 Miquel Sánchez-Osuna
Miquel Sánchez-Osuna Pilar Cortés
Pilar Cortés Jordi Barbé
Jordi Barbé Ivan Erill
Ivan Erill- 1Departament de Genètica i de Microbiologia, Universitat Autònoma de Barcelona, Bellaterra, Spain
- 2Department of Biological Sciences, University of Maryland, Baltimore County, Baltimore, MD, United States
Sulfonamides are synthetic chemotherapeutic agents that work as competitive inhibitors of the di-hydro-pteroate synthase (DHPS) enzyme, encoded by the folP gene. Resistance to sulfonamides is widespread in the clinical setting and predominantly mediated by plasmid- and integron-borne sul1-3 genes encoding mutant DHPS enzymes that do not bind sulfonamides. In spite of their clinical importance, the genetic origin of sul1-3 genes remains unknown. Here we analyze sul genes and their genetic neighborhoods to uncover sul signature elements that enable the elucidation of their genetic origin. We identify a protein sequence Sul motif associated with sul-encoded proteins, as well as consistent association of a phosphoglucosamine mutase gene (glmM) with the sul2 gene. We identify chromosomal folP genes bearing these genetic markers in two bacterial families: the Rhodobiaceae and the Leptospiraceae. Bayesian phylogenetic inference of FolP/Sul and GlmM protein sequences clearly establishes that sul1-2 and sul3 genes originated as a mobilization of folP genes present in, respectively, the Rhodobiaceae and the Leptospiraceae, and indicate that the Rhodobiaceae folP gene was transferred from the Leptospiraceae. Analysis of %GC content in folP/sul gene sequences supports the phylogenetic inference results and indicates that the emergence of the Sul motif in chromosomally encoded FolP proteins is ancient and considerably predates the clinical introduction of sulfonamides. In vitro assays reveal that both the Rhodobiaceae and the Leptospiraceae, but not other related chromosomally encoded FolP proteins confer resistance in a sulfonamide-sensitive Escherichia coli background, indicating that the Sul motif is associated with sulfonamide resistance. Given the absence of any known natural sulfonamides targeting DHPS, these results provide a novel perspective on the emergence of resistance to synthetic chemotherapeutic agents, whereby preexisting resistant variants in the vast bacterial pangenome may be rapidly selected for and disseminated upon the clinical introduction of novel chemotherapeuticals.
Introduction
Antibiotic resistance is a pressing problem in modern healthcare (Carlet et al., 2014; Rossolini et al., 2014). Bacterial cells present several mechanisms to cope with exposure to antibiotics or chemotherapeutic agents, which may be acquired through mutation or, most frequently, via lateral gene transfer on mobile genetic elements (Davies and Davies, 2010). These mechanisms include modification of the antimicrobial target, degradation or chemical modification of the antimicrobial molecule, targeted reduction of antimicrobial uptake, active export of the antimicrobial through efflux pumps and use of alternate pathways and enzymes (Davies and Davies, 2010).
It is widely accepted that many antibiotic resistance genes present today in pathogenic bacteria originated from homologs evolved over eons in either the microbes that naturally produce the antibiotics or their natural competitors (Aminov and Mackie, 2007). When coupled with the high plasticity of bacterial genomes and their co-existence with a large variety of genetic mobile elements, the availability of a readily evolved pool of antibiotic resistance genes set the stage for the rapid proliferation of multi-resistant strains in the clinical setting shortly after the commercial introduction of antibiotics (Aminov and Mackie, 2007). In contrast, the origins of resistance against chemotherapeutic agents are harder to pinpoint. Since these were designed in vitro, it seems unlikely that a large pool of genes conferring resistance to chemotherapeutic agents existed before their introduction. After their discovery in the 1960’s, resistance to quinolones was initially rare and limited to chromosomal mutations in DNA gyrase, topoisomerase IV or efflux pumps (Hooper, 1999). However, in the early 2000’s plasmid-borne qnr genes were first detected and spread rapidly to clinical pathogens. Qnr is a member of the pentapeptide repeat family and was shown to confer resistance by binding to DNA gyrase and limiting the effect of quinolone drugs. The origin of plasmid-borne qnr genes has been traced to environmental homologs and these are thought to have derived from genes originally targeting antibiotics, such as microcin B17 (Tran and Jacoby, 2002).
Aryl sulfonamides are synthetic antibacterial compounds presenting a structure similar to para-amino benzoic acid (PABA), and containing a sulfonamide group linked to an aromatic group. Commonly referred to as sulfonamides or sulfa drugs due to their clinical relevance, synthetic aryl sulfonamides function as competitive inhibitors of the di-hydro-pteroate synthase (DHPS) enzyme, encoded in bacteria by the folP gene (Sköld, 2000). DHPS participates in folate synthesis using PABA as a substrate, and the competitive inhibition of DHPS by sulfonamides results in growth arrest (Mitsuhashi, 1993; Sköld, 2000). Experiments in mice in the 1930’s demonstrated the effectiveness of sulfonamide against bacteria, and sulfonamide became the first antibacterial chemotherapeutic to be used systemically (Domagk, 1935; Aminov, 2010). It remained in use throughout World War II, but by the end of the 1940’s resistant strains started to emerge and sulfonamides were rapidly displaced in favor of the newly discovered antibiotics (Sköld, 2000; Davenport, 2012).
Resistance to sulfonamide through increased production of PABA was reported in the early 1940’s (Landy et al., 1943), but the most commonly reported mechanism of sulfonamide resistance are mutations to the chromosomal folP gene (Huovinen et al., 1995; Sköld, 2000). Mutations to the chromosomal folP gene have been shown to provide varying degrees of trade-off between resistance and efficient folate synthesis, decreasing DHPS affinity for sulfonamide while maintaining or increasing its affinity for PABA (Sköld, 2000). These mutations have occurred independently in multiple bacterial genera and target multiple conserved areas of the DHPS protein (Sköld, 2000). However, similar mutational profiles, such as two-amino acid insertions in Neisseria meningitidis and Streptococcus pneumoniae, have been reported (Rådström et al., 1992; Haasum et al., 2001), and in both these genera there is evidence of extensive recombination within folP genes (Fermer et al., 1995; Swedberg et al., 1998).
In spite of the multiple instances of chromosomal folP resistant variants, clinical resistance to sulfonamides is predominantly plasmid-borne and mediated by sul genes encoding alternative sulfonamide-resistant DHPS enzymes (Sköld, 2000). Four different sul genes have been described to date, with sul1 and sul2 being the predominant forms in clinical isolates (Rådström et al., 1991). The sul1 gene is typically found in class 1 integrons and linked to other resistance genes (Rådström et al., 1991), whereas sul2 is usually associated to non-conjugative plasmids of the IncQ group (Enne et al., 2001) and to large transmissible plasmids like pBP1 (Treeck et al., 1981). The sul3 gene was characterized in the Escherichia coli conjugative plasmid pVP440. It was shown to be flanked by two copies of the insertion element IS15Δ/26 and to be widespread in E. coli isolates from pigs in Switzerland (Perreten and Boerlin, 2003). Recently, a sul4 gene was identified in a systematic prospection of class 1 integron-borne genes in Indian river sediments, but this sul variant has not yet been detected in clinical isolates. Genomic context analyses revealed that the sul4 gene had been recently mobilized and phylogenetic inference pinpointed its putative origin as part of the folate synthesis cluster in the Chloroflexi phylum (Razavi et al., 2017).
Despite the importance of sulfonamides in human and animal therapy, the putative origin of the three sul genes that account for the vast majority of reported clinical resistance to sulfonamide remains to be elucidated. In this work we leverage comparative genomics, phylogenetic analysis and in vitro determination of minimal inhibitory concentrations (MIC) of sulfamethoxazole to unravel the origin of the sul1, sul2, and sul3 genes. Our analysis indicates that chromosomally encoded folP genes conferring resistance to sulfonamide originated in members of the Leptospiraceae family and were transferred to the Alphaproteobacteria Rhodobiaceae family more than 500 million years ago. These isolated sources of chromosomally encoded sulfonamide-resistant DHPS were mobilized independently, leading to the broadly disseminated sul1, sul2, and sul3 resistance genes. Our results hence indicate that resistance to synthetic chemotherapeutic agents may be available in the form of chromosomally encoded variants among the extremely diverse bacterial domain, and can be rapidly disseminated upon the release of novel synthetic drugs.
Materials and Methods
Data Collection
FolP, GlmM, and Sul1-3 homologs were identified in complete GenBank sequences (GenBank, RRID:SCR_002760) through BLASTP (BLASTP, RRID:SCR_001010) (Altschul et al., 1997) using the E. coli FolP (WP_000764731) and GlmM (WP_000071134) proteins as the query. Putative homologs were detected as BLASTP hits passing stringent e-value (<1e-20) and query coverage (75%) thresholds. FolP and GlmM chromosomally encoded proteins were identified on a representative genome of all bacterial orders with complete genome assemblies on RefSeq (RefSeq, RRID:SCR_003496), of each bacterial family for the Proteobacteria, of any bacterial species where chromosomally encoded sulfonamide resistance mutants had been reported, and on all available complete genomes for clades of interest (Rhodobiaceae, Spirochaetes, and Chlamydiae) (Supplementary Table S1). All protein coding gene sequences for these genomes were downloaded for %GC analysis. Sul proteins encoded by mobile sul genes were identified on complete plasmid, transposon, and integron GenBank sequences (GenBank, RRID:SCR_002760).
Identification and Visualization of Sul-Like Signatures in FolP Sequences
To identify sequence motifs associated with Sul proteins, we performed a CLUSTALW alignment (Clustal Omega, RRID:SCR_001591) using a non-redundant (<99% identity) subset of the Sul1-3 homologous sequences detected previously and FolP sequence sampled from each bacterial clade. Following visual inspection of the resulting alignment, a Sul-like motif conserved in several chromosomally encoded FolP proteins was visualized using iceLogo (iceLogo, RRID:SCR_012137) (Colaert et al., 2009) and a consensus motif was derived and encoded into a PROSITE-format pattern (PROSITE, RRID:SCR_003457). The inferred PROSITE pattern was used to seed a Pattern Hit Initiated BLAST (NCBI PHI-BLAST; RRID:SCR_004870) search against the NCBI non-redundant Protein database (Protein, RRID:SCR_003257) using as a query the protein sequences of Sul1-3 reported in the literature (WP_001336346, WP_010890159, and WP_000034420) and conservative e-value (<1e-20) and query coverage (75%) limits. Only chromosomal hits with the identified signature characteristic of sul gene products were retained for further analysis.
Multiple Sequence Alignment and Phylogenetic Inference
For phylogenetic inference, multiple sequence alignments of identified FolP/Sul1-3 and GlmM homologous sequences were performed with CLUSTALW (Clustal Omega, RRID:SCR_001591) (Thompson et al., 1994) using variable (5, 10, and 25) gap opening penalties. These alignments were then integrated with local LALIGN alignments with T-COFFEE (T-Coffee, RRID:SCR_011818) (Notredame et al., 2000), and the resulting alignment was trimmed using the “less stringent selection” parameters of the Gblocks online service (Castresana, 2000; Dereeper et al., 2008). Bayesian phylogenetic inference on trimmed alignments was performed with MrBayes (MrBayes, RRID:SCR_012067) (Ronquist and Huelsenbeck, 2003). Four Metropolis-Coupled Markov Chain Monte Carlo runs with four independent chains were carried out for 30,000,000 generations, and the resulting consensus tree was plotted with FigTree (FigTree, RRID:SCR_008515).
DNA Sequence Analyses
Analysis of %GC in synonymous and non-synonymous patterns and Ka/Ks divergence were performed according to the Nei–Gojobori computation method (Nei and Gojobori, 1986) and the standalone PAL2NAL program for codon-based alignments (Suyama et al., 2006), using custom Python scripts for pipelining. Analyses of %GC content were performed on all sampled bacterial genomes, computing genome-wide %GC statistics and comparing them to folP estimates. Analyses of Ka/Ks divergence were performed on pair-wise alignments of the N- and C-terminal ends of the glmM gene sequence of all sampled bacterial groups. One-sided Mann–Whitney U-tests were performed using GraphPad Prism (GraphPad, RRID:SCR_002798) to determine whether differences between folP and chromosomal %GC content were significantly different in the presence and absence of Sul-like signature motifs, and whether the N- and C-terminal regions presented different mutational profiles. The scripts used for the analysis are available at the GitHub ErillLab repository. Nucleotide sequence identities percentages were computed on gapless positions of PAL2NAL codon-based alignments using a custom Python script. Amelioration times were estimated using the Ameliorator program (Lawrence and Ochman, 1997) under different selection modes. Ka and Ks values were estimated from pairwise alignments of orthologs between the Parvibaculum lavamentivorans and Leptospira interrogans genomes as determined by the OMA Orthology database (OMA Orthology database, RRID:SCR_016425) (Altenhoff et al., 2018) and species divergence times were inferred from published molecular clock phylogenies (Battistuzzi et al., 2004).
Cloning, Transformation and Complementation of the folP Gene for Broth Microdilution Assays
The L. interrogans serovar Lai str. 56601 folP and Chlamydia trachomatis D/UW-3/CX folKP gene were synthesized and adapted to E. coli codon usage at ATG:biosynthetics GmbH, Germany; whereas P. lavamentivorans DS-1 (DSMZ 13023) and Rhodobacter sphaeroides 2.4.1 (gently provided by Professor S. Kaplan; Health Science Center. University of Texas) folP genes were amplified from genomic DNA. The sul2 gene was amplified from the RSF1010 plasmid (Kushner, 1978; Honda et al., 1991) and used as a positive control. The folP/sul genes were amplified using suitable primers (Supplementary Table S2), purified PCR products were then digested with NdeI and BamHI (New England Biolabs, RRID:SCR_013517) and cloned into a dephosphorylated pUA1108 vector (Mayola et al., 2014), previously cut with the same restriction enzymes. The ligation was introduced by transformation into competent E. coli DH5α cells and recombinant plasmids were extracted with the NZYMiniprep kit (NZYTech, RRID:SCR_016772), sequenced (Macrogen, RRID:SCR_014454) and introduced by transformation in competent E. coli K-12 (CGSC 5073) (CGSC, RRID:SCR_002303). Susceptibility to sulfamethoxazole (Sigma-Aldrich, RRID:SCR_008988) for the strains containing the folP/sul genes was determined using broth microdilution tests in Mueller-Hinton broth (MH) with half serial dilutions of sulfamethoxazole ranging from 512 to 0.125 mg/L, following the CLSI guidelines (Clinical and Laboratory Standards Institute, 2003). Colonies were grown on Luria-Bertani (LB) agar for 18 h and then suspended in sterile 0.9% NaCl solution to a McFarland 0.5 turbidity level. These suspensions were diluted at 10-2 in Mueller-Hinton (MH) broth, and 50 μl (5.104 cells) were inoculated onto microtiter plates containing 50 μl of MH broth supplemented with 1024–0.250 mg/L of sulfamethoxazole. To determine growth, absorbance at 550 nm was measured after 24 h incubation at 37°C (Sunrise plate reader; Tecan Life Sciences, RRID:SCR_016771).
Results
Identification of Putative Chromosomal Origins for sul1-3 Genes
To identify putative chromosomal homologs of sul1-3 genes, we performed a multiple sequence alignment including any protein sequences with at most 90% similarity to those encoded by sul1-3 genes reported in the literature and by chromosomal folP genes from a representative of each bacterial order. Inspection of the resulting alignment (Figure 1A and Supplementary Data 1) revealed the presence of a two-amino acid insertion in proteins encoded by sul1-3 genes that is not present in those encoded by sul4 or the analyzed chromosomal folP genes. This two-amino acid insertion is located in a conserved region of the FolP protein (residues R171-N211 of the E. coli FolP protein [WP_000764731]) that presents other signature changes in sul-encoded proteins with respect to chromosomally encoded FolP proteins (Figures 1A,B and Supplementary Data 1) (Rådström and Swedberg, 1988; Morgan et al., 2011). We derived a PROSITE-format pattern (PROSITE, RRID:SCR_003457) (Supplementary Data 2) of the identified Sul motif to seed a Pattern Hit Initiated BLAST (NCBI PHI-BLAST; RRID:SCR_004870) search against the NCBI non-redundant (NR) protein database (NCBI, RRID:SCR_006472). This search identified several proteins encoded by Rhodobiaceae family members that presented a similar insertion pattern. BLASTP (BLASTP, RRID:SCR_001010) searches with these Rhodobiaceae FolP sequences matched proteins in several members of the Leptospiraceae and the Chlamydiae. However, analysis of the resulting multiple sequence alignment showed that only the Leptospiraceae FolP protein sequences displayed the identified two-amino acid insertion pattern (Supplementary Data 3). Heretofore, we refer to these chromosomally encoded FolP proteins containing the signature Sul motif as FolP∗, and to their encoding gene as folP∗.
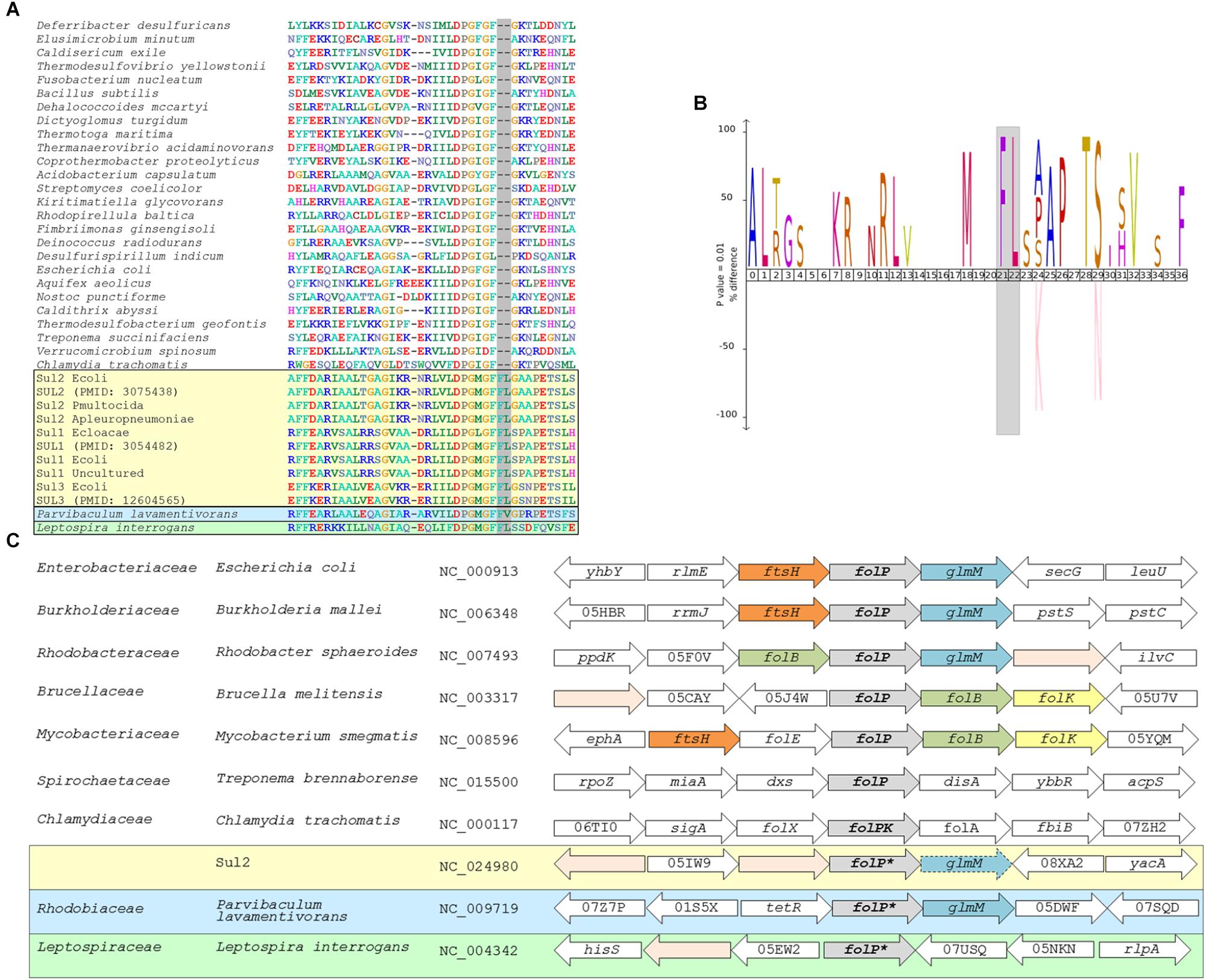
Figure 1. (A) Segment of the multiple sequence alignment including any sul genes with at most 90% similarity to reported sul genes and a representative chromosomal folP gene for all bacterial phyla with complete genomes available in NCBI RefSeq. (B) IceLOGO highlighting the difference in amino acid frequency at each position of the region of the folP protein sequence containing the identified insertion between the multiple sequence alignment of sul gene products and the chromosomally encoded FolP proteins. The upper part of the iceLOGO plot shows residues overrepresented in the sul-encoded FolP proteins; the bottom part shows residues overrepresented in chromosomally encoded FolP proteins for all bacterial phyla with complete genomes available. Only differences with significant z-score under a confidence interval of 0.01 are shown. (C) Schematic representation of the genetic environment of sul2 genes, similar arrangements in chromosomally encoded folP genes of the Gammaproteobacteria, Betaproteobacteria, and Alphaproteobacteria, and arrangements in other major phyla. Arrow boxes indicate coding regions. When available, gene names or NOG identifiers are provided. Boxes for folP genes containing the two-amino acid insertion are designated as folP∗.
In order to gain further insight into the possible chromosomal origins of sul genes, we performed tBLASTX searches against the NCBI RefSeq Genome Database (RefSeq, RRID:SCR_003496) using the genetic surroundings (5,000 bp) of sul1, sul2, and sul3 genes with at most 90% similarity to those reported in the literature (Supplementary Table S3). This search did not return consistent results for the sul1 and sul3 genetic surroundings, but it identified a conserved gene fragment encoding the N-terminal region of the phosphoglucosamine mutase GlmM protein downstream of sul2 in multiple plasmids harboring this resistance gene. These sul2-associated GlmM sequences lack the entire GlmM C-terminal region, including three of its functional domains (Mehra-Chaudhary et al., 2011), and it can therefore be safely assumed that they are not functional as phosphoglucosamine mutases. This genetic arrangement has been reported previously as a feature of sul2 isolates (Kehrenberg and Schwarz, 2005; Hu et al., 2016), and it is strongly conserved in the genomic surroundings of chromosomal folP genes in the Gammaproteobacteria, the Betaproteobacteria and several Alphaproteobacteria lineages (Figure 1C). Analysis of the folP genetic surroundings in complete genomes of the Spirochaetes and the Alphaproteobacteria shows clear differences between the genes coding for the identified Rhodobiaceae and Leptospiraceae FolP∗ proteins harboring the two-amino acid insertion pattern and those without it (Figure 1C). The Leptospiraceae show a conserved arrangement with folP∗ flanked by a peptidoglycan-associated lipoprotein and a tetratricopeptide repeat-containing domain protein, whereas in most other Spirochaetes folP is flanked by a 1-deoxy-D-xylulose-5-phosphate synthase and a diadenylate cyclase. In contrast, the Alphaproteobacteria yield several distinct syntenic regions for folP. In the Rhodobiaceae, folP∗ is flanked by genes coding for either a FtsH-family metallopeptidase or a TetR-family transcriptional repressor and the phosphoglucosamine mutase glmM. In the Rhodobacterales, folP is flanked by a dihydroneopterin aldolase and glmM, but in the Rhizobiales it is flanked by a Zn-dependent protease and the dihydroneopterin aldolase. This last arrangement, in which the dihydroneopterin aldolase is followed by a 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase is also part of the genetic surroundings of folP in most Actinobacteria (Figure 1C).
Phylogenetic Analysis of sul/folP and glmM Genes
The presence of a signature two-amino acid insertion characteristic of sul gene products in chromosomally encoded FolP∗ proteins and the identification of a genetic environment for sul2 genes that is conserved in multiple bacterial genomes suggested that it might be possible to pinpoint the evolutionary origin of sul genes. To further investigate this possibility, we performed a rigorous phylogenetic analysis of FolP/Sul protein sequences. We sampled a representative genome of all bacterial orders with complete genome assemblies, of each bacterial family for the Proteobacteria and all available complete genomes for clades of interest (Rhodobiaceae, Spirochaetes, and Chlamydiae), and we identified chromosomally encoded FolP homologs in each of these genomes using BLASTP (BLASTP, RRID:SCR_001010) with the E. coli FolP protein as a query. We used a distance tree generated with CLUSTALW (Clustal Omega, RRID:SCR_001591) to identify and discard a set of protein sequences from duplicated folP genes in the Actinobacteria (Supplementary Data 4), and we performed multiple sequence alignment and Bayesian phylogenetic reconstruction of the remaining FolP/Sul sequences with T-COFFEE (T-Coffee, RRID:SCR_011818) and MrBayes (MrBayes, RRID:SCR_012067) (Supplementary Data 5).
The resulting tree (Figure 2) provides strong support for the hypothesis that sul1-3 genes originated in the Rhodobiaceae and Leptospiraceae families. In particular, the topology inferred by MrBayes suggests that the Leptospiraceae folP∗ gene gave rise to both sul3 and the folP∗ gene encountered in the Rhodobiaceae, most likely through a lateral gene transfer event in an ancestor of this Alphaproteobacteria family. According to the reconstructed FolP phylogeny, the Rhodobiaceae folP∗ gene was subsequently mobilized as sul2, and later evolved into the integron-borne sul1 gene (Orman et al., 2002). The fact that the Leptospiraceae FolP∗ sequences branch independently of other Spirochaetes sequences and immediately after the Chlamydiae suggests that the Leptospiraceae folP∗ gene might have originated as a result of lateral gene transfer event from the Chlamydiae, and that it subsequently incorporated the signature two-amino acid insert present in sul-encoded DHPS proteins. Importantly, the trimmed multiple sequence alignment used for FolP/Sul Bayesian phylogenetic inference (Supplementary Data 5) does not incorporate the two-amino acid insertion of the Sul motif, indicating that the joint branching of Sul1-3 sequences with chromosomally encoded Rhodobiaceae and Leptospiraceae FolP proteins is based on sequence similarity beyond this insertion and its immediate vicinity (Figures 1A,B).
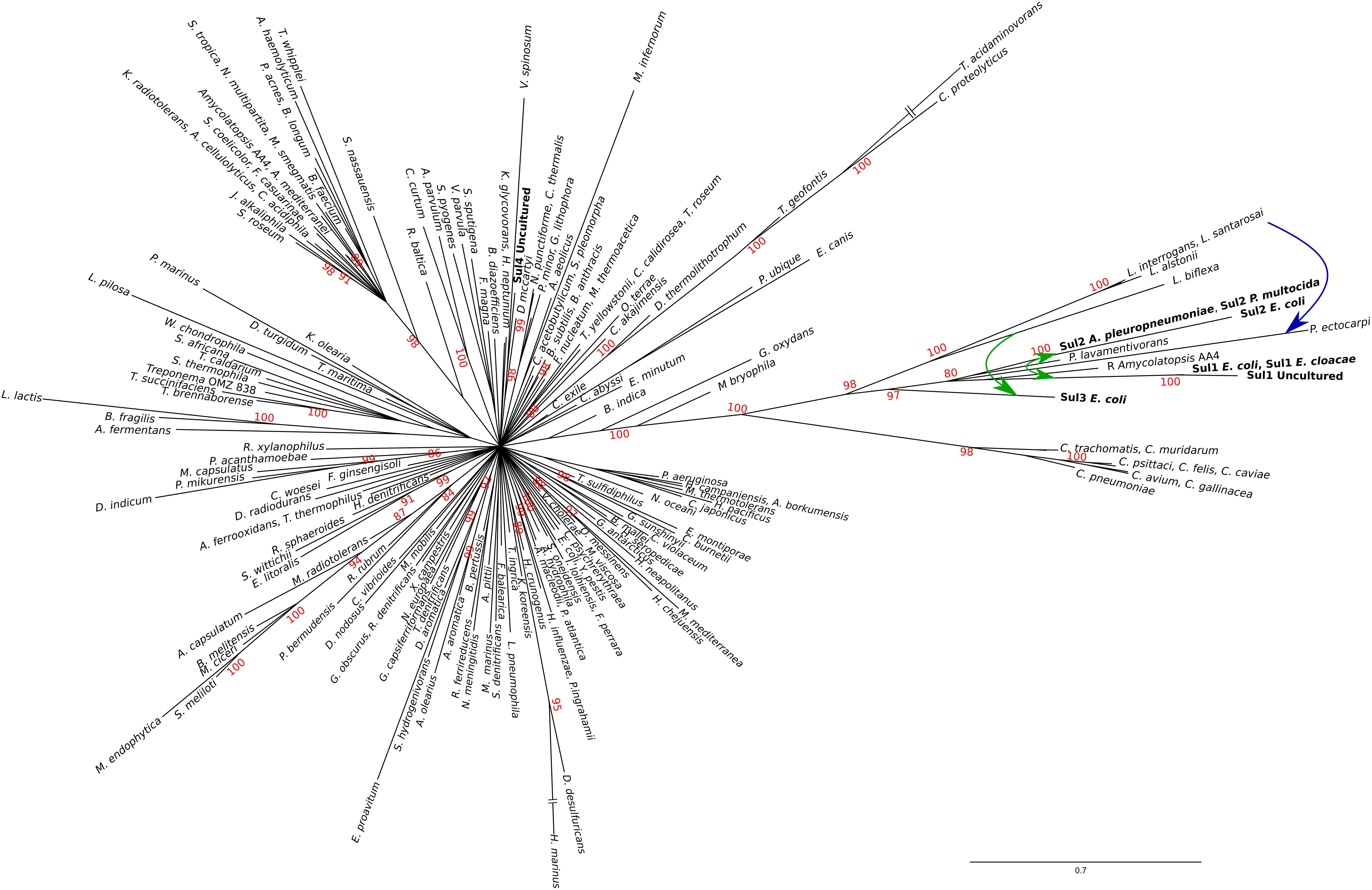
Figure 2. Consensus tree of Sul/FolP protein sequences. Branch support values are provided as Bayesian posterior probabilities. For clarity, only posterior probability values higher than 0.8 are displayed. Proposed lateral gene transfer and mobilization events are shown by means of superimposed continuous and dotted, respectively, arrows.
The existence of a genetic environment for sul2 genes conserved in bacterial chromosomes provides the means to independently assess the likelihood of the evolutionary scenario inferred from the FolP phylogeny. Using the same sampling methods utilized for sul/folP protein products, we collected protein sequences for phosphoglucosamine mutase (GlmM) homologs and performed Bayesian phylogenetic inference on the aligned N-terminal regions. The resulting GlmM tree (Figure 3) provides further support for a Rhodobiaceae origin of the sul2 gene, with the sul2-associated GlmM sequences branching with the Rhodobiaceae GlmM protein sequences deep within an otherwise monophyletic Alphaproteobacteria clade. Taken together, the consistent branching with the Rhodobiaceae of the protein sequences encoded by both sul2 and its accompanying glmM gene fragment firmly establish this Alphaproteobacteria family as the chromosomal origin for the sul2 gene. The phylogenetic evidence thus indicates that the sul2 gene was excised with the N-terminal fragment of the glmM gene during the mobilization event that led to their incorporation into plasmid vectors. Given that the folP-glmM arrangement is only seen in the Proteobacteria, this also excludes the possibility that the sul2 gene was mobilized directly from a Leptospiraceae background, where the folP gene presents an unrelated, yet conserved, genomic environment (Figure 1C).

Figure 3. Consensus tree of N-terminal GlmM protein sequences. Branch support values are provided as Bayesian posterior probabilities. For clarity, only posterior probability values higher than 0.8 are displayed. The placement of sul2-encoded proteins is indicated by a shaded ellipse.
Analysis of sul/folP and glmM Gene Sequences
The phylogenetic analysis of FolP and GlmM sequences puts forward an evolutionary scenario wherein the Leptospiraceae folP∗ was transferred to the members of the Rhodobiaceae family before being mobilized independently into the sul3- and sul1/2-harboring mobile genetic elements reported in sulfonamide-resistant clinical isolates. To further investigate this hypothesis, we undertook a systematic analysis of folP and glmM coding sequences. We compiled folP gene sequences for all the FolP proteins included in the phylogenetic analysis (Figure 2), as well as any sul gene sequences with less than 90% identity to those reported in the literature and any chromosomal folP∗ genes encoding a DHPS with the signature Sul motif (Figure 1A) for which there were at least 1 Mbp of whole genome shotgun sequence data (Supplementary Data 6). We computed the overall and codon-position %GC content on both the folP/sul coding sequences and all the available coding sequences in their respective genome assembly (Supplementary Table S4). The %GC content data (Figure 4A) reveals that sul1/2 sequences have a high %GC content (60.76 SD ± 1.42) that is consistent with their origin as mobilized Rhodobiaceae folP∗ sequences (%GC content: 62.02 SD ± 2.22). Similarly, sul3 sequences display a %GC content (38.14 SD ± 0.55) consistent with their mobilization from a Leptospiraceae folP∗ background (39.39 SD ± 4.17). This similarity in %GC content precludes the dating of these mobilization events through analysis of DNA amelioration rates. However, examination of DNA sequence similarity (Table 1) reveals identity values of 50–60% between the posited chromosomal donors and their mobile counterparts. These values are in the lower range of sequence identities for previously described mobilization events (Table 1), and hence point to an early mobilization of folP∗ genes that is consistent with the almost universal association of sul genes with class 1 mobile integrons (Lévesque et al., 1995). Together with the phylogenetic inference results, these data provide strong support for an independent mobilization of sul1/2 and sul3 genes from, respectively, Rhodobiaceae and Leptospiraceae family chromosomal backgrounds.
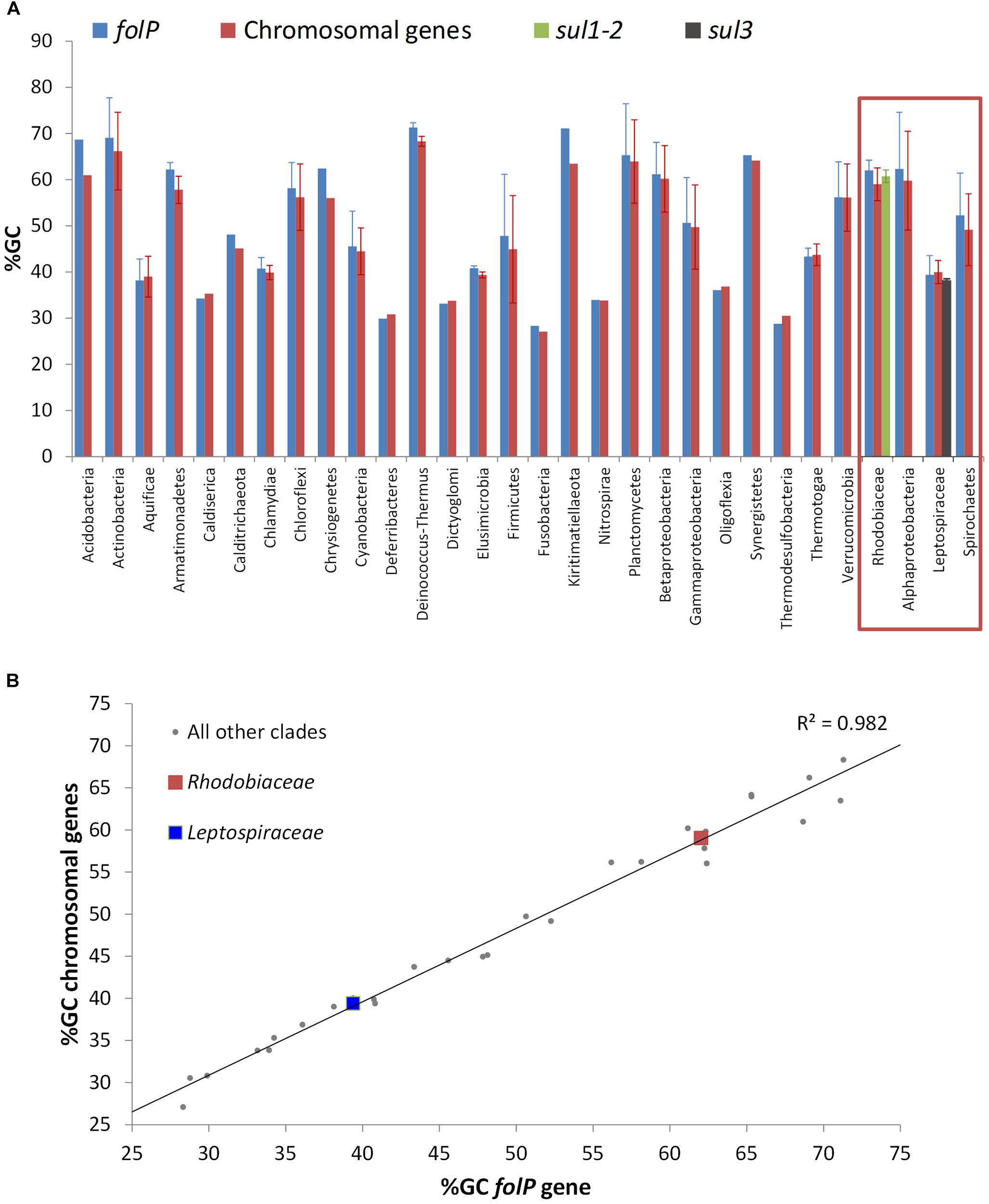
Figure 4. (A) %GC content of folP and all other chromosomal coding sequences in different clades. The %GC content of sul1/2 and sul3 genes is shown adjacent to that of the Rhodobiaceae and the Leptospiraceae. (B) Correlation between the %GC content of folP genes and that of all other coding sequences in their respective genomes. The data points corresponding to folP∗ genes from the Rhodobiaceae and the Leptospiraceae are shown as squares.
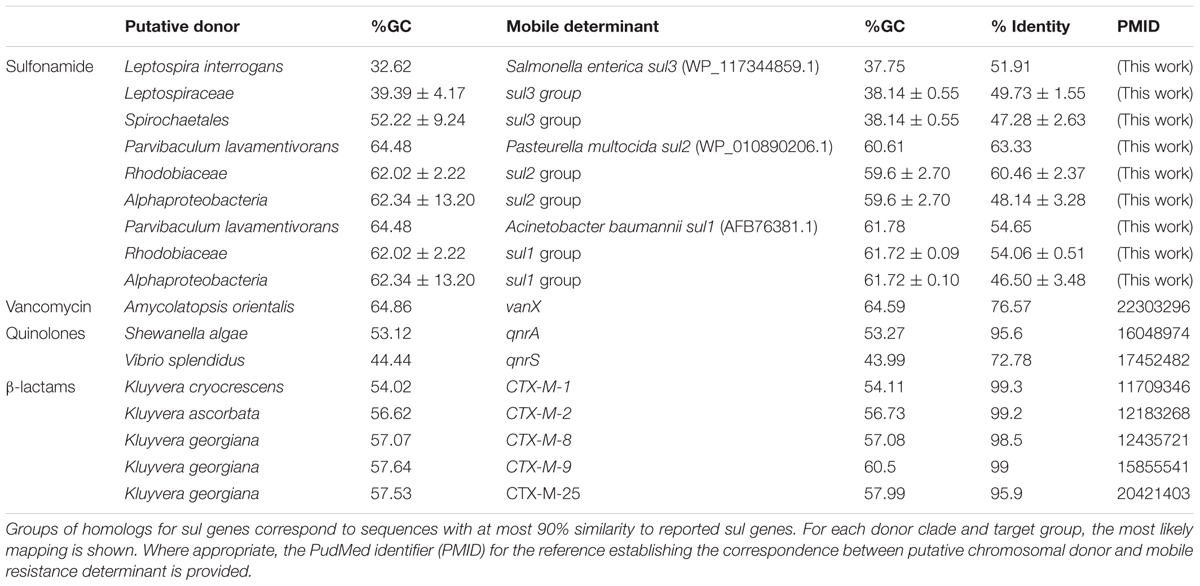
Table 1. Percent GC content and nucleotide identity between posited chromosomal donors and the corresponding mobilized resistance genes for a number of antibiotics.
The independent mobilization of sul1/2 and sul3 is underpinned by a preceding lateral gene transfer of folP∗ from the Leptospiraceae into a Rhodobiaceae ancestor. In this context, the substantial divergence in %GC content between the chromosomal folP∗ genes of both clades indicates a long process of amelioration. In fact, statistical analysis of the differences in codon position %GC content between folP genes and all available coding sequences in their respective genomes shows that Leptospiraceae and Rhodobiaceae folP∗ genes encoding proteins with the Sul motif cannot be distinguished from other folP genes (one-sided Mann–Whitney U-test p > 0.05 for GC1, GC2, and GC3) (Figure 4B) (Supplementary Table S4). We used Ameliorator (Lawrence and Ochman, 1997) to estimate the time required for the observed amelioration via forward simulation from Leptospiraceae codon position %GC values. Even under assumptions of fast evolutionary change, the software provides a lower bound of 476 million years for the observed amelioration of the Leptospiraceae folP∗ gene into the Rhodobiaceae one. Statistical analysis of synonymous and non-synonymous mutation patterns in the N- and C-terminal regions of the glmM gene also shows that mutation patterns in each region of the Rhodobiaceae glmM gene are indistinguishable from those observed in other glmM genes (one-sided Mann–Whitney U-test p > 0.05). Since the glmM gene fragment associated to sul2 genes is likely to be non-functional and subject to genetic drift, the absence of diverging substitution patterns between the N- and C-terminal regions of Rhodobiaceae glmM sequences indicates that the glmM and sul2 genes were transferred from the Rhodobiaceae to sul2-harboring vectors, and not vice versa (Supplementary Table S5). Lastly, given that gene loss is much more likely than gain (Kannan et al., 2013), the absence of glmM fragments in sul1 isolates supports in turn the notion that sul1 derived from sul2. This is consistent with the branching pattern observed in the FolP/Sul tree (Figure 2) and the observed DNA identity values (Table 1), which define a scenario of independent mobilization of sul3 from the Leptospiraceae and sul2 from the Rhodobiaceae, with the subsequent uptake of sul1 by class 1 integrons.
Sulfonamide Resistance of Chromosomal folP Genes
Phylogenetic and sequence analysis results indicate that chromosomal folP∗ genes encoding proteins with the signature Sul motif were independently mobilized into the sul1-3-harboring mobile elements found in sulfonamide-resistant clinical isolates, but they do not address whether the presence of this motif is associated with sulfonamide resistance. To investigate this possibility, we cloned the folP gene coding for DHPS in the Rhodobiaceae P. lavamentivorans DS-1 (WP_012111048), the Leptospiraceae L. interrogans serovar Lai str. 56601 (WP_000444207), the Rhodobacteraceae R. sphaeroides 2.4.1 (WP_011337038) and the Chlamydiae C. trachomatis D/UW-3/CX (WP_009871981). Following Clinical and Laboratory Standards Institute (CLSI) guidelines (Clinical and Laboratory Standards Institute, 2003), we then performed broth microdilution assays to determine the minimal inhibitory concentration (MIC) of sulfamethoxazole. The results shown in Table 2 reveal that both P. lavamentivorans and L. interrogans chromosomal folP∗ genes confer resistance to sulfamethoxazole in an E. coli strain sensitive to sulfonamides. These data are in agreement with previous reporting of sulfonamide resistance in multiple L. interrogans strains (Chakraborty et al., 2010, 2011; Wuthiekanun et al., 2015), and suggest that the observed resistance was likely due to mutations in the Leptospiraceae chromosomal folP∗ gene rather than to the presence of plasmid-borne sul genes. Moreover, our results show that complementation with folP genes from another Alphaproteobacteria family lacking the Sul motif, the Rhodobacteraceae, does not confer resistance to sulfamethoxazole. These results reveal that the chromosomal folP∗ genes that gave rise to sul genes are capable of conferring resistance to sulfonamide in E. coli. In contrast with the Leptospiraceae and the Rhodobiaceae folP∗ genes, the chromosomal folKP gene of the Chlamydiae, which encodes a DHPS lacking the Sul motif, does not confer resistance to sulfamethoxazole (Table 2). This is in agreement with abundant reports of sulfonamide susceptibility in several Chlamydia species (Hammerschlag, 1982; Fan et al., 1992; Sandoz and Rockey, 2010; Marti et al., 2018). Since the Chlamydiae folKP gene is the most closely related chromosomal folP gene to the cluster encompassing the sul genes and the Leptospiraceae and the Rhodobiaceae folP∗ (Figure 2), the lack of resistance in Chlamydiae folKP genes strongly suggests that changes in the region encompassing the Sul motif may be responsible for the observed resistance. This region is located in a connector loop within the N-terminal ‘pole’ of the eight-stranded α/β barrel of DHPS, which is involved in sulfonamide recognition (Rådström and Swedberg, 1988; Morgan et al., 2011). The two-amino acid insertion might hence result in decreased affinity for sulfonamide by locally disrupting folding, as has been proposed previously for similar insertions in chromosomal folP genes (Achari et al., 1997).

Table 2. Broth microdilution assays.
Discussion
Prevalence of Sulfonamide Resistance in Ancestral Bacteria
The evidence presented here converges toward an evolutionary scenario in which sul1-3 genes from clinical isolates derive from ancestral chromosomal mutations in the folP∗ gene of the Leptospiraceae and the Rhodobiaceae (Figure 5). The emergence and maintenance of a sulfonamide-resistant folP∗ gene in the Leptospiraceae and its subsequent transfer to the Rhodobiaceae suggests that it might have conveyed some selective advantage prior to the introduction of sulfonamides, but the advent of mutations providing significant resistance to sulfonamide and their subsequent spread could also have been fortuitous. In both scenarios, formally variants of an evolutionary exaptation process (Gould and Vrba, 1982), a resistance-causing mutation may arise and be maintained in the population in the absence of direct antibiotic selection. Upon the clinical introduction of the relevant antibiotic, selection favors the rapid spread of the resistance determinant.
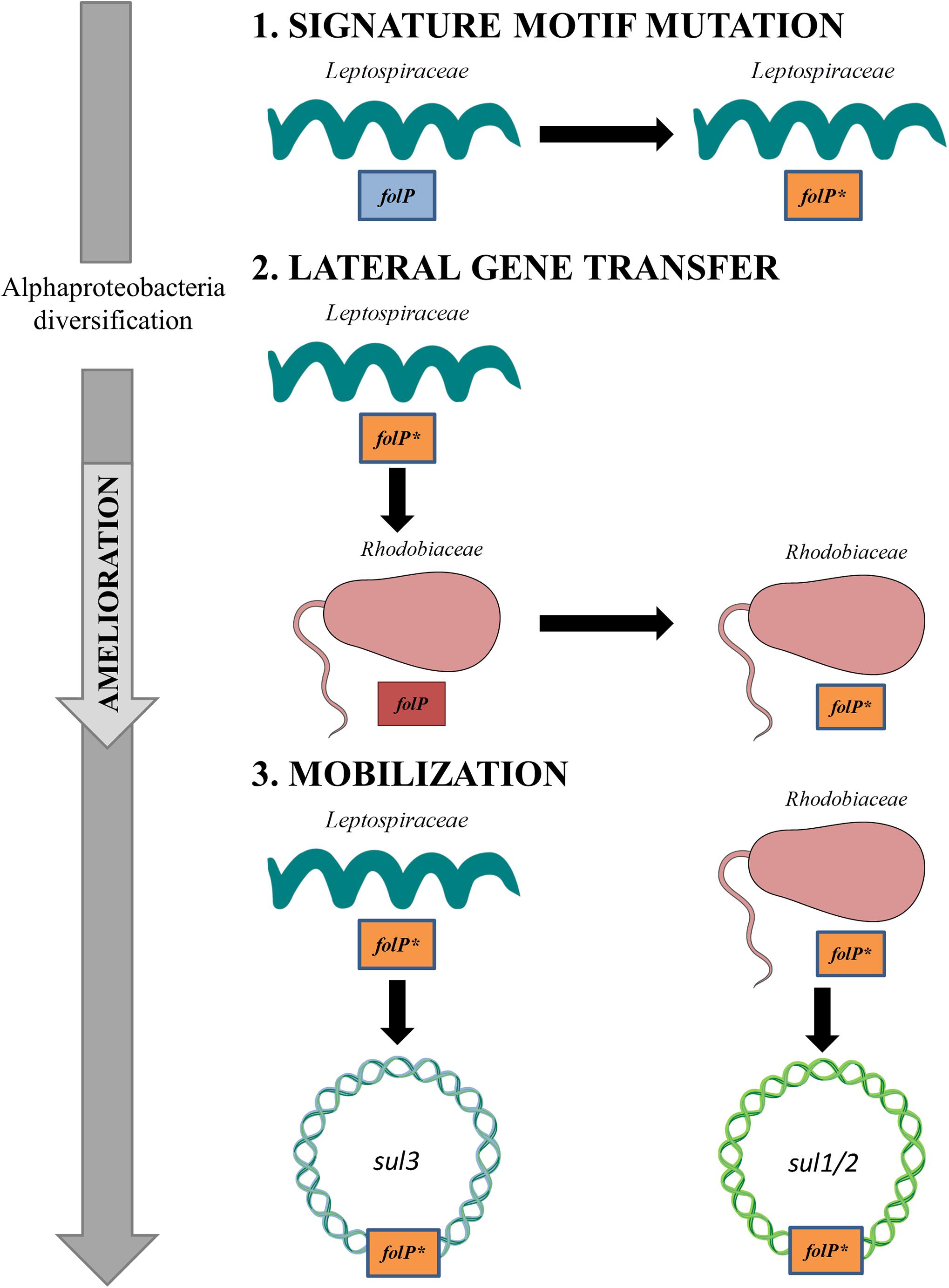
Figure 5. Schematic diagram of the evolutionary process leading to the emergence of sul1/2- and sul3-harboring mobile genetic elements. (1) A set of mutagenic events in the Leptospiraceae folP gene generates the signature motif observed in folP∗ and sul genes. (2) Following the diversification of the Alphaproteobacteria, the Leptospiraceae folP∗ gene is transferred to the Rhodobiaceae. (3) Upon the clinical and agricultural introduction of sulfonamides, folP∗ genes from the Leptospiraceae and the Rhodobiaceae are independently mobilized, giving rise to the sul–containing mobile elements reported in clinical isolates. This figure was constructed using some Servier Medical Art templates, which are licensed under a Creative Commons - Attribution Unported License.
The emergence and maintenance of resistance against synthetic chemotherapeutic agents prior to their clinical deployment may hence reside in the evolution of resistance as a side-effect of mutations providing some other fitness benefit. A case in point is the evolution and rapid mobilization via plasmid-borne qnr genes of resistance to quinolones (Nordmann and Poirel, 2005). Qnr proteins bind to DNA gyrase, the target of quinolones, early in the gyrase catalytic cycle and decrease binding of quinolones to the enzyme-DNA complex (Tran et al., 2005). Other members of the pentapeptide repeat family are known to bind DNA gyrase and provide some level of quinolone resistance (Hegde et al., 2005). Conversely, qnr-encoded proteins can provide cross-protection against naturally occurring antibiotics and synthetic agents targeting DNA gyrase (Hooper and Jacoby, 2016). The likely chromosomal origins of qnrA and qnrS genes have been traced to water-borne isolates of, respectively, Shewanella algae and Vibrio splendidus (Cantón, 2009) (Table 1). It has been hypothesized that these ancestral qnr genes and other pentapeptide repeat family members may have evolved to provide resistance against other compounds targeting DNA gyrase, such as the plasmid-borne microcin B17 (Heddle et al., 2001), as elements facilitating or modulating the normal function of DNA gyrase (Aminov and Mackie, 2007) or as functional components of the adaptation of Shewanella species to cold environments (Kim et al., 2011). In this context, the sequence determinants associated with chromosomal folP∗ genes may have originally enhanced PABA binding or another aspect of folate biosynthesis (Sköld, 2000), provided protection against hitherto unknown antibiotics targeting DHPS, or fulfilled an unrelated function.
The emergence and persistence of mutations conferring resistance against chemotherapeutic agents prior to their discovery may also have taken place in the absence of selection for the resistance-conferring mutations. The appearance of sulfonamide-resistance mutations in chromosomal folP genes has been amply documented (Huovinen et al., 1995; Sköld, 2000), and these were in fact the primary drivers of sulfonamide resistance following the introduction of sulfa drugs (Sköld, 2000). Furthermore, it has been documented that the presence of sulfonamide resistant DHPS does not necessarily impose a fitness cost on bacteria (Enne et al., 2004). Structural studies have suggested that most sulfonamide resistance mutations act by modulating accessibility of sulfonamides to the PABA-binding pocket without hindering PABA binding (Baca et al., 2000; Morgan et al., 2011). It is hence conceivable that naturally occurring mutations conferring resistance to sulfonamide might not be selected against in the absence of this chemotherapeutic agent. Subsequent complementary changes to adjust the affinity for PABA of the altered DHPS molecule may have resulted in fixation of the original mutations conferring resistance to sulfonamide (Andersson, 2006).
Alternatively, sulfonamide resistance mutations in folP may have arisen and persisted in response to naturally occurring sulfonamides produced by competing organisms. Sulfonamides are rare in nature, with only eight known natural sulfonamides reported to date (Petkowski et al., 2018). Of these, only two naturally occurring sulfonamides are aryl sulfonamides, produced in very small amounts by recombinant Streptomyces species harboring the complete xiamycin biosynthesis gene cluster (Baunach et al., 2015). Although these sulfonamides show potent antimicrobial activity, their bulky substitution pattern suggests that their mode of action and molecular target are likely different from synthetic aryl sulfonamides (Baunach et al., 2015).
Mobilization of Ancestral Resistance Reservoirs
The phylogenetic inference and genomic analysis results reported in this work uphold an evolutionary scenario wherein chromosomally encoded sulfonamide resistant folP variants were independently mobilized from Leptospiraceae and Rhodobiaceae backgrounds, and that mobile folP∗ genes subsequently spread very rapidly following the clinical introduction of synthetic aryl sulfonamides, giving rise to the sul1/2 and sul3 genes routinely detected in clinical isolates (Figure 5). The rapid mobilization and dissemination of genes conferring resistance to antibiotic and chemotherapeutic agents upon the clinical or agricultural use of these compounds has been amply documented (Aminov and Mackie, 2007; Stokes and Gillings, 2011). Mobilization and spread may be mediated by plasmids encoding transposons and integrons, as well as integrative and conjugative elements, mobile pathogenicity islands and bacteriophages, but the common tenet is that sustained exposure of bacterial populations to antibiotics or chemotherapeutic agents induces a strong selective pressure to elicit the mobilization of resistance determinants (Stokes and Gillings, 2011).
Together with penicillin and tetracycline, sulfonamides have been the antibacterial agents most frequently used at sub-therapeutic levels in livestock production (Franco et al., 1990), and it has been reported that sulfonamides have higher mobility, lower removal efficiency and deeper environmental penetration than most other antibacterial agents (Kumar et al., 2005). The widespread and intensive use of sulfonamides in agriculture, aquaculture and animal husbandry since the mid 1960’s, and their persistence in soil, sediments and subterranean aquatic communities where Leptospiraceae and Rhodobiaceae abound, provides an ample window of opportunity for the spread of chromosomally encoded or already mobilized folP∗ genes within these bacterial communities and the subsequent transfer of these mobile resistance determinants to other bacterial clades.
Recent mobilization from a Chloroflexi chromosomal folP background has been postulated as the likely origin of the sul4 gene (Razavi et al., 2017), and this result is in agreement with the phylogenetic analysis reported here (Figure 2). In the case of the chromosomal folP∗ genes identified here and their mobilization into sul-harboring resistance vectors, several sources of evidence provide additional support for the frequent mobilization of chromosomal folP genes. For instance, phylogenetic evidence (Figure 2) indicates that the Rhodobiaceae folP∗ was incorporated at some point by the Actinobacterium Amycolatopsis, which harbors three folP orthologs (Supplementary Data 7). Similarly, a plasmid broadly distributed among Azospirillum isolates (e.g., AP010951, FQ311873), a member of the Rhodospirillaceae Alphaproteobacteria family, contains a folP gene flanked by genes coding for a flagellar export pore protein (FlhB) and the full length phosphoglucosamine mutase (GlmM) (Supplementary Data 7). This folP does not contain the signature two-amino acid insertion, indicating that its mobilization occurred independently of those leading to sul1/2 genes.
More significantly, a partial genomic sequence from a Pseudomonas aeruginosa isolate (LLMY01000073.1) harbors a folP∗ gene with high sequence and genetic neighborhood similarity to the Rhodobiaceae P. lavamentivorans DS-1 (van Belkum et al., 2015). The genes immediately upstream and downstream of this P. aeruginosa folP∗, which contains the Sul motif, encode a TetR family regulator and a partial phosphoglucosamine mutase (GlmM) protein (Supplementary Data 7). These three genes are flanked by IS91 and ISL3 family transposases. Importantly, the IS91 transposase contains similar sequence motifs and shares termini identity with ISCR elements, which are present in both sul1 and sul2-harboring plasmids (Toleman et al., 2006; Toleman and Walsh, 2011). Moreover, this P. aeruginosa folP∗ presents 72.76% nucleotide identity with the P. lavamentivorans folP∗, which is significantly higher than the one observed between P. lavamentivorans folP∗ and sul2 (Table 1). It is hence highly likely that the P. aeruginosa folP∗ represents an intermediate step or an independent mobilization of the Rhodobiaceae folP∗.
Taken together, the protein sequence phylogeny and genomic context evidence (Figures 1–3), the absence of Sul-containing motifs in any other chromosomal folP genes (Figure 1), the tight alignment in %GC content between the different chromosomal folP∗ and mobile sul genes (Figure 4), and the identification of multiple folP mobilization events consistently point toward an independent mobilization from Leptospiraceae and Rhodobiaceae chromosomal backgrounds that gave rise to, respectively, sul3 and sul2/1 genes. The close similarity in %GC content between the putative donor and mobilized sequences (Figure 4), and the lack of suitable models for sequence evolution in mobile elements make it difficult to estimate precisely the timing of this mobilization using sequence analysis methods. The relatively low nucleotide sequence identity between the Leptospiraceae and Rhodobiaceae chromosomal folP∗ genes and sul genes (Table 1) suggests that folP∗ genes were mobilized and diversified long before the clinical introduction of sulfonamides. Nonetheless, extensive recombination and unusual selection pressures on mobile elements could in principle also account for the observed sequence divergence within a shorter timescale. What seems abundantly clear is that shortly after the clinical introduction of sulfonamides, sul genes spread rapidly on a variety of mobile elements, as attested by the well-established association between sul genes and integrons (Lévesque et al., 1995; Davies, 2007).
Metagenomics analysis and prospective studies of preserved ancient environments, such as permafrost and remote cave habitats, have largely displaced the notion that antibiotic resistance emerges in response to anthropogenic antibiotic use (D’Costa et al., 2011; Bhullar et al., 2012; Perron et al., 2015; Crofts et al., 2017). These studies have conclusively shown that antibiotic resistance predates the use of antibiotics by humans, and that it is widely distributed across the bacterial pangenome. In a few isolated cases, resistance determinants for synthetic chemotherapeutic agents that predate or have rapidly arisen upon human use has been documented, but their existence can be attributed to cross-resistance to naturally occurring antibiotics [e.g., microcin B17 for quinolones (Tran and Jacoby, 2002), sisomicin for amikacin (Perron et al., 2015)]. The identification in this work of ancient chromosomal mutations in folP conferring resistance to sulfonamide as the likely origins of the sul1-3 genes present in sulfonamide-resistant clinical isolates puts forward an alternative scenario. Given the absence of known naturally occurring aryl sulfonamides targeting DHPS, our results suggest that resistance to novel synthetic chemotherapeutic agents may be already available in the vast microbial pangenome, and that its global dissemination can take place in a very short amount of time upon the clinical introduction of novel chemotherapeutic compounds.
Data Availability Statement
The datasets used in this study can all be freely accessed at the NCBI GenBank/RefSeq databases (https://www.ncbi.nlm.nih.gov/). All scripts used for analysis can be obtained at the GitHub ErillLab repository (https://github.com/ErillLab/).
Author Contributions
MS-O and IE performed the in silico analyses. MS-O and IE developed scripts for genomic analyses and ran phylogenetic inference methods. MS-O and PC performed the in vitro analyses. IE and JB conceived the experiments and coordinated the research. IE and MS-O drafted the manuscript. All authors discussed the findings and interpreted the results.
Funding
This work was supported by grant BIO2016-77011-R from the Spanish Ministerio de Economia y Competitividad to JB. MS-O was the recipient of a predoctoral fellowship from the Ministerio de Educación, Cultura y Deporte de España.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Joan Ruiz and Susana Escribano for their technical support during some of the experimental procedures, as well as Júlia López for her continued support. A preprint version of this manuscript prior to peer review is available on bioRxiv (https://doi.org/10.1101/472472).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03332/full#supplementary-material
References
Achari, A., Somers, D. O., Champness, J. N., Bryant, P. K., Rosemond, J., and Stammers, D. K. (1997). Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat. Struct. Biol. 4, 490–497. doi: 10.1038/nsb0697-490
Altenhoff, A. M., Glover, N. M., Train, C.-M., Kaleb, K., Warwick Vesztrocy, A., Dylus, D., et al. (2018). The OMA orthology database in 2018: retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 46, D477–D485. doi: 10.1093/nar/gkx1019
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Aminov, R. I. (2010). A brief history of the antibiotic era: lessons learned and challenges for the future. Front. Microbiol. 1:134. doi: 10.3389/fmicb.2010.00134
Aminov, R. I., and Mackie, R. I. (2007). Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 271, 147–161. doi: 10.1111/j.1574-6968.2007.00757.x
Andersson, D. I. (2006). The biological cost of mutational antibiotic resistance: any practical conclusions? Curr. Opin. Microbiol. 9, 461–465. doi: 10.1016/j.mib.2006.07.002
Baca, A. M., Sirawaraporn, R., Turley, S., Sirawaraporn, W., and Hol, W. G. (2000). Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J. Mol. Biol. 302, 1193–1212. doi: 10.1006/jmbi.2000.4094
Battistuzzi, F. U., Feijao, A., and Hedges, S. B. (2004). A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 4:44. doi: 10.1186/1471-2148-4-44
Baunach, M., Ding, L., Willing, K., and Hertweck, C. (2015). Bacterial synthesis of unusual sulfonamide and sulfone antibiotics by flavoenzyme-mediated sulfur dioxide capture. Angew. Chem. Int. Ed Engl. 54, 13279–13283. doi: 10.1002/anie.201506541
Bhullar, K., Waglechner, N., Pawlowski, A., Koteva, K., Banks, E. D., Johnston, M. D., et al. (2012). Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7:e34953. doi: 10.1371/journal.pone.0034953
Cantón, R. (2009). Antibiotic resistance genes from the environment: a perspective through newly identified antibiotic resistance mechanisms in the clinical setting. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 15(Suppl. 1), 20–25. doi: 10.1111/j.1469-0691.2008.02679.x
Carlet, J., Rambaud, C., and Pulcini, C. (2014). Save antibiotics: a call for action of the world alliance against antibiotic resistance (WAAAR). BMC Infect. Dis. 14:436. doi: 10.1186/1471-2334-14-436
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chakraborty, A., Miyahara, S., Villanueva, S. Y. A. M., Gloriani, N. G., and Yoshida, S. (2010). In vitro sensitivity and resistance of 46 leptospira strains isolated from rats in the philippines to 14 antimicrobial agents. Antimicrob. Agents Chemother. 54, 5403–5405. doi: 10.1128/AAC.00973-10
Chakraborty, A., Miyahara, S., Villanueva, S. Y. A. M., Saito, M., Gloriani, N. G., and Yoshida, S. (2011). A novel combination of selective agents for isolation of Leptospira species. Microbiol. Immunol. 55, 494–501. doi: 10.1111/j.1348-0421.2011.00347.x
Clinical and Laboratory Standards Institute (2003). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically –Approved Standard, 6 Edn. Wayne, PA: Clinical and Laboratory Standards Institute.
Colaert, N., Helsens, K., Martens, L., Vandekerckhove, J., and Gevaert, K. (2009). Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787. doi: 10.1038/nmeth1109-786
Crofts, T. S., Gasparrini, A. J., and Dantas, G. (2017). Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 15, 422–434. doi: 10.1038/nrmicro.2017.28
Davenport, D. (2012). The war against bacteria: how were sulphonamide drugs used by Britain during World War II? Med. Human. 38, 55–58. doi: 10.1136/medhum-2011-010024
Davies, J. (2007). Microbes have the last word. A drastic re-evaluation of antimicrobial treatment is needed to overcome the threat of antibiotic-resistant bacteria. EMBO Rep. 8, 616–621. doi: 10.1038/sj.embor.7401022
Davies, J., and Davies, D. (2010). Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433. doi: 10.1128/MMBR.00016-10
D’Costa, V. M., King, C. E., Kalan, L., Morar, M., Sung, W. W. L., Schwarz, C., et al. (2011). Antibiotic resistance is ancient. Nature 477:457. doi: 10.1038/nature10388
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. doi: 10.1093/nar/gkn180
Domagk, G. (1935). Ein beitrag zur chemotherapie der bakteriellen infektionen. Dtsch. Med. Wochenschr. 61, 250–253. doi: 10.1055/s-0028-1129486
Enne, V. I., Bennett, P. M., Livermore, D. M., and Hall, L. M. C. (2004). Enhancement of host fitness by the sul2-coding plasmid p9123 in the absence of selective pressure. J. Antimicrob. Chemother. 53, 958–963. doi: 10.1093/jac/dkh217
Enne, V. I., Livermore, D. M., Stephens, P., and Hall, L. M. (2001). Persistence of sulphonamide resistance in Escherichia coli in the UK despite national prescribing restriction. Lancet 357, 1325–1328. doi: 10.1016/S0140-6736(00)04519-0
Fan, H., Brunham, R. C., and McClarty, G. (1992). Acquisition and synthesis of folates by obligate intracellular bacteria of the genus Chlamydia. J. Clin. Invest. 90, 1803–1811. doi: 10.1172/JCI116055
Fermer, C., Kristiansen, B. E., Sköld, O., and Swedberg, G. (1995). Sulfonamide resistance in Neisseria meningitidis as defined by site-directed mutagenesis could have its origin in other species. J. Bacteriol. 177, 4669–4675. doi: 10.1128/jb.177.16.4669-4675.1995
Franco, D. A., Webb, J., and Taylor, C. E. (1990). Antibiotic and sulfonamide residues in meat: implications for human health. J. Food Prot. 53, 178–185. doi: 10.4315/0362-028X-53.2.178
Gould, S. J., and Vrba, E. S. (1982). Exaptation—a missing term in the science of form. Paleobiology 8, 4–15. doi: 10.1017/S0094837300004310
Haasum, Y., Ström, K., Wehelie, R., Luna, V., Roberts, M. C., Maskell, J. P., et al. (2001). Amino acid repetitions in the dihydropteroate synthase of Streptococcus pneumoniae lead to sulfonamide resistance with limited effects on substrateKm. Antimicrob. Agents Chemother. 45, 805–809. doi: 10.1128/AAC.45.3.805-809.2001
Hammerschlag, M. R. (1982). Activity of trimethoprim-sulfamethoxazole against Chlamydia trachomatis in vitro. Rev. Infect. Dis. 4, 500–505. doi: 10.1093/clinids/4.2.500
Heddle, J. G., Blance, S. J., Zamble, D. B., Hollfelder, F., Miller, D. A., Wentzell, L. M., et al. (2001). The antibiotic microcin B17 is a DNA gyrase poison: characterisation of the mode of inhibition. J. Mol. Biol. 307, 1223–1234. doi: 10.1006/jmbi.2001.4562
Hegde, S. S., Vetting, M. W., Roderick, S. L., Mitchenall, L. A., Maxwell, A., Takiff, H. E., et al. (2005). A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308, 1480–1483. doi: 10.1126/science.1110699
Honda, Y., Sakai, H., Hiasa, H., Tanaka, K., Komano, T., and Bagdasarian, M. (1991). Functional division and reconstruction of a plasmid replication origin: molecular dissection of the oriV of the broad-host-range plasmid RSF1010. Proc. Natl. Acad. Sci. U.S.A. 88, 179–183. doi: 10.1073/pnas.88.1.179
Hooper, D. C. (1999). Mechanisms of fluoroquinolone resistance. Drug Resist. Update 2, 38–55. doi: 10.1054/drup.1998.0068
Hooper, D. C., and Jacoby, G. A. (2016). Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 6:a025320. doi: 10.1101/cshperspect.a025320
Hu, L. F., Xu, X. H., Yang, H. F., Ye, Y., and Li, J. B. (2016). Role of sul2 gene linked to transposase in resistance to trimethoprim/sulfamethoxazole among Stenotrophomonas maltophilia isolates. Ann. Lab. Med. 36, 73–75. doi: 10.3343/alm.2016.36.1.73
Huovinen, P., Sundström, L., Swedberg, G., and Sköld, O. (1995). Trimethoprim and sulfonamide resistance. Antimicrob. Agents Chemother. 39, 279–289. doi: 10.1128/AAC.39.2.279
Kannan, L., Li, H., Rubinstein, B., and Mushegian, A. (2013). Models of gene gain and gene loss for probabilistic reconstruction of gene content in the last universal common ancestor of life. Biol. Direct 8:32. doi: 10.1186/1745-6150-8-32
Kehrenberg, C., and Schwarz, S. (2005). dfrA20, A novel trimethoprim resistance gene from Pasteurella multocida. Antimicrob. Agents Chemother. 49, 414–417. doi: 10.1128/AAC.49.1.414-417.2005
Kim, H. B., Park, C. H., Gavin, M., Jacoby, G. A., and Hooper, D. C. (2011). Cold shock induces qnrA expression in shewanella algae. Antimicrob. Agents Chemother. 55, 414–416. doi: 10.1128/AAC.00991-10
Kumar, K., Gupta, S. C., Chander, Y., and Singh, A. K. (2005). Antibiotic use in agriculture and its impact on the terrestrial environment. Adv. Agron. 87, 1–54. doi: 10.1016/S0065-2113(05)87001-4
Kushner, S. R. (1978). “An improved method for transformation of. Escherichia coli with ColE 1 derived plasmids,” in Genetic Engineering, eds H. W. Boyer and S. Nicosia (Amsterdam: Elsevier), 17–23.
Landy, M., Larkum, N. W., Oswald, E. J., and Streightoff, F. (1943). Increased synthesis of p-aminobenzoic acid associated with the development of sulfonamide resistance in Staphylococcus aureus. Science 97, 265–267. doi: 10.1126/science.97.2516.265
Lawrence, J. G., and Ochman, H. (1997). Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 44, 383–397. doi: 10.1007/PL00006158
Lévesque, C., Piché, L., Larose, C., and Roy, P. H. (1995). PCR mapping of integrons reveals several novel combinations of resistance genes. Antimicrob. Agents Chemother. 39, 185–191. doi: 10.1128/AAC.39.1.185
Marti, H., Borel, N., Dean, D., and Leonard, C. A. (2018). Evaluating the antibiotic susceptibility of chlamydia – New approaches for in vitro assays. Front. Microbiol. 9:1414. doi: 10.3389/fmicb.2018.01414
Mayola, A., Irazoki, O., Martínez, I. A., Petrov, D., Menolascina, F., Stocker, R., et al. (2014). RecA protein plays a role in the chemotactic response and chemoreceptor clustering of Salmonella enterica. PLoS One 9:e105578. doi: 10.1371/journal.pone.0105578
Mehra-Chaudhary, R., Mick, J., and Beamer, L. J. (2011). Crystal structure of Bacillus anthracis phosphoglucosamine mutase, an enzyme in the peptidoglycan biosynthetic pathway? J. Bacteriol. 193, 4081–4087. doi: 10.1128/JB.00418-11
Mitsuhashi, S. (1993). Drug resistance in bacteria: history, genetics and biochemistry. J. Int. Med. Res. 21, 1–14. doi: 10.1177/030006059302100101
Morgan, R. E., Batot, G. O., Dement, J. M., Rao, V. A., Eadsforth, T. C., and Hunter, W. N. (2011). Crystal structures of Burkholderia cenocepacia dihydropteroate synthase in the apo-form and complexed with the product 7,8-dihydropteroate. BMC Struct. Biol. 11:21. doi: 10.1186/1472-6807-11-21
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426. doi: 10.1093/oxfordjournals.molbev.a040410
Nordmann, P., and Poirel, L. (2005). Emergence of plasmid-mediated resistance to quinolones in Enterobacteriaceae. J. Antimicrob. Chemother. 56, 463–469. doi: 10.1093/jac/dki245
Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217. doi: 10.1006/jmbi.2000.4042
Orman, B. E., Piñeiro, S. A., Arduino, S., Galas, M., Melano, R., Caffer, M. I., et al. (2002). Evolution of multiresistance in nontyphoid salmonella serovars from 1984 to 1998 in Argentina. Antimicrob. Agents Chemother. 46, 3963–3970. doi: 10.1128/AAC.46.12.3963-3970.2002
Perreten, V., and Boerlin, P. (2003). A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob. Agents Chemother. 47, 1169–1172. doi: 10.1128/AAC.47.3.1169-1172.2003
Perron, G. G., Whyte, L., Turnbaugh, P. J., Goordial, J., Hanage, W. P., Dantas, G., et al. (2015). Functional characterization of bacteria isolated from ancient arctic soil exposes diverse resistance mechanisms to modern antibiotics. PLoS One 10:e0069533. doi: 10.1371/journal.pone.0069533
Petkowski, J. J., Bains, W., and Seager, S. (2018). Natural products containing a Nitrogen–Sulfur bond. J. Nat. Prod. 81, 423–446. doi: 10.1021/acs.jnatprod.7b00921
Rådström, P., Fermér, C., Kristiansen, B. E., Jenkins, A., Sköld, O., and Swedberg, G. (1992). Transformational exchanges in the dihydropteroate synthase gene of Neisseria meningitidis: a novel mechanism for acquisition of sulfonamide resistance. J. Bacteriol. 174, 6386–6393. doi: 10.1128/jb.174.20.6386-6393.1992
Rådström, P., and Swedberg, G. (1988). RSF1010 and a conjugative plasmid contain sulII, one of two known genes for plasmid-borne sulfonamide resistance dihydropteroate synthase. Antimicrob. Agents Chemother. 32, 1684–1692. doi: 10.1128/AAC.32.11.1684
Rådström, P., Swedberg, G., and Sköld, O. (1991). Genetic analyses of sulfonamide resistance and its dissemination in gram-negative bacteria illustrate new aspects of R plasmid evolution. Antimicrob. Agents Chemother. 35, 1840–1848. doi: 10.1128/AAC.35.9.1840
Razavi, M., Marathe, N. P., Gillings, M. R., Flach, C.-F., Kristiansson, E., and Joakim Larsson, D. G. (2017). Discovery of the fourth mobile sulfonamide resistance gene. Microbiome 5:160. doi: 10.1186/s40168-017-0379-y
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rossolini, G. M., Arena, F., Pecile, P., and Pollini, S. (2014). Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 18, 56–60. doi: 10.1016/j.coph.2014.09.006
Sandoz, K. M., and Rockey, D. D. (2010). Antibiotic resistance in chlamydiae. Future Microbiol. 5, 1427–1442. doi: 10.2217/fmb.10.96
Sköld, O. (2000). Sulfonamide resistance: mechanisms and trends. Drug Resist. Updat. 3, 155–160. doi: 10.1054/drup.2000.0146
Stokes, H. W., and Gillings, M. R. (2011). Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 35, 790–819. doi: 10.1111/j.1574-6976.2011.00273.x
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Swedberg, G., Ringertz, S., and Sköld, O. (1998). Sulfonamide resistance in streptococcus pyogenes is associated with differences in the amino acid sequence of its chromosomal dihydropteroate synthase. Antimicrob. Agents Chemother. 42, 1062–1067. doi: 10.1128/AAC.42.5.1062
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Toleman, M. A., Bennett, P. M., and Walsh, T. R. (2006). ISCR elements: novel gene-capturing systems of the 21st century? Microbiol. Mol. Biol. Rev. 70, 296–316. doi: 10.1128/MMBR.00048-05
Toleman, M. A., and Walsh, T. R. (2011). Combinatorial events of insertion sequences and ICE in Gram-negative bacteria. FEMS Microbiol. Rev. 35, 912–935. doi: 10.1111/j.1574-6976.2011.00294.x
Tran, J. H., and Jacoby, G. A. (2002). Mechanism of plasmid-mediated quinolone resistance. Proc. Natl. Acad. Sci. U.S.A. 99, 5638–5642. doi: 10.1073/pnas.082092899
Tran, J. H., Jacoby, G. A., and Hooper, D. C. (2005). Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase. Antimicrob. Agents Chemother. 49, 118–125. doi: 10.1128/AAC.49.1.118-125.2005
Treeck, U., van Schmidt, F., and Wiedemann, B. (1981). Molecular nature of a streptomycin and sulfonamide resistance plasmid (pBP1) prevalent in clinical Escherichia coli strains and integration of an ampicillin resistance transposon (TnA). Antimicrob. Agents Chemother. 19, 371–380. doi: 10.1128/AAC.19.3.371
van Belkum, A., Soriaga, L. B., LaFave, M. C., Akella, S., Veyrieras, J.-B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio 6:e01796-15. doi: 10.1128/mBio.01796-15
Keywords: sulfonamide, resistance, antibiotic, phylogeny, mobile element, integron, plasmid
Citation: Sánchez-Osuna M, Cortés P, Barbé J and Erill I (2019) Origin of the Mobile Di-Hydro-Pteroate Synthase Gene Determining Sulfonamide Resistance in Clinical Isolates. Front. Microbiol. 9:3332. doi: 10.3389/fmicb.2018.03332
Received: 14 September 2018; Accepted: 21 December 2018;
Published: 10 January 2019.
Edited by:
Laurent Poirel, Université de Fribourg, SwitzerlandReviewed by:
Jose L. Martinez, Spanish National Research Council (CSIC), SpainRune Henning Sørum, Norwegian University of Life Sciences, Norway
Copyright © 2019 Sánchez-Osuna, Cortés, Barbé and Erill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordi Barbé, jordi.barbe@uab.cat Ivan Erill, erill@umbc.edu