 Natalia Olivero-Deibe1*
Natalia Olivero-Deibe1* Lorena Tomé-Poderti1,2†Federico Carrión1†Sergio Bianchi1,3Martín Fló1,4
Lorena Tomé-Poderti1,2†Federico Carrión1†Sergio Bianchi1,3Martín Fló1,4 Daniel Prieto5,6
Daniel Prieto5,6 Florencia Rammauro1,4Andrés Addiego1Natalia Ibañez1Madelón Portela7
Florencia Rammauro1,4Andrés Addiego1Natalia Ibañez1Madelón Portela7 Rosario Duran8Mabel Berois9
Rosario Duran8Mabel Berois9 Otto Pritsch1,4*
Otto Pritsch1,4*- 1Laboratorio de Inmunovirología, Institut Pasteur de Montevideo, Montevideo, Uruguay
- 2Morphogenèse et Antigénicité du VIH et des Virus des Hépatites (MAVIVH), UMR Inserm U1259, UFR de Médecine, Tours, France
- 3Laboratorio de Biomarcadores Moleculares, Departamento de Fisiopatología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
- 4Departamento de Inmunobiología, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
- 5Departamento de Biología de Neurodesarrollo, Instituto de Investigaciones Biológicas Clemente Estable, Montevideo, Uruguay
- 6Unidad de Bioimagenología Avanzada, Institut Pasteur de Montevideo, Montevideo, Uruguay
- 7Unidad de Bioquímica y Proteómica Analíticas, Institut Pasteur de Montevideo, Facultad de Ciencias, Montevideo, Uruguay
- 8Unidad de Bioquímica y Proteómica Analíticas, Institut Pasteur de Montevideo, Instituto de Investigaciones Biológicas Clemente Estable (IIBCE), Montevideo, Uruguay
- 9Sección Virología, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay
Bovine leukemia virus (BLV) is an oncogenic deltaretrovirus that infects cattle worldwide. In Uruguay, it is estimated that more than 70% of dairy cattle are infected, causing serious economic losses due to decreased milk production, increased calving interval, and livestock losses due to lymphosarcoma. Several attempts to develop vaccine candidates that activate protective immune responses against BLV were performed, but up to date, there is no vaccine that ensures efficient protection and/or decreased viral transmission. The development and application of new vaccines that effectively control BLV infection represent a major challenge for countries with a high prevalence of infection. In this study, we generated two Drosophila melanogaster S2 stable cell lines capable of producing BLV virus-like particles (BLV-VLPs). One of them, BLV-VLP1, expressed both Gag and Env wild-type (Envwt) full-length proteins, whereas BLV-VLP2 contain Gag together with a mutant form of Env non-susceptible to proteolytic maturation by cellular furin type enzymes (EnvFm). We showed that Envwt is properly cleaved by cellular furin, whereas EnvFm is produced as a full-length gp72 precursor, which undergoes some partial cleavage. We observed that said mutation does not drastically affect its expression or its entry into the secretory pathway of S2 insect cells. In addition, it is expressed on the membrane and retains significant structural motifs when expressed in S2 insect cells. Morphology and size of purified BLV-VLPs were analyzed by transmission electron microscopy and dynamic light scattering, showing numerous non-aggregated and approximately spherical particles of variable diameter (70–200 nm) as previously reported for retroviral VLPs produced using different expression systems. Furthermore, we identified two N-glycosylation patterns rich in mannose in EnvFm protein displayed on VLP2. Our results suggest that the VLPs produced in Drosophila S2 cells could be a potential immunogen to be used in the development of BLV vaccines that might contribute, in conjunction with other control strategies, to reduce the transmission of the virus.
Introduction
Enzootic bovine leukosis, caused by the bovine leukemia virus (BLV), infects cattle worldwide. Two-thirds of the infected animals are asymptomatic, around 30% displays persistent lymphocytosis, and between 5 and 10% develop an aggressive tumor pathology called lymphosarcoma, which frequently causes the death of these animals (1, 2).
Horizontal BLV transmission between herd animals depends on the transfer of infected cells from blood, semen, or mucous fluids. This cellular transfer is carried out by direct contact, by iatrogenic veterinary procedures, or by insect bites. Vertical BLV transmission from cow to calf depends on the transfer of infected cells through the placenta, by contact with infected blood during calving, and through feeding with colostrum and milk from infected cows (3, 4).
BLV infection leads to high economic losses in dairy and beef industries, related to mortality caused by lymphosarcoma (5); negative impact on production parameters (6, 7); immunological alteration and secondary infections (8); and restriction on the international trade of live cattle, semen, and infected embryos (3, 4, 9). European and Oceanian countries have developed efficient programs to control and eradicate BLV infection to reduce economic losses and to control the spread of this disease (3). However, in the Americas region, most countries display high enzootic bovine leukosis prevalence.
In this context, the control strategy designed to fight against BLV infection includes a set of actions aimed at reducing viral transmission. These include improving veterinary practices, physical separation of infected and non-infected animals, and genetic selection of animals resistant to infection. Likewise, the use of vaccines that generate protective immunity has been proposed as an essential tool to reduce BLV infection (2).
Several attempts to develop vaccine candidates that activate protective immune responses against BLV were performed, but up to date, there is no vaccine that ensures efficient protection and/or decreased viral transmission (4). Inactivated virus vaccines (10, 11), cell lysates from BLV-infected cells (12), recombinant viral antigens (gp51 and p24 proteins) (13, 14), and synthetic peptides were tested for immunization. These vaccine candidates demonstrated immunogenicity, but they did not display efficient protection from the BLV challenge. Other vaccines, such as recombinant vaccinia virus expressing BLV envelope protein, only conferred partial protection in sheep but were ineffective in cows. Novel DNA-based vaccines containing env and tax genes elicited strong immune responses but did not prevent later infection or generate an effective immunological memory (15–17). Recently, a new viral-attenuated vaccine platform based on a BLV provirus with multiple deletions in oncogenic genes (tax, G4, microRNAs) and genes responsible for infectivity or replication was reported (env and G3). This modified provirus is infectious in cattle but displays deficient levels of replication (18, 19). This approach is auspicious, but several regulatory aspects must be solved before its massive use.
The use of virus-like particle (VLP)-based vaccines on preventing different viral diseases appears as an alternative strategy to traditional formulations (20, 21). In general, VLPs mimic the natural virus while lacking the viral genetic material, thus making them safe (without the risk of virus replication) and are highly immunogenic (22, 23). Due to their particulate nature, it is very efficiently uptaken by antigen-presenting cells leading to potent activation of humoral and cellular immune responses (20). VLPs are produced in different cellular systems, including yeast, bacteria, mammalian, plants, and insect cells (24–26). Drosophila melanogaster Schneider (S2) cells have been used in the efficient production of several VLP types (27–30). S2 cells display several advantages as a eukaryotic system for protein expression: simple glycosylation profiles, fast-growing in a serum-free medium, very high cell densities without aggregation or toxic metabolite issues, and lack of adventitious agents that could infect humans (27).
Enveloped VLPs usually include scaffold proteins that can be coated by immunogenic proteins to enhance the immune response. In this regard, Yang et al. (28) produced human immunodeficiency virus (HIV) VLPs in S2 cells, including Gag and Env proteins, and demonstrated that HIV Env glycoprotein was properly cleaved, glycosylated, and incorporated into VLPs (28).
BLV Env glycoprotein trimer binds to the cellular receptor cationic amino acid transporter-1/solute carrier family 7 member 1 (31), promoting the fusion of the viral and host cell membranes and plays a crucial role in determining viral infectivity (32, 33). BLV Env is synthesized as a glycosylated polypeptide precursor (gp72) that is cleaved by cellular furin proteases into surface gp51 (SU) and the transmembrane gp30 (TM) subunits; both proteins are associated through a disulfide bond. This cleavage gives rise to a metastable pre-fusion conformation that has the necessary energy to carry out the fusion process once the recognition with the cellular receptor occurs (34). Env is a major target of antiviral immunity, as indicated by the rapid emergence of neutralizing antibodies after viral inoculation (35). The presence of helper CD4+ and cytotoxic T-lymphocyte CD8+ epitopes in gp51 and gp30 indicates that Env also stimulates a T-cell response (32, 35).
The Gag protein plays important structural roles responsible for intracellular transport, direct assembly of virus particles, and catalyzing the budding process (33). Expression of the Gag polyprotein alone can lead to the formation of VLPs (36). BLV Gag is initially synthesized as a polyprotein that is cleaved into major structural domains [matrix (MA, p15), capsid (CA, p24), and nucleocapsid (NC, p12)] (37). The CA protein is also a major target of antiviral immunity with high antibody titers found in the sera of infected animals and contains two CD4+ epitopes recognized by specific T lymphocytes (38).
BLV VLPs have been produced in different systems by using mammalian, silkworm, or other insect cells. These BLV VLPs were only composed of Gag polyprotein, and none of them contained Env surface glycoprotein (37, 39–41).
In this study, we describe for the first time the production and characterization of BLV VLPs containing Env and Gag immunogenic proteins expressed in Drosophila melanogaster S2 stable cell lines. Purified VLP morphology was analyzed by negative staining transmission electron microscopy (TEM) and dynamic light scattering (DLS), showing a polydisperse population with shape and sizes compatible with previously reported data for another retrovirus VLPs (42–49). Gag and Env in VLPs were detected by Western blot and further confirmed by mass spectrometry.
Materials and Methods
Construction Design for Protein Expression in Drosophila Cells
DNA sequences of BLV (GenBank, access number: EF600696) encoding the precursor Gag protein (amplified from FLK-BLV cells, DSMZ #ACC 153) and Env glycoprotein (synthesized by Genscript) were cloned by RF cloning in pMT/BiP/V5-His expression vector (Invitrogen). All constructs were cloned under the control of Drosophila metal-inducible metallothionein promoter (MT), followed by a stop codon immediately after its C-terminus. All sequences were confirmed by automated sequencing (Macrogen, Korea).
Two different constructs (483 aa) were designed to express BLV-Env protein: the native form, pMT/BiP/Envwt, which is processed by cellular furin proteases, giving rise to SU and TM subunits and the furin-mutated form, pMT/BiP/EnvFm, containing a double substitution at positions R301N and R302N to avoid its proteolytic processing (50), leading to a stable Env precursor protein. The Drosophila BiP secretory sequence replaced the native signal peptide to ensure its entry into the secretory pathway.
In the case of pMT/Gag, the N-terminus of the Gag protein (393 aa) corresponds to its first native amino acid (lacking the BiP signal from the expression vector) and giving rise to a cytosolic protein.
Stable Transfection of S2 Drosophila Cell Line
Twenty-four hours before transfection, S2 cells were seeded in T25 flasks (1 × 106 cells/ml) and grown at 28°C in Schneider's culture medium supplemented with 10% fetal bovine serum (Invitrogen) and 50-unit penicillin/50-μg streptomycin per milliliter of medium. Co-transfection was performed with 0.1-μg pCoPuro plasmid (Invitrogen; conferring resistance to puromycin), 1-μg of pMT/Gag, and 1-μg pMT/Bip/Envwt or pMT/Bip/EnvFm using Effectene Transfection Reagent (Qiagen) according to the manufacturer's instructions. Three days post-transfection, 6 μg/ml puromycin was added, and resistant cells were selected and gradually adapted to the protein-free culture medium (Insect Xpress, Lonza) for 4 weeks to generate two stable cell lines Gag/Envwt-S2 and Gag/EnvFm-S2 capable of expressing Gag and Envwt or EnvFm proteins upon induction with 5-μM CdCl2 (Sigma).
Immunofluorescence Assay
The expression of recombinant proteins was evidenced by indirect immunofluorescence and confocal microscopy. Briefly, 50 μl of induced cells were fixed for 30 min with 4% (v/v) paraformaldehyde in PBS, washed three times with PBS, and subsequently blocked for 15 min with PBS-3%BSA-0.1% Triton X-100. Cells were further incubated 1 h at RT with monoclonal antibodies anti-p24 (BLV3, 1/200) or anti-gp51 (BLV1, 1/500) (VMRD, USA). After three washes with PBS, cells were incubated for 1 h at RT with Alexa Fluor®594 conjugated goat anti-mouse IgG (Invitrogen) diluted 1/1,000 in PBS added with 3% (w/v) BSA and methyl green 1/5,000, washed three times with PBS and mounted in 70% (v/v) glycerol pH 8.8. Cell images were acquired on a Leica TCS SP5 microscope with a 63 × oil immersion lens (NA = 1.4), pinhole set at 1 Airy unit. Stacks were deconvolved with FIJI using DeconvolutionLab2 software (51), applying a Fast Iterative Shrinkage-Thresholding algorithm (52) and using a theoretical PSF estimated with the Born and Wolf 3D optical model.
Cell Surface Env Protein Expression Analysis by Flow Cytometry Analysis
Induced or non-induced EnvFm/Gag-S2 cells were incubated with a monoclonal antibody anti-gp51 (BLV1, 1/50) (VMRD, USA) and subsequently stained with a goat anti-mouse IgG Alexa-594 secondary antibody (Invitrogen). Cells were immediately acquired, without permeabilization or fixation, using a BD FACSAriaTM Fusion Cell Sorter (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed with FlowJo software (Tree Star, Inc.). Unstained cells were used as a control of fluorescence background.
Production and Purification of Virus-Like Particles
Envwt/Gag-S2 and EnvFm/Gag-S2 cells were cultured at 28°C with moderate agitation in glass Erlenmeyer flasks and induced with 5-μM CdCl2 upon reaching a density of 7.5 × 106 cell/ml. Seven days post-induction, cells were harvested by centrifugation at 6,000 × g for 30 min at 4°C, and supernatant was filtered through 0.45 μm. Clarified supernatant was further ultra-centrifuged in a 20% sucrose cushion (72,000 × g for 1.5 h at 4°C), and the pellet containing VLPs was resuspended in PBS and stored at −80°C (28).
Western Blot
The expression of Envwt, EnvFm, and Gag was evidenced by Western blot analysis from induced cells culture supernatant and purified VLP fractions. Samples were migrated in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) 12% under reducing or non-reducing conditions and electroblotted onto nitrocellulose membrane (Hybond ECL, Amersham-GE Healthcare) for 2 h at 300 mA and 4°C. Membranes were blocked overnight at 4°C with PBS + 3% (w/v) BSA, incubated for 1 h at RT with monoclonal antibodies: anti-p24 (BLV3, 1/2,000) or anti-gp51 (BLV2, 1/2,000) (VMRD, USA) and vigorously washed three times for 5 min with PBS + 0.3% (v/v) Tween 20. Membranes were then incubated with goat anti-mouse monoclonal antibody conjugated to peroxidase (1/5,000) (Santa Cruz Biotechnology) for 1 h at RT and washed as previously described. Both primary and secondary antibodies were diluted in PBS + 3 % (w/v) BSA and 0.3% (v/v) Tween 20. Finally, membranes were incubated 5 min at RT with the enhanced chemiluminescence enzyme substrate kit (Thermo Fisher Scientific, USA), exposed to Hyperfilm enhanced chemiluminescence (GE Healthcare), and manually developed.
Identification of EnvFm and Gag Proteins by Mass Spectrometry
Purified VLPs were migrated in reducing 12% SDS-PAGE and stained with Coomassie blue, and the fraction corresponding to molecular weights between 35 and 80 kDa was cut under keratin-free conditions. Proteins were in-gel digested with trypsin (Sequencing grade Promega) at 37°C overnight. Peptide mixtures extracted from geles were desalted using micro-columns (C18 ZipTip®, Merck, Millipore), eluted in 60% ACN/0.1% formic acid, dried and resuspended in 0.1% formic acid, before nano-LC mass spectrometry (MS)/MS analysis. Tryptic peptides were separated using a nano-HPLC (UltiMate 3000, Thermo Scientific) coupled online with a hybrid quadrupole-Orbitrap mass spectrometer (Q-Exactive Plus, Thermo Scientific). Peptide mixtures were injected into a trap column (Acclaim PepMap 100, C18, Thermo Scientific) and separated into a PepMapTM RSLC C18 column (2 μm, 100A,75 μm ID, 50 cm length) at a flow rate of 200 nl/min. Elution was performed using the following gradient: 1–55% B over 86 min and 55–99% B over 9 min at a constant flow rate of 200 nl/min [A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile (ACN)]. The mass spectrometer was operated in data-dependent acquisition mode with automatic switching between MS and MS/MS scans. The full MS scans were acquired at 70 K resolution, and MS/MS was performed using a data-dependent top 12 method at a resolution of 17.5 K using a dynamic exclusion list. Bioinformatics data analysis was performed using PatternLab for Proteomics software, v 4.0.0.84 (http://patternlabforproteomics.org). A target-decoy database was generated using Drosophila melanogaster UP000000803 from Uniprot (https://www.uniprot.org) 2018/10/31 added with the amino acidic sequences of Gag, EnvFm, and 127 most common contaminants. Search parameters were set as follows: tryptic peptides; methionine oxidation and asparagine glycosylation (Hex3HexNAc2 and Hex3HexNAc2dHex) as variable modifications. Peptide spectrum matches were filtered using the Search Engine Processor (SEPro) using the following parameters: precursor mass tolerance 10 ppm; FDR < 1% at the protein level. The mass spectrometry proteomics raw data have been deposited to the ProteomeXchange Consortium via the PRIDE (53) partner repository with the dataset identifier PXD027973.
Analysis of Virus-Like Particles by Transmission Electron Microscopy
Purified VLPs were deposited onto copper grids covered with commercial carbon/Formvar film, stained with 1–3% uranyl acetate, washed with Milli-Q water or 0.1-M KCl, and the excess liquid was removed by absorption with regular tissue paper. TEM images were obtained in a JEOL JEM-1010 microscope operated at 80 kV.
Analysis of Transfected S2 Cells by Transmission Electron Microscopy
Induced cells producing VLPs were obtained by centrifugation at 250 × g for 5 min, fixed with 2.5% (v/v) glutaraldehyde in PBS, pH 7.2–7.4 and further washed with PBS. A post-fixation step with 1% osmium tetroxide (OsO4) in distilled water was carried out, washed with distilled water and finally dehydrated through a graded [25, 50, 75, 95, and 100% (v/v)] ethanol-water series. The material was infiltrated with Araldite epoxy resin and after polymerization blocks were semi-thin (about 300 nm) and ultra-thin (~70 nm) sectioned using a RMC MT-X ultramicrotome. Ultra-thin cuts were mounted on formvar-coated copper grids, stained in 2% aqueous uranyl acetate followed by Reynold's lead citrate, and visualized in a Jeol JEM-1010 transmission electron microscope operated at 80 kV.
Dynamic Light Scattering
Purified VLPs were analyzed by DLS using a Zetasizer Nano S (Malvern Panalytical). Samples were pre-incubated at 25°C for 10 min and measured with automatic parameter using disposable plastic cuvettes (UVette, Eppendorf). Data from triplicate measurements were averaged and analyzed with Zetasizer Software v.7.13 (Malvern Panalytical) to obtain size distribution of the samples.
Results
Expression of Envwt, EnvFm, and Gag Protein in Drosophila S2 Cells
S2 Drosophila cells were co-transfected with plasmids pMT/Gag and pMT/BiP/Envwt or pMT/BiP/EnvFm to express Gag and Envwt or EnvFm proteins, respectively (Figure 1A). Transfected cells were selected with puromycin and progressively adapted to grow in protein-free culture medium, generating stable polyclonal cell lines Gag/Envwt-S2 and Gag/EnvFm-S2 to produce BLV-VLPs identified as VLP1 and VLP2, respectively (Figure 1B).
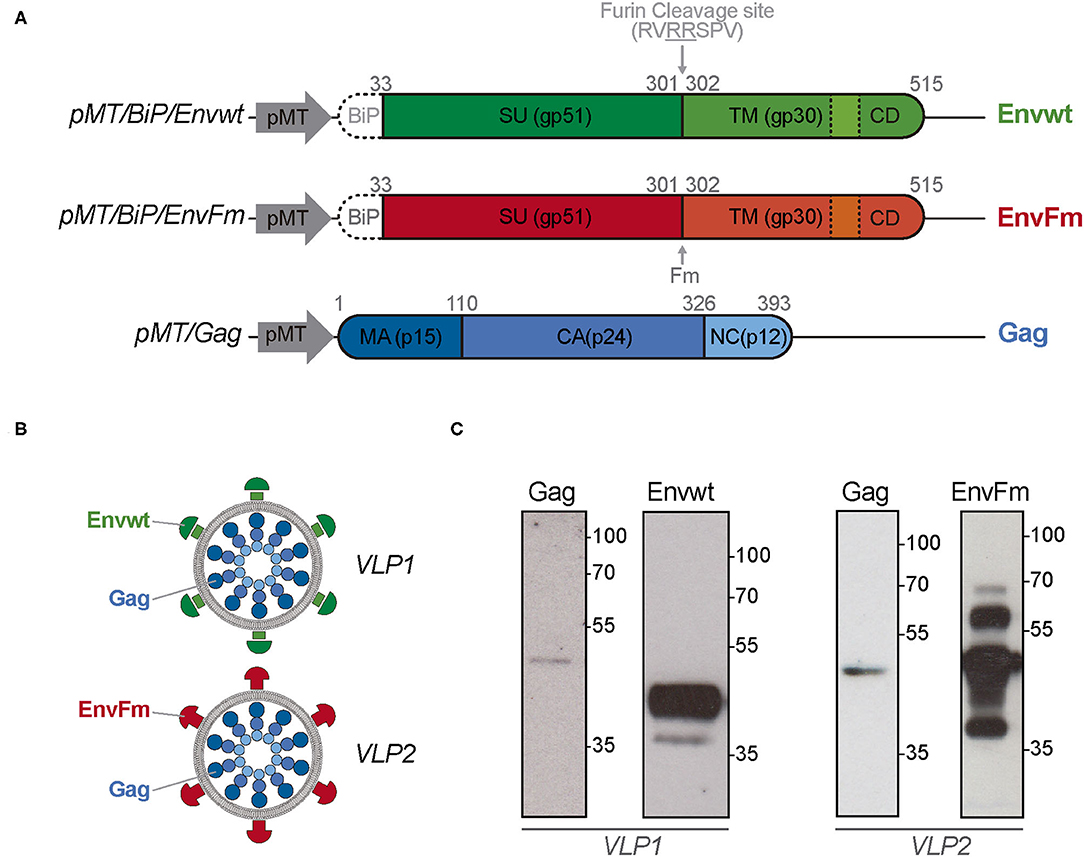
Figure 1. Schematic representation of the expression vectors, produced BLV VLPs, and expression of Gag, Envwt, and EnvFm proteins in transfected Drosophila S2 cells. (A) Representative vectors used to express BLV Envwt, EnvFm, or Gag proteins in S2 cells. pMT, metal-inducible metallothionein promoter; BiP, Drosophila secretory sequence; SU, Surface subunit; TM, Transmembrane subunit; CD, cytosolic domain; Fm, mutated furin cleavage site; MA, matrix; CA, capsid; NC, nucleocapsid. Dotted lines represent the membrane spanning domain present in Envwt and EnvFm. (B) Representative scheme of the BLV VLPs (VLP1 and VLP2) produced in S2 cells. (C) S2 Drosophila cells were co-transfected with plasmids pMT/Gag and pMT/BiP/Envwt or pMT/BiP/EnvFm to generating stable Gag/Envwt-S2 and Gag/EnvFm-S2 cell lines to produce BLV VLP1 and VLP2. Gag, Envwt and EnvFm proteins expression were analyzed by western blot in reducing conditions, in the culture supernatant after 7 days post-induction. Gag protein, with an expected size of 42.8 kDa was detected with an anti-p24 monoclonal antibody (BLV3) in both cell culture supernatants. Properly cleaved Envwt protein was detected as major band with a size between 35 and 55 kDa (left panel). Full-length EnvFm (~65 kDa) and proteolyzed lower fragments were detected (right panel). Envwt and EnvFm were detected using monoclonal antibody anti-gp51 (BLV2) which recognizes the linear D epitope belonging to gp51 subunit. Protein size were indicated by Page Ruler Prestained Protein Ladder, 10–180 kDa (Thermo Fisher Scientific).
The presence of Gag, Envwt, and EnvFm proteins was first confirmed by Western blot analysis in reducing conditions, in the culture supernatant after 7 days post-induction (Figure 1C). Gag protein, with an expected size of 42.8 kDa, was detected with an anti-p24 monoclonal antibody (BLV3) in both cell culture supernatants. Because Gag protein is devoid of secretory signal peptides, its presence in the culture medium could be associated with its incorporation into VLPs (Figure 1C). In the case of Envwt, a major band with a size between 35 and 55 kDa was detected with monoclonal antibody BLV2, which recognizes the linear D epitope belonging to gp51 subunit. This result is in agreement with proteolytic processing of Envwt by cellular furin endopeptidases, giving rise to gp51 subunit (~40 kDa) detected by BLV2 antibody and the gp30 subunit (~24 kDa) not detected by this antibody.
On the other hand, the BLV2 antibody detects a band with a size according to the expected full-length protein (~65 kDa) in EnvFm cell culture supernatant, along with bands of lower sizes (probably related to proteolysis over 7 days of induction) (Figure 1C). Expression of Gag, Envwt, and EnvFm was detected in transfected S2 cells after 7 days of induction by immunofluorescence using monoclonal antibodies BLV1 directed against the conformational epitope G of Env (gp51) and BLV3 against the capsid protein, p24 (Figure 2A and Supplementary Figure 1A). In both cases, Gag and Env proteins were exclusively detected in transfected cells, and no expression was detected in untransfected S2 cells. These results show that both Gag and Env proteins were expressed in transfected S2 cells and despite the modification of the highly conserved furin cleavage site (in EnvFm), this mutant may be successfully expressed representing a suitable candidate to stabilize a prefusion-like version of Env in BLV VLPs, which could be used as an immunogen. To gain insight into the feasibility of this alternative, we further characterized VLP2.
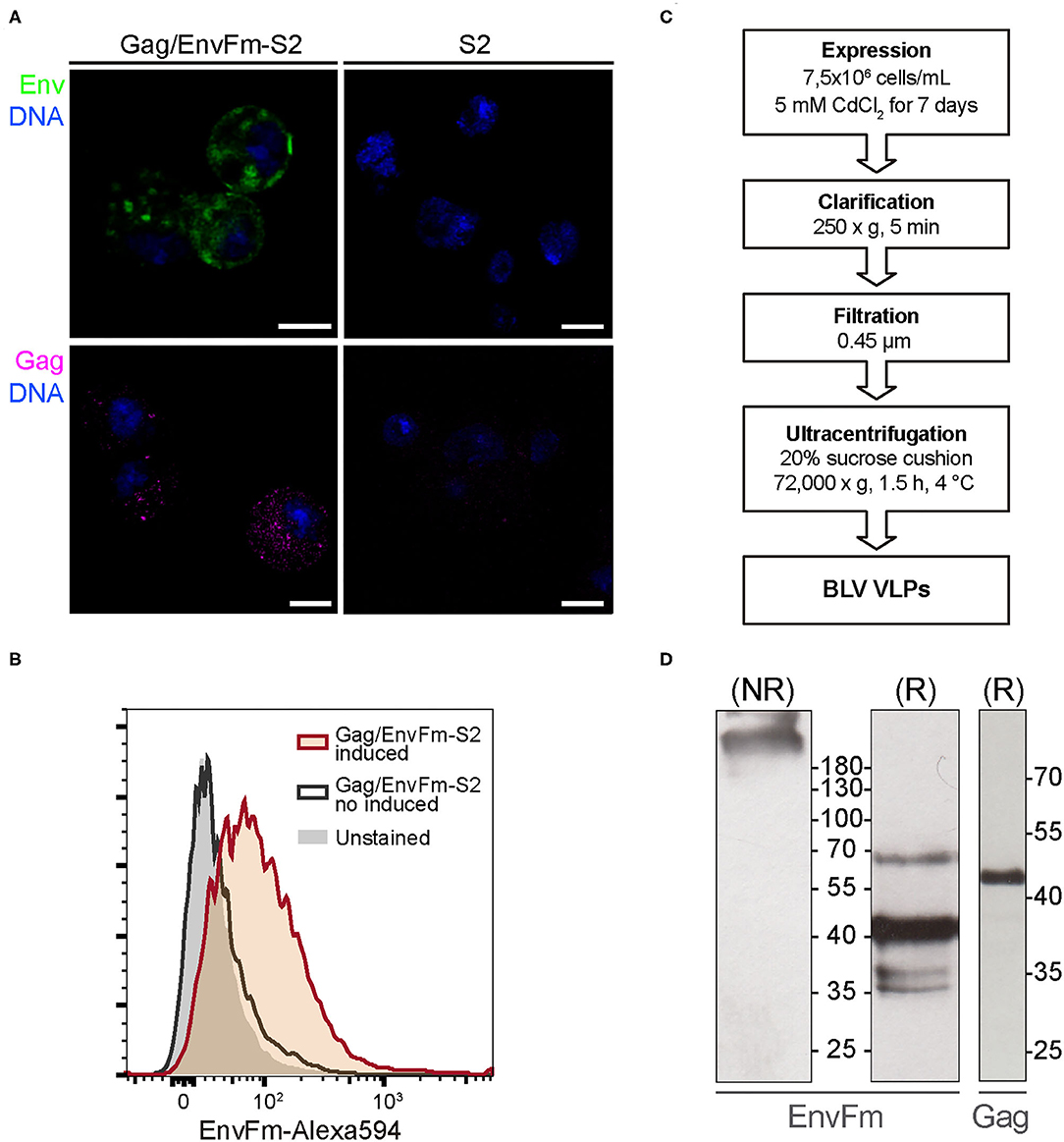
Figure 2. (A) Confocal microscopy of cytoplasmatic Gag and EnvFm expression in Gag/EnvFm-S2 cells. A strong EnvFm and Gag signal is observed in the cytoplasm of CdCl2-induced cells (left), green and magenta respectively, while being absent in untransfected S2 cells (right). EnvFm and Gag proteins were detected using anti-gp51 conformational monoclonal antibody (BLV-1) and anti-p24 monoclonal antibody (BLV-3), respectively. Alexa 594 goat anti-mouse antibody (Invitrogen) was used to reveal both proteins and DNA was stained with methyl green (blue). Scale bar: 5 μm. (B) Flow cytometry histograms to analyze cell surface expression of EnvFm in CdCl2-induced Gag/EnvFm-S2 cells. Unstained control is shaded in gray. The increase in EnvFm cell surface expression in induced cells (red line) is shown compared to the non-induced S2 cells (gray line). EnvFm was detected using a monoclonal antibody anti-gp51 (BLV1) and stained with a goat anti-mouse IgG Alexa-594 secondary antibody (Invitrogen). (C) Schematic representation of the experimental strategy used to express and purify VLPs. (D) Western blot analysis of Gag and EnvFm in purified VLPs. Supernatants from cultures of induced S2 cell line were harvested 7 days post induction and sedimented through a sucrose cushion. Individual proteins expressed in the VLPs were analyzed in western blot. On the left and center panels, western blot analysis of EnvFm protein using anti-gp51 monoclonal antibody (BLV-2) in non-reducing (NR) or in reducing (R) conditions, respectively. On the right, analysis of Gag protein using anti-p24 monoclonal antibody (BLV-3). Protein size were indicated by Page Ruler Prestained Protein Ladder, 10–180 kDa (Thermo Fisher Scientific).
The proper display of EnvFm protein on the membrane of Gag/EnvFm-S2 cells upon induction, which is a prerequisite for its incorporation into VLPs, was demonstrated by flow cytometry (Figure 2B). When non-induced unpermeabilized Gag/EnvFm-S2 cells were treated/stained with BLV1 monoclonal antibody, no change in the median fluorescence intensity (MFI) compared with unstained control cells was observed. Regarding the induced culture, an MFI shift was detected in those cells expressing EnvFm, showing that abrogation of proteolytic processing by furin does not hamper its transport and final anchoring on the membrane of S2 cells, allowing its specific detection with a conformational anti-BLV gp51 antibody.
Incorporation of Gag and Env Proteins Into Virus-Like Particles
Previous results allowed us to produce and purify BLV VLPs according to the protocol schematized in Figure 2C to produce particles displaying wild-type Env (VLP1) and pre-fusion like Env (VLP2). Purified VLPs were analyzed by Western blot using anti-p24 and anti-gp51 monoclonal antibodies. For VLP1, Gag was detected as a unique band with the size according to a molecular weight of 42.8 kDa (Supplementary Figure 1B), whereas Envwt was evidenced as a ~40 kDa band corresponding to fully processed gp51 subunit, as described before in the Western blot results from cell culture supernatant. For VLP2 under non-reducing conditions (NR), EnvFm was detected as species with sizes >180 kDa, in agreement with the expected size of the Env trimer (~200 kDa) or even multimers probably stabilized by disulfide bridges (Figure 2D). To evaluate this, we also analyzed VLP2 by Western blot under reducing conditions, revealing the presence of a band corresponding to EnvFm size (~70 kDa) along with other bands of lower sizes ~35–40 kDa, which are not detected under NR conditions (Figure 2D). These results indicate that part of the EnvFm protein is found as a full-length precursor gp72 (64.5 kDa, theoretical mass) together with smaller size species held together by disulfide bridges. The presence of Gag in VLP2 was also evidenced by Western blot under reducing conditions showing the same results as seen for VLP1 (Figure 2D).
Identification of Gag and EnvFm Proteins by Mass Spectrometry
The identity of EnvFm and Gag proteins from VLP2 was assessed by LC-MS/MS analysis after SDS-PAGE band digestion with trypsin. The sequence of both proteins with peptides identified from three independent replicates highlighted (Supplementary Table 1) are shown in Figure 3A. In the case of Gag, the total sequence coverage was 17.5 %, including the PPPY motif located in the MA domain, which was fully identified with a 100% sequence coverage. In the case of EnvFm, the BiP sequence was not detected in any case as expected for a fully processed protein entering the secretory pathway, so it is not included starting at amino acid position 33. Sequence identification coverage for EnvFm was 25%, allowing the confirmation of both linear and conformational epitopes. Conformational H and F epitopes, linear E epitope and conserved CX6CC motif were identified with 100% coverage, whereas for conformational G epitope, 4 of 5 amino acid residues were also confirmed. Other sequences, such as the neutralization domain, the CD4+ and CD8+ T cell epitopes, 4-3 heptad repeat domain and the immunosuppressive peptide, were partially identified. A unique glycosylated peptide in EnvFm was identified by MS/MS (SWALLLNQTAR) with two N-glycan residues, Man3GlcNAc2Fuc (m/z = 1,038.38 Da) (Figure 3B) or Man3GlcNAc2 (m/z = 892,32 Da) (Supplementary Figure 2).

Figure 3. Identification of amino acid sequence Gag and EnvFm expressed VLP2 by mass spectrometry. (A) Amino acid sequence of Gag (up) and EnvFm (down) of BLV VLP2. Those peptides identified by MS in three replicates are shown in bold. The sequence of the different proteins in the construction are depicted with different shades of grays. Conformational epitopes G, H, and F, and CX6CC motif are marked above the text. The remaining motifs are indicated below the text (54). (B) Representative MS/MS spectrum of the EnvFm N-glycosylated peptide (sequence SWALLLNQTAR modified with Man3GlcNAc2Fuc doubly charged ion, m/z: 1,156.546. The N-terminal and C-terminal fragment ions, including those with partial loss of the glycan structure are labeled.
Morphology and Size of BLV Virus-Like Particles
Gag/EnvFm-S2 cells were analyzed by TEM, showing budding structures on its surface with size and shape resembling VLPs (Figure 4A). Purified VLPs were also analyzed by negatively stained TEM showing numerous non-aggregated and approximately spherical particles of variable diameter (70–200 nm). Within this polydisperse population, VLPs were observed as particles surrounded by a lipid membrane (shiny in appearance) with an electrondense nucleus (Figure 4B and Supplementary Figure 1C) as previously reported retroviral VLPs produced using different expression systems (42–49). Size distribution of purified VLPs was analyzed by DLS revealing the presence of monomodal size distribution with a unique peak by intensity, with a polydisperse population characterized by a z-average hydrodynamic radius (z-ave RH ± sd) of 102.1 ± 50.04 nm for VLP1 (Supplementary Figure 1D) and 93.02 ± 36.72 nm for VLP2 (Figure 4C) with polydispersity index (PdI) values of 0.156 and 0.240 (polydisperse), respectively. DLS results confirmed the polydisperse and non-aggregated nature of purified VLPs in agreement with TEM results.
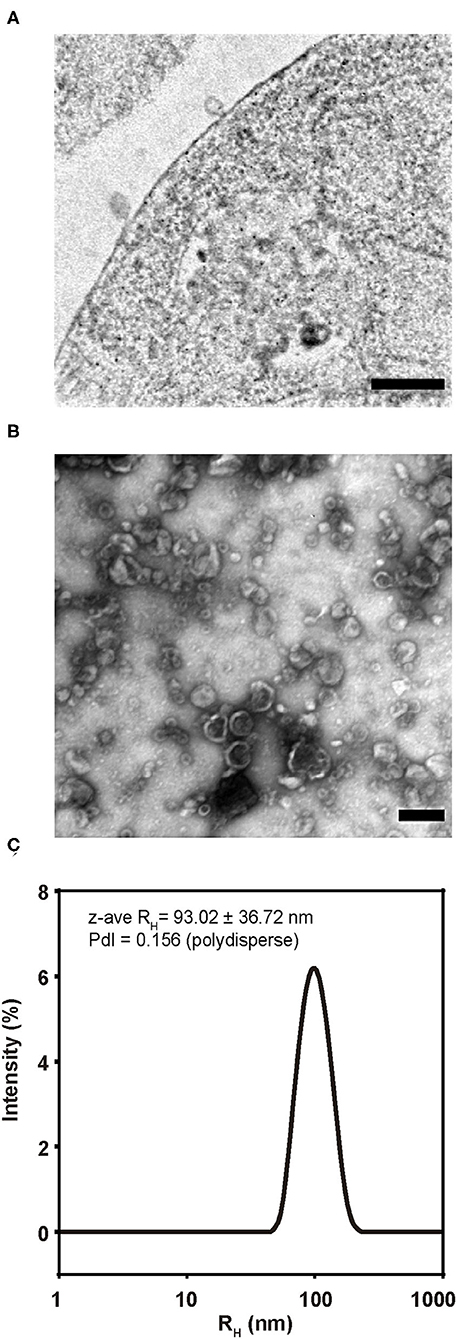
Figure 4. Morphology and size of BLV VLP2. (A) TEM image of an ultrathin section showing budding structures from Gag/EnvFm-S2 cells. Bar 100 nm. (B) TEM image of purified VLP2 negatively stained. Numerous non-aggregated and approximately spherical particles of variable diameter (70–200 nm) were observed. Bar 100 nm. (C) Size distribution of purified VLP2 analyzed by DLS. Monomodal size distribution with a unique peak by intensity, with a polydisperse population was observed. The size distribution is shown as the average of triplicate measurements and mean size represented as z-average RH (z-ave RH) (in nm with its standard deviation) and the polydispersity index (PdI) are shown.
Discussion
Trimeric Env glycoprotein is displayed on the surface of retroviral particles as a metastable conformer. After being processed by furin protease, SU and TM subunits are held together through inter-subunit disulfide bond among other molecular contacts. Despite this mechanism is not fully understood, disulfide isomerization leading to establishment of intra-subunit bonds and dissociation of SU and TM subunits generates a dramatic conformational change resulting in viral and cellular membranes fusion.
This conformational change leads to exposure of non-neutralizing regions of Env to the immune system, giving rise to an immune evasion mechanism known for several retroviruses such as HTLV (55) and HIV (56). Moreover, gp120 from HIV (analogous to BLV gp51 subunit) was found to be very flexible (57), resulting in an immune system presentation that may involve different structural conformations, thus diverting attention from functional Env. This conformational diversity also imposes significant barriers in the development of efficient vaccines aimed to induce broadly neutralizing antibodies, and several strategies have been proposed to stabilize Env trimers in the pre-fusion state (58–61). Recently, Gonelli et al. (62) generated VLPs displaying stabilized HIV Env trimers by introducing inter-subunit disulfides and a proline substitution (SOSIP) mutation. The resulting modified VLPs can efficiently activate B cells by displaying epitopes recognized by broadly neutralizing antibodies while minimizing exposure of non-neutralizing antibody sites (62).
Our group has previously expressed soluble ectodomain of BLV Env proteins in S2 insect cells, giving rise to unstable gp51-gp30 complexes (for ectoEnvwt), which show a high tendency to form aggregates; meanwhile, a proteolytic processing defective mutant (ectoEnvFm) showed higher stability (63). Previous works intended to produce HIV VLPs in insect cells showed Env maturation probably driven as a consequence of endogenous furin proteases present in S2 Drosophila cells (28, 43, 64), so we decided to mutate the proteolytic processing site of Env, to keep both gp51 and gp30 epitopes bound to the VLP surface and, at the same time, lock the protein in a more stable pre-fusion-like conformation, thus reducing immune evasion. The inclusion of other sequence modifications may also be further considered, but the lack of tridimensional structural data of BLV Env trimer does not allow us to step forward into a rational design. The newly reported Alpha Fold artificial intelligence program, which performs predictions of protein 3D structures from its aminoacid sequences may be a potentially helpful alternative to solve this problem (65).
In this work, we transfected S2 Drosophila cells with expression plasmids encoding BLV Gag and Env proteins to generate stable cell lines capable of producing two types of BLV VLPs. The expression of Gag, Envwt, and EnvFm proteins in both stable cell lines was confirmed by Western blot and immunofluorescence and its antigenicity evidenced using monoclonal antibodies recognizing linear and conformational epitopes. Furthermore, the incorporation of the EnvFm protein into the membrane of transfected cells was confirmed by flow cytometry. Altogether, these results show that the mutation of Env does not drastically affect neither its expression nor its entry into the secretory pathway of S2 insect cells. In addition, its detection by BLV1 monoclonal antibody (recognizing a conformational epitope) suggests that EnvFm retain important structural motifs when expressed in S2 insect cells.
The presence of Gag, Envwt, and EnvFm in purified VLPs was also demonstrated by Western blot analysis. For VLP1, only one band with molecular weight corresponding to gp51 is detected, indicating that Envwt is fully processed by cellular furin proteases as expected. EnvFm displayed on VLP2 is produced as a full-length gp72 precursor that undergoes some partial cleavage, along with additional fragments showing smaller and discrete sizes, which remains covalently bound by disulfide bonds and displayed on BLV VLPs. The genesis of this partial proteolysis remains elusive to us, being the recognition of other sequences in a less efficient way by different Drosophila furin proteases, a plausible explanation for this (66). On the other hand, we cannot exclude the possibility that a proportion of the EnvFm protein is cleaved by other intracellular proteases. In fact, cellular proteases play crucial roles by regulating the conformation of major viral proteins (67, 68). The existence of undescribed hydrolysis sites for other cellular proteases cannot be ruled out (69).
The identity of EnvFm and Gag proteins from purified VLP2 was further confirmed by mass spectrometry. In the case of EnvFm, several peptides were detected, allowing the identification of distinctive motifs, some of them containing conformational and linear epitopes of gp51 in agreement with results obtained by immunofluorescence and flow cytometry assays.
Regarding the glycosylation of EnvFm, we identified two paucimannosidic residues at asparagine 203, in agreement with previous results obtained with the recombinant soluble ectodomain of BLV-Env expressed in S2 Drosophila cells in our laboratory (63) and in accordance with the type of glycosylation described in insect cells (70, 71). The presence of an N-glycosylation site at this position, which has been demonstrated to be essential for BLV in vitro infection (72), remains as an important feature in our attempt to produce non-infectious particles showing epitopes mimicking those found in native virions. N-glycosylation of Env protein is essential for BLV infectivity and could be responsible for antigenic determinants (72, 73). In this regard, taking into account the low coverage of sequence identification obtained by MS, along with previous results obtained in our group (63) reflects that as in native BLV virions, Env protein present in VLPs is highly glycosylated even if N-glycans are paucimannosidic thus less complex than in mammalian cells. Moreover, the electrophoretic migration of EnvFm identified by western blot agrees with the presence of several modifications wich increases the overall mass of the protein. In this regard, antigen paucimannosylation may represent a promising mechanism to increase vaccine immunogenicity (74).
In the case of Gag, one of the identified peptides on the MA domain (p15) contains the PPPY motif which is essential in the assembly and budding of the BLV particle (37, 75).
Structures with size and shape resembling budding VLPs were visualized in TEM images obtained from ultrathin sections of transfected S2 cells. TEM analysis of purified VLPs showed the presence of non-aggregated spherical vesicles with variable size and DLS results confirmed the polydisperse and non-aggregated nature of this fraction. This polydispersity could be associated with the existence of VLPs with variable size, however we cannot rule out the coexistence of membrane-covered vesicles with similar density (exosomes, microvesicles) which could be copurified in VLPs fraction (76).
A series of attempts have been performed before to develop vaccines capable of generating protective responses against BLV; however, until now, no vaccine candidate ensures efficient protection and/or decreased transmission of BLV infection in cattle (15, 16, 77–82).
Th1 responses, particularly involving cytotoxic T lymphocytes (CTL), are proposed to mediate immunity against retroviruses, including BLV (79). Vaccine trials in sheep have shown efficacy in protection against BLV challenge, and this has been correlated with a Th1-polarized cellular immune response (80, 81). However, in cattle, vaccine trials shown a lack of protection efficacy that was correlated with a Th2 polarized response (77). These results indicate that it would be important to include adjuvants in vaccine formulations that can polarize the type of response toward Th1 to improve anti-viral protection. The presence of adequate neutralizing antibody levels and a strong CTL-mediated cellular response would be desirable characteristics for an efficient anti-BLV vaccine (83).
The use of an attenuated vaccine composed of a provirus with deletions and mutations located in BLV genomic regions involved in viral pathogenesis represent a very promising approach. This live-attenuated vaccine is infectious in cattle but replicates at reduced levels in cows and is able to efficiently stimulate the humoral and cellular immune response in cattle (84). However, risks as genetic drift, antigenic change or acquisition of mutations that increase pathogenicity, such as substituting by N230E of an N-linked envelope glycosylation site, must be considered significant concerns (18).
In this work, we developed BLV VLPs containing full-length Env and Gag proteins. As mentioned by Bai et al. (32), one of the major problems of using only BLV gp51 protein as the immunogen is due to the high variability of CTL epitopes within this subunit. Therefore, the best candidate for a vaccine against BLV would contain the full-length Env protein, as gp30 CTL epitopes appear to be less polymorphic (32) and will contribute better in the development of an efficient immune response. Our results suggest that the VLPs produced in Drosophila S2 cells could be a potential immunogen to be used in the development of a BLV vaccine that contributes, in conjunction with other control strategies, to reduce the transmission of the virus. In vivo studies are still needed to evaluate its ability to stimulate effective immune responses to evaluate its possible application in the rational design of an effective vaccine. Considering the different proven strategies for the development of a vaccine against BLV infection (18, 85), in addition to the characteristics of the presentation of antigens in a particulate form (22), the reduced biological risk associated to the use of non-infectious material, and the immunogenic features of proteins with N-glycosylation patterns rich in mannose, BLV VLPs produced in S2 Drosophila cells could be a potential immunogen. Future trials designed to evaluate immunogenicity and protective efficacy against viral challenges of this VLP platform in cattle may provide information on its potential advantages to be incorporated into new veterinary vaccine formulations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/pride/archive/projects/PXD027973. And I detected the following words and expressions: Global list: GenBank, Orbitrap, ProteomeXchange, UniProt.
Author Contributions
NO-D and OP: conceptualization and designed the project. NO-D, LT-P, FC, MF, SB, AA, and NI: performed experiments. MP and RD: performed mass spectrometric measurements. FR: performed flow cytometry. DP: image processing of confocal microscopy and MET. NO-D: data analyses and original draft preparation. NO-D, LT-P, FC, MB, and OP: review and editing manuscript. All authors approved the final version of the manuscript.
Funding
This work was partially funded by Comisión Sectorial de Investigación Científica (CSIC I+D 2014), Agencia Nacional de Investigación e Innovación ANII (ALI_1_2016_2_129851), and Programa de Desarrollo de las Ciencias Básicas (PEDECIBA) and FOCEM - Fondo para la Convergencia Estructural del Mercosur (COF 03/11). NO-D was recipient of a postgraduate fellow ship from Agencia Nacional de Investigación e Innovación (ANII) (POS_NAC_2015_1_109471) and a postgraduate fellow ship from Comisión Académica de Posgrado (cap) (BFPD_2020_1#28143834).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge the Advanced Bioimaging Unit at the Institut Pasteur Montevideo and Universidad de la República for their support and assistance in the present work. The authors gratefully acknowledge the Cell Biology Unit at the Institut Pasteur de Montevideo for their support and assistance in the present work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2021.756559/full#supplementary-material
References
1. Kettmann R, Portetelle D, Mammerickx M, Cleuter Y, Dekegel D, Galoux M, et al. Bovine leukemia virus: an exogenous RNA oncogenic virus. Proc Natl Acad Sci USA. (1976) 73:1014–8. doi: 10.1073/pnas.73.4.1014
2. Rodríguez SM, Florins A, Gillet N, de Brogniez A, Sánchez-Alcaraz MT, Boxus M, et al. Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Viruses. (2011) 3:1210–48. doi: 10.3390/v3071210
3. EFSA AHAW Panel (EFSA Panel on Animal Health and Welfare). Scientific opinion on enzootic bovine leukosis. EFSA J. (2015) 13:4188. doi: 10.2903/j.efsa.2015.4188.
4. Barez P-Y, de Brogniez A, Carpentier A, Gazon H, Gillet N, Gutiérrez G, et al. Recent advances in BLresearch V. Viruses. (2015) 7:6080–8. doi: 10.3390/v7112929
5. Emanuelson U, Scherling K, Pettersson H. Relationships between herd bovine leukemia virus infection status and reproduction, disease incidence, and productivity in Swedish dairy herds. Prev Vet Med. (1992) 12:121–31. doi: 10.1016/0167-5877(92)90075-Q
6. Brunner MA, Lein DH, Dubovi EJ. Experiences with the New York state bovine leukosis virus eradication and certification program. Vet Clin North Am Food Anim Pract. (1997) 13:143–50. doi: 10.1016/S0749-0720(15)30369-8
7. Ott SL, Johnson R, Wells SJ. Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev Vet Med. (2003) 61:249–62. doi: 10.1016/j.prevetmed.2003.08.003
8. Frie MC, Coussens PM. Bovine leukemia virus: a major silent threat to proper immune responses in cattle. Vet Immunol Immunopathol. (2015) 163:103–14. doi: 10.1016/j.vetimm.2014.11.014
9. Vanleeuwen JA, Haddad JP, Dohoo IR, Keefe GP, Tiwari A, Tremblay R. Associations between reproductive performance and seropositivity for bovine leukemia virus, bovine viral-diarrhea virus, Mycobacterium avium subspecies paratuberculosis, and Neospora caninum in Canadian dairy cows. Prev Vet Med. (2010) 94:54–64. doi: 10.1016/j.prevetmed.2009.11.012
10. Fukuyama S, Kodama K, Hirahara T, Nakajima N, Takamura K, Sasaki O, et al. Protection against bovine leukemia virus infection by use of inactivated vaccines in cattle. J Vet Med Sci. (1993) 55:99–106. doi: 10.1292/jvms.55.99
11. Parfanovich MI, Zhdanov VM, Lazarenko AA, Nömm EM, Simovart YA, Parakin VK, et al. The possibility of specific protection against bovine leukaemia virus infection and bovine leukaemia with inactivated blv. Br Vet J. (1983). doi: 10.1016/S0007-1935(17)30537-7
12. Altaner C, Ban J, Altanerova V, Janik V. Protective vaccination against bovine leukaemia virus infection by means of cell-derived vaccine. Vaccine. (1991) 9:889–95. doi: 10.1016/0264-410X(91)90009-U
13. Merza M, Söber J, Sundquist B, Toots I, Morein B. Characterization of purified gp 51 from bovine leukemia virus integrated into iscom. Arch Virol. (1991) 120:219–31. doi: 10.1007/BF01310477
14. Kabeya H, Ohashi K, Ohishi K, Sugimoto C, Amanuma H, Onuma M. An effective peptide vaccine to eliminate bovine leukaemia virus (BLV) infected cells in carrier sheep. Vaccine. (1996) 14:1118–22. doi: 10.1016/0264-410X(96)00047-3
15. Kerkhofs P, Gatot JS, Knapen K, Mammerickx M, Burny A, Portetelle D, et al. Long-term protection against bovine leukaemia virus replication in cattle and sheep. J Gen Virol. (2000) 81:957–63. doi: 10.1099/0022-1317-81-4-957
16. Usui T, Konnai S, Tajima S, Watarai S, Aida Y, Ohashi K, et al. Protective effects of vaccination with bovine leukemia virus (BLV) Tax DNA against BLV infection in sheep. J Vet Med Sci. (2003). doi: 10.1292/jvms.65.1201
17. Reichert M, Cantor GH, Willems L, Kettmann R. Protective effects of a live attenuated bovine leukaemia virus vaccine with deletion in the R3 and G4 genes. J Gen Virol. (2000) 81:965–9. doi: 10.1099/0022-1317-81-4-965
18. Abdala A, Alvarez I, Brossel H, Calvinho L, Carignano H, Franco L, et al. BLV: lessons on vaccine development. Retrovirology. (2019) 16:26. doi: 10.1186/s12977-019-0488-8
19. Rodríguez SM, Gutiérrez G, Gillet N, Trono K, Willems L A. recombinant attenuated candidate vaccine that efficiently and persistently protects against bovine leukemia virus in herds. Retrovirology. (2014) 2014:10. doi: 10.1186/1742-4690-11-S1-O10
20. Nooraei S, Bahrulolum H, Sadat Hoseini Z, Katalani C, Hajizade A, Andrew EJ, et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. Nanobiotechnol J. (2021) 19:59. doi: 10.1186/s12951-021-00806-7
21. Jeong H, Seong BL. Exploiting virus-like particles as innovative vaccines against emerging viral infections. Microbiol J. (2017) 55:220–30. doi: 10.1007/s12275-017-7058-3
22. Kushnir N, Streatfield SJ, Yusibov V. Virus-like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine. (2012) 31:58–83. doi: 10.1016/j.vaccine.2012.10.083
23. Pushko P, Pumpens P, Grens E. Development of virus-like particle technology from small highly symmetric to large complex virus-like particle structures. Intervirology. (2013) 56:141–65. doi: 10.1159/000346773
24. Crisci E, Bárcena J, Montoya M. Virus-like particles: the new frontier of vaccines for animal viral infections. Vet Immunol Immunopathol. (2012) 48:211–25. doi: 10.1016/j.vetimm.2012.04.026
25. Liu F, Ge S, Li L, Wu X, Liu Z, Wang Z. Virus-like particles: potential veterinary vaccine immunogens. Res Vet Sci. (2012) 93:553–9. doi: 10.1016/j.rvsc.2011.10.018
26. Fuenmayor J, Gòdia F, Cervera L. Production of virus-like particles for vaccines. N Biotechnol. (2017) 10:174–180. doi: 10.1016/j.nbt.2017.07.010
27. de Jongh AW, Salgueiro S, Dyring C. The use of Drosophila S2 cells in R&D and bioprocessing. Pharm Bioprocess. (2013). 1:197–213. doi: 10.4155/pbp.13.18
28. Yang L, Song Y, Li X, Huang X, Liu J, Ding H, et al. HIV-1 virus-like particles produced by stably transfected Drosophila S2 cells: a desirable vaccine component. Virol J. (2012) 2012:11. doi: 10.1128/JVI.07164-11
29. Park EM, Park SW, Lee YJ, Lee WJ, Choi W. Production of Ebola virus-like particles in Drosophila melanogaster Schneider 2 cells. J Virol Methods. (2018) 261:156–9. doi: 10.1016/j.jviromet.2018.08.016
30. Lee JM, Chung HY, Kim KI, Yoo KH, Hwang-Bo J, Chung IS. Synthesis of double-layered rotavirus-like particles using internal ribosome entry site vector system in stably-transformed Drosophila melanogaster. Biotechnol Lett. (2011) 33:41–6. doi: 10.1007/s10529-010-0390-x
31. Bai L, Sato H, Kubo U, Wada S, Aida Y, et al. CAT1/SLC7A1 acts as a cellular receptor for bovine leukemia virus infection. FASEJ B. (2019) 2019:666958. doi: 10.1101/666958
32. Bai L, Takeshima SN, Isogai E, Kohara J, Aida Y. Novel CD8+ cytotoxic T cell epitopes in bovine leukemia virus with cattle. Vaccine. (2015) 10:128. doi: 10.1016/j.vaccine.2015.10.128
33. Freed EO. HIV-1 Gag proteins: diverse functions in the virus life cycle. Virology. (1998) 251:1–15. doi: 10.1006/viro.1998.9398
34. Johnston ER, Albritton LM, Radke K. Envelope proteins containing single amino acid substitutions support a structural model of the receptor-binding domain of bovine leukemia virus surface protein. Virol J. (2002) 76:10861–72. doi: 10.1128/JVI.76.21.10861-10872.2002
35. de Brogniez A, Mast J, Willems L. Determinants of the bovine leukemia virus envelope glycoproteins involved in infectivity, replication and pathogenesis. Viruses. (2016) 8:88. doi: 10.3390/v8040088
36. Jacobs E, Gheysen D, Thines D, Francotte M, de Wilde M. The HIV-1 Gag precursor Pr55gag synthesized in yeast is myristoylated and targeted to the plasma membrane. Gene. (1989) 1989:90093. doi: 10.1016/0378-1119(89)90093-0
37. Kakker NK, Mikhailov MV, Nermut MV, Burny A, Roy P. Bovine leukemia virus Gag particle assembly in insect cells: formation of chimeric particles by domain-switched leukemia/lentivirus Gag polyprotein. Virology. (1999) 265:308–18. doi: 10.1006/viro.1999.0007
38. Mager A, Masengo R, Mammerickx M, Letesson J-J. T cell proliferative response to bovine leukaemia virus (BLV): identification of T cell epitopes on the major core protein (p24) in BLV-infected cattle with normal haematological values. J Gen Virol. (1994) 75:2223–31. doi: 10.1099/0022-1317-75-9-2223
39. Otsuki H, Takeshima S, Aida Y. Generation of virus-like particle as a vaccine strategy against bovine leukemia virus. Retrovirology. (2015) 12:P47. doi: 10.1186/1742-4690-12-S1-P47
40. Wang H, Norris KM, Mansky LM. Analysis of bovine leukemia virus Gag membrane targeting and late domain function. Virol J. (2002) 76:8485–93. doi: 10.1128/JVI.76.16.8485-8493.2002
41. Hertig C, Pye AD, Hyatt AD, Boyle DB. Retrovirus-like particles produced by vaccinia viruses expressing gag-pro-pol region genes of bovine leukaemia virus. J Gen Virol. (1994) 75:2213–21. doi: 10.1099/0022-1317-75-9-2213
42. González-Domínguez I, Gutiérrez-Granados S, Cervera L, Gòdia F, Domingo N. Identification of HIV-1–based virus-like particles by multifrequency atomic force microscopy. Biophys J. (2016) 111:1173–9. doi: 10.1016/j.bpj.2016.07.046
43. Tagliamonte M, Visciano ML, Tornesello ML, De Stradis A, Buonaguro FM, Buonaguro L. HIV-Gag VLPs presenting trimeric HIV-1 gp140 spikes constitutively expressed in stable double transfected insect cell line. Vaccine. (2011) 29:4913–22. doi: 10.1016/j.vaccine.2011.05.004
44. Cervera L, Gutiérrez-Granados S, Martínez M, Blanco J, Gòdia F, Mercedes Segura M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. Biotechnol J. (2013) 166:152–65. doi: 10.1016/j.jbiotec.2013.05.001
45. Gutiérrez-Granados S, Cervera L, Gòdia F, Carrillo J, Mercedes Segura M. Development and validation of a quantitation assay for fluorescently tagged HIV-1 virus-like particles. J Virol Methods. (2013) 193:85–95. doi: 10.1016/j.jviromet.2013.05.010
46. González-Domínguez I, Puente-Massaguer E, Cervera L, Gòdia F. Quality assessment of virus-like particles at single particle level: a comparative study. Viruses. (2020) 12:1–24. doi: 10.3390/v12020223
47. Lynch A, Meyers AE, Williamson AL, Rybicki EP. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol J. (2012) 9:1. doi: 10.1186/1743-422X-9-210
48. Valley-Omar Z, Ann Meyers E, Enid Shephard G, Williamson AL, Edward Rybicki P. Abrogation of contaminating RNA activity in HIV-1 Gag VLPs. Virol J. (2011) 8:1–12. doi: 10.1186/1743-422X-8-462
49. Steppert P, Burgstaller D, Klausberger M, Berger E, Aguilar P, Tobias Schneider A, et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J Chromatogr A. (2016) 1455:93–101. doi: 10.1016/j.chroma.2016.05.053
50. González-Reyes L, Ruiz-Argüello MB, García-Barreno B, Calder L, López JA, Albar JP, et al. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc Natl Acad Sci USA. (2001) 98:9859–64. doi: 10.1073/pnas.151098198
51. Sage D, Donati L, Soulez F, Fortun D, Schmit G, Seitz A, et al. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods. (2017) 115:28–41. doi: 10.1016/j.ymeth.2016.12.015
52. Beck A, Teboulle M. A fast iterative shrinkage-thresholding algorithm. SIAM J Imaging Sci. (2009) 2:183–202. doi: 10.1137/080716542
53. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Deepti Kundu J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. (2019) 47:D442–50. doi: 10.1093/nar/gky1106
54. Callebaut I, Vonèche V, Mager A, Fumière O, Krchnak V, Merza M, et al. Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. Virol J. (1993) 67:5321–7. doi: 10.1128/jvi.67.9.5321-5327.1993
55. Gelderblom HR, Reupke H, Pauli G. Loss of envelope antigens of htlv-III/LAV, a factor in aids pathogenesis? Lancet. (1985) 2:1016–7. doi: 10.1016/S0140-6736(85)90570-7
56. McKeating JA, McKnight A, Moore JP. Differential loss of envelope glycoprotein gp120 from virions of human immunodeficiency virus type 1 isolates: effects on infectivity and neutralization. Virol J. (1991) 74:860. doi: 10.1128/jvi.65.2.852-860.1991
57. Pan Y, Ma B, Keskin O, Nussinov R. Characterization of the conformational state and flexibility of HIV-1 glycoprotein gp120 core domain. J Biol Chem. (2004) 279:30523–30. doi: 10.1074/jbc.M404364200
58. Rutten L, Lai YT, Blokland S, Truan D, Ilona Bisschop JM, Nika Strokappe M, et al. A universal approach to optimize the folding and stability of prefusion-closed HIV-1 envelope trimers. Cell Rep. (2018) 23:584–95. doi: 10.1016/j.celrep.2018.03.061
59. Sliepen K, Woo Han B, Bontjer I, Mooij P, Garces F, Behrens AJ, et al. Structure and immunogenicity of a stabilized HIV-1 envelope trimer based on a group-M consensus sequence. Nat Commun. (2019) 10:1–16. doi: 10.1038/s41467-019-10262-5
60. Rawi R, Rutten L, Lai YT, Adam Olia S, Blokland S, Juraszek J, et al. Automated design by structure-based stabilization and consensus repair to achieve prefusion-closed envelope trimers in a wide variety of HIStrains V. Cell Rep. (2020) 33:108432. doi: 10.1016/j.celrep.2020.108432
61. Wang Q, Ma B, Liang Q, Zhu A, Wang H, Fu L, et al. Stabilized diverse HIV-1 envelope trimers for vaccine design. Emerg Microbes Infect. 9:775–86. doi: 10.1080/22221751.2020.1745093
62. Gonelli CA, King HAD, Mackenzie C, Sonza S, Center RJ, Damian F, et al. Immunogenicity of HIV-1-based virus-like particles with increased incorporation and stability of membrane-bound Env. Vaccines. (2021) 9:239. doi: 10.3390/vaccines9030239
63. Tomé Poderti L. Estudio de la interacción entre el virus de la leucemia bovina y la célula hospedera. Universidad de la República (Uruguay). Facultad de Ciencias - PEDECIBA (2014). Available online at: https://www.colibri.udelar.edu.uy/jspui/handle/20.500.12008/24048 (accessed 15 October 2021).
64. Wang BZ, Liu W, Kang SM, Alam M, Huang C, Ye L, et al. Incorporation of high levels of chimeric human immunodeficiency virus envelope glycoproteins into virus-like particles. Virol J. (2007) 81:10869–78. doi: 10.1128/JVI.00542-07
65. Andrew W, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved protein structure prediction using potentials from deep learning. Nature. (2020) 577:706–10. doi: 10.1038/s41586-019-1923-7
66. Künnapuu J, Björkgren I, Shimmi O. The Drosophila DPP signal is produced by cleavage of its proprotein at evolutionary diversified furin-recognition sites. Proc Natl Acad Sci USA. (2009). doi: 10.1073/pnas.0809885106
67. Kido H, Niwa Y, Beppu Y, Towatari T. Cellular proteases involved in the pathogenicity of enveloped animal viruses, human immunodeficiency virus, influenza virus A and sendal virus. Adv Enzyme Regul. (1996) 36:325–47. doi: 10.1016/0065-2571(95)00016-X
68. Seidah NG, Pasquato A, Andreo U. How do enveloped viruses exploit the secretory proprotein convertases to regulate infectivity and spread? Viruses. (2021) 7:1229. doi: 10.3390/v13071229
69. Li SW, Wright M, Healey JF, Hutchinson JM, O'Rourke S, Mesa KA, et al. Gene editing in CHO cells to prevent proteolysis and enhance glycosylation: Production of HIV envelope proteins as vaccine immunogens. PLoS ONE. (2020) 2020:233866. doi: 10.1371/journal.pone.0233866
70. Rendić D, Wilson IBH, Paschinger K. The glycosylation capacity of insect cells. Croatica Chemica Acta. (2008) 81:7–21. doi: 10.1002/chin.200831267
71. Wilson IBH. Glycosylation of proteins in plants and invertebrates. Curr Opin Struct Biol. (2002) 12:569–77. doi: 10.1016/S0959-440X(02)00367-6
72. Rizzo G, Forti K, Serroni A, Cagiola M, Baglivo S, Scoccia E, et al. Single N -glycosylation site of bovine leukemia virus SU is involved in conformation and viral escape. Vet Microbiol. (2016) 197:21–6. doi: 10.1016/j.vetmic.2016.10.024
73. de Brogniez A, Bouzar AB, Jacques JR, Cosse JP, Gillet N, Callebaut I, et al. Mutation of a single envelope N-linked glycosylation site enhances the pathogenicity of bovine leukemia virus. Virol J. (2015) 261:15. doi: 10.1128/JVI.00261-15
74. Urbanowicz RA, Wang R, Schiel JE, Keck ZY, Kerzic MC, Lau P, et al. Antigenicity and immunogenicity of differentially glycosylated hepatitis C virus E2 envelope proteins expressed in mammalian and insect cells. Virol J. (2019) 93:18. doi: 10.1128/JVI.01403-18
75. Gillet N, Florins A, Boxus M, Burteau C, Nigro A, Vandermeers F, et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology. (2007) 4:18. doi: 10.1186/1742-4690-4-18
76. Reiter K, Aguilar PP, Wetter V, Steppert P, Tover A, Jungbauer A. Separation of virus-like particles and extracellular vesicles by flow-through and heparin affinity chromatography. J Chromatogr A. (2019) 12:35. doi: 10.1016/j.chroma.2018.12.035
77. Cherney TM, Schultz RD. Viral status and antibody response in cattle inoculated with recombinant bovine leukemia virus-vaccinia virus vaccines after challenge exposure with bovine leukemia virus-infected lymphocytes. Am J Vet Res. (1996) 57:812–8.
78. Onuma M, Hodatsu T, Yamamoto S, Higashihara M, Masu S, Mikami T, et al. Protection by vaccination against bovine leukemia virus infection in sheep. Am J Vet Res. (1984) 45:1212–5.
79. Ohishi K, Kabeya H, Amanuma H, Onuma M. Peptide-based bovine leukemia virus (BLV) vaccine that induces BLV-Env specific Th-1 type immunity. Leukemia. (1997) 11(Suppl.3):223–6.
80. Okada K, Sonoda K, Koyama M, Yin S, Ikeda M, Goryo M, et al. Delayed-type hypersensitivity in sheep induced by synthetic peptides of bovine leukemia virus encapsulated in mannan-coated liposome. J Vet Med Sci. (2003) 65:515–8. doi: 10.1292/jvms.65.515
81. Portetelle D, Limbach K, Burny A, Mammerickx M, Desmettre P, Riviere M, et al. Recombinant vaccinia virus expression of the bovine leukaemia virus envelope gene and protection of immunized sheep against infection. Vaccine. (1991) 9:194–200. doi: 10.1016/0264-410X(91)90153-W
82. Ohishi K, Suzuki H, Yasutomi Y, Onuma M, Okada K, Numakunai S, et al. Augmentation of bovine leukemia virus (BLV)-specific lymphocyte proliferation responses in ruminants by inoculation with BLV env-recombinant vaccinia virus: their role in the suppression of BLReplication V. Microbiol Immunol. (1992) 36:1317–23. doi: 10.1111/j.1348-0421.1992.tb02133.x
83. Von Beust BR, Brown WC, Estes DM, Zarlenga DS, McElwain TF, Palmer GH. Development and in vitro characterization of recombinant vaccinia viruses expressing bovine leukemia virus gp51 in combination with bovine IL4 or IL12. Vaccine. (1999) 17:384–95. doi: 10.1016/S0264-410X(98)00208-4
84. Gutiérrez G, Rodríguez SM, Vilor A, Gillet N, DeBrogniez A, Trono K, et al. An efficient vaccine against bovine leukemia virus. Retrovirology. (2015) 12:1. doi: 10.1186/1742-4690-12-S1-P3
Keywords: bovine leukemia virus (BLV), virus-like particles (VLP), furin cleavage site, immunogens, Gag, Env, retrovirus
Citation: Olivero-Deibe N, Tomé-Poderti L, Carrión F, Bianchi S, Fló M, Prieto D, Rammauro F, Addiego A, Ibañez N, Portela M, Duran R, Berois M and Pritsch O (2021) Expression, Purification, and Characterization of Bovine Leukemia Virus-Like Particles Produced in Drosophila S2 Cells. Front. Virol. 1:756559. doi: 10.3389/fviro.2021.756559
Received: 10 August 2021; Accepted: 28 September 2021;
Published: 03 November 2021.
Edited by:
Karen Duus, Touro University Nevada, United StatesReviewed by:
Hyesun Jang, J. Craig Venter Institute, United StatesWolfgang Ernst, University of Natural Resources and Life Sciences, Austria
Copyright © 2021 Olivero-Deibe, Tomé-Poderti, Carrión, Bianchi, Fló, Prieto, Rammauro, Addiego, Ibañez, Portela, Duran, Berois and Pritsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Otto Pritsch, pritsch@pasteur.edu.uy; Natalia Olivero-Deibe, nolivero@pasteur.edu.uy
†These authors have contributed equally to this work