 Pooja Malhotra1
Pooja Malhotra1 Ravinder K. Gill
Ravinder K. Gill Seema Saksena
Seema Saksena Waddah A. Alrefai
Waddah A. Alrefai- 1Division of Gastroenterology and Hepatology, Department of Medicine, University of Illinois at Chicago, Chicago, IL, United States
- 2Jesse Brown VA Medical Center, Chicago, IL, United States
Non-alcoholic fatty liver disease (NAFLD) is a major health problem associated with obesity and other features of the metabolic syndrome including insulin resistance and dyslipidemia. The accumulation of lipids in hepatocytes causes liver damage and triggers inflammation, fibrosis, and cirrhosis. Beside fatty acids and triglycerides, evidence showed an increased accumulation of free cholesterol in the liver with subsequent toxic effects contributing to liver damage. The maintenance of cholesterol homeostasis in the body requires a balance between several pathways responsible for cholesterol synthesis, transport and conversion into bile acids. Intestinal absorption is also one of the major determinants of cholesterol homeostasis. The nature of changes in cholesterol homeostasis associated with NAFLD has been a subject of extensive investigations. In this article, we will attempt to provide a brief overview of the current knowledge about the disturbances in cholesterol metabolism associated with NAFLD and discuss how certain molecular targets of these pathways could be exploited for the treatment of this multifactorial disease.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a common chronic hepatic disorder associated with obesity and metabolic syndrome (1, 2). Histologically, NAFLD encompasses a broad range of liver injury including simple steatosis or non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH) and subsequent fibrosis as well as cirrhosis (3). The key feature is lipid accumulation in the liver that is not related to alcohol in hepatocytes (4). The accumulation of lipids in hepatocytes as a result of dyslipidemia and insulin resistance leads to liver damage and triggers elaborate response causing hepatic inflammation and fibrosis (2). It is widely accepted that increased flux of fatty acids associated with insulin resistance and/or increased de novo lipogenesis in the liver lead to fatty acid accumulation in the liver with associated lipotoxicity (5). However, emerging evidence also suggested that the increased level of cellular cholesterol contributes to the development of NAFLD and the progression of the disease (6). Understanding the roles of cholesterol in the pathophysiology of NAFLD is of a particular importance as it may unravel novel molecular targets for effective therapeutic interventions for NAFLD. In this review, we will give a brief summary about processes involved in the maintenance of cholesterol homeostasis and discuss their potential roles in NAFLD. A synopsis of current knowledge regarding the benefits of cholesterol lowering drugs in the management of patients with NAFLD is also included.
Cholesterol Homeostasis
Maintaining cholesterol pool in the body is achieved by balancing between input and output pathways of cholesterol metabolism (7). Components of input pathways include endogenous de novo synthesis and intestinal absorption of cholesterol, whereas the output pathways include the excretion of free cholesterol in bile, the conversion into bile acids, and the non-biliary trans-epithelial cholesterol efflux (TICE) in the intestine (8, 9). Cholesterol is secreted into the circulation in the very low-density lipoprotein (VLDL) from the liver, and its clearance from the blood involves the uptake of low-density lipoprotein (LDL) by the LDL receptor (LDLr). Cholesterol is transported from the peripheral tissues to liver in the high-density lipoproteins (HDL) particles by the process of reverse cholesterol transport (8).
Intestinal cholesterol absorption represents one of the major determinants of cholesterol homeostasis. The total amount of cholesterol in the intestinal lumen is derived from multiple sources including diet (~300–500 mg/day from western-type of diet) as well as the epithelial cells that are shed into the lumen (~300 mg/day) and biliary cholesterol (~800–1,200 mg/day). A total amount of ~1,400–1,700 mg of cholesterol is presented in the intestine on a daily basis for absorption (10). The efficiency of cholesterol absorption exhibits wide inter-individual variations (28–80%) and depends on several luminal (emulsifiers: bile acids, phospholipids) and epithelial factors (10). Cholesterol uptake by enterocytes is mainly mediated by the specific transporter protein Niemann-Pick C1 Like 1 (NPC1L1), which is abundantly expressed on the brush-border membrane of small-intestinal enterocytes (8, 11) (Figure 1). Cholesterol is esterified by acetyl-CoA cholesterol acyltransferase 2 (ACAT-2) and incorporated along with triglycerides and apolipoprotein B-48 into chylomicrons. Triglycerides are hydrolyzed by lipoprotein lipase and chylomicrons are transformed into chylomicron remnants that are taken up by the liver (10).
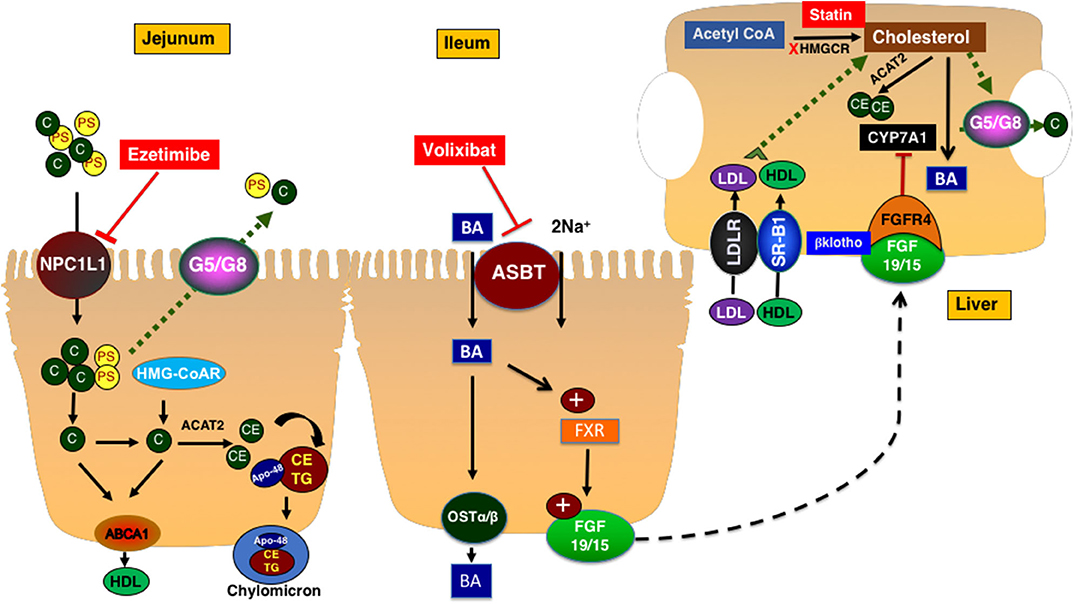
Figure 1. Cholesterol metabolism and transport. NPC1L1 mediates the uptake of cholesterol and plant sterols into small intestinal epithelial cells. The majority of plant sterols are secreted back into the lumen by the action of ABCG5/G8 heterodimer. Cholesterol is esterified and packaged into chylomicrons along with triglycerides. Ezetimibe inhibits NPC1L1 and decreases the intestinal cholesterol absorption. Cholesterol is synthesized in the liver and intestine. ABCA1 transporter mediates the efflux of cholesterol to a nascent HDL particle. Cholesterol enters the hepatocytes via LDLr or the HDL receptor SR-B1. In hepatocytes, cholesterol is esterified, secreted as free cholesterol into bile via ABCG5/G8 transporters, or converted to bile acids with a process in which CYP7A1 mediates the rate-limiting step. Bile acids are reabsorbed by ASBT in the ileum and activate FXR which increased the expression and secretion of FGF19 in humans (15 is rodents). FGF19/15 circulates back to the liver to inhibit CYP7A1 via FGF4 receptor and β-klotho co-receptor. ASBT, apical sodium-dependent bile acid transporter; BA, bile acid; CYP7A1, cytochrome P450 7A1; FGF15/19, fibroblast growth factor 15/19; FXR, farnesoid X receptor; Ost a & β, organic solute transporter alpha and beta; HMGCR, 3-Hydroxy-3-methylglutaryl-CoA Reductase; C, cholesterol; PS, plant sterols; NPC1L1, Niemann-pick C1 Like 1; G5/G8, ATP binding casette transporter G5 and G8; SR-B1, scavenger receptor B1; ABCA1, ATP-binding cassette transporter ABCA1; ACAT2, Acetyl-Coenzyme A Acetyltransferase 2; HDL, High density lipoprotein; CE, cholesterol ester; TG, triglyceride; FGFR4, fibroblast growth factor receptor 4; LDLR, Low density lipoprotein receptor; LDL, low density lipoprotein.
The de novo synthesis of cholesterol occurs in all cells in the body. However, the liver represents the main site for cholesterol synthesis and storage (8). Cholesterol synthesis starts with acetyl-CoA and involves multiple reactions. The two rate-limiting steps in this complex process are mediated by 3-hydroxy-3-methyl-glutaryl CoA reductase (HMGCoAr) and squalene monooxygenase. Cholesterol, the newly synthesized in the liver and the one derived from the intestinal absorption, is then packaged along with triglycerides and apolipoprotein B-100 into the VLDL particles that are assembled by the microsomal transfer protein. The VLDL loses its load of triglycerides while circulating in the tissues by lipoprotein lipase and transforms into LDL that represents the main vehicle of cholesterol transport to the peripheral tissues. Other processes involving cholesterol in the liver include esterification by ACAT2 for storage, excretion into bile by the function of heterodimer of ATP-binding cassette transporters ABCG5/G8 found on the canalicular membrane of hepatocytes, or the conversion to bile acids by a complex process in which the cytochrome P450 7A1 (CYP7A1) enzyme mediates the rate-limiting reaction (8). The expression of CYP7A1 is mainly regulated by a signal generated from the ileum represented by fibroblast growth factor 19 in human and 15 in rodents (FGF19/15) (12). The synthesis and secretion of FGF19/15 are induced by the uptake of bile acids by enterocytes in the distal ileum that is mediated by the action of the apical sodium dependent bile acid transporter ASBT. The inhibition of ASBT reduces FGF19/15 expression and promotes hepatic bile acid synthesis and therefore, decreases levels of hepatic cholesterol (Figure 1).
The transport of cholesterol from the peripheral tissues back to liver is initiated by cholesterol efflux from cells via the ATP-binding cassette transporter ABCA1 to the nascent apolipoprotein A containing HDL particles. Cholesterol could be transferred from HDL in the circulation to LDL by cholesterol-ester transfer protein (CETP). HDL is taken up by hepatocytes via the scavenger receptor B1 (SR-B1) (8) (Figure 1).
The expression of genes responsible for cholesterol transport and metabolism are tightly regulated by coordinated actions of transcription factors. For example, the decrease in the level of cellular cholesterol activates the ER membrane-bound transcription factor Sterol regulatory element-binding protein isoform 2 (SREBP-2) that induces the expression of HMGCoAr and LDLr (8). On the other hand, the increase in cellular cholesterol elevates the levels of the oxygenated cholesterol intermediates oxysterols that trigger Liver X receptors (LXRs) transcription factors to stimulate the pathways of cholesterol efflux and to promote cholesterol elimination from the liver (8).
Cholesterol and the Pathophysiology of NAFLD
The interest in understanding the relationship between cholesterol metabolism and the development of NAFLD and NASH was incited by observational studies linking liver damage with the risk for the development of cardiovascular disease (CVD) (13). In fact, the increased prevalence of metabolic syndrome in patients with NAFLD was to such an extent that some authors suggested that NALFD represents the hepatic manifestation in a spectrum of metabolic disorders (14). The metabolic syndrome refers to a cluster of metabolic disturbances including obesity, insulin resistance, hypertension, and atherogenic dyslipidemia that increase the risk for cardiovascular disease, stroke and diabetes mellitus (14). Meta-analysis of observational retrospective and prospective studies demonstrated an increased risk for CVD of both fatal and non-fatal events of CVD in patients with NAFLD as compared to patients without NAFLD (15). Such a link between NAFLD and CVD provided a compelling rationale to closely examine cholesterol metabolism in patients with NAFLD. The features of dyslipidemia associated with NALFD include hypertriglyceridemia and a decrease in HDL cholesterol (2). Although there is no evidence for an increase in total LDL cholesterol, patients with NAFLD were shown to have elevated levels of highly atherogenic subpopulations of LDL such as the oxidized particles (2). Furthermore, feeding high cholesterol atherogenic diets was able to induce lesions of early NAFLD as well as atherosclerosis in several animal models.
It should be noted that animal species have remarkable differences in cholesterol metabolism and respond differently to cholesterol rich diet. In this regard, it is well-established that rabbits have features of cholesterol metabolism that are closer to those in humans (16). For example, the predominant plasma lipoproteins in both rabbits and humans is LDL whereas it the HDL in mice. This difference is due to the fact that the CETP is lacking in the plasma of mice (17). More importantly, rabbits are sensitive to dietary cholesterol and develop severe hypercholesterolemia in response to high dietary cholesterol with prominent atherosclerosis (16). A recent study showed that diet-induced hypercholesterolemia in rabbits fed with diet containing 1% cholesterol caused liver injury that resembles early lesions of NAFLD (changes in fatty acid and sporadic fibrosis) concomitant with the development of atherosclerosis in the aorta (18). This study generated evidence linking blood cholesterol with NAFLD and provided novel insights into the roles of free cholesterol in inducing liver damage. In the mouse model, hypercholesterolemia and atherosclerosis can be produced only in genetically modified animals such as apoE and LDLr knockout mice (19). In this regard, steatohepatitis was shown in apoE knockout mice in response to high fat high cholesterol feeding (20). Combined transcriptomics and metabolomics analysis demonstrated that high dose of dietary cholesterol in apoE knockout mice triggered hepatic inflammation (21).
An interesting observation was made in wildtype mice (but not in the transgenic mice models of hypercholesterolemia) showing that high cholesterol in the diet was essential to elicit steatohepatitis and fibrosis after 30 weeks of feeding, whereas high fat diet alone failed to produce the same degree of liver injury (22). This role of high dose of dietary cholesterol in the induction of liver damage in wildtype mice was also illustrated in other species. For example, a study showed that feeding Ossabaw pigs atherogenic diet containing high fat along with 2% cholesterol caused severe metabolic syndrome along with lesions in the liver with features resembling human steatohepatitis including microvesicular and macrovesicular steatosis, fatty Kupffer cells, hepatocyte ballooning and fibrosis (23). Collectively, the evidence from preclinical animal models strongly indicate that a high dose of cholesterol is an independent risk factor for liver damage and the development of atherosclerosis. A conclusion can be drawn from these studies linking NAFLD with atherosclerosis implicating the accumulation of cholesterol in the liver and arteries as a trigger for inflammation and subsequent manifestations of liver disease and CVD.
Beside the studies in animal models, the results of a landmark study in humans also supported the role of cholesterol in NAFLD/NASH. Puri et al. (24) showed high levels of free cholesterol in patients with NAFLD (simple steatosis) that were more pronounced in cases with progressive form of the disease, NASH. Cholesterol esters remained unchanged despite the increase in free cholesterol suggesting an impaired function and/or expression of the enzyme responsible for cholesterol esterification (ACAT). Collectively, the evidence from animal models and humans indicates that cholesterol is one of the toxic lipids that accumulates in the liver contributing to liver damage during the course of NAFLD and the progression to NASH.
Recent studies investigated the molecular mechanisms underlying the toxic effects of high cholesterol in the liver. The findings of Mari et al. (25) provided evidence showing that accumulation of free cholesterol but not triglycerides or fatty acids makes hepatocytes more sensitive to TNFa and Fas-mediated apoptosis due to depletion of mitochondrial glutathione. Other studies suggested that the accumulated free cholesterol in damaged hepatocytes precipitates as crystals which in turn induce inflammatory response by interacting with NLRP3 inflammasomes of Kupffer cells (26). Free cholesterol was shown also to directly accumulates in lysosomes of Kupffer cells triggering an inflammatory response (27). Such inflammatory cascade was blocked by 27-hyroxycholesterol that prevented the accumulation of cholesterol in Kupffer cells and reduced NASH. The accumulation of cholesterol in hepatic stellate cells (HSCs) was also implicated in the progression to NASH and fibrosis (28). For example, studies provided evidence showing that the accumulation of free cholesterol in HSCs resulted in an increase in the expression of TLR4 receptor and an increase in the sensitivity of HSCs to TGFb with subsequent fibrosis (29). It is apparent therefore, that increased cellular levels of cholesterol in different cell types in the liver may be responsible for liver damage and the progression from simple steatosis to NASH.
Molecular Pathways Underlying Cholesterol Accumulation in NAFLD
The accumulation of free cholesterol in NAFLD may occur due to an upregulation in the input pathways or a decrease in the elimination of cholesterol. Studies showed that the expression of HMGCoAr are increased in patients with NAFLD/NASH (30). There is evidence to suggest that changes in microRNAs (miRs) might be responsible for the observed increase in HMGCoAr. Several studies demonstrated the roles of several miRs in the development of liver diseases including NAFLD. The roles of these miRs including miR-122, miR-33, and miR-24a are discussed in details in other excellent review articles (31, 32). Studies have also demonstrated the regulation of several genes involved in cholesterol metabolism such as HMGCoAr by miRs and these findings are nicely summarized in other review articles (33, 34).
With respect to cholesterol metabolism as it relates to NAFLD/NASH, miR-34a levels were shown to be elevated in patients with NAFLD/NASH causing a decrease in hepatic NAD-dependent deacetylase Sirtuin1 with subsequent dephosphorylation and an increase in HMGCoAr (30). The roles of miR-29a was also suggested by a supporting evidence showing a negative correlation between miR-29a and HMGCoAr expression after 3 weeks feeding of methionine-choline deficient diet (MCD) to mice (35). The study revealed that 3′ un-translated region (3′UTR) of HMGCoAr was targeted by miR-29a (35). The decrease in miR-29a was concomitant with a decrease in hepatic Dicer1 enzyme that is essential for the maturation of microRNAs. In fact, liver-specific knockout of Dicer1 enzyme resulted in an increase in HMGCoAr associated with accumulation of free cholesterol in the liver (35). These findings revealed an important molecular pathway involving hepatic Dicer1 and microRNAs in the increase in cholesterol synthesis and accumulation of free cholesterol in the liver contributing to the development of NAFLD/NASH.
The expression of SREBP2 transcription factor was also increased in patients with NAFLD/NASH (30). Since SREBP2 induces the expression HMGCoAr, the observed increase in active SERBP2 represents an additional molecular pathway that might be responsible for the increases in hepatic levels of free cholesterol. Mari et al. (25) demonstrated that SREBP2 overexpression was not found in obese patients and patients with other chronic liver diseases such as hepatitis C, but was rather a specific feature associated with NAFLD/NASH. It should be noted that the accumulation of free cholesterol in hepatocytes is expected to inhibit the activation of SREBP2 (36). It is possible that SREBP2 is activated in NAFLD/NASH by a mechanism independent from the canonical pathway related to changes in cellular cholesterol (37). Indeed, a previous study showed that the injection of cytokines in C57BL mice resulted in an increase in hepatic SREBP2 and HMGCoAr expression (38). Similar findings were obtained in response to incubation of human hepatic HepG2 cells with cytokines. Although loading of HepG2 cells with cholesterol inhibited the SREBP2 pathways as expected, such a negative feedback inhibition was overridden by inflammatory stress caused by the cytokines (38). It is also reported that miR-122 stabilizes the inactive form of SREBP2 (39). Interestingly, miR-122 was shown to be significantly decreased in patients with NASH, thus providing a potential explanation for the increase in SREBP2 (39). It is possible also that hyperinsulinemia resulted from increased insulin resistance may also cause an increase in the active form of SREBP2 (40). Collectively, the inflammatory stress, the decrease in miR-122 and associated hyperinsulinemia in NASH presents a reasonable explanation for the constant activation of SREBP2 bypassing the canonical negative feedback inhibitory effect of high levels of cellular cholesterol (37). Beside the role of hepatic SREBP2 in the pathophysiology of NAFLD/NASH, our studies in transgenic mouse model of intestine-specific overactivation of SREBP2, demonstrated hepatic steatosis, and increased susceptibility of the animals to severe liver damage and profound inflammation and fibrosis in response to a high fat/high cholesterol diet (41). These observations strongly suggest that the activation of SREBP2 not only in the liver but also in the intestine plays a critical role in the development of NAFLD/NASH.
Beside the increase in cholesterol synthesis, changes in the pathways involved in the elimination of cholesterol were also noted in patients with NASH. Among these observations is the decrease in the expression of CYP7A1 implicating reduced bile acid synthesis from cholesterol (30). Also, a decrease in the expression of cholesterol transporters ABCG5/G8 responsible for cholesterol excretion into bile was reported (30). It is clear therefore, that the accumulation of hepatic free cholesterol in NAFLD/NASH patients is multifactorial with underlying increase in synthesis and decrease in the elimination of cholesterol.
With respect to dietary cholesterol and liver disease in humans, an epidemiological study suggested that dietary cholesterol is an independent risk factor for liver cirrhosis and hepatic cancer (42). These studies provide support to the observations made in animal models demonstrating the role of cholesterol in the induction of liver damage (43, 44). Since dietary cholesterol represents only a small portion of the total amount of luminal cholesterol presented for absorption (10), one could argue that the relation between the efficiency of cholesterol absorption and the development of liver diseases including NALFD/NASH should be carefully investigated. One study by Simonen et al. (45) evaluated serum surrogate markers for cholesterol synthesis (cholestenol, desmosterol, and lathosterol) and absorption (plant sterols). Individuals with NALFD were identified as those having liver fat ≥ 5.56% as judged by with proton magnetic resonance spectroscopy. The authors found that the content of liver fat was positively correlated with markers of cholesterol synthesis and inversely correlated with markers of cholesterol absorption (45). It is possible that there is an interrelationship between these two processes so that the decrease in cholesterol absorption may represent a homeostatic response to an increase in cholesterol synthesis (45). It will be important to investigate a possible difference in the efficiency of cholesterol absorption in patients with NASH as compared to those with NAFLD. With respect to the expression of intestinal NPC1L1, previous studies showed an increase in the expression of this intestinal in patients with diabetes mellitus (46). It is of central importance to investigate the expression of NPC1L1 in patients with NAFLD and NASH to a make a conclusion regarding potential changes in cholesterol absorption. Also, the studies mentioned above assessed the efficiency of cholesterol absorption indirectly by measuring serum plant sterols as surrogate markers. A direct measurement of cholesterol absorption in patients with NAFLD/NASH is warranted.
Cholesterol Lowering Drugs in the Management of NAFLD/NASH
According to current guidelines, decreasing the levels of plasma cholesterol to stringent low levels is highly recommended in patients with high risk for CVD (47, 48). Patients with NAFLD/NASH have high risk of developing CVD (15, 49). Therefore, it is of central importance to address treatment options available to lower plasma cholesterol in this patient population. Table 1 summarizes the results of selected studies related to cholesterol-based therapy for NAFLD.
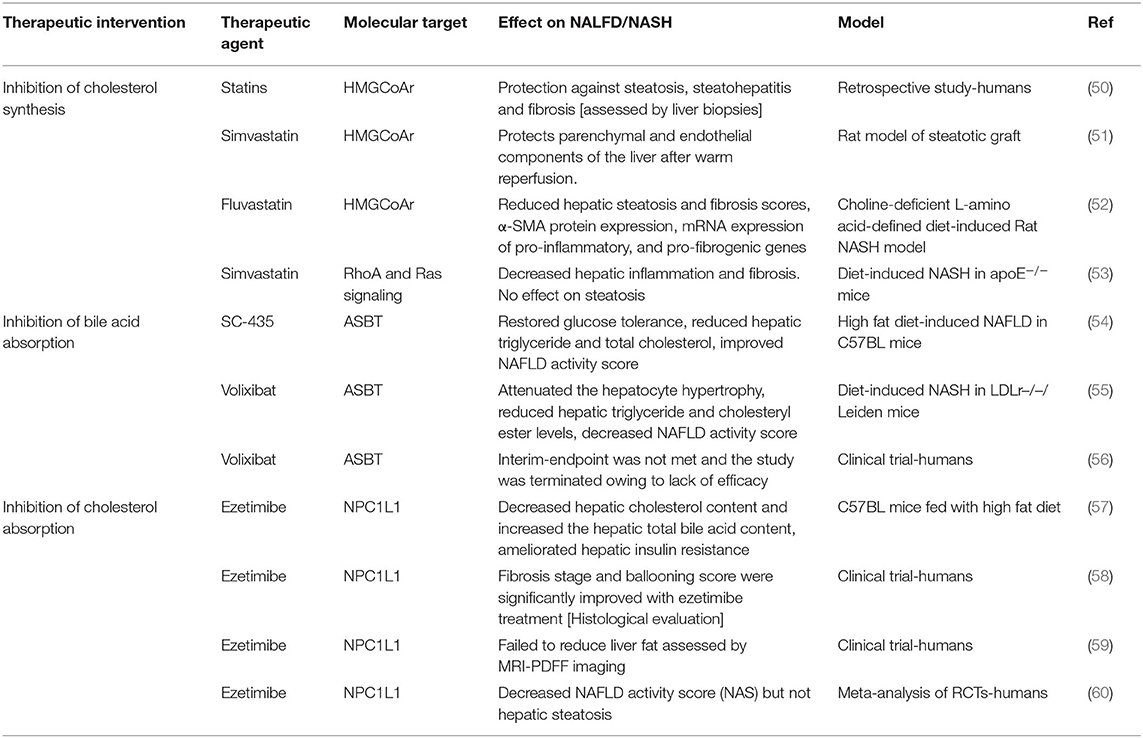
Table 1. Therapeutic interventions for NAFLD/NASH.
Inhibitors of Cholesterol Synthesis
Statins, the inhibitors of HMGCoAr, are the widely used drugs to lower plasma cholesterol (61). It is well-established that statins reduce LDL cholesterol and significantly decrease the risk for CVD. Thus, it is logical to use statins in NAFLD patients who are at risk for CVD (62). The use of statins was noted to be associated with an increase in the levels of serum transaminases raising concerns about their safety especially in patients with NAFLD/NASH (63). However, recent studies in a cohort of ~1,200 European patients provided strong evidence demonstrating the beneficial effects of statins in the protection against steatosis, steatohepatitis, and fibrosis as assessed by liver biopsies (50). Several other meta-analysis of available data suggested that atorvastatin improved liver injury in NAFLD/NASH patients and was more potent in decreasing CVD risk as compared to individuals with normal liver function (64).
The effects of statins on several aspects of liver injury was shown in animal models (51). Statins were shown to decrease experimentally induced fibrosis in rats by inhibiting the activation of hepatic stellate cells (52). Recent study also showed that simvastatin reduced liver inflammation and fibrosis in ApoE knockout mice fed with western-type of diet (53). These effects were attributed to the inhibition of RhoA and Ras signaling (53). It is possible therefore, that the amelioration of liver injury by statins is due to other effects beside the inhibition of cholesterol synthesis (65). Statins may also affect cholesterol metabolism in other organs and indirectly influence liver function. For example, it was shown that statin treatment in humans increased the expression of intestinal SREBP2 (66). In this regard, our studies demonstrated that the intestine-specific overexpression of SREBP2 caused hepatic steatosis and profound diet-induced liver injury in mice (41). It is possible therefore, that statins may negatively affect liver function by increasing intestinal SREBP2 expression. In light of the complexity underlying the global effects of statins, comprehensive studies are warranted to delineate the molecular pathways involved in the effects of statins on NAFLD/NASH.
Inhibitors of Bile Acid Absorption
Blocking the absorption of bile acids eliminates the negative feedback inhibition on their hepatic synthesis and promotes cholesterol degradation (12). ASBT mediates the first and rate limiting step in bile acid absorption and its inhibition represents an attractive therapeutic target to reduce hepatic cholesterol toxicity in NAFLD/NASH (12). Recent studies provided a compelling evidence showing that the lack of ASBT in knockout mice protected against diet-induced liver injury and steatosis (54). Further, these studies showed that the pharmacological inhibition of ASBT significantly decreased hepatic triglycerides and total cholesterol and improved NAFLD activity score (54). Additional studies using the ASBT inhibitor volixibat in LDLr leiden knockout mice supported that conclusion and demonstrated a significant decrease in diet-induced Non-alcoholic fatty liver disease activity score by the inhibition of ASBT (55). In humans however, a phase 2 clinical trial to assess the effects of 48 weeks treatment with the ASBT inhibitor, volixibat, was terminated after mid-term assessment due to lack of improvement in NASH score as judged by MRI-PDFF imaging (56). It should be noted that the histological evaluations including the scores for ballooning and inflammation in this clinical trial were lacking. Such histological analysis in response to treatment with volixibat is crucial as it may provide more insights on the potential effects of inhibiting bile acid absorption on liver injury in NAFLD/NASH. Also, additional studies in preclinical models seem essential to unravel the molecular basis for the observed improvement of NAFLD/NASH in the animal models. Enhanced understanding of these pathways at the molecular level may unravel novel targets that could be more efficacious in the treatment of NALFD/NASH in humans.
Inhibitors of Cholesterol Absorption
Studies showed that the inhibitor of cholesterol absorption ezetimibe reduces plasma cholesterol and decreases the risk of CVD when used alone or in combination with statins (67, 68). Earlier studies in animal models showed that ezetimibe alleviated liver steatosis in mice fed with high fat diet and had promising effects on NAFLD/NASH (57). However, the studies about the beneficial effects of ezetimibe in patients with NAFLD/NASH generated conflicting results (69). Takeshita et al. (58) showed in a randomized clinical trial in a cohort of 32 patients with NAFLD that ezetimibe improved hepatic fibrosis. However, the clinical trial by Loomba et al. (59) with 50 patients showed that ezetimibe failed to significantly reduce liver fat in NAFLD as assessed by MRI-PDDF imaging. A recent meta-analysis, which exclusively included studies assessing NAFLD/NASH based on biopsy, with only one study that was based MRI-PDFF imaging, showed that ezetimibe decreased NAFLD activity score (NAS) but not hepatic steatosis (60). It was noted that current available data about ezetimibe and NAFLD/NASH were generated from limited number of clinical trials with small sample size (69). Large scale randomized clinical trials are needed for better assessment of the outcome of ezetimibe on the disease activity in patients with NAFLD/NASH. It is noteworthy to mention that the inter-individual variations in the efficiency in cholesterol absorption may influence the results of these studies. We suggest that this potential confounding factor be taken into consideration when assessing the effects of ezetimibe on NAFLD/NASH.
Conclusion
Disturbances in cholesterol metabolism contribute to the pathophysiology of NAFLD/NASH. An increase in cholesterol synthesis and a decrease in the pathways responsible for the elimination of cholesterol lead to accumulation of free cholesterol in the liver. The toxicity of high cellular levels of cholesterol in hepatocytes is one of the major factors causing inflammation and fibrosis leading to liver damage. The pathways involved in cholesterol metabolism are potential targets for the treatment of NAFLD/NASH. One challenging area is the complex nature of the processes involved in cholesterol homeostasis. For example, cholesterol absorption exhibits wide interindividual variations that may explain the conflicting results when evaluating the efficacy of cholesterol lowering drugs in the treatment of NAFLD/NASH. Such variations evoke the need for precision medicine approaches and individualized treatment. It is possible that individuals with naturally occurring low efficiency of cholesterol absorption may not benefit from the use of cholesterol absorption blocker ezetimibe. It will be interesting in future studies to stratify patients into relevant subgroups in a rigorous manner and take into consideration the inter-individual variations in cholesterol metabolism when assessing the outcome of cholesterol lowering drugs in the treatment of NAFLD/NASH. Precision medicine approaches necessitate the development of novel and simple methods to directly measure dynamic processes such as cholesterol absorption when individualized therapeutic interventions are to be considered for NAFLD/NASH treatment.
Author Contributions
WA and PM wrote the initial draft. RG and SS edited the initial manuscript. PM and RG made the figure. RG and WA made the table. PM, RG, SS, and WA edited and approved the final draft. All authors contributed to the article and approved the submitted version.
Funding
The studies in our laboratories were supported by the Veterans Affairs Research Career Scientist Award (IK6BX005243) and a Merit Award (BX000152) to WA, and by NIH/NIDDK grant DK109709 (WA). The studies were also partially supported by NIH/NIDDK grants: DK98170 (RG), and VA Merit Award BX002867 (SS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. (2009) 2:231–7. doi: 10.1242/dmm.001180
2. Chatrath H, Vuppalanchi R, Chalasani N. Dyslipidemia in patients with nonalcoholic fatty liver disease. Semin Liver Dis. (2012) 32:22–9. doi: 10.1055/s-0032-1306423
3. Kerr TA, Davidson NO. Cholesterol and NAFLD: renewed focus on an old villain. Hepatol Baltim Md. (2012) 56:1995–8. doi: 10.1002/hep.26088
4. Zhang QQ, Lu LG. Nonalcoholic fatty liver disease: dyslipidemia, risk for cardiovascular complications, and treatment strategy. J Clin Transl Hepatol. (2015) 3:78–84. doi: 10.14218/JCTH.2014.00037
5. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. (2018) 75:3313–27. doi: 10.1007/s00018-018-2860-6
6. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. (2016) 27:84–95. doi: 10.1016/j.tem.2015.11.008
7. Cook RP. Cholesterol, Chemistry, Biochemistry And Pathology. New York, NY: Academic Press, Inc. (1958) p. xii–542.
8. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. (2020) 21:225–45. doi: 10.1038/s41580-019-0190-7
9. Nemes K, Åberg F, Gylling H, Isoniemi H. Cholesterol metabolism in cholestatic liver disease and liver transplantation: from molecular mechanisms to clinical implications. World J Hepatol. (2016) 8:924–32. doi: 10.4254/wjh.v8.i22.924
10. Wang DQH. Regulation of intestinal cholesterol absorption. Annu Rev Physiol. (2007) 69:221–48. doi: 10.1146/annurev.physiol.69.031905.160725
11. Alrefai WA, Annaba F, Sarwar Z, Dwivedi A, Saksena S, Singla A, et al. Modulation of human niemann-Pick C1-like 1 gene expression by sterol: role of sterol regulatory element binding protein 2. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G369–76. doi: 10.1152/ajpgi.00306.2006
12. Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Intestinal absorption of bile acids in health and disease. Compr Physiol. (2019) 10:21–56. doi: 10.1002/cphy.c190007
13. Mantovani A, Ballestri S, Lonardo A, Targher G. Cardiovascular disease and myocardial abnormalities in nonalcoholic fatty liver disease. Dig Dis Sci. (2016) 61:1246–67. doi: 10.1007/s10620-016-4040-6
14. Neuschwander-Tetri BA. Nonalcoholic steatohepatitis and the metabolic syndrome. Am J Med Sci. (2005) 330:326–35. doi: 10.1097/00000441-200512000-00011
15. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol. (2016) 65:589–600. doi: 10.1016/j.jhep.2016.05.013
16. Fan J, Kitajima S, Watanabe T, Xu J, Zhang J, Liu E, et al. Rabbit models for the study of human atherosclerosis: from pathophysiological mechanisms to translational medicine. Pharmacol Ther. (2015) 146:104–19. doi: 10.1016/j.pharmthera.2014.09.009
18. Kim EJ, Kim B, Seo HS, Lee YJ, Kim HH, Son HH, et al. Cholesterol-induced non-alcoholic fatty liver disease and atherosclerosis aggravated by systemic inflammation. PLoS ONE. (2014) 9:e97841. doi: 10.1371/journal.pone.0097841
19. Knowles JW, Maeda N. Genetic modifiers of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2000) 20:2336–45. doi: 10.1161/01.ATV.20.11.2336
20. Schierwagen R, Maybüchen L, Zimmer S, Hittatiya K, Bäck C, Klein S, et al. Seven weeks of western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci Rep. (2015) 5:12931. doi: 10.1038/srep12931
21. Kleemann R, Verschuren L, van Erk MJ, Nikolsky Y, Cnubben NH, Verheij ER, et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol. (2007) 8:R200. doi: 10.1186/gb-2007-8-9-r200
22. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatol Baltim Md. (2013) 57:81–92. doi: 10.1002/hep.25789
23. Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, et al. Nutritional model of steatohepatitis and metabolic syndrome in the ossabaw miniature swine. Hepatol Baltim Md. (2009) 50:56–67. doi: 10.1002/hep.22904
24. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatol Baltim Md. (2007) 46:1081–90. doi: 10.1002/hep.21763
25. Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and fas-mediated steatohepatitis. Cell Metab. (2006) 4:185–98. doi: 10.1016/j.cmet.2006.07.006
26. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. (2017) 58:1067–79. doi: 10.1194/jlr.M072454
27. Bieghs V, Hendrikx T, van Gorp PJ, Verheyen F, Guichot YD, Walenbergh SMA, et al. The cholesterol derivative 27-hydroxycholesterol reduces steatohepatitis in mice. Gastroenterology. (2013) 144:167–78.e1. doi: 10.1053/j.gastro.2012.09.062
28. Tirosh O. Hypoxic signaling and cholesterol lipotoxicity in fatty liver disease progression. Oxid Med Cell Longev. (2018) 2018:2548154. doi: 10.1155/2018/2548154
29. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. (2014) 61:98–106. doi: 10.1016/j.jhep.2014.03.018
30. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. (2012) 15:665–74. doi: 10.1016/j.cmet.2012.04.004
31. McDaniel K, Herrera L, Zhou T, Francis H, Han Y, Levine P, et al. The functional role of microRNAs in alcoholic liver injury. J Cell Mol Med. (2014) 18:197–207. doi: 10.1111/jcmm.12223
32. Kerr TA, Korenblat KM, Davidson NO. MicroRNAs and liver disease. Transl Res. (2011) 157:241–52. doi: 10.1016/j.trsl.2011.01.008
33. Aryal B, Singh AK, Rotllan N, Price N, Fernández-Hernando C. microRNAs and lipid metabolism. Curr Opin Lipidol. (2017) 28:273–80. doi: 10.1097/MOL.0000000000000420
34. Moore KJ, Rayner KJ, Suárez Y, Fernández-Hernando C. The role of microRNAs in cholesterol efflux and hepatic lipid metabolism. Annu Rev Nutr. (2011) 31:49–63. doi: 10.1146/annurev-nutr-081810-160756
35. Liu M, Gao M, Li C, Yu C, Yan H, Peng C, et al. Dicer 1 /miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta Pharmacol Sin. (2017) 38:660–71. doi: 10.1038/aps.2016.158
36. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem. (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
37. Rooyen DMV, Farrell GC. SREBP-2: a link between insulin resistance, hepatic cholesterol, and inflammation in NASH. J Gastroenterol Hepatol. (2011) 26:789–92. doi: 10.1111/j.1440-1746.2011.06704.x
38. Zhao L, Chen Y, Tang R, Chen Y, Li Q, Gong J, et al. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J Gastroenterol Hepatol. (2011) 26:875–83. doi: 10.1111/j.1440-1746.2010.06560.x
39. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatol Baltim Md. (2008) 48:1810–20. doi: 10.1002/hep.22569
40. Xie X, Liao H, Dang H, Pang W, Guan Y, Wang X, et al. Down-regulation of hepatic HNF4alpha gene expression during hyperinsulinemia via SREBPs. Mol Endocrinol Baltim Md. (2009) 23:434–43. doi: 10.1210/me.2007-0531
41. Malhotra P, Aloman C, Ankireddy A, Khadra H, Ooka K, Gill RK, et al. Overactivation of intestinal sterol response element-binding protein 2 promotes diet-induced nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. (2017) 313:G376–85. doi: 10.1152/ajpgi.00174.2017
42. Ioannou GN, Morrow OB, Connole ML, Lee SP. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatol Baltim Md. (2009) 50:175–84. doi: 10.1002/hep.22941
43. Subramanian S, Goodspeed L, Wang S, Kim J, Zeng L, Ioannou GN, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res. (2011) 52:1626–35. doi: 10.1194/jlr.M016246
44. Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology. (2011) 141:1393–403. doi: 10.1053/j.gastro.2011.06.040
45. Simonen P, Kotronen A, Hallikainen M, Sevastianova K, Makkonen J, Hakkarainen A, et al. Cholesterol synthesis is increased and absorption decreased in non-alcoholic fatty liver disease independent of obesity. J Hepatol. (2011) 54:153–59. doi: 10.1016/j.jhep.2010.05.037
46. Lally S, Tan CY, Owens D, Tomkin GH. Messenger RNA levels of genes involved in dysregulation of postprandial lipoproteins in type 2 diabetes: the role of niemann-pick C1-like 1, ATP-binding cassette, transporters G5 and G8, and of microsomal triglyceride transfer protein. Diabetologia. (2006) 49:1008–16. doi: 10.1007/s00125-006-0177-8
47. Jarcho JA, Keaney JF. Proof that lower is better — ldl cholesterol and IMPROVE-IT. N Engl J Med. (2015) 372:2448–50. doi: 10.1056/NEJMe1507041
48. Kastelein JJP, Akdim F, Stroes ESG, Zwinderman AH, Bots ML, Stalenhoef AFH, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. (2008) 358:1431–43. doi: 10.1056/NEJMoa0800742
49. Munteanu MA, Mircea PA. From NAFLD to cardiovascular disease. Is it (still) the metabolic syndrome? Clujul Med. (2014) 87:80–6. doi: 10.15386/cjmed-277
50. Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. (2015) 63:705–12. doi: 10.1016/j.jhep.2015.05.006
51. Gracia-Sancho J, García-Calderó H, Hide D, Marrone G, Guixé-Muntet S, Peralta C, et al. Simvastatin maintains function and viability of steatotic rat livers procured for transplantation. J Hepatol. (2013) 58:1140–6. doi: 10.1016/j.jhep.2013.02.005
52. Chong LW, Hsu YC, Lee TF, Lin Y, Chiu YT, Yang KC, et al. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. (2015) 15:22. doi: 10.1186/s12876-015-0248-8
53. Schierwagen R, Maybüchen L, Hittatiya K, Klein S, Uschner FE, Braga TT, et al. Statins improve NASH via inhibition of RhoA and Ras. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G724–33. doi: 10.1152/ajpgi.00063.2016
54. Rao A, Kosters A, Mells JE, Zhang W, Setchell KDR, Amanso AM, et al. Inhibition of ileal bile acid uptake protects against non-alcoholic fatty liver disease in high fat diet-fed mice. Sci Transl Med. (2016) 8:357ra122. doi: 10.1126/scitranslmed.aaf4823
55. Salic K, Kleemann R, Wilkins-Port C, McNulty J, Verschuren L, Palmer M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/-.Leiden mice. PLoS ONE. (2019) 14:e0218459. doi: 10.1371/journal.pone.0218459
56. Newsome PN, Palmer M, Freilich B, Sheikh MY, Sheikh A, Sarles H, et al. Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J Hepatol. (2020) 73:231–40. doi: 10.1016/j.jhep.2020.03.024
57. Muraoka T, Aoki K, Iwasaki T, Shinoda K, Nakamura A, Aburatani H, et al. Ezetimibe decreases SREBP-1c expression in liver and reverses hepatic insulin resistance in mice fed a high-fat diet. Metabolism. (2011) 60:617–28. doi: 10.1016/j.metabol.2010.06.008
58. Takeshita Y, Takamura T, Honda M, Kita Y, Zen Y, Kato K, et al. The effects of ezetimibe on non-alcoholic fatty liver disease and glucose metabolism: a randomised controlled trial. Diabetologia. (2014) 57:878–90. doi: 10.1007/s00125-013-3149-9
59. Loomba R, Sirlin CB, Ang B, Bettencourt R, Jain R, Salotti J, et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial). Hepatol Baltim Md. (2015) 61:1239–50. doi: 10.1002/hep.27647
60. Lee HY, Jun DW, Kim HJ, Oh H, Saeed WK, Ahn H, et al. Ezetimibe decreased nonalcoholic fatty liver disease activity score but not hepatic steatosis. Korean J Intern Med. (2019) 34:296–304. doi: 10.3904/kjim.2017.194
61. Kreisberg RA, Oberman A. Medical management of hyperlipidemia/dyslipidemia. J Clin Endocrinol Metab. (2003) 88:2445–61. doi: 10.1210/jc.2003-030388
62. Vuppalanchi R, Chalasani N. Statins for hyperlipidemia in patients with chronic liver disease: are they safe? Clin Gastroenterol Hepatol. (2006) 4:838–9. doi: 10.1016/j.cgh.2006.04.020
63. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatol Baltim Md. (2005) 41:690–5. doi: 10.1002/hep.20671
64. Athyros VG, Boutari C, Stavropoulos K, Anagnostis P, Imprialos KP, Doumas M, et al. Statins: an under-appreciated asset for the prevention and the treatment of NAFLD or NASH and the related cardiovascular risk. Curr Vasc Pharmacol. (2018) 16:246–53. doi: 10.2174/1570161115666170621082910
65. Pose E, Trebicka J, Mookerjee RP, Angeli P, Ginès P. Statins: old drugs as new therapy for liver diseases? J Hepatol. (2019) 70:194–202. doi: 10.1016/j.jhep.2018.07.019
66. Tremblay AJ, Lamarche B, Lemelin V, Hoos L, Benjannet S, Seidah NG, et al. Atorvastatin increases intestinal expression of NPC1L1 in hyperlipidemic men. J Lipid Res. (2011) 52:558–65. doi: 10.1194/jlr.M011080
67. Bohula EA, Giugliano RP, Cannon CP, Zhou J, Murphy SA, White JA, et al. Achievement of dual low-density lipoprotein cholesterol and high-sensitivity C-reactive protein targets more frequent with the addition of ezetimibe to simvastatin and associated with better outcomes in IMPROVE-IT. Circulation. (2015) 132:1224–33. doi: 10.1161/CIRCULATIONAHA.115.018381
68. Assmann G, Kannenberg F, Ramey DR, Musliner TA, Gutkin SW, Veltri EP. Effects of ezetimibe, simvastatin, atorvastatin, and ezetimibe-statin therapies on non-cholesterol sterols in patients with primary hypercholesterolemia. Curr Med Res Opin. (2008) 24:249–59. doi: 10.1185/030079908X253663
Keywords: NPC1L1, ezetimibe, statins, bile acids, fatty liver
Citation: Malhotra P, Gill RK, Saksena S and Alrefai WA (2020) Disturbances in Cholesterol Homeostasis and Non-alcoholic Fatty Liver Diseases. Front. Med. 7:467. doi: 10.3389/fmed.2020.00467
Received: 02 June 2020; Accepted: 13 July 2020;
Published: 02 September 2020.
Edited by:
Gianfranco Danilo Alpini, Indiana University, United StatesReviewed by:
Lixian Chen, Indiana University, United StatesChaodong Wu, Texas A&M University, United States
Copyright © 2020 Malhotra, Gill, Saksena and Alrefai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Waddah A. Alrefai, d2FscmVmYWlAdWljLmVkdQ==