 Miriam Gagliardi
Miriam Gagliardi Maria Strazzullo
Maria Strazzullo Maria R. Matarazzo
Maria R. Matarazzo- 1Institute of Genetics and Biophysics “Adriano Buzzati Traverso”, CNR, Naples, Italy
- 2Max Planck Institute of Psychiatry, Munich, Germany
DNA methylation plays important roles in gene expression regulation and chromatin structure. Its proper establishment and maintenance are essential for mammalian development and cellular differentiation. DNMT3B is the major de novo DNA methyltransferase expressed and active during the early stage of embryonic development, including implantation. In addition to its well-known role to methylate centromeric, pericentromeric, and subtelomeric repeats, recent observations suggest that DNMT3B acts as the main enzyme methylating intragenic regions of active genes. Although largely studied, much remains unknown regarding how these specific patterns of de novo CpG methylation are established in mammalian cells, and which are the rules governing DNMT3B recruitment and activity. Latest evidence indicates that DNMT3B recruitment is regulated by numerous mechanisms including chromatin modifications, transcription levels, non-coding RNAs, and the presence of DNA-binding factors. DNA methylation abnormalities are a common mark of human diseases involving chromosomal and genomic instabilities, such as inherited disease and cancer. The autosomal recessive Immunodeficiency, Centromeric instability and Facial anomalies syndrome, type I (ICF-1), is associated to hypomorphic mutations in DNMT3B gene, while its altered expression has been correlated with the development of tumors. In both cases, this implies that abnormal DNA hypomethylation and hypermethylation patterns affect gene expression and genomic architecture contributing to the pathological states. We will provide an overview of the most recent research aimed at deciphering the molecular mechanisms by which DNMT3B abnormalities are associated with the onset and progression of these pathologies.
Introduction
Three DNA methyltransferase, DNMT1, DNMT3A, and DNMT3B, are the enzymatic players of DNA methylation (Leppert and Matarazzo, 2014). They cooperatively act to transfer a methyl group from S-adenosyl methionine (SAM) to the fifth carbon of cytosines (Cs). The methylation mark concerns about 70–80% of CpGs in the mammalian genome, but it also may involve Cs outside the dinucleotide CpG in tissue-specific gene regulation (Arand et al., 2012; Jeltsch and Jurkowska, 2014).
De novo methylation occurs largely during the early embryogenesis and is faithfully copied following DNA replication at each cell cycle (Iurlaro et al., 2017). DNA methylation is a regulatory mechanism involved in numerous biological processes, embryonic development, cell differentiation, parental imprinting, transposon silencing, and X inactivation (Scarano et al., 2005). Alterations in DNA methylation pattern are observed at different extent in rare genetic diseases, in common and complex diseases and in cancer (Strazzullo et al., 2003; Robertson, 2005; Scarano et al., 2005). A proper distribution of DNA methylation is the result of a fine equilibrium between methylation “writers” and “erasers” signals, mediated by appropriate “readers” proteins, contributing to the overall epigenetic regulation of cell homeostasis.
Our current view of DNA methylation mechanism and its functional meaning is rapidly changing. This review will focus on new concepts emerged over the last few years that transcended the conventional distinction between de novo and maintenance DNA methylation, the general association of DNA methylation with transcriptional gene silencing and the unspecific recruitment of DNA methylation machinery to genomic targets.
A particular emphasis will be given to traditional and novel functions of DNMT3B that is the major de novo DNA methyltransferase active during implantation and is impaired in human diseases with chromosomal and genomic instabilities, including inherited disease and cancer.
Traditional and Novel Functions of DNMT3B in DNA Methylation
Traditionally, DNMT1 was considered responsible for the maintenance methylation of hemimethylated DNA after replication, whereas DNMT3A and DNMT3B for the de novo methylation during early development. In contrast to this original view, multiple evidence support that a crosstalk exists between the de novo and maintenance DNA methylation machinery acting in the establishment and inheritance of methyl-CpGs patterns in the genome (Kim et al., 2002). DNMT3A/B gene knockdown provokes a failure in the maintenance of methylation at specific loci and a progressive global DNA hypomethylation in mouse embryonic stem cells (Liang et al., 2002; Chen et al., 2003). From a mechanistic point of view, it has been suggested that a reserve of DNMT3A/B stably associated to CG-rich regions (i.e., those present at the heterochromatic sequences), might integrate the methylation of hemimethylated CpGs failed by the maintenance activity of DNMT1 (Jeong et al., 2009). Indeed, a close cooperation between the maintenance and de novo methylating activities has been reported in several cellular contexts, including many cancer cells (Rhee et al., 2002; Ting et al., 2004).
The DNA methylation-mediated transcriptional repression occurring at CpG islands (CGI) associated promoters and repetitive sequences is the most canonical activity described for DNMT3B. This activity mainly contributes to long-term gene silencing, which requires to be preserved in certain tissues for the lifespan of the organism.
Besides the centromeric, pericentromeric, and subtelomeric repeats, the germline genes are well-known genomic targets of DNMT3B. Notably, the maintenance of methylation at CGIs of certain germlines specific genes in somatic cells is fully dependent on DNMT3B activity (Walton et al., 2011, 2014). Here, the enzyme binds to promoters and protects somatic cells from their illegitimate transcription through the interaction with the transcriptional repressor E2F6 (Velasco et al., 2010). The ectopic expression of these genes is a common feature in ICF syndrome and cancer cells showing DNMT3B dysfunction (Jeanpierre et al., 1993; Matarazzo et al., 2009). Interestingly, the Dnmt3b-mediated DNA methylation controls the expression of some germline genes acting against transposable elements in developing germ cells (Hackett et al., 2012).
Additional unique features of the DNA methylation, in particular at non-promoter CGIs, have been identified with the advent of novel methodologies of DNA methylation profiling characterized by highest resolution and coverage (Ball et al., 2009; Rauch et al., 2009). It has been clearly stated that DNA methylation plays an opposite role in the regulation of gene expression when associated with intragenic CGIs.
Interestingly, a recent study showed that many CGIs within gene bodies undergo methylation during development and differentiation (Jeziorska et al., 2017). Here, the transcription through CGIs, together with specific histone marks, is a primary determinant for DNMT3B activity in methylation of these intragenic CGIs. DNA methylation of these regions promotes their silencing and reduces the transcriptional noise within the gene (Jeziorska et al., 2017). Similarly, in mouse embryonic stem cells the Dnmt3b-mediated DNA methylation at intragenic regions inhibits the access of RNA polymerase II and cryptic transcription initiation. Maintaining the fidelity of transcription initiation is exclusively ascribed to the catalytic activity of Dnmt3b that is recruited to the gene body by the H3K36me3 histone mark (Neri et al., 2017).
These observations are in line with a large block of evidence supporting the view that DNA methylation at gene bodies of highly expressed genes is dependent on the activity of DNMT3B rather than of DNMT3A (Yang et al., 2014; Baubec et al., 2015; Duymich et al., 2016). This specific role might be associated to the transcriptional repression of alternative promoters, transcription factor binding sites, and retrotransposon elements (LINEs, SINEs, LTRs, and other retroviruses) to preserve the function of the canonical transcriptional start site (TSS) (Maunakea et al., 2010; Wolff et al., 2010; Kulis et al., 2012).
Our work in cells derived from ICF1 patients is compatible with this view since we showed that DNMT3B mutations alter the transcriptional regulation at intragenic level, impairing the proper TSS usage and causing spurious transcription from intragenic cryptic TSS. Indeed, the aberrant hypomethylation at alternative or cryptic TSSs caused by DNMT3B dysfunction leads to their illegitimate activation interfering with the transcription and elongation of the appropriate mRNA. Switching to an alternative intragenic TSS may be caused by the hypermethylation of the canonical TSS following an altered recruitment of DNMT3A/3B proteins (Figure 1A; Gatto et al., 2017).
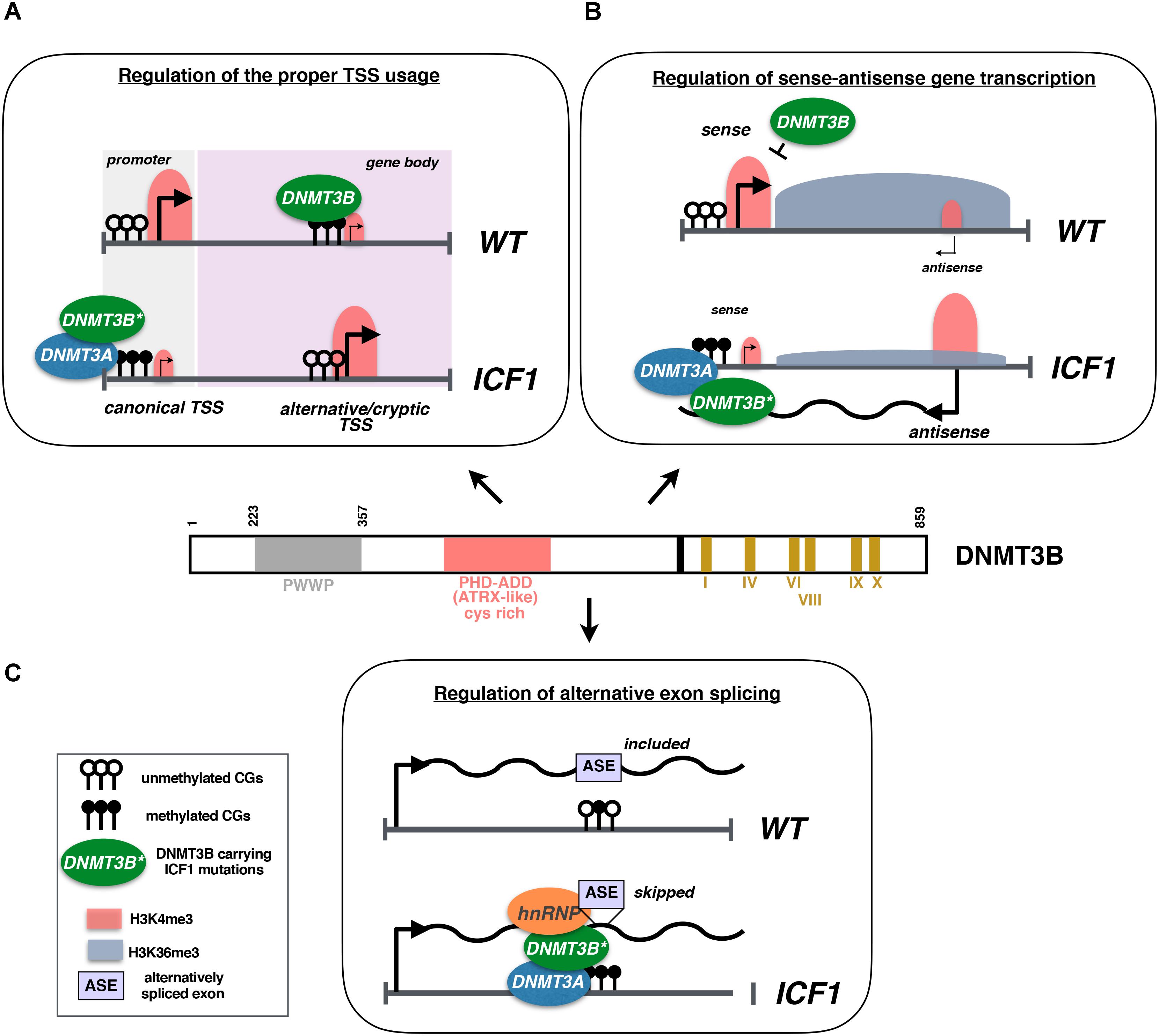
FIGURE 1. (A) DNMT3B methylates gene bodies to promote a repressive chromatin environment that inhibits the activity of Pol II at alternative and/or cryptic TSSs. In ICF1 cells, DNMT3B dysfunction associates with CpG hypomethylation and leads to illegitimate transcription from these TSSs. At certain loci, the usage of intragenic alternative TSS is mediated by the hypermethylation of the canonical TSS caused by mistargeting of DNMT3A in complex with the mutated DNMT3B protein. (B) In wild-type cells, the high expression of sense gene associates with the spreading of a transcription-induced accumulation of H3K36me3. In turn, H3K36me3 enrichment at the antisense TSS promotes a repressive chromatin environment inhibiting the transcription of antisense transcript. Conversely, in ICF1 cells the epigenetic silencing of antisense promoter (CpG methylation and H3K4me3 level) is altered, fostering its transcription. The antisense transcript interacts with DNMT3B/3A proteins inducing their recruitment to sense TSS, which in turn acquires CpG hypermethylation and loses H3K4me3 mark. The sense gene is then silenced and this results in a reduced H3K36me3 level at antisense TSS, thereby sustaining the antisense transcription. (C) In disease cells, DNMT3B promotes aberrant exon skipping during alternative splicing, acting as adaptor protein able to interact with DNA and pre-mRNA, and by recruiting hnRNPs to the pre-mRNA.
Furthermore, we highlighted that DNMT3B plays a key role in the control of sense-antisense gene expression and the splicing of alternative exons (Gatto et al., 2017). Generally, the expression of natural antisense RNAs (NATs) induces a threshold-dependent turn on/off of sense-gene expression, representing a fine-tuned regulatory mechanism of transcription (Pelechano and Steinmetz, 2013). In ICF1, the lack of DNMT3B proper activity disturbs the epigenetic silencing of the antisense CD27AS TSS (located at 3′ of the memory B-cell marker CD27 gene locus) and promotes its transcription. The upregulated antisense transcript recruits the complex DNMT3B/3A to the sense TSS, inducing CpG hypermethylation and CD27 gene silencing (Figure 1B).
Alternative exons inclusion in the mature mRNA during the co-transcriptional splicing may also be influenced by mutations in DNMT3B. In patient-derived lymphoblastoid cell lines (LCLs), cells, three alternative exons of the trans-membrane protein tyrosine phosphatase (PTPRC) gene are abnormally spliced giving rise to a shorter mature mRNA compared to control cells. Here, the mutant DNMT3B protein would contribute to the exon skipping acting as an adaptor protein, by interacting with both heterogeneous nuclear ribonucleoproteins (hnRNPs) and the pre-mRNA (Figure 1C). The evidence that DNA methylation may have an essential role in the alternative splicing is increasingly growing (Chodavarapu et al., 2010; Anastasiadou et al., 2011; Gelfman et al., 2013). First, exon sequences tend to be hypermethylated compared to the flanking introns and alternative exons with higher DNA methylation level exhibit a higher inclusion rate (Shukla et al., 2011; Yearim et al., 2015). Also, exonic DNA methylation might influence alternative splicing by gathering MeCP2 and HDACs proteins to foster exon recognition and their inclusion in the mature mRNA (Maunakea et al., 2013). Remarkably, the functional inactivation of DNA methyltransferase 3 (dnmt3) in honeybee suggests that the regulation of alternative splicing might represent a specific and evolutionary conserved activity for DNMT3B (Li-Byarlay et al., 2013).
Two working models have been proposed to explain how epigenetic marks (histone modifications and DNA methylation) may influence the alternative splicing. The first is a kinetic mechanism involving the modulation of the transcriptional elongation rate, the second implies a recruitment mechanism, in which adaptor proteins recognize the epigenetic marks and tether splicing factors to pre-mRNAs (Iannone and Valcarcel, 2013). Our findings point to a role of adapter protein for the mutant DNMT3B that forms an aberrant complex including both the hnRNP and the abnormally spliced pre-mRNA (Gatto et al., 2017).
Regulation of DNMT3B Recruitment to Genomic Targets
DNMT3B recruitment to target sites is tightly regulated by the crosstalk between the de novo methyltransferase and chromatin remodeling complexes, histone modifications and transcription factors (Hervouet et al., 2018). It has been reported that DNMT3B has both distinct and overlapping functions with its paralog DNMT3A, with which it shares a high degree of similarity for the catalytic domain. The binding peculiarity might be associated with the N-terminal of the protein. This region is less conserved and contains the regulatory domains ADD and PWWP responsible for the protein–protein and DNA–protein interactions, respectively (Jurkowska et al., 2011).
In particular, the ADD domain enables DNMT3B to recognize and preferentially bind histone 3 tails not methylated in lysine 4 (Ooi et al., 2007; Zhang et al., 2010; Morselli et al., 2015), and prevents the binding to active TSS. Recent reports highlighted that PWWP domain is responsible for the recruitment of DNMT3B protein at the gene body through the recognition of H3K36me3 enrichment in those regions (Baubec et al., 2015). The interaction of DNMT3B with H3K36me3 was also observed in human epidermal stem cells, where DNMT3B interacts with the body of cell-type specific actively transcribed enhancers and catalyzes their hypermethylation (Rinaldi et al., 2016).
Moreover, the positioning of DNMT3B at pericentromeric regions is associated with its ability to interact with the H3K9me3 specific methyltransferase SETDB1 and is stabilized through HP1 recognition (Lehnertz et al., 2003). Apparently, this mechanism does not involve the centromeres where the DNMT3B recruitment is favored by the interaction with CENP-C through the PWWP domain (Gopalakrishnan et al., 2009). Similarly, the polycomb group protein EZH2 mediates the recruitment of DNMT3B to H3K27me3 enriched regions through a direct physical interaction (Vire et al., 2006).
DNMT3B recruitment and activity are also regulated by transcription factors. It was indeed reported that several transcription factors, such as E2F6 (Velasco et al., 2010), MIZ1, and PU.1 (de la Rica et al., 2013) also act as positive regulators of the DNMT3B recruitment, leading to silencing of their target genes (Brenner et al., 2005; Suzuki et al., 2006; de la Rica et al., 2013). In vitro studies suggested that DNMT3B might be enrolled at the Oct-3/4 promoter by NR6A1 transcription factors, repressing the expression of this pluripotency gene during murine differentiation (Sato et al., 2006). However, transcription factors can also act as negative regulators of DNMT3B binding. CTCF and SP1 were shown to block de novo DNA methylation events at the target regions (Brandeis et al., 1994; Wang et al., 2012).
Non-coding RNA (ncRNA) may also influence the activity of DNMT3B. pRNA, a small ncRNA transcribed from the rDNA promoter, interacting with the TTF1 transcription factors forms a DNA:RNA triplex that stabilizes DNMT3B binding to the promoter, inducing the rDNA silencing (Schmitz et al., 2010).
The GC composition of the RNA molecules interacting with DNMT3B is an important factor regulating the DNMT3B activity. Indeed, RNAs transcribed from regions with high GC skew form DNA:RNA hybrids named R-loop that inhibit DNMT3B binding to DNA (Santos-Pereira and Aguilera, 2015). These structures are associated with the maintenance of the not methylated status of promoter CGIs that were shown to have higher GC skew compared to intragenic CGIs (Ginno et al., 2012). Recently, Sagie et al. (2017) demonstrated that the majority of telomeric repeat-containing RNAs (TERRAs) are GC skew rich and thus able to form strong R-loops. The RNA:DNA hybrid formation in TERRA upregulated contexts, as described in ICF patients LCLs, might contribute to the maintenance of the abnormal open chromatin status at the telomeric regions involved in telomeres shortening (Deng et al., 2010; Toubiana and Selig, 2018).
Alteration of DNMT3B Function in Human Disease
An in-depth knowledge of the specific role that each component of the DNA methylation machinery may play in the determination of pathological phenotypes is fundamental to untangle the complex network of epigenetic regulators in health and pathology. In Table 1 are summarized some molecular alterations in DNMT3B gene associated with different classes of human diseases.
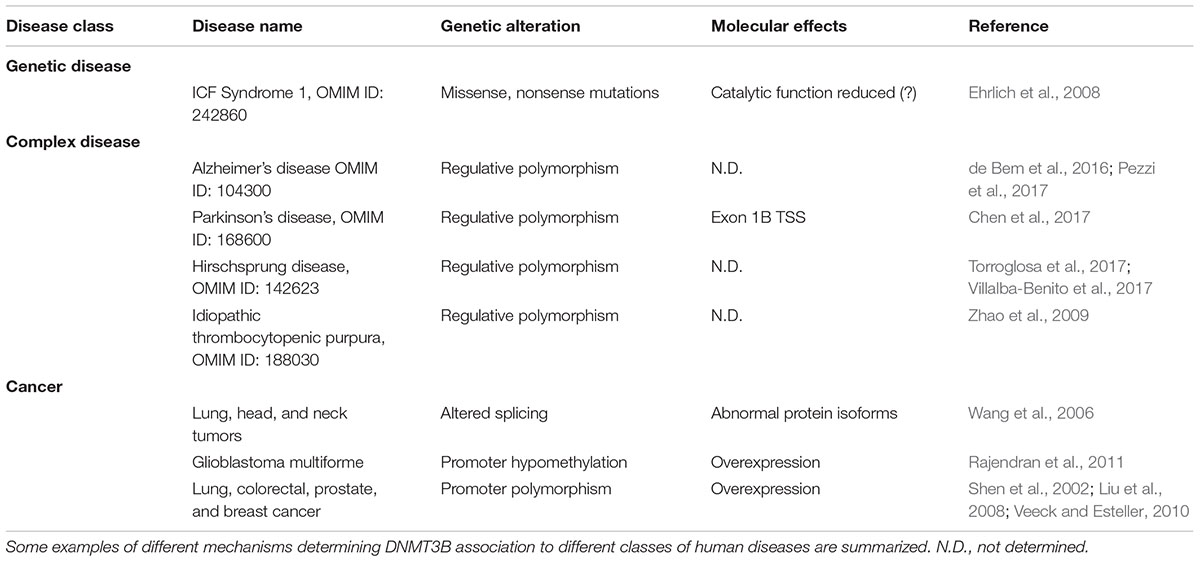
TABLE 1. DNMT3B alterations in human diseases.
In humans, germinal mutations of DNMT3B have been observed only in a condition of partial preservation of the protein expression and function. This is the case of ICF syndrome, the extremely rare genetic syndrome that is, as reported above, a fundamental source of the molecular findings about DNMT3B function.
A comprehensive evaluation of the genetic contribution of DNMT3B to the onset of complex disorders needs the identification of disease-predisposing polymorphisms. In this case, the general difficulty in dissecting complex traits is further complicated by the effect of the partial redundancy existing in DNMTs activity (Liao et al., 2015). Thus, the definition of a robust genotype/phenotype correlation is often missing.
As mentioned above, DNMT3B protein contains several functional domains connecting the protein to a wider network of regulative complexes. The PWWP and the cysteine-rich domains recognize specific histone post-translational modifications that, in a combinatory code, contribute to the overall regulative pattern. In this light, polymorphisms modifying the functional integrity may be found in the whole DNMT3B protein. Further, predisposing polymorphisms may be also recognized in the regulative regions, influencing the transcriptional and post-transcriptional behavior of DNMT3b locus (Gianfrani et al., 2018).
Recently, DNMT3B polymorphisms have been associated with higher susceptibility to develop nervous system degenerative pathologies and immune disorders.
The association between DNMT3B polymorphisms and Alzheimer’s disease (AD) is controversial. A particular DNMT3B haplotype (TGG: rs2424913/rs998382/rs2424932) shows an increased protein expression (over 30%) and was associated with higher susceptibility risk (de Bem et al., 2016). Notably, rs2424913, rs998382, and rs2424932 are localized in non-coding regions of the gene, suggesting a regulative effect of these polymorphisms.
The non-coding polymorphism rs2424913 (the T allele) was also associated with Parkinson’s Disease (PD) (Chen et al., 2017; Pezzi et al., 2017) and with a decreased risk of developing colon cancer in Asian population (Duan et al., 2015). Epidemiological studies (Driver, 2014) suggest an inverse correlation between neurodegenerative disorders and cancer risk. A similar mechanism, the hyper-methylation of the promoters of specific genes (neuro-specific genes and oncogenes, respectively), due to DNMT3B abnormal activity, may contribute to explain these shared phenotypes (Pezzi et al., 2017).
The onset of the intestinal Hirschsprung disease was associated with the overexpression of recognized DNMT3B target genes in enteric nervous system development (Torroglosa et al., 2017; Villalba-Benito et al., 2017). Moreover, a promoter polymorphism (-579G > T) was associated with idiopathic thrombocytopenic purpura (Zhao et al., 2009) and with a higher risk of thymomas in patients with myasthenia gravis (Coppede et al., 2013).
In cancer, hypo- and hyper-methylation constitute a fundamental molecular hallmark, widely analyzed in term of pathogenetic and progression mechanisms, diagnosis and prognosis markers and therapeutic targets. In a fine-tuned cross-talk with other epigenetic mechanisms, such as chromatin remodeling factors, genomic hypermethylation is one of the mechanisms inducing tumor suppressor silencing while genomic hypomethylation contributes to oncogene overexpression and genomic instability. Specific patterns of hypo- and hyper-methylation have been identified in specific tumor types and subtypes. In this scenario, the emerging role of DNMT3B is quite complex. Altered DNMT3B expression levels result into variation in the targeting efficiency and abnormal catalytic activity contributing to cancer development and progression. This enzyme is highly expressed during early embryonic development and then down-regulated in most tissues. Overexpression of DNMT3B in tumors is a frequent observation (DNMT3B is overexpressed in 30% of breast cancers) (Bishop and Ferguson, 2015), associated with the down-regulation of its targets. Therefore, DNMT3B appears to act primarily as an oncogene, and in some tumor types, its overexpression is an unfavorable prognostic marker (Hayette et al., 2012).
DNMT3B is overexpressed in glioblastoma as a consequence of the hypomethylation of its own promoter (Rajendran et al., 2011) and represents a marker for tumor staging and prognosis (Purkait et al., 2016).
In other contexts, data support the idea that the reduced expression of a normal DNMT3B contributes to accelerate tumorigenesis acting as a haploinsufficient tumor suppressor. Furthermore the overexpression of truncated variants and other catalytically inactive isoforms or those derived from abnormal alternative splicing, gives a significant contribution to cancer evolution.
A large number of different isoforms (over 30) of DNMT3B have been described, produced from alternative splicing and/or alternative promoter usage (Ostler et al., 2007; Gopalakrishnan et al., 2009; Gordon et al., 2013).
In myc-induced lymphomas, prompted tumorigenesis seems to be associated to the expression of a truncated isoform, DNMT3B7, acting as dominant negative and commonly expressed in human cancer (Vasanthakumar et al., 2013).
Genetic mutations in DNMT3B locus are observed with low frequency also in cancer. Cancer risk predisposing polymorphisms in regulatory and coding regions have been observed in a large number of studies (Chen et al., 2017; Feng et al., 2018). For example, polymorphisms determining upregulation of gene transcription have been associated with an increased risk of developing lung cancer and head and neck carcinoma (Shen et al., 2002; Liu et al., 2008). Based on meta-analysis data, a polymorphism in a DNMT3B regulative region has also been associated with reduced risk of cancer (Zhu et al., 2012) but this point is still debated (Xia et al., 2015).
As described above, DNMT3B is involved in the regulation of DNA methylation in gene bodies. The intragenic DNA methylation, in cooperation with H3K36me3 and Pol II, contributes to the preservation of correct transcript initiation, counteracting intragenic spurious transcription within the gene (Neri et al., 2017). Aberrant DNA methylation occurring at the gene bodies in cancer, might account for stochastic aberrant transcription and promote tumor cell heterogeneity (Teneng et al., 2015). A published study investigates the effect of demethylating agents (i.e., 5-aza-2′-deoxycytidine), as a potential therapeutic agent in cancer not only contributing to oncosuppressor reactivation but also determining specific oncogenes repression (Yang et al., 2014). The authors suggest that hypomethylation at the gene bodies in specific genomic and cellular context could also result in, at least partial, restoration of their appropriate expression levels.
Conclusion
Recent genome-wide studies have revealed previously unrecognized functions of DNMT3B, providing unprecedented mechanistic insights into how DNA methylation contributes to gene expression and cell identity. These studies have shown that in addition to the traditional silencing role at CpG-associated promoters, DNA methylation plays multiple regulatory functions. Having such a functional flexibility, it is plausible that DNMT3B protein plays different roles in transcriptional regulation that are context-dependent. Prominent examples are the fine-tuning of gene expression by preventing spurious activation from cryptic internal promoters and by regulating the alternative exons inclusion during mRNA splicing. The evidence that multiple molecular factors are able to influence the recruitment of DNMT3B to genomic targets provides additional levels of complexity.
The traditional and new functions ascribed to DNMT3B enzyme reflect the intricate role of DNA methylation in the regulation of the overall gene expression in disease pathogenesis. Synergistic effects of transcriptional and post-transcriptional alterations of a subset of target genes, determine, at a different extent, the perturbation of cell homeostasis. A better understanding of all the DNMTs functions will allow deciphering the molecular basis of pathological phenotypes associated with abnormal patterns of DNA methylation and how these defects contribute to them.
Strategies integrating genome editing and genome-wide identification of methylation sites are expected to provide further evidence into the mechanisms driving the recruitment and the activity of DNMT3B and the other DNMTs. To improve our knowledge of the biological roles of DNA methylation, it will also be important to distinguish the catalytic function of the DNA methyltransferases from the other biochemical properties of these proteins. Further studies of the context-specific functions of DNMT3B will be thus required to better understand how this enzyme integrates the methylation signals with the chromatin into the network of epigenetic regulation.
Author Contributions
MM and MS conceived and wrote the review. MG wrote the review.
Funding
This work was financially supported by the “Epigenomics Flagship Project (EPIGEN) Italian Ministry of Education, University and Research-CNR” and the Telethon grant GGP15209 to MM and the CNR-DSB Flagship Project “InterOmics” 2017 to MS. Funding for open access charge was provided by Epigenomics Flagship Project (EPIGEN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anastasiadou, C., Malousi, A., Maglaveras, N., and Kouidou, S. (2011). Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 30, 267–275. doi: 10.1089/dna.2010.1094
Arand, J., Spieler, D., Karius, T., Branco, M. R., Meilinger, D., Meissner, A., et al. (2012). In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 8:e1002750. doi: 10.1371/journal.pgen.1002750
Ball, M. P., Li, J. B., Gao, Y., Lee, J. H., Leproust, E. M., Park, I. H., et al. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368. doi: 10.1038/nbt.1533
Baubec, T., Colombo, D. F., Wirbelauer, C., Schmidt, J., Burger, L., Krebs, A. R., et al. (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. doi: 10.1038/nature14176
Bishop, K. S., and Ferguson, L. R. (2015). The interaction between epigenetics, nutrition and the development of cancer. Nutrients 7, 922–947. doi: 10.3390/nu7020922
Brandeis, M., Frank, D., Keshet, I., Siegfried, Z., Mendelsohn, M., Nemes, A., et al. (1994). Sp1 elements protect a CpG island from de novo methylation. Nature 371, 435–438. doi: 10.1038/371435a0
Brenner, C., Deplus, R., Didelot, C., Loriot, A., Vire, E., De Smet, C., et al. (2005). Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 24, 336–346. doi: 10.1038/sj.emboj.7600509
Chen, T., Ueda, Y., Dodge, J. E., Wang, Z., and Li, E. (2003). Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003
Chen, X., Xiao, Y. S., Wei, L., Wu, Y. J., Lu, J. J., Guo, W. Y., et al. (2017). Association of DNMT3b gene variants with sporadic Parkinson’s disease in a Chinese Han population. J. Gene Med. 19, 360–365. doi: 10.1002/jgm.2991
Chodavarapu, R. K., Feng, S., Bernatavichute, Y. V., Chen, P. Y., Stroud, H., Yu, Y., et al. (2010). Relationship between nucleosome positioning and DNA methylation. Nature 466, 388–392. doi: 10.1038/nature09147
Coppede, F., Ricciardi, R., Denaro, M., De Rosa, A., Provenzano, C., Bartoccioni, E., et al. (2013). Association of the DNMT3B -579G > T polymorphism with risk of thymomas in patients with myasthenia gravis. PLoS One
de Bem, C. M., Pezzi, J. C., Borba, E. M., Chaves, M. L., De Andrade, F. M., Fiegenbaum, M., et al. (2016). The synergistic risk effect of apolipoprotein epsilon4 and DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B) haplotype for Alzheimer’s disease. Mol. Biol. Rep. 43, 653–658. doi: 10.1007/s11033-016-3999-6
de la Rica, L., Rodriguez-Ubreva, J., Garcia, M., Islam, A. B., Urquiza, J. M., Hernando, H., et al. (2013). PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 14:R99. doi: 10.1186/gb-2013-14-9-r99
Deng, Z., Campbell, A. E., and Lieberman, P. M. (2010). TERRA, CpG methylation and telomere heterochromatin: lessons from ICF syndrome cells. Cell Cycle 9, 69–74. doi: 10.4161/cc.9.1.10358
Driver, J. A. (2014). Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology 15, 547–557. doi: 10.1007/s10522-014-9523-2
Duan, F., Cui, S., Song, C., Dai, L., Zhao, X., and Zhang, X. (2015). Systematic evaluation of cancer risk associated with DNMT3B polymorphisms. J. Cancer Res. Clin. Oncol. 141, 1205–1220. doi: 10.1007/s00432-014-1894-x
Duymich, C. E., Charlet, J., Yang, X., Jones, P. A., and Liang, G. (2016). DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 7:11453. doi: 10.1038/ncomms11453
Ehrlich, M., Sanchez, C., Shao, C., Nishiyama, R., Kehrl, J., Kuick, R., et al. (2008). ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 41, 253–271. doi: 10.1080/08916930802024202
Feng, X., Wang, J., Gu, X., Zhang, J., Li, X., Tao, Z., et al. (2018). Association of DNMT3B -283T > C polymorphism with risk of lung and gastric cancer: a case-control study and a meta-analysis. Int. J. Biol. Markers 33, 195–200. doi: 10.5301/ijbm.5000306
Gatto, S., Gagliardi, M., Franzese, M., Leppert, S., Papa, M., Cammisa, M., et al. (2017). ICF-specific DNMT3B dysfunction interferes with intragenic regulation of mRNA transcription and alternative splicing. Nucleic Acids Res. 45, 5739–5756. doi: 10.1093/nar/gkx163
Gelfman, S., Cohen, N., Yearim, A., and Ast, G. (2013). DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 23, 789–799. doi: 10.1101/gr.143503.112
Gianfrani, C., Pisapia, L., Picascia, S., Strazzullo, M., and Del Pozzo, G. (2018). Expression level of risk genes of MHC class II is a susceptibility factor for autoimmunity: new insights. J. Autoimmun. 89, 1–10. doi: 10.1016/j.jaut.2017.12.016
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., and Chedin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 45, 814–825. doi: 10.1016/j.molcel.2012.01.017
Gopalakrishnan, S., Van Emburgh, B. O., Shan, J., Su, Z., Fields, C. R., Vieweg, J., et al. (2009). A novel DNMT3B splice variant expressed in tumor and pluripotent cells modulates genomic DNA methylation patterns and displays altered DNA binding. Mol. Cancer Res. 7, 1622–1634. doi: 10.1158/1541-7786.MCR-09-0018
Gordon, C. A., Hartono, S. R., and Chedin, F. (2013). Inactive DNMT3B splice variants modulate de novo DNA methylation. PLoS One 8:e69486. doi: 10.1371/journal.pone.0069486
Hackett, J. A., Reddington, J. P., Nestor, C. E., Dunican, D. S., Branco, M. R., Reichmann, J., et al. (2012). Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 139, 3623–3632. doi: 10.1242/dev.081661
Hayette, S., Thomas, X., Jallades, L., Chabane, K., Charlot, C., Tigaud, I., et al. (2012). High DNA methyltransferase DNMT3B levels: a poor prognostic marker in acute myeloid leukemia. PLoS One 7:e51527. doi: 10.1371/journal.pone.0051527
Hervouet, E., Peixoto, P., Delage-Mourroux, R., Boyer-Guittaut, M., and Cartron, P. F. (2018). Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 10:17. doi: 10.1186/s13148-018-0450-y
Iannone, C., and Valcarcel, J. (2013). Chromatin’s thread to alternative splicing regulation. Chromosoma 122, 465–474. doi: 10.1007/s00412-013-0425-x
Iurlaro, M., Von Meyenn, F., and Reik, W. (2017). DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 43, 101–109. doi: 10.1016/j.gde.2017.02.003
Jeanpierre, M., Turleau, C., Aurias, A., Prieur, M., Ledeist, F., Fischer, A., et al. (1993). An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 2, 731–735. doi: 10.1093/hmg/2.6.731
Jeltsch, A., and Jurkowska, R. Z. (2014). New concepts in DNA methylation. Trends Biochem. Sci. 39, 310–318. doi: 10.1016/j.tibs.2014.05.002
Jeong, S., Liang, G., Sharma, S., Lin, J. C., Choi, S. H., Han, H., et al. (2009). Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 29, 5366–5376. doi: 10.1128/MCB.00484-09
Jeziorska, D. M., Murray, R. J. S., De Gobbi, M., Gaentzsch, R., Garrick, D., Ayyub, H., et al. (2017). DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. U.S.A. 114, E7526–E7535. doi: 10.1073/pnas.1703087114
Jurkowska, R. Z., Jurkowski, T. P., and Jeltsch, A. (2011). Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–222. doi: 10.1002/cbic.201000195
Kim, G. D., Ni, J., Kelesoglu, N., Roberts, R. J., and Pradhan, S. (2002). Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 21, 4183–4195. doi: 10.1093/emboj/cdf401
Kulis, M., Heath, S., Bibikova, M., Queiros, A. C., Navarro, A., Clot, G., et al. (2012). Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 44, 1236–1242. doi: 10.1038/ng.2443
Lehnertz, B., Ueda, Y., Derijck, A. A., Braunschweig, U., Perez-Burgos, L., Kubicek, S., et al. (2003). Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200. doi: 10.1016/S0960-9822(03)00432-9
Leppert, S., and Matarazzo, M. R. (2014). De novo DNMTs and DNA methylation: novel insights into disease pathogenesis and therapy from epigenomics. Curr. Pharm. Des. 20, 1812–1818. doi: 10.2174/13816128113199990534
Liang, G., Chan, M. F., Tomigahara, Y., Tsai, Y. C., Gonzales, F. A., Li, E., et al. (2002). Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell. Biol. 22, 480–491. doi: 10.1128/MCB.22.2.480-491.2002
Liao, J., Karnik, R., Gu, H., Ziller, M. J., Clement, K., Tsankov, A. M., et al. (2015). Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 47, 469–478. doi: 10.1038/ng.3258
Li-Byarlay, H., Li, Y., Stroud, H., Feng, S., Newman, T. C., Kaneda, M., et al. (2013). RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc. Natl. Acad. Sci. U.S.A. 110, 12750–12755. doi: 10.1073/pnas.1310735110
Liu, Z., Wang, L., Wang, L. E., Sturgis, E. M., and Wei, Q. (2008). Polymorphisms of the DNMT3B gene and risk of squamous cell carcinoma of the head and neck: a case-control study. Cancer Lett. 268, 158–165. doi: 10.1016/j.canlet.2008.03.034
Matarazzo, M. R., De Bonis, M. L., Vacca, M., Della Ragione, F., and D’esposito, M. (2009). Lessons from two human chromatin diseases, ICF syndrome and Rett syndrome. Int. J. Biochem. Cell Biol. 41, 117–126. doi: 10.1016/j.biocel.2008.07.026
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
Morselli, M., Pastor, W. A., Montanini, B., Nee, K., Ferrari, R., Fu, K., et al. (2015). In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. eLife 4:e06205. doi: 10.7554/eLife.06205
Neri, F., Rapelli, S., Krepelova, A., Incarnato, D., Parlato, C., Basile, G., et al. (2017). Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77. doi: 10.1038/nature21373
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. doi: 10.1038/nature05987
Ostler, K. R., Davis, E. M., Payne, S. L., Gosalia, B. B., Exposito-Cespedes, J., Le Beau, M. M., et al. (2007). Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 26, 5553–5563. doi: 10.1038/sj.onc.1210351
Pelechano, V., and Steinmetz, L. M. (2013). Gene regulation by antisense transcription. Nat. Rev. Genet. 14, 880–893. doi: 10.1038/nrg3594
Pezzi, J. C., De Bem, C. M., Da Rocha, T. J., Schumacher-Schuh, A. F., Chaves, M. L., Rieder, C. R., et al. (2017). Association between DNA methyltransferase gene polymorphism and Parkinson’s disease. Neurosci. Lett. 639, 146–150. doi: 10.1016/j.neulet.2016.12.058
Purkait, S., Sharma, V., Kumar, A., Pathak, P., Mallick, S., Jha, P., et al. (2016). Expression of DNA methyltransferases 1 and 3B correlates with EZH2 and this 3-marker epigenetic signature predicts outcome in glioblastomas. Exp. Mol. Pathol. 100, 312–320. doi: 10.1016/j.yexmp.2016.02.002
Rajendran, G., Shanmuganandam, K., Bendre, A., Muzumdar, D., Goel, A., and Shiras, A. (2011). Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 104, 483–494. doi: 10.1007/s11060-010-0520-2
Rauch, T. A., Wu, X., Zhong, X., Riggs, A. D., and Pfeifer, G. P. (2009). A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 671–678. doi: 10.1073/pnas.0812399106
Rhee, I., Bachman, K. E., Park, B. H., Jair, K. W., Yen, R. W., Schuebel, K. E., et al. (2002). DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556. doi: 10.1038/416552a
Rinaldi, L., Datta, D., Serrat, J., Morey, L., Solanas, G., Avgustinova, A., et al. (2016). Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 19, 491–501. doi: 10.1016/j.stem.2016.06.020
Robertson, K. D. (2005). DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610. doi: 10.1038/nrg1655
Sagie, S., Toubiana, S., Hartono, S. R., Katzir, H., Tzur-Gilat, A., Havazelet, S., et al. (2017). Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat. Commun. 8:14015. doi: 10.1038/ncomms14015
Santos-Pereira, J. M., and Aguilera, A. (2015). R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597. doi: 10.1038/nrg3961
Sato, N., Kondo, M., and Arai, K. (2006). The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochem. Biophys. Res. Commun. 344, 845–851. doi: 10.1016/j.bbrc.2006.04.007
Scarano, M. I., Strazzullo, M., Matarazzo, M. R., and D’esposito, M. (2005). DNA methylation 40 years later: its role in human health and disease. J. Cell. Physiol. 204, 21–35. doi: 10.1002/jcp.20280
Schmitz, K. M., Mayer, C., Postepska, A., and Grummt, I. (2010). Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 24, 2264–2269. doi: 10.1101/gad.590910
Shen, H., Wang, L., Spitz, M. R., Hong, W. K., Mao, L., and Wei, Q. (2002). A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res. 62, 4992–4995.
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., et al. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. doi: 10.1038/nature10442
Strazzullo, M., Cossu, A., Baldinu, P., Colombino, M., Satta, M. P., Tanda, F., et al. (2003). High-resolution methylation analysis of the hMLH1 promoter in sporadic endometrial and colorectal carcinomas. Cancer 98, 1540–1546. doi: 10.1002/cncr.11651
Suzuki, M., Yamada, T., Kihara-Negishi, F., Sakurai, T., Hara, E., Tenen, D. G., et al. (2006). Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 25, 2477–2488. doi: 10.1038/sj.onc.1209272
Teneng, I., Tellez, C. S., Picchi, M. A., Klinge, D. M., Yingling, C. M., Snider, A. M., et al. (2015). Global identification of genes targeted by DNMT3b for epigenetic silencing in lung cancer. Oncogene 34, 621–630. doi: 10.1038/onc.2013.580
Ting, A. H., Jair, K. W., Suzuki, H., Yen, R. W., Baylin, S. B., and Schuebel, K. E. (2004). CpG island hypermethylation is maintained in human colorectal cancer cells after RNAi-mediated depletion of DNMT1. Nat. Genet. 36, 582–584. doi: 10.1038/ng1365
Torroglosa, A., Villalba-Benito, L., Fernandez, R. M., Moya-Jimenez, M. J., Antinolo, G., and Borrego, S. (2017). Dnmt3b knock-down in enteric precursors reveals a possible mechanism by which this de novo methyltransferase is involved in the enteric nervous system development and the onset of Hirschsprung disease. Oncotarget 8, 106443–106453. doi: 10.18632/oncotarget.22473
Toubiana, S., and Selig, S. (2018). DNA:RNA hybrids at telomeres - when it is better to be out of the (R) loop. FEBS J. 285, 2552–2566. doi: 10.1111/febs.14464
Vasanthakumar, A., Lepore, J. B., Zegarek, M. H., Kocherginsky, M., Singh, M., Davis, E. M., et al. (2013). Dnmt3b is a haploinsufficient tumor suppressor gene in Myc-induced lymphomagenesis. Blood 121, 2059–2063. doi: 10.1182/blood-2012-04-421065
Veeck, J., and Esteller, M. (2010). Breast cancer epigenetics: from DNA methylation to microRNAs. J. Mammary Gland Biol. Neoplasia 15, 5–17. doi: 10.1007/s10911-010-9165-1
Velasco, G., Hube, F., Rollin, J., Neuillet, D., Philippe, C., Bouzinba-Segard, H., et al. (2010). Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl. Acad. Sci. U.S.A. 107, 9281–9286. doi: 10.1073/pnas.1000473107
Villalba-Benito, L., Torroglosa, A., Fernandez, R. M., Ruiz-Ferrer, M., Moya-Jimenez, M. J., Antinolo, G., et al. (2017). Overexpression of DNMT3b target genes during Enteric Nervous System development contribute to the onset of Hirschsprung disease. Sci. Rep. 7:6221. doi: 10.1038/s41598-017-06539-8
Vire, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., et al. (2006). The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874. doi: 10.1038/nature04431
Walton, E. L., Francastel, C., and Velasco, G. (2011). Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics 6, 1373–1377. doi: 10.4161/epi.6.11.17978
Walton, E. L., Francastel, C., and Velasco, G. (2014). Dnmt3b prefers germ line genes and centromeric regions: lessons from the ICF syndrome and cancer and implications for diseases. Biology 3, 578–605. doi: 10.3390/biology3030578
Wang, H., Maurano, M. T., Qu, H., Varley, K. E., Gertz, J., Pauli, F., et al. (2012). Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 22, 1680–1688. doi: 10.1101/gr.136101.111
Wang, L., Wang, J., Sun, S., Rodriguez, M., Yue, P., Jang, S. J., et al. (2006). A novel DNMT3B subfamily, DeltaDNMT3B, is the predominant form of DNMT3B in non-small cell lung cancer. Int. J. Oncol. 29, 201–207.
Wolff, E. M., Byun, H. M., Han, H. F., Sharma, S., Nichols, P. W., Siegmund, K. D., et al. (2010). Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 6:e1000917. doi: 10.1371/journal.pgen.1000917
Xia, Z., Duan, F., Jing, C., Guo, Z., Nie, C., and Song, C. (2015). Quantitative assessment of the association between DNMT3B-579G > T polymorphism and cancer risk. Cancer Biomark. 15, 707–716. doi: 10.3233/CBM-150512
Yang, X., Han, H., De Carvalho, D. D., Lay, F. D., Jones, P. A., and Liang, G. (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590. doi: 10.1016/j.ccr.2014.07.028
Yearim, A., Gelfman, S., Shayevitch, R., Melcer, S., Glaich, O., Mallm, J. P., et al. (2015). HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 10, 1122–1134. doi: 10.1016/j.celrep.2015.01.038
Zhang, Y., Jurkowska, R., Soeroes, S., Rajavelu, A., Dhayalan, A., Bock, I., et al. (2010). Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253. doi: 10.1093/nar/gkq147
Zhao, H., Du, W., Gu, D., Wang, D., Xue, F., Ge, J., et al. (2009). DNMT3B 579G > T promoter polymorphism and the risk for idiopathic thrombocytopenic purpura in a Chinese population. Acta Haematol. 122, 31–35. doi: 10.1159/000235616
Keywords: DNMT3B, DNA methylation, ICF syndrome, cancer, epigenetics, gene expression, human disease
Citation: Gagliardi M, Strazzullo M and Matarazzo MR (2018) DNMT3B Functions: Novel Insights From Human Disease. Front. Cell Dev. Biol. 6:140. doi: 10.3389/fcell.2018.00140
Received: 30 June 2018; Accepted: 02 October 2018;
Published: 22 October 2018.
Edited by:
Beatrice Bodega, Istituto Nazionale Genetica Molecolare (INGM), ItalyReviewed by:
Gokul Gopinath, Texas A&M University College of Dentistry, United StatesNicoletta Landsberger, Università degli Studi di Milano, Italy
Copyright © 2018 Gagliardi, Strazzullo and Matarazzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Strazzullo, bWFyaWEuc3RyYXp6dWxsb0BpZ2IuY25yLml0 Maria R. Matarazzo, bWFyaWEubWF0YXJhenpvQGlnYi5jbnIuaXQ=