 Guohong Xiao1
Guohong Xiao1 Yanhong Xiao
Yanhong Xiao- 1Jilin Provincial Key Laboratory of Animal Resource Conservation and Utilization, Northeast Normal University, Changchun, China
- 2Institute of Resources and Environment, Henan Polytechnic University, Jiaozuo, China
- 3School of Life Sciences, Jilin Agricultural University, Changchun, China
A large number of microorganisms colonize the intestines of animals. The gut microbiota plays an important role in nutrient metabolism and affects a number of physiological mechanisms in the host. Studies have shown that seasonal changes occur in the intestinal microbes of mammals that hibernate seasonally. However, these studies only focused on ground squirrels and bears. It remains unclear how hibernation might affect the intestinal microbes of bats. In this study, we measured microbial diversity and composition in the gut of Rhinolophus ferrumequinum in different periods (early spring, early summer, late summer, torpor, and interbout arousal) using 16S ribosomal RNA gene amplicon sequencing and PICRUSt to predict functional profiles. We found seasonal changes in the diversity and composition of the gut microbes in R. ferrumequinum. The diversity of gut microbiota was highest in the late summer and lowest in the early summer. The relative abundance of Proteobacteria was highest in the early summer and significantly lower in other periods. The relative abundance of Firmicutes was lowest in the early summer and significantly increased in the late summer, followed by a significant decrease in the early winter and early spring. The relative abundance of Tenericutes was significantly higher in the early spring compared with other periods. The results of functional prediction by PICRUSt showed seasonal variations in the relative abundance of metabolism-related pathways, including lipid metabolism, carbohydrate metabolism, and energy metabolism. Functional categories for carbohydrate metabolism had significantly lower relative abundance in early winter-torpor compared with late summer, while those associated with lipid metabolism had significantly higher relative abundance in the early winter compared with late summer. Overall, our results show that seasonal physiological changes associated with hibernation alter the gut microbial community of R. ferrumequinum. Hibernation may also alter the metabolic function of intestinal microbes, possibly by converting the gut microflora from carbohydrate-related to lipid-related functional categories. This study deepens our understanding of the symbiosis between hibernating mammals and gut microbes.
Introduction
The continuous co-evolution of mammals and their gut microbes forms a complex relationship that provides benefits for both partners (Ley et al., 2008a, b). The gut microbiome of animals is intimately linked to host physiology, nutrition, and metabolism (Hooper et al., 2012; Human Microbiome Project Consortium, 2012; Sommer and Backhed, 2013). The microbiome enhances resistance to pathogen colonization, affects the structure and function of the gut, drives the development of the immune system, and increases energy harvested from the diet (McFall-Ngai et al., 2013). In turn, the host provides sufficient nutrients and suitable living conditions for the microorganisms, and affects the structure and composition of microorganisms in many ways, including genetics, immune status, intestinal environment, and diet (Faith et al., 2011; David et al., 2014). The type of diet and amount of food consumed have been shown to have a considerable impact on the composition of intestinal microbes (Ley et al., 2008a; Muegge et al., 2011). Although host diet provides the major source of substrates that support microbial growth, microbes can also use host-derived substrates, including mucus glycans, nutrients in sloughed epithelial cells, and biliary secretions (Leser et al., 2000; Hooper et al., 2002; Sonnenburg et al., 2005). When the host is fasting, however, there are few or no dietary substrates available for intestinal microbes. Prolonging the fasting period of the host may result in the selective development of gut microbes toward microbial communities capable of decomposing host-derived substrates (e.g., mucin).
Hibernation is a typical example of extended fasting, and is a key feature in the biology of many hibernating mammals (Lyman et al., 1982; Carey et al., 2003; Florant and Healy, 2012). In these species, excessive diets lead to a large accumulation of fat stores in the summer and early fall, followed by voluntary fasting, which can last for up to 8 months. Despite this, these animals manage to maintain the integrity of their organ systems and protect their bodies from damage. Therefore, hibernating mammals provide a unique, natural model system for studying the effects of dietary changes on gut microbiota and host-microbiota symbiosis. Previous studies have shown that hibernation changes the structure and function of intestinal microbes (Carey et al., 2013; Dill-McFarland et al., 2014, 2016). Phylum-level changes in gut microbiota composition over the hibernation cycle are common in herbivores (e.g., small rodents, such as Arctic ground squirrels, thirteen-line ground squirrels) and in carnivores (e.g., large mammals, such as brown bears) (Carey et al., 2013; Dill-McFarland et al., 2014). In these studies, the relative abundance of Firmicutes decreased, while Bacteroidetes increased, in both animals; however, the relative abundance of Verrucomicrobia decreased in brown bears and increased in ground squirrels (Carey et al., 2013; Dill-McFarland et al., 2014, 2016). The taxonomic shifts in the microbiotas of ground squirrels correlate well with known substrate preferences of some of the major taxa of the mammalian gut. The structure of the gut microflora in winter is mainly composed of the Verrucomicrobia taxa (Derrien et al., 2004), which can decompose host-derived substrates, or a taxonomic group such as Bacteroidetes, which have the ability to switch their complement of carbohydrate-degrading enzymes depending on the availability of dietary vs. host-derived substrates (Sonnenburg et al., 2005). Conversely, the hibernator microbiota is generally depleted in bacteria that prefer dietary polysaccharides, which include many Firmicutes species (Flint et al., 2012). Are there similar changes in the intestinal microbes of other hibernating mammals? Answering this question will require studies on more hibernating species, such as insectivores.
Bats, of which a large number of species are seasonal hibernators, are one of the representative groups of hibernating mammals (Whitaker and Rissler, 1992; Geluso, 2007). Unlike the hibernating rodents, which feed on plants, most hibernating bats are insectivores, as well as being small mammals (Burton and Burton, 2002; Carey et al., 2013; Stevenson et al., 2014). An important energy-saving strategy is the decline in metabolic rate during hibernation of many mammals, which plays a vital role in surviving harsh winter environments (Storey and Storey, 2004). The metabolic rate during torpor can be reduced to 4.3% of the basal metabolic rate (Ruf and Geiser, 2015). Moreover, it is commonly known that the major energy source in small mammals switches from carbohydrate to lipid during hibernation (Hampton et al., 2011; Xiao et al., 2015). In addition, studies on the transcriptomes of small hibernating mammals found that the differential expression of genes related to lipid metabolism in hibernating bats is consistent with other small mammals (Williams et al., 2005; Xiao et al., 2015). However, it is unclear whether the changes in gut microbiota are similar in bats and other small hibernating mammals. Furthermore, recent studies of the gastrointestinal flora in bats have mainly focused on the effects of different types of diet (Phillips et al., 2012). Maliničová et al. (2017) utilized traditional culture methods for a dynamic study of the effects of hibernation on the intestinal microbes of bats; however, their results were limited because most intestinal bacteria cannot be cultivated. Using new high-throughput sequencing technologies, such as 16S ribosomal RNA (rRNA) amplicon sequencing, we expect that far more information on the effects of hibernation on intestinal microbiota can be obtained compared with traditional methods. Importantly, seasonal changes in the intestinal microbes of hibernating bats have not been studied.
The greater horseshoe bat (Rhinolophus ferrumequinum), a typical hibernating insectivore, is widely distributed in Europe, Africa, South Asia, Australia, and China (Burton and Burton, 2002). In China, its habitat ranges from the northeastern to southwestern regions (Wang, 2003). During hibernation, R. ferrumequinum enters a state of torpor, with a lower body temperature (Tb) of about 10°C, and spontaneous interbout arousals (IBAs) (Park and Ransome, 2000; Burton and Burton, 2002). It has become a model species in hibernation studies of bats. In this study, 16S rRNA amplicon sequencing was used to investigate the effects of hibernation on the intestinal microbes of R. ferrumequinum, and PICRUSt was applied to generate a simple prediction of functional gut microbiota. The results of this study enhance our understanding of the symbiotic and adaptive evolution of hibernating mammals and their intestinal microbes.
Materials and Methods
Animals
All bats were collected from an artificial canal in Jiyuan City, Henan Province, between September 2017 and July 2018. There were five collection time points over the annual hibernation cycle (Table 1): early spring (n = 8; mid-April, AA); early summer (n = 7; end of July, JA); late summer (n = 8; mid-September, SA); early winter-torpor (n = 8; end of January, JT); and early winter-IBA (n = 8; end of January, JIBA).
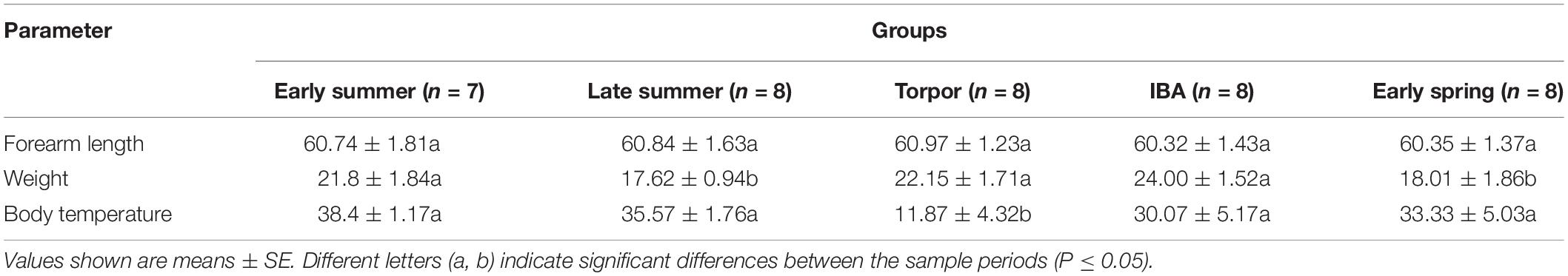
Table 1. Summary of sample collection information for the study bats.
During active seasons, bats were collected using mist nets and taken back to a temporary laboratory in the field, where sampling processes immediately followed. During the hibernating season, bats were collected from a cave wall using hand nets and then taken back to the temporary laboratory in the field. Eight active bats were immediately sacrificed to constitute the IBA group. For the torpor group, another eight bats were placed in a small refrigerator at about 10°C to mimic the environmental temperature and allow them to re-enter torpor (∼24 h). To eliminate the effects of age, all of the bats collected for this study were adults. Age was estimated according to the degree of ossification of the epiphyseal spacing (Kunz and Anthony, 1982).
Sample Collection
Before sampling, we measured the weight and forearm length of the bats (Table 1). Bats were then sacrificed by cervical dislocation and immediate decapitation. Body temperature (Tb) was promptly measured and recorded (Table 1) by insertion of a thermal probe into the body cavity. The intact guts were rapidly removed for collection of intestinal contents, which were immediately frozen in liquid nitrogen and stored at −80°C until DNA extraction.
Sample collection was carried out with the permission of the local forestry department. All studies in this experiment were approved by the National Animal Research Authority of Northeast Normal University, China (approval number: NENU-20080416).
DNA Extraction
The intestinal contents of each sample were homogenized. We then used 0.1 g per sample for the extraction of microbial genomic DNA using the DNeasy PowerWater kits (QIAGEN, Inc., Netherlands), following the manufacturer’s instructions. DNA samples were stored at −80°C until further analysis. The quantity of extracted DNA was measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and the quality was assessed using agarose gel electrophoresis.
16S rDNA Amplicon Pyrosequencing
PCR amplification of the V3–V4 region of bacterial 16S rRNA genes was performed using forward primer 308F (5′-ACTCCTACGGGAGGCAGCA-3′) and reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Dennis et al., 2013). Sample-specific 7-bp barcodes were incorporated into the primers for multiplex sequencing. A single-step 30-cycle PCR using the HotStarTaq Plus master mix kit (Qiagen, Valencia, CA, United States) was performed under the following conditions: 98°C for 2 min, followed by 25 cycles consisting of denaturation at 98°C for 15 s, annealing at 55°C for 30 s, extension at 72°C for 30 s, and a final elongation step at 72°C for 5 min. PCR amplicons were purified with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN, United States) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, United States). After the individual quantification step, amplicons were pooled in equal amounts and pair-end 2 × 300-bp sequencing was performed using the Illlumina MiSeq platform with MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
Sequence Data Processing
Sequence processing was performed using the QIIME v1.8.0 program (Caporaso et al., 2010). Briefly, raw sequencing reads with exact matches to the barcodes were assigned to respective samples and identified as valid sequences. All sequences were trimmed and de-noised prior to alignment against the Greengenes 16S rRNA gene reference alignment database (DeSantis et al., 2006). Sequences were screened (filter.seqs) and reduced using unique.seqs and pre.cluster. Chimera detection (chimera.uchime) and removal was performed and the remaining high-quality sequences were clustered into operational taxonomic units (OTUs) at 97% sequence identity by UCLUST (Edgar, 2010). A representative sequence was selected from each OTU using default parameters. OTU taxonomic classification was conducted by BLAST searching of the representative sequences set against the Greengenes Database using the best hit (DeSantis et al., 2006).
Statistical Analysis
Sequence data analyses were mainly performed using QIIME (v1.8.0) and R packages (v3.5.1). OTU-level ranked abundance curves were generated to compare the richness and evenness of OTUs among samples. The Shannon diversity index and Chao1 richness index were calculated using the OTU table in QIIME to determine alpha diversity for the seasonal groups of bats. We then used the rank sum test to calculate significant differences. Beta diversity analysis was performed to investigate structural variation in microbial communities of the different group samples using unweighted and weighted UniFrac distance metrics (Lozupone and Knight, 2005; Lozupone et al., 2007). The significance of differentiation of microbiota structure among groups was assessed by PERMANOVA (Permutational multivariate analysis of variance) using R package “vegan.” Using QIIME, and according to the sample grouping, the unweighted UniFrac distance matrix was used to statistically compare the mean sample distances of different groups. Differences in the Unifrac distances for pairwise comparisons among groups were determined using the Student’s t-test and the Monte Carlo permutation test with 1,000 permutations and then visualized through box-and-whisker plots. Taxonomies were grouped at the phylum, class, order, family, and genus levels. Taxa abundances at the phylum, class, order, family and genus levels were statistically compared among groups by Metastats (White et al., 2009). Microbial functions were predicted by PICRUSt (Phylogenetic investigation of communities by reconstruction of unobserved states), based on high-quality sequences (Langille et al., 2013).
Results
Microbial Diversity
After quality processing, a total of 1,327,632 16S rRNA gene reads (average 34,041 reads/sample) were obtained, from which 1,750 OTUs were identified to be from gut microbiotas of 39 bats. The rarefaction curve analysis indicated that sequencing captured the majority of microbial diversity in our samples (Supplementary Figure 1).
Alpha diversity of gut microbiotas differed significantly among sample periods (Figure 1). The alpha diversity value was highest in the late summer, followed by early spring and early winter, and lowest in the early summer (Figure 1 and Supplementary Figure 1). Alpha diversity (for two indices) was significantly higher in the late summer compared with the early summer (P < 0.01) and early winter (P < 0.05) (Figure 1). There was no significant difference in the alpha diversity of gut microbiotas between the IBA and torpor groups in early winter. The alpha diversity of gut microbiotas in early winter was not significantly different from that in early spring. However, we found that the Shannon index of gut microbiotas in early summer was significantly lower than those during torpor (P = 0.015) and IBA (P = 0.037) in early winter (Figure 1B).
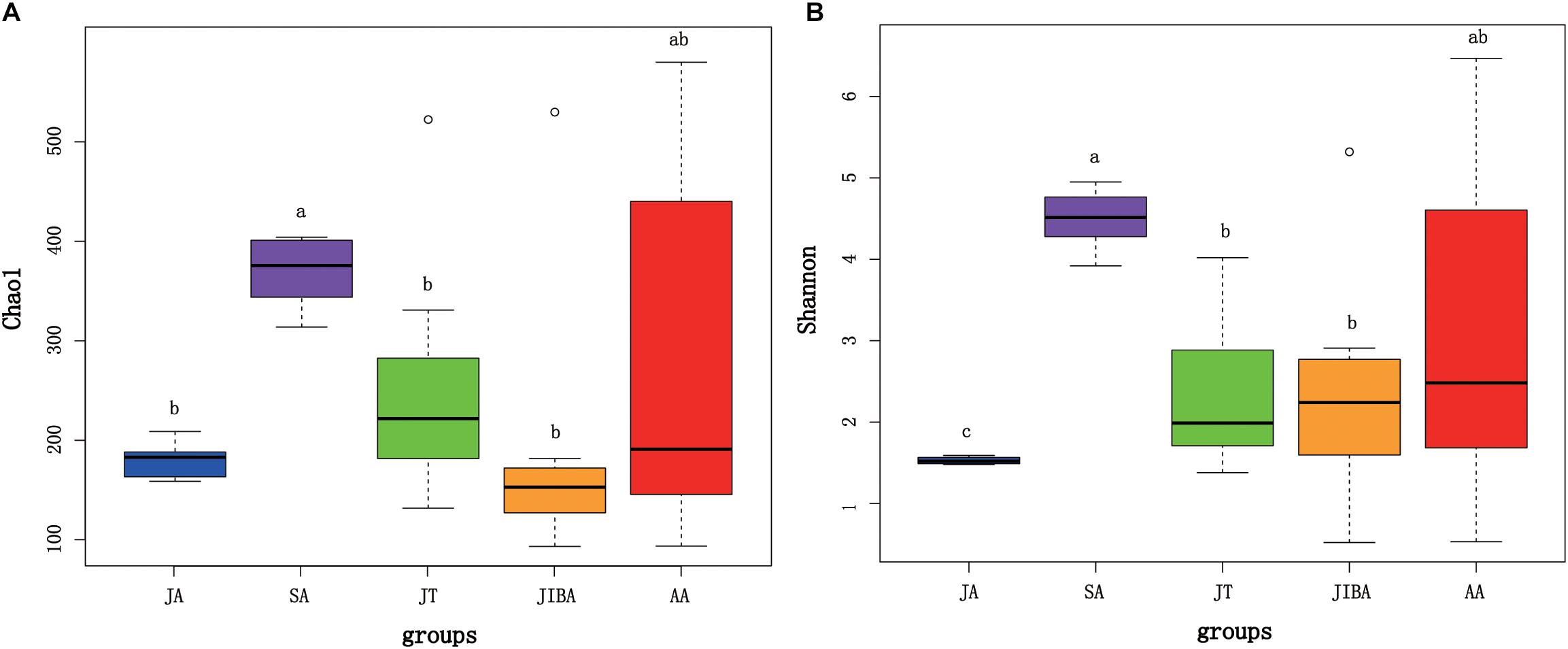
Figure 1. Alpha diversity of microbial communities in the bat gut. Data plotted show the last means ± SE of the Chao1 (A) and Shannon (B) indexes from rarefaction plots (Supplementary Figure 1). Significant differences in alpha diversity among the groups are indicated by different letters according to Kruskal–Wallis rank sum test (P ≤ 0.05). JA, early summer; SA, late summer; JT, early winter-torpor; JIBA, early winter IBA; AA, early spring.
Cluster Analysis of Microbiotas
Comparison of individual bat microbiotas, using principal coordinate analysis (PCoA) based on unweighted UniFrac metric data, showed distinct clustering by season, with most of the variation explained by the first three coordinates (Figure 2). Individuals from early summer and late summer groups clustered separately. Most individuals from early spring and early winter groups (torpor and IBA) clustered together. The thermal and metabolic state of hibernating bats at the time of sampling had little effect on microbiota clustering in either winter group, because individuals in torpor are clustered together with individuals in IBA (Figure 2). The results of PERMANOVA showed significant differences between two groups at least (P ≤ 0.001) (Supplementary Table 1). Differences in the UniFrac distances for pairwise comparisons among groups were determined using Student’s t-test and the Monte Carlo permutation test with 1,000 permutations to clarify significant differences between two groups. The results showed that the community structure of intestinal microbes in early summer and late summer were significantly different, and that these two groups were also significantly different from other groups. There was no significant difference in the microbial community structure among winter torpor, IBA, and early spring groups (Figure 3 and Table 2).
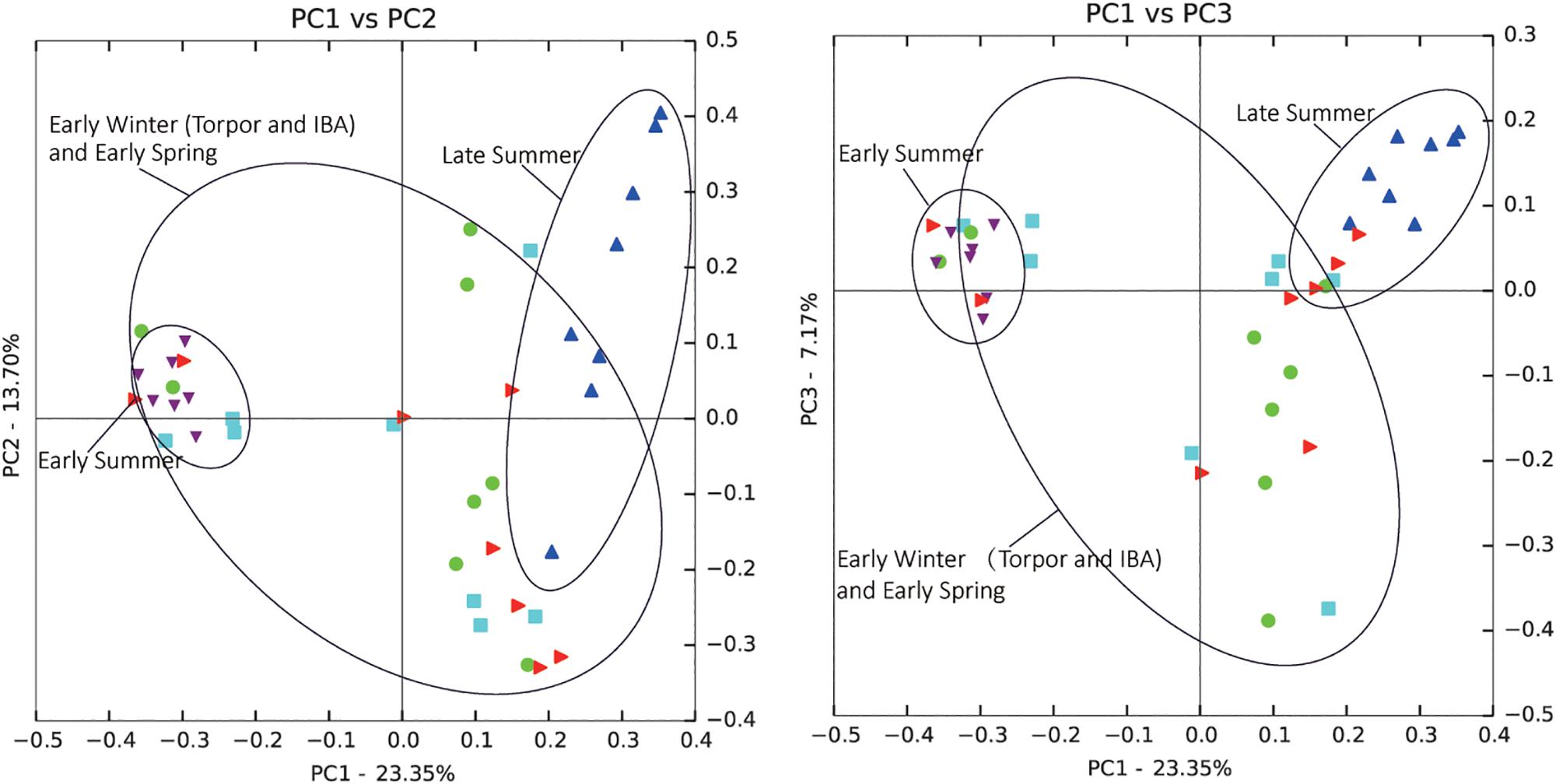
Figure 2. Principal coordinate analysis (PCoA) plots of unweighted UniFrac metrics for bat microbiotas. Each dot represents individual microbiota samples obtained from early spring (red triangles), early summer (purple triangles), late summer (blue triangles), early winter-torpor (light blue squares), and early winter-IBA (green circles). Left, PCA1 vs. PCA2; Right, PCA1 vs. PCA3. Numbers of bats in each seasonal group are shown in Table 1.
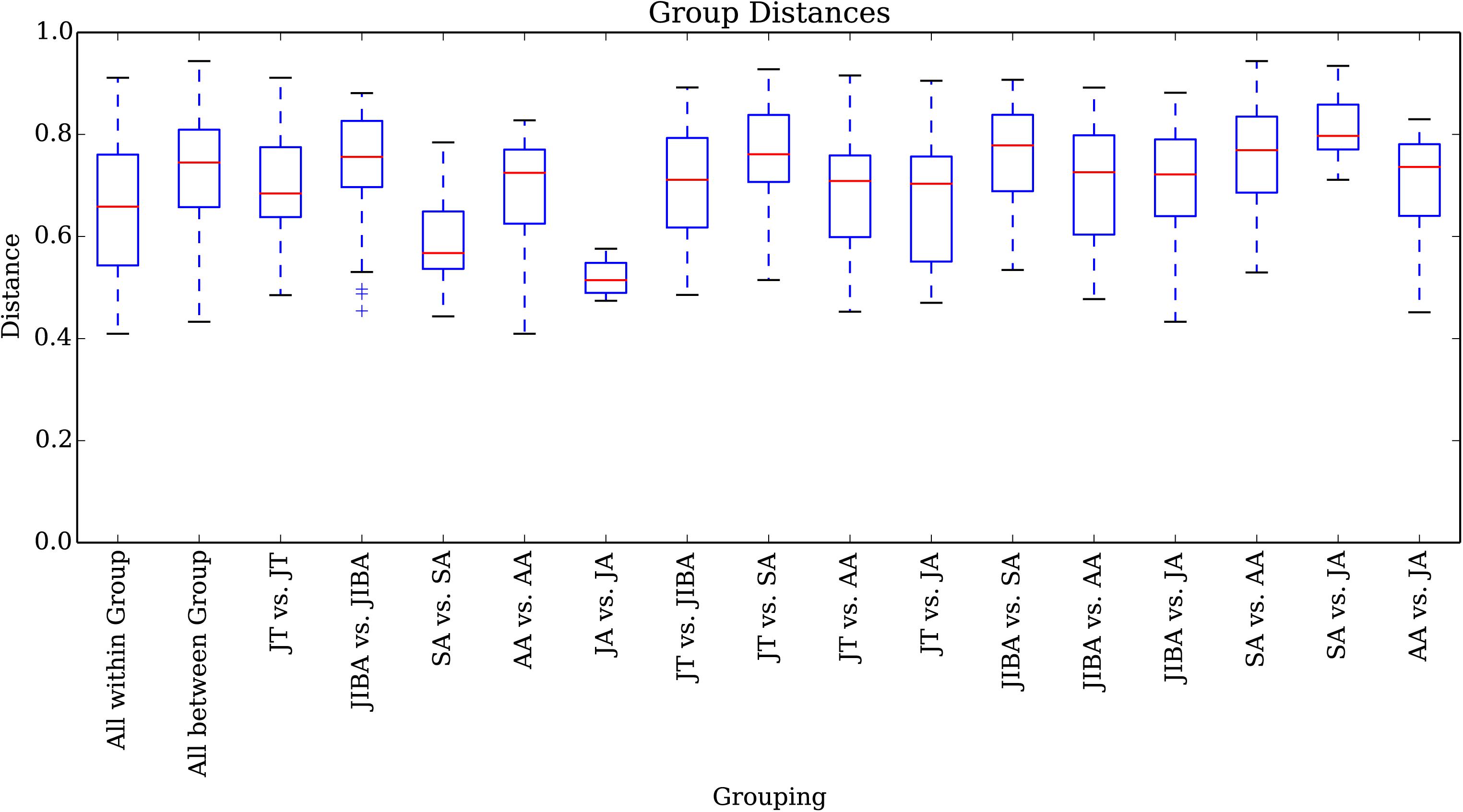
Figure 3. UniFrac distances for pairwise comparisons among groups. The X-axis shows pairwise comparisons among groups; the Y-axis shows UniFrac distances. The box plot border represents the upper and lower quartile spacing, the horizontal line represents the median value, and the upper and lower tentacles represent the 1.5 × IQR range outside the upper and lower quartiles, respectively. Significant differences in the UniFrac distances for pairwise comparisons among groups are shown in Table 2. If the distance between two groups was significantly greater than that within the groups, the difference between these groups was considered significant.
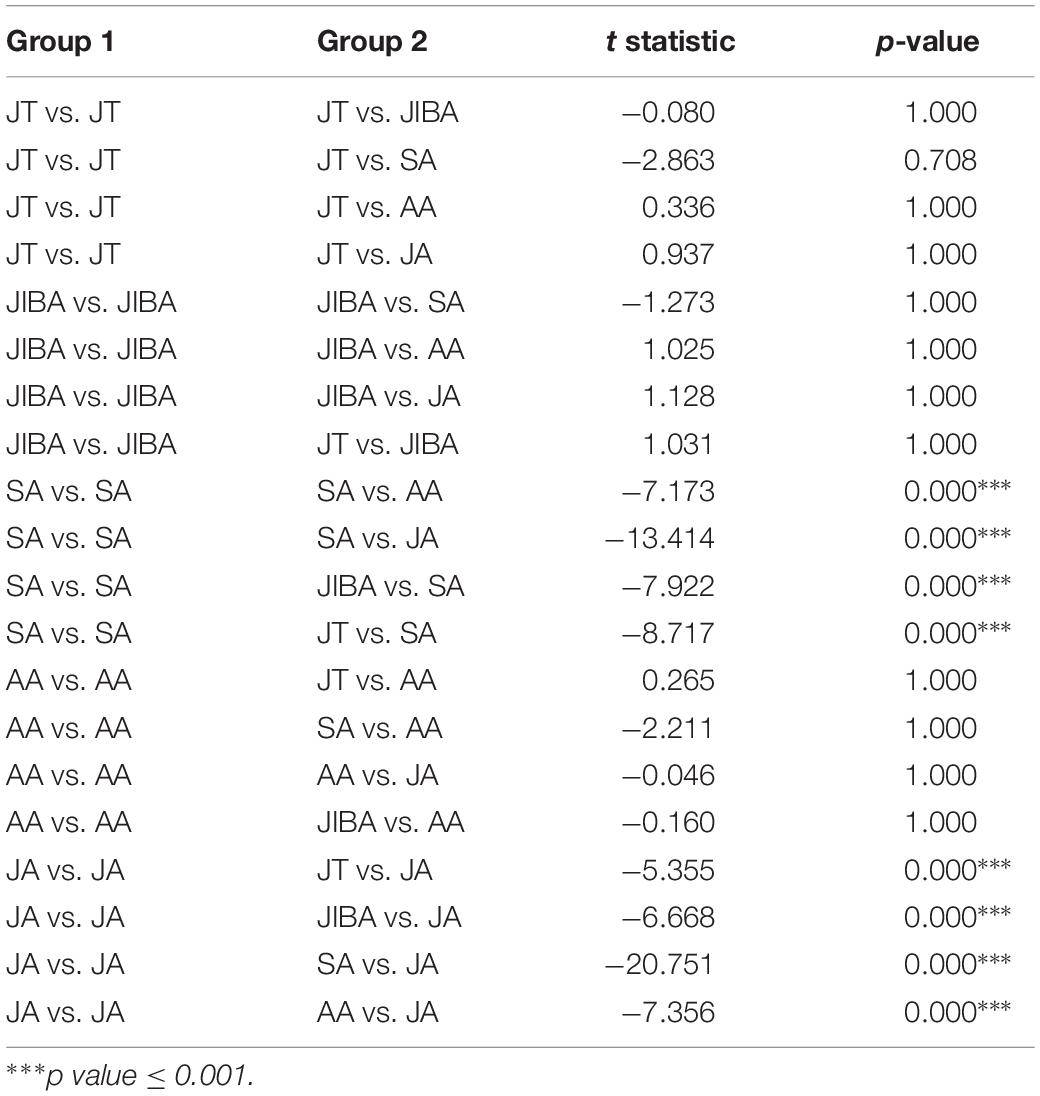
Table 2. Results of Student’s t-test and the Monte Carlo permutation test of differences in the UniFrac distances for pairwise comparisons among groups.
Taxonomic Composition of Bat Microbiotas
Four major bacterial phyla were identified in the R. ferrumequinum microbiota, with the majority of sequences classified as Proteobacteria (∼74.6%), followed by Firmicutes (∼15.9%), Tenericutes (∼4.8%), and some Bacteroidetes (∼1.6%) (Figure 4 and Supplementary Table 2). The other phyla were Fusobacteria, Chlamydiae, and Actinobacteria, but were not abundant (<1%). An unclassified group of sequences accounted for the remainder of the bacterial diversity of the bat microbiota (Supplementary Table 2).
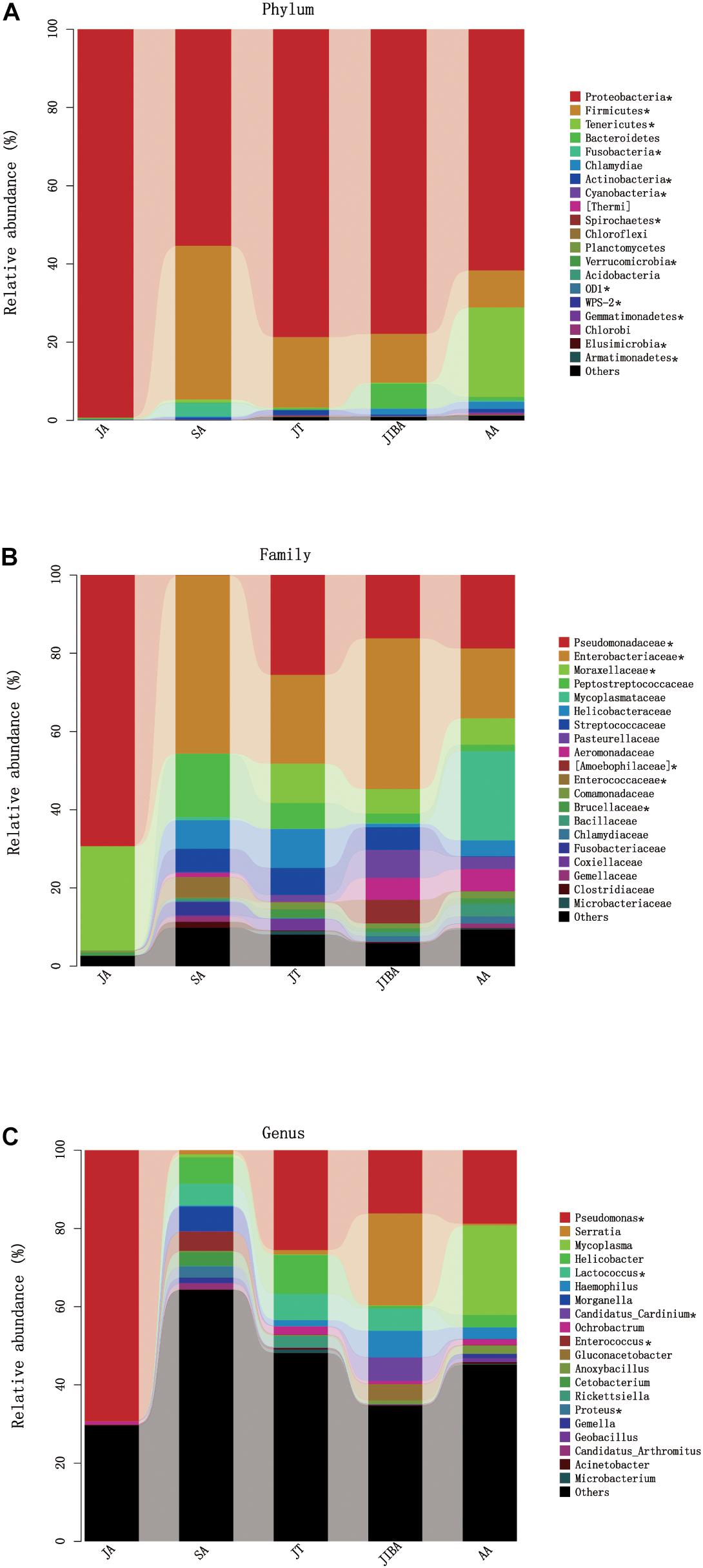
Figure 4. Relative abundance of major taxa in microbiotas of the bat gut. (A) Phylum; (B) Family; (C) Genus. Asterisks represent microbial taxa with significant differences in each seasonal period. The numbers of bats in each seasonal group are shown in Table 1.
The majority of Proteobacteria sequences identified in the bat microbiota matched the order Pseudomonadales and Enterobacteriates. The order Pseudomonadales were represented primarily by the family Pseudomonadaceae and Moraxellaceae. At the genus level, dominant groups were Pseudomonas. The order Enterobacteriates were represented primarily by the family Enterobacterlaceae. There were some unclassified Enterobacteriaceae and Serratia, and less abundant Proteus at the genus level (Supplementary Table 2). Firmicutes were dominated by Lactobacillales and Clostridiales. Lactobacillus were represented by Streptococcaceae, Enterococcaceae, and some unclassified Lactobacillales, with most OTUs matching to the genera Lactococcus and Enterococcus. Clostridiales was mainly represented by Peptostreptococcaceae, which contained some taxa of the unclassified Peptostreptococcaceae. The sequences of Tenericutes mainly matched the order Mycoplasmatales, which was primarily represented by the family Mycoplasmataceae. The taxonomic group at the genus level was Mycoplasma (Figure 4 and Supplementary Table 2).
At the phylum level, the relative abundance of Proteobacteria was highest in early summer and significantly lower in other periods. The relative abundance of Firmicutes was lowest in early summer and significantly increased in late summer, followed by significant decreases in subsequent periods. The relative abundance of Tenericutes was significantly higher in early spring than in other periods and decreased to undetectable levels in early summer (Figure 4 and Table 3). At the family level, the relative abundance of Enterobacteriaceae significantly increased from early summer to early winter, and the IBA group had double that of the torpor period (Figure 4 and Table 3). The changes in the relative abundance of Pseudomonadaceae and Moraxellaceae were consistent, with significantly higher values in early summer than in other periods, the lowest being in late summer, and followed by significant increases in subsequent periods. Enterococcaceae was significantly higher in late summer than in other periods, reduced in early winter, and reduced to undetectable levels in early summer (Figure 4 and Table 3). At the genus level, Pseudomonas was obviously affected by season; the relative abundance was highest in the early summer group and reduced to the lowest abundance in the late summer group, then increased during hibernation, but was still significantly lower than in early summer. The relative abundances of Enterococcus, Lactococcus, and Proteus were highest in late summer, reduced in early winter, and almost undetectable in early summer. Seasonal changes in diet also influenced the relative abundance of some of the less common taxa (Figure 4 and Table 3).
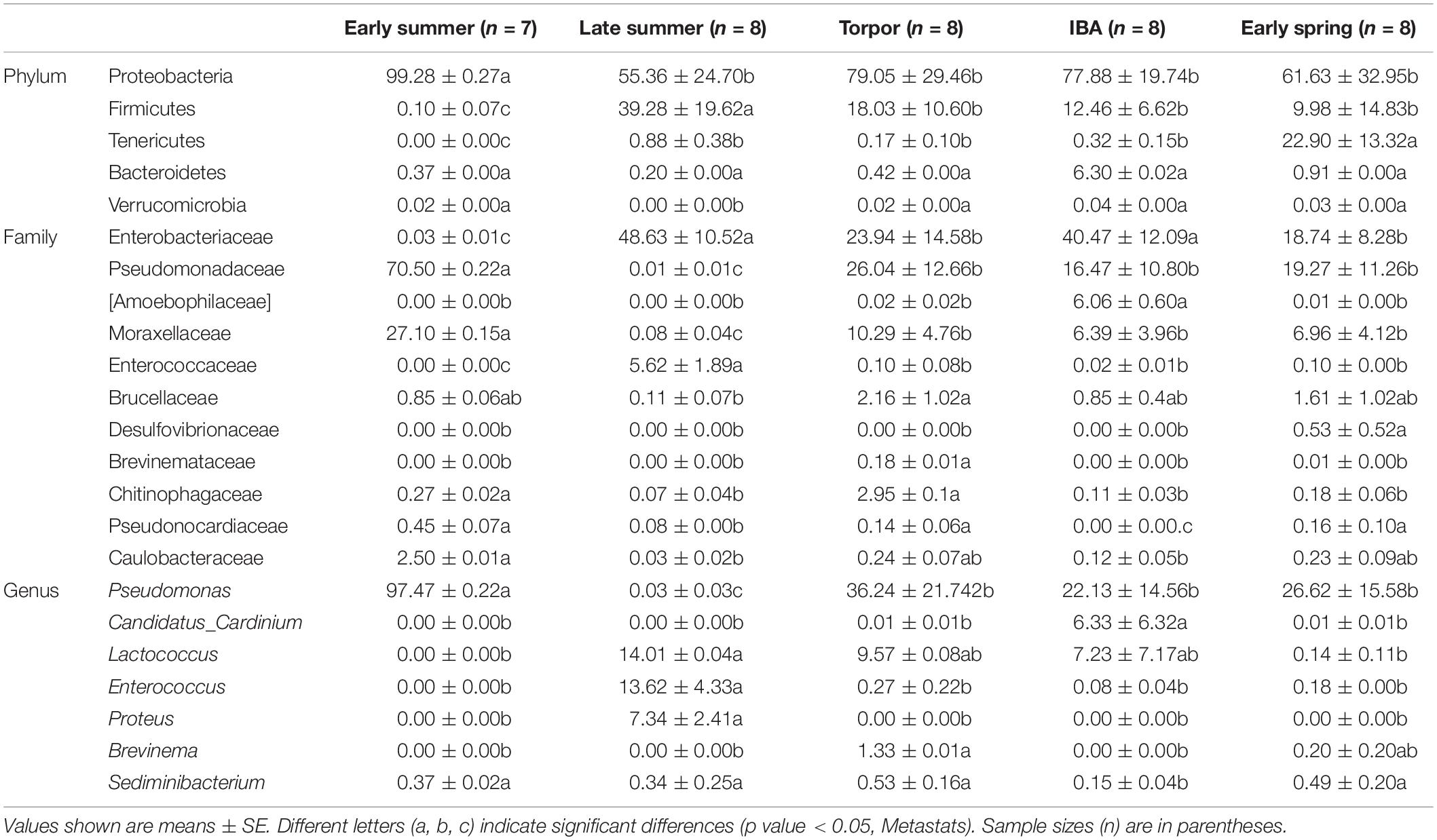
Table 3. Relative abundances of taxa with seasonal differences in bat gut microbiotas.
Significant Seasonal Changes in Gut Microbial Functions Predicted via PICRUSt
We used PICRUSt to perform functional predictions on the results of 16S rDNA sequencing. Based on the KEGG pathway database, analysis of the second level of functional categories of the KEGG pathway suggested that the relative abundance of most predicted functional categories changed significantly among the different groups (Kruskal–Wallis test, P < 0.05) (Supplementary Table 3), including metabolism of lipids, carbohydrates, energy, and amino acids (Figure 5 and Supplementary Table 3). The relative abundance of functional categories related to lipid metabolism was highest in early summer and lowest in late summer, and significantly increased in early winter and early spring compared with late summer, but was still significantly lower than that in early summer. The relative abundance of carbohydrate-related functional categories was lowest in the early summer and significantly increased in the late summer, followed by a significant decrease in torpor in the early winter. The relative abundance of energy metabolism was lowest in early summer, and in late summer and early winter torpor increased significantly. The relative abundance of functional categories associated with amino acid metabolism was significantly higher in early summer than in other periods (Figure 5 and Supplementary Table 3). These results indicated that the metabolic function of gut microbes in hibernating bats are altered the most by the changes in season.
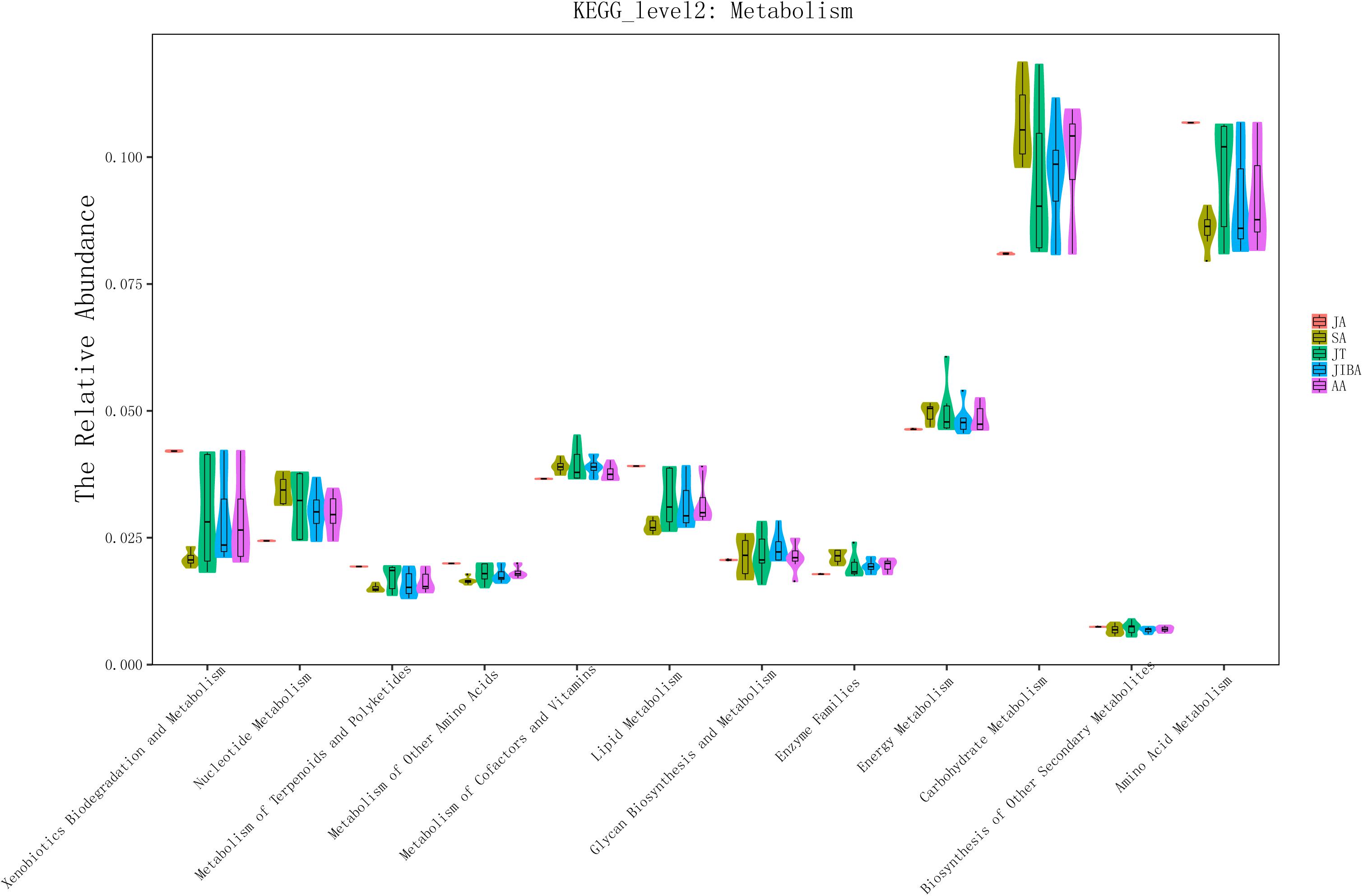
Figure 5. 16S rRNA gene sequencing of bat gut microbiota samples analyzed using PICRUSt. The abscissa shows all of the metabolic-related functional groups of the KEGG second level. The relative abundance of all functional taxa had significant differences between the groups for at least two periods (Kruskal–Wallis’s test, p < 0.01), except for Biosynthesis of Other Secondary Metabolites and Glycan Biosynthesis and Metabolism (Supplementary Table 3).
Discussion
Like other fat-storing hibernators, R. ferrumequinum stores large amounts of adipose tissue during active seasons, uses stored lipids as the main source of energy during hibernation, and begins refeeding in spring (Park and Ransome, 2000; Xiao et al., 2015). During hibernation, the Tb drops to about 10°C (slightly higher than the ambient temperature) with spontaneous awakening during hibernation (Park and Ransome, 2000). During IBA periods, the Tb returns to the active level of about 34–38°C. This hibernation-related physiological change causes a great alteration in the intestinal environment of bats, which greatly impacts the intestinal microbes. In this investigation, the microbial diversity, composition, and function of the intestines of R. ferrumequinum were each found to undergo seasonal changes.
Seasonal Changes in Diet Is the Main Factor Driving Intestinal Microbial Variation
The diversity in gut microbes gradually decreased sequentially: in the order of late summer, early spring, early winter and early summer, which was consistent with the seasonal changes in the diet of R. ferrumequinum. It has been reported that their diet is most diverse in autumn (Jones, 1990). Thus, the higher diversity of gut microbes in late summer may be associated with a more diverse diet at that time, which is close to autumn. Effects of hibernation were also evident in the gut microbial diversity, which was lower during hibernation than in early spring and late summer. This may be due to the bats fasting during hibernation. These changes in diversity are consistent with the findings in brown bears and ground squirrels (Carey et al., 2013; Stevenson et al., 2014; Sommer et al., 2016). Similarly to the results in ground squirrels (Carey et al., 2013), the diversity of bat gut microbes in early summer was lower than that in early winter, which suggested that the intestinal microbes become more stable and less diverse in the summer, as the time of re-feeding increases. The summer period may be dominated by species that have a competitive advantage in the presence of abundant and continuously available dietary substrates.
The PCoA based on the unweighted UniFrac metric showed that the community structure of bat gut microbes significantly clustered by seasons, with significant differences between samples from early summer and late summer. R. ferrumequinum mainly feeds on Lepidopteran and Coleopteran insects, and there are significant seasonal changes in food composition (Jones, 1990; Wang et al., 2010). Studies have shown that the proportions of Lepidoptera in the diet are higher in the early summer, while in the late summer, the proportion of Coleoptera is larger (Wang et al., 2010). Changes in food composition may be the main reason for the separate clustering of intestinal microflora in early summer and late summer. Most individuals from early spring and early winter (torpor and IBA) clustered together. The individuals in the torpor group clustered with the individuals in the IBA group, indicating that the euthermic and metabolic status of the hibernating bats during sampling had little effect on the microbial clustering of the winter group, which is consistent with a study of the ground squirrel (Stevenson et al., 2014). In the early winter hibernation period, bats have fasted, so that the early winter-torpor and IBA groups may have been dominated by some microbial communities capable of decomposing host-derived substrates. The individuals in early spring and early winter clustered, indicating that the microbial community structure changed slightly during the spring. That may have been because the bats were just starting to awaken from hibernation. During early spring, when some individuals may eat a little food, the gut microbe diversity was higher than that in winter, although not significantly. This observation indicated that the diversity of the bats’ gut microbiota was recovering. Ecosystem function is often positively correlated with species richness (Hooper et al., 2005; Maestre et al., 2012; Cardinale et al., 2013; Wagg et al., 2014). Significantly decreased gut microbial diversity during early winter indicates that hibernation, accompanied by slower metabolism, may weaken microbial ecosystem function in the gastrointestinal tract.
Changes in Intestinal Microbes of Hibernating Bats Are the Same as Those of Other Small Hibernating Mammals
Our taxonomic composition analysis across different seasonal periods found that, unlike other mammals, the gut microbiota of R. ferrumequinum was dominated by the Proteobacteria, while Firmicutes, Tenericutes, and Bacteroidetes occupied smaller parts. Proteobacteria was also found to be the primary bacteria in other studies on the microbial composition of the bat gut (Carrillo-Araujo et al., 2015; Nishida and Ochman, 2017; Hughes et al., 2018; Ingala et al., 2018). In other mammals, such as ground squirrels and brown bears, as well as humans, Bacteroides and Firmicutes dominate, while the Proteobacteria account for only a small part of the microbiota. In the human intestinal tract, Proteobacteria comprise less than 5% of the total (Shin et al., 2015). Therefore, our data indicate that the dominant species of gut microbiota differs between bats and other hibernating mammals.
In our comparison of the composition of bat gut microbes at different seasonal times, we found significant differences in the relative abundance of major bacterial phyla. The relative abundance of Proteobacteria was higher in early winter than in late summer, the relative abundance of Firmicutes was significantly lower in early winter than in late summer, and the relative abundance of Tenericutes was significantly higher in the early spring than in other periods. In the study of ground squirrels, the relative abundance of Firmicutes decreased, while Bacteroidetes and Verrucomicrobia increased during hibernation (Carey et al., 2013; Stevenson et al., 2014). In brown bears, the relative abundance of Firmicutes decreased and Bacteroides increased during hibernation, but the relative abundance of Verrucomicrobia decreased (Sommer et al., 2016). Although the dominant intestinal microbe differed between the bats in our study and some other mammals, there were similar changes in the gut microbiota of bats and ground squirrels. During hibernation in our bats, the relative abundance of Firmicutes decreased, and the relative abundance of Bacteroidetes and Verrucomicrobia increased, although the changes in Bacteroidetes were not significant. These results suggested that the composition of the gut microbiome of small mammals, including bats and squirrels, tend to be similarly altered in response to hibernation.
Seasonal Changes in the Metabolic Function of Bat Intestinal Microbes
The relative abundances of most of the predicted functional taxa of bat gut microbials in the KEGG pathway changed significantly by season. The primary changes occurred in the relative abundance of metabolism-related functional taxa. In seasonal comparisons of functional categories, those associated with carbohydrate metabolism had significantly lower relative abundances in early winter-torpor compared with late summer, while the reverse was true for those categories associated with lipid metabolism. These results suggested that the metabolic function of the gut microflora is converted from carbohydrate-related to lipid-related functional categories during hibernation. This may be related to the transformation in substrate utilization involved in energy metabolism during hibernation, which involves an important physiological shift from carbohydrate oxidation to lipid catabolism (Carey et al., 2003). Studies of transcriptomes in small hibernating mammals have revealed that many genes involved in metabolism are differentially expressed during hibernation. Specifically, the genes involved in carbohydrate catabolism are downregulated during hibernation, while genes responsible for lipid catabolism are upregulated (Carey et al., 2013; Stevenson et al., 2014; Han et al., 2015; Xiao et al., 2015; Sommer et al., 2016). We also found that the relative abundance of functional categories associated with amino acid metabolism increased in the early winter compared with the late summer, consistent with other studies showing that hibernating bats have elevated levels of amino acid metabolism during hibernation (Pan et al., 2013). We propose that such an increase in amino acid metabolism may produce the very metabolic substrates that are required to alter the relative abundance of functional categories of gut microbes involved in amino acid metabolism. Overall, such results are consistent with seasonal changes in intestinal microbial metabolic function that support the co-evolution of gut microbes and hibernating hosts. Studies of ground squirrels and brown bears also found that intestinal microbes can regulate metabolic function (Carey et al., 2013; Stevenson et al., 2014; Sommer et al., 2016). Seasonal differences in gut microbes may contribute to the metabolic changes that occur in hibernating animals. The interaction between the host and its gut microbes appears to be vital in promoting the adaptation of the hibernating host to the extreme environment of fasting in winter.
Seasonal Variation in Intestinal Microbes and Adaptation to Extreme Environments in Bats
In this study, we also predicted the functions of some microbial species. For example, the relative abundance of Firmicutes was highest in late summer, which may be associated with fattening before entering hibernation. Some studies on non-hibernating mammals have suggested that changes in the relative abundance of the Firmicutes phylum are associated with fat accumulation and the potential for obesity (Ley et al., 2008a; Turnbaugh and Gordon, 2009), although not all studies support this opinion (Duncan et al., 2007; Schwiertz et al., 2010). In our study, the weight of bats in late summer was significantly lower than that in early summer and early winter, and not significantly different from that in spring, indicating that our late-summer sampling occurred before the fattening period. Indeed, the relative abundance of Firmicutes increased sharply before the bats entered the fattening period in autumn and decreased after entering hibernation. In addition, the relative abundance of Pseudomonas was significantly increased during hibernation. Bacteria of this genus are able to grow at a wide range of temperatures, from 5° to 42°C (Termine and Michel, 2009). Moreover, fasting hosts use lipids as the main energy source during hibernation, and most species of Pseudomonas secrete a low-temperature lipase that can break down lipids. The optimum temperature for the low-temperature lipase is 7–10°C, and it also has certain catalytic activity at 0°C (Roh and Villatte, 2008; Ji et al., 2015; Jain et al., 2017). The low Tb and shift in energy source from glucose to lipids in hibernating bats may be responsible for the significant increase in the relative abundance of Pseudomonas during hibernation. Studies in Rhinolophus euryale have shown that Pseudomonas is also predominant in the stool samples of hibernating R. euryale (Maliničová et al., 2017). Our study revealed that the gut microbiomes of bats and ground squirrels tend to have similar changes in response to hibernation. In a study of hibernating ground squirrels, it was shown that hibernation increased the relative abundance of Bacteroidetes and Verrucomicrobia (phyla which contain species capable of surviving on host-derived substrates such as mucins), and reduced the relative abundance of Firmicutes (many of which prefer dietary polysaccharides). The ground squirrel microbiota is restructured each year in a manner that reflects differences in the abilities of different bacterial taxa to survive in the changing environment of the hibernator gut (Carey et al., 2013). Therefore, seasonal changes in the intestinal microbial composition of hibernating bats may reflect compatibility with microbial functions. Seasonal changes in diet alter the structure of the gut microbiota; such rebuilding of microbial communities in the bat gut may alter the interactions between microbial species and the overall function of the ecosystem. These may be important factors for host adaptation to extreme winter environments.
Conclusion
Seasonal changes occurred in the diversity and composition of the intestinal microbes of hibernating R. ferrumequinum. The alpha diversity value was highest in the late summer, followed by early spring and early winter, and lowest in the early summer. Seasonal changes in diet strongly affected taxonomic representation of the dominant phyla. The relative abundance of Proteobacteria was highest in the early summer and significantly lower in other periods. The relative abundance of Firmicutes was lowest in the early summer and significantly increased in the late summer, followed by a significant decrease in the early winter and early spring. Comparison of changes in gut microbiota between bats and other hibernating mammals suggested that gut microbiota tend to show similar responses to hibernation in some small mammals (i.e., bats and ground squirrels). The results of functional prediction by PICRUSt showed that the relative abundance of metabolism-related pathways changed in different seasons. This indicates the important role of gut microbes in the adaptation of hibernating animals to the extreme environment of fasting in winter. More work is needed to understand the functions of gut microorganisms in hibernating mammals during different periods. For example, we plan to investigate whether seasonal changes in the intestinal microbiota of hibernating mammals reprograms the metabolic functions of the microorganisms using a combination of metagenomics and metabolomics.
Data Availability Statement
The datasets generated for this study can be accessed from NCBI Sequence Read Archive (SRA), SRR8756041–SRR8756079.
Ethics Statement
According to the regulations of Wildlife Conservation of the People’s Republic of China (Chairman Decree [2004] No. 24), permits are required only for species included in the list of state-protected and region-protected wildlife species. R. ferrumequinum is not an endangered or region-protected animal species, so no specific permission was required. Sampling was conducted outside the protected areas, with permission of the local forestry department. All experimental procedures carried out in this study were approved by the Forestry Bureau of Jilin Province of China (approval number: [2006]178). And all efforts were made to minimize suffering of animals in the process of collecting samples.
Author Contributions
YX and GX contributed to the design of the study. GX, YZ, and SL collected the gut content samples in all period. YX collected samples in late summer. AL and ZL collected samples in winter. GX analyzed the data. GX, YX, SL, HZ, and JF contributed to the writing of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31700319 and 31870354), the Fundamental Research Funds for the Central Universities (Grant No. 2412017QD026), and the project funded by China Postdoctoral Science Foundation (Grant No. 2018M631852).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Shuxin Yuan, Chuanyu Chen, Haixia Leng, and Wentao Dai for their great contributions to samples collection. Sequencing service was provided by Personal Biotechnology Co., Ltd., Shanghai, China. We thank Elizabeth Kelly, MSc, and Michelle Kahmeyer-Gabbe, Ph.D., from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02247/full#supplementary-material
References
Burton, M., and Burton, R. (2002). International Wildlife Encyclopedia. Tarrytown, NY: Marshall Cavendish Corp.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of highthroughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth0510-335
Cardinale, B., Gross, K., Fritschie, K., Flombaum, P., Fox, J., Rixen, C., et al. (2013). Biodiversity simultaneously enhances the production and stability of community biomass, but the effects are independent. Ecology 94, 1697–1707. doi: 10.1890/12-1334.1
Carey, H. V., Andrews, M. T., and Martin, S. L. (2003). Mammalian hibernation: cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 83, 1153–1181. doi: 10.1152/physrev.00008.2003
Carey, H. V., Walters, W. A., and Knight, R. (2013). Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R33–R42. doi: 10.1152/ajpregu.00387.2012
Carrillo-Araujo, M., Tas, N., Alcantara-Hernandez, R. J., Gaona, O., Schondube, J. E., Medellin, R. A., et al. (2015). Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Front. Microbiol. 6:447. doi: 10.3389/fmicb.2015.00447
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Dennis, K. L., Wang, Y., Blatner, N. R., Wang, S., Saadalla, A., Trudeau, E., et al. (2013). Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 73, 5905–5913. doi: 10.1158/0008-5472.CAN-13-1511
Derrien, M., Vaughan, E. E., Plugge, C. M., and de Vos, W. M. (2004). Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 54(Pt 5), 1469–1476. doi: 10.1099/ijs.0.02873-0
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dill-McFarland, K. A., Neil, K. L., Zeng, A., Sprenger, R. J., Kurtz, C. C., Suen, G., et al. (2014). Hibernation alters the diversity and composition of mucosa-associated bacteria while enhancing antimicrobial defence in the gut of 13-lined ground squirrels. Mol. Ecol. 23, 4658–4669. doi: 10.1111/mec.12884
Dill-McFarland, K. A., Suen, G., and Carey, H. V. (2016). Bears arouse interest in microbiota’s role in health. Trends Microbiol. 24, 245–246. doi: 10.1016/j.tim.2016.01.011
Duncan, S. H., Belenguer, A., Holtrop, G., Johnstone, A. M., Flint, H. J., and Lobley, G. E. (2007). Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 73, 1073–1078. doi: 10.1128/AEM.02340-06
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Faith, J. J. M., McNulty, N. P., Rey, F. E., and Gordon, J. I. (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. doi: 10.1126/science.1206025
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P., and Forano, E. (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. doi: 10.4161/gmic.19897
Florant, G. L., and Healy, J. E. (2012). The regulation of food intake in mammalian hibernators: a review. J. Comp. Physiol. B 182, 451–467. doi: 10.1007/s00360-011-0630-y
Geluso, K. (2007). Winter activity of bats over water and along flyways in New Mexico. Southwest. Nat. 52, 482–492. doi: 10.1894/0038-4909(2007)52%5B482:waobow%5D2.0.co;2
Hampton, M., Melvin, R. G., Kendall, A. H., Kirkpatrick, B. R., and Peterson, N. (2011). Deep sequencing the transcriptome reveals seasonal adaptive mechanisms in a hibernating mammal. PLoS One 6:e27021. doi: 10.1371/journal.pone.0027021
Han, Y., Zheng, G., Yang, T., Zhang, S., Dong, D., and Pan, Y. H. (2015). Adaptation of peroxisome proliferator-activated receptor alpha to hibernation in bats. BMC Evol. Biol. 15:88. doi: 10.1186/s12862-015-0373-6
Hooper, D., Chapin, I. I. I. F., Ewel, J., Hector, A., Inchausti, P., Lavorel, S., et al. (2005). Effects of biodiversity on ecosystem functioning: a consensus of current knowledge. Ecol. Monogr. 75, 3–35. doi: 10.1890/04-0922
Hooper, L., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Hughes, G. M., Leech, J., Puechmaille, S. J., Lopez, J. V., and Teeling, E. C. (2018). Is there a link between aging and microbiome diversity in exceptional mammalian longevity? PeerJ 6:e4174. doi: 10.7717/peerj.4174
Human Microbiome Project Consortium. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Ingala, M. R., Simmons, N. B., Wultsch, C., Krampis, K., Speer, K. A., and Perkins, S. L. (2018). Comparing microbiome sampling methods in a wild mammal: fecal and intestinal samples record different signals of host ecology, evolution. Front. Microbiol. 9:803. doi: 10.3389/fmicb.2018.00803
Jain, R., Pandey, A., Pasupuleti, M., and Pande, V. (2017). Prolonged production and aggregation complexity of cold-active lipase from Pseudomonas proteolytica (GBPI_Hb61) isolated from cold Desert Himalaya. Mol. Biotechnol. 59, 34–45. doi: 10.1007/s12033-016-9989-z
Ji, X., Li, S., Lin, L., Zhang, Q., and Wei, Y. (2015). Gene cloning, sequence analysis and heterologous expression of a novel cold-active lipase from Pseudomonas sp. PF16. Technol. Health Care 23(Suppl. 1), S109–S117. doi: 10.3233/thc-150941
Jones, G. (1990). Prey sellection by the grater horseshoe bat(Rhnolophus ferrumequinum): optimal foraging by echolocation? J. Anim. Ecol. 59, 587–602.
Kunz, T. H., and Anthony, A. E. L. P. (1982). Age estimation and postnatal-growth in the Bat Myotis lucifugus. J. Mammal. 63, 23–32. doi: 10.2307/1380667
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Leser, T. D., Lindecrona, R. H., Jensen, T. K., Jensen, B. B., and Moller, K. (2000). Changes in bacterial community structure in the colon of pigs fed different experimental diets and after infection with Brachyspira hyodysenteriae. Appl. Environ. Microbiol. 66, 3290–3296. doi: 10.1128/aem.66.8.3290-3296.2000
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, B. J., et al. (2008a). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Lozupone, C. A., Hamady, M. K., Knight, R., and Gordon, J. I. (2008b). Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788. doi: 10.1038/nrmicro1978
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Lyman, C., Willis, J. S., Malan, A., and Wang, L. (1982). Hibernation and Torpor in Mammals and Birds. New York, NY: AcademicPress.
Maestre, F., Castillo-Monroy, A. P., Bowker, M. A., and Ochoa-Hueso, R. (2012). Species richness effects on ecosystem multifunctionality depend on evenness, composition and spatial pattern. J. Ecol. 100, 317–330. doi: 10.1111/j.1365-2745.2011.01918.x
Maliničová, L., Hrehová, L., Maxinová, E., Uhrin, M., and Pristaš, P. (2017). The dynamics of mediterranean horseshoe bat (Rhinolophus euryale, Chiroptera) gut microflora during hibernation. Acta Chiropt. 19, 211–218. doi: 10.3161/15081109acc2017.19.1.017
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Loso, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Muegge, B., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
Nishida, A. H., and Ochman, H. (2017). Rates of gut microbiome divergence in mammals. Mol. Ecol. 27, 1884–1897. doi: 10.1111/mec.14473
Pan, Y. H., Zhang, Y., Cui, J., Liu, Y., McAllan, B. M., Liao, C. C., et al. (2013). Adaptation of phenylalanine and tyrosine catabolic pathway to hibernation in bats. PLoS One 8:e62039. doi: 10.1371/journal.pone.0062039
Park, G. J., and Ransome, R. D. (2000). Torpor, arousal and activity of hibernating greater horseshoe bats (Rhinolophus ferrumequinum). Funct. Ecol. 14, 580–588. doi: 10.1046/j.1365-2435.2000.t01-1-00460.x
Phillips, C. D., Phelan, G., Dowd, S. E., McDonough, M. M., Ferguson, A. W., Delton Hanson, J., et al. (2012). Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627. doi: 10.1111/j.1365-294X.2012.05568.x
Roh, C., and Villatte, F. (2008). Isolation of a low-temperature adapted lipolytic enzyme from uncultivated micro-organism. J. Appl. Microbiol. 105, 116–123. doi: 10.1111/j.1365-2672.2007.03717.x
Ruf, T., and Geiser, F. (2015). Daily torpor and hibernation in birds and mammals. Biol. Rev. Camb. Philos. Soc. 90, 891–926. doi: 10.1111/brv.12137
Schwiertz, A., Taras, D., Schafer, K., Beijer, S., Bos, N. A., Donus, C., et al. (2010). Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18, 190–195. doi: 10.1038/oby.2009.167
Shin, N. R., Whon, T. W., and Bae, J. W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Sommer, F., and Backhed, F. (2013). The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Sommer, F., Stahlman, M., Ilkayeva, O., Arnemo, J. M., Kindberg, J., Josefsson, J., et al. (2016). The gut microbiota modulates energy metabolism in the hibernating brown bear ursus arctos. Cell Rep. 14, 1655–1661. doi: 10.1016/j.celrep.2016.01.026
Sonnenburg, J. L., Xu, J., Leip, D. D., Chen, C. H., Westover, B. P., Weatherford, J., et al. (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959. doi: 10.1126/science.1109051
Stevenson, T., Duddleston, K. N., and Loren Buck, C. (2014). Effects of season and host physiological state on the diversity, density, and activity of the arctic ground squirrel cecal microbiota. Appl. Environ. Microbiol. 80, 5611–5622. doi: 10.1128/AEM.01537-14
Storey, K. B., and Storey, J. M. (2004). Metabolic rate depression in animals: transcriptional and translational controls. Biol. Rev. Camb. Philos. Soc. 79, 207–233. doi: 10.1017/S1464793103006195
Termine, E., and Michel, G. P. F. (2009). Transcriptome and secretome analyses of the adaptive response of Pseudomonas aeruginosa to suboptimal growth temperature. Int. Microbiol. 12, 7–12. doi: 10.2436/20.1501.01.76
Turnbaugh, P. J., and Gordon, J. I. (2009). The core gut microbiome, energy balance and obesity. J. Physiol. 587(Pt 17), 4153–4158. doi: 10.1113/jphysiol.2009.174136
Wagg, C., Bender, S. F., Widmer, F., and van der Heijden, M. G. A. (2014). Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. U.S.A. 111, 5266–5270. doi: 10.1073/pnas.1320054111
Wang, J., Jiang, T., Wang, L., Lu, G., You, Y., Liu, Y., et al. (2010). Relationships between foraging activity of greater horseshoe bat (Rhinolophusferrumequinum) and prey resources. Acta Theriol. Sin. 30, 157–162.
Wang, Y. (2003). A Complete Checklist of Mammal Species and Subspecies in China. Beijing: China Forestry Pubblishing House.
Whitaker, J. O., and Rissler, L. J. (1992). Winter activity of bats at a mine entrance in vermillion county, Indiana. Am. Midl. Nat. 127, 52–59. doi: 10.2307/2426321
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Williams, D., Epperson, L. E., Li, W., Hughes, M., Taylor, R., and Rogers, J. (2005). Seasonally hibernating phenotype assessed through transcript screening. Physiol. Genom. 24, 13–22. doi: 10.1152/physiolgenomics.00301.2004
Keywords: hibernation, gut microbiota, fasting, diet, Rhinolophus ferrumequinum
Citation: Xiao G, Liu S, Xiao Y, Zhu Y, Zhao H, Li A, Li Z and Feng J (2019) Seasonal Changes in Gut Microbiota Diversity and Composition in the Greater Horseshoe Bat. Front. Microbiol. 10:2247. doi: 10.3389/fmicb.2019.02247
Received: 20 April 2019; Accepted: 13 September 2019;
Published: 01 October 2019.
Edited by:
Paula García-Fraile, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Yi-Hsuan Pan, East China Normal University, ChinaLixin Luo, South China University of Technology, China
Copyright © 2019 Xiao, Liu, Xiao, Zhu, Zhao, Li, Li and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhong Xiao, xiaoyh767@nenu.edu.cn; Jiang Feng, fengj@nenu.edu.cn