
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Bioeng. Biotechnol. , 09 October 2020
Sec. Nanobiotechnology
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.585363
 Valentina Parodi1*
Valentina Parodi1* Emanuela Jacchetti1
Emanuela Jacchetti1 Roberto Osellame2,3
Roberto Osellame2,3 Giulio Cerullo2,3
Giulio Cerullo2,3 Dario Polli2,3†
Dario Polli2,3† Manuela Teresa Raimondi1†
Manuela Teresa Raimondi1†A recent challenge in the field of bioimaging is to image vital, thick, and complex tissues in real time and in non-invasive mode. Among the different tools available for diagnostics, nonlinear optical (NLO) multi-photon microscopy allows label-free non-destructive investigation of physio-pathological processes in live samples at sub-cellular spatial resolution, enabling to study the mechanisms underlying several cellular functions. In this review, we discuss the fundamentals of NLO microscopy and the techniques suitable for biological applications, such as two-photon excited fluorescence (TPEF), second and third harmonic generation (SHG-THG), and coherent Raman scattering (CRS). In addition, we present a few of the most recent examples of NLO imaging employed as a label-free diagnostic instrument to functionally monitor in vitro and in vivo vital biological specimens in their unperturbed state, highlighting the technological advantages of multi-modal, multi-photon NLO microscopy and the outstanding challenges in biomedical engineering applications.
One of the most fascinating advancements in bioengineering is the possibility to observe and control the microscopic universe of cells in order to understand the biological mechanisms involved in physiological and pathological phenomena of life (Vo-Dinh, 2003). Since researchers are concerned to find high speed, effective, and non-invasive diagnostic tools to identify the state of biological samples, several micro-scale imaging techniques have been developed recently, aiming at applications usable by clinicians on human patients (König et al., 2019). The power of optical microscopy is to provide a non-invasive morphological and functional characterization of the observed living specimen over time (Stephens, 2003). Among the existing microscopy techniques, brightfield microscopy is suitable to observe unlabelled vital and thin samples, and it is the easiest to use. However, monolayered cells appears transparent and contrast methods, like phase contrast microscopy, differential interference contrast microscopy or digital holography, are necessary to distinguish cells or parts of them (Murphy, 2001). Nevertheless, these methods provide low amount of information from live samples, being limited to qualitative estimation on cell morphology and growth (e.g., % area occupied by the cells over time in the field of view) and being capable of simply distinguishing endoplasmic reticulum, nucleus and vesicles. To gain more details, biological specimens can be characterized by means of cyto-histopathological assays, which however, require fixative procedure to color specific cellular structures, thus affecting vitality. Another common technique to enrich the information content achievable from a vital biological sample is fluorescence microscopy (Combs and Shroff, 2017). In this technique, highly specific light-emitting probes (dye molecules, semiconductor nanoparticles, or fluorescent proteins) chemically bound to specific biological targets (i.e., DNA, phospholipids, and proteins) are administered to the live or fixed samples. By exciting the fluorescent species with a lamp or a laser, it is possible to filter the longer wavelength (Stokes shifted) emitted signal, detecting it in either a widefield or a point scanning approach and reconstructing images (Murphy, 2001; Lichtman and Conchello, 2005). Technological advances consist in the addition of automated scanners, vertical stages, high numerical aperture objectives (oil or water immersion), and corrective pinholes and filters to detect fluorescence over the volume of the specimen (<100 μm thick) (Combs and Shroff, 2017). Furthermore, systems equipped with incubator chambers allow performing live imaging and time-lapse microscopy. However, fluorescence-based techniques show limits related to the required short excitation wavelengths with continuous lasers that cause photobleaching of fluorophores, scattering and absorption affecting the signal collection and the penetration depth in thick samples. Moreover, the use of fluorescent probes can influence the vitality of biological specimens and it often involves complex preparatory treatments to ensure a proper binding with their biological target, thus resulting toxic over time (Liu et al., 1999). The advent of nonlinear optical (NLO) microscopy techniques allowed to overcome these limitations exploiting multi-photon processes stimulated by pulsed lasers with infrared wavelengths. NLO microscopy offers deep tissue penetration (>500 μm) since the infrared excitation wavelengths inherently provide a reduced light scattering and absorption (Konig, 2000; Sanderson et al., 2016). Furthermore, NLO processes based on multi-photon excitation overcome the use of staining, allowing one to obtain rich morphological/structural/molecular information from a sample, which shows nonlinear properties and/or distinctive chemical composition. NLO-based microscopy offers inherent advantages with respect to single-photon fluorescence (SPF), such as label-free observations, 3D-sectioning capability, small focal volume and greater penetration depth, thus making functional imaging possible (Helmchen and Denk, 2005; Hoover and Squier, 2013). Despite NLO microscopy comprises several techniques, the most relevant for biological investigations are: two-photon excited fluorescence (TPEF) (W Denk et al., 1990), second and third harmonic generation (SHG and THG) (Barad et al., 1997; P. J. Campagnola et al., 2001) and coherent Raman scattering (CRS) (Evans and Xie, 2008) microscopy.
Two-photon fluorescence allows the visualization of both exogenous (dye molecules, semiconductor quantum dots, and fluorescent proteins such as GFP, RFP, and YFP) (Xu et al., 1996; Drobizhev et al., 2011) and endogenous fluorophores (such as nicotinamide adenine dinucleotide phosphate-NAD(P) H-, flavin adenine dinucleotide - FAD-, flavoprotein -FP-), and it is often exploited in fluorescence lifetime imaging (FLIM) studies, resulting suitable for metabolic and hybrid investigations. SHG microscopy enables the observation of non-centrosymmetric structures in unperturbed biological specimen, for example collagen fibers, myosin filaments and microtubules and their spatial distribution (Campagnola et al., 2001; Chu et al., 2003). THG microscopy is sensitive to refractive index mismatch between structures in the focal plane, allowing to image interfaces for example between aqueous interstitial fluids and lipid-rich structures, such as cellular membranes, lipid droplets and calcified bone (Weigelin et al., 2016). Finally, CRS techniques are based on the coherent excitation of vibrational modes related to specific chemical bonds of the molecules present in the focal volume, such as lipids, proteins, and DNA (Yue et al., 2011; Zumbusch et al., 2013). Coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) microscopies benefit from the fact that the signal provides information on the chemical composition and the molecular structure, identified in the peaks of the Raman spectrum (Krafft, 2012).
The growing interest in NLO microscopy is testified by the variety of biological studies performed using the previously mentioned techniques in their individual (single-modality), combined (multi-modality) and multi-spectral (multi-color) conditions, resulting in continuously improved acquisition systems. In order to underline the importance of live imaging in the investigation of fundamental biological processes, we provide a background on the physical principles of the different NLO microscopies and we discuss the state of the art and new advanced applications of label-free multi-modal microscopy in living organisms.
Generally, in SPF microscopy the light sources used to probe the specimen are in the visible wavelength range. As shown in the left side of the panel in Figure 1A, the energy of the incident photon is absorbed by promoting the electron from the ground to the excited state. After non-radiative relaxation in the excited state, the electron returns to the ground state releasing a photon with lower energy (longer wavelength) with respect to the incoming one, thus emitting fluorescence (Williams et al., 1994). On the other hand, when an electron absorbs the energy of multiple photons, it will decay by emitting a photon with higher energy (shorter wavelength) with respect to the excitation photons (Zinselmeyer et al., 2009).
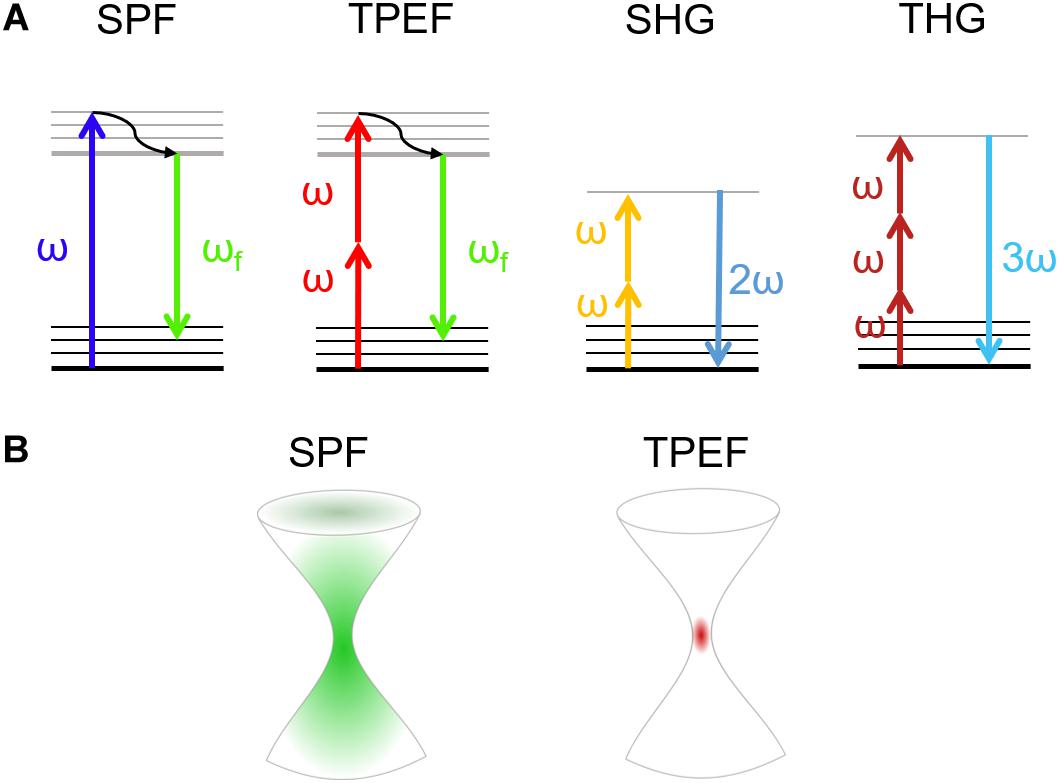
Figure 1. (A) Jablonski diagrams involving (from left to right) single-photon (SPF) and two-photon excited (TPEF) fluorescence, and second harmonic (SHG) and third harmonic (THG) generation. (B) Size of the excited volume in single-photon (left) and multi-photon (right) fluorescence.
Multi-photon absorption phenomena, exploited in NLO microscopy, are typically achieved with ultrashort-pulsed lasers in the near-infrared wavelength range. NLO processes require a high spatial and temporal density of low-energy photons (Figure 1A; Sanderson et al., 2016). To increase the probability of multi-photon absorption and to gain sufficient signal intensity in NLO microscopy, tightly focused ultrashort pulses are typically used, for which high peak power can be obtained, while keeping the average power comparatively low, thus minimizing the risk of photodamage (Chen et al., 2018).
In NLO microscopy, the strong electric fields achievable in a laser pulse can drive the motion of electrons and atoms to create a polarization which depends nonlinearly on the driving light field, expressed by high-order terms in the electric susceptibility [χ(n) where n > 1] (Bloembergen, 1982; Shen, 1985; Di Bartolo and Collins, 2013; Gupta, 2017; Kumar et al., 2018). Due to the strong intensity dependence of the signal, NLO microscopy allows to confine the generation to a very small volume, localized at the laser focus, with respect to the extended excitation obtainable with single photon absorption (Figure 1B). The use of NLO-based techniques is therefore advantageous due to the intrinsic capability of sectioning the specimens. In this way, one can obtain a 3D high-resolution reconstruction of the sample without background noise with just one laser source (Piston et al., 1994).
Two-photon fluorescence is a third order NLO process based on the principle of two-photon absorption established in 1931 by Maria Göppert-Mayer in her doctoral thesis (Göppert-Mayer, 1931) and firstly validated 30 years later with the advent of lasers (Kaiser and Garrett, 1961). Here the combined action of two or more photons, which simultaneously interact with the matter, induces an electronic transition from the ground to an excited electronic state. This process is possible when the sum of the energies of these photons is enough to match the energy gap between the fundamental and the excited state. Hence, the most intense excitation is confined within a small volume (voxel) and no out-of-focus light is generated, removing the need for spatial filters (confocal pinholes) to block the unwanted background (Figure 1B; Rubart, 2004). As a matter of fact, since the voxel is small (less than femtoliter for high numerical aperture objectives) and the trajectories of emitted photons are close to the collection angle of objective lens, the TPEF microscopy enables the pulse to penetrate scattering samples in depth, enabling to perform 3D scanning (Denk et al., 1995). Moreover, since the risk of photodamage is limited to the focal volume, TPEF is preferable with respect to single photon fluorescence. The absorption efficiency depends on the physical properties of the fluorophore, the so called two-photon absorption cross-section, and on the excitation light (Helmchen and Denk, 2005; Drobizhev et al., 2011). The unit used to identify this phenomenon is the Göppert-Mayer (GM per photon) that corresponds to 10–50 cm4 s per photons. Intrinsic molecules such as NAD(P)H are characterized by small cross-sections (<10–4 GM), while common fluorescent dyes show absorption cross-sections in the range of 1-300 GM. Hence, the higher is the two-photon absorption cross-section, the higher is the loss in resolution while increasing the peak power (Zipfel et al., 2003a). However, under normal operation the photodamage is very low in TPEF (Hopt and Neher, 2001).
The harmonic generation is an instantaneous process of coherent nonlinear light scattering, which involves two or three photons in phase matching conditions (Zipfel et al., 2003b). SHG is a second-order NLO process, which happens when two low-energy photons at the same frequency interacting with a nonlinear material are annihilated to generate a new photon with twice the energy of the excitation photons (Campagnola, 2012; Figure 1A). The second-order symmetry of SHG imposes restrictions on active molecular arrays (called “harmonophores”) (Tokarz et al., 2012) and requires the environment to be non-centrosymmetric at the excitation wavelength scale, otherwise the SHG signal vanishes (Campagnola et al., 2001). Since SHG signals occur as a consequence of induced nonlinear polarization instead of absorption phenomena, they exploit the excitation of endogenous factors. The SHG signal has a well-defined polarization, whose anisotropy can be used to determine the hierarchical organization of proteins in tissues. Therefore, the scattering nature of SHG signal drives its intensity emission within a preferential direction. Forward SHG signal is more intense when thin samples are imaged, instead, only backscattered photons can be collected from thick specimens (Mertz and Moreaux, 2001).
In THG microscopy, a third-order NLO process occurs when the fundamental wavelength, typically above 1200 nm, irradiates a medium with intrinsic inhomogeneities due to refractive index mismatch (Yelin and Silberberg, 1999; Rehberg et al., 2011). In THG the combined energy of three photons is converted into a photon emitted at the triple of the energy (a third of the excitation wavelength) (Barad et al., 1997; Figure 1A). In THG microscopy, due to the Guoy phase shift of the fundamental beam, the nonlinear signals generated at symmetric planes with respect to the focus interfere destructively, resulting in zero signal from a homogeneous medium. The THG signal thus reveals the optical heterogeneities of the material illuminated by the laser beam, such as those occurring at the interface between two optically different materials. In contrast to SHG microscopy, no specific symmetries in the material are required to produce a THG signal (Meshulach et al., 1997; Müller et al., 1998).
Raman scattering is a powerful technique for label-free identification of a molecule/material based on the characteristic vibrational spectrum. In spontaneous Raman (SR) microscopy, a monochromatic laser at frequency ωp (“pump”) excites the molecules to a virtual state, which then relax to the ground state scattering photons with lower frequency ωS (“Stokes”). The inelastic frequency shifts Ω = ωp – ωS match the molecular vibrations, which in turn reflect the molecular structure. The resulting SR spectrum provides a detailed picture of the biochemical composition of the measured cells/tissues (Cantarero, 2015). SR microscopy has been translated to clinics e.g., to obtain direct local information from intraoperative analysis of cancer detection (Santos et al., 2016) in human brain (Jermyn et al., 2015; Santos et al., 2017). Despite its advantages, SR microscopy suffers from the drawback of the very weak scattering cross section, due to its incoherent nature, which is 10–12 orders of magnitude lower than fluorescence. This results in low acquisition speed, with pixel dwell times of ≈ 1 s (up to several hours for an image), preventing the acquisition of high spatial resolution images of cells and tissues.
Coherent Raman scattering microscopy overcomes this limitation by generating the Raman signal from a coherent superposition of the molecules in the sample, illuminated by two synchronized ultrashort laser pulses of different color, the pump (at frequency ωp) and the Stokes (at frequency ωS). When the difference between pump and Stokes frequencies matches a vibrational frequency Ω, i.e., ωp – ωS = Ω, then all the molecules in the focal volume are resonantly excited and vibrate in phase. This vibrational coherence enhances the Raman response by many orders of magnitude with respect to the incoherent SR process, decreasing the acquisition times from seconds to microseconds per pixel. Like other NLO microscopies, CRS provides further advantages: (i) the signal is generated only in the focal volume, allowing three-dimensional imaging; (ii) working out of electronic resonance, it minimizes photo-damage to biological samples.
The two most widely employed CRS techniques are coherent anti-Stokes Raman scattering (CARS) (Cheng and Xie, 2004) and stimulated Raman scattering (SRS) (Nandakumar et al., 2009). In CARS (Figure 2A) the vibrational coherence is read by a further interaction with the pump beam, generating a coherent radiation at the anti-Stokes frequency ωaS = ωp + Ω. In SRS (Figure 2B) the coherent interaction with the sample induces stimulated emission from a virtual state of the sample to the investigated vibrational state, resulting in a Stokes-field amplification (Stimulated Raman Gain, SRG) and in a simultaneous pump-field attenuation (Stimulated Raman Loss, SRL). By comparing CARS and SRS, a delicate balance of advantages and drawbacks emerges: CARS benefits from being a background-free process, since the emitted signal has a frequency ωaS differing from those of pump and Stokes (Figure 2C). In this sense, it is similar to TPEF, SHG, and THG microscopies. On the other hand, it suffers from the so-called non-resonant background (NRB) generated both by the molecular species under study and by the surrounding medium, according to a four-wave mixing scheme. The NRB does not carry any chemically specific information and, when the concentrations of the target molecules are low, can distort and even overwhelm the resonant signal of interest. In addition, the CARS signal scales as N2, where N is the number of oscillators in the focal volume, so that its sensitivity rapidly drops with decreasing oscillator concentration, making it difficult to detect the less abundant biomolecules, and in particular their characteristic signatures in the fingerprint region. The SRS signal is proportional to the imaginary part of the third-order susceptibility tensor χ(3): Since the NRB is a real quantity, SRS is inherently free from NRB. Furthermore, SRS scales linearly with N, thus allowing the detection of weakly concentrated species. On the other hand, SRS requires the detection of a weak differential transmission signal (SRG/SRL) sitting on the large background given by the Stokes (pump) light. Extraction of this signal calls for the use of sophisticated techniques, involving high-speed modulation and lock-in detection, to overcome the laser fluctuations and achieve shot-noise limited detection. Such techniques are particularly challenging to implement at high speeds, with integration times of few tens of microseconds, and over a broad spectrum, required to extract the entire Raman fingerprint of the molecules (Wang et al., 2007; Gupta, 2017; Kumar et al., 2018). Briefly, CARS microscopy can be preferentially performed on high molecular concentration specimens, while SRS with high-sensitive and high-speed acquisition systems can be performed on less chemically dense samples (Kumar et al., 2018).
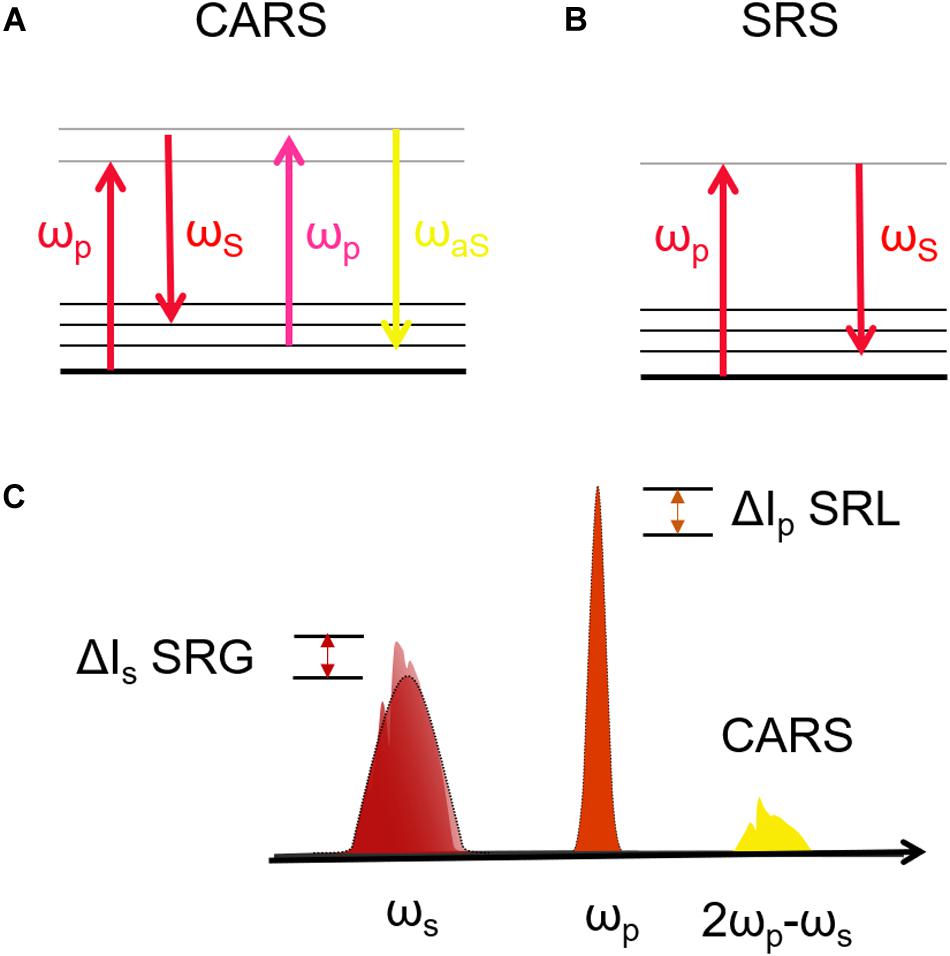
Figure 2. (A,B) Jablonski diagrams of CARS and SRS energy transitions. (C) Schematic representation of the excitation and emission frequencies involved in SRS and CARS: in evidence the SRS signal in terms of gain (SRG) on the Stokes pulse or the loss (SRL) on the pump pulse.
Table 1 summarizes the physical principles, the main advantages and limitations that still affect the above-described techniques within biophysical application.

Table 1. Summary of the main properties of the label-free NLO microscopy techniques from physical principles to advantages and limitations.
Nowadays, there is an ever-growing need to develop diagnostic techniques capable to reveal and quantify at the cellular level the expression of pathological processes that affect human health. A valid solution is represented by NLO microscopy techniques which allow to perform label-free, vital and three-dimensional observations in biological specimens, from in vitro samples to preclinical in vivo models and, potentially, to clinical applications. In the last 30 years, NLO microscopy techniques have been applied to a broad range of biomedical problems, such as tumor infiltration and growth (Lu et al., 2015; Gavgiotaki et al., 2017; De Bortoli et al., 2018; You et al., 2018; Sun et al., 2019), stem cell differentiation and proliferation (Quinn et al., 2013; Meleshina et al., 2016, 2017), tissue regeneration (Yu et al., 2012) and repair (Huemer et al., 2017; Rico-Jimenez et al., 2020). In Figure 3A a schematic representation of a NLO microscope in inverted configuration, in which the excitation light irradiates the sample from the bottom to the top. The collection of the emitted signal can be performed both in reflection than in transmission modality, depending on the sample thickness. In biological applications, several specimens can be imaged, as shown in Figure 3B. As described in the following sections, in vitro monolayer cell models are easy to investigate since thick and transparent media facilitates the orientation and the focusing. Within that, the detection of the generated signal can be dual, especially in case of fixed specimens, without taking care of sterility in case of contact with immersion objectives and environmental air. In vivo analyses, on the other hand, are more difficult since they can be preferentially performed in reflection. Hence, imaging windows for long-term time-dependent studies are often created in specific sites of the animal, thus resulting highly invasive. For this reason, an intermediate application of NLO microscopy to both replicate the heterogenous in vivo environmental conditions without involving direct animal experimentations, is from bioengineering cellular constructs. Thus, 3D, vital and functional imaging of a more realistic patho-physiological condition with respect to cell monolayer and less invasive than preclinical model, is guaranteed.
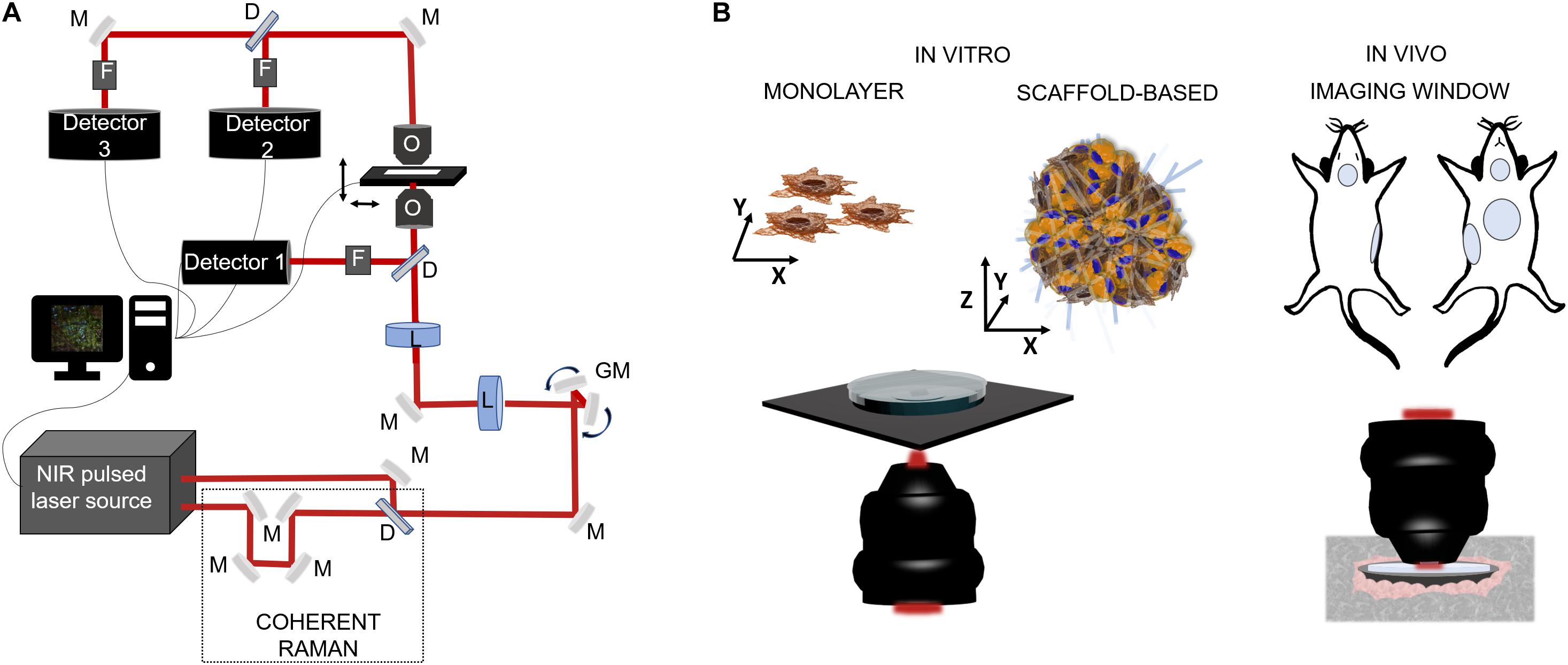
Figure 3. Schematic representations of NLO microscopy application in biology. (A) Example of a NLO set-up characterized by a NIR laser source (two in the case of coherent Raman), a set of fixed (M) and mobile (galvanometric mirrors-GM) mirrors and lenses (L) to drive the light toward the focalization objective (O) and then to the sample which is positioned on a motorized stage. The collection of the emitted light can be in reflection by the use of a dichroic mirror (D), a set of filters (F) (to exclude the excitation component from the reflected light) and, a photodetector, or by transmission thanks to a collection objective or a condenser positioned above the sample. (B) On the left side, cell model for in vitro inspections: single cell analysis on cellular monolayer or 3D cellular constructs composed by heterogenous cell population and a scaffold matrix. The microscope configuration can be in reflection modality, to maintain sample sterility, or in transmission, exposing sample to immersion objective and contaminants. On the right side, intravital microscopy for preclinical analysis is usually made by the use of imaging windows for repetitive observations on the same site for small rodent models. In vivo imaging is made in reflection modality using of cover-glasses sealed on the skin of the animal in specific locations, i.e., mammary glands, cranial, abdominal, and tracheal sites.
The majority of label-free, vital NLO microscopy techniques in biology are applied in vitro using cell culture models that allow to reproduce and to control complex phenomena such as lineage differentiation of stem cells (Quinn et al., 2013; Meleshina et al., 2016, 2017), cell response to drugs (Fu et al., 2014; De Bortoli et al., 2018), synthesis or inhibition of single cellular structures (Zhang et al., 2012; Van Steenbergen et al., 2019). Table 2 summarizes a selection of representative biological studies on vital cultured cells through NLO microscopy published during the last decade. Several works involve the use of multi-frequency SRS microscopy, since it is the most recently developed technique able to provide molecular information of less chemically concentrated biological specimens without the signal impairments typical of CARS, i.e., NRB. Moreover, SRS microscopy is frequently applied to the study of tumor microenvironment, cancer cell composition and dynamics (Zhang et al., 2012; Lu et al., 2015) since the outcomes are often comparable with their histopathological counterparts using conventional hematoxylin and eosin staining (Ji et al., 2013; Chan, 2014). Fa-Ke Lu published in 2015 (Lu et al., 2015) an innovative study based on label-free DNA imaging in cancer cells during their mitotic phase by the use of multi-frequency SRS microscopy (Figure 4A). Both the fingerprint region and the C-H-stretch region of the Raman spectrum were investigated. The best result was reached by linear decomposition of three distinctive peaks at different wavenumbers in the C-H-stretching region thus subtracting the strong protein background and enhancing the Raman signal of the distinct cellular structures (lipids, proteins and nucleic acids) (Figure 4B). Another example of NLO imaging in biology is represented by the study of Meleshina et al. (2016, 2017) based on TPEF-FLIM and SHG microscopy. FLIM was employed to evaluate the metabolic rate of human mesenchymal stem cells (MSCs) during controlled differentiation toward adipogenic, chondrogenic and osteogenic phenotypes at different time-points. SHG microscopy, allowed to monitor collagen formation during chondrogenesis. While differentiation occurred, a metabolic shift was estimated from redox ratio of FAD/NAD(P)H and the lifetimes of bound and free state of NADH in all the phenotypes investigated. The application of THG microscopy is testified by the work of Wu et al. in 2016 (Wu et al., 2016) since different type of white blood cells from the immune system showed distinctive morphological features and intrinsic organelles distribution. Similarly, Gavgiotaki et al. (2017) demonstrated that a discrimination among several sub-types of breast cancer cells was possible by the use of TPEF and THG imaging comparing primary, immortalized cells with peripheral mononuclear cells (control). CARS microscopy is a powerful technique to follow lipid production because the large number of C-H bonds in lipid molecular tail corresponds to high Raman signal intensity (Cheng and Xie, 2004). Saarinen et al. (2017) applied CARS microscopy to a transwell culture configuration on an intestinal epithelium model of Caco-2 cells to evaluate drug permeability throughout the cell layer over 21 days of culture monitoring lipid formation. Another interesting study at the cell scale is from Van Steenbergen et al. (2019) where SHG microscopy is used to observe microtubular structures, a cytoskeletal component involved in protein intracellular transport and mitosis, in neural cells axons derived from different regions of the nervous system. This unusual application of SHG was aimed to validate label-free microscopy as a promising alternative to immunofluorescence staining, since microtubules can be imaged in their native and dynamic configuration. Moreover, this group demonstrated that cell fixation and dehydration with acetone, methanol and common aldehydes highly impaired SHG emission in microtubules, suggesting the potentialities of this technique for imaging untreated live cells. Although Van Steenberg et al. demonstrated that SGH is suitable for investigating in label-free condition single cell process, limitations due to the weakness of the generated signal and the microtubular concentration-dependence of the cell type need to be overcome.
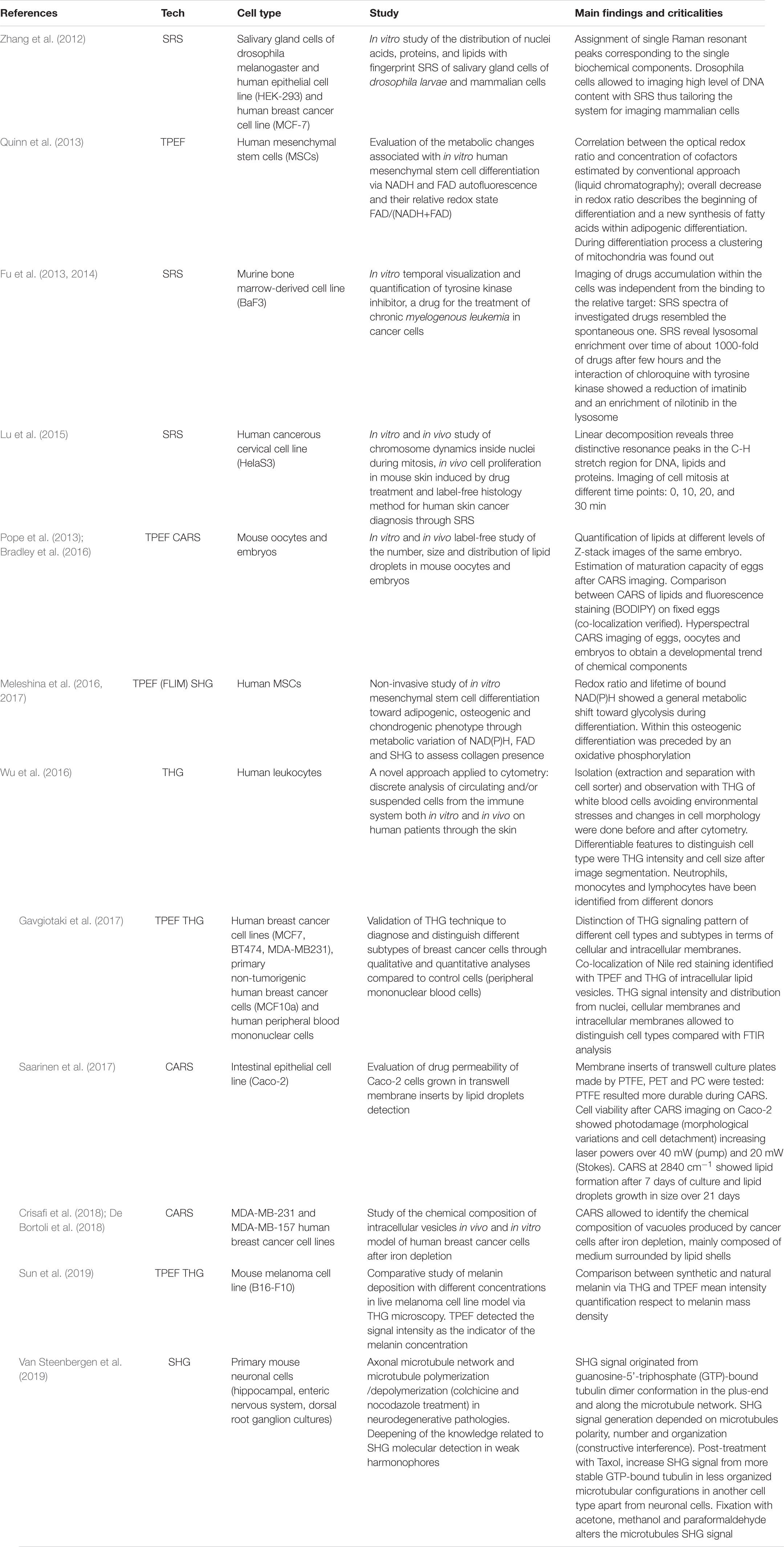
Table 2. Most recent in vitro studies where NLO imaging had been applied in vital and label-free conditions.
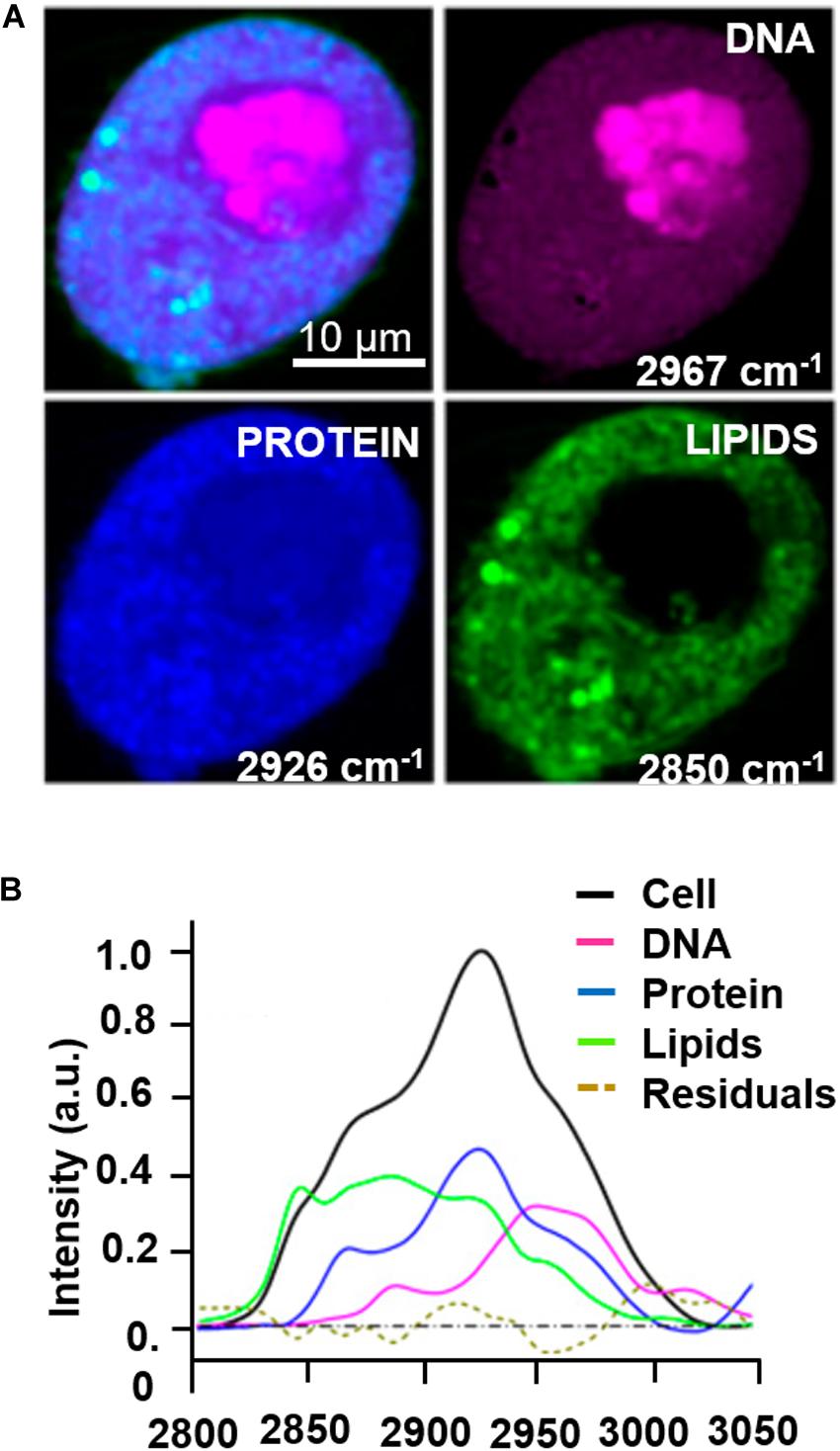
Figure 4. Example of multi-color SRS microscopy on living tumor cells during prophase: (A) DNA (magenta) at 2967 cm–1, proteins (blue) at 2926 cm–1 and lipids (green) at 2850 cm–1. (B) Raman spectrum extracted from the cell pellet showing the signatures of the different species. Scale bar = 10 μm. Figures adapted from (Lu et al., 2015) for kind permission of PNAS.
Completely label-free and vital NLO microscopy applications in vivo are less available in literature since these acquisition techniques are often hybridized with fluorescent tags in situ or directly ex vivo on freshly/cryopreserved tissue slices to improve the orientation within heterogenous samples. Adapting the design of a microscope for animal or human imaging often requires reflection configuration (epi-detection) coupled with immersion objectives and fixative stages to reduce image blurring, aberrations and resolution losses. Table 3 collects in vivo applications of NLO microscopy on vital and unlabeled animal models. Imaging small organism such as zebrafishes (Yu et al., 2012; Huemer et al., 2017), is relatively common in developmental and regenerative time-lapse microscopy thanks to the small size and the low cost related to animal growth. However, the mouse model is still the most used animal in biology to resemble human physiopathology. In NLO microscopy, this is especially employed to reproduce the tumor infiltration dynamics. In fact, You et al. (2018) presented an innovative NLO multi-modal imaging system based on four photodetectors to speed up the acquisition time by sequentially collecting autofluorescence signals of FAD with TPEF, NAD(P)H with three-photon fluorescence, extracellular matrix (ECM) with SHG, and interfaces through THG microscopy on intravital mouse model of mammary gland tumor (Figure 5). Despite the high quality and the good level of interpretability of the results, the intravital approach is highly invasive since a direct access to the imaging site is necessary. The animal underwent surgery to expose the tumor mass and at the end it was euthanized. Similarly, Alonzo et al. (2016) performed a metabolic study by TPEF microscopy coupled with time-resolved analysis of NAD(P)H and FAD of adipose cells from white, brown and induced-beige adipose tissue, to assess the thermoregulatory property of fat. In vivo observations confirmed the estimations previously obtained ex vivo: time-variant metabolic profiles corresponded to a different function of tissue, thus offering more significant results with respect to gene expression analysis. Instead, Lu et al. (2015) studied the proliferation kinetics of tumor cells via multi-color SRS microscopy through a dorsal skinfold chamber. This system is based on a thin layer of skin stretched and encaged between two glasses, thus creating a window that allows optical accessibility to monitor tissue growth over time, and minimizing animal movement and ensuring repetitive analysis in the same area of the sample. A time-dependent analysis of scar tissue formation after superficial dorsal wounding, has been presented by Rico-Jimenez et al. (2020). A comparative preclinical analysis of diabetic mouse model exposed to placebo, low and high dose of drug (hypoxia-inducible factor coupled with prolyl hydroxylase inhibitor) allowed to reveal, via TPEF-FLIM and SHG microscopy, wound healing, angiogenesis, and metabolic behavior differences among cell populations in the injured site at different time points. In this work, the animal survival was guaranteed for the whole duration of the experiments, allowing repetitive observations of the same area without invasive surgical intervention and imaging window implant.
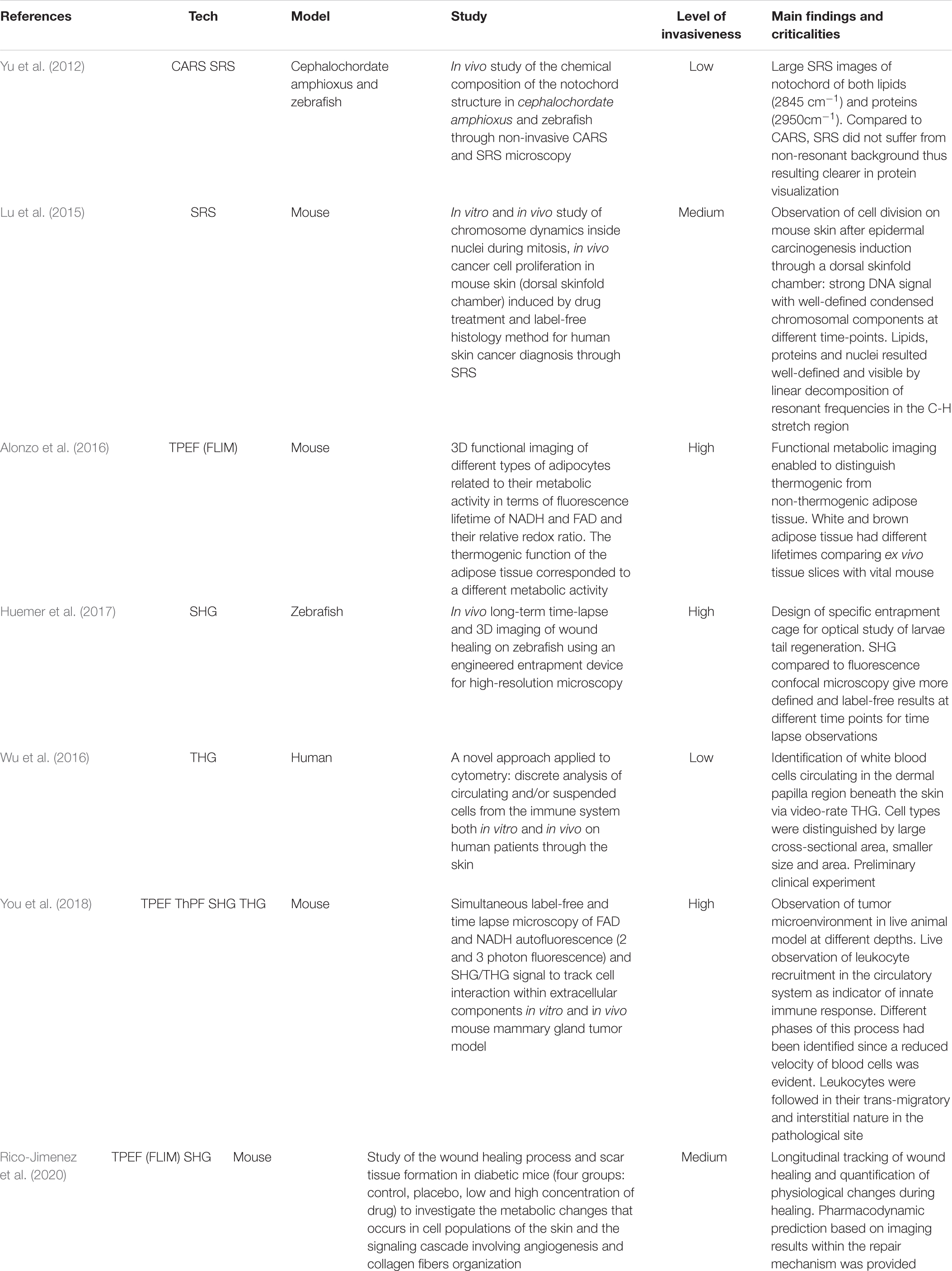
Table 3. Intravital and in vivo NLO imaging in label-free and vital conditions.
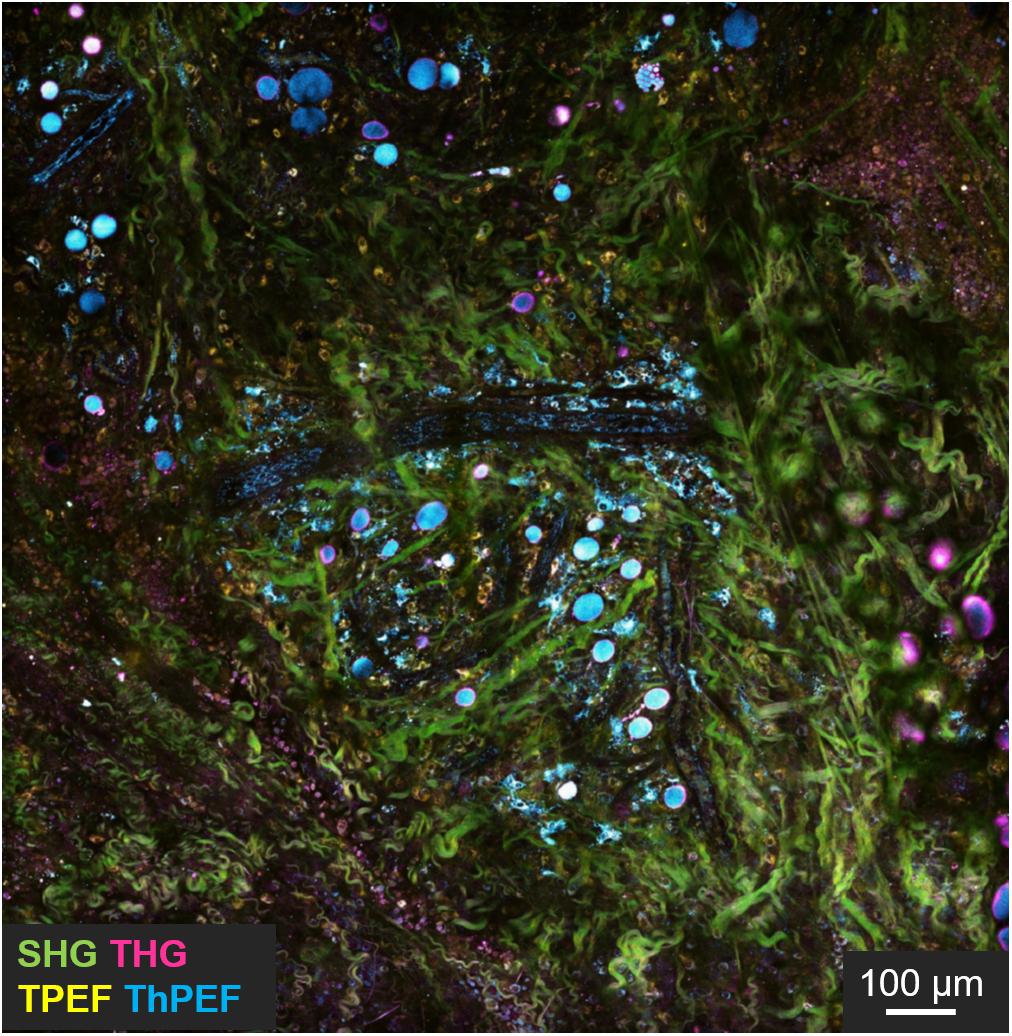
Figure 5. Intravital imaging of mouse tumor microenvironment showing SHG (green), THG (magenta), TPEF (yellow), and three photon fluorescence (cyan) signals. In evidence the green signal from collagen fibers, cyan from NADH, yellow from FAD and magenta from interfaces. Scale bar = 100 μm. Figure reproduced from You et al. (2018) with kind permission from Springer-Nature Communications and licensed by s100.copyright. com\AppDispatchServlet.
Nowadays, although the NLO techniques presented have allowed a wide range of label-free observations with increasingly high-performance, they are still limited in transferring their application from the bi-dimensional in vitro model to complex in vivo samples. Hence, the big challenge of label-free imaging of vital animal models remains to be addressed due to the high level of difficulty when observing several tissue layers and multiple packed populations of cells. A step in between, would be to characterize bioengineered cellular models aimed at emulating in vitro the biological environment in its structural, physical and chemical compositions for modeling tissue regeneration or for local tissue repair. Therefore, the advantages offered by the imaging of in vitro systems are combined with the study of systems near to the preclinical model without incurring the costs and limits of animal experimentation and allowing to obtain equally useful data for scientific research.
In the following section, we will present exemplificative applications of nonlinear imaging in tissue engineering and our preliminary steps in multi-modal NLO microscopy based on a 3D synthetic scaffold for cell culture.
In the last decade, it has been demonstrated that non-invasive nonlinear microscopy represents a promising strategy for label-free live imaging of 3D engineered tissues. In fact, several studies had been performed to characterize morphology (Hofemeier et al., 2016; Nguyen et al., 2017; Syverud et al., 2017; Costa Moura et al., 2018; Kaushik et al., 2019; Moura et al., 2019), functionality (Hofemeier et al., 2016; Li et al., 2017; Syverud et al., 2017; Cong et al., 2019; Okkelman et al., 2020), composition and distribution of chemicals (Hofemeier et al., 2016; Li et al., 2017; Syverud et al., 2017; Costa Moura et al., 2018; Moura et al., 2019; Sood et al., 2019), invasion, infiltration and mechano-regulation of cellular constructs (Hofemeier et al., 2016; Nguyen et al., 2017; Syverud et al., 2017; Costa Moura et al., 2018; Kaushik et al., 2019; Moura et al., 2019; Sood et al., 2019), which have been collected in Table 4. A representative study made by Hofemeier et al. (2016) carried out a multi-spectral CARS and SHG imaging on an engineered bone tissue from stem cell differentiation toward osteogenic phenotype within 3 weeks of culture. Similarly, Syverud et al. (2017) generated a fully-differentiated musculoskeletal tissue and exploited NLO microscopy with TPEF for imaging the autofluorescence of NAD(P)H, and SHG to identify collagen and myosin fibers. This work established label-free NLO microscopy as a valid alternative to collagen and myosin immunostaining, even if the tissue construct was fixed and sliced before the acquisitions, and only monolayered cells were imaged in their entirety and vitality. Contrarily, Moura et al. (Costa Moura et al., 2018; Moura et al., 2019) investigated the composition and the distribution of collagen, proteins and lipids with bioengineered cartilage constructs by the use of multi-modal TPEF, SHG and CARS microscopy both in the fingerprint than in the C-H stretch region of the Raman spectrum. To date, Moura provided a comparison between SHG and CARS imaging of collagen to highlight the difference in signal distribution and observed the synthetic constructs in their thickness. Another innovative example of label-free, vital NLO imaging in tissue engineering is the study of Kaushik et al. (2019) which observed blood vessels formation in a dynamic bioreactor providing multi-well plates with media-flow re-circulation circuits. By monitoring the endogenous TPEF from the cells encapsulated and differentiated within a 3D hydrogel, they were able to reconstruct the network in its three-dimensionality. Furthermore, diameter, density, branch nodes amount and anisotropy of self-assembled blood vessel networks in static, recirculating and continuous flow conditions were estimated from image post-processing. Unfortunately, this study was limited by the duration of the acquisition which prevented the analysis over multiple days. The biological investigations described previously, performed on vital adherent and suspended cells, spheroids/organoids, cellular constructs and small-sized animals, exclusively by the use of label-free NLO microscopy, provided functional information otherwise difficult to obtain through conventional methods. Limited versatility of the acquisition systems, poor optical accessibility of the engineered tissue construct and slow acquisition speed may require further technological improvements in this field. In our group, we built a customize transmission inverted multi-photon NLO microscope designed to perform parallel CARS and SRS microscopy (Crisafi et al., 2017, 2018) and recently adapted also for SHG and TPEF microscopy with pulses < 100 fs. The advantage offered from our personalized acquisition system relies in the possibility to perform in series and on the same area of a sample, CARS, TPEF, SHG, and SRS imaging by filtering properly the emitted signals. We applied these technologies on an optically accessible in vitro experimental model to investigate the behavior of rat bone marrow-derived MSCs growth in a microfabricated 3D scaffold, named Nichoid (Nava et al., 2012; Raimondi et al., 2013; Zandrini et al., 2019) with the aim to perform serial 3D scanning multi-modal NLO imaging over time (Figure 6A). For example, MSCs were cultured inside Nichoids and differentiated toward adipogenic and chondrogenic phenotypes and they were characterized with both conventional biochemical assays and NLO microscopy. Conventional results on the cellular adipogenic phenotype were obtained from bright field imaging of cyto-histological oil red-O staining of lipid droplets produced inside cells after cell fixation and staining. This technique is effective and fast but, unfortunately, returns only qualitative results. On the other hand, CARS microscopy provided the chemical selectivity for lipids, resonant at 2845 cm–1 Raman shift, in vital and unperturbed adipocytes, also allowing to distinguish the droplets volumetric distribution with higher spatial resolution (less than 1 μm of axial resolution) (Figure 6B). Similarly, conventional approach to recognize chondrogenic phenotype, revealed the ECM formation within Nichoids after cell fixation and staining with toluidine blue staining. Conversely, SHG and CARS imaging (2940 cm–1) enable to observe the distribution and quantify the level of collagen, in vital cultured MSC growth inside Nichoids (Figure 6C). In conclusion, our multi-modal set-up, just switching among different techniques (TPEF, SHG, and multi-color CARS/SRS) is able to perform functional imaging on several tissue and on optically accessible 3D device to investigate the phenomena governing the healthy and pathological conditions of cells.

Table 4. Examples of NLO microscopy techniques applied to 3D bioengineered systems in vital and label-free conditions.

Figure 6. (A) Schematic representation of the experimental model employed in this work: on the left, SEM image of a single element of Nichoid 3D synthetic scaffold (Scale bar = 20 μm) used for MSCs culture. Nichoids design was made of three lattice parallel grids with graded pore size and an overall thickness of 30 μm to enable optical accessibility. On the right a rendering of the transmission inverted NLO microscope for multi-modal TPEF, SHG, CARS, and SRS microscopy. (B) Adipogenic differentiation of MSCs after 2 weeks of culture. On the left, oil red-O colorimetric assay of fixed cells, showed lipid droplets formation in red, while in the center, CARS image, of the same sample acquired before fixation and oil red-O staining, showing lipid droplets, resonant at 2845 cm–1 and indicated by white arrows. On the right a 3D rendering of CARS signal acquired by sequential imaging at variable depth inside the sample and stacking the images. (C) Chondrogenic MSCs differentiation after 3 weeks of culture. On the left, toluidine blue assays performed on fixed specimen, revealed production of acidic component of ECM stained in blue. In the center, CARS imaging at 2940 cm–1 Raman shift, resonant with collagen (highlighted by arrows), in untreated, live, and differentiated sample. On the right side, SHG image acquired on vital stem cells after 21 days of differentiation, white arrows indicate collagen fibrils. Autofluorescence signal from the Nichoids material is present and more evident in SHG modality.
This first NLO imaging assessment is a starting point for tissue engineering, oncological research and drug testing applications since conventional methods commonly used to assess phenotypic expression are often disruptive (gene expression analyses) and perturbative (immuno-histochemical staining) thus limiting long-term studies. In this review we presented the main NLO techniques for functional observation of label-free and vital biological specimens, such as TPEF, SHG, THG, CARS, and SRS microscopy and their physical principles. Each of these methods exploits different properties of the light-matter interaction and it is useful to image characteristics of biological specimens in unlabeled conditions. TPEF is recognized as the main NLO techniques capable to excite endogenous molecules and co-factors, and exogenous fluorophores, reaching a great penetration depth (>500 μm). If combined with a fast detector such as a time-correlated single photon counter, it is possible to perform TPEF-FLIM for lifetime measurements of auto-fluorescent molecules, such as NADH and flavoproteins. SHG microscopy, instead, is very useful to reveal label-free signal from a non-centrosymmetric medium such as collagen fibrils, myosin and microtubules. THG, instead, enables the observation of interfaces within media with different refraction indexes, for example, enabling the identification of lipid-water interfaces in cellular vesicles. Vibrational-based CRS techniques excite the vibrational resonance frequency of the main chemical bonds with a specific and distinguishable fingerprint to investigate the chemical composition of the specimens. Finally, CARS and SRS allow the characterization of the biochemical composition of living samples both at the single-cell and at the tissue levels by measuring the vibrational spectrum of the specimen at every position, thus quantifying the concentration of e.g., DNA, lipids and proteins. Despite, all the advantages related to these techniques i.e., 3D sectioning capability, vital imaging and label-free acquisitions, they present a resolution close to the one achievable with confocal microscopy (lateral 200–300 nm and axial 600–900 nm). Currently, single-photon microscopy techniques are used for super resolution microscopy (SRM) such as stimulated emission depletion (STED) microscopy, thus enabling to image molecules with dimension under the diffraction limit (<100 nm) thanks to specific fluorophores or combinations of techniques to extrapolate signals. However, even if the development of SRM-NLO is recent, we believe that this combination results in efficient functional microscopy enabling the observation of dynamic processes at the molecular level in the absence of dyes. Multi-modal and multi-spectral nonlinear imaging will be of a great interest for tissue engineering applications thanks to the variety of biological information observable, from single cell components to heterogenous tissues, without the use of staining in unperturbed and non-destructive way. Hence, future challenging applications would be in clinic since the need for rapid and label-free diagnostic tools is far from being satisfied. In fact, pre-operatory assessments and surgical observations at the cellular level would highly reduce the risk of tissue damage due to unprecise excisions and uncertainty in the identification of injured/pathological sites.
VP wrote the manuscript and selected published scientific articles. EJ, DP, and MR structured the manuscript. EJ, RO, GC, DP, and MR reviewed the work. DP and MR provided financial support. All authors contributed to the article and approved the submitted version.
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (grant agreements Nos. 646990 – NICHOID, 754467 – NICHOIDS, 648615 – VIBRA, and GRAPHENE CORE 3- 881603), the Regione Lombardia project NEWMED (Grant No. POR FESR 2014-2020), the Italian FARE-MIUR project CH2ROME project (Grant No. R164WYYR8N), and BEYOND project (Grant No. R16ZNN2R9K).
MR, GC, and RO were co-founders of a university spin-off company, MOAB S.r.l., and hold shares.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2D, bi-dimensional; 3D, three-dimensional; Caco-2, Caucasian colon adenocarcinoma cells; CARS, coherent anti-Stokes Raman scattering; C-H, carbon-hydrogen; CRS, coherent Raman scattering; DNA, deoxyribonucleic acid; ECM, extracellular matrix; FAD, flavin-adenine dinucleotide; FLIM, fluorescent lifetime imaging; FP, flavoprotein; FRAP, fluorescence recovery after photobleaching; FRET, florescence resonance energy transfer; FTIR, Fourier transform infrared spectroscopy; GFP, green fluorescent protein; GTP, guanosine-5’-triphosphate; HEK, human embryonic kidney cells; HelaS3, Henrietta Lacks cervical cancer cells; iPSC, induced pluripotent stem cells; MCF, Michigan cancer foundation; MDA-MB, M. D. Anderson metastasis breast cancer; MSCs, mesenchymal stem cells; NAD(P)H, nicotinamide adenine dinucleotide phosphate; NLO, nonlinear optical; NRB, non-resonant background; PC, polycarbonate; PET, polyethylene; PTFE, polytetrafluoroethylene; RFP, red fluorescent protein; SEM, scanning electron microscopy; SHG, second harmonic generation; SR, spontaneous Raman; SRG, stimulated Raman gain; SRL, stimulated Raman loss; SRM, super resolution microscopy; SRS, stimulated Raman scattering; STED, stimulated emission depletion; THG, third harmonic generation; TPEF, two-photon excited fluorescence; YFP, yellow fluorescent protein.
Alonzo, C. A., Karaliota, S., Pouli, D., Liu, Z., Karalis, K. P., and Georgakoudi, I. (2016). Two-photon excited fluorescence of intrinsic fluorophores enables label-free assessment of adipose tissue function. Sci. Rep. 6:31012. doi: 10.1038/srep31012
Barad, Y., Eisenberg, H., Horowitz, M., and Silberberg, Y. (1997). Nonlinear scanning laser microscopy by third harmonic generation. Appl. Phys. Lett. 70, 922–924. doi: 10.1063/1.118442
Bradley, J., Pope, I., Masia, F., Sanusi, R., Langbein, W., Swann, K., et al. (2016). Quantitative imaging of lipids in live mouse oocytes and early embryos using CARS microscopy. Development 143, 2238–2247. doi: 10.1242/dev.129908
Campagnola, P. (2012). Second harmonic generation imaging microscopy: applications to diseases diagnostics. Anal. Chem. 83, 3224–3231. doi: 10.1021/ac1032325.Second
Campagnola, P. J., Clark, H. A., Mohler, W. A., Lewis, A., and Loew, L. M. (2001). Second-harmonic imaging microscopy of living cells. J. Biomed. Opt. 6:277. doi: 10.1117/1.1383294
Cantarero, A. (2015). Raman scattering applied to materials science. Proc. Mat. Sci. 9, 113–122. doi: 10.1016/j.mspro.2015.04.014
Chan, J. K. C. (2014). The wonderful colors of the hematoxylin–eosin stain in diagnostic surgical pathology. Int. J. Surg. Pathol. 22, 12–32. doi: 10.1177/1066896913517939
Chen, X.-D., Li, S., Du, B., Dong, Y., Wang, Z.-H., Guo, G.-C., et al. (2018). High-resolution multiphoton microscopy with a low-power continuous wave laser pump. Opt. Lett. 43:699. doi: 10.1364/OL.43.000699
Cheng, J.-X., and Xie, X. S. (2004). Coherent anti-stokes raman scattering microscopy: instrumentation, theory, and applications. J. Phys. Chem. B 108, 827–840. doi: 10.1021/jp035693v
Chu, S.-W., Chen, S.-Y., Tsai, T.-H., Liu, T.-M., Lin, C.-Y., Tsai, H.-J., et al. (2003). In vivo developmental biology study using noninvasive multi-harmonic generation microscopy. Opt. Express 11, 3093–3099. doi: 10.1364/OE.11.003093
Combs, C. A., and Shroff, H. (2017). Fluorescence microscopy: a concise guide to current imaging methods. Curr. Prot. Neurosci. 79, 2.1.1–2.1.25. doi: 10.1002/cpns.29
Cong, A., Pimenta, R. M. L., Lee, H. B., Mereddy, V., Holy, J., and Heikal, A. A. (2019). Two-photon fluorescence lifetime imaging of intrinsic NADH in three-dimensional tumor models: 2P FLIM of intrinsic NADH in 3D tumor models. Cytometry Part A 95, 80–92. doi: 10.1002/cyto.a.23632
Costa Moura, C., Lanham, S. A., Monfort, T., Bourdakos, K. N., Tare, R. S., Oreffo, R. O. C., et al. (2018). Quantitative temporal interrogation in 3D of bioengineered human cartilage using multimodal label-free imaging. Integr. Biol. 10, 635–645. doi: 10.1039/C8IB00050F
Crisafi, F., Kumar, V., Perri, A., Marangoni, M., Cerullo, G., and Polli, D. (2018). Multimodal nonlinear microscope based on a compact fiber-format laser source. Spectrochim. Acta A 188, 135–140. doi: 10.1016/j.saa.2017.06.055
Crisafi, F., Kumar, V., Scopigno, T., Marangoni, M., Cerullo, G., and Polli, D. (2017). In-line balanced detection stimulated Raman scattering microscopy. Sci. Rep. 7:10745. doi: 10.1038/s41598-017-09839-1
De Bortoli, M., Taverna, E., Maffioli, E., Casalini, P., Crisafi, F., Kumar, V., et al. (2018). Lipid accumulation in human breast cancer cells injured by iron depletors. J. Exp. Clin. Cancer Res. 37:75. doi: 10.1186/s13046-018-0737-z
Denk, W., Piston, D. W., and Webb, W. W. (1995). “Two-photon molecular excitation in laser-scanning microscopy,” in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley (Cham: Springer), 445–458. doi: 10.1007/978-1-4757-5348-6_28
Denk, W., Strickler, J., and Webb, W. (1990). Two-photon laser scanning fluorescence microscopy. Science 248, 73–76. doi: 10.1126/science.2321027
Di Bartolo, B., and Collins, J. (2013). Nano-Optics for Enhancing Light-Matter Interactions on a Molecular Scale Plasmonics, Photonic Materials and Sub-Wavelength Resolution. Cham: Springer.
Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E., and Rebane, A. (2011). Two-photon absorption properties of fluorescent proteins. Nat. Methods 8, 393–399. doi: 10.1038/nmeth.1596
Evans, C. L., and Xie, X. S. (2008). Coherent anti-stokes raman scattering microscopy: chemical imaging for biology and medicine. Annu. Rev. Anal. Chem. 1, 883–909. doi: 10.1146/annurev.anchem.1.031207.112754
Fu, D., Holtom, G., Freudiger, C., Zhang, X., and Xie, X. S. (2013). Hyperspectral imaging with stimulated raman scattering by chirped femtosecond lasers. J. Phys. Chem. B 117, 4634–4640. doi: 10.1021/jp308938t
Fu, D., Zhou, J., Zhu, W. S., Manley, P. W., Wang, Y. K., Hood, T., et al. (2014). Imaging the intracellular distribution of tyrosine kinase inhibitors in living cells with quantitative hyperspectral stimulated Raman scattering. Nat. Chem. 6, 614–622. doi: 10.1038/nchem.1961
Gavgiotaki, E., Filippidis, G., Markomanolaki, H., Kenanakis, G., Agelaki, S., Georgoulias, V., et al. (2017). Distinction between breast cancer cell subtypes using third harmonic generation microscopy. J. Biophot. 10, 1152–1162. doi: 10.1002/jbio.201600173
Göppert-Mayer, M. (1931). Über Elementarakte mit zwei quantensprüngen. Annal. Phys. 401, 273–294. doi: 10.1002/andp.19314010303
Gupta, V. P. (2017). Molecular and Laser Spectroscopy: Advances and Applications. Amsterdam: Elsevier.
Helmchen, F., and Denk, W. (2005). Deep tissue two-photon microscopy. Nat. Methods 2, 932–940. doi: 10.1038/nmeth818
Hofemeier, A. D., Hachmeister, H., Pilger, C., Schürmann, M., Greiner, J. F. W., Nolte, L., et al. (2016). Label-free nonlinear optical microscopy detects early markers for osteogenic differentiation of human stem cells. Sci. Rep. 6:26716. doi: 10.1038/srep26716
Hoover, E. E., and Squier, J. A. (2013). Advances in multiphoton microscopy technology. Nat. Photonics 7, 93–101. doi: 10.1038/nphoton.2012.361
Hopt, A., and Neher E., (2001). Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys. J. 80, 2029–2036. doi: 10.1016/S0006-3495(01)76173-5
Huemer, K., Squirrell, J. M., Swader, R., LeBert, D. C., Huttenlocher, A., and Eliceiri, K. W. (2017). zWEDGI: wounding and entrapment device for imaging live Zebrafish larvae. Zebrafish 14, 42–50. doi: 10.1089/zeb.2016.1323
Jermyn, M., Mok, K., Mercier, J., Desroches, J., Pichette, J., Saint-Arnaud, K., et al. (2015). Intraoperative brain cancer detection with Raman spectroscopy in humans. Sci. Transl. Med. 7:274ra19. doi: 10.1126/scitranslmed.aaa2384
Ji, M., Orringer, D. A., Freudiger, C. W., Ramkissoon, S., Liu, X., Lau, D., et al. (2013). Rapid, label-free detection of brain tumors with stimulated raman scattering microscopy. Sci. Transl. Med. 5:201ra119. doi: 10.1126/scitranslmed.3005954
Kaiser, W., and Garrett, C. G. B. (1961). Two-photon excitation in Ca F 2: Eu 2 +. Phys. Rev. Lett. 7, 229–231. doi: 10.1103/PhysRevLett.7.229
Kaushik, G., Gil, D. A., Torr, E., Berge, E. S., Soref, C., Uhl, P., et al. (2019). Quantitative label-free imaging of 3D vascular networks self-assembled in synthetic hydrogels. Adv. Healthcare Mat. 8:1801186. doi: 10.1002/adhm.201801186
Konig, K. (2000). Multiphoton microscopy in life sciences. J. Microsc. 200, 83–104. doi: 10.1046/j.1365-2818.2000.00738.x
König, T. T., Goedeke, J., and Muensterer, O. J. (2019). Multiphoton microscopy in surgical oncology- a systematic review and guide for clinical translatability. Surg. Oncol. 31, 119–131. doi: 10.1016/j.suronc.2019.10.011
Krafft, C. (2012). Raman and coherent anti-Stokes Raman scattering microspectroscopy for biomedical applications. J. Biomed. Opt. 17:040801. doi: 10.1117/1.JBO.17.4.040801
Kumar, V., Coluccelli, N., and Polli, D. (2018). “Coherent optical spectroscopy/microscopy and applications,” in Molecular and Laser Spectroscopy, Chap. 5. 87–115. doi: 10.1016/B978-0-12-849883-5.00005-X
Li, J., Condello, S., Thomes-Pepin, J., Ma, X., Xia, Y., Hurley, T. D., et al. (2017). Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 20, 303–314.e5. doi: 10.1016/j.stem.2016.11.004
Lichtman, J. W., and Conchello, J.-A. (2005). Fluorescence microscopy. Nat. Methods 2, 910–919. doi: 10.1038/nmeth817
Liu, H.-S., Jan, M.-S., Chou, C.-K., Chen, P.-H., and Ke, N.-J. (1999). Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 260, 712–717. doi: 10.1006/bbrc.1999.0954
Lu, F.-K., Basu, S., Igras, V., Hoang, M. P., Ji, M., Fu, D., et al. (2015). Label-free DNA imaging in vivo with stimulated Raman scattering microscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 11624–11629. doi: 10.1073/pnas.1515121112
Meleshina, A. V., Dudenkova, V. V., Bystrova, A. S., Kuznetsova, D. S., Shirmanova, M. V., and Zagaynova, E. V. (2017). Two-photon FLIM of NAD(P)H and FAD in mesenchymal stem cells undergoing either osteogenic or chondrogenic differentiation. Stem Cell Res. Ther. 8:15. doi: 10.1186/s13287-017-0484-7
Meleshina, A. V., Dudenkova, V. V., Shirmanova, M. V., Shcheslavskiy, V. I, Becker, W., Bystrova, A. S., et al. (2016). Probing metabolic states of differentiating stem cells using two-photon FLIM. Sci. Rep. 6:21853. doi: 10.1038/srep21853
Mertz, J., and Moreaux, L. (2001). Second-harmonic generation by focused excitation of inhomogeneously distributed scatterers. Opt. Commun. 196, 325–330. doi: 10.1016/S0030-4018(01)01403-1
Meshulach, D., Barad, Y., and Silberberg, Y. (1997). Measurement of ultrashort optical pulses by third-harmonic generation. J. Opt. Soc. Am. B 14, 2122–2125. doi: 10.1117/1.3477535
Moura, C. C., Bourdakos, K. N., Tare, R. S., Oreffo, R. O. C., and Mahajan, S. (2019). Live-imaging of bioengineered cartilage tissue using multimodal non-linear molecular imaging. Sci. Rep. 9:5561. doi: 10.1038/s41598-019-41466-w
Müller, M., Squier, J., Wilson, K. R., and Brakenhoff, G. J. (1998). 3D microscopy of transparent objects using third-harmonic generation. J. Microsc. 191, 266–274. doi: 10.1046/j.1365-2818.1998.00399.x
Murphy, D. B. (2001). Fundamentals of Light Microscopy and Electronic Imaging. New York, NY: Wiley-Liss.
Nandakumar, P., Kovalev, A., and Volkmer, A. (2009). Vibrational imaging based on stimulated Raman scattering microscopy. New J. Phys. 11:033026. doi: 10.1088/1367-2630/11/3/033026
Nava, M. M., Raimondi, M. T., and Pietrabissa, R. (2012). Controlling self-renewal and differentiation of stem cells via mechanical cues. J. Biomed. Biotech. 2012:797410. doi: 10.1155/2012/797410
Nguyen, D., Hägg, D. A., Forsman, A., Ekholm, J., Nimkingratana, P., Brantsing, C., et al. (2017). Cartilage tissue engineering by the 3D bioprinting of iPS cells in a nanocellulose/alginate bioink. Sci. Rep. 7:658. doi: 10.1038/s41598-017-00690-y
Okkelman, I. A., Neto, N., Papkovsky, D. B., Monaghan, M. G., and Dmitriev, R. I. (2020). A deeper understanding of intestinal organoid metabolism revealed by combining fluorescence lifetime imaging microscopy (FLIM) and extracellular flux analyses. Redox Biol. 30:101420. doi: 10.1016/j.redox.2019.101420
Piston, D. W., Kirby, M. S., Cheng, H., Lederer, W. J., and Webb, W. W. (1994). Two-photon-excitation fluorescence imaging of three-dimensional calcium-ion activity. Appl. Opt. 33:662. doi: 10.1364/AO.33.000662
Pope, I., Langbein, W., Watson, P., and Borri, P. (2013). Simultaneous hyperspectral differential-CARS, TPF and SHG microscopy with a single 5 fs Ti:Sa laser. Opt. Express 21:7096. doi: 10.1364/OE.21.007096
Quinn, K. P., Sridharan, G. V., Hayden, R. S., Kaplan, D. L., Lee, K., and Georgakoudi, I. (2013). Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 3:3432. doi: 10.1038/srep03432
Raimondi, M. T., Eaton, S. M., Laganà, M., Aprile, V., Nava, M. M., Cerullo, G., et al. (2013). Three-dimensional structural niches engineered via two-photon laser polymerization promote stem cell homing. Acta Biomat. 9, 4579–4584. doi: 10.1016/j.actbio.2012.08.022
Rehberg, M., Krombach, F., Pohl, U., and Dietzel, S. (2011). Label-free 3D visualization of cellular and tissue structures in intact muscle with second and third harmonic generation microscopy. PLoS One 6:e28237. doi: 10.1371/journal.pone.0028237
Rico-Jimenez, J., Lee, J. H., Alex, A., Musaad, S., Chaney, E., Barkalifa, R., et al. (2020). Non-invasive monitoring of pharmacodynamics during the skin wound healing process using multimodal optical microscopy. BMJ Open Diabetes Res. Care 8:e000974. doi: 10.1136/bmjdrc-2019-000974
Rubart, M. (2004). Two-photon microscopy of cells and tissue. Circ. Res. 95, 1154–1166. doi: 10.1161/01.RES.0000150593.30324.42
Saarinen, J., Sözeri, E., Fraser-Miller, S. J., Peltonen, L., Santos, H. A., Isomäki, A., et al. (2017). Insights into Caco-2 cell culture structure using coherent anti-Stokes Raman scattering (CARS) microscopy. Int. J. Pharmac. 523, 270–280. doi: 10.1016/j.ijpharm.2017.03.015
Sanderson, M. J., Smith, I., Parker, I., and Bootman, M. D. (2016). Flourescence microscopy. Cold Spring Harb. Protoc. 2014:36. doi: 10.1101/pdb.top071795.Fluorescence
Santos, I. P., Barroso, E. M., Bakker Schut, T. C., Caspers, P. J., van Lanschot, C. G. F., Choi, D.-H., et al. (2017). Raman spectroscopy for cancer detection and cancer surgery guidance: translation to the clinics. Analyst 142, 3025–3047. doi: 10.1039/C7AN00957G
Santos, I. P., Caspers, P. J., Bakker Schut, T. C., van Doorn, R., Noordhoek Hegt, V., Koljenoviæ, S., et al. (2016). Raman spectroscopic characterization of melanoma and benign melanocytic lesions suspected of melanoma using high-wavenumber Raman spectroscopy. Anal. Chem. 88, 7683–7688. doi: 10.1021/acs.analchem.6b01592
Shen, Y. R. (1985). “The principles of nonlinear optics,” in Optics Laser Technology, ed. A. Cusano (Cham: Springer), 17. doi: 10.1016/0030-3992(85)90011-8
Sood, D., Tang-Schomer, M., Pouli, D., Mizzoni, C., Raia, N., Tai, A., et al. (2019). 3D extracellular matrix microenvironment in bioengineered tissue models of primary pediatric and adult brain tumors. Nat. Commun. 10:4529. doi: 10.1038/s41467-019-12420-1
Stephens, D. J. (2003). Light microscopy techniques for live cell imaging. Science 300, 82–86. doi: 10.1126/science.1082160
Sun, C.-K., Liu, W.-M., and Liao, Y.-H. (2019). Study on melanin enhanced third harmonic generation in a live cell model. Biomed. Opt. Express 10:5716. doi: 10.1364/BOE.10.005716
Syverud, B. C., Mycek, M.-A., and Larkin, L. M. (2017). Quantitative, label-free evaluation of tissue-engineered skeletal muscle through multiphoton microscopy. Tissue Eng. Part C 23, 616–626. doi: 10.1089/ten.tec.2017.0284
Tokarz, D., Cisek, R., Prent, N., Fekl, U., and Barzda, V. (2012). Measuring the molecular second hyperpolarizability in absorptive solutions by the third harmonic generation ratio technique. Anal. Chim. Acta 755, 86–92. doi: 10.1016/j.aca.2012.09.049
Van Steenbergen, V., Boesmans, W., Li, Z., de Coene, Y., Vints, K., Baatsen, P., et al. (2019). Molecular understanding of label-free second harmonic imaging of microtubules. Nat. Commun. 10:3530. doi: 10.1038/s41467-019-11463-8
Wang, H., Fu, Y., and Cheng, J.-X. (2007). Experimental observation and theoretical analysis of Raman resonance-enhanced photodamage in coherent anti-Stokes Raman scattering microscopy. J. Opt. Soc. Am. B 24:544. doi: 10.1364/JOSAB.24.000544
Weigelin, B., Bakker, G.-J., and Friedl, P. (2016). Third harmonic generation microscopy of cells and tissue organization. J. Cell Sci. 129, 245–255. doi: 10.1242/jcs.152272
Williams, R. M., Piston, D. W., and Webb, W. W. (1994). Two−photon molecular excitation provides intrinsic 3−dimensional resolution for laser−based microscopy and microphotochemistry. FASEB J. 8, 804–813. doi: 10.1096/fasebj.8.11.8070629
Wu, C.-H., Wang, T.-D., Hsieh, C.-H., Huang, S.-H., Lin, J.-W., Hsu, S.-C., et al. (2016). Imaging cytometry of human leukocytes with third harmonic generation microscopy. Sci. Rep. 6:37210. doi: 10.1038/srep37210
Xu, C., Zipfel, W., Shear, J. B., Williams, R. M., and Webb, W. W. (1996). Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. U.S.A. 93, 10763–10768. doi: 10.1073/pnas.93.20.10763
Yelin, D., and Silberberg, Y. (1999). Laser scanning third-harmonic-generation microscopy in biology. Opt. Express 5:169. doi: 10.1364/OE.5.000169
You, S., Tu, H., Chaney, E. J., Sun, Y., Zhao, Y., Bower, A. J., et al. (2018). Intravital imaging by simultaneous label-free autofluorescence-multiharmonic microscopy. Nat. Commun. 9:2125. doi: 10.1038/s41467-018-04470-8
Yu, Z., Chen, T., Zhang, X., Fu, D., Liao, X., Shen, J., et al. (2012). Label-free chemical imaging in vivo: three-dimensional non-invasive microscopic observation of amphioxus notochord through stimulated Raman scattering (SRS). Chem. Sci. 3:2646. doi: 10.1039/c2sc20316b
Yue, S., Slipchenko, M. N., and Cheng, J.-X. (2011). Multimodal nonlinear optical microscopy. Laser Phot. Rev. 5, 496–512. doi: 10.1002/lpor.201000027
Zandrini, T., Shan, O., Parodi, V., Cerullo, G., Raimondi, M. T., and Osellame, R. (2019). Multi-foci laser microfabrication of 3D polymeric scaffolds for stem cell expansion in regenerative medicine. Sci. Rep. 9:11761. doi: 10.1038/s41598-019-48080-w
Zhang, X., Roeffaers, M. B. J., Basu, S., Daniele, J. R., Fu, D., Freudiger, C. W., et al. (2012). Label-free live-cell imaging of nucleic acids using stimulated raman scattering microscopy. Chem. Phys. Chem. 13, 1054–1059. doi: 10.1002/cphc.201100890
Zinselmeyer, B. H., Dempster, J., Wokosin, D. L., Cannon, J. J., Pless, R., Parker, I., et al. (2009). Chapter 16 two−photon microscopy and multidimensional analysis of cell dynamics. Methods Enzymol. 461, 349–378. doi: 10.1016/S0076-6879(09)05416-0
Zipfel, W. R., Williams, R. M., Christie, R., Nikitin, A. Y., Hyman, B. T., and Webb, W. W. (2003a). Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. U.S.A. 100, 7075–7080. doi: 10.1073/pnas.0832308100
Zipfel, W. R., Williams, R. M., and Webb, W. W. (2003b). Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 21, 1369–1377. doi: 10.1038/nbt899
Keywords: nonlinear microscopy, stem cells, tissue engineering, label-free microscopy, live imaging
Citation: Parodi V, Jacchetti E, Osellame R, Cerullo G, Polli D and Raimondi MT (2020) Nonlinear Optical Microscopy: From Fundamentals to Applications in Live Bioimaging. Front. Bioeng. Biotechnol. 8:585363. doi: 10.3389/fbioe.2020.585363
Received: 20 July 2020; Accepted: 16 September 2020;
Published: 09 October 2020.
Edited by:
Paolo Bianchini, Italian Institute of Technology (IIT), ItalyReviewed by:
Jayaramakrishnan Velusamy, University of Guanajuato, MexicoCopyright © 2020 Parodi, Jacchetti, Osellame, Cerullo, Polli and Raimondi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Parodi, dmFsZW50aW5hLnBhcm9kaUBwb2xpbWkuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.