 Gundeep Kaur
Gundeep Kaur Srajan Kapoor
Srajan Kapoor Soni Kaundal
Soni Kaundal Dipak Dutta
Dipak Dutta Krishan Gopal Thakur
Krishan Gopal Thakur- 1Structural Biology Laboratory, G. N. Ramachandran Protein Centre, Council of Scientific and Industrial Research-Institute of Microbial Technology, Chandigarh, India
- 2Molecular Microbiology Laboratory, Council of Scientific and Industrial Research-Institute of Microbial Technology, Chandigarh, India
The mycobacterial RNA polymerase (RNAP) is an essential and validated drug target for developing antibacterial drugs. The β-subunit of Mycobacterium tuberculosis (Mtb) RNAP (RpoB) interacts with an essential and global transcription factor, CarD, and confers antibiotic and oxidative stress resistance to Mtb. Compromising the RpoB/CarD interactions results in the killing of mycobacteria, hence disrupting the RpoB/CarD interaction has been proposed as a novel strategy for the development of anti-tubercular drugs. Here, we describe the first approach to rationally design and test the efficacy of the peptide-based inhibitors which specifically target the conserved PPI interface between the bacterial RNAP β/transcription factor complex. We performed in silico protein-peptide docking studies along with biochemical assays to characterize the novel peptide-based inhibitors. Our results suggest that the top ranked peptides are highly stable, soluble in aqueous buffer, and capable of inhibiting transcription with IC50 > 50 μM concentration. Using peptide-based molecules, our study provides the first piece of evidence to target the conserved RNAP β/transcription factor interface for designing new inhibitors. Our results may hence form the basis to further improve the potential of these novel peptides in modulating bacterial gene expression, thus inhibiting bacterial growth and combating bacterial infections.
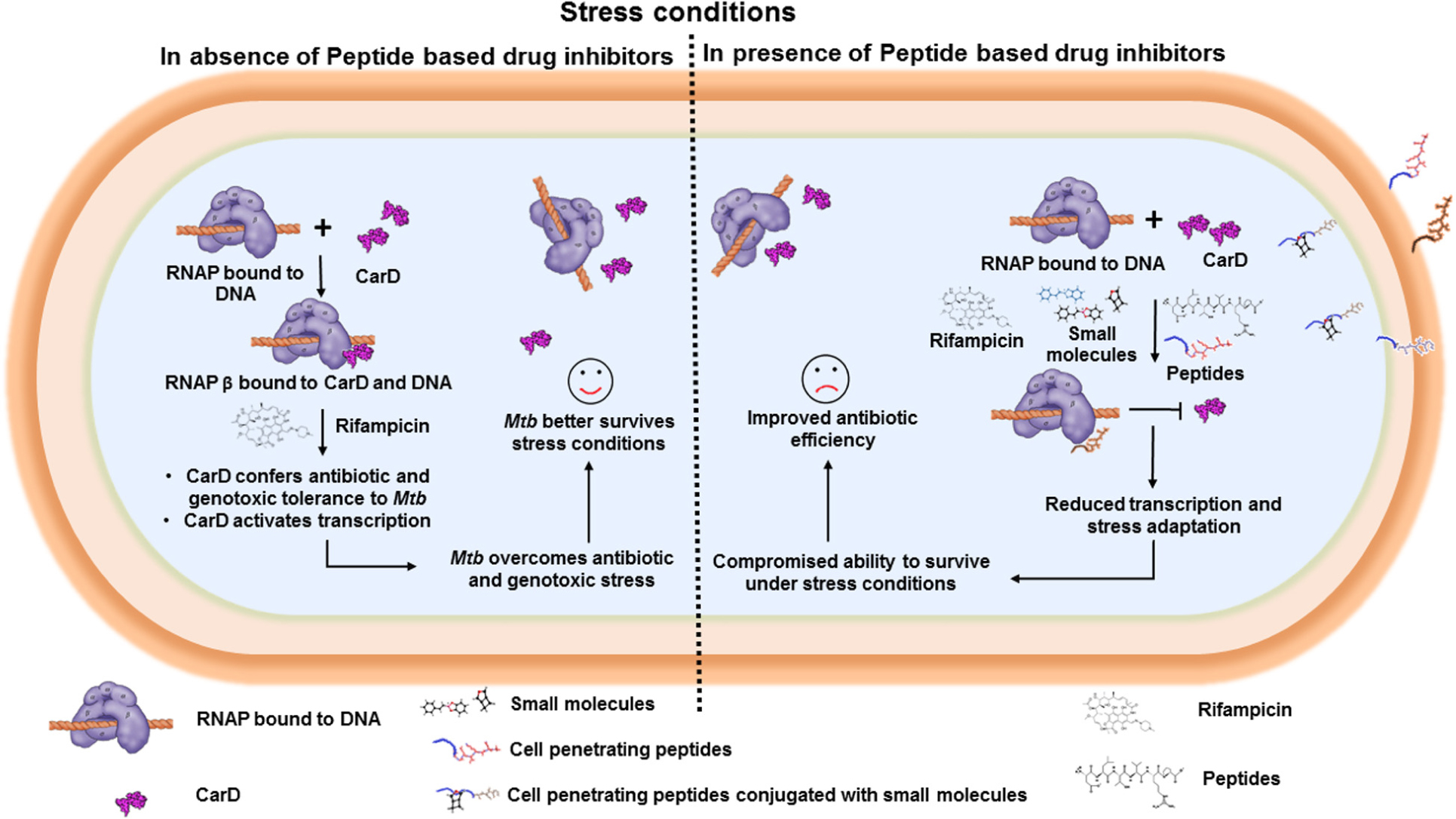
Graphical Abstract. Bacterial RNAP interacts with transcription factors, such as CarD and TRCF, which stabilizes the transcription initiation complex and activates transcription. Interface peptide-based inhibitors target and interact with RNAP (β and interfere with RNAP/transcription factor interaction, and thus affect the bacterial transcription.
Introduction
Tuberculosis (TB) remains a major cause of death in the world (WHO, 2016). Rifampicin, the lynchpin for TB, targets bacterial RNA Polymerase (RNAP) (Mcclure and Cech, 1978; Campbell et al., 2001), a key and essential enzyme involved in transcription. RNAP is conserved in structure and function in all bacteria (Werner, 2008; Werner and Grohmann, 2011). Bacterial RNAP is a proven and validated target for developing antibacterial drugs (Chopra, 2007; Ho et al., 2009; Ma et al., 2016). But due to the emergence of the multi-drug and extremely drug-resistant strains of pathogenic bacteria (Lushniak, 2014; Alos, 2015; Ventola, 2015a), the treatment of infectious diseases is becoming challenging. Thus, there is an urgent need to develop novel drug molecules against newer targets or newer sites in the established targets in bacteria (Gould, 2010; Mendelson, 2015; Ventola, 2015b). So, the potent novel antibacterial drugs should ideally target bacterial RNAP in such a way that they do not show cross-resistance with already approved drugs, such as rifampicin.
Bacterial RNAP is a multi-subunit complex comprised of five subunits (α2ββ′ω) which associate reversibly with an σ-factor to form a holoenzyme (Helmann and Chamberlin, 1988). Different subunits of RNAP interact with several transcription factors, like CarD (Stallings et al., 2009), WhiB7 (Burian et al., 2013), UvrD helicase (Epshtein et al., 2014), RNA polymerase binding protein A (RbpA) (Newell et al., 2006), Transcription Repair Coupling Factor (TRCF) (Selby and Sancar, 1995a, b), Rho (Das et al., 1978), etc., at various stages of transcription. Some of the transcription regulators, such as CarD (Weiss et al., 2012) and TRCF (Westblade et al., 2010), interact exclusively with β-subunit of RNAP and modulate transcription. Mtb and Escherichia coli RNAP differ in their abilities to form an open promoter complex (Rammohan et al., 2015, 2016). Mtb RNAP, being a relatively slow and unstable enzyme, requires the association with the essential accessory factors like CarD and RbpA (Hu et al., 2012; Davis et al., 2015) to increase the rate of transcript production in vitro and stabilize the unstable RNAP open complex (Davis et al., 2015) by preventing transcription bubble collapse (Rammohan et al., 2015).
CarD has been proposed as an attractive drug target, as Mycobacterium smegmatis lacking CarD has been shown to be susceptible to killing by Reactive Oxygen Species (ROS), ciprofloxacin, and starvation (Stallings et al., 2009). Although absent in E. coli and eukaryotes, CarD is widely present across actinomycetes. Weakening the interaction between RNAP and CarD makes the mycobacteria susceptible to antibiotic and oxidative stress, thereby killing the bug (Weiss et al., 2012). Recent structural studies on Mtb CarD and its homologs (Gallego-Garcia et al., 2012, 2014; Gulten and Sacchettini, 2013; Srivastava et al., 2013; Kaur et al., 2014; Bernal-Bernal et al., 2015; Kaur et al., 2018) suggests that CarDNTD (N-Terminal Domain of CarD), also known as RNAP Interacting Domain (RID), adopts a Tudor-like fold, similar to E. coli and Tth TRCFNTD (N-Terminal Domain of transcription-repair coupling factor, involved in coupling transcription to DNA repair)(Westblade et al., 2010). Both CarDNTD and TRCFNTD interact with β1 β2 domain (RpoBtr) and β1 domain of β-subunit of RNAP, respectively (PDBID’s: 4KBM and 3MLQ) (Westblade et al., 2010; Weiss et al., 2012).
Targeting and inhibiting PPIs using peptide-based inhibitors is an attractive strategy to block a number of important targets involved in pathological conditions (Fry, 2015; Bakail and Ochsenbein, 2016). However, it is quite challenging to design the peptide-based inhibitors. It is also difficult to characterize the protein-peptide interactions since the interface peptides are quite flexible and may adopt multiple conformations in solution (Pelay-Gimeno et al., 2015; Lamiable et al., 2016). Nonetheless, the market for peptide-based drugs is expanding day-by-day (Craik et al., 2013; Uhlig et al., 2014) and they are emerging as attractive candidates for disrupting PPI (Fosgerau and Hoffmann, 2015). Recently, Shanmuga Priya et al. (2018), reported a virtual screening based strategy to identify and target small molecules against CarD.
In this study, we describe the first approach to rationally design and test the efficacy of the peptide-based inhibitors which specifically target the conserved RpoBtr/transcription factor PPI interface (Supplementary Figure 1). We hypothesized that binding of the peptide-based inhibitors to the solvent exposed protein interface of Mtb RpoBtr might interfere with the binding of an essential transcription factor, CarD, consequently affecting the stability of the open promoter complex and reducing the amount of transcript formed. Our results suggest that one of the rationally designed peptide-based inhibitors was able to reduce the CarD mediated transcription activation, hence demonstrating the potential of targeting this conserved PPI interface for developing improved molecules to combat mycobacterial infections in the future.
Materials and Methods
Identification of the Novel Druggable Site in Bacterial RNAP β and Structure-Guided Designing of Novel Synthetic Interface Peptides
Four different peptides were designed manually after analyzing the structures of RNAP β/CarD (PDBID: 4KBM) and Tth TRCF (PDBID: 3MLQ). Taking into account the residues involved in the interaction between Mtb RNAP β/CarD and Tth RNAP/TRCF, the interface peptide approach was used to design the peptides (7–8 amino acids in length), which could target and bind bacterial RNAP (Table 1).
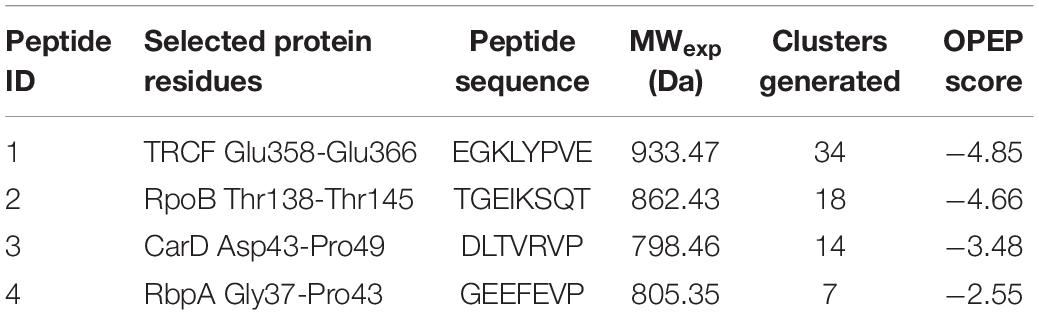
Table 1. The details of the designed interface peptides and the parameters obtained after modeling peptides using PEP-FOLD3.1 web server (Lamiable et al., 2016).
Peptide Synthesis and Peptide Modeling
The designed peptides were commercially synthesized using solid phase peptide synthesis strategy and purchased from GL Biosciences, China. The 3D structures of the designed peptides were predicted and modeled using the PEP-FOLD3.1 web server (Lamiable et al., 2016). PEP-FOLD 3.1 predicts the structure of peptides on the basis of Hidden Markov model conformations.
LC-MS Analysis of Peptides
The stability of all the designed peptides was analyzed using Liquid chromatography-mass spectrometry (LC-MS) using Agilent Technologies (model G6550A). Peptide 1, 2, and 4 were injected in the C18 column equilibrated in a negative mode at a flow rate of 0.5 mL/min using water as a mobile phase. Peptide 3 was injected in the C18 column equilibrated in a positive mode at a flow rate of 0.5 mL/min using water as a mobile phase.
Molecular Docking Analyses of Designed Peptides and RpoB
The crystal structure of the β1β2 domain of β-subunit of RNAP (RpoBtr) in complex with CarD (PDBID 4KBM) was retrieved from Protein Data Bank1 and pre-processed using the Protein Preparation wizard 2.2 of the Maestro, Schrödinger software suite (Schrödinger, 2017). The CarD was removed at the protein preparation stage in order to prepare RpoBtr as the receptor. H-atoms were added and the bond orders were assigned. This was followed by the creation of zero-order bonds to metals and disulfide bonds. Missing side chains of the RpoBtr were modeled and the loops were refined using Prime 3.0 (Jacobson et al., 2002; Jacobson et al., 2004) module of Schrödinger. Water molecules were deleted, and the ionization and tautomeric states in the pH range 7 ± 2 were generated by Epik (Shelley et al., 2007). The H-bonds were assigned and optimized by selecting PROPKA at pH 7. The structure was finally restrained and minimized using OPLS3 force field (Harder et al., 2016). All the peptides were prepared using LigPrep (Schrödinger, 2017) module of Schrödinger for generating all possible energetically minimum conformers. Molecular docking was initiated by defining the prepared RNAP β as a receptor and providing the binding site information required to dock the peptides on the receptor followed by the generation of the grid file. Thereafter, the generated grid file was used for molecular docking using the SP-peptide module (Friesner et al., 2004) of Maestro. The sampling of the peptides was kept flexible throughout the process of docking. The docking results were analyzed for the best-docked pose for each peptide-RNAP β on the basis of Glide SP score. All the molecular graphics figures were prepared using Pymol software (Delano, 2002).
Physiochemical Properties and Biological Assessment of Lead Peptides
The lead peptide-based inhibitors were assessed for their physiochemical properties. The molecular weight, theoretical isoelectric point (pI), overall charge, aliphatic index value, instability index, and average hydropathy (GRAVY) were calculated using Protparam server (Wilkins et al., 1999). The subcellular localization of the lead peptide-based inhibitor was determined using the CELLO prediction server v.2.5 (Yu et al., 2004) which helps to predict subcellular localization in eukaryotes.
Cloning, Over-Expression, and Purification of Recombinant Proteins
The cloning, over-expression, and purification of Mtb CarD was carried out as per the procedures mentioned in Kaur et al. (2014, 2018). Mtb RpoBtr was cloned in pNIC28-Bsa4 between NdeI and HindIII sites. The over-expression and purification of RpoBtr was carried out in a similar way as described for CarD (Kaur et al., 2014), except the buffer used for purification for both CarD and RpoB in this study was 25 mM Tris-HCl pH 7.5, 100 mM NaCl, and 2 mM β-Mercaptoethanol (Buffer A). The over-expression and purification of Mtb RNAP holoenzyme was carried out as per the reported procedures (Banerjee et al., 2014).
In vitro Transcription Assays
In vitro transcription assays were performed as mentioned in Banerjee et al. (2014) with slight modifications. sinP3 promoter DNA was PCR amplified and further gel extracted using Thermo Fischer Gel extraction kit. Transcription initiation complex was incubated with varying concentrations of each peptide (10 μM − 1 mM) followed by incubation with CarD (1 μM). The reaction was initiated with the addition of NTP mixture (final concentration 250 mM ATP, GTP, UTP, and 50 mM CTP containing 0.5 mCi α32P-CTP) and allowed to proceed for 60 s. The transcription was terminated by the addition of 2X Formamide loading dye (80% formamide, 10 mM EDTA, 0.01% Bromophenol Blue, 0.01% Xylene Cyanol, and 0.1%SDS). The terminated reactions were heated at 95°C for 5 min and loaded on the 10% Urea denaturing gel at 25 W for 2 h. The images were scanned used Fujifilm Phoshoimager (FLA 5000). Densitometry analysis was performed using Image J software (Perez and Pascau, 2013). The data presented is the average of three independent experiments.
Results
Structural Analysis of the Conserved RNAP β Subunit/Transcription Factor Binding Interface
In order to analyze the conserved bacterial RNAP β/transcription factor binding interface, we retrieved and analyzed the available X-ray crystal structures of bacterial RNAP β and the transcription factor complexes from the Protein Data Bank (PDB). The structural analysis of Mtb RpoBtr/CarD complex (PDB ID 4KBM) reveals that CarD binds RpoBtr in 1:1 stoichiometry, burying a relatively small surface area of about 500 Å2 (Figure 1A). The PPI interface is stabilized by eight stranded antiparallel β-sheets, where four-stranded antiparallel β-sheet are from the CarDNTD and the four-stranded antiparallel β-sheet from the RpoBtr and eight hydrogen bonds (Gulten and Sacchettini, 2013). The PPI analysis suggests the role of “β-strand addition,” especially β-stand augmentation (Remaut and Waksman, 2006), which involves the addition of β-strand in CarD to β-strand in RNAP β or vice versa, which ultimately results in the formation of a continuous β-sheet. Similarly, the structure analysis of Tth RNAP β1/TRCF-RID complex (PDB ID 3MLQ) reveals that TRCF also binds β1 domain of β-subunit of RNAP in 1:1 stoichiometry (Figure 1A). Though CarD and TRCF do not share any significant sequence similarities, their secondary structure segments are more conserved (Figure 1B). Structural superposition of TRCF-RID and CarDNTD yields a root mean square deviation (RMSD) of 1.19 Å, thus suggesting that both the transcription regulators containing tudor-like domains share the same interface for interaction with RNAP β. In addition, the RMSD between Cα atoms of RNAP β and Tth RNAP β1 is 1.14 Å, thus suggesting the conserved site of interaction between RNAP β/transcription factor-RID. Since the interface residues in RNAP involved in PPI in Tth RNAP β1/TRCF-RID and Mtb RpoBtr/CarD complexes are structurally conserved in both gram-positive and gram-negative bacteria (Supplementary Table 1 and Figures 1B,C), and absent in eukaryotes, targeting and inhibiting these PPIs might help in developing novel broad-spectrum antibacterial inhibitors.
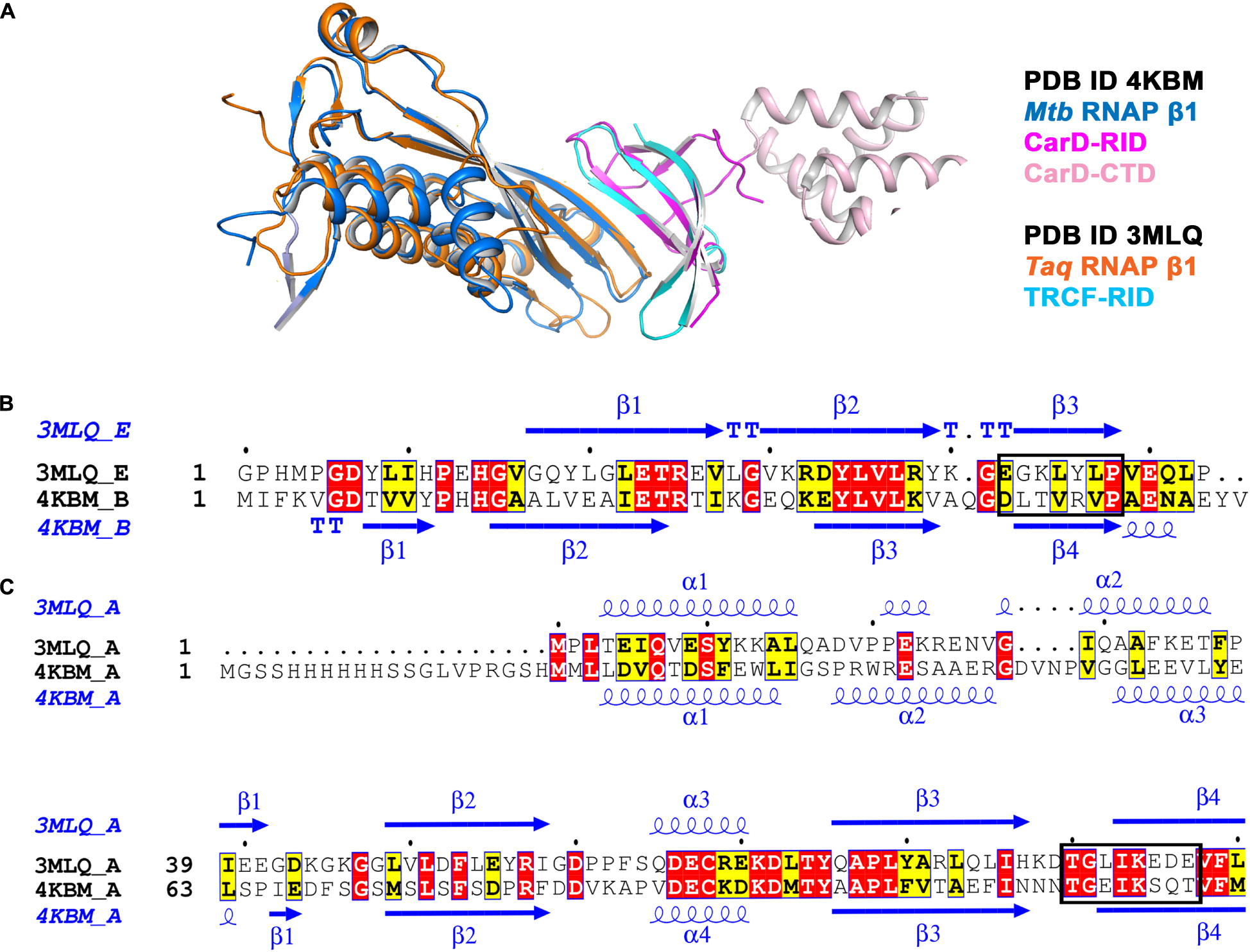
Figure 1. (A) Structural superposition of Mtb RNAP β1 (marine)/CARD-RID (magenta) [PDB ID 4KBM] with Tth RNAP β1 (orange)/TRCF-RID complex (cyan) [PDB ID 3MLQ] shows the solvent-exposed conserved protein-protein interface. (B) Structural and sequence alignment of Mtb CarD-RID (4KBM chain B) with Tth TRCF-RID (3MLQ chain E). Mtb CarD-RID with Tth TRCF-RID do not share significant sequence similarity but do share the same site for interaction with RNAP β1 (shown in the rectangular box). The figure was generated using ESPript 3 server (Robert and Gouet, 2014). (C) Structural and sequence alignment of Mtb RNAP β1β2 (4KBM chain A) with Tth RNAP β1 suggests that both Mtb RNAP β1β2 and Tth RNAP β1 (3MLQ chain A) share the conserved site for interaction with transcription factor-RID (shown in the rectangular box). The figure was generated using ESPript 3 server (Robert and Gouet, 2014).
Sequence-Based Analysis for the Conservation of the RNAP β/CarD Binding Interface
To analyze the conservation of the binding interface of RpoBtr and CarD, we retrieved the whole genome sequencing data of several clinical multi-drug resistant (MDR) and extensively drug resistant (XDR) Mtb strains and performed BLASTp (Mcginnis and Madden, 2004) analysis [selected Mycobacterium tuberculosis complex (taxid:77643) as organism in drop down menu, max target sequences were set to 10,000, and the rest of the parameters were set as default] for both RpoB and CarD. We analyzed sequences of CarD and RpoB in several MDR and XDR strains of Mtb and observed that the residues involved in the CarD/RpoB interaction are highly conserved (Supplementary Figure 2), thus suggesting that CarD/RNAP β interactions are crucial for the survival of the bug. This prompted us to design inhibitors targeting this highly conserved RpoBtr/CarD interface which can potentially inhibit growth of even MDR and XDR Mtb strains.
Design, 3D-Structure Prediction, and Analyses of Peptide-Based RNAP β/Transcription Factor Inhibitors
As the interaction interface between RNAP β1β2 domain and the transcription factor (CarDNTD and TRCF-RID) (Westblade et al., 2010; Gulten and Sacchettini, 2013) is relatively large, flat, solvent-exposed, and devoid of the binding pocket, it is challenging to design small molecule-based inhibitors. We therefore aimed to design the peptide-based RNAP β/transcription factor inhibitors by selecting the key residues involved in the interaction at the conserved PPI interface between RNAP β and transcription factor complex. The sequence and molecular weight of the designed peptides are mentioned in Table 1. PEP-FOLD3.1 (Lamiable et al., 2016), an online server, was used to predict the de novo structures of the designed peptides by running 100 simulations for each peptide. PEP-FOLD3.1 provides the information in the form of models which are generated by OPEP (Optimized Potential for Efficient structure Prediction) energy. The topmost model with the lowest OPEP score having minimum energy and maximum stability for each peptide was selected for further computational analysis. The detailed parameters derived from PEP-FOLD3.1 are summarized in Table 1. The designed peptides were 7–8 amino acids in length and were commercially synthesized. The peptides were analyzed for their stability and solubility. All the peptides were soluble in water as well as in aqueous buffer with a solubility of > 10 mM. The LC-MS analysis of all the designed peptides shows that the peptides were stable in water even when stored for over a year at 4°C as the experimentally observed molecular weight of all peptides matched well with their theoretically calculated molecular weights (Supplementary Figure 3).
Identification of the Potential Lead Peptide-Based RNAP β Inhibitors Using Molecular Docking
To examine the interaction between RpoBtr and the designed interface peptides, we performed molecular docking experiments where we docked the peptides on the RpoBtr (receptor) using the SP-peptide module of Glide in Maestro 9.8 of the Schrödinger software suite. The top ranking RpoBtr-docked peptide conformations were selected and analyzed for their glide score, docking score, and low binding energy values. The parameters obtained after molecular docking are listed in Table 2. All the protein-peptide complexes exhibited a multipose binding. The top scoring poses of the protein-peptide complexes were further analyzed manually (Figures 2A–D). Molecular docking results suggest that peptides 1 and 2 have a comparable binding affinity and docking scores and are relatively better than peptides 3 and 4. This is also in agreement with the interaction energy analysis performed using PEP-FOLD3.1 web server (Lamiable et al., 2016).
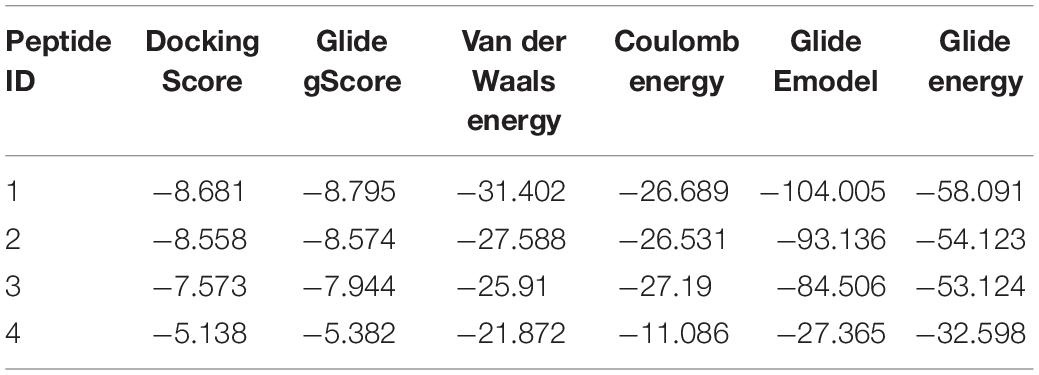
Table 2. Comparison of docking and binding energy scores (Kcal/mol) of potential shortlisted interface peptides targeting conserved RNAP β/transcription factor interface.
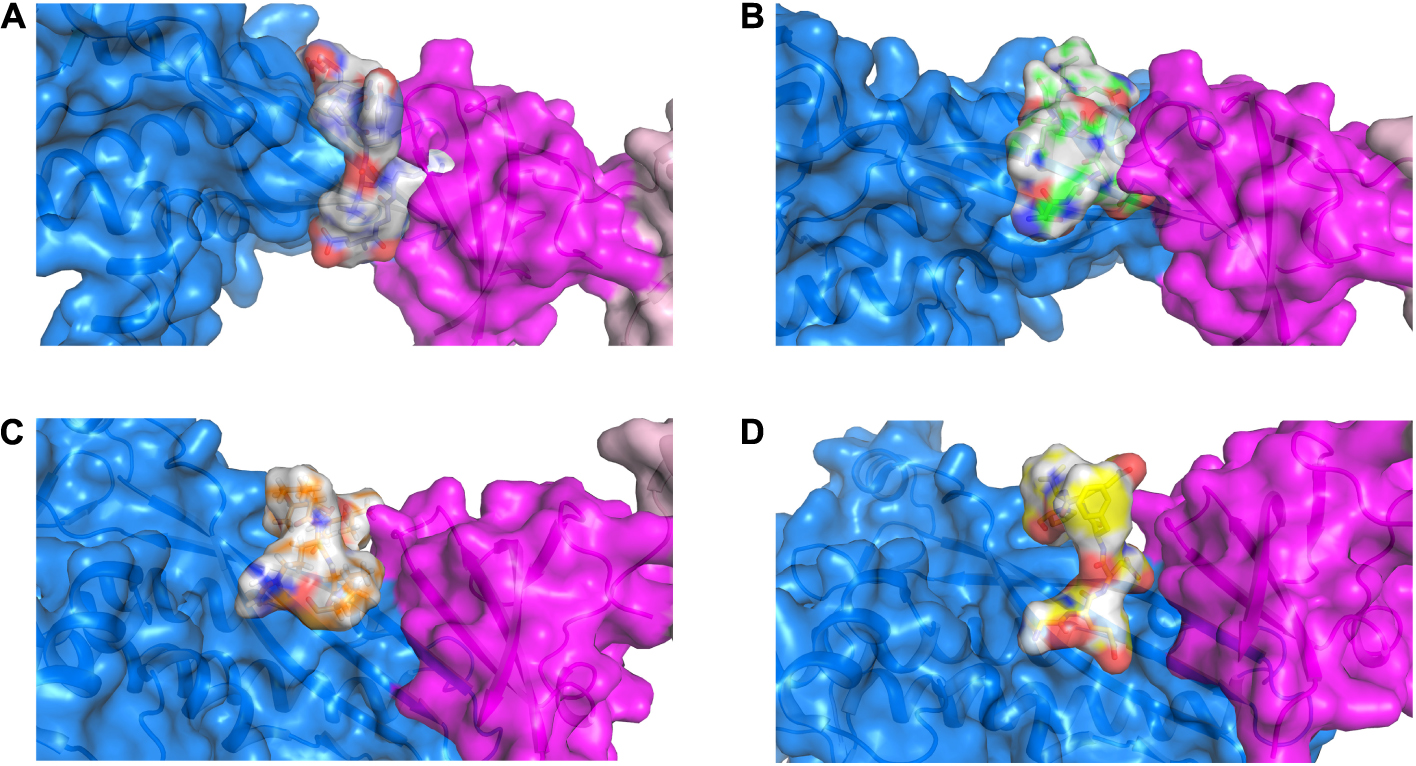
Figure 2. 3D representation of the RNAP β (marine), CarD (magenta) and peptide-based inhibitor binding interface displaying the top ranked pose for each peptide (gray) when docked onto RNAP β. (A) RNAP β and peptide 1; (B) RNAP β and peptide 2; (C) RNAP β and peptide 3; and (D) RNAP β and peptide 4.
The key residues involved in the interaction between RpoBtr-docked peptides (1–4) were determined using the 2D peptide interaction diagrams generated by Ligand Interaction script using Maestro (Schrödinger Inc.)2 (Supplementary Figures 4–7). The binding mode and the docking results suggest that the designed peptides 1 and 2 interact and bind at a similar binding site of RpoBtr/transcription factor, whereas the peptides 3 and 4 bind near the proximity of the binding site. Next, we examined the key residues involved in the interaction between RpoBtr-peptide 1 and RpoBtr-peptide 2 complexes. Eight intermolecular hydrogen bonds formed between RNAP β (Thr130, Glu132, Ser143, Thr145, Phe147, and Gln409) and peptide 1, which help in stabilizing the RpoBtr-peptide 1 complex. In addition, peptide 1 Asp1 forms a salt bridge with the RNAP β Glu132. Six intermolecular hydrogen bonds formed between RNAP β (Arg105, Asp107, Thr145, Gln409, Arg415) and peptide 2, which help in stabilizing the RpoBtr-peptide 2 complex. Furthermore, RNAP β Asp107 forms two salt bridges with peptide 2 Thr1 and Lys5. The surface representation showing the binding mode and the key residues involved in the interactions between RpoBtr and the peptides 1–4 are shown in Supplementary Figures 4–7.
Physiochemical Properties and Biological Assessment of Lead Peptide
Protparam server (Wilkins et al., 1999) was used to assess the physiological properties of the lead RpoB/transcription factor complex peptide-based inhibitors. Peptides 1 and 2 have low molecular weights with an acidic nature and are highly stable with an instability index of 27.03 and 26.25, respectively. The aliphatic index values for both peptide 1 and 2 were relatively higher, suggesting these peptides are less hydrophobic. The subcellular localization in eukaryotes of all the designed peptides was predicted using the CELLO prediction server v.2.5 (Yu et al., 2004) which predicted the localization of all the peptides in mitochondrial and nuclear organelle with a high reliability score (2.044 and 1.057, respectively).
In vitro Testing of the Peptide-Based RNAP β Inhibitors
Our in silico docking experiments suggested that peptide-based inhibitors 1 and 2 have a higher affinity for bacterial RpoBtr as compared to other peptides. As reported earlier, CarD increases the rate of transcript production from sinP3 promoter (Srivastava et al., 2013). We hypothesized that the binding of peptide-based inhibitors to RpoBtr might compete with CarD for the binding site. As a result, transcription activation would be affected and only the basal level transcription will be observed. In order to test our hypothesis, we performed the in vitro transcription assays. In agreement with previously published studies (Garner et al., 2014; Davis et al., 2015; Rammohan et al., 2015), we were able to observe about a 3.5-fold increase in the transcription product in the presence of CarD when compared to RNAP alone (Figure 3). To test the inhibitory activity of the peptides, we incubated Mtb RNAP holoenzyme with varying concentrations (10 μM – 1 mM) of the peptides (1–3) in the presence of CarD and initiated the transcription with the addition of the radiolabeled CTP. The densitometric analysis of in vitro transcription products suggested that peptide 2 showed the maximum inhibitory activity and was able to reduce transcription by approximately twofold at ∼50 μM concentration (Figures 3A,B). The calculated IC50 values of all the evaluated peptides are mentioned in Supplementary Table 2. In agreement with the computational studies, peptides 1 and 2 were most efficient in inhibiting CarD mediated transcription activation. However, in contrast to docking scores, peptide 2 showed higher in vitro CarD-mediated inhibitory activity. Based on our hypothesis, binding of the designed peptides will inhibit CarD/RNAP interactions, hence resulting in reduced CarD mediated activation. However, these peptides will not be able to inhibit the transcription completely as RNAP can perform basal transcription even in the absence of CarD.
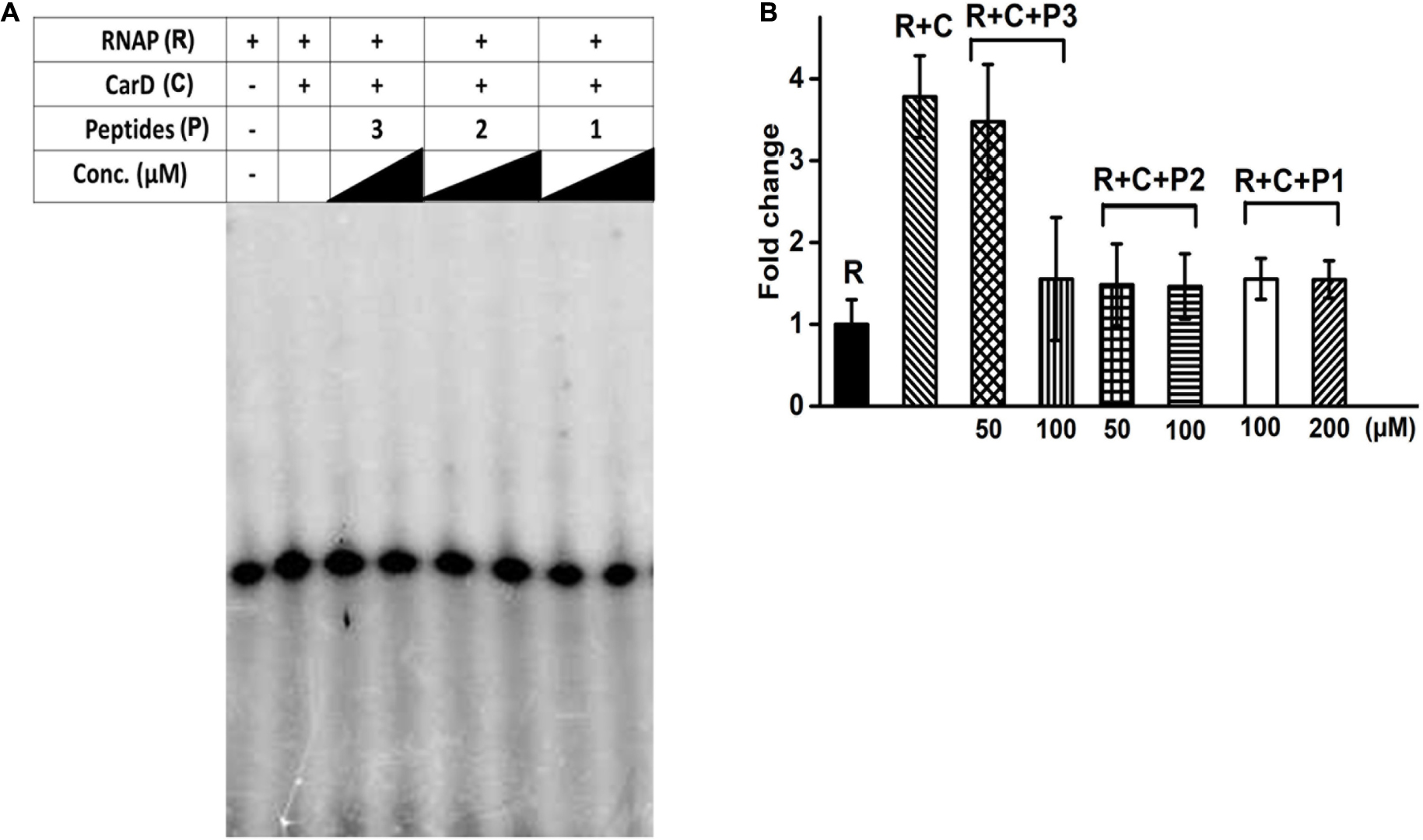
Figure 3. (A) In vitro transcription assay in the presence of varying concentrations of designed interface peptides (10 μM – 1 mM) and Mtb CarD. (B) Densitometry analysis of the transcripts obtained in the denaturing gel shown in (A) in the presence of peptides 1–3. Abbreviations: R, RNAP; C, CarD; P1, Peptide 1; P2, Peptide 2; P3, Peptide 3.
Discussion
In the current study, we report the rationally designed novel peptide-based inhibitors targeting the conserved PPI interface between the bacterial RpoBtr/transcription factor complexes. The target site investigated in our study has several advantages. Our results demonstrate that the target site is unique, does not share cross-resistance with the commercially available mycobacterial drugs, is highly conserved in both gram-positive and gram-negative bacteria, and no mutations were observed either in MDR or XDR strains of Mtb. Our in silico protein-peptide docking studies suggest that peptides 1 and 2 have a higher binding affinity for RpoBtr, thus highlighting their potential as the lead peptide-based RNAP β inhibitors. Our in vitro experiments suggest that the top ranked peptide 2 is highly stable, soluble in aqueous buffer, and capable of reducing CarD-mediated transcription activation.
Several studies have shown that peptides can be used as potential inhibitors to disrupt protein-protein interactions as they are highly selective, less toxic, and are effective against a broad range of targets (Wang et al., 2016). Hüsecken et al. (2013), designed the peptides mimicking σ2.2 which could be used for targeting E. coli RNAP σ70/core interface effectively (Hüsecken et al., 2013). Antisense peptide nucleic acids have been designed and demonstrated to inhibit the production of σ70 as an alternative therapeutic strategy against methicillin-resistant Staphylococcus aureus (Bai et al., 2012). Jeong et al. (2006), designed peptides mimicking Mtb RshA, an anti-σ factor, which could specifically inhibit the initiation of transcription by SigH, an alternative σ-factor in vitro. GE23077, a heptapeptide antibiotic target, binds to i and i + 1 site of the active center of RNAP (Ciciliato et al., 2004; Zhang et al., 2014). Microcin J25, a 21-mer peptide, binds near the secondary channel of RNAP and inhibits the transcription by competitively preventing the uptake of NTP (Mukhopadhyay et al., 2004). Besides several advantages offered by the peptide-based drugs, there are a few limitations (Otvos and Wade, 2014). However, the peptides designed in our study offer additional advantages as they were highly soluble in both water and aqueous buffers, and highly stable when stored over a long period of time at 4°C. We propose that conjugating the lead peptide 2 to antibodies, small molecules, or oligo ribonucleotides, incorporating non-native amino acids, cyclization, chemical stapling, or altering its chirality (Wang et al., 2016) may further improve its efficacy and selectivity. Our study suggests that targeting the newer site in the RpoB/transcription factor interface could be used as a potential site for developing novel drug molecules to tackle the increasing threat of MDR and XDR Mtb strains. Our study demonstrates the potential of using small molecules to target unexplored PPI sites on the transcription machinery of Mtb for possibly developing drugs for even MDR and XDR strains.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
GK and KT proposed the concepts, designed the experiments, and wrote the manuscript. GK, SKap, and SKau carried out the experiments under the supervision of DD and KT. GK and KT analyzed and interpreted the results. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to KT by the Council of Scientific and Industrial Research, India. KT was a recipient of the Innovative Young Biotechnologist Award 2011, Department of Biotechnology, India. GK was a recipient of the Newton-Bhabha Award, 2016, Department of Science and Technology (DST), India and British Council, UK and DST-Innovation in Science Pursuit for Inspired Research fellowship-India. SKap was a recipient of a DBT fellowship, India. SKau was a recipient of a UGC fellowship, India. DD was a recipient of a Ramanujan fellowship and research grant supported by SERB-DST.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
KT and GK would like to thank Dr. Ravi, Schrödinger, India for assisting in protein-peptide docking experiments. We also thank Mr. Deepak Bhatt and Mr. Mohit Paul, CSIR-IMTECH, for helping in DNA sequencing and mass spectrometry studies, respectively.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00797/full#supplementary-material
Footnotes
References
Alos, J. I. (2015). [Antibiotic resistance: a global crisis]. Enferm. Infecc. Microbiol. Clin. 33, 692–699.
Bai, H., Sang, G., You, Y., Xue, X., Zhou, Y., Hou, Z., et al. (2012). Targeting RNA polymerase primary σ70 as a therapeutic strategy against methicillin-resistant Staphylococcus aureus by antisense peptide nucleic acid. PLoS One 7:e29886. doi: 10.1371/journal.pone.0029886
Bakail, M., and Ochsenbein, F. (2016). Targeting protein–protein interactions, a wide open field for drug design. Comptes Rendus Chim. 19, 19–27. doi: 10.1016/j.crci.2015.12.004
Banerjee, R., Rudra, P., Prajapati, R. K., Sengupta, S., and Mukhopadhyay, J. (2014). Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis 94, 397–404. doi: 10.1016/j.tube.2014.03.008
Bernal-Bernal, D., Gallego-Garcia, A., Garcia-Martinez, G., Garcia-Heras, F., Jimenez, M. A., Padmanabhan, S., et al. (2015). Structure-function dissection of Myxococcus xanthus CarD N-terminal domain, a defining member of the CarD_CdnL_TRCF family of RNA polymerase interacting proteins. PLoS One 10:e0121322. doi: 10.1371/journal.pone.0121322
Burian, J., Yim, G., Hsing, M., Axerio-Cilies, P., Cherkasov, A., Spiegelman, G. B., et al. (2013). The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res. 41, 10062–10076. doi: 10.1093/nar/gkt751
Campbell, E. A., Korzheva, N., Mustaev, A., Murakami, K., Nair, S., Goldfarb, A., et al. (2001). Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 104, 901–912. doi: 10.1016/s0092-8674(01)00286-0
Chopra, I. (2007). Bacterial RNA polymerase: a promising target for the discovery of new antimicrobial agents. Curr. Opin. Investig. Drugs 8, 600–607.
Ciciliato, I., Corti, E., Sarubbi, E., Stefanelli, S., Gastaldo, L., Montanini, N., et al. (2004). Antibiotics GE23077, novel inhibitors of bacterial RNA polymerase. I. Taxonomy, isolation and characterization. J. Antibiot. 57, 210–217. doi: 10.7164/antibiotics.57.210
Craik, D. J., Fairlie, D. P., Liras, S., and Price, D. (2013). The future of peptide-based drugs. Chem. Biol. Drug Des. 81, 136–147. doi: 10.1111/cbdd.12055
Das, A., Merril, C., and Adhya, S. (1978). Interaction of RNA polymerase and rho in transcription termination: coupled ATPase. Proc. Natl. Acad. Sci. U.S.A. 75, 4828–4832. doi: 10.1073/pnas.75.10.4828
Davis, E., Chen, J., Leon, K., Darst, S. A., and Campbell, E. A. (2015). Mycobacterial RNA polymerase forms unstable open promoter complexes that are stabilized by CarD. Nucleic Acids Res. 43, 433–445. doi: 10.1093/nar/gku1231
Epshtein, V., Kamarthapu, V., Mcgary, K., Svetlov, V., Ueberheide, B., Proshkin, S., et al. (2014). UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505, 372–377. doi: 10.1038/nature12928
Fosgerau, K., and Hoffmann, T. (2015). Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128. doi: 10.1016/j.drudis.2014.10.003
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi: 10.1021/jm0306430
Fry, D. C. (2015). Targeting protein-protein interactions for drug discovery. Methods Mol. Biol. 1278, 93–106. doi: 10.1007/978-1-4939-2425-7_6
Gallego-Garcia, A., Mirassou, Y., Elias-Arnanz, M., Padmanabhan, S., and Jimenez, M. A. (2012). NMR structure note: N-terminal domain of Thermus thermophilus CdnL. J. Biomol. NMR 53, 355–363. doi: 10.1007/s10858-012-9648-z
Gallego-Garcia, A., Mirassou, Y., Garcia-Moreno, D., Elias-Arnanz, M., Jimenez, M. A., and Padmanabhan, S. (2014). Structural insights into RNA polymerase recognition and essential function of Myxococcus xanthus CdnL. PLoS One 9:e108946. doi: 10.1371/journal.pone.0108946
Garner, A. L., Weiss, L. A., Manzano, A. R., Galburt, E. A., and Stallings, C. L. (2014). CarD integrates three functional modules to promote efficient transcription, antibiotic tolerance, and pathogenesis in mycobacteria. Mol. Microbiol. 93, 682–697. doi: 10.1111/mmi.12681
Gould, I. M. (2010). Coping with antibiotic resistance: the impending crisis. Int. J. Antimicrob. Agents 36(Suppl. 3), S1–S2.
Gulten, G., and Sacchettini, J. C. (2013). Structure of the Mtb CarD/RNAP beta-lobes complex reveals the molecular basis of interaction and presents a distinct DNA-binding domain for Mtb CarD. Structure 21, 1859–1869. doi: 10.1016/j.str.2013.08.014
Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., et al. (2016). OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. doi: 10.1021/acs.jctc.5b00864
Helmann, J. D., and Chamberlin, M. J. (1988). Structure and function of bacterial sigma factors. Annu. Rev. Biochem. 57, 839–872. doi: 10.1146/annurev.bi.57.070188.004203
Ho, M. X., Hudson, B. P., Das, K., Arnold, E., and Ebright, R. H. (2009). Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 19, 715–723. doi: 10.1016/j.sbi.2009.10.010
Hu, Y., Morichaud, Z., Chen, S., Leonetti, J. P., and Brodolin, K. (2012). Mycobacterium tuberculosis RbpA protein is a new type of transcriptional activator that stabilizes the sigma A-containing RNA polymerase holoenzyme. Nucleic Acids Res. 40, 6547–6557. doi: 10.1093/nar/gks346
Hüsecken, K., Negri, M., Fruth, M., Boettcher, S., Hartmann, R. W., and Haupenthal, J. (2013). Peptide-Based Investigation of the Escherichia coli RNA Polymerase σ70:Core Interface As Target Site. ACS Chem. Biol. 8, 758–766.
Jacobson, M. P., Friesner, R. A., Xiang, Z., and Honig, B. (2002). On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608. doi: 10.1016/s0022-2836(02)00470-9
Jacobson, M. P., Pincus, D. L., Rapp, C. S., Day, T. J., Honig, B., Shaw, D. E., et al. (2004). A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367. doi: 10.1002/prot.10613
Jeong, E. H., Son, Y. M., Hah, Y. S., Choi, Y. J., Lee, K. H., Song, T., et al. (2006). RshA mimetic peptides inhibiting the transcription driven by a Mycobacterium tuberculosis sigma factor SigH. Biochem. Biophys. Res. Commun. 339, 392–398. doi: 10.1016/j.bbrc.2005.11.032
Kaur, G., Dutta, D., and Thakur, K. G. (2014). Crystal structure of Mycobacterium tuberculosis CarD, an essential RNA polymerase binding protein, reveals a quasidomain-swapped dimeric structural architecture. Proteins 82, 879–884. doi: 10.1002/prot.24419
Kaur, G., Kaundal, S., Kapoor, S., Grimes, J. M., Huiskonen, J. T., and Thakur, K. G. (2018). Mycobacterium tuberculosis CarD, an essential global transcriptional regulator forms amyloid-like fibrils. Sci. Rep. 8:10124.
Lamiable, A., Thevenet, P., Rey, J., Vavrusa, M., Derreumaux, P., and Tuffery, P. (2016). PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 44, W449–W454.
Lushniak, B. D. (2014). Antibiotic resistance: a public health crisis. Public Health Rep. 129, 314–316. doi: 10.1177/003335491412900402
Ma, C., Yang, X., and Lewis, P. J. (2016). Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 80, 139–160. doi: 10.1128/mmbr.00055-15
Mcclure, W. R., and Cech, C. L. (1978). On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 253, 8949–8956.
Mcginnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25.
Mendelson, M. (2015). Practical solutions to the antibiotic resistance crisis. S. Afr. Med. J. 105:413. doi: 10.7196/samj.9642
Mukhopadhyay, J., Sineva, E., Knight, J., Levy, R. M., and Ebright, R. H. (2004). Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol. Cell. 14, 739–751. doi: 10.1016/j.molcel.2004.06.010
Newell, K. V., Thomas, D. P., Brekasis, D., and Paget, M. S. (2006). The RNA polymerase-binding protein RbpA confers basal levels of rifampicin resistance on Streptomyces coelicolor. Mol. Microbiol. 60, 687–696. doi: 10.1111/j.1365-2958.2006.05116.x
Otvos, L. Jr., and Wade, J. D. (2014). Current challenges in peptide-based drug discovery. Front. Chem. 2:62. doi: 10.3389/fchem.2014.00062
Pelay-Gimeno, M., Glas, A., Koch, O., and Grossmann, T. N. (2015). Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem. Int. Ed. Engl. 54, 8896–8927. doi: 10.1002/anie.201412070
Rammohan, J., Ruiz Manzano, A., Garner, A. L., Prusa, J., Stallings, C. L., and Galburt, E. A. (2016). Cooperative stabilization of Mycobacterium tuberculosis rrnAP3 promoter open complexes by RbpA and CarD. Nucleic Acids Res. 44, 7304–7313.
Rammohan, J., Ruiz Manzano, A., Garner, A. L., Stallings, C. L., and Galburt, E. A. (2015). CarD stabilizes mycobacterial open complexes via a two-tiered kinetic mechanism. Nucleic Acids Res. 43, 3272–3285. doi: 10.1093/nar/gkv078
Remaut, H., and Waksman, G. (2006). Protein-protein interaction through beta-strand addition. Trends Biochem. Sci. 31, 436–444. doi: 10.1016/j.tibs.2006.06.007
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324.
Selby, C. P., and Sancar, A. (1995a). Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J. Biol. Chem. 270, 4882–4889. doi: 10.1074/jbc.270.9.4882
Selby, C. P., and Sancar, A. (1995b). Structure and function of transcription-repair coupling factor. II. Catalytic properties. J. Biol. Chem. 270, 4890–4895. doi: 10.1074/jbc.270.9.4890
Shanmuga Priya, V. G., Swaminathan, P., Muddapur, U. M., Fandilolu, P. M., Parulekar, R. S., and Sonawane, K. D. (2018). Peptide similarity search based and virtual screening based strategies to identify small molecules to inhibit CarD–RNAP Interaction in M. tuberculosis. Int. J. Peptide Res. Ther. 25, 697–709. doi: 10.1007/s10989-018-9716-7
Shelley, J. C., Cholleti, A., Frye, L. L., Greenwood, J. R., Timlin, M. R., and Uchimaya, M. (2007). Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691. doi: 10.1007/s10822-007-9133-z
Srivastava, D. B., Leon, K., Osmundson, J., Garner, A. L., Weiss, L. A., Westblade, L. F., et al. (2013). Structure and function of CarD, an essential mycobacterial transcription factor. Proc. Natl. Acad. Sci. U.S.A. 110, 12619–12624. doi: 10.1073/pnas.1308270110
Stallings, C. L., Stephanou, N. C., Chu, L., Hochschild, A., Nickels, B. E., and Glickman, M. S. (2009). CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138, 146–159. doi: 10.1016/j.cell.2009.04.041
Uhlig, T., Kyprianou, T., Martinelli, F. G., Oppici, C. A., Heiligers, D., Hills, D., et al. (2014). The emergence of peptides in the pharmaceutical business: from exploration to exploitation. EuPA Open Proteomics 4, 58–69. doi: 10.1016/j.euprot.2014.05.003
Ventola, C. L. (2015a). The antibiotic resistance crisis: part 1: causes and threats. PT 40, 277–283.
Ventola, C. L. (2015b). The antibiotic resistance crisis: part 2: management strategies and new agents. PT 40, 344–352.
Wang, Z. A., Ding, X. Z., Tian, C.-L., and Zheng, J.-S. (2016). Protein/peptide secondary structural mimics: design, characterization, and modulation of protein-protein interactions. RSC Adv. 6, 61599–61609. doi: 10.1039/c6ra13976k
Weiss, L. A., Harrison, P. G., Nickels, B. E., Glickman, M. S., Campbell, E. A., Darst, S. A., et al. (2012). Interaction of CarD with RNA polymerase mediates Mycobacterium tuberculosis viability, rifampin resistance, and pathogenesis. J. Bacteriol. 194, 5621–5631. doi: 10.1128/jb.00879-12
Werner, F. (2008). Structural evolution of multisubunit RNA polymerases. Trends Microbiol. 16, 247–250. doi: 10.1016/j.tim.2008.03.008
Werner, F., and Grohmann, D. (2011). Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 9, 85–98. doi: 10.1038/nrmicro2507
Westblade, L. F., Campbell, E. A., Pukhrambam, C., Padovan, J. C., Nickels, B. E., Lamour, V., et al. (2010). Structural basis for the bacterial transcription-repair coupling factor/RNA polymerase interaction. Nucleic Acids Res. 38, 8357–8369. doi: 10.1093/nar/gkq692
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., et al. (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552. doi: 10.1385/1-59259-584-7:531
Yu, C. S., Lin, C. J., and Hwang, J. K. (2004). Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13, 1402–1406. doi: 10.1110/ps.03479604
Keywords: RNA polymerase, CarD, peptide-based inhibitors, Mycobacterium tuberculosis, bacterial transcription, protein-protein interaction
Citation: Kaur G, Kapoor S, Kaundal S, Dutta D and Thakur KG (2020) Structure-Guided Designing and Evaluation of Peptides Targeting Bacterial Transcription. Front. Bioeng. Biotechnol. 8:797. doi: 10.3389/fbioe.2020.00797
Received: 27 February 2020; Accepted: 22 June 2020;
Published: 08 September 2020.
Edited by:
Yusuf Akhter, Babasaheb Bhimrao Ambedkar University, IndiaReviewed by:
Zhiyun Guo, Southwest Jiaotong University, ChinaIntekhab Alam, Case Western Reserve University, United States
Santhi Devasundaram, National Cancer Institute at Frederick, United States
Copyright © 2020 Kaur, Kapoor, Kaundal, Dutta and Thakur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krishan Gopal Thakur, a3Jpc2hhbmdAaW10ZWNoLnJlcy5pbg==
†Present address: Gundeep Kaur, Department of Epigenetics and Molecular Carcinogenesis, University of Texas, MD Anderson Cancer Center, Houston, TX, United States; Soni Kaundal, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, United States