The Intense World Syndrome – an alternative hypothesis for autism
- Brain Mind Institute, Ecole Polytechnique Fédérale de Lausanne Switzerland
Autism is a devastating neurodevelopmental disorder with a polygenetic predisposition that seems to be triggered by multiple environmental factors during embryonic and/or early postnatal life. While significant advances have been made in identifying the neuronal structures and cells affected, a unifying theory that could explain the manifold autistic symptoms has still not emerged. Based on recent synaptic, cellular, molecular, microcircuit, and behavioral results obtained with the valproic acid (VPA) rat model of autism, we propose here a unifying hypothesis where the core pathology of the autistic brain is hyper-reactivity and hyper-plasticity of local neuronal circuits. Such excessive neuronal processing in circumscribed circuits is suggested to lead to hyper-perception, hyper-attention, and hyper-memory, which may lie at the heart of most autistic symptoms. In this view, the autistic spectrum are disorders of hyper-functionality, which turns debilitating, as opposed to disorders of hypo-functionality, as is often assumed. We discuss how excessive neuronal processing may render the world painfully intense when the neocortex is affected and even aversive when the amygdala is affected, leading to social and environmental withdrawal. Excessive neuronal learning is also hypothesized to rapidly lock down the individual into a small repertoire of secure behavioral routines that are obsessively repeated. We further discuss the key autistic neuropathologies and several of the main theories of autism and re-interpret them in the light of the hypothesized Intense World Syndrome.
Introduction
Autism as a syndrome was first described by Leo Kanner, a child psychologist, in 1943. His initial description, based on 11 case studies emphasized “…an innate inability to form the usual, biologically provided affective contact with other people.” For a long time, autism was thought to be a consequence of bad parenting and the “refrigerator mother” theory (Bettelheim, 1967 ) lasted from the 1950s well beyond the 1970s. Bernard Rimland (Rimland, 1964 ) and Michael Rutter (Rutter, 1968 ) established empirically that the parents of autistic children were not different in their parenting from the parents of non-autistic controls and helped building a case for a neurobiological basis of autism. Autism is now recognized as a neurodevelopmental disorder manifesting within the first 3 years after birth and progressively worsening in the course of life. The core symptoms are impairments of sociability, communicative skills and imagination, together with stereotypic behaviors and repetitive tendencies (DSM-IV, 1994 ). At the cognitive level, all autistic children seem to display some form of abnormality in perception, attention, and memory (Ben Shalom, 2003 ; Dakin and Frith, 2005 ; Sanders et al., 2007 ).
Genetic analyses have revealed that autism is a polygenetic disorder where any one or more set of genes can predispose toward, but no one gene has been found to cause autism (Bonora et al., 2006 ; Cook, 2001 ; Lamb et al., 2000 ; Persico and Bourgeron, 2006 ). The primary cause of autism is most likely a form of epigenetic alteration during development (Beaudet and Zoghbi, 2006 ), which triggers a cascade of diverse neuropathologies depending on the timing of the epigenetic attack. A number of toxic insults have been implicated as the key triggers of autism (Bandim et al., 2003 ; Chess, 1971 ; Moore et al., 2000 ; Nanson, 1992 ; Rasalam et al., 2005 ; Stromland et al., 1994 ) (Table 1 ) probably with a higher probability in the genetically predisposed.
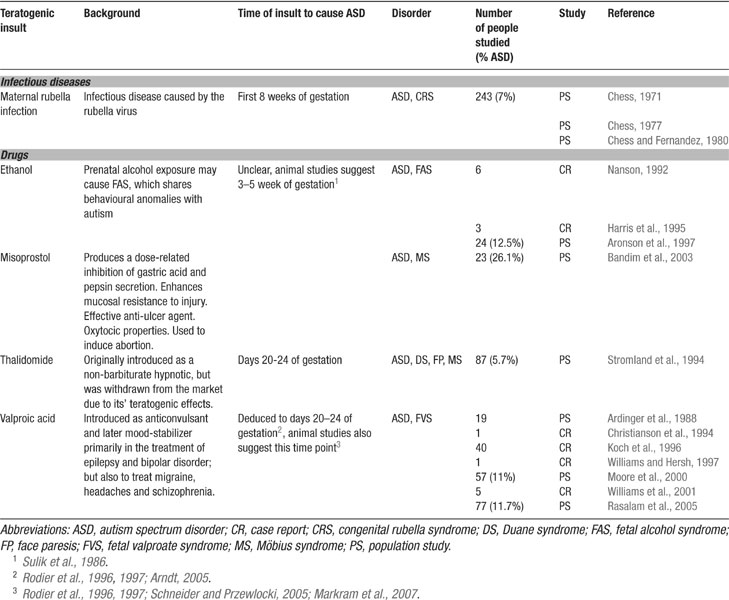
Table 1. Potential environmental triggers for autism.
Autism encompasses a spectrum of disorders ranging from severe mental retardation to high functioning Asperger's and “idiots savants” with many brain regions implicated making it difficult to develop a unified theory of autism. High functioning autism has been viewed as the exception to the mainstream view that autism is a severe form of mental retardation with poor cognitive capabilities (Lord and Spence, 2006 ; Pring, 2005 ). Indeed, of several hundred patents filed on “shot in the dark” treatments for autism, most are aimed at enhancing rather than reducing cognitive processes. However, a quite opposite scenario could be the case, in which the handicap may be resulting from excessive, rather than poor cognitive functioning.
Based on the recent multi-screening results obtained on the valproic acid (VPA) rat model of autism, we propose here a unifying hypothesis of autism where the core neurophysiological pathology is excessive neuronal information processing and storage in local circuits of the brain, which gives rise to hyper-functioning of the brain regions most affected. Such hyper-functioning in different brain regions is proposed to cause hyper-perception, hyper-attention, and hyper-memory that could potentially explain the full spectrum of symptoms in autism.
We propose that a common molecular syndrome is activated in autism that produces hyper-functioning in a coordinated manner by forming hyper-reactive and hyper-plastic microcircuits in different brain areas. The vast autism spectrum could be explained by the specific degree to which this hyper-functional molecular syndrome is active in different brain areas, which could depend on the precise stage of development that the brain is exposed to a triggering insult, the type of toxic insult, and the presence of any predisposing genes.
We propose that these super-charged microcircuits render aspects of the world painfully intense and aversive, and autism is therefore proposed as an Intense World Syndrome. We present recent molecular, cellular, synaptic, circuit, and behavioral evidence to support this new hypothesis and re-interpret the symptomology and pathology in the light of the proposed syndrome in which the world is aversively intense.
Heterogeneity in the Autistic Syndrome
The major problem in developing a unifying theory of autism is the large number of variations of the disorder. The heterogeneity is so extreme that at least five behavioral subtypes are classified as part of the autism spectrum disorders (ASD) – autistic disorder, Asperger's syndrome, Rett's syndrome, disintegrative disorder, and pervasive developmental disorder (PDD) not otherwise specified. According to the DSM-IV, a triad of symptoms, impaired social interactions, communication deficits as well as stereotypic, restricted, and repetitive behaviors, characterizes all these subtypes of autism.
Heterogeneity further manifests within each of the three core symptoms. In the social domain, inter-individual variability may range from a complete absence of interest in interacting with others, to more subtle dysfunctions in managing complex social interactions, in which other peoples’ intentions or the social context need to be taken into account. Communication impairments may range from a complete absence of spoken language over mild impairment, with the use of idiosyncratic vocabulary, to hyper-linguism in some of the Asperger cases. Stereotyped behaviors may also range from simple motor stereotypies and a preference for sameness to more complex rituals, which may be accompanied by considerable distress and aggression when they cannot be fulfilled.
Intellectual capabilities also vary across the entire IQ spectrum with the majority of autistic individuals displaying very low IQs and the high functioning savants coming out on the other extreme high end of the IQ spectrum. The communication handicap, however, confounds the accurate assessment of IQ in autists and numerous anecdotal reports suggest that when the communication deficits are solved in some way (e.g., through communication devices), unusually high IQ's are revealed. Moreover, some individuals exhibit truly high intellectual capabilities and excellent achievements in highly specialized fields of interest (Asperger, 1944 ; Pring, 2005 ).
Comorbidities
Austistic symptoms are found in association with many other disorders. Roughly 30% of autistic individuals develop clinically apparent seizures, suggesting certain imbalances in cortical circuits. Other comorbidities include neurofibromatosis, congenital rubella, hydrocephalus, ambulation, fetal alcohol syndrome and Fragile X syndrome, (Lord and Spence, 2006 ).
Estimates of the frequency of such problems and conclusions about the nature of the association have differed from one research group to another (Gillberg and Coleman, 1996 ; Rutter et al., 1994 ). Studies that consider these comorbidities as subtypes of autism and use these disorders as the basis for developing genetic models of autism have contributed conflicting results (Hayashi et al., 2007 ; Tabuchi et al., 2007 ), which have confounded a coherent picture of the core pathologies underlying autism.
Understanding the Common Cause of Autism
Autism is recognized as one of the most heritable neuropsychiatric disorders, since the concordance rates of autism in monozygotic twins are above 60%, whereas sibling risk is around 2–7%, which is much higher than in the general population (0.01–0.08%) (Bailey et al., 1995 ; Bonora et al., 2006 ; Hallmayer et al., 2002 ; Le Couteur et al., 1996 ; Rutter, 2000 ; Spiker et al., 1994 ). The mode of inheritance seems to be complex, since up to 15 different chromosomal loci have been identified in producing the risk (Bonora et al., 2006 ; Cook, 2001 ; Lamb et al., 2000 ; Persico and Bourgeron, 2006 ). Many of these loci encode molecules and proteins, which have been linked to neurodevelopment and synaptic function (Cook, 2001 ; Lamb et al., 2000 ; Persico and Bourgeron, 2006 ). Not all children with predisposing genes develop autism indicating that the genetic alterations should not be seen as the cause of autism, but as a major predisposing factor.
Autism is a neurodevelopmental disorder suggesting that there is a relatively narrow time window during embryogenesis (perhaps extending into early postnatal life), during which the normal unfolding of the genome can be sabotaged by an epigenetic attack. Evidence has accumulated that toxic exposure during early embryogenesis and/or very early after birth can trigger the onset of autism. In the extreme view, autism may even result from such epigenetic insults without predisposing genes, but with a lower probability. Some potent teratogens include maternal rubella infection (Chess, 1971 ), ethanol (Nanson, 1992 ), misoprostol (Bandim et al., 2003 ), thalidomide (Stromland et al., 1994 ), and VPA (Moore et al., 2000 ; Rasalam et al., 2005 ) (summarized in Table 1 ). The most vulnerable period of exposure seems to be the first trimester of gestation (Arndt et al., 2005 ).
Understanding the ultimate cause of autism lies in understanding the nature of the epigenetic attack and developing the ultimate cure for autism lies in being able to prevent this attack and reverse its’ effects once it has occurred. The particular form of the attack may give rise to a syndrome with very diverse outcomes and reconciling these diverse outcomes under a common cascade of effects (if one such cascade exists) is crucial to both understanding the cause and developing the for autism.
Animal Models of Autism
Autism is generally considered a human disorder because of the high level cognitive symptomology in the domain of social interaction, communication, and theory of mind. This conclusion is, however, unjustified and there is no reason to believe that the core neurophysiological pathology that occurs in humans cannot also occur in other mammals. Monkeys are more easily related to the human condition, but rodents also exhibit complex social systems and rich social interactions, and many cognitive functions can be studied such as perception, attention, memory, and emotions. Specific tasks targeted at unraveling “autistic” symptoms have been designed and tested in several rodent models (Crawley, 2004 ). It is of course not possible to study all the subtleties of the disorder using animal models, but they provide a scope of research just not possible in humans and in many cases also not possible in monkeys. Rodent models in particular allow an extensive multi-omics approach to autism with a spectrum of non-invasive and invasive approaches at the genetic, molecular, cellular, synaptic, local circuit, circuit, systems, and behavioral levels. Ultimately, all neuropsychiatric and neurological disorders are due to some type of dysfunction in the manner in which neurons process information and to understand this dysfunction requires invasive electrophysiological experiments, which cannot be studied in humans.
There are three types of animal models of autism – genetically based, insult-based, and lesion-based models. Genetically based models assume that a specific gene can cause the disorder and the hypothesis is explored typically in mice by knocking out the gene or by engineering subtle mutations. An example of a genetically based model of autism is the oxytocin knockout mouse (Winslow and Insel, 2002 ). Insult-based models such as the VPA model (Rodier et al., 1997 ), examine the teratogenic effect with the assumption that the insult alone is sufficient to trigger the disorder. Lesion-based models damage a specific part of the brain hypothesized to be involved in an aspect of causing autism and an example is amygdala lesions in monkeys (Bachevalier, 1994 ). Future models may begin to combine genetically predisposed models with insult-based approaches. The advantage of the insult-based models is that the neurodevelopmental disorder which emerges is independent of the genetic predisposition allowing the systematic study of core abnormal brain and behavior developmental cascade that is triggered from the moment of the epigenetic attack to the matured animal.
The next chapter concentrates on the insult-based VPA rat model of autism, as this model is one of the best studied and validated models, and has allowed us to perform extensive multi-omics studies that yield a more comprehensive view of the induced disorder.
VPA Exposure in Humans
Clinically, VPA was first introduced in 1964 in France as an anticonvulsant and later as a mood-stabilizing drug, primarily in the treatment of epilepsy and bipolar disorder, but also used for migraine headaches and schizophrenia. In epileptics, VPA is used to control absence seizures, tonic-clonic seizures, complex partial seizures, and the seizures associated with Lennox-Gastaut syndrome. VPA use during pregnancy has been linked to autism.
Studies Implicating VPA in Autism
The first indications for VPA to cause autism stems from seven case studies of kids with fetal valproate syndrome (Christianson et al., 1994 ; Williams and Hersh, 1997 ; Williams et al., 2001 ), of which all exhibited a full diagnosis of autism. Moore and colleagues conducted the first population study on 57 children with various fetal anticonvulsant syndromes (caused by a variety of anticonvulsant drugs) in Scotland (Moore et al., 2000 ). These all children had been exposed to either VPA alone (60%), VPA in combination with another anticonvulsant drug (21%), or another anticonvulsant drug (carbamezepine or phenytoin) alone or in combination with each other (19%). They reported 46 (81%) kids with speech delays and 34 (60%) kids with two or more autistic features, of which 6 (11%) had a diagnosis of ASD. Furthermore, 46 (81%) had behavioral problems, 22 (39%) displayed hyper-activity or poor concentration, of which 4 (7%) had a diagnosis of attention deficit/hyper-activity disorder. Forty-four (77%) kids had learning difficulties, 34 (60%) had gross motor delay, and 24 (42%) had fine motor delay. These findings confirmed the association between fetal valproate syndrome and autism as suggested in the prior case reports. A more recent longitudinal population study spanning a period of 20 years examined 292 children whose mothers were exposed to antiepileptic drugs during pregnancy (Rasalam et al., 2005 ). Out of this population, 56 children were exposed to VPA alone and 77 children to VPA alone or in combination with another antiepileptic drug. Among these two sub-populations, 5 (8.9%) and 9 (11.7%) children fulfilled the criteria for ASD, respectively (Rasalam et al., 2005 ). Both population studies indicate that the rate of autism in the prenatally VPA-exposed population is much higher than the estimated rate of ASD in the general population of approximately 10 to 16 cases per 10.000 (Fombonne, 2006 ).
VPA, Thalidomide, and the Early Brain-stem Injury Hypothesis
The malformations caused by VPA and thalidomide, another autism causing teratogen, indicate an early insult during embryogenesis and, more specifically, around the time of neural tube closure, which led to the hypothesis that autism may be caused by a brain-stem injury during embryonic development (Arndt et al., 2005 ; Rodier et al., 1996 ; Rodier et al., 1997 ; Stromland et al., 1994 ). First indications for this hypothesis stem form a Swedish thalidomide study (Stromland et al., 1994 ) in which 87 patients were examined with the initial purpose of evaluating ophthalmologic effects, but a psychiatric evaluation was also performed. Five cases with autism were found in this study. All of these cases were from a group of 15 patients where thalidomide exposure occurred between the 20th–24th days of gestation while no autistic cases were reported for any other exposure times. The probability of autism after thalidomide exposure during this time period is, therefore, extremely high.
This period of gestation is when the neural tube closes and the first neurons are produced. These neurons are part of the motor nuclei of the cranial nerves and an insult affecting these neurons, therefore, is associated with abnormalities in facial features - indeed observed in all of the five autistic thalidomide cases. Three patients had Duane syndrome (failure of the VIth/abducens cranial nerve to innervate the lateral rectus muscle by the eye with subsequent reinnervation of the muscle by the IIIrd/oculomotor cranial nerve); one patient had face paresis (oculomotor palsy); four had Möbius syndrome (failure of the VIIth/facial cranial nerve to innervate the facial muscles); two had abnormal lacrimation (due to a failure of the neurons of the superior salivatory nucleus – cranial nerve VII – to innervate the lacrimal apparatus). All five patients had ear malformations and hearing deficits. Ear malformations (Walker, 1977 ), eye motility problems (Scharre and Creedon, 1992 ), and Möbius syndrome (Gillberg and Steffenburg, 1989 ) had previously been associated with autism. In fact, external ear malformation is the most common physical abnormality observed in autism and the one which best distinguishes between autism and mental retardation (Walker, 1977 ). The conclusion from this thalidomide study was that autism is associated with a brainstem injury at a very specific time during embryogenesis.
Some of the teratogentic effects of VPA resemble those of thalidomide. These include the same neural tube closure defects such as facial dysmorphy and ear abnormalities. Even though VPA, as a remedy for epilepsy, is usually taken throughout the entire pregnancy, the time point of injury can be deduced on the basis of these physical malformations. Since these are very similar to the thalidomide-induced autistic cases and the exact time period for thalidomide to cause autism is known to be between embryonic days (EDs) 20–24 (Stromland et al., 1994 ), it has been argued that the time point of VPA to cause autism is the same as for thalidomide (Rodier et al., 1996 ; Rodier et al., 1997 ).
In order to test the brain-stem hypothesis of autism, the brain of an autistic person, never exposed to thalidomide or VPA, was examined for brain-stem injuries and compared to a healthy brain (Rodier et al., 1996 ). It turned out that the brain stem of the autistic brain exhibited a severe loss of motor neurons in the facial nucleus. Whereas the facial nucleus in the healthy brain contained more than 9000 neurons, in the autistic brain only 400 neurons were present in this area. The superior olive, an auditory relay nucleus, was also missing completely, further supporting the brain-stem association in autism. This study further indicated that brain-stem injuries indeed occur in autism.
The early brain-stem hypothesis of autism states that all other brain defects observed in autism must be a consequence of this one early brain-stem injury – a big bang – since no other brain regions are yet developed (Rodier et al., 1996 ). It is, however, possible that progenitor cells for other brain regions may also be affected and the damage produced would only become obvious once these regions begin to develop. It is also not yet clear whether other brain regions may be vulnerable to a VPA insult at later stages of development, i.e., whether each brain region has its own vulnerability to VPA exposure at the moment of its first differentiation.
VPA Exposure in Rats
In order to prove the hypothesis that an early brain-stem injury may provoke the same pattern of overall brain anomalies as observed in autism, an animal model was developed (Rodier et al., 1996 ). VPA was the drug of choice, since thalidomide has different effects in rodents than in humans (Schumacher et al., 1972 ). VPA, on the other hand, is a powerful teratogen in rodents and produces many of the malformations observed in humans (Binkerd et al., 1988 ; Collins et al., 1991 ; Ehlers et al., 1992 ). The time of neural tube closure in the rat occurs on day 11.5 and within the 12th day of gestation, production of the motor nuclei of trigeminal, abducens, and hypoglossal nerves is completed (Altman and Bayer, 1980 ). A single dose of VPA (350 mg/kg) administered to pregnant dams on ED 11.5 resulted in a reduction of the trigeminal and hypoglossal motor nuclei. Exposure on ED12 caused an additional loss of neurons in the abducens nucleus and on ED12.5 in all the previous and additionally in the oculomotor nucleus (Table 2 ; Rodier et al., 1996 ).
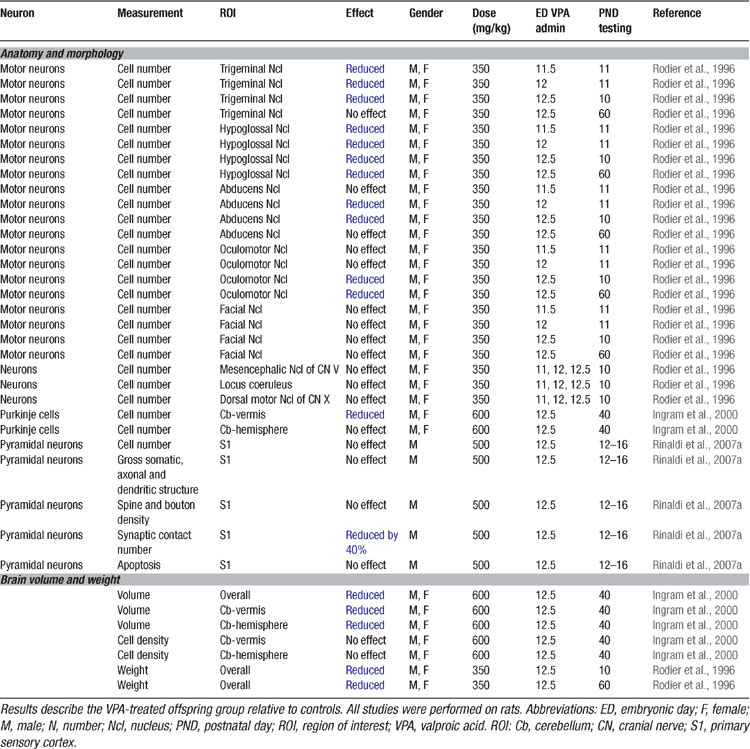
Table 2. Anatomical and morphological alterations in the VPA rat of autism.
Follow-up anatomical studies showed that VPA exposure on ED12.5 also results in a loss of cerebellar neurons (Ingram et al., 2000 ; Rodier et al., 1997 ), one of the most prominent features in the autistic brain (Kemper and Bauman, 1998 ; Palmen et al., 2004 ; Ritvo et al., 1986 ). Purkinje cells were particularly reduced in the lobules VI–VIII and IX, but not the anterior lobules (IV and V) of the vermis. Moreover, the nucleus interpositus (corresponding to the globose and emboliform nuclei in humans) was smaller. These early experimental studies were able to prove that a single dose of VPA may cause the same neural tube closure injuries as observed after thalidomide and VPA exposure in humans as well as in autism.
More recently, the serotonergic system was studied in VPA-treated rat offspring (Miyazaki et al., 2005 ; Narita et al., 2002 ; Tsujino et al., 2007 ) (Table 3 ). Administration of a single dose of VPA on ED9 (neural plate stage) has also been found to dramatically increase the serotonin levels in the blood as well as the frontal cortex, hippocampus, and cerebellum (Narita et al., 2002 ; Tsujino et al., 2007 ). VPA administration also irreversibly altered serotonergic neuronal differentiation and migration in the dorsal Raphé nucleus (Miyazaki et al., 2005 ; Tsujino et al., 2007 ). These results are strikingly similar to the data obtained on the serotonergic system in human autism (Lam et al., 2006 ), discussed further below.

Table 3. Neurochemical and genetic alterations in the VPA rat model of autism.
At the behavioral level, it was already known for some time that VPA may cause severe and selective alterations in the offspring when administered throughout pregnancy (Vorhees, 1987b , 1987a ; Wagner et al., 2006 ), summarized in Table 4 . However, these behavioral alterations may also reflect other teratogenic effects induced by prolonged VPA exposure. Therefore, it is more advantageous to test animals which received a single injection at ED12.5 as proposed by Rodier et al. (1996) , since this model offers a specific hypothesis about the genesis of autism and has been validated on anatomical and proteomic grounds. Offspring of pregnant dams exposed to VPA during this period exhibited decreased social interactions, increased repetitive behaviors, enhanced anxiety, locomotor hyper-activity, lower sensitivity to pain, higher sensitivity to non-painful sensory stimulation, impaired pre-pulse inhibition, and enhanced eye-blink conditioning (Markram et al., 2007 ; Schneider and Przewlocki, 2005 ; Schneider et al., 2001 ; Schneider et al., 2006 ; Stanton et al., 2007 ) – symptoms described in the DSM-IV and in the autism literature (DSM-IV, 1994 ; McAlonan et al., 2002 ; Muris et al., 1998 ; Perry et al., 2007 ; Sears et al., 1994 ). All results obtained on the VPA rodent model of autism are summarized in Tables 2 3 4 5 .
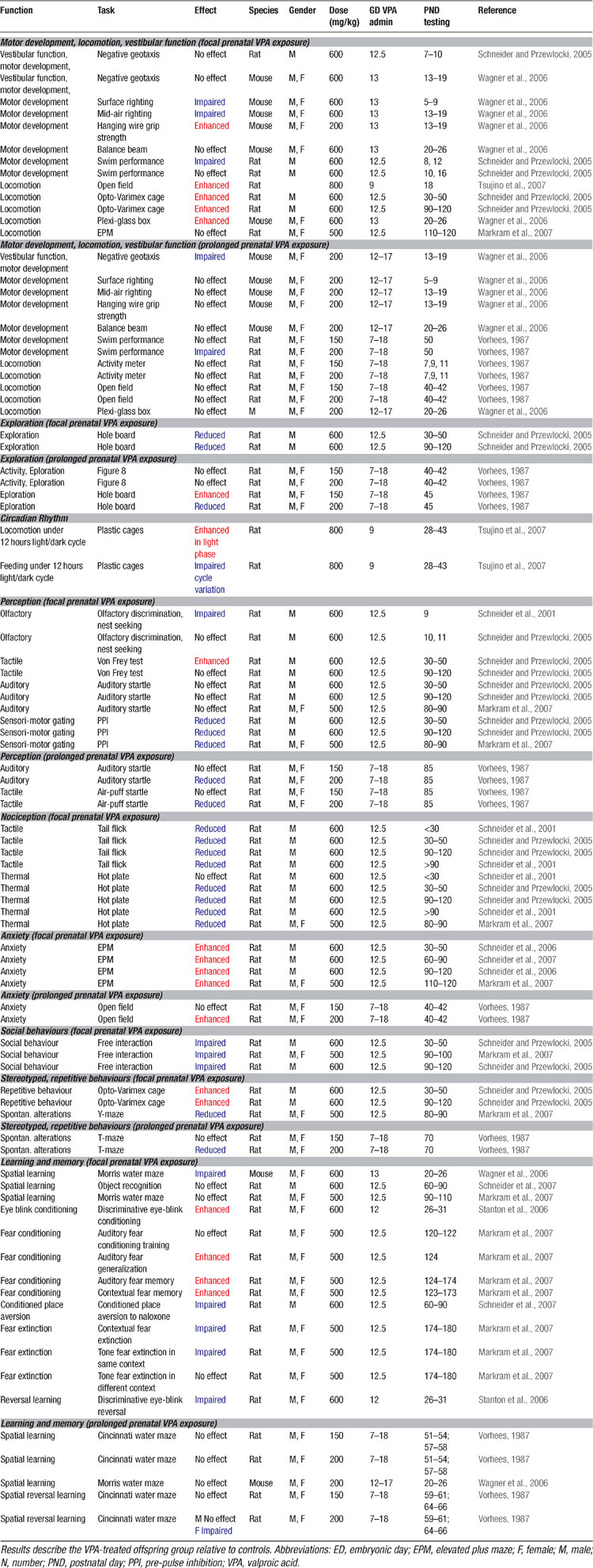
Table 4. Behavioural alterations in the VPA rat model of autism.
In summary, the data obtained on the VPA rat model proposed by Rodier et al. (1996) clearly indicates a strong concordance with some of the main symptoms and pathologies of autism. For this reason, we chose this model for a multi-omics approach to study the molecular, cellular, synaptic, local circuit as well as further behavioral alterations possibly underlying autism. The following subchapters summarize the main findings.
Hyper-reactivity in Neocortical Microcircuits
Since high cognitive functions are affected in autism, we examined alterations in the neocortex using the VPA rat model. The results are summarized in Table 5 . The somatosensory cortex microcircuitry of two-week old VPA-treated offspring (Rinaldi et al., 2007a ) were examined after exposure on ED12.5. Brain slices were placed on a multi-electrode array (MEA) stimulator and the stimulation current was gradually increased to examine the stimulus–response relationship of the microcircuitry. The microcircuitry reacted much stronger to the same stimulus in the VPA-treated rats with nearly twice the response of the normal microcircuit. This extreme hyper-reactivity was observed in both layer 5 and in layer 2/3 indicating that the alteration is not layer specific. Recently we found that the microcircuits of the prefrontal cortex (T. Rinaldi, and H. Makram, unpublished data) and the amygdala (Markram et al., 2007 ) are also hyper-reactive indicating that the alterations are not specific to a single neocortical region and are also found in subcortical brain regions.
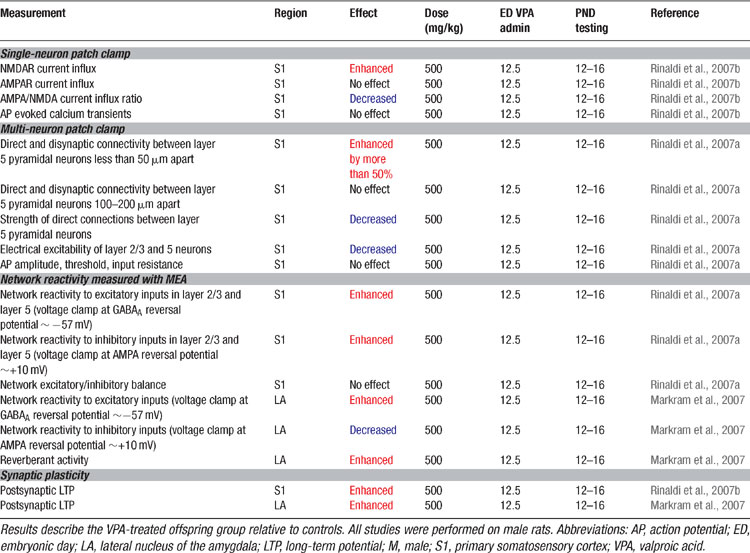
Table 5. Microcircuit alterations in the VPA rat model of autism.
Enhanced reactivity of microcircuits could be caused by larger synaptic currents, hyper-excitable neurons due to changes in active conductance, lack of proper inhibitory control, alterations in neuron numbers and morphology or by excessive recurrent circuitry. Excitatory synaptic responses studied in paired recordings revealed that the AMPA-mediated synaptic responses of connections between neurons were weaker rather than stronger in the VPA-treated rat offspring, which could not explain the hyper-reactivity. Anatomical reconstructions revealed that less synapses are deployed in each synaptic connection, which could account for these weaker synaptic connections.
Passive and active properties of the excitatory pyramidal neurons were examined. The pyramidal neurons required much more current to drive their voltage to spiking threshold and the number of spikes generated for a series of current injections was much lower than in controls. There were no differences in the passive properties indicating that it is the active excitability of pyramidal neurons that is depressed in this animal model of autism. This hypo-excitability of pyramidal neurons can clearly not cause hyper-reactivity to stimulation, and may in fact be an attempt to counter the hyper-reactivity as a compensatory mechanism.
The inhibitory component of the excessive response of the microcircuitry was also greater as compared to control rats. The balance between excitation and inhibition was not affected indicating that inhibition is increased proportionately to excitation. The increased delivery of inhibition is most likely due to matching recruitment of inhibitory neurons by the stronger excitatory response in the treated rats. Hyper-reactivity of the microcircuitry also cannot be caused by excessive inhibition in the neocortical microcircuitry.
Morphological examination of 3D reconstructions of biocytin stained pyramidal neurons did not show any significant differences in the extent of axonal or dendritic arbors, in the spine or bouton densities, and in the size of pyramidal neuron somata. There was also no change in the number of pyramidal neurons. Hyper-reactivity of the neocortical microcircuitry is therefore not caused by larger or more elaborate neurons, more excitable neurons, an increase in neuron numbers, more powerful synapses between neurons, nor by a loss of inhibition. Indeed, changes in these parameters seem to act in the opposite direction, perhaps part of a compensatory strategy.
Hyper-connectivity in Neocortical Microcircuits
We examined the number of direct connections established between pyramidal neurons and found an increase of more than 50% in the number of neurons connected in VPA-treated offspring (Rinaldi et al., 2007a ). This hyper-connectivity was only found for very close neighboring neurons confined within the typical dimensions of a neocortical minicolumn (∼50 μm somatic distance, and not for pairs of pyramidal neurons 100–200 μm apart). The probability of activating inhibitory interneurons by studying a disynaptic inhibitory connection between pyramidal neurons (Silberberg and Markram, 2007 ) also revealed a significant increase of more than 50%. This indicates that activating pyramidal neurons can indeed recruit proportionately more interneurons due to hyper-connectivity which extends beyond pyramidal neurons. Hyper-connectivity in microcircuits can lead to exaggerated recruitment of neurons when presented with a stimulus and could therefore account for the hyper-reactivity found in these local circuits. The results are summarized in Table 5 .
An intriguing aspect of this hyper-connectivity is the finding that pyramidal neurons target more neurons even at the expense of deploying less synapses per connection. This form of hyper-connectivity is, therefore, not a general hypertrophy of synapse formation, but rather a hypertrophy of connectivity between neurons. We, therefore, propose that a novel molecular mechanism involved in “target dominance” is enhanced in the VPA-treated neocortex and this molecular syndrome triggered by VPA exposure causes hyper-reactivity.
Hyper-plasticity in Neocortical Microcircuits
Since memory processes are also altered in autism, we examined whether synaptic plasticity is affected in the VPA-treated neocortex (Rinaldi et al., 2007b ). The results are summarized in Table 5 . Synaptic responses recorded in pyramidal neurons following a Hebbian pairing stimulation protocol caused more than a two-fold increase in the subsequent synaptic responses (i.e., enhanced long term potentiation). The results were also found for both layer 2/3 and in layer 5 pyramidal neurons and a closer examination revealed that the boosted plasticity was a postsynaptic form of plasticity. The presynaptic form of plasticity that is normally observed between these neurons was normal in the VPA treated slices. These results indicate that glutamatergic synapses are hyper-plastic in this animal model of autism.
Hyper-expression of NMDA Receptors in the Neocortex
To better understand the molecular syndrome underlying the hyper-plasticity, we tested for protein expression alterations (Rinaldi et al., 2007b ). The results are summarized in Table 3 . We found that the AMPA receptor subunits GluR1, GluR2, and GluR3 and the obligatory subunit of the NMDA receptor, NR1, were unaltered in the VPA-treated neocortex. However, the expression level of the NMDA receptor subunits NR2A and NR2B were massively over-expressed (more than 100%). We did not find evidence that this enhanced NMDA receptor subunit levels rendered neurons more vulnerable to neurotoxicity.
We also found a large increase in the level of CaMKII protein expression (more than 60%), which is a key signaling enzyme associated with NMDA receptor-mediated synaptic plasticity. In contrast, the expression level of extracellular signal-regulated kinase (ERK) and cAMP response element binding protein (CREB), some phosphorylated forms of signaling proteins (pCREB-S133, pCaMKII-T286/287, GluR1-S831, pGluR1-S845, pNR1-S896, pNR1-S897, pNR2B-S1303), as well as the main metabotropic glutamate receptor subunits (mGluR1, mGluR5, mGluR4, mGluR6/7) and the kainite receptor subunits (GluR6/7), were unaffected. This indicated that NMDA receptor subunit levels are excessively expressed and that this is a highly selective abnormality within the glutamatergic system of this animal model of autism.
We performed multi-neuron patch-clamp experiments on slices of the somatosensory cortex to directly examine the synaptic currents mediated by AMPA and NMDA receptors between pairs of pyramidal neurons. Indeed, we found that much more charge entered the postsynaptic cells through the NMDA receptors consistent with the increased expression of the NMDA receptors. The decay-time courses of the NMDA receptor mediated currents were, however, unaffected suggesting that the proportions of subunits used in the assembly of NMDA receptors were not affected. We do not however know whether the normal developmental switches in sub-units are affected at a later stage.
Hyper-fear Memories
Kanners’ original case studies (1943) suggested that some of the autistic children he observed exhibited abnormal anxiety levels and phobias. More recent studies have also suggested abnormally high anxiety levels and phobias in children with ASD (Evans et al., 2005 ; Gillott et al., 2001 ; Muris et al., 1998 ) and their relatives (Micali et al., 2004 ). We found that VPA-treated offspring exhibited greatly amplified conditioned cued and contextual fear memories when tested up to 3 months after conditioning. Fear memories were not only amplified, but also more generalized to other stimulus configurations (Markram et al., 2007 ) (Table 4 ). These data indicated that VPA-treated animals are not only overtly anxious, but also acquire fear memories in an exaggerated manner. It is, however, not clear if enhanced fears are common across the autistic spectrum since two recent studies indicate that some high functioning autists, in particular patients diagnosed with Asperger disorder, do not seem to display abnormal conditioned fear (Bernier et al., 2005 ; Gaigg and Bowler, 2007 ).
Impaired Fear Extinction
Extinction is usually defined as a learning process in which a new (probably inhibitory) association is built, namely that the conditioned stimulus no longer predicts danger (Quirk, 2002 ; Rescorla, 2004 ). Extinction training has been shown to involve a network of interactive brain regions, with connections between the medial prefrontal cortex and the amygdala playing a prominent role (Barad, 2006 ; Quirk and Beer, 2006 ; Sotres-Bayon et al., 2004 ; Sotres-Bayon et al., 2006 ). Autistic children are known to have impairments in extinction learning and to display strong perseveration tendencies (Coldren and Halloran, 2003 ; Mullins and Rincover, 1985 ; Sears et al., 1994 ), but it was not known whether this applies to the fear domain as well. We found that once fear memories were acquired by VPA-treated rat offspring, they were exceedingly more resistant to extinction than in control animals (Markram et al., 2007 ) (Table 4 ).
Hyper-reactivity in the Amygdala Microcircuitry
The amygdala is a crucial brain structure mediating generalized anxiety and fear conditioning (Davis and Whalen, 2001 ; LeDoux, 2003 ). We, therefore, attempted to understand the microcircuit changes in the amygdala that could underlie the increased fear memories, over-generalization of conditioned fear and resistance to fear extinction which we observed in the VPA-treated offspring (Markram et al., 2007 ). We found that the lateral amygdala microcircuit was also extremely hyper-reactive when stimulated with progressively stronger electrical stimulations using the MEA stimulator (Table 5 ). The amygdala responded differently from the neocortex in that stimulation easily produced prolonged episodes of up-state-like bursts. The number, frequency and duration of these evoked up-states were all greatly enhanced in slices from VPA-treated animals. When we isolated the inhibition, we found that, unlike in the neocortex, inhibition was greatly reduced. We have not yet been able to obtain sufficient data to determine whether the excitatory cells of the amygdala are hyper-connected as in the neocortex, but the cellular and synaptic alterations are such that they also result in hyper-reactivity.
Hyper-plasticity in the Amygdala Microcircuitry
We examined whether synaptic plasticity was affected in the amygdala and found an equally significant enhancement of long-term potentiation (Markram et al., 2007 ) (Table 5 ) as in the neocortex. Interestingly amygdala disinhibition induced either by GABAergic blockade (Isoardi et al., 2004 ), genetic knockout of the GABAB(1a)-receptor subtype (Shaban et al., 2006 ), benzodiazepine withdrawal (Isoardi et al., 2004 ), dopamine receptor activation (Bissiere et al., 2003 ), or stress (Rodriguez Manzanares et al., 2005 ), can result in amygdaloid hyper-excitability (Isoardi et al., 2004 ; Rodriguez Manzanares et al., 2005 ) and facilitate LTP induction (Bissiere et al., 2003 ; Rodriguez Manzanares et al., 2005 ; Shaban et al., 2006 ), which has been associated not only with enhanced fear memories (Isoardi et al., 2004 ; Rodriguez Manzanares et al., 2005 ), but also with over-generalization of conditioned fear to neutral stimuli (Shaban et al., 2006 ).
Autism as an Intense World Syndrome
Autism is a polygenetically predisposed neurodevelopmental disorder that is most likely triggered by an epigenetic attack from multiple potential sources to set in motion a genetic and molecular cascade – a molecular syndrome - that unfolds a spectrum of disorders. All forms of autism have a vague commonality and the question is whether there is a common molecular syndrome producing common neurophysiological, cognitive, and behavioral pathologies. We propose here a potentially unifying hypothesis of autism in which a common molecular syndrome causes excessive neuronal information processing and storage in the microcircuits of the brain. Such excessive information handling is proposed to produce hyper-perception, hyper-attention, and hyper-memory, which could become the fundamental cognitive handicap in all cases of autism. We further propose that the core molecular syndrome that causes excessive information processing and storage does so by causing hyper-reactivity and hyper-plasticity of the brains’ microcircuits. In the neocortex, this core pathology is caused by hyper-connecting neighbouring neurons to produce excessive excitation and by hyper-expressing NMDA receptors to produce excessive plasticity. On the other hand, in the amygdala, the hyper-reactivity seems to additionally involve diminished inhibitory synaptic transmission. Hyper-reactivity may therefore be caused additionally by reducing inhibition in those brain regions with high numbers of inhibitory neurons. Indeed, a reduction in the inhibitory Purkinje cells in the cerebellum is well known (Bailey et al., 1998 ; Kemper and Bauman, 1998 ; Ritvo et al., 1986 ; Rodier et al., 1996 ). The full spectrum of autism may be explained by the varying degrees of expression of the molecular syndrome that drives hyper-reactivity and hyper-plasticity in different brain regions, depending on the precise moment that the developing brain was exposed to a triggering insult, the type of insult, and the presence of any predisposing genes.
This core hyper-functioning pathology is proposed to cause the spectrum of autistic symptoms by rendering local neural circuits hyper-sensitive to novel and past stimulation, and once activated, these microcircuits could become autonomous, difficult to control and coordinate with the activity in other microcircuits. Hyper-reactivity and hyper-plasticity are therefore proposed to cause exaggerated perception to fragments of a sensory world that are normally holistically correlated and multimodal, and furthermore to cause hyper-focusing on fragment of the sensory world with exaggerated and persistent attention. Such hyper-attention could become difficult to shift to new stimuli due to the difficulty for top-down mechanisms to coordinate the overly autonomous microcircuits. The hyper-plasticity component may also drive exaggerated memories to amplify further hyper-attention towards the same stimulus and drive over-generalization of attention to all related forms of the stimulus. The positive consequences are exceptional capabilities for specific tasks while the negative consequences are a rapid lock down of behavioral routines to a minute fraction of possibilities, which are then repeated excessively.
The intense world that the autistic person faces could also easily become aversive if the amygdala and related emotional areas are affected with hyper-reactivity and hyper-plasticity. The lack of social interaction in autism may therefore not be because of deficits in the ability to process social and emotional cues as previously thought, but because a subset of cues are overly intense, compulsively attended to, excessively processed and remembered with frightening clarity and intensity. Autistic people may, therefore, neither at all be mind-blind nor lack empathy for others, but be hyper-aware of selected fragments of the mind, which may be so intense that they avoid eye contact, withdraw from social interactions and stop communicating. In such a scenario, the world may become painfully intense for autistics and we, therefore, propose autism as an Intense World Syndrome. We now review some of the key findings in past autism research and provide a possible alternative interpretation of the results in the light of the hypothesized syndrome.
Core Pathologies in Autism in the Light of the Intense World Syndrome
The neurobiological research on autism is about 35 years old with a large number of findings many of which are controversial. Amongst the most consistent findings are the paucity of Purkinje cells in the cerebellar hemispheres (Palmen et al., 2004 ), increased platelet serotonin levels (Lam et al., 2006 ) and accelerated brain growth in early infancy (Courchesne et al., 2003 ). Recent advances made with functional neuroimaging techniques have contributed greatly to the understanding of the autistic brain.
Accelerated Brain Growth
The human brain continues to develop considerably throughout the first years of life. In the normal brain this development follows a specific hierarchical scheme where basic sensory areas mediating perceptual function, mature earlier than higher order association areas such as the frontal cortex. Such progressive neurodevelopment underlies the later development of refined skills and higher cognitive, emotional, social, and communication functions.
One of the most striking and reliable observations in the autistic brain is its abnormal development. Newborn autistic infants usually exhibit a normal (Gillberg and de Souza, 2002 ; Lainhart et al., 1997 ; Stevenson et al., 1997 ) or even slightly smaller than normal brain size (Courchesne et al., 2003 ). However, within the first year of life there is an accelerated growth (Dementieva et al., 2005 ), such that by the age of 2–3 years the overall volume is about 10% greater than normal (Courchesne et al., 2001 ; Sparks et al., 2002 ). The accelerated growth takes place in a more or less reverse hierarchical order with the frontal and temporal lobes, the limbic system and the cerebellum leading the development (Courchesne, 2004 ). In the neocortex, for example, the white matter increase is most pronounced in the frontal, followed by the temporal and parietal lobes, whereas occipital lobes remain normal (Carper et al., 2002 ). In 2–3 years old autistic kids, gray matter is increased most in the frontal followed by the temporal lobes (Carper et al., 2002 ). In the limbic system the amygdala and hippocampus are enlarged in children ranging from 3 to 13 years of age (Schumann et al., 2004 ; Sparks et al., 2002 ), while in older subjects, the amygdala volume seems comparable to normal people or even smaller (Courchesne et al., 1993 ; Dziobek et al., 2006 ; Haznedar et al., 2000 ; Nowell et al., 1990 ; Palmen et al., 2006 ; Schumann et al., 2004 ), but the results are not always consistent (Abell et al., 1999 ; Howard et al., 2000 ). The abnormally accelerated growth early in childhood is followed by an abnormally slow or arrested growth in later childhood. In other words, the autistic brain outruns the normal brain within the first 4 years of life, reaching mature levels in higher brain regions too soon and then ceases to develop further. Thus, throughout childhood the normal rate of growth declines and the normal brain catches up until the size of the autistic brain is only 1–2% above normal in adolescence (Redcay and Courchesne, 2005 ).
The excessive growth could be due to neuronal, synaptic, and/or connection hypertrophy. The question is whether there is a link between the regions that grow too fast and their normal local circuit properties that could explain the reverse development of some brain regions. The level of reactivity of local circuits is finely tuned to allow all brain regions to act in an orchestrated manner. This level reflects a balance between the number of synapses each neuron uses to contact neighbors and the number of neighbours that can be contacted as well as the manner in which inhibition counters the excitation of any neuron. This balance determines the impact of each neuron on the microcircuit, which is normally very small, but varies for different brain regions. For example, the higher neocortical regions such as the prefrontal cortex displays higher local connectivity in the normal case (Wang et al., 2006 ) which makes the prefrontal cortex normally more reactive than sensory areas allowing this region to more easily display sustained states of activity (Goldman-Rakic, 1995 ; Miller et al., 1996 ). The prefrontal cortex is also hyper-reactive in the VPA animal model (T. Rinaldi and H. Markram, unpublished data). It may, therefore, be possible that while the hierarchy of brain region development is normally inverse to how reactive the local circuits are set to become, this negative correlation switches to a positive one in the autistic brain upsetting the normal sequence of first developing rudimentary sensory and motor processing abilities. Since hyper-reactivity may be caused in a number of ways (hyper-connectivity, hyper-excitable excitatory neurons, hypo-excitable inhibitory neurons, upsetting the excitation-inhibitory balance, lowering long-range control pathways into microcircuits, etc.), each region may switch their developmental sequences differently depending on the predominant mechanisms setting the reactivity level. Furthermore, while the core outcome of the molecular syndrome of autism is proposed to cause hyper-reactivity and hyper-plasticity at the microcircuit level, the syndrome may also act at the system level to render the whole brain hyper-reactive and hyper-plastic and system level factors may also determine which brain areas develop more rapidly than others in autism.
Cellular Alterations
Postmortem neuropathology on autopsied autistic brains (Bailey et al., 1998 ; Coleman et al., 1985 ; Kemper and Bauman, 1998 ; Rodier et al., 1996 ; Williams et al., 1980 ) revealed alterations in neuronal anatomy within frontal (Bailey et al., 1998 ; Kemper and Bauman, 1998 ), temporal (Bailey et al., 1998 ), parietal (Bailey et al., 1998 ), limbic (Bailey et al., 1998 ; Kemper and Bauman, 1998 ; Raymond et al., 1996 ) and cerebellar (Bailey et al., 1998 ; Kemper and Bauman, 1998 ; Ritvo et al., 1986 ; Rodier et al., 1996 ) regions. Bailey et al. (1998) reported irregular laminar patterns in the frontal lobe, ectopic neurons in the white matter, thickened areas in the parietal, temporal, frontal, and cingulate lobes, and increased neuronal density and subplial gliosis in the right cerebral hemisphere in four out of six autistic subjects with low IQs. Kemper and Bauman (1998) investigated the brains of nine autistic subjects. In eight out of nine subjects they found abnormally small neurons and increased cell packing in the anterior cingulated gyrus, amygdala, hippocampus, subiculum, enthorinal cortex, mamillary body, and medial septum. Higher numbers of smaller neurons in the neocortex of humans may be part of the strategy of the autistic molecular syndrome to increase the number of target neurons for neurons to contact. Neurons in the CA1 and CA4 subregion of the hippocampus exhibited reduced complexity and less extensive dendritic arbors (Raymond et al., 1996 ). Interestingly, local connectivity in these hippocampal regions is normally extremely low perhaps consistent with the hypothesized positive correlation between local connectivity and regional brain development in autism.
The cerebellum is a powerful inhibitory brain region and one of the most consistent findings in autism is a reduced number of its’ principal inhibitory neurons – the Purkinje cells (Courchesne et al., 2005 ; Kemper and Bauman, 1998 ; Rodier et al., 1996 ; Ritvo et al., 1986 ). These changes in the cerebellum, may be part of the manifestations of the molecular syndrome driving hyper-reactivity at the level of brain regions – a systems wide manifestation.
Modern stereological counts of neuron number mostly confirm the above studies and reveal an excess number of neurons in the cerebrum and a decreased amount of neurons in the cerebellum (Courchesne et al., 2005 ). However, contrary to earlier studies, modern stereological counts on the autistic amygdala revealed fewer neurons overall and in particular in the lateral nucleus (Schumann and Amaral, 2006 ).
Further evidence for altered neuronal anatomy and circuitry stems from recent studies on minicolumnar arrangements in the neocortex (Casanova et al., 2002 ). The minicolumn is thought to be the smallest computational circuit in the brain (Mountcastle, 1997 ). It consists of a core line of vertically ascending pyramidal and inhibitory neurons, their connections and input/output axons. A minicolumn is 30–60 μm in diameter and contains around 120 neurons and is relatively consistent in size in different species and neocortical regions (with some exceptions). Minicolumns in nine autistic brains were abnormally narrow, both in the column core as well as in the neurophil, in the frontal and temporal lobes (Casanova et al., 2002 ). This suggests that the autistic brain exhibits an increased number of minicolumns, thus more processing units (Casanova et al., 2002 ). One should however ask, why minicolumns are not just larger to accommodate the excess number of neurons? Hyper-connectivity found in the animal model is restricted to the minicolumnar dimensions, which may not only render these minicolumns more tightly coupled internally, but may also facilitate the parcellation of neurons into more minicolumns.
Hyper-connectivity leading to hyper-reactivity could render minicolumns more independent from surrounding minicolumns and autonomous once activated, but more difficult to collectively coordinate activity across multiple minicolumns and in concert with the rest of the brain. The neurophil space around the mincolumn core in humans is also reduced (Casanova et al., 2002 ), which contains important types of interneurons and such a deficit in inhibition could further add to the autonomy of the minicolumns caused by the molecular syndrome driving hyper-reactivity.
Hyper-seretonemia
Many neurotransmitter systems have been studied in autism including serotonin (5-HT), dopamine, norepinephrine, acetylcholine, glutamate, gamma-aminobutyric acid, endogenous opiods, oxytocin, and cortisol (reviewed in Lam et al., 2006 ). The most studied neurotransmitter is 5-HT as it proves to be the best bio-chemical marker of autism so far. The behavioral effects of 5-HT are complex, as it regulates mood, eating, body temperature, arousal, and modulates pain sensitivity, sexual behavior, and hormone release. Serotonin blood levels are highly elevated in a significant number of autistic children (Anderson et al., 1987 ; Betancur et al., 2002 ; Cook et al., 1993 ; Schain and Freedman, 1961 ). Higher rates of autism also occur in children exposed in utero to drugs known to increase 5-HT levels such as cocaine (Kramer et al., 1994 ). Direct in vivo measurements using positron emission tomography (PET) demonstrated asymmetries of 5-HT synthesis in the frontal cortex, thalamus and cerebellum in autistic boys, but not in autistic girls or in normal siblings (Chugani, 2002 ; Chugani et al., 1997 ). Also, while 5-HT synthesis is usually high in young children and then gradually declines, the levels are persistently high in autistic children (Chugani, 2002 ; Chugani et al., 1999a ; Chugani et al., 1999b ). A number of studies have attempted to elucidate the causes of mean level elevation seen for platelet 5-HT in autism, but metabolism, catabolism and transport mechanisms for 5-HT do not seem to be affected in autism (Anderson et al., 1990 ; Anderson et al., 2002 ; Cook et al., 1988 ), which may suggest that the elevated 5-HT levels are more secondary in the disorder to excessive stimulation of synthesis and release.
The main problem with the 5-HT theory of autism as a primary cause of autism is that treatments which further increase 5-HT levels seem to improve some symptoms of autism, such as obsessionality and social relatedness (McBride et al., 1989 ), while depletion of tryptophan, a serotonin precursor, seems to exacerbate autistic symptoms such as flapping, banging and self-hitting, rocking and increase anxiety (McDougle et al., 1996 ).
5-HT is not only a neurotransmitter, but also regulates the development of target brain areas, such as the neocortex, hippocampus, and cerebellum. Depletion of serotonin results in a significant delay in maturation of the somatosensory cortex (Bennett-Clarke et al., 1994 ). In contrast, excessive serotonin during early development results in hyper-innervation and expansion of cortical architecture (Cases et al., 1996 ). Embryonic exposure to VPA in the rat was also found to dramatically increase the serotonin levels in the blood as well as the frontal cortex, hippocampus, and cerebellum (Narita et al., 2002 ; Tsujino et al., 2007 ). 5-HT excess may therefore participate in the molecular syndrome that drives the altered developmental patterns and perhaps also plays a role in driving hyper-reactivity and hyper-plasticity early in development.
Altered Brain Activity
Advances made with functional neuroimaging techniques have contributed greatly to the understanding of the autistic brain. The most pronounced being the reduced activity in higher order association cortices, the frontal and temporal regions, as well as in the cerebellum while activity in lower order sensory regions are normal or even slightly increased (Di Martino and Castellanos, 2003 ). These findings have been interpreted to reflect a lack of functionality, impaired long-range connections, and reduced top–down control of primary areas
Courchesne et al. (2005) summarized the functional neuroimaging data which indicates reduced activation of the frontal cortex in a theory of mind task (Castelli et al., 2002 ), in response to socially familiar faces (Pierce et al., 2004 ), in face recognition (Hubl et al., 2003 ), in a working memory task (Luna et al., 2002 ), in an embedded figures task (Ring et al., 1999 ), in visual spatial attention tasks (Belmonte and Yurgelun-Todd, 2003 ) and during sentence comprehension (Just et al., 2004 ; Muller et al., 1998 ). Additionally, EEG studies consistently found reduced or absent electrical responses from the frontal cortex in several auditory and visual attention and orienting tasks (Ciesielski et al., 1990 ; Courchesne et al., 1984 ; Townsend et al., 1999 ). Temporal lobe activation was also found to be reduced during processing of vocal sounds (Gervais et al., 2004 ), speech sounds (Boddaert et al., 2003 ; Muller et al., 1999 ), face recognition (Pierce et al., 2001 ; Pierce et al., 2004 ; Schultz et al., 2000 ), and theory of mind tasks (Castelli et al., 2002 ). Impaired amygdala activation was reported in tasks of face perception and evaluation of facial expressions (Baron-Cohen et al., 1999 ; Critchley et al., 2000 ; Pierce et al., 2001 ). In striking contrast to the hypo-activation of the frontal and temporal lobes is the normal or even hyper-activation of the occipital lobe in response to visual stimulation (Belmonte and Yurgelun-Todd, 2003 ; Hadjikhani et al., 2004 ; Hubl et al., 2003 ; Ring et al., 1999 ).
These imaging results seem to be consistent with cognitive theories (see following chapters) built around autistic people's apparent inability to empathize with other peoples feelings and thoughts (Frith and Happe, 1994 ), deficits in executive function (Russell, 1997 ), deficits in holistic (or Gestalt processing) with a simultaneous preference for details (Frith, 1989 ; Happe and Frith, 2006 ), and deficits in face perception and evaluation of social cues from facial expressions (Schultz, 2005 ).
It is, however, not trivial to interpret functional brain imaging results in the autistic brain since highly reactive and autonomous microcircuits may be difficult to activate in a coherent manner with normal stimuli and tasks. They may, therefore, be hypo-active, but hyper-reactive to a highly selected set of stimuli. Indeed, cerebellar responses can be reduced, normal and increased depending on the task. Reduced activation was reported in attention tasks (Allen and Courchesne, 2003 ), speech recognition and generation (Muller et al., 1998 ; Muller et al., 1999 ) and judgement of facial expressions and normal to increased activation was observed during motor tasks (Allen and Courchesne, 2003 ; Allen et al., 2004 ; Muller et al., 2001 ). Normal stimulus paradigms may, therefore, not be optimal for the stimulation of the autistic brain and reduced activity for normal stimulation may not be indicative of lower functionality. There is also the additional problem of potentially higher baseline levels of activation in the autistic brain, which may render the difference responses apparently lower.
Reduced Functional Connectivity
Functional connectivity studies have suggested reduced connectivity between occipital and frontal or temporal lobes (Castelli et al., 2002 ), superior temporal to inferior frontal lobes (Just et al., 2004 ), and parietal to frontal lobes (Horwitz et al., 1988 ). This has been interpreted as support for the reduced long-distance connectivity hypothesis of autism. While reduced long-range connectivity may go hand in hand with enhanced local connectivity, the later alone may also be sufficient to produce results that could be interpreted as reduced functional long-range connectivity since the microcircuits would be more difficult to coordinate and engage when they can easily become autonomously active.
Overall, functional imaging studies seem to suggest that higher order brain areas are not fully activated, are disconnected from lower order sensory areas and that lower order sensory areas may even be hyper-activated. We hypothesise that it is easier to apply the appropriate stimulation to observe the hyper-reactivity in lower areas while much more complex stimulation patterns would be required to observe the hyper-reactivity in higher brain areas. Nevertheless, the cognitive consequences are perhaps similar in that information from one area might not be easily integrated with information from another area, thus leaving the autistic person in a world of “bits and pieces” that may seem isolated and feel chaotic and confusing.
Theories of Autism Re-interpreted in the Light of the Intense World Syndrome
Weak Central Coherence Theory
Autistic individuals do display abnormally weak central coherence required to integrate sensory information in a holistic (Gestalt) manner (Frith, 1989 ; Happe and Frith, 2006 ) and display “piece meal perception” which seems to arise because they are easily trapped into processing spurious stimuli with extreme detail. These assumptions are summarized in the “weak central coherence theory of autism” (Frith, 1989 ; Happe and Frith, 2006 ) and have been supported by experimental tasks, in which weak central coherence would be expected to have a task advantage over integrative, Gestalt perception or tasks in which integrative information processing would give an advantage over detailistic feature processing. For example, autistic subjects tend to perform better than controls on the Wechsler Block Design task, which is due to a greater ability to segment the whole design into its component parts (Shah and Frith, 1993 ) and in the Embedded Figures Test (Shah and Frith, 1983 ). In contrast, in a homograph disambiguation task which specifically requires the processing of information in context, autistic individuals fail to use preceding sentence context to determine the correct pronunciation of the homographs (Happe and Frith, 1997 ).
Support for piece-meal perception comes also from fMRI studies on face perception (Pierce et al., 2001 ). In normal subjects, faces consistently activate the fusiform face area in the fusiform gyrus while in autistic subjects, these regions exhibit abnormally weak or no activation at all (Schultz et al., 2003 ). Interestingly, in autistic subjects the activation pattern evoked by faces is rather distributed over several cortical regions (e.g., frontal, primary visual, cerebellum, etc.) and is different from subject to subject, suggesting each autistic individual activates different distributed neural systems (Pierce et al., 2001 ), which may be due to different degrees of hyper-reactivity in affected regions. We propose that piece meal perception is primarily due to hyper-functionality of local microcircuits.
Executive Function Theory
Data supporting hypo-functionality of the frontal lobes and loss of top–down control has also supported the “executive function theory of autism” (Russell, 1997 ) which proposes that the strong repetitive routines and preference for sameness in autism is due to impairments in executive functions. The term “executive functions” encompass many kinds of mental operations which enable an individual to disengage from the immediate context in order to guide behavior based on mental models or future goals, a function which is highly dependent on the integrity of the prefrontal lobes. These interpretations are further supported by findings that patients with frontal lobe lesions also exhibit symptoms of perseveration and the inability to shift attention and autistic subjects do perform badly on tests of executive function, such as the classic Wisconsin card sorting test (Ozonoff et al., 1991 ; Rumsey and Hamburger, 1990 ; Sandson and Albert, 1984 ) or the Tower of Hanoi planning task (Ozonoff et al., 1991 ).
We propose that the frontal cortices become functionally impaired because of the hyper-functionality of the microcircuits, which excessively process and remember fragments of information, and further amplify the hyper-attention to the same details. The theory predicts that these regions are not hypo-functional and that presentation of stimuli or tasks that the autistic child prefers will produce elevated responses above controls. Furthermore, impairments of higher cognitive functions observed in autism (Minshew et al., 1997 ), which are currently either attributed to a hypo-functioning of higher order brain regions or disconnection phenomena, could also be due to a failure to control the autonomous hyper-functional microcircuits. Hyper-functionality is also consistent with the hypertrophy of these regions during development.
Mind Blindness and Emphasizing Deficit Theory
Autistic people are thought to be severely impaired in empathising with other people and “reading their mind”, which is captured in the “theory of mind” or “mind-blindness theory” of autism (Baron-Cohen et al., 1985 ; Frith and Happe, 1994 ). The theory involves two elements: (1) the ability to attribute mental states to oneself and others, to be able to distinguish between oneself and others and realize that others have independent minds and may pursue different goals from oneself; (2) the ability to express an appropriate emotional reaction to the other person's mental state, thus to be able to empathize with the others’ mind. The proposed deficits in reading other people's feelings and thoughts and the lack in emphasizing with other people has been commonly used to explain the impairments in social interactions and communication as well as inappropriate responses in social encounters and a few functional imaging studies suggest that these deficits may be mediated by a hypo-active amygdala (Baron-Cohen et al., 1999 ; Critchley et al., 2000 ; Pierce et al., 2001 ). Indeed the amygdala plays a pivotal role in modulating and regulating emotional responses (Adolphs, 2006 ; Davis and Whalen, 2001 ; LeDoux, 2003 ; McGaugh, 2004 ; Zald, 2003 ) and a malfunctioning in this particular brain region has been proposed to underlie the social deficits in autism (Amaral et al., 2003 ; Bachevalier and Loveland, 2006 ; Baron-Cohen et al., 2000 ; Schultz, 2005 ; Sweeten et al., 2002 ). Indeed we and other groups found social impairments after embryonic exposure to VPA in rats as well (Schneider et al., 2005, Markram et al., 2007 ). However, because we were able to use invasive techniques we observed a hyper-reactive and hyper-plastic amygdala, as expressed in terms of hyper-reactivity to electrical stimulation as well as amplified fear memories (Markram et al., 2007 ). Hyper-reactivity and hyper-plasticity were also found in the primary somatosensory cortex (Rinaldi et al., 2007a ).
We, therefore, propose that the autistic person may perceive its surroundings not only as overwhelming intense due to hyper-reactivity of primary sensory areas, but also as aversive and highly stressful due to a hyper-reactive amygdala, which also makes quick and powerful fear associations with usually neutral stimuli. The autistic person may well try to cope with the intense and aversive world by avoidance. Thus, impaired social interactions and withdrawal may not be the result of a lack of compassion, incapability to put oneself into some else's position or lack of emotionality, but quite to the contrary a result of an intensely if not painfully aversively perceived environment.
Since enhanced emotionality (along with enhanced perception) is a crucial point in the suggested Intense World Syndrome of autism we dedicate a separate chapter to the amygdala theory of autism below.
The Amygdala Theory of Autism
The amygdala has many functional roles such as detecting and interpreting signs of emotional and social significance in the environment, modulating memory storage across multiple brain sites, establishing fear memories, anxiety and the regulation of autonomic and hormonal responses (reviewed in Adolphs, 2006 ; Davis and Whalen, 2001 ; LeDoux, 2003 ; McGaugh, 2004 ; Zald, 2003 ). Dysfunction of the amygdala has been related to disorders of fear processing, anxiety, and social behaviors (reviewed in Blair et al., 2006 ; Cottraux, 2005 ; Damsa et al., 2005 ; Hajek et al., 2005 ; Shayegan and Stahl, 2005 ). The particular interest in the amygdala in autism research stems from the role it plays in the processing and interpretation of socio-emotional cues and the influence on social behaviors. The amygdala theory of autism postulates that a dysfunction in this particular brain region underlies the cardinal disturbances observed in autism, that is the difficulty in relating to others and incapability to from appropriate social interactions (Amaral et al., 2003 ; Bachevalier and Loveland, 2006 ; Baron-Cohen et al., 2000 ; Schultz, 2005 ; Sweeten et al., 2002 ). Support was provided from cellular and structural lines of investigation, which showed cellular alterations in the amygdala of autistic brains (Bauman and Kemper, 1985 ; Kemper and Bauman, 1998 ; Schumann and Amaral, 2006 ) as well as abnormal structural amygdaloid development in autism (Schumann et al., 2004 ; Sparks et al., 2002 ). We contrast the current version of the amygdala theory of autism, which implies a hypo-functioning of the amygdala, with our own version, which explicitly postulates a hyper-reactive and hyper-plastic amygdala in autism.
The “Hypo-functioning” Amygdala Theory of Autism
The current version of the amygdala theory of autism implies a hypo-functioning of the amygdala. This is a rather implicit assumption, born mostly by the methodologies used. Amygdala lesions in non-human primates, parallels between amygdala-damaged human patients and autistic patients, as well as a few neuroimaging studies have led to this “hypo-active” amygdala theory of autism (Amaral et al., 2003 ; Bachevalier and Loveland, 2006 ; Baron-Cohen et al., 2000 ; Schultz, 2005 ; Sweeten et al., 2002 ).
Support from lesions in non-human primates. Amygdala lesions in monkeys were the first, and for a long time the only animal model of autism available, and was based on observations that amygdala damage may lead to severe disturbances in social behavior (Bachevalier, 1994 ). The famous Kluver-Bucy syndrome is – among other symptoms – characterized by psychic blindness and emotional alterations, including changes or absence of anger and fear, lack of social behavior, and abnormal sexual behaviors and can be caused by bilateral damage to the amygdala (Aggleton and Passingham, 1981 ; Kluver and Bucy, 1937 ; Rosvold et al., 1954 ; Schreiner and Kling, 1956 ; Weiskrantz, 1956 ; Zola-Morgan et al., 1991 ) or the inferior temporal cortex (Horel et al., 1975 ). The cumulated results of a manifold of studies in monkeys indicated that amygdala lesions disrupt social hierarchies, species-specific social behaviors and lead to social isolation and passivity in social encounters (Bachevalier, 1994 ; Kling and Brothers, 1992 ). These early lesion studies and further neuropsychological data led Brothers to propose that the amygdala must be an essential part of “the social brain” in which the prefrontal (orbitofrontal cortex and cingulate gyrus) and temporal (inferotemporal and superior temporal sulcus) cortex are also involved (Brothers, 1990 ). However, more recently the role of the amygdala as an essential part of the “social brain” has been questioned (Amaral and Corbett, 2003 ; Amaral et al., 2003 ; Emery et al., 2001 ; Prather et al., 2001 ).
Support from comparisons between autism and amygdala damage in humans. Even though people with amygdala damage may not be autistic in the classical sense of the DMS-IV, they do exhibit a few striking similarities with autistic patients regarding face perception and evaluation.
In a series of experiments, brain-damaged, amygdala-lesioned, and control subjects were compared in terms of their ability to recognize emotions conveyed through facial expressions. Adolphs and colleagues found that patients with either bilateral or unilateral damage to the amygdala exhibited impaired recognition of fear and in some cases also negative emotions such as anger or disgust when compared to controls or other brain-damaged subjects. The recognition of happy emotions was not impaired (Adolphs and Tranel, 2003 ; Adolphs et al., 1994 ; Adolphs et al., 1999 ). These patients were also impaired when they had to judge the trustworthiness of faces or when they had to identify more complex social emotions and mental states from facial expressions or merely the eye region, such as arrogance, guilt, admiration or flirtatiousness (Adolphs et al., 1998 ; Adolphs et al., 2002 ). Furthermore, amygdala-lesioned patients exhibited severe deficits in attributing mental states to others particularly when they acquired the lesion early in life, but not during adulthood (Shaw et al., 2004 ). Amygdala lesions also produce a deficit in gazing at the eyes of another person and instructing the patient to look directly at the eyes could restore the deficits in interpreting the information conveyed by the eyes (Adolphs et al., 2005 ).
It was argued that in particular an early insult to the amygdala might have severe consequences for the subsequent development of social networks in the brain, therefore social development in general und thus lead to autism (Bachevalier and Loveland, 2006 ; Schultz, 2005 ). For example, proper face perception and correct interpretation of emotional and mental states from face expressions is crucial for the successful navigation through the social world and virtually all normal human beings are experts in effortlessly recognizing faces and interpreting facial expressions (Carey, 1992 ; Diamond and Carey, 1986 ). This face expertise could be due to an innate tendency to attend to emotionally significant stimuli mediated by the amygdala. Already newborns show a natural tendency to preferentially attend to faces over other stimuli (Goren et al., 1975 ; Simion et al., 1998 ). This innate preference for faces has been postulated to be mediated by a subcortical visual system that passes information from the retina to the superior colliculus, the pulvinar nucleus of the thalamus, and from there to the amygdala (Pasley et al., 2004 ). Schultz (2005) proposed that an insult to this system, and in particular to the amygdala alone, may profoundly interfere with socio-emotional development, because emotional significance might not be properly assigned. In fact, autistic infants display deviant eye gaze (resemblend to amygdala damage) and fail to attend to faces as early as in the first 6 months of life (Maestro et al., 2002 ). As a consequence they might never acquire normal face perception expertise.
Indeed, autistic people are selectively impaired in recognizing faces (Boucher and Lewis, 1992 ; Braverman et al., 1989 ; Davies et al., 1994 ; Hobson, 1986b ; Hobson et al., 1988a ; Klin et al., 1999 ; Langdell, 1978 ; ) and are also impaired in correctly recognizing facial expressions (Adolphs et al., 2001 ; Braverman et al., 1989 ; Hobson, 1986a ; Hobson et al., 1988b ,a; Howard et al., 2000 ; Tantam et al., 1989 ; Weeks and Hobson, 1987 ). Direct comparisons on the same tests revealed that a few autistic subjects had similar impairments to amygdala-damaged people when rating faces expressing fear, disgust, and surprise (Adolphs et al., 2001 ). Furthermore, autistic people were reported to have severe problems in interpreting more complex social information such as judging the trustworthiness of others or interpreting the mental states of other people conveyed through both, the whole face or only the eyes, a pattern resembling amygdala damage (Adolphs et al., 2001 ; Baron-Cohen et al., 1997 ).
The amygdala also seems to be important in detecting and attributing social meaning in a much broader range of stimuli than just faces. In a now classical study, Heider and Simmel (1944) demonstrated that normal subjects, when viewing animations that depict geometrical shapes on a plain, white background, spontaneously attribute social significance to the shapes (Heider and Simmel, 1944 ). In contrast, patients with amygdala lesions do not make such attributions, but describe the shapes in purely geometric terms (Heberlein and Adolphs, 2004 ) alike to autistic subjects who are also severely impaired in this task (Klin, 2000 ).
In summary, autistic subjects and amygdala-damaged patients do share some common features, most pronounced an impairment in correctly judging complex emotions and mental states from other peoples faces and in some cases also an impairment of recognizing simple negative emotions such as fear. Both patient groups have problems in correctly applying eye gaze to relevant stimuli (Adolphs et al., 2005 ; Howard et al., 2000 ). Finally, both patient groups have problems in performing spontaneous anthropomorphizations (Heberlein and Adolphs, 2004 ; Klin, 2000 ).
Support from imaging studies. Three important fMRI studies have evaluated the involvement of the amygdala in autism focussing on face perception and evaluation of facial expressions. All of these studies consistently reported a hypo-activation of the amygdala (Baron-Cohen et al., 1999 ; Critchley et al., 2000 ; Pierce et al., 2001 ). For example, in the study of Baron-Cohen et al. (1999) six subjects with autism were examined on a test of judging from the eye expression what another person might be feeling or thinking. While normal subjects showed increased activity in the prefrontal cortex, superior temporal temporal gyrus and amygdala on this task, autistic subjects did also activate fronto-temporal regions, but failed to activate the amygdala, thus giving rise to the term “amygdala theory of autism”, but basically meaning “hypo-active amygdala theory of autism”.
The “Hyper-functioning” Amygdala Theory of Autism
The current version of the amygdala theory of autism assumes a hypo-functional amydala, which leads to lack or inappropriateness of social behavior in autism. In this view, autists fail to assign emotional significance to their environment and for this reason are not interested in others, do not attend to faces, and fail to engage in normal social interaction. However, based on the result in the VPA model of autism and observations obtained in autistic humans, we propose that this view may be not correct and that quite to the contrary, the amygdala in the autistic individual may be hyper-reactive which leads to rapid excessive responses to socio-emotional stimuli. In this view, the autistic person would be overwhelmed with emotional significance and salience. As a consequence, the subject would want to avoid this emotional overload and would have to withdraw from situations, such as social encounters, which are rich in complex stimuli.
Support from the VPA animal model and humans. Our studies on the VPA model of autism indicate that the amygdala is hyper-reactive, hyper-plastic and mediates hyper-fear, excessive fear memory generalization, and resistance to fear extinction (Markram et al., 2007 ). The current theory is also inconsistent with high anxiety levels in autism as well as the early developmental hypertrophy of the amygdala. Increased anxiety and phobias were not only noted by Kanner in 1943, but also in more recent studies in children with ASD (Evans et al., 2005 ; Gillott et al., 2001 ; Muris et al., 1998 ) and their relatives (Micali et al., 2004 ). For example, Muris et al. (1998) investigated 44 children diagnosed with ASD and found 84.1% of the children met the criteria for at least one anxiety disorder, such as simple phobia (28%), social phobia (9%), agoraphobia (20%), panic disorder (4%), separation anxiety disorder (12%), avoidant disorder (8%), overanxious disorder (10%), and obsessivecompulsive disorder (5%). Rather than solely mediating the social deficits observed in autistic individuals, excessive and highly associate processing in the amygdala might be causal for the enhanced anxiety and fear so often reported in the autistic population, a view which has been brought forward recently (Amaral et al., 2003 ). Indeed enhanced levels of amygdala activation, possibly due to reduced inhibition, have been associated with increased anxiety and fear conditioning (Rodriguez Manzanares et al., 2005 ).
The hyper-fear hypothesis. We suggest that enhanced fear and anxiety levels, mediated by a hyper-reactive and hyper-plastic amygdala, might underlie a core symptom of autism-impaired social behavior. A person daunted by anxiety and fears will normally not tend to interact with other people and will not dare to explore new situations and environments in the way a normal person does. A recent study screening for autism-like symptoms in children with mood and anxiety disorders, found that up to 62% of these kids fall into the autistic spectrum and might qualify for a possible ASD diagnosis (Towbin et al., 2005 ). Thus, increased fear processing might cause some of the autistic impairments in social and non-social situations. In further support of this view is that decreased amygdala activation has been linked to genetic hyper-sociability (Meyer-Lindenberg et al., 2005 ), whereas increased activation is observed in social avoidance and phobia (Stein et al., 2002 ), which further contradicts the current (hypo-active) version of amygdala theory of autism. Moreover, autistic children exhibit increased autonomic responses, indicative of enhanced amygdala activity (Corbett et al., 2006 ; Hirstein et al., 2001 ; Tordjman et al., 1997 ). Interestingly, we do also observe enhanced levels of the stress hormone coricosterone in the VPA-exposed rat offspring in the blood stream (K. Markram, unpublished data), suggesting increased stress levels. Post-traumatic stress disorder, in which the amygdala is hyper-active and hyper-responsivee to traumatic triggers, is accompanied by enhanced amygdala volumes (Damsa et al., 2005 ). Indeed, structural imaging studies revealed an accelerated amygdala growth during early infancy (Schumann et al., 2004 ; Sparks et al., 2002 ), suggesting that the amygdala of autistic children reaches adult size before adolescence, whereas the amygdala of typically developing children undergoes a progressive growth throughout adolescence. A hyper-trophism in the amygdala is also supported by some (but not all) structural imaging studies in autism (Abell et al., 1999 ; Howard et al., 2000 ; Schumann et al., 2004 ; Sparks et al., 2002 ).
It is, therefore, possible that the autistic person may perceive its surroundings not only as overwhelmingly intense due to hyper-reactivity of primary sensory areas, but also as aversive and highly stressful due to an overly reactive amygdala, which also makes quick and powerful fear associations with usually neutral stimuli. The autistic person may well try to cope with the intense and aversive world by avoidance.
Conclusion
While many of the neuropathologies and symptoms observed in autism seem to resemble pieces of information in a scrambled puzzle, we proposed here a unifying hypothesis, which makes the first attempt to assemble the pieces into a coherent picture centered on excessive local circuit functionality. It has already been proposed that the autistic brain may be hyper-excitable due to excessive excitation or reduced inhibition (Hussman, 2001 ; Rubenstein and Merzenich, 2003 ), but our findings suggest that a more fundamental pathology is hype-reactivity which may be caused in a number of different ways. We reviewed here experimental findings obtained on the VPA rat model of autism that provide the first direct evidence for a unifying hypothesis. We propose that the core pathology of the autistic brain may be hyper-reactivity and hyper-plasticity of local neuronal circuits. On a perceptual and cognitive level, this excessive functioning of neuronal circuits may lead to an intensely perceived world, which may turn aversive if the amygdaloid complex is also affected. Many of the observed neuropathologies can be viewed as a consequence of hyper-reactive and hyper-plastic neural circuits, while many of the autistic symptoms may be re-interpreted in the light of an aversively intense world.
The Intense World Syndrome suggests that the autistic person is an individual with remarkable and far above average capabilities due to greatly enhanced perception, attention and memory. In fact it is this hyper-functionality, which could render the individual debilitated. This perspective of hyper-functionality offers new hope for pharmacological as well as behavioral treatments. For example, while most the commonly prescribed medication try to increase neuronal and cognitive functioning, we conclude that the autistic brain needs to be calmed down, learning needs to be slowed, and cognitive functions need to be diminished in order to re-instate proper functionality. In terms of behavioral treatments, the hyper-plasticity offers an immense scope for rehabilitation therapies that are based on excessive positive reward and comforting approaches and that avoid direct punishment, which may lead to a lockdown of behavioral routines. It may well turn out that successful treatments could expose truly capable and highly gifted individuals.
Conflict of Interest Statement
The research from this group that is cited here was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
The research from this group that is cited here was supported by the National Alliance for Autism Research and by a grant from the European Commission for the EUSynapse project.
References
Abell, F., Krams, M., Ashburner, J., Passingham, R., Friston, K., Frackowiak, R., Happe, F., Frith, C., and Frith, U. (1999). The neuroanatomy of autism: a voxel-based whole brain analysis of structural scans. Neuroreport 10, 1647-1651.
Adolphs, R. (2006). How do we know the minds of others? Domain-specificity, simulation, and enactive social cognition. Brain Res. 1079, 25-35.
Adolphs, R., and Tranel, D. (2003). Amygdala damage impairs emotion recognition from scenes only when they contain facial expressions. Neuropsychologia 41, 1281-1289.
Adolphs, R., Tranel, D., and Damasio, A. R. (1998). The human amygdala in social judgment. Nature 393, 470-474.
Adolphs, R., Sears, L., and Piven, J. (2001). Abnormal processing of social information from faces in autism. J. Cogn. Neurosci. 13, 232-240.
Adolphs, R., Baron-Cohen, S., and Tranel, D. (2002). Impaired recognition of social emotions following amygdala damage. J. Cogn. Neurosci. 14, 1264-1274.
Adolphs, R., Tranel, D., Damasio, H., and Damasio, A. (1994). Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature 372, 669-672.
Adolphs, R., Gosselin, F., Buchanan, T. W., Tranel, D., Schyns, P., and Damasio, A. R. (2005). A mechanism for impaired fear recognition after amygdala damage. Nature 433, 68-72.
Adolphs, R., Tranel, D., Hamann, S., Young, A.W., Calder, A.J., Phelps, E. A., Anderson, A., Lee, G.P., and Damasio, A. R. (1999). Recognition of facial emotion in nine individuals with bilateral amygdala damage. Neuropsychologia 37, 1111-1117.
Aggleton, J.P., and Passingham, R.E. (1981). Syndrome produced by lesions of the amygdala in monkeys (Macaca mulatta). J. Comp. Physiol. Psychol. 95, 961-977.
Allen, G., and Courchesne, E. (2003). Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: an fMRI study of autism. Am. J. Psychiatry 160, 262-273.
Allen, G., Muller, R.A., and Courchesne, E. (2004). Cerebellar function in autism: functional magnetic resonance image activation during a simple motor task. Biol. Psychiatry 56, 269-278.
Altman, J., and Bayer, S.A. (1980). Development of the brain stem in the rat. I. Thymidine-radiographic study of the time of origin of neurons of the lower medulla. J. Comp. Neurol. 194, 1-35.
Amaral, D. G., and Corbett, B. A. (2003). The amygdala, autism and anxiety. Novartis Found. Symp. 251, 177-187; discussion 187–197, 281–197.
Amaral, D.G., Bauman, M.D., and Schumann, C.M. (2003). The amygdala and autism: implications from non-human primate studies. Genes Brain Behav. 2, 295-302.
Anderson, G.M., Horne, W.C., Chatterjee, D., and Cohen, D. J. (1990). The hyperserotonemia of autism. Ann. N.Y. Acad. Sci. 600, 331-340; discussion 341-332.
Anderson, G. M., Gutknecht, L., Cohen, D. J., Brailly-Tabard, S., Cohen, J.H., Ferrari, P., Roubertoux, P. L., and Tordjman, S. (2002). Serotonin transporter promoter variants in autism: functional effects and relationship to platelet hyperserotonemia. Mol. Psychiatry 7, 831-836.
Anderson, G. M., Freedman, D. X., Cohen, D. J., Volkmar, F. R., Hoder, E. L., McPhedran, P., Minderaa, R.B., Hansen, C. R., and Young, J. G. (1987). Whole blood serotonin in autistic and normal subjects. J. Child Psychol. Psychiatry 28, 885-900.
Ardinger, H. H., Atkin, J. F., Blackston, R. D., Elsas, L. J., Clarren, S. K., Livingstone, S., Flannery, D. B., Pellock, J. M., Harrod, M. J., Lammer, E. J., and et al. (1988). Verification of the fetal valproate syndrome phenotype. Am. J. Med. Genet. 29: 171-185.
Arndt, T. L., Stodgell, C. J., and Rodier, P.M. (2005). The teratology of autism. Int. J. Dev. Neurosci. 23, 189-199.
Aronson, M., Hagberg, B., and Gillberg, C. (1997). Attention deficits and autistic spectrum problems in children exposed to alcohol during gestation: a follow-up study. Dev. Med. Child Neurol. 39: 583-587./a>
Asperger, H. (1944). Die “Autistischen Psyhophaten” im Kindesalter. Arch. Psychiatr. Nervenkr. 117, 76-136.
Bachevalier, J. (1994). Medial temporal lobe structures and autism: a review of clinical and experimental findings. Neuropsychologia 32, 627-648.
Bachevalier, J., and Loveland, K. A. (2006). The orbitofrontal-amygdala circuit and self-regulation of social-emotional behavior in autism. Neurosci. Biobehav. Rev. 30: 97-117.
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., and Rutter, M. (1995). Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 25, 63-77.
Bailey, A., Luthert, P., Dean, A., Harding, B., Janota, I., Montgomery, M., Rutter, M., and Lantos, P. (1998). A clinicopathological study of autism. Brain 121, (Pt 5), 889-905.
Bandim, J. M., Ventura, L. O., Miller, M. T., Almeida, H. C., and Costa, A. E. (2003). Autism and Mobius sequence: an exploratory study of children in northeastern Brazil. Arq. Neuropsiquiatr. 61, 181-185.
Barad, M. (2006). Is extinction of fear erasure or inhibition? Why both, of course. Learn. Mem. 13, 108-109.
Baron-Cohen, S., Leslie, A. M., and Frith, U. (1985). Does the autistic child have a “theory of mind”? Cognition 21, 37-46.
Baron-Cohen, S., Jolliffe, T., Mortimore, C., and Robertson, M. (1997). Another advanced test of theory of mind: evidence from very high functioning adults with autism or asperger syndrome. J. Child Psychol. Psychiatry 38, 813-822.
Baron-Cohen, S., Ring, H. A., Bullmore, E. T., Wheelwright, S., Ashwin, C., and Williams, S. C. (2000). The amygdala theory of autism. Neurosci. Biobehav. Rev. 24, 355-364.
Baron-Cohen, S., Ring, H.A., Wheelwright, S., Bullmore, E.T., Brammer, M.J., Simmons, A., and Williams, S.C. (1999). Social intelligence in the normal and autistic brain: an fMRI study. Eur. J. Neurosci. 11, 1891-1898.
Bauman, M., and Kemper, T. L. (1985). Histoanatomic observations of the brain in early infantile autism. Neurology 35, 866-874.
Beaudet, A., and Zoghbi, H. Y. (2006). A mixed epigenetic and genetic and mixed de novo and inherited model for autism. In Understandin Autism, S. O., Moldin, and J. L., Rubenstein, eds. (Boca Raton: Taylor & Francis Group, LCC), PP. 95-111.
Belmonte, M. K., and Yurgelun-Todd, D. A. (2003). Functional anatomy of impaired selective attention and compensatory processing in autism. Brain Res. Cogn. Brain Res. 17, 651-664.
Bennett-Clarke, C. A., Leslie, M. J., Lane, R. D., and Rhoades, R. W. (1994). Effect of serotonin depletion on vibrissa-related patterns of thalamic afferents in the rat's somatosensory cortex. J. Neurosci. 14, 7594-7607.
Bernier, R., Dawson, G., Panagiotides, H., and Webb, S. (2005). Individuals with autism spectrum disorder show normal responses to a fear potential startle paradigm. J. Autism Dev. Disord. 35, 575-583.
Betancur, C., Corbex, M., Spielewoy, C., Philippe, A., Laplanche, J. L., Launay, J. M., Gillberg, C., Mouren-Simeoni, MC, Hamon, M, Giros, B, Nosten-Bertrand, M., and Leboyer, M. (2002). Serotonin transporter gene polymorphisms and hyperserotonemia in autistic disorder. Mol. Psychiatry 7, 67-71.
Bettelheim, B. (1967). The empty fortress: infantile autism and the birth of the self. (New York: Free Press).
Binkerd, P. E., Rowland, J. M., Nau, H., and Hendrickx, A. G. (1988). Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague-Dawley rats. Fundam. Appl. Toxicol. 11, 485-493.
Bissiere, S., Humeau, Y., and Luthi, A. (2003). Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 6, 587-592.
Blair, R. J., Peschardt, K. S., Budhani, S., Mitchell, D.G., and Pine, D.S. (2006). The development of psychopathy. J. Child Psychol. Psychiatry 47, 262-276.
Boddaert, N., Belin, P., Chabane, N., Poline, J. B., Barthelemy, C., Mouren-Simeoni, M. C., Brunelle, F., Samson, Y., and Zilbovicius, M. (2003). Perception of complex sounds: abnormal pattern of cortical activation in autism. Am. J. Psychiatry 160, 2057-2060.
Bonora, E., Lamb, J. A., Barnby, G., Bailey, A. J., and Monaco, A. P. (2006). Genetic Basis of Autism. In: Understanding Autism, S. O. Moldin, J. L., Rubenstein eds., (Boca Raton, CRC Press, Taylor and Francis Group), PP. 49-74.
Boucher, J., and Lewis, V. (1992). Unfamiliar face recognition in relatively able autistic children. J. Child Psychol. Psychiatry 33, 843-859.
Braverman, M., Fein, D., Lucci, D., and Waterhouse, L. (1989). Affect comprehension in children with pervasive developmental disorders. J. Autism Dev. Disord. 19, 301-316.
Brothers, L. (1990). The social brain: a project for integrating primate behaviour and neurophysiology in a new domain. Concepts Neurosci. 1, 27-51.
Carey, S. (1992). Becoming a face expert. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 335, 95-102; discussion 102-103.
Carper, R. A., Moses, P., Tigue, Z. D., and Courchesne, E. (2002). Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage 16, 1038-1051.
Casanova, M. F., Buxhoeveden, D. P., Switala, A. E., and Roy, E. (2002). Minicolumnar pathology in autism. Neurology 58, 428-432.
Cases, O., Vitalis, T., Seif, I., De Maeyer, E., Sotelo, C., and Gaspar, P. (1996). Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron 16, 297-307.
Castelli, F., Frith, C., Happe, F., and Frith, U. (2002). Autism, Asperger syndrome and brain mechanisms for the attribution of mental states to animated shapes. Brain 125, 1839-1849.
Chess, S. (1977). Follow-up report on autism in congenital rubella. J. Autism Child Schizophr. 7, 69-81.
Chess, S., and Fernandez, P. (1980). Neurologic damage and behavior disorder in rubella children. Am. Ann. Deaf. 125: 998-1001.
Christianson, A. L., Chesler, N., and Kromberg, J. G. (1994). Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev. Med. Child Neurol. 36, 361-369.
Chugani, D. C. (2002). Role of altered brain serotonin mechanisms in autism. Mol. Psychiatry 7, Suppl 2, S16-S17.
Chugani, D. C., Sundram, B. S., Behen, M., Lee, M.L., and Moore, G. J. (1999a). Evidence of altered energy metabolism in autistic children. Prog. Neuropsychopharmacol. Biol. Psychiatry 23, 635-641.
Chugani, D. C., Muzik, O., Behen, M., Rothermel, R., Janisse, J. J., Lee, J., and Chugani, H. T. (1999b). Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann. Neurol. 45, 287-295.
Chugani, D. C., Muzik, O., Rothermel, R., Behen, M., Chakraborty, P., Mangner, T., da Silva, E. A., and Chugani, H. T. (1997). Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann. Neurol. 42, 666-669.
Ciesielski, K. T., Courchesne, E., and Elmasian, R. (1990). Effects of focused selective attention tasks on event-related potentials in autistic and normal individuals. Electroencephalogr. Clin. Neurophysiol. 75, 207-220.
Coldren, J. T., and Halloran, C. (2003). Spatial reversal as a measure of executive functioning in children with autism. J. Genet. Psychol. 164, 29-41.
Coleman, P. D., Romano, J., Lapham, L., and Simon, W. (1985). Cell counts in cerebral cortex of an autistic patient. J. Autism Dev. Disord. 15, 245-255.
Collins, M. D., Walling, K. M., Resnick, E., Scott, W. J., Jr., and (1991). The effect of administration time on malformations induced by three anticonvulsant agents in C57BL/6J mice with emphasis on forelimb ectrodactyly. Teratology 44, 617-627.
Cook, E. H., Jr., Leventhal, B. L., and Freedman, D. X. (1988). Free serotonin in plasma: autistic children and their first-degree relatives. Biol. Psychiatry 24, 488-491.
Cook, E. H., Jr., Arora, R. C., Anderson, G. M., Berry-Kravis, E. M., Yan, S. Y., Yeoh, H. C., Sklena, P. J., Charak, D. A., and Leventhal, B. L. (1993). Platelet serotonin studies in hyperserotonemic relatives of children with autistic disorder. Life Sci. 52, 2005-2015.
Corbett, B. A., Mendoza, S., Abdullah, M., Wegelin, J. A., and Levine, S. (2006). Cortisol circadian rhythms and response to stress in children with autism. Psychoneuroendocrinology 31, 59-68.
Cottraux, J. (2005). Recent developments in research and treatment for social phobia (social anxiety disorder). Curr. Opin. Psychiatry 18, 51-54.
Courchesne, E. (2004). Brain development in autism: early overgrowth followed by premature arrest of growth. Ment. Retard. Dev. Disabil. Res. Rev. 10, 106-111.
Courchesne, E., Press, G.A., and Yeung-Courchesne, R. (1993). Parietal lobe abnormalities detected with MR in patients with infantile autism. AJR Am. J. Roentgenol. 160, 387-393.
Courchesne, E., Carper, R., and Akshoomoff, N. (2003). Evidence of brain overgrowth in the first year of life in autism. JAMA 290, 337-344.
Courchesne, E., Kilman, B. A., Galambos, R., and Lincoln, A. J. (1984). Autism: processing of novel auditory information assessed by event-related brain potentials. Electroencephalogr. Clin. Neurophysiol. 59, 238-248.
Courchesne, E., Redcay, E., Morgan, J. T., and Kennedy, D. P. (2005). Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev. Psychopathol. 17, 577-597.
Courchesne, E., Karns, C. M., Davis, H. R., Ziccardi, R., Carper, R. A., Tigue, Z. D., Chisum, H. J., Moses, P., Pierce, K., Lord, C., Lincoln, A. J., Pizzo, S., Schreibman, L., Haas, R. H., Akshoomoff, N. A., and Courchesne, R. Y. (2001). Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology 57, 245-254.
Crawley, J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment. Retard. Dev. Disabil. Res. Rev. 10, 248-258.
Critchley, H. D., Daly, E. M., Bullmore, E. T., Williams, S. C., Van Amelsvoort, T., Robertson, D. M., Rowe, A., Phillips, M., McAlonan, G., Howlin, P., and Murphy, D. G. (2000). The functional neuroanatomy of social behaviour: changes in cerebral blood flow when people with autistic disorder process facial expressions. Brain 123, (Pt 11), 2203-2212.
Damsa, C., Maris, S., and Pull, C. B. (2005). New fields of research in posttraumatic stress disorder: brain imaging. Curr. Opin. Psychiatry 18, 55-64.
Davies, S., Bishop, D., Manstead, A. S., and Tantam, D. (1994). Face perception in children with autism and Asperger's syndrome. J. Child. Psychol. Psychiatry 35, 1033-1057.
Dementieva, Y. A., Vance, D. D., Donnelly, S. L., Elston, L. A., Wolpert, C. M., Ravan, S. A., DeLong, G. R., Abramson, R. K., Wright, H. H., and Cuccaro, M. L. (2005). Accelerated head growth in early development of individuals with autism. Pediatr. Neurol. 32, 102-108.
Di Martino, A., and Castellanos, F. X. (2003). Functional neuroimaging of social cognition in pervasive developmental disorders: a brief review. Ann. N. Y. Acad. Sci. 1008, 256-260.
Diamond, R., and Carey, S. (1994). (1986). Why faces are and are not special: an effect of expertise. J. Exp. Psychol. Gen. 115, 107-117.
Dziobek, I., Fleck, S., Rogers, K., Wolf, O. T., and Convit, A. (2006). The ’amygdala theory of autism’ revisited: Linking structure to behavior. Neuropsychologia.
Ehlers, K., Sturje, H., Merker, H. J., and Nau, H. (1992). Spina bifida aperta induced by valproic acid and by all-trans-retinoic acid in the mouse: distinct differences in morphology and periods of sensitivity. Teratology 46, 117-130.
Emery, N. J., Capitanio, J. P., Mason., W. A., Machado, C. J., Mendoza, S. P., and Amaral, D.G. (2001). The effects of bilateral lesions of the amygdala on dyadic social interactions in rhesus monkeys (Macaca mulatta). Behav. Neurosci. 115: 515-544.
Evans, D. W., Canavera, K., Kleinpeter, F. L., Maccubbin, E., and Taga, K. (2005). The fears, phobias and anxieties of children with autism spectrum disorders and down syndrome: comparisons with developmentally and chronologically age matched children. Child Psychiatry Hum. Dev. 36: 3-26.
Fombonne, E. (2006). Past and Future Perspectives on Autism Epidemiology. In Understanding Autism: From Basic Neuroscience to Treatment, S. O. Moldin, and J. R. L. Rubenstein, eds., (Boca Raton, CRC Press, Taylor & Francis Group), PP. 25-48.
Gaigg, S. B., and Bowler, D. M. (2007). Differential fear conditioning in Asperger's syndrome: implications for an amygdala theory of autism. Neuropsychologia 45: 2125-2134.
Gervais, H., Belin, P., Boddaert, N., Leboyer, M., Coez, A., Sfaello, I., Barthelemy, C., Brunelle, F., Samson, Y., and Zilbovicius, M. (2004). Abnormal cortical voice processing in autism. Nat. Neurosci. 7: 801-802.
Gillberg, C., and Steffenburg, S. (1989). Autistic behaviour in Moebius syndrome. Acta Paediatr. Scand. 78: 314-316.
Gillberg, C., and Coleman, M. (1996). Autism and medical disorders: a review of the literature. Dev. Med. Child Neurol. 38: 191-202.
Gillberg, C., and de Souza, L. (2002). Head circumference in autism, Asperger syndrome, and ADHD: a comparative study. Dev. Med. Child Neurol. 44: 296-300.
Gillott, A., Furniss, F., and Walter, A. (2001). Anxiety in high-functioning children with autism. Autism 5: 277-286.
Goren, C. C., Sarty, M., and Wu, P. Y. (1975). Visual following and pattern discrimination of face-like stimuli by newborn infants. Pediatrics 56: 544-549.
Hadjikhani, N., Chabris, C. F., Joseph, R. M., Clark, J., McGrath, L., Aharon, I., Feczko, E., Tager-Flusberg, H., and Harris, G. J. (2004). Early visual cortex organization in autism: an fMRI study. Neuroreport 15: 267-270.
Hajek, T., Carrey, N., and Alda, M. (2005). Neuroanatomical abnormalities as risk factors for bipolar disorder. Bipolar Disord. 7: 393-403.
Hallmayer, J., Glasson, E. J., Bower, C., Petterson, B., Croen, L., Grether, J., and Risch, N. (2002). On the twin risk in autism. Am. J. Hum. Genet 71: 941-946.
Happe, F., and Frith, U. (1997). Central Coherence and Theory of Mind in Autism: Reading Homographs in Contex. British Journal of Developmental Psychology 15: 1-12.
Happe, F., and Frith, U. (2006). The Weak Coherence Account: Detail-focused Cognitive Style in Autism Spectrum Disorders. J. Autism Dev. Disord. 36: 5-25.
Harris, S. R., MacKay, L. L., and Osborn, J. A. (1995). Autistic behaviors in offspring of mothers abusing alcohol and other drugs: a series of case reports. Alcohol. Clin. Exp. Res. 19: 660-665.
Hayashi, M. L., Rao, B. S., Seo, J. S., Choi, H. S., Dolan, B. M., Choi, S. Y., Chattarji, S., and Tonegawa, S. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. USA 104: 11489-11494.
Haznedar, M. M., Buchsbaum, M. S., Wei, T. C., Hof, P. R., Cartwright, C., Bienstock, C. A., and Hollander, E. (2000). Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am. J. Psychiatry 157: 1994-2001.
Heberlein, A. S., and Adolphs, R. (2004). Impaired spontaneous anthropomorphizing despite intact perception and social knowledge. Proc. Natl. Acad. Sci. USA 101: 7487-7491.
Heider, F., and Simmel, M. (1944). An Experimental Study of Apparant Behavior. Am. J. of Psychol. 57: 243-259.
Hirstein, W., Iversen, P., and Ramachandran, V.S. (2001). Autonomic responses of autistic children to people and objects. Proc. Biol. Sci. 268: 1883-1888.
Hobson, R. P. (1986a). The autistic child's appraisal of expressions of emotion: a further study. J. Child Psychol. Psychiatry 27: 671-680.
Hobson, R. P. (1986b). The autistic child's appraisal of expressions of emotion. J. Child Psychol. Psychiatry 27: 321-342.
Hobson, R. P., Ouston, J., and Lee, A. (1988a). What's in a face? The case of autism. Br. J. Psychol. 79, (Pt 4), 441-453.
Hobson, R. P., Ouston, J., and Lee, A. (1988b). Emotion recognition in autism: coordinating faces and voices. Psychol. Med. 18: 911-923.
Horel, J. A., Keating, E. G., and Misantone, L. J. (1975). Partial Kluver-Bucy syndrome produced by destroying temporal neocortex or amygdala. Brain Res. 94: 347-359.
Horwitz, B., Rumsey, J. M., Grady, C. L., and Rapoport, S. I. (1988). The cerebral metabolic landscape in autism. Intercorrelations of regional glucose utilization. Arch. Neurol. 45: 749-755.
Howard, M. A., Cowell, P. E., Boucher, J., Broks, P., Mayes, A., Farrant, A., and Roberts, N. (2000). Convergent neuroanatomical and behavioural evidence of an amygdala hypothesis of autism. Neuroreport 11: 2931-2935.
Hubl, D., Bolte, S., Feineis-Matthews, S., Lanfermann, H., Federspiel, A., Strik, W., Poustka, F., and Dierks, T. (2003). Functional imbalance of visual pathways indicates alternative face processing strategies in autism. Neurology 61: 1232-1237.
Hussman, J. P. (2001). Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J. Autism Dev. Disord. 31: 247-248.
Ingram, J. L., Peckham, S. M., Tisdale, B., and Rodier, P. M. (2000). Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol. 22: 319-324.
Isoardi, N. A., Martijena, I. D., Carrer, H. F., and Molina, V. A. (2004). Increased fear learning coincides with neuronal dysinhibition and facilitated LTP in the basolateral amygdala following benzodiazepine withdrawal in rats. Neuropsychopharmacology 29: 1852-1864.
Just, M. A., Cherkassky, V. L., Keller, T. A., and Minshew, N. J. (2004). Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain 127: 1811-1821.
Kemper, T. L., and Bauman, M. (1998). Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol. 57: 645-652.
Klin, A. (2000). Attributing social meaning to ambiguous visual stimuli in higher-functioning autism and Asperger syndrome: The Social Attribution Task. J. Child Psychol. Psychiatry 41: 831-846.
Klin, A., Sparrow, S. S., de Bildt, A., Cicchetti, D. V., Cohen, D. J., and Volkmar, F. R. (1999). A normed study of face recognition in autism and related disorders. J. Autism Dev. Disord. 29: 499-508.
Kling, A., and Brothers, L. (1992). The amygdala and social behavior. In J. P. Aggleton, ed. Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction (New York, Wiley).
Kluver, H., and Bucy, P. (1937). “Psychic Blindness” ad other symptoms following bilateral temporal lobectomy in rhesus monkeys. Am. J. Physiol. 119: 352-353.
Koch, S., Jager-Roman, E., Losche, G., Nau, H., Rating, D., and Helge, H. (1996). Antiepileptic drug treatment in pregnancy: drug side effects in the neonate and neurological outcome. Acta Paediatr. 85: 739-746.
Kramer, K., Azmitia, E. C., and Whitaker-Azmitia, P. M. (1994). In vitro release of [3H5-hydroxytryptamine from fetal and maternal brain by drugs of abuse. Brain Res. Dev. Brain Res. 78: 142-146.
Lainhart, J. E., Piven, J., Wzorek, M., Landa, R., Santangelo, S. L., Coon, H., and Folstein, S. E. (1997). Macrocephaly in children and adults with autism. J. Am. Acad. Child Adolesc. Psychiatry 36: 282-290.
Lam, K. S., Aman, M. G., and Arnold, L. E. (2006). Neurochemical correlates of autistic disorder: A review of the literature. Res Dev Disabil. 27: 254-289
Lamb, J. A., Moore, J., Bailey, A., and Monaco, A. P. (2000). Autism: recent molecular genetic advances. Hum. Mol. Genet. 9: 861-868.
Langdell, T. (1978). Recognition of faces: an approach to the study of autism. J. Child Psychol. Psychiatry 19: 255-268.
Le Couteur, A., Bailey, A., Goode, S., Pickles, A., Robertson, S., Gottesman, I., and Rutter, M. (1996). A broader phenotype of autism: the clinical spectrum in twins. J. Child Psycho.l Psychiatry 37: 785-801.
Lord, C., and Spence, S. (2006). Autism sepctrum disorder: phenotype and diagnosis. In Understanding Autism: From Basic Neuroscience to Treatment, S.O. Moldin, J. L. Rubenstein, eds., (Boca Raton, CRC Press, Taylor & Francis Group) PP. 1-23.
Luna, B, Minshew, N. J., Garver, K. E., Lazar, N. A., Thulborn, K. R., Eddy, W. F., and Sweeney, J. A. (2002). Neocortical system abnormalities in autism: an fMRI study of spatial working memory. Neurology 59: 834-840.
Maestro, S., Muratori, F., Cavallaro, M. C., Pei, F., Stern, D., Golse, B., and Palacio-Espasa, F. (2002). Attentional skills during the first 6 months of age in autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 41: 1239-1245.
Markram, K., Rinaldi, T., Mendola, D. L., Sandi, C., and Markram, H. (2007). Abnormal Fear Conditioning and Amygdala Processing in an Animal Model of Autism. Neuropsychopharmacology. doi:10.1038/sj.npp.1301453.
McAlonan, G. M., Daly, E., Kumari, V., Critchley, H. D., van Amelsvoort, T., Suckling, J., Simmons, A., Sigmundsson, T., Greenwood, K., Russell, A., Schmitz, N., Happe, F., Howlin, P., and Murphy, D. G. (2002). Brain anatomy and sensorimotor gating in Asperger's syndrome. Brain 125: 1594-1606.
McBride, P. A., Anderson, G. M., Hertzig, M. E., Sweeney, J. A., Kream, J., Cohen, D. J., and Mann, J. J. (1989). Serotonergic responsivity in male young adults with autistic disorder. Results of a pilot study. Arch. Gen. Psychiatry 46: 213-221.
McDougle, C. J., Naylor, S. T., Cohen, D. J., Aghajanian, G. K., Heninger, G. R., and Price, L. H. (1996). Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 53: 993-1000.
McGaugh, J. L. (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 27: 1-28.
Meyer-Lindenberg, A., Hariri, A. R., Munoz, K. E., Mervis, C. B., Mattay, V. S., Morris, C. A., and Berman, K. F. (2005). Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat. Neurosci. 8: 991-993.
Micali, N., Chakrabarti, S., and Fombonne, E. (2004). The broad autism phenotype: findings from an epidemiological survey. Autism 8: 21-37.
Miller, E. K., Erickson, C. A., and Desimone, R. (1996). Neural mechanisms of visual working memory in prefrontal cortex of the macaque. J. Neurosci. 16: 5154-5167.
Minshew, N. J., Goldstein, G., and Siegel, D. J. (1997). Neuropsychologic functioning in autism: profile of a complex information processing disorder. J. Int. Neuropsychol. Soc. 3: 303-316.
Miyazaki, K., Narita, N., and Narita, M. (2005). Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int. J. Dev. Neurosci. 23: 287-297.
Moore, S. J., Turnpenny, P., Quinn, A., Glover, S., Lloyd, D. J., Montgomery, T., and Dean, J. C. (2000). A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 37: 489-497.
Muller, R. A., Pierce, K., Ambrose, J. B., Allen, G., and Courchesne, E. (2001). Atypical patterns of cerebral motor activation in autism: a functional magnetic resonance study. Biol. Psychiatry 49: 665-676.
Muller, R. A., Chugani, D. C., Behen, M. E., Rothermel, R. D., Muzik, O., Chakraborty, P. K., and Chugani, H. T. (1998). Impairment of dentato-thalamo-cortical pathway in autistic men: language activation data from positron emission tomography. Neurosci. Lett. 245: 1-4.
Muller, R. A., Behen, M. E., Rothermel, R. D., Chugani, D. C., Muzik, O., Mangner, T. J., and Chugani, H. T. (1999). Brain mapping of language and auditory perception in high-functioning autistic adults: a PET study. J. Autism Dev. Disord. 29: 19-31.
Mullins, M., and Rincover, A. (1985). Comparing autistic and normal children along the dimensions of reinforcement maximization, stimulus sampling, and responsiveness to extinction. J. Exp. Child Psychol. 40: 350-374.
Muris, P., Steerneman, P., Merckelbach, H., Holdrinet, I., and Meesters, C. (1998). Comorbid anxiety symptoms in children with pervasive developmental disorders. J. Anxiety Disord. 12: 387-393.
Nanson, J. L. (1992). Autism in fetal alcohol syndrome: a report of six cases. Alcohol. Clin. Exp. Res. 16: 558-565.
Narita, N., Kato, M., Tazoe, M., Miyazaki, K., Narita, M., and Okado, N. (2002). Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr. Res. 52: 576-579.
Nowell, M. A., Hackney, D. B., Muraki, A. S., and Coleman, M. (1990). Varied MR appearance of autism: fifty-three pediatric patients having the full autistic syndrome. Magn. Reson. Imaging 8: 811-816.
Ozonoff, S., Pennington, B. F., and Rogers, S. J. (1991). Executive function deficits in high-functioning autistic individuals: relationship to theory of mind. J. Child Psychol. Psychiatry 32: 1081-1105.
Palmen, S. J., van Engeland, H., Hof, P. R., and Schmitz, C. (2004). Neuropathological findings in autism. Brain 127: 2572-2583.
Palmen, S. J., Durston, S., Nederveen, H., and Vaneh, H. (2006). No evidence for preferential involvement of medial temporal lobe structures in high-functioning autism. Psychol. Med. 36: 827-834.
Pasley, B. N., Mayes, L. C., and Schultz, R. T. (2004). Subcortical discrimination of unperceived objects during binocular rivalry. Neuron 42: 163-172.
Perry, W., Minassian, A., Lopez, B., Maron, L., and Lincoln, A. (2007). Sensorimotor gating deficits in adults with autism. Biol. Psychiatry 61: 482-486.
Persico, A. M., and Bourgeron, T. (2006). Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 29: 349-358.
Pierce, K., Haist, F., Sedaghat, F., and Courchesne, E. (2004). The brain response to personally familiar faces in autism: findings of fusiform activity and beyond. Brain 127: 2703-2716.
Pierce, K., Muller, R. A., Ambrose, J., Allen, G., and Courchesne, E. (2001). Face processing occurs outside the fusiform ’face area’ in autism: evidence from functional MRI. Brain 124: 2059-2073.
Prather, M. D., Lavenex, P., Mauldin-Jourdain, M. L., Mason, W. A., Capitanio, J. P., Mendoza, S. P., and Amaral, D. G. (2001). Increased social fear and decreased fear of objects in monkeys with neonatal amygdala lesions. Neuroscience 106: 653-658.
Quirk, G. J. (2002). Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn. Mem. 9: 402-407.
Quirk, G. J., and Beer, J. S. (2006). Prefrontal involvement in the regulation of emotion: convergence of rat and human studies. Curr. Opin. Neurobiol. 6: 723-727.
Rasalam, A. D., Hailey, H., Williams, J. H., Moore, S. J., Turnpenny, P. D., Lloyd, D. J., and Dean, J. C. (2005). Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol. 47: 551-555.
Raymond, G. V., Bauman, M. L., and Kemper, T. L. (1996). Hippocampus in autism: a Golgi analysis. Acta Neuropathol. (Berl) 91: 117-119.
Redcay, E., and Courchesne, E. (2005). When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol. Psychiatry 58: 1-9.
Rimland, B. (1964). Infantile autism: the syndrome and its implications for a neural theory of behavior (New York, Appleton-Centuring-Crofts).
Rinaldi, T., Silberberg, G., and Markram, H. (2007a). Hyperconnectivity of Local Neocortical Microcircuitry Induced by Prenatal Exposure to Valproic Acid. Cereb Cortex. doi:10.1093/cercor/bhm117.
Rinaldi, T., Kulangara, K., Antoniello, K., and Markram, H. (2007b). Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proceedings of the National Academy of Sciences 104: 13501-13506.
Ring, H. A., Baron-Cohen, S., Wheelwright, S., Williams, S. C., Brammer, M., Andrew, C., and Bullmore, E. T. (1999). Cerebral correlates of preserved cognitive skills in autism: a functional MRI study of embedded figures task performance. Brain 122, (Pt 7), 1305-1315.
Ritvo, E. R., Freeman, B. J., Scheibel, A. B., Duong, T., Robinson, H., Guthrie, D., and Ritvo, A. (1986). Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC Autopsy Research Report. Am. J. Psychiatry 143: 862-866.
Rodier, P. M., Ingram, J. L., Tisdale, B., and Croog, V. J. (1997). Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol. 11: 417-422.
Rodier, P. M., Ingram, J. L., Tisdale, B., Nelson, S., and Romano, J. (1996). Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 370: 247-261.
Rodriguez Manzanares, P. A., Isoardi, N. A., Carrer, H. F., and Molina, V. A. (2005). Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J. Neurosci. 25: 8725-8734.
Rosvold, H. E., Mirsky, A. F., and Pribram, K. H. (1954). Influence of amygdalectomy on social behavior in monkeys. J. Comp. Physiol. Psychol. 47: 173-178.
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2: 255-267.
Rumsey, J. M., and Hamburger, S. D. (1990). Neuropsychological divergence of high-level autism and severe dyslexia. J. Autism Dev. Disord. 20: 155-168.
Rutter, M. (2000). Genetic studies of autism: from the 1970s into the millennium. J. Abnorm. Child Psychol. 28: 3-14.
Rutter, M., Bailey, A., Bolton, P., and Le Couteur, A. (1994). Autism and known medical conditions: myth and substance. J. Child Psychol. Psychiatry 35: 311-322.
Sanders, J., Johnson, K. A., Garavan, H., Gill, M., and Gallagher, L. (2007). A review of neuropsychological and neuroimaging research in autistic spectrum disorders: Attention, inhibition and cognitive flexibility. Res. Autism Spectr. Disord. doi:10.1016/j.rasd.2007.03.005.
Schain, R. J., and Freedman, D. X. (1961). Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J. Pediatr. 58: 315-320.
Scharre, J. E., and Creedon, M. P. (1992). Assessment of visual function in autistic children. Optom. Vis. Sci. 69: 433-439.
Schneider, T., and Przewlocki, R. (2005). Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30: 80-89.
Schneider, T., Labuz, D., and Przewlocki, R. (2001). Nociceptive changes in rats after prenatal exposure to valproic acid. Pol. J. Pharmacol. 53: 531-534.
Schneider, T., Turczak, J., and Przewlocki, R. (2006). Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic Acid: issues for a therapeutic approach in autism. Neuropsychopharmacology 31: 36-46.
Schultz, R. T. (2005). Developmental deficits in social perception in autism: the role of the amygdala and fusiform face area. Int. J. Dev. Neurosci. 23: 125-141.
Schultz, R. T., Grelotti, D. J., Klin, A., Kleinman, J., Van der Gaag, C., Marois, R., and Skudlarski, P. (2003). The role of the fusiform face area in social cognition: implications for the pathobiology of autism. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 358: 415-427.
Schultz, R. T., Gauthier, I., Klin, A., Fulbright, R. K., Anderson, A. W., Volkmar, F., Skudlarski, P., Lacadie, C., Cohen, D. J., and Gore, J. C. (2000). Abnormal ventral temporal cortical activity during face discrimination among individuals with autism and Asperger syndrome. Arch. Gen. Psychiatry 57: 331-340.
Schumacher, H. J., Terapane, J., Jordan, R. L., and Wilson, J. G. (1972). The teratogenic activity of a thalidomide analogue, EM 12 in rabbits, rats, and monkeys. Teratology 5: 233-240.
Schumann, C. M., and Amaral, D. G. (2006). Stereological analysis of amygdala neuron number in autism. J. Neurosci. 26: 7674-7679.
Schumann, C. M., Hamstra, J., Goodlin-Jones, B. L., Lotspeich, L. J., Kwon, H., Buonocore, M. H., Lammers, C. R., Reiss, A. L., and Amaral, D. G. (2004). The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci. 24: 6392-6401.
Sears, L. L., Finn, P. R., and Steinmetz, J. E. (1994). Abnormal classical eye-blink conditioning in autism. J. Autism Dev. Disord. 24: 737-751.
Shaban, H., Humeau, Y., Herry, C., Cassasus, G., Shigemoto, R., Ciocchi, S., Barbieri, S., van der Putten, H., Kaupmann, K., Bettler, B., and Luthi, A. (2006). Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat. Neurosci. 9: 1028-1035.
Shah, A., and Frith, U. (1983). An islet of ability in autistic children: a research note. J. Child Psychol. Psychiatry 24: 613-620.
Shah, A., and Frith, U. (1993). Why do autistic individuals show superior performance on the block design task? J. Child Psychol. Psychiatry 34: 1351-1364.
Shaw, P., Lawrence, E. J., Radbourne, C., Bramham, J., Polkey, C. E., and David, A. S. (2004). The impact of early and late damage to the human amygdala on ’theory of mind’ reasoning. Brain 127: 1535-1548.
Shayegan, D. K., and Stahl, S. M. (2005). Emotion processing, the amygdala, and outcome in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 29: 840-845.
Silberberg, G., and Markram, H. (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53: 735-746.
Simion, F., Valenza, E., Umilta, C., and Dalla Barba, B. (1998). Preferential orienting to faces in newborns: a temporal-nasal asymmetry. J. Exp. Psychol. Hum. Percept Perform. 24: 1399-1405.
Sotres-Bayon, F., Bush, D. E., and LeDoux, J. E. (2004). Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn. Mem. 11: 525-535.
Sotres-Bayon, F., Cain, C. K., and LeDoux, J. E. (2006). Brain mechanisms of fear extinction: historical perspectives on the contribution of prefrontal cortex. Biol. Psychiatry. 60: 329-336.
Sparks, B. F., Friedman, S. D., Shaw, D. W., Aylward, E. H., Echelard, D., Artru, A. A., Maravilla, K. R., Giedd, J. N., Munson, J., Dawson, G., and Dager, S. R. (2002). Brain structural abnormalities in young children with autism spectrum disorder. Neurology 59: 184-192.
Spiker, D., Lotspeich, L., Kraemer, H. C., Hallmayer, J., McMahon, W., Petersen, P. B., Nicholas, P., Pingree, C., Wiese-Slater, S., Chiotti, C., and et al. (1994). Genetics of autism: characteristics of affected and unaffected children from 37 multiplex families. Am. J. Med. Genet. 54: 27-35.
Stanton, M. E., Peloso, E., Brown, K. L., and Rodier, P. (2007). Discrimination learning and reversal of the conditioned eyeblink reflex in a rodent model of autism. Behav. Brain. Res. 176: 133-140.
Stein, M. B., Goldin, P. R., Sareen, J., Zorrilla, L. T., and Brown, G. G. (2002). Increased amygdala activation to angry and contemptuous faces in generalized social phobia. Arch. Gen. Psychiatry 59: 1027-1034.
Stevenson, R. E., Schroer, R. J., Skinner, C., Fender, D., and Simensen, R. J. (1997). Autism and macrocephaly. Lancet 349: 1744-1745.
Stodgell, C. J., Ingram, J. L., O’Bara, M., Tisdale, B. K., Nau, H., and Rodier, P. M. (2006). Induction of the homeotic gene Hoxa1 through valproic acid's teratogenic mechanism of action. Neurotoxicol. Teratol. 28: 617-624.
Stromland, K., Nordin, V., Miller, M., Akerstrom, B., and Gillberg, C. (1994). Autism in thalidomide embryopathy: a population study. Dev. Med. Child Neurol. 36: 351-356.
Sweeten, T. L., Posey, D. J., Shekhar, A., and McDougle, C. J. (2002). The amygdala and related structures in the pathophysiology of autism. Pharmacol. Biochem. Behav. 71: 449-455.
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., and Sudhof, T. C. (2007). A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science: 1146221
Tantam, D., Monaghan, L., Nicholson, H., and Stirling, J. (1989). Autistic children's ability to interpret faces: a research note. J. Child Psychol. Psychiatry 30: 623-630.
Tordjman, S., Anderson, G. M., McBride, P. A., Hertzig, M. E., Snow, M. E., Hall, L. M., Thompson, S. M., Ferrari, P., and Cohen, D. J. (1997). Plasma beta-endorphin, adrenocorticotropin hormone, and cortisol in autism. J. Child Psychol. Psychiatry 38: 705-715.
Towbin, K. E., Pradella, A., Gorrindo, T., Pine, D. S., and Leibenluft, E. (2005). Autism spectrum traits in children with mood and anxiety disorders. J. Child Adolesc. Psychopharmacol. 15: 452-464.
Townsend, J., Courchesne, E., Covington, J., Westerfield, M., Harris, N. S., Lyden, P., Lowry, T. P., and Press, G. A. (1999). Spatial attention deficits in patients with acquired or developmental cerebellar abnormality. J. Neurosci. 19: 5632-5643.
Tsujino, N., Nakatani, Y., Seki, Y., Nakasato, A., Nakamura, M., Sugawara, M., and Arita, H. (2007). Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci. Res. 57: 289-295.
Vorhees, C. V. (1987a). Teratogenicity and developmental toxicity of valproic acid in rats. Teratology 35: 195-202.
Vorhees, C. V. (1987b). Behavioral teratogenicity of valproic acid: selective effects on behavior after prenatal exposure to rats. Psychopharmacology (Berl) 92: 173-179.
Wagner, G. C., Reuhl, K. R., Cheh, M., McRae, P., and Halladay, A. K. (2006). A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord. 36: 779-793.
Walker, H. A. (1977). Incidence of minor physical anomaly in autism. J. Autism Child Schizophr. 7: 165-176.
Wang, Y., Markram, H., Goodman, P. H., Berger, T. K., Ma, J., and Goldman-Rakic, P. S. (2006). Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nat. Neurosci. 9: 534-542.
Weeks, S. J., and Hobson, R. P. (1987). The salience of facial expression for autistic children. J. Child Psychol. Psychiatry 28: 137-151.
Weiskrantz, L. (1956). Behavioral changes associated with ablation of the amygdaloid complex in monkeys. J. Comp. Physiol. Psychol. 49: 381-391.
Williams, G., King, J., Cunningham, M., Stephan, M., Kerr, B., and Hersh, J. H. (2001). Fetal valproate syndrome and autism: additional evidence of an association. Dev. Med. Child Neurol. 43: 202-206.
Williams, P. G., and Hersh, J. H. (1997). A male with fetal valproate syndrome and autism. Dev. Med. Child Neurol. 39: 632-634.
Williams, R. S., Hauser, S. L., Purpura, D. P., DeLong, G. R., and Swisher, C. N. (1980). Autism and mental retardation: neuropathologic studies performed in four retarded persons with autistic behavior. Arch. Neurol. 37: 749-753.
Winslow, J. T., and Insel, T. R. (2002). The social deficits of the oxytocin knockout mouse. Neuropeptides 36: 221-229.
Zald, D. H. (2003). The human amygdala and the emotional evaluation of sensory stimuli. Brain Res. Brain Res. Rev. 41: 88-123.
Keywords: autism, microcircuit, connectivity, plasticity, neocortex, amygdala, valproic acid
Citation: Henry Markram, Tania Rinaldi and Kamila Markram (2007). The Intense World Syndrome – an alternative hypothesis for autism. Front. Neurosci. 1: 1. 77-96. doi: 10.3389/neuro.01/1.1.006.2007
Received: 15 August 2007; Paper pending published: 01 September 2007;
Accepted: 01 September 2007;
Published online: 15 October 2007.
Edited by:
Idan Segev, Hebrew University, IsraelReviewed by:
Joseph LeDoux, New York University, USAJacqueline N. Crawley, National Institute of Mental Health, USA
Copyright: © 2007 Markram, Rinaldi and Markram. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Dr. Kamila Markram, Brain Mind Institute, EPFL SV BMI LNEP, Building AAB - Office 201 - Station 15, 1015 Lausanne, Switzerland. Tel.: +41 21 6931642; fax: +41 21 6939635. e-mail: kamila.markram@epfl.ch