes, H. M., et al. (2006). Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J. Clin. Endocrinol. Metab. 91, 4006–4012. doi: 10.1210/jc.2005-2793
Truwit, C. L., Barkovich, A. J., Grumbach, M. M., and Martini, J. J. (1993). MR imaging of Kallmann syndrome, a genetic disorder of neuronal migration affecting the olfactory and genital systems. AJNR Am. J. Neuroradiol. 14, 827–838.
Tsang, M., Friesel, R., Kudoh, T., and Dawid, I. B. (2002). Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol. 4, 165–169. doi: 10.1038/ncb749
Tsuji, L., Yamashita, T., Kubo, T., Madura, T., Tanaka, H., Hosokawa, K., et al. (2004). FLRT3, a cell surface molecule containing LRR repeats and a FNIII domain, promotes neurite outgrowth. Biochem. Biophys. Res. Commun. 313, 1086–1091. doi: 10.1016/j.bbrc.2003.12.047
Tucker, E. S., Lehtinen, M. K., Maynard, T., Zirlinger, M., Dulac, C., Rawson, N., et al. (2010). Proliferative and transcriptional identity of distinct classes of neural precursors in the mammalian olfactory epithelium. Development 137, 2471–2481. doi: 10.1242/dev.049718
Turan, I., Hutchins, B. I., Hacihamdioglu, B., Kotan, L. D., Gurbuz, F., Ulubay, A., et al. (2017). CCDC141 mutations in idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 102, 1816–1825. doi: 10.1210/jc.2016-3391
Urness, L. D., Li, C., Wang, X., and Mansour, S. L. (2008). Expression of ERK signaling inhibitors Dusp6, Dusp7, and Dusp9 during mouse ear development. Dev. Dyn. 237, 163–169. doi: 10.1002/dvdy.21380
Valdes-Socin, H., Rubio Almanza, M., Tome Fernandez-Ladreda, M., Debray, F. G., Bours, V., and Beckers, A. (2014). Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front. Endocrinol. 5:109. doi: 10.3389/fendo.2014.00109
Wang, Y. Z., Yamagami, T., Gan, Q., Wang, Y., Zhao, T., Hamad, S., et al. (2011). Canonical Wnt signaling promotes the proliferation and neurogenesis of peripheral olfactory stem cells during postnatal development and adult regeneration. J. Cell Sci. 124(Pt 9), 1553–1563. doi: 10.1242/jcs.080580
Watanabe, Y., Inoue, K., Okuyama-Yamamoto, A., Nakai, N., Nakatani, J., Nibu, K., et al. (2009). Fezf1 is required for penetration of the basal lamina by olfactory axons to promote olfactory development. J. Comp. Neurol. 515, 565–584. doi: 10.1002/cne.22074
Wegenke, J. D., Uehling, D. T., Wear, J. B. Jr., Gordon, E. S., Bargman, J. G., Deacon, J. S., et al. (1975). Familial Kallmann syndrome with unilateral renal aplasia. Clin. Genet. 7, 368–381. doi: 10.1111/j.1399-0004.1975.tb00344.x
Whitlock, K. E. (2004). Development of the nervus terminalis: origin and migration. Microsc. Res. Tech. 65, 2–12. doi: 10.1002/jemt.20094
Whitlock, K. E. (2005a). A role for foxd3 and sox10 in the differentiation of gonadotropin-releasing hormone (GnRH) cells in the zebrafish Danio rerio. Development 132, 5491–5502. doi: 10.1242/dev.02158
Whitlock, K. E. (2005b). Origin and development of GnRH neurons. Trends Endocrinol. Metab. 16, 145–151. doi: 10.1016/j.tem.2005.03.005
Whitlock, K. E., Illing, N., Brideau, N. J., Smith, K. M., and Twomey, S. (2006). Development of GnRH cells: setting the stage for puberty. Mol. Cell. Endocrinol. 254-255, 39–50. doi: 10.1016/j.mce.2006.04.038
Wong, S. T., Trinh, K., Hacker, B., Chan, G. C. K., Lowe, G., Gaggar, A., et al. (2000). Disruption of the Type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron 27, 487–497. doi: 10.1016/s0896-6273(00)00060-x
Wray, S. (2002a). Development of gonadotropin-releasing hormone-1 neurons. Front. Neuroendocrinol. 23:292–316. doi: 10.1016/S0091-3022(02)00001-8
Wray, S. (2002b). Molecular mechanisms for migration of placodally derived GnRH neurons. Chem. Senses 27, 569–572. doi: 10.1093/chemse/27.6.569
Wray, S. (2010). From nose to brain: development of gonadotrophin-releasing hormone-1 neurones. J. Neuroendocrinol. 22, 743–753. doi: 10.1111/j.1365-2826.2010.02034.x
Wray, S., Fueshko, S. M., Kusano, K., and Gainer, H. (1996). GABAergic neurons in the embryonic olfactory pit/vomeronasal organ: maintenance of functional GABAergic synapses in olfactory explants. Dev. Biol. 180, 631–645. doi: 10.1006/dbio.1996.0334
Wray, S., Grant, P., and Gainer, H. (1989a). Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progeniotr cells in the olfactory placode. Proc. Natl. Acad. Sci. U.S.A. 86, 8132–8136. doi: 10.1073/pnas.86.20.8132
Wray, S., Nieburgs, A., and Elkabes, S. (1989b). Spatiotemporal cell expression of luteinizing hormone-releasing hormone in the prenatal mouse: evidence for an embryonic origin in the olfactory placode. Brain Res. Dev. Brain Res. 46, 309–318. doi: 10.1016/0165-3806(89)90295-2
Wray, S., Key, S., Qualls, R., and Fueshko, S. M. (1994). A subset of peripherin positive olfactory axons delineates the luteinizing hormone releasing hormone neuronal migratory pathway in developing mouse. Dev. Biol. 166, 349–354. doi: 10.1006/dbio.1994.1320
Xu, C., Messina, A., Somm, E., Miraoui, H., Kinnunen, T., Acierno, J. Jr., et al. (2017). KLB, encoding beta-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. EMBO Mol. Med. 9, 1379–1397. doi: 10.15252/emmm.201607376
Xu, H., Li, Z., Wang, T., Wang, S., Liu, J., and Wang, D. W. (2015). Novel homozygous deletion of segmental KAL1 and entire STS cause Kallmann syndrome and X-linked ichthyosis in a Chinese family. Andrologia 47, 1160–1165. doi: 10.1111/and.12397
Xu, N., Kim, H. G., Bhagavath, B., Cho, S. G., Lee, J. H., Ha, K., et al. (2011). Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 95, e1611–e1617.
Yan, H., Nie, X., and Kocsis, J. D. (2001). Hepatocyte growth factor is a mitogen for olfactory ensheathing cells. J. Neurosci. Res. 66, 698–704. doi: 10.1002/jnr.10009
Yang, H. C., Farooqui, A. A., and Horrocks, L. A. (1994). Effects of sialic acid and sialoglycoconjugates on cytosolic phospholipases A2 from bovine brain. Biochem. Biophys. Res. Commun. 199, 1158–1166. doi: 10.1006/bbrc.1994.1352
Yoshida, K., Tobet, S. A., Crandall, J. E., Jimenez, T. P., and Schwarting, G. A. (1995). The migration of luteinizing hormone-releasing hormone neurons in the developing rat is associated with a transient, caudal projection of the vomeronasal nerve. J. Neurosci. 15, 7769–7777. doi: 10.1523/jneurosci.15-12-07769.1995
Young, J., Metay, C., Bouligand, J., Tou, B., Francou, B., Maione, L., et al. (2012). SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 27, 1460–1465. doi: 10.1093/humrep/des022
Yu, X., Nieman, B. J., Sudarov, A., Szulc, K. U., Abdollahian, D. J., Bhatia, N., et al. (2011). Morphological and functional midbrain phenotypes in Fibroblast Growth Factor 17 mutant mice detected by Mn-enhanced MRI. Neuroimage 56, 1251–1258. doi: 10.1016/j.neuroimage.2011.02.068
Zenaty, D., Bretones, P., Lambe, C., Guemas, I., David, M., Leger, J., et al. (2006). Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1). Mol. Cell. Endocrinol. 254-255, 78–83. doi: 10.1016/j.mce.2006.04.006
Zhang, S., Lin, Y., Itaranta, P., Yagi, A., and Vainio, S. (2001). Expression of Sprouty genes 1, 2, and 4 during mouse organogenesis. Mech. Dev. 109, 367–370. doi: 10.1016/s0925-4773(01)00526-3
Zhao, Y., Lin, M. C., Farajzadeh, M., and Wayne, N. L. (2013). Early development of the gonadotropin-releasing hormone neuronal network in transgenic zebrafish. Front. Endocrinol. 4:107. doi: 10.3389/fendo.2013.00107
Keywords: GnRH, neurodevelopment, olfactory placode, neuronal migration, olfactory system, Kallmann syndrome
Citation: Cho H-J, Shan Y, Whittington NC and Wray S (2019) Nasal Placode Development, GnRH Neuronal Migration and Kallmann Syndrome. Front. Cell Dev. Biol. 7:121. doi: 10.3389/fcell.2019.00121
Received: 21 March 2019; Accepted: 14 June 2019;
Published: 11 July 2019.
Edited by:
Kazuhito Toyo-oka, Drexel University, United StatesReviewed by:
Imre Kalló, Institute of Experimental Medicine (MTA), HungaryCopyright © 2019 Cho, Shan, Whittington and Wray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Wray, d3JheXNAbmluZHMubmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.