
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 March 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00449
This article is part of the Research Topic Etiopathogenesis of Systemic Sclerosis: An Update View all 11 articles
 Marzia Dolcino1†
Marzia Dolcino1† Andrea Pelosi2†
Andrea Pelosi2† Piera Filomena Fiore2
Piera Filomena Fiore2 Giuseppe Patuzzo1
Giuseppe Patuzzo1 Elisa Tinazzi1
Elisa Tinazzi1 Claudio Lunardi1*†
Claudio Lunardi1*† Antonio Puccetti2,3†
Antonio Puccetti2,3†
Systemic sclerosis (SSc) is a rare connective tissue disease characterized by three pathogenetic hallmarks: vasculopathy, dysregulation of the immune system, and fibrosis. A particular feature of SSc is the increased frequency of some types of malignancies, namely breast, lung, and hematological malignancies. Moreover, SSc may also be a paraneoplastic disease, again indicating a strong link between cancer and scleroderma. The reason of this association is still unknown; therefore, we aimed at investigating whether particular genetic or epigenetic factors may play a role in promoting cancer development in patients with SSc and whether some features are shared by the two conditions. We therefore performed a gene expression profiling of peripheral blood mononuclear cells (PBMCs) derived from patients with limited and diffuse SSc, showing that the various classes of genes potentially linked to the pathogenesis of SSc (such as apoptosis, endothelial cell activation, extracellular matrix remodeling, immune response, and inflammation) include genes that directly participate in the development of malignancies or that are involved in pathways known to be associated with carcinogenesis. The transcriptional analysis was then complemented by a complex network analysis of modulated genes which further confirmed the presence of signaling pathways associated with carcinogenesis. Since epigenetic mechanisms, such as microRNAs (miRNAs), are believed to play a central role in the pathogenesis of SSc, we also evaluated whether specific cancer-related miRNAs could be deregulated in the serum of SSc patients. We focused our attention on miRNAs already found upregulated in SSc such as miR-21-5p, miR-92a-3p, and on miR-155-5p, miR 126-3p and miR-16-5p known to be deregulated in malignancies associated to SSc, i.e., breast, lung, and hematological malignancies. miR-21-5p, miR-92a-3p, miR-155-5p, and miR-16-5p expression was significantly higher in SSc sera compared to healthy controls. Our findings indicate the presence of modulated genes and miRNAs that can play a predisposing role in the development of malignancies in SSc and are important for a better risk stratification of patients and for the identification of a better individualized precision medicine strategy.
Systemic sclerosis (SSc) is a rare, chronic disease characterized by three pathogenetic hallmarks: vasculopathy, dysregulation of the immune system, and increased extracellular matrix deposition in the skin and internal organs, leading to extended fibrosis and to a remarkable heterogeneity in clinical features and course of the disease, resulting in high morbidity and mortality (1).
Several environmental factors alongside genetic susceptibility and epigenetic mechanisms contribute to the onset of the disease (2–5).
Increased frequency of a few types of cancer, namely breast, lung, and hematologic malignancies in SSc has been reported (6), and it seems to be associated with the presence of particular autoantibodies (7). Moreover, it is worthwhile mentioning that SSc may also be a paraneoplastic disease (8, 9), indicating a strong link between cancer and scleroderma.
Given the high incidence of tumors in patients with SSc, we wanted to assess whether this phenomenon may be supported by a particular gene modulation that can favor cancer development in patients with SSc. Thus, we analyzed transcriptional profiles of peripheral blood mononuclear cells (PBMCs) obtained from patients with SSc to evaluate the presence of modulated genes that are involved in signaling pathways associated with malignancy. Moreover, through a complex network analysis, we verified which pathways could play a pivotal role in cancer development in SSc patients.
Besides environmental and genetic factors, epigenetic mechanisms, such as microRNAs (miRNAs), are believed to play a central role in the pathogenesis of the disease.
microRNAs are a class of small non-coding RNAs that bind 3’ untranslated region of messenger RNAs (mRNAs), negatively regulating target gene expression by either the repression of translation or degradation of target mRNAs (10). miRNAs are considered an important class of epigenetics regulators in many basic cellular processes as well as in the vast majority of diseases, including cancer and autoimmunity (11, 12). Little is known about the role of dysregulated expression of miRNAs in the pathogenesis of SSc (13). An altered expression of profibrotic and/or antifibrotic miRNAs has been suggested as an important factor in the development of fibrosis in SSc patients (14). Moreover, increased evidence suggests that serum miRNA levels may be promising biomarkers for the diagnosis, prognosis, and therapeutic approach in SSc (15). Serum levels of miR-21 and miR-92a have been found significantly higher in SSc samples compared to normal controls (16, 17). Their function is implicated in inflammation, in regulation of immune cells (18) and in favoring fibrosis (16, 17).
Since particular miRNAs have been associated with malignancies, we aimed at evaluating whether transcriptional profiles and dysregulation of particular miRNAs are shared by cancer and SSc.
We enrolled 30 patients affected by SSc, attending the Unit of Autoimmune Disease at the University Hospital of Verona, and 30 sex and age matched healthy controls. All patients fulfilled the ACR/Eular classification criteria for SSc (19). The distinction between limited (lSSc) and diffuse cutaneous SSc (dSSc) was performed according to the criteria proposed by LeRoy et al. (20). Fifteen of the 30 patients were affected by lSSc and 15 by dSSc. The clinical and demographic features of the patients enrolled in the study are summarized in Table 1.
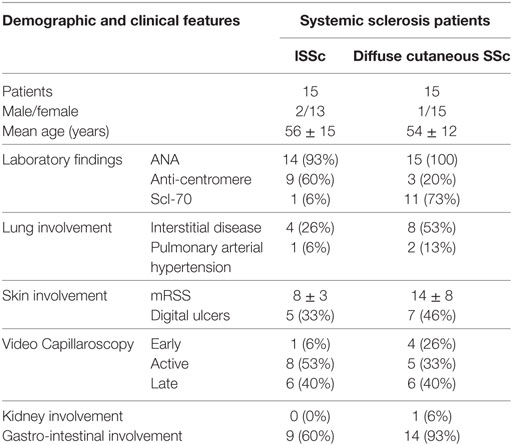
Table 1. Clinical and demographic features of the patients enrolled in the study.
Samples obtained from 10 patients with lSSc, 10 patients with dSSc, and 10 healthy controls were used for the gene expression analysis, whereas samples obtained from all the patients and controls were used for the microRNA study. Blood samples were collected from patients with active disease and in the absence of immunosuppressive therapies.
A written informed consent was obtained by all the participants to the study and the study protocol was approved by the Ethical Committee of the Azienda Ospedaliera Universitaria Integrata di Verona. All the investigations have been performed according to the principles contained in the Helsinki declaration.
Blood samples were collected in BD Vacutainer K2EDTA tubes using a 21-gage needle. PBMC were obtained upon stratification on Lympholyte® cell separation density gradient (Cedarlane, Burlington, Canada). Total RNA extraction from PBMC was performed with miRNeasy mini kit following manufacturer’s protocol (Qiagen GmbH, Hilden, Germany). cRNA preparation, samples hybridization, and scanning were performed following the Affymetrix (Affymetrix, Santa Clara, CA, USA) provided protocols, by Cogentech Affymetrix microarray unit (Campus IFOM IEO, Milan, Italy). All samples were hybridized on Human Clariom D (Affymetrix) gene chip and were analyzed using the Transcriptome Analysis Console (TAC) 4.0 software (Applied Biosystem, Foster City, CA USA by Thermo Fisher Scientific, Waltham, MA, USA). The Signal Space Transformation (SST)-Robust Multi-Array Average algorithm (RMA) were applied to background-adjust, normalize, and log-transform signals intensity.
Relative gene expression levels of each transcript were validated applying a one-way analysis of variance (ANOVA) (p ≤ 0.01) and multiple testing correction. Genes that displayed an expression level at least 1.5 fold different in the test sample versus control sample (p ≤ 0.01) were submitted both to functional classification, using the Gene Ontology (GO) annotations, and to Pathway analysis, employing the Panther expression analysis tools1 (21). The enrichment of all pathways and functional classes associated to the differentially expressed genes compared to the distribution of genes included on the Clariom D microarray was analyzed and p values ≤0.05, calculated by the binomial statistical test, were considered as significant enrichment.
The Search Tool for the Retrieval of Interacting Genes (STRING version 10.52) is a web-based database which comprises experimental as well as predicted interaction and covers >1,100 completely sequenced organisms (22). DEGs were mapped to the STRING database to detect protein–protein interactions (PPI) pairs that were validated by experimental studies (23) and the corresponding PPI network was constructed. A score of ≥0.7 for each PPI pair was selected to design the PPI network.
To detect high-flow areas (highly connected regions) of the network, we used the CFinder software tool, based on the Clique Percolation Method (CPM) (24). Finally, Cytoscape software (25) was used to define the topology of the built networks.
To isolate cell-free circulating miRNA (cf-miRNA) in serum, we used miRNeasy serum/plasma kit (Qiagen, GmbH, Hilden, Germany). For all the samples, RNA was extracted from 200 µL of serum in accordance with manufacturer’s instructions. In order to minimize the technical variation between samples in down-stream PCR analysis we added, for all isolations, 0.90 fmol of spike-in non-human synthetic miRNA cel-39-3p into the sample after the addition of the lysis/denaturant buffer to the serum. After extraction, RNA was eluted in 14 µL of nuclease-free water.
Mature miRNA expression was assayed by TaqMan® Advanced miRNA assays chemistry (Applied Biosystems, Foster City, CA, USA). Briefly, 2 µL of serum RNA was reverse transcribed and pre-amplified with TaqMan® Advanced miRNA cDNA synthesis kit following manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Pre-amplified cDNA was diluted 1/10 in nuclease-free water and 5 µL of diluted cDNA for each replicate were loaded in PCR. 20 µL PCR reactions were composed by 2× Fast Advanced Master Mix and TaqMan® Advanced miRNA assays for hsa-miR-126-3p (477887_mir), hsa-miR-21-5p (477975_mir), hsa-miR-92a-3p (477827_mir), hsa-miR-155a-5p (477927_mir), and hsa-miR-16-5p (477860_mir). Since established consensus house-keeping miRNAs for data normalization are lacking for serum miRNAs, we used cel-39-3p expression (478293_mir) to normalize miRNA expression. Real time PCR were carried out in triplicate on a QuantStudio 6 Flex instrument (Applied Biosystems, Foster City, CA, USA). Expression values were obtained by ΔCt method using QuantStudio Real-time PCR system software v. 1.3.
In order to gain new insights into the pathogenesis of SSc, we compared the gene expression profiles of PBMC samples obtained from 10 lSSc and 10 dSSc patients with 10 PBMC samples obtained from age and sex matched healthy donors. Raw data have been deposited in the ArrayExpress database at EMBL-EBI3 under accession number E-MTAB-6531.
When we analyzed lSSc samples, we found that 829 differently expressed genes (DEGs) satisfied the Bonferroni-corrected p value criterion (p ≤ 0.01) and the fold change criterion (FC ≥ |1.5|), displaying robust and statistically significant variation between SSc and healthy controls samples. In particular, 736 and 93 transcripts resulted to be over- and underexpressed, respectively. The complete list of modulated genes can be found in Table S1 in Supplementary Material.
When the same Bonferroni-corrected p value and fold change criteria were used, 456 transcripts were differentially modulated in dSSc patients compared to healthy controls, 327 and 129 transcripts resulted to be up- and downregulated, respectively (Table S2 in Supplementary Material).
The Gene Ontology analysis of modulated genes in lSSc patients, showed that a large number of the modulated transcripts can be ascribed to biological processes that may play a role in SSc, including: apoptotic process, cell proliferation, growth factor and growth factor binding, inflammatory response, immune response, angiogenesis, endothelial cell activation, cell adhesion, and extracellular matrix organization process.
Moreover, a large number of modulated genes were ascribed to well-known signaling pathways encompassing Type I interferon, epidermal growth factor (EGF) receptor, transforming growth factor (TGF)-beta, interleukin, Wnt, glycolysis, platelet derived growth factor (PDGF), FAS, and Phosphoinositide 3 (PI3)-kinase signaling pathway.
Table 2 shows a selection of DEGs within the above-mentioned categories and also includes public gene accession numbers and fold changes.
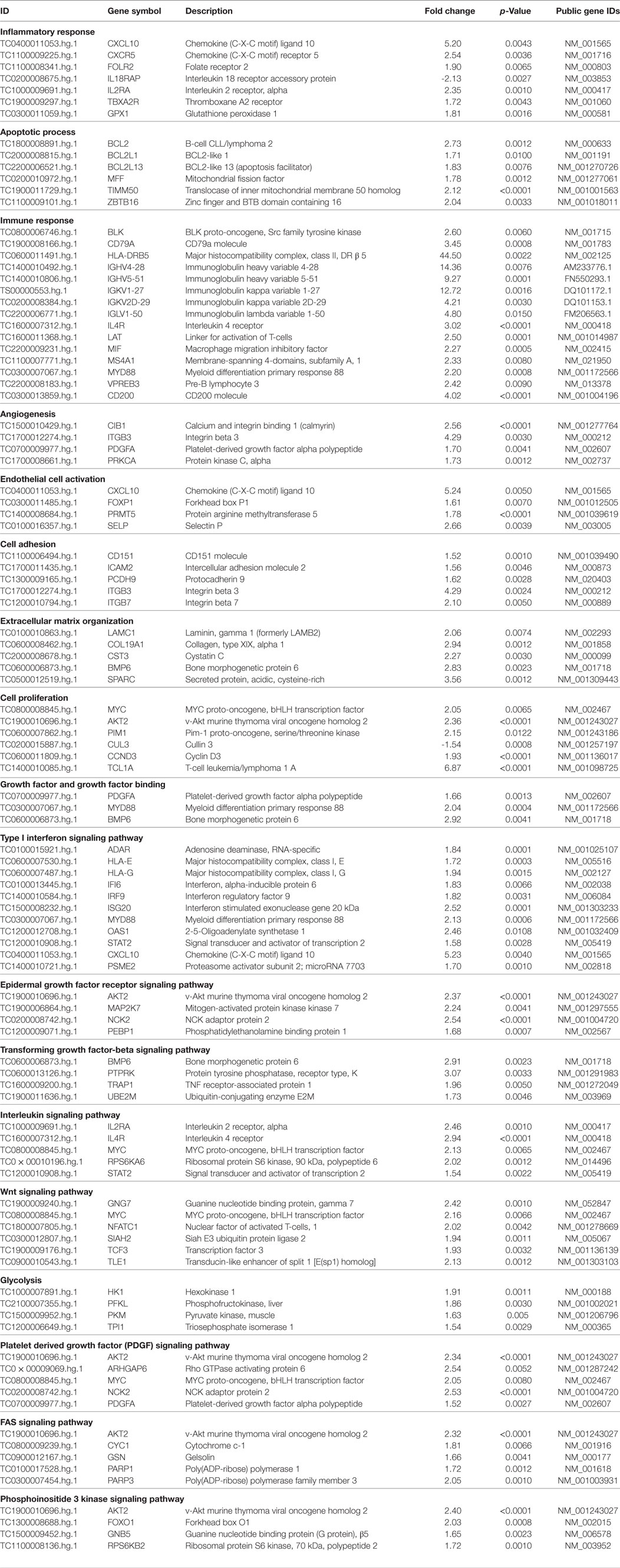
Table 2. Selection of modulated genes in lSSc patients versus healthy controls.
Among genes involved in apoptosis, we found the upregulation of B-cell lymphoma 2 (BCL2); BCL2-like 1 (BCL2L1); BCL2-like 13 (BCL2L13); mitochondrial fission factor (MFF); translocase of inner mitochondrial membrane 50 homolog (TIMM50), and zinc finger and BTB domain containing 16 (ZBTB16). Interestingly, TIMM50 is an anti-apoptotic gene that has been involved in breast cancer cell proliferation (26).
The upregulation of apoptotic molecules was paralleled by the overexpression of genes involved in cell proliferation including MYC proto-oncogene, bHLH transcription factor (MYC); v-akt murine thymoma viral oncogene homolog 2 (AKT2); Pim-1 proto-oncogene, serine/threonine kinase (PIM); cyclin D3 (CCND3), and T-cell leukemia/lymphoma 1 A (TCL1A).
Interestingly, all these molecules have been previously associated to different type of cancer (27–30). Moreover, we found downregulation for cullin 3 (CUL3) a tumor suppressor molecule (31). All these observations may suggest that a dysregulation of proliferative genes in patients with SSc may predispose to the development of malignancies.
We also found upregulation of growth factors encoding molecules such as platelet-derived growth factor alpha polypeptide (PDGFA), bone morphogenetic factor 6 (BM6), and myeloid differentiation primary response 88 (MYD88) a fibrogenic molecule that controls pericyte migration and trans-differentiation of endothelial cells to myofibroblasts (32).
Chronic inflammation is a feature of SSc; therefore, the upregulation of genes involved in the inflammatory response, including chemokine (C-X-C motif) ligand 10 (CXCL10), chemokine (C-X-C motif) receptor 5 (CXCR5), folate receptor 2 (FOLR2), interleukin 2 receptor, alpha (IL2RA), thromboxane A2 receptor (TBXA2R), and glutathione peroxidase 1 (GPX1), is not surprising.
Scleroderma, like other autoimmune connective tissue diseases, is characterized by immune system alterations and the results of our analysis also highlighted this aspect, showing a modulation of a large number of genes involved in immune response. A selection of these molecules is reported in Table 2 and includes: BLK proto-oncogene, Src family tyrosine kinase; CD79a molecule, immunoglobulin-associated alpha (CD79A); major histocompatibility complex, class II, DR beta 5 (HLA-DRB5); immunoglobulin heavy variable 4-28 (IGHV4-28); immunoglobulin heavy variable 5-51 (IGHV5-51); (IGKV1-27); immunoglobulin kappa variable 2D-29; immunoglobulin lambda variable 1-50 (IGLV1-50); interleukin 4 receptor (IL4R), linker for activation of T-cells (LAT); macrophage migration inhibitory factor (MIF); membrane-spanning 4-domains, subfamily A, member 1 (MS4A1); myeloid differentiation primary response 88 (MYD88); pre-B lymphocyte 3 (VPREB3), and CD200 molecule (CD200). Among the abovementioned molecules, MIF has been described as a lung metastasis inducer (33) and CD200 has been involved in the development of breast cancer metastasis (34) and of myeloid leukemia (35).
The initial stages of SSc are accompanied by an angiogenic response to tissue ischemia and vascular damage, that is later replaced by a deficient wound healing and by fibrosis (36). Indeed, several genes involved in angiogenesis were upregulated in lSSc samples including calcium and integrin binding 1 (CIB1); integrin beta 3 (ITGB3), platelet-derived growth factor alpha polypeptide (PDGFA), and protein kinase C, alpha (PRKCA). Interestingly, PDGF stimulates tumor cells, promotes angiogenesis, and the development of cancer associated fibroblasts (37) leading to tumor progression. In addition, aberrant expression of PKCA is associated with a range of malignancies and has recently become a target for anti-cancer therapies (38).
The upregulation of genes such as chemokine (C-X-C motif) ligand 10 (CXCL10), forkhead box P1(FOXP1), protein arginine methyltransferase 5(PRMT5), and selectin P (SELP) is consistent with the endothelial cells activation that is typical of SSc and is accompanied by the overexpression of cell adhesion molecules involved in endothelial cells and leukocytes interactions as well as in blood cells extravasation. We indeed found upregulation for intercellular adhesion molecule 2 (ICAM2), protocadherin 9 (PCDH9), integrin beta 3 (ITGB3), integrin beta 7 (ITGB7), and CD151 molecule that, importantly, is an emerging possible poor prognostic factor for solid tumors (39).
Systemic sclerosis is characterized by connective tissue fibrosis of skin and internal organs that is sustained by extracellular matrix (ECM) remodeling (40). Accordingly, we found an increased expression of transcripts that play a role in ECM organization including laminin gamma 1 (LAMC1), collagen, type XIX (COL19A1), bone morphogenetic protein 6 (BMP6), cystatin C (CST3), and secreted protein, acidic, cysteine-rich (SPARC), a secreted protein that is overexpressed in the fibroblasts of skin biopsy from patients with SSc (41). Noteworthy, remodeling of the stromal ECM by cancer-associated fibroblasts is crucial for tumor cell migration and invasion (42) and SPARC has been associated to these events (43). Interestingly, an elevated preoperative CST3 level was demonstrated to be related with worse survival in patients with renal cell carcinoma (44).
The pathway enrichment analysis that we performed to find signaling network that were overrepresented by modulated genes in lSSc samples, showed an enrichment in apoptosis, glycolysis, PDGF, 5HT4 type receptor, Fas cell surface death receptor (FAS), histamine H2 receptor, serine glycine biosynthesis, beta 3 adrenergic receptor, angiotensin (through G proteins and beta-arrestin), and interleukin signaling pathways (Table 3).

Table 3. Pathways enriched in genes modulated in lSSc samples.
Interestingly, several of the abovementioned enriched pathways are involved in cancer development.
Other modulated transcripts belong to the epidermal growth factor (EGF), transforming growth factor (TGF) beta, Wnt, and PI3 kinase signalings, all involved not only in the development of the SSc associated fibrosis (45, 46) but also in cancer development (47–50). In particular, we observed overexpression of the AKT2 member of the EGF signaling pathway and of the FOXO1 component of the PI3K pathway. The deregulation of these genes has been associated with tumor progression and metastatic spread (29, 51).
The functional classification of DEGs in dSSc samples shows that they reflects the gene modulation observed in lSSC samples (see Table 4).
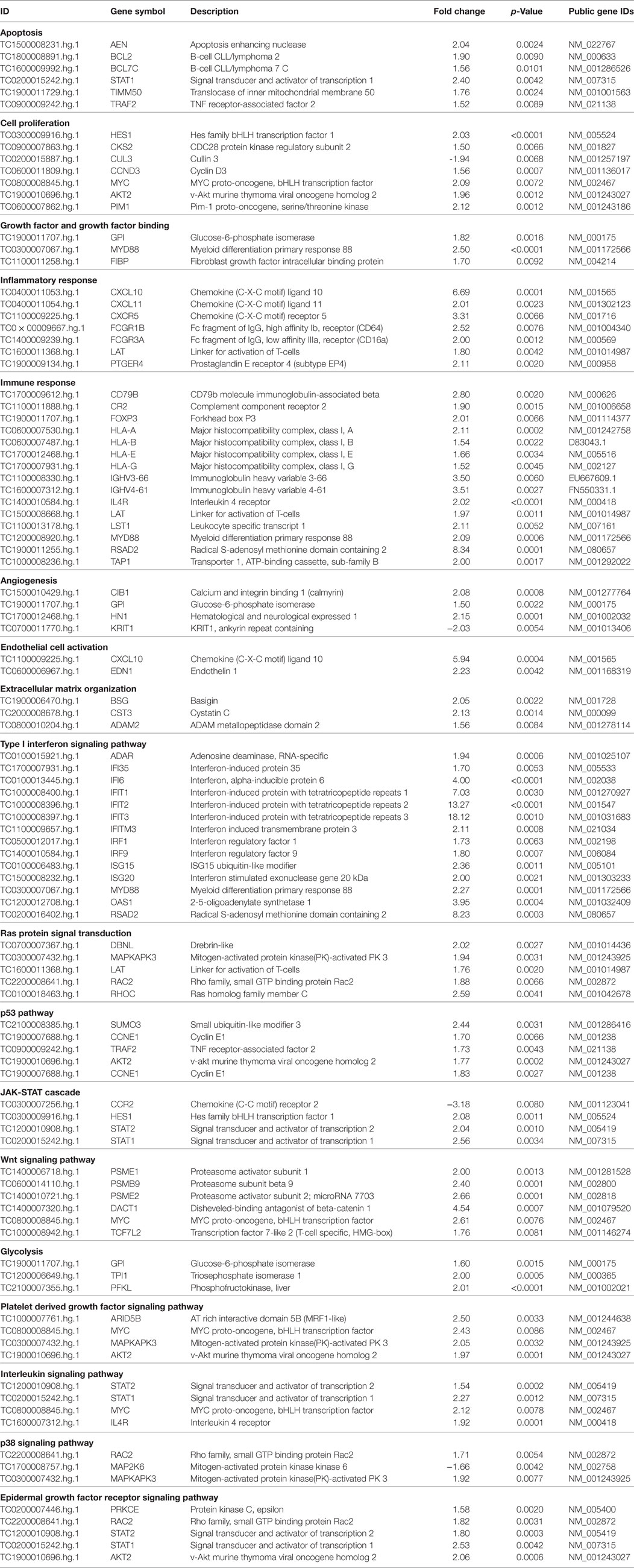
Table 4. Selection of modulated genes in diffuse SSc patients versus healthy controls.
The apoptosis functional class accounted for several upregulated transcripts namely apoptosis enhancing nuclease (AEN), B-cell CLL/lymphoma 2 (BCL2), B-cell CLL/lymphoma 7 C (BCL7C), signal transducer and activator of transcription 1 (STAT1), TNF receptor-associated factor 2 (TRAF2), and translocase of inner mitochondrial membrane 50 homolog (TIMM50). In addition to genes that were also modulated in lSSc samples (i.e., MYC, AKT2, and PIM1), other genes involved in cell proliferation and also in tumor development were overexpressed. These genes include: hes family bHLH transcription factor 1(HES1), CDC28 protein kinase regulatory subunit 2 (CKS2), and cyclin D3 (CCND3) (52–54). Moreover, three upregulated transcripts were ascribed to the growth factor binding gene category: glucose-6-phosphate isomerase (GPI), fibroblast growth factor intracellular binding protein (FIBP), and MYD88 (also modulated in lSSc samples as mentioned above). Noteworthy, also in dSSc samples, we observed the down-modulation of the tumor suppressor CUL3.
Several modulated genes encode for inflammatory molecules including chemokine (C-X-C motif) ligand 11 (CXCL11), Fc fragment of IgG, high affinity Ib, receptor (FCGR1B), Fc fragment of IgG, low affinity IIIa, receptor (FCGR3A), linker for activation of T-cells (LAT), prostaglandin E receptor 4 (PTGER4), CXCL10, and CXCR5. These last two transcripts were also overexpressed in lSSc samples.
Genes involved in the immune response were modulated also in dSSc samples and, besides the genes overexpressed in lSSc samples (i.e., IL4R, LAT, and MYD88), we found upregulation of CD79b molecule, immunoglobulin-associated beta (CD79B), complement component receptor 2 (CR2), forkhead box P3 (FOXP3), major histocompatibility complex, class I, A (HLA-A), major histocompatibility complex, class I, B (HLA-B), major histocompatibility complex, class I, E (HLA-E), major histocompatibility complex, class I, G (HLA-G), immunoglobulin heavy variable 3-66 (IGHV3-66), immunoglobulin heavy variable 4-61 (IGHV4-61), radical S-adenosyl methionine domain containing 2 (RSAD2), and transporter 1, ATP-binding cassette, sub-family B (TAP1).
Among DEGs involved in angiogenesis there are: glucose-6-phosphate isomerase (GPI), hematological and neurological expressed 1 (HN1), KRIT1, ankyrin repeat containing (KRIT1) and CIB1 (also modulated in lSSc samples).
Endothelial activation was well represented in dSSc samples by the overexpression of gene encoding for endothelin 1 (EDN1) that is associated with diseases characterized by endothelial dysfunction and fibrosis (55).
Finally, we found upregulation of three transcripts that play a role in the ECM organization including ADAM metallopeptidase domain 2 (ADAM2), CST3 (increased also in lSSc samples), and basigin (BSG). BSG, also named ECM metalloproteinase inducer (EMMPRIN), is expressed on the surface of tumor cells and induces fibroblasts to synthesize matrix metalloproteinases (56).
Pathways enrichment analysis showed that pathways also overrepresented in lSSc samples were enriched in dSSc samples (i.e., glycolysis, apoptosis, and interleukin signaling pathway). Besides these signaling pathways, we found an enrichment in pathways involved in tumor development including: inflammation mediated by chemokine and cytokine, cell cycle, Ras (57), oxidative stress response (58), p53 (59), ubiquitin proteasome (60), JAK/STAT (61), p38 MAPK (62), angiogenesis, and EGF receptor signaling pathway (Table 5). Among modulated genes belonging to these pathways, we mention ras homolog family member C (RHOC), the overexpression of which indicates poor prognosis in breast cancer cells (63), and the abovementioned AKT2 and HES1.

Table 5. Pathways enriched in genes modulated in diffuse SSc samples.
As found in lSSc samples, among other DEGs involved in signal transduction, several transcripts were involved in Wnt, PDGF, and type I interferon signaling pathway and, their role in cancer development has been already stressed in our dissection of genes modulated in lSSc. Not surprisingly, type I interferon pathway accounted for a large amount of DEGs (14) and the evidence of the modulation of this molecular signaling both in lSSc and in dSSc further underlines its role in the pathogenesis of the disease.
The gene expression profiling of SSc patients was complemented with a network analysis. With this purpose, by a bioinformatic analysis, we selected all the functional and experimentally validated interactions between the protein products of modulated genes and we constructed the two protein–protein interaction (PPI) networks that were representative of lSSc and dSSc dataset.
The lSSc-PPI network comprised 440 genes (nodes) and 1351 pairs of interactions (edges) (Figure 1A), whereas the dSSc included 225 genes and 870 pairs of interactions (Figure 1B).
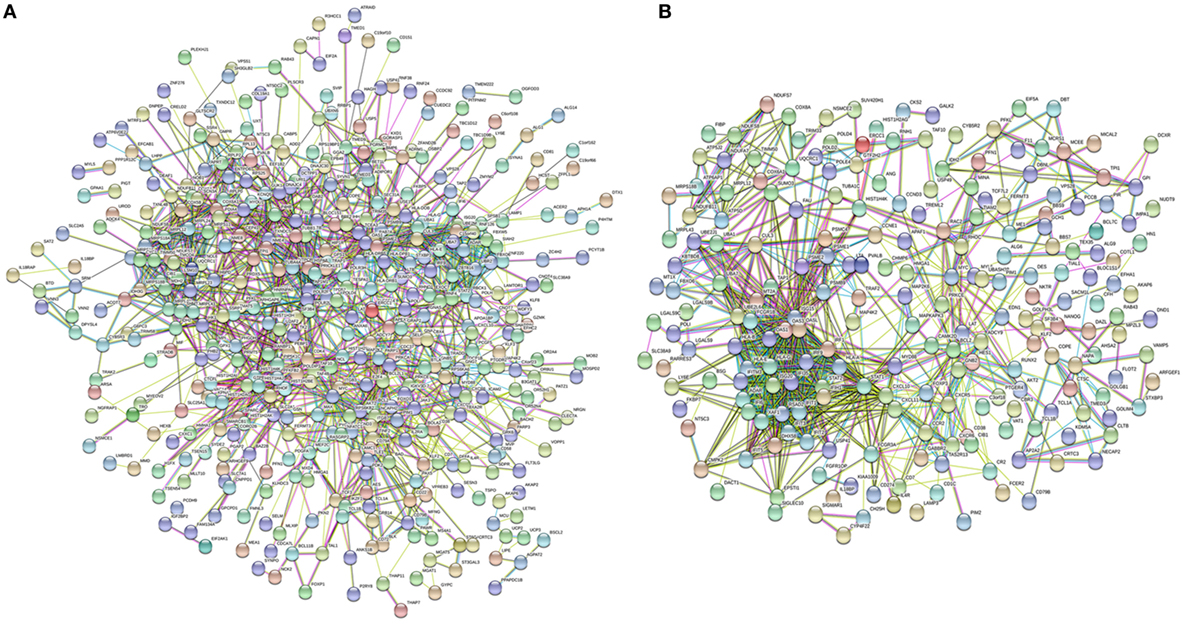
Figure 1. Network analysis of modulated genes in systemic sclerosis (SSc) patients. Protein–protein interaction network of differently expressed genes in lSSc (A) and in diffuse cutaneous SSc (B) samples.
The PPI networks were then submitted to a modular analysis to find set of highly interconnected nodes (modules) that participate in multiple activities in a coordinated manner and that are expected to play a prominent role in the development of biological phenomena.
In the lSSc-PPI network, we identified nine modules that are graphically represented in Figure 2. Moreover, a functional enrichment analysis was applied to find the associations between each module and relevant enriched “GO terms” and pathways. All the significantly enriched (p < 0.05) biological processes (BPs) and pathways in each module are showed in Table S3 in Supplementary Material and Table 6 shows a selection of the most relevant terms. We observed that the most enriched BPs in module M0, were the signaling of G-protein coupled receptors (GPCRs) and mediated by chemokine. Interestingly, the GPCRs pathway is involved in cancer initiation and progression and GPCRs are emerging as anti-cancer drug targets (64). In the same module M0, we could highlight, among other, an enrichment in inflammation mediated by chemokine and cytokine, PI3 kinase and endothelin signaling pathway. Module M1 showed an enrichment in the positive regulation of type I interferon production BP whereas, among different enriched pathways, we found the glycolysis pathway and again, the endothelin signaling pathway. Module M2 and M4 were the most representative of the type I interferon response. In M2, the GO terms associated to the type I interferon signaling were the most enriched biological processes, followed by terms associated to the innate immune response (i.e., innate immune response, defense response to virus, interferon gamma mediate signaling) and to the adaptive immune response (i.e., antigen processing and presentation of exogenous peptides). Interestingly, the enriched pathways in M2 were related to the Jak/Stat and Interleukin signaling, both associated to tumor development as previously remarked.
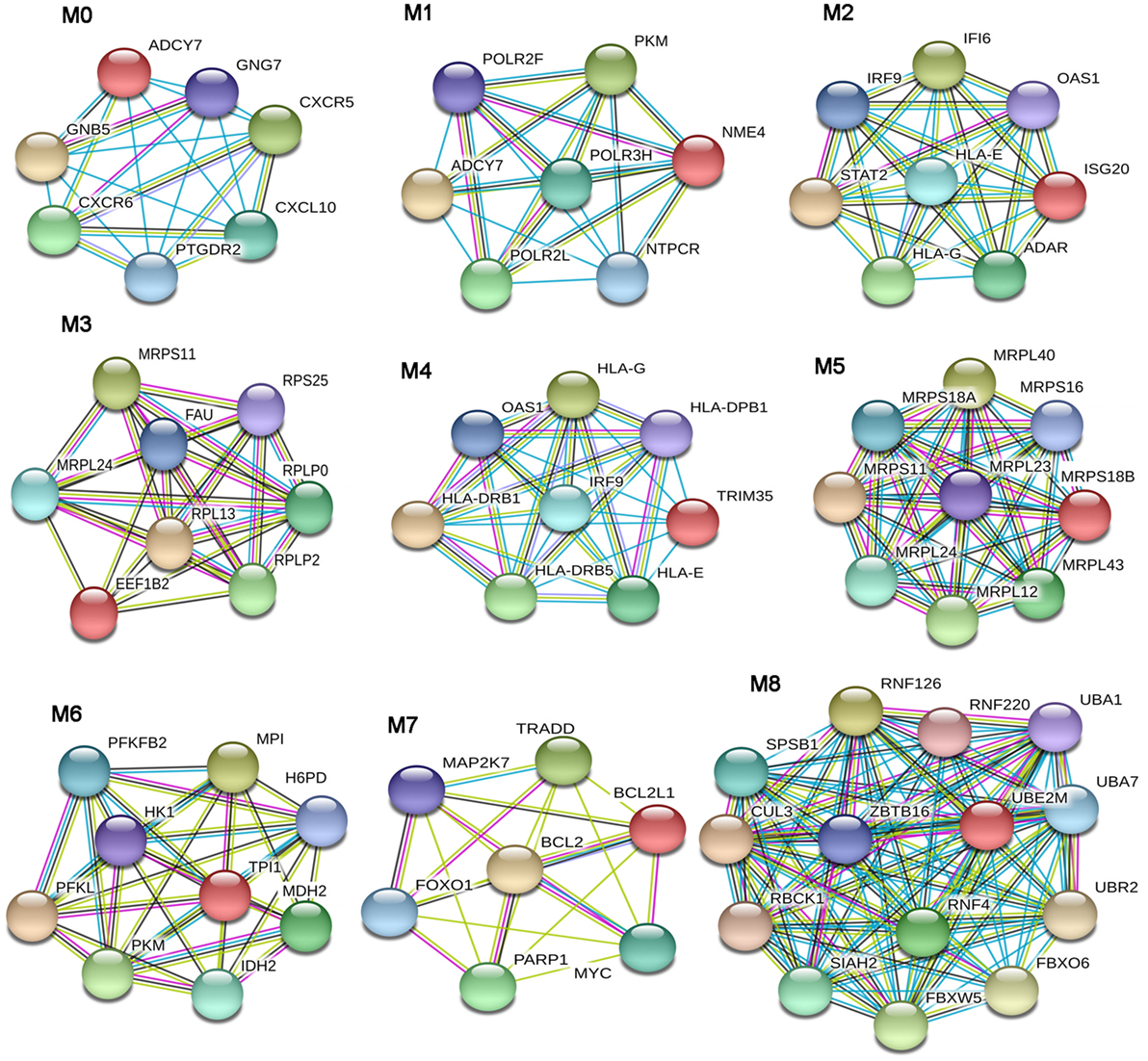
Figure 2. Modular analysis of the lSSc-protein–protein interaction network. Modules originated from the interaction network of modulated genes in lSSc samples.
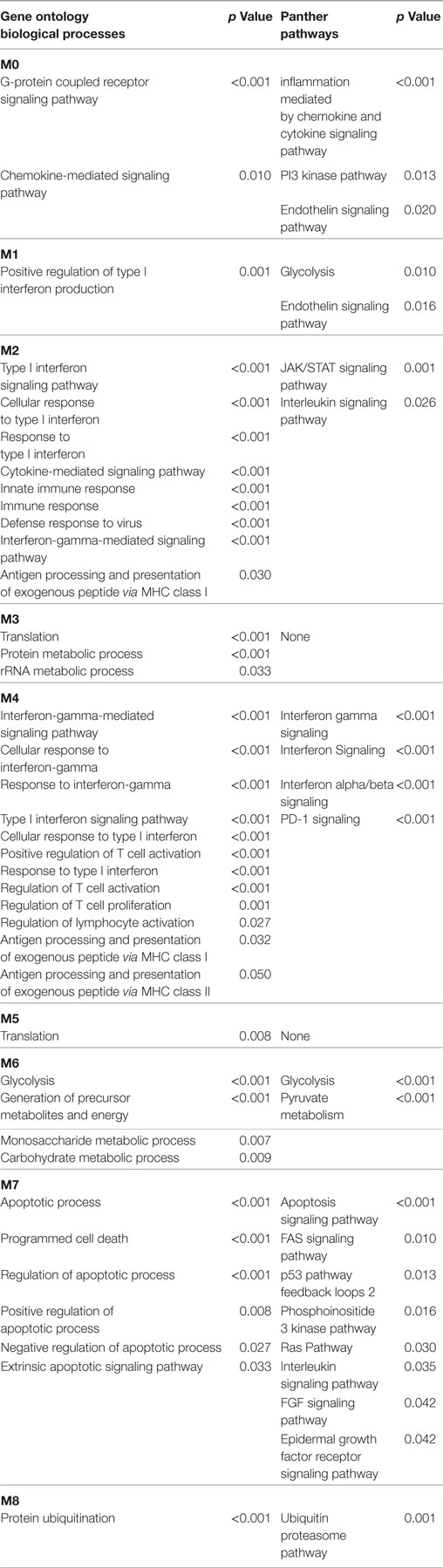
Table 6. Most relevant biological processes and pathways enriched in lSSc modules.
In M4, the most enriched BPs were interferon-gamma-mediated signaling pathway and cellular response to interferon-gamma. Besides the BPs associated to type I interferon response, we found an enrichment in terms referred to the lymphocyte-associated immune response (i.e., regulation of lymphocyte activation, regulation of T cell activation, regulation of T cell proliferation and antigen processing, and presentation of peptide antigen via MHC class I and II). Most enriched pathways in M4 were referred to interferons (alpha, beta and gamma) pathways. Interestingly, in M4, we observed an enrichment in the PD-1 signaling, an immune-inhibitory-checkpoint that acts as crucial mediator for the escape phase of cancer immune editing (65). Module M3 and M5 were enriched in terms related to translation and other metabolic processes of proteins and, in module M6 the most enriched BPs and pathways were referred to the glycolytic pathway. In module M7, we observed an enrichment of many BPs associated to positive and negative regulation of apoptosis and concordantly, an enrichment in the apoptotic FAS and p53 pathways signaling. Moreover, in M7, several cancer-associated signaling pathways were included, including PI3 kinase, Ras, Interleukin, FGF, and EGF receptors signaling pathways. Finally, in module M8, the protein ubiquitination BP and the ubiquitin proteasome pathway were the most over-represented. Interestingly, tumor cells have a high dependency on the proteasome for survive and proteasome deregulation is frequently induced by many types of tumors (66).
From the dSSC-PPI network, we could extract seven modules (Figure 3) that were studied by a functional enrichment analysis. The results of this analysis are fully presented in Table S4 in Supplementary Material and the most relevant terms are showed in Table 7.
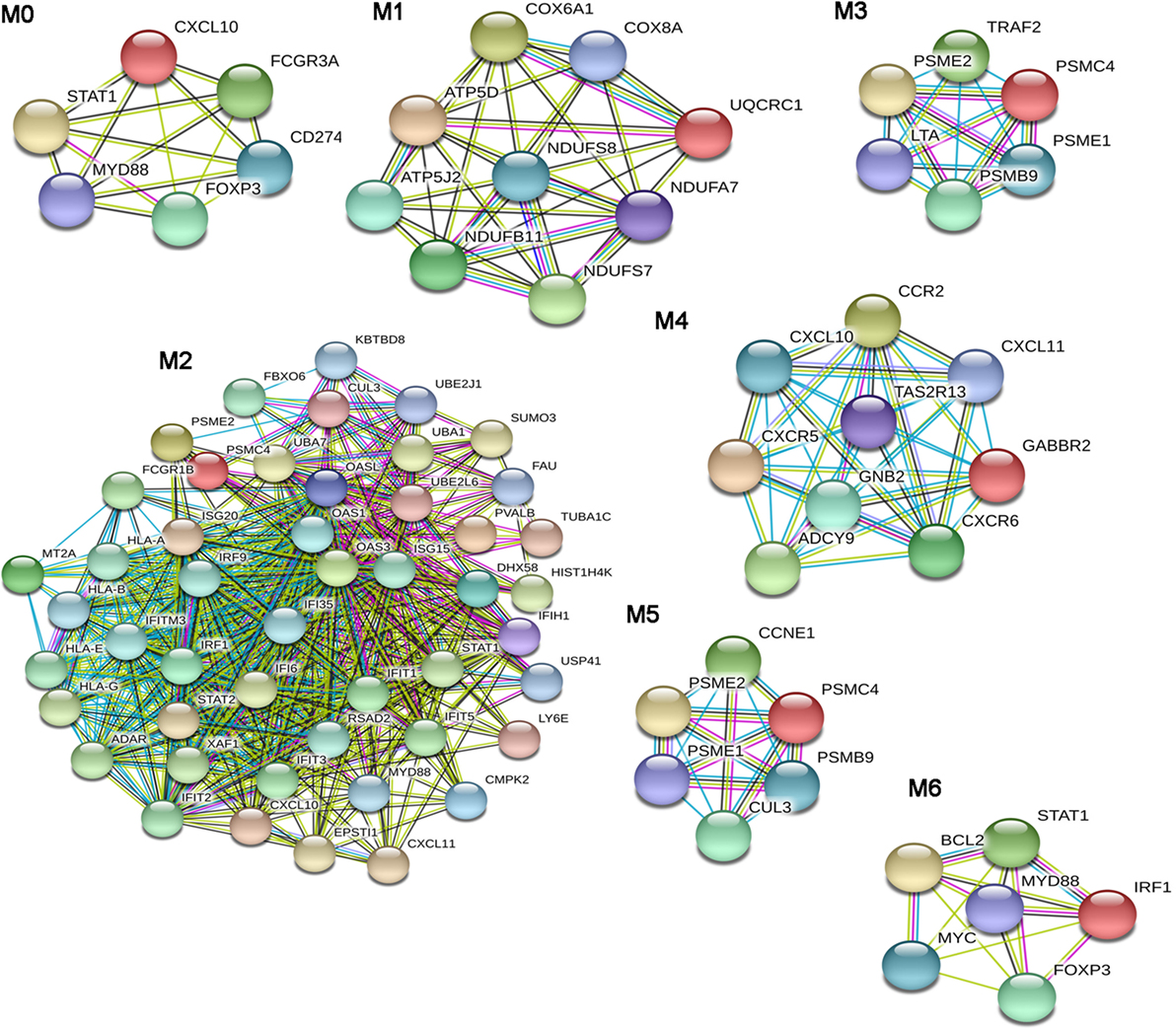
Figure 3. Modular analysis of the diffuse SSc (dSSc)-protein–protein interaction network. Modules originated from the interaction network of modulated genes in dSSc samples.

Table 7. Most relevant biological processes and pathways enriched in diffuse SSc modules.
The BPs that we found enriched in the module M0 were mainly referred to the immune response. In particular, we observed an enrichment in the regulation of tumor necrosis factor superfamily cytokine production process. Moreover, the over-represented pathways in M0 reassumed nearly all signalings that were enriched in the entire dSSc dataset, including inflammation mediated by chemokine and cytokine, JAK/STAT, p53, oxidative stress response, Ras, interleukin, EGF receptor, and angiogenesis signaling pathway. Module M1 was largely enriched in ATP and purine ribo/nucleoside metabolic processes, whereas M2 was mostly represented by BPs referred to the type I interferon signaling followed by BPs involved in both innate and adaptive immune response. In addition, DEGs in M2 mainly play a role in ubiquitin proteasome, Jak/Stat, interleukin, EGF receptor, and PDGF signaling pathways. In M3, the most enriched BP was cellular response to TNF and, not surprising, the most over-represented pathway was the apoptosis signaling. Other pathways, statistically representative of module M3 were toll-like receptor (TLR), ubiquitin proteasome, and p53 pathway. In particular, the enrichment of TLR signaling is interesting since it is well known that inducing pro-inflammatory cytokines and co-stimulatory molecules, it contributes to the development of an excessive inflammatory response, leading to both autoimmune disorders and tumor growth (67). Module M4 was mostly enriched in G-protein coupled receptor signaling, and we also observed enriched BPs related to leukocyte/lymphocyte chemotaxis, whereas the most over-represented pathway was inflammation mediated by chemokine and cytokine signaling pathways. The Wnt signaling pathway was preeminent in module M5. In addition, DEGs in this module were also primarily involved in the p53 pathway. Response to virus was the most representative BP of module M6: is worthwhile mentioning that numerous viruses have been proposed as possible triggering factors in SSc (68), and indeed, it has been estimated that up to a quarter of human tumors are connected to infection or infection-associated chronic inflammation (69).
A large number of BPs involved in immune cells activation, proliferation, and differentiation and in type I interferon signaling, were prevalent in M6. The over-represented pathways in this module were again signalings also involved in cancer and already mentioned in this analysis (i.e., p53, interleukin, PDGF, Jak/Stat, TLR, Ras, and apoptosis signaling pathway).
Systemic sclerosis is associated with an increased risk of malignancies, and in the present study, we found a modulation of genes encoding for molecules that have been previously associated to different types of cancers. An important role of miRNAs in human cancers is well established (12). The higher incidence of cancer in SSc patients prompted us to investigate whether specific cancer-related miRNAs could be deregulated in the serum of SSc patients as compared to healthy controls.
Since SSc patients are mainly affected by breast, lung, or hematological malignancies (70), we selected miRNAs with a solid evidence in literature for deregulation in these kind of cancers. We focused on miR-155-5p, miR-126-3p, and miR-16-5p. Furthermore, we decided to analyze miR-21-5p and miR-92a, since they play key roles in many cancers, and to confirm their upregulation in our cohort of SSc patients (Table 8). Cell-free miRNAs (cf-miRNA) in limited and diffuse SSc and in healthy sera was evaluated by real time PCR, as represented in Figure 4. A significant higher expression of 4/5 of the miRNAs tested in SSc sera was found as compared to healthy controls. miR-126-3p expression also showed a trend of upregulation in SSc samples although it did not reach statistically significant differences in the samples tested compared to controls. Moreover, no significant differences between limited and diffuse SSc were found in our analysis. miR-21-5p and miR-92a-3p were upregulated, as reported in literature, further supporting the hypothesis of their involvement in SSc pathogenesis. To our knowledge, upregulation of miR-155-5p and miR-16-5p in SSc sera has not been reported yet, and it suggests a role of these miRNAs in the disease.
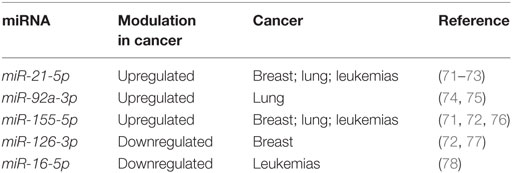
Table 8. Cancer-related miRNAs selected for expression analysis in SSc serum.
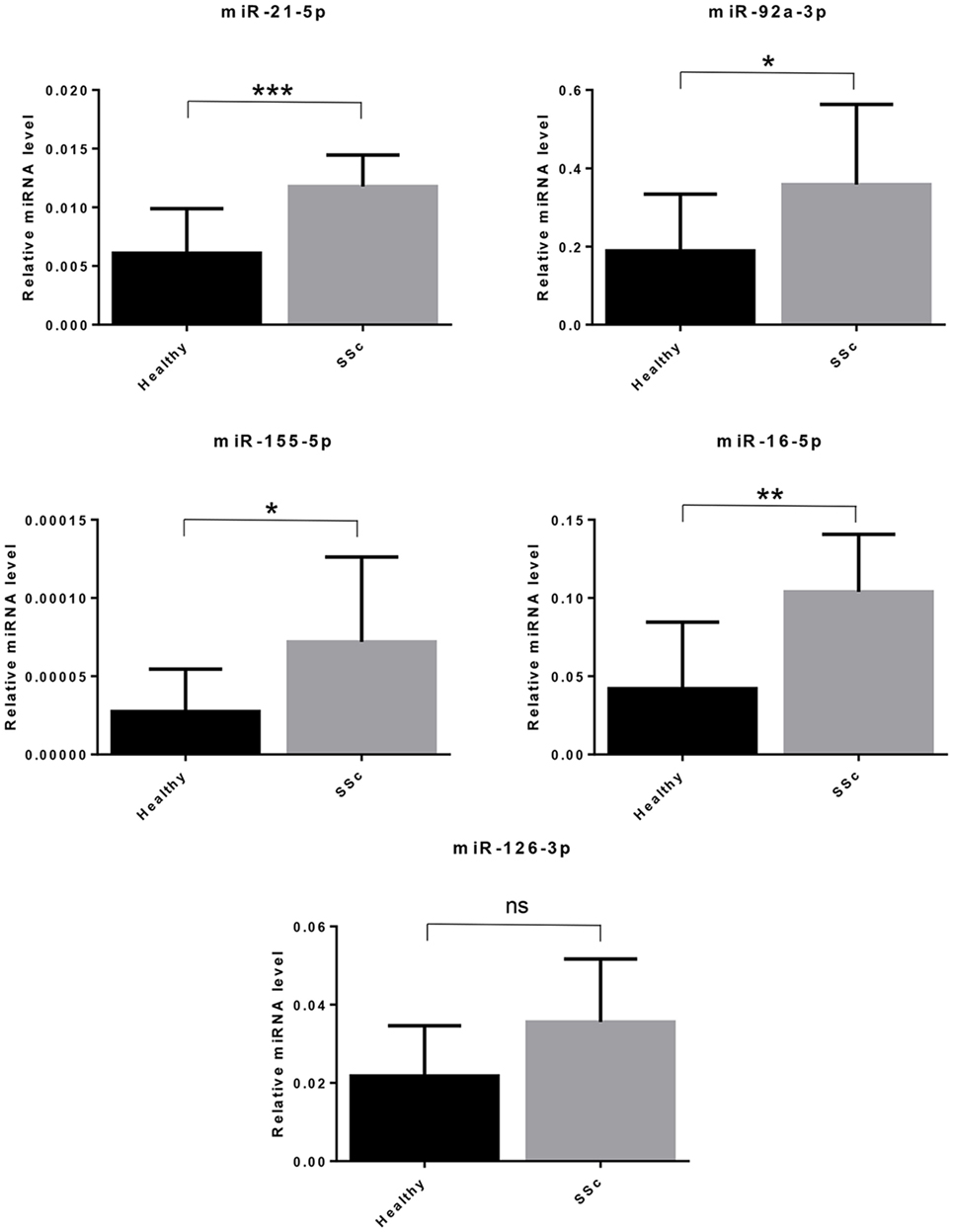
Figure 4. Cell-free circulating miRNA expression in systemic sclerosis (SSc) sera. The expression of the indicated miRNAs was evaluated by real-time PCR in serum of systemic sclerosis patients (SSc; n = 30) and of healthy controls (Healthy; n = 30). Expression values of mature miRNAs were calculated using the comparative ΔCt method and normalized to spike-in cel-39-3p. Histograms represent mean ± SD. p-values (ns = not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001) were determined using the Mann–Whitney rank sum test.
Systemic sclerosis represents a major medical challenge and many unmet needs in the treatment of the disease still remain. However, advances have been made thanks to the increased understanding of the pathogenesis of SSc and the overall patients’ survival is improved.
The increased risk of malignancies in SSc is a major source of concern and the identification of risk factors for both disorders may have implications for the prognosis and for the treatment.
We therefore wanted to carry out a gene expression profile and an epigenetic analysis in scleroderma patients to verify the presence of common bases for the development of malignancies and SSc.
We have noticed that the functional classes, to which the genes modulated in limited and diffuse forms of SSc belong, were virtually overlapping and, in both cases, we observed the modulation of genes and pathways previously associated with malignancy. Although it has been reported a higher incidence of some tumors in patients with dSSc than in those with lSSc, our data seem to suggest that in both forms of the disease there is a genetic modulation that may be linked to the onset of a neoplastic transformation. Indeed, from our analysis has emerged that the various classes comprising genes potentially linked to the pathogenesis of SSc (such as apoptosis, endothelial cell activation, extracellular matrix remodeling, immune response, and inflammation), include genes that directly participate in the development of malignancies or that are involved in pathways known to be associated with carcinogenesis.
In this regard, several pathways enriched in lSSc and/or dSSc (apoptosis, glycolysis, PDGF, Fas cell surface death receptor, FAS, angiogenesis, interleukin, Ras, Jak/Stat, and EGFR signaling pathways) are involved in cancer development.
Indeed, altered apoptosis has a crucial role in the induction of a malignant phenotype, and it is well known that some oncogenic mutations block apoptosis, leading to cancer progression (79). Moreover, some cancer cells express FAS ligand (FasL) and the activation of FAS pathway may favor immune privilege to tumors by inducing apoptosis of anti-tumor lymphocytes (80).
The modulation of genes involved in the glycolytic pathway is not surprising, given that upregulation of glycolysis is a well-documented property of cancers, and this mechanism confers to tumor cells a significant growth advantage (81).
Platelet derived growth factors and PDGF receptors have substantial role in regulating cell growth during embryonal development. An enhancement of PDGF receptor signaling, may also sustain tumor cell growth. Moreover, fibroblasts, and myofibroblasts of solid tumors stroma express PDGF receptors, and PDGF stimulates these cells promoting tumorigenesis (82). Noteworthy, PDGF enhances c-myc expression and stimulates the c-myc promoter (83). This gene, found overexpressed in our lSSc dataset, coordinates cell growth and cell proliferation and its deregulation is a near-universal property of primary and metastatic cancers (84).
Ras proteins are key elements in malignant transformation; moreover, the Jak/stat signaling pathway sustains epithelial mesenchymal transition and generates a pro-tumorigenic microenvironment. The EGF receptor signaling pathway modulates migration and survival of cancer cells.
It has been established that inflammation caused by either chronic disease or infection is an important risk factor in cancer development and that a variety of interleukins are involved in both inflammation and carcinogenesis (85). In particular, among genes involved in the interleukin signaling pathways that was enriched in lSSc modulated genes, we found overexpression of the interleukin 2 receptor alpha (IL2RA) gene. The soluble form of this molecule is released from neoplastic cells and is expressed on the surface of both lymphoid and non-lymphoid cancer cells (86).
Pathway analysis also highlighted the modulation of a large number of genes (11) involved in the Type 1 interferon signaling pathway. This pathway is a hallmark of many systemic autoimmune diseases (87) including SSc (88) and has also been considered as a “double-edged sword” in cancer. Indeed, in cancer, it promotes both T cell responses and a negative feedback leading to immunosuppression. Tumor cells can take advantage from this counter-regulatory effects induced by IFN type I to avoid immune cell killing (89).
This suggests that there are multiple points of contact between the signaling pathways leading to scleroderma and those that predispose to the development of a malignancy. However, these pathways may lead to different outcomes in different tissues in which they are expressed, and indeed in patients with scleroderma, some malignancies are more frequent such as those affecting breast, lung, and lymphoid tissue.
Endothelial cell apoptosis is among the first manifestations of vasculopathy associated with the development of SSc, and fibroblasts activation and proliferation lead to the fibrotic characteristics of the disease. Gene expression profiles obtained from the PBMC of patients with SSc indicate an altered modulation of genes involved in apoptosis and in cell proliferation that could, at tissue level, exert a mitogenic effect on some cellular populations or lead to down-modulation of oncosuppressor genes, as suggested by the downregulation of CUL3 (31) in both lSSc and dSSc.
Our data also suggest the presence of a genetic modulation that can favor angiogenesis, a common feature associated to the development and progression of any type of cancer.
Modulation of many genes involved in the immune response, including genes inducible by type I interferon, reflects the strong immune system dysregulation associated with excessive antigenic stimulation typical of autoimmune diseases. Such dysregulation can either induce proliferation of cells of the immune system, leading to malignancy in susceptible individuals or modify the immune system’s regulatory mechanisms that inhibit the development of naturally occurring cancers.
Several modulated genes participate in the remodeling process of the extracellular tissue matrix. Excessive deposition of ECM promotes the development of fibrosis, hallmark of SSc, and the fibrotic and inflammatory processes of the lung have been considered the basis for the eventual onset of lung cancer in SSc. On the other hand, within these functional classes, we have also observed over-expression of metalloproteases (i.e., ADAM2) or ECM metalloproteases inducers (i.e., BSG) typically induced by several tumor cells (56, 90). Moreover, genes associated with the migration of tumor cells such as SPARC (43), a glycoprotein associated with ECM, are involved in the development of non-small cell lung cancer. Therefore, while ECM deposition and related profibrotic events may favor carcinogenesis through different mechanisms such as blocking lymphatic channels and creating niches in which carcinogens may eventually accumulate, on the other hand, there is a genetic modulation that favors cancer cells spreading.
A crucial issue in basic and clinical research is to understand gene modulation in terms of biological networks since proteins usually function in protein–protein interacting networks. We therefore wanted to dissect meaningful relationships among modulated genes in SSc, analyzing the PPI network in which their protein products can be involved. Moreover, since it is known that the deregulation of protein expression may provoke more drastic biological effects when genes/proteins with more interacting partners are involved, we focused our attention on the most connected genes/proteins that “as a matter of fact” (by definition) are included in the network areas called modules. This is particularly important when studying gene regulation in certain diseases in order to identify the molecular pathways that are most relevant in disease pathogenesis. The pathway enrichment analysis of modulated genes included in the modules confirmed the enrichment of signaling pathways (i.e., the aforementioned Jak/stat, glycolysis, Ras, PDGF, EGF receptor, Wnt, and type I interferon signaling pathways) associated with carcinogenesis in both datasets.
The genes participating to these pathways are indeed comprised in network areas (modules), whereas gene interactions are concentrated and generally underline significant biological processes.
These molecular pathways also emerged from our first global analysis of the two datasets, but the network analysis further underlined their involvement in SSc. Indeed, members of these signaling networks are concentrated in modules were the molecules that are supposed to play a prominent role in shaping the typical features of the disease, are positioned.
Another aspect we have investigated is the expression of microRNAs in SSc. Altered expression of miRNAs in SSc, as well as their involvement in inflammation and fibrosis, has been described (14, 91). Similarly, the implication of miRNAs in human cancers is well established (12). A fascinating hypothesis is that a dysregulated epigenetic control mediated by miRNAs in SSc could promote tumorigenesis. Deregulated expression of miRNAs has been found in blood of SSc patients and it could exert oncogenic effects at distant sites from SSc lesions. Indeed, the upregulation of miR-21 and miR-92a in SSc (16, 92) may support this hypothesis, since these miRNAs play a role in many tumors, repressing important oncosuppressor genes (74, 93). Thus, the higher incidence of breast, lung and hematological malignancies in SSc patients (70) prompted us to investigate whether specific cancer-related miRNAs could be deregulated in the serum of SSc patients. We found that miR-21-5p, miR-92a-3p, miR-155-5p, and miR-16-5p expression was significantly higher in SSc sera compared to healthy controls. miR-21 can play a role in SSc since it is also upregulated in SSc fybroblasts and it is implicated in TGF-β-regulated fibrosis (17). Interestingly, miR-21 upregulation promotes proliferation, migration, and invasion in lung cancer (71). Elevated miR-92a levels in serum and in dermal fibroblasts of SSc patients have also been reported (16), and this miRNA is implicated in angiogenesis and proliferation in lung cancer and it can promote leukemogenesis (74, 75). MiR-155-5p expression was also increased in SSc sera. Notably, miR-155 was found upregulated in SSc fybroblasts, and it was associated to the progression of lung fibrosis in dSSc patients (94, 95). miR-155 upregulation also sustains survival and proliferation in hematological disorders and breast cancer (72, 76). We found increased miR-16-5p levels in SSc sera despite this miRNA is frequently associated to oncosuppressor functions and it is frequently absent in human leukemias (78). Since this miRNA inhibits cell proliferation and promotes apoptosis (96), it could participate to the apoptotic process of endothelial cells, considered the first pathogenic event in SSc. A release of miR-16 in the bloodstream from apoptotic endothelial cells could explain the increased levels of the miRNA in SSc sera. We also decided to evaluate miR-126 in SSc sera since it was found downregulated in breast and lung cancer (76, 77). Moreover, miR-126 plays an important role in angiogenic signaling and in vascular integrity (97), and we found several genes involved in angiogenesis upregulated in SSc by microarray analysis. We did not find statistically significant differences in the miR-126 expression levels between SSc patients and healthy controls, although we observed a trend toward upregulation in SSc samples. In conclusion, we describe here interesting findings on deregulated cancer-related miRNAs in SSc patients. However, further studies are needed to elucidate the potential role of these miRNA in SSc. In particular, since the levels of circulating microRNAs can be affected by different cell types, interesting points will be to elucidate which cells mainly contribute to these circulating oncogenic miRNAs and whether they can play an active role in the increased tumorigenesis associated to the disease.
Taken together, our data suggest the presence of modulated genes and miRNAs that can play a predisposing role in the development of malignancies in SSc. The findings of genetic and epigenetic features that are shared by SSc and cancer shed new light on the pathogenesis of the disease and strengthen the idea that autoimmunity plays a central role in the initiation and progression of SSc, since the presence or development of malignancies is associated with particular autoantibodies. These aspects are central to a better risk stratification of patients and to develop an individualized precision medicine strategy.
This study was carried out in accordance with the recommendations of “local ethical committee Azienda integrata Università di Verona e Ospedale Borgo Roma Verona, Italy” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the “name of committee.”
APu, CL, MD, APe, and APu conceived and designed the experiments. MD, APe, and PF performed the experiments. MD, APe and APu analyzed the data. GP and ET contributed reagents. ET, GP, and CL contributed materials. MD and APe wrote the paper.
The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SV and handling editor declared their shared affiliation.
The work was funded by the University of Verona and by donation of the Italian Association of Sclerodermic Patients (AILS).
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00449/full#supplementary-material.
1. Denton CP, Khanna D. Systemic sclerosis. Lancet (2017) 390:1685–99. doi:10.1016/S0140-6736(17)30933-9
2. Dolcino M, Puccetti A, Barbieri A, Bason C, Tinazzi E, Ottria A, et al. Infections and autoimmunity: role of human cytomegalovirus in autoimmune endothelial cell damage. Lupus (2015) 24:419–32. doi:10.1177/0961203314558677
3. Lunardi C, Bason C, Navone R, Millo E, Damonte G, Corrocher R, et al. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med (2000) 6:1183–6. doi:10.1038/80533
4. Luo Y, Wang Y, Wang Q, Xiao R, Lu Q. Systemic sclerosis: genetics and epigenetics. J Autoimmun (2013) 41:161–7. doi:10.1016/j.jaut.2013.01.012
5. Murdaca G, Contatore M, Gulli R, Mandich P, Puppo F. Genetic factors and systemic sclerosis. Autoimmun Rev (2016) 15:427–32. doi:10.1016/j.autrev.2016.01.016
6. Bernal-Bello D, de Tena JG, Guillen-Del Castillo A, Selva-O’Callaghan A, Callejas-Moraga EL, Marin-Sanchez AM, et al. Novel risk factors related to cancer in scleroderma. Autoimmun Rev (2017) 16:461–8. doi:10.1016/j.autrev.2017.03.012
7. Shah AA, Xu G, Rosen A, Hummers LK, Wigley FM, Elledge SJ, et al. Brief report: anti-RNPC-3 antibodies as a marker of cancer-associated scleroderma. Arthritis Rheumatol (2017) 69:1306–12. doi:10.1002/art.40065
8. Monfort JB, Lazareth I, Priollet P. Paraneoplastic systemic sclerosis: about 3 cases and review of literature. J Mal Vasc (2016) 41:365–70. doi:10.1016/j.jmv.2016.07.001
9. Shah AA, Casciola-Rosen L. Cancer and scleroderma: a paraneoplastic disease with implications for malignancy screening. Curr Opin Rheumatol (2015) 27:563–70. doi:10.1097/BOR.0000000000000222
10. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136:215–33. doi:10.1016/j.cell.2009.01.002
11. Ceribelli A, Satoh M, Chan EK. MicroRNAs and autoimmunity. Curr Opin Immunol (2012) 24:686–91. doi:10.1016/j.coi.2012.07.011
12. Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med (2009) 60:167–79. doi:10.1146/annurev.med.59.053006.104707
13. Miao CG, Xiong YY, Yu H, Zhang XL, Qin MS, Song TW, et al. Critical roles of microRNAs in the pathogenesis of systemic sclerosis: new advances, challenges and potential directions. Int Immunopharmacol (2015) 28:626–33. doi:10.1016/j.intimp.2015.07.042
14. Zhu H, Luo H, Zuo X. MicroRNAs: their involvement in fibrosis pathogenesis and use as diagnostic biomarkers in scleroderma. Exp Mol Med (2013) 45:e41. doi:10.1038/emm.2013.71
15. Babalola O, Mamalis A, Lev-Tov H, Jagdeo J. The role of microRNAs in skin fibrosis. Arch Dermatol Res (2013) 305:763–76. doi:10.1007/s00403-013-1410-1
16. Sing T, Jinnin M, Yamane K, Honda N, Makino K, Kajihara I, et al. microRNA-92a expression in the sera and dermal fibroblasts increases in patients with scleroderma. Rheumatology (2012) 51:1550–6. doi:10.1093/rheumatology/kes120
17. Zhu H, Luo H, Li Y, Zhou Y, Jiang Y, Chai J, et al. MicroRNA-21 in scleroderma fibrosis and its function in TGF-beta-regulated fibrosis-related genes expression. J Clin Immunol (2013) 33:1100–9. doi:10.1007/s10875-013-9896-z
18. Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol (2011) 8:706–13. doi:10.4161/rna.8.5.16154
19. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum (2013) 65:2737–47. doi:10.1002/art.38098
20. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol (1988) 15:202–5.
21. Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol (2009) 563:123–40. doi:10.1007/978-1-60761-175-2_7
22. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res (2013) 41:D808–15. doi:10.1093/nar/gks1094
23. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res (2009) 37:D412–6. doi:10.1093/nar/gkn760
24. Palla G, Derenyi I, Farkas I, Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature (2005) 435:814–8. doi:10.1038/nature03607
25. Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc (2007) 2:2366–82. doi:10.1038/nprot.2007.324
26. Gao SP, Sun HF, Jiang HL, Li LD, Hu X, Xu XE, et al. Loss of TIM50 suppresses proliferation and induces apoptosis in breast cancer. Tumour Biol (2016) 37:1279–87. doi:10.1007/s13277-015-3878-0
27. Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN. MYC-driven pathways in breast cancer subtypes. Biomolecules (2017) 7:E53. doi:10.3390/biom7030053
28. Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med (2016) 22:1321–9. doi:10.1038/nm.4213
29. Pereira L, Horta S, Mateus R, Videira MA. Implications of Akt2/Twist crosstalk on breast cancer metastatic outcome. Drug Discov Today (2015) 20:1152–8. doi:10.1016/j.drudis.2015.06.010
30. Pignataro L, Sambataro G, Pagani D, Pruneri G. Clinico-prognostic value of D-type cyclins and p27 in laryngeal cancer patients: a review. Acta Otorhinolaryngol Ital (2005) 25:75–85.
31. Dorr C, Janik C, Weg M, Been RA, Bader J, Kang R, et al. Transposon mutagenesis screen identifies potential lung cancer drivers and CUL3 as a tumor suppressor. Mol Cancer Res (2015) 13:1238–47. doi:10.1158/1541-7786.MCR-14-0674-T
32. Leaf IA, Nakagawa S, Johnson BG, Cha JJ, Mittelsteadt K, Guckian KM, et al. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J Clin Invest (2017) 127:321–34. doi:10.1172/JCI87532
33. Wang C, Zhou X, Li W, Li M, Tu T, Ba X, et al. Macrophage migration inhibitory factor promotes osteosarcoma growth and lung metastasis through activating the RAS/MAPK pathway. Cancer Lett (2017) 403:271–9. doi:10.1016/j.canlet.2017.06.011
34. Podnos A, Clark DA, Erin N, Yu K, Gorczynski RM. Further evidence for a role of tumor CD200 expression in breast cancer metastasis: decreased metastasis in CD200R1KO mice or using CD200-silenced EMT6. Breast Cancer Res Treat (2012) 136:117–27. doi:10.1007/s10549-012-2258-3
35. Damiani D, Tiribelli M, Raspadori D, Sirianni S, Meneghel A, Cavalllin M, et al. Clinical impact of CD200 expression in patients with acute myeloid leukemia and correlation with other molecular prognostic factors. Oncotarget (2015) 6:30212–21. doi:10.18632/oncotarget.4901
36. Mulligan-Kehoe MJ, Simons M. Current concepts in normal and defective angiogenesis: implications for systemic sclerosis. Curr Rheumatol Rep (2007) 9:173–9. doi:10.1007/s11926-007-0013-2
37. Heldin CH, Lennartsson J, Westermark B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J Intern Med (2017) 283(1):16–44. doi:10.1111/joim.12690
38. Michie AM, Nakagawa R. The link between PKCalpha regulation and cellular transformation. Immunol Lett (2005) 96:155–62. doi:10.1016/j.imlet.2004.08.013
39. Zeng P, Wang YH, Si M, Gu JH, Li P, Lu PH, et al. Tetraspanin CD151 as an emerging potential poor prognostic factor across solid tumors: a systematic review and meta-analysis. Oncotarget (2017) 8:5592–602. doi:10.18632/oncotarget.13532
40. Leask A. Matrix remodeling in systemic sclerosis. Semin Immunopathol (2015) 37:559–63. doi:10.1007/s00281-015-0508-2
41. Zhou X, Tan FK, Guo X, Arnett FC. Attenuation of collagen production with small interfering RNA of SPARC in cultured fibroblasts from the skin of patients with scleroderma. Arthritis Rheum (2006) 54:2626–31. doi:10.1002/art.21973
42. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans (2017) 45:229–36. doi:10.1042/BST20160387
43. Hung JY, Yen MC, Jian SF, Wu CY, Chang WA, Liu KT, et al. Secreted protein acidic and rich in cysteine (SPARC) induces cell migration and epithelial mesenchymal transition through WNK1/snail in non-small cell lung cancer. Oncotarget (2017) 8:63691–702. doi:10.18632/oncotarget.19475
44. Guo S, Xue Y, He Q, He X, Guo K, Dong P, et al. Preoperative serum cystatin-C as a potential biomarker for prognosis of renal cell carcinoma. PLoS One (2017) 12:e0178823. doi:10.1371/journal.pone.0178823
45. Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Phosphatidylinositol 3-kinase is involved in alpha2(I) collagen gene expression in normal and scleroderma fibroblasts. J Immunol (2004) 172:7123–35. doi:10.4049/jimmunol.172.11.7123
46. Bergmann C, Distler JH. Canonical Wnt signaling in systemic sclerosis. Lab Invest (2016) 96:151–5. doi:10.1038/labinvest.2015.154
47. Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: from laboratory to patients. Cancer Treat Rev (2017) 59:93–101. doi:10.1016/j.ctrv.2017.07.005
48. Li J, Ji L, Chen J, Zhang W, Ye Z. Wnt/beta-Catenin signaling pathway in skin carcinogenesis and therapy. Biomed Res Int (2015) 2015:964842. doi:10.1155/2015/964842
49. Sasaki T, Hiroki K, Yamashita Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed Res Int (2013) 2013:546318. doi:10.1155/2013/546318
50. Shen W, Tao GQ, Zhang Y, Cai B, Sun J, Tian ZQ. TGF-beta in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci (2017) 7:39. doi:10.1186/s13578-017-0168-0
51. Kim CG, Lee H, Gupta N, Ramachandran S, Kaushik I, Srivastava S, et al. Role of Forkhead Box Class O proteins in cancer progression and metastasis. Semin Cancer Biol (2017) S1044-579X:30092–5. doi:10.1016/j.semcancer.2017.07.007
52. Liu ZH, Dai XM, Du B. Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol Ther (2015) 16:353–9. doi:10.1080/15384047.2015.1016662
53. Wang J, Xu L, Liu Y, Chen J, Jiang H, Yang S, et al. Expression of cyclin kinase subunit 2 in human breast cancer and its prognostic significance. Int J Clin Exp Pathol (2014) 7:8593–601.
54. Weng AP, Aster JC. No T without D3: a critical role for cyclin D3 in normal and malignant precursor T cells. Cancer Cell (2003) 4:417–8. doi:10.1016/S1535-6108(03)00305-2
55. Ohura N, Yamamoto K, Ichioka S, Sokabe T, Nakatsuka H, Baba A, et al. Global analysis of shear stress-responsive genes in vascular endothelial cells. J Atheroscler Thromb (2003) 10:304–13. doi:10.5551/jat.10.304
56. Guo H, Majmudar G, Jensen TC, Biswas C, Toole BP, Gordon MK. Characterization of the gene for human EMMPRIN, a tumor cell surface inducer of matrix metalloproteinases. Gene (1998) 220:99–108. doi:10.1016/S0378-1119(98)00400-4
57. Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer (2011) 2:344–58. doi:10.1177/1947601911411084
58. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med (2010) 49:1603–16. doi:10.1016/j.freeradbiomed.2010.09.006
59. Ozaki T, Nakagawara A. Role of p53 in cell death and human cancers. Cancers (Basel) (2011) 3:994–1013. doi:10.3390/cancers3010994
60. Devoy A, Soane T, Welchman R, Mayer RJ. The ubiquitin-proteasome system and cancer. Essays Biochem (2005) 41:187–203. doi:10.1042/EB0410187
61. Pencik J, Pham HT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, et al. JAK-STAT signaling in cancer: from cytokines to non-coding genome. Cytokine (2016) 87:26–36. doi:10.1016/j.cyto.2016.06.017
62. Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle (2006) 5:824–8. doi:10.4161/cc.5.8.2685
63. Xu XD, Shen HB, Zhu L, Lu JQ, Zhang L, Luo ZY, et al. Anti-RhoC siRNAs inhibit the proliferation and invasiveness of breast cancer cells via modulating the KAI1, MMP9, and CXCR4 expression. Onco Targets Ther (2017) 10:1827–34. doi:10.2147/OTT.S93164
64. Nieto Gutierrez A, McDonald PH. GPCRs: emerging anti-cancer drug targets. Cell Signal (2017) 41:65–74. doi:10.1016/j.cellsig.2017.09.005
65. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi:10.3389/fphar.2017.00561
66. Chen Y, Zhang Y, Guo X. Proteasome dysregulation in human cancer: implications for clinical therapies. Cancer Metastasis Rev (2017) 36:703–16. doi:10.1007/s10555-017-9704-y
67. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol (2018) 37:20–36. doi:10.1080/08830185.2017.1380200
68. Randone SB, Guiducci S, Cerinic MM. Systemic sclerosis and infections. Autoimmun Rev (2008) 8:36–40. doi:10.1016/j.autrev.2008.07.022
69. Goldszmid RS, Dzutsev A, Trinchieri G. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe (2014) 15:295–305. doi:10.1016/j.chom.2014.02.003
70. Zeineddine N, Khoury LE, Mosak J. Systemic sclerosis and malignancy: a review of current data. J Clin Med Res (2016) 8:625–32. doi:10.14740/jocmr2606w
71. Xue X, Liu Y, Wang Y, Meng M, Wang K, Zang X, et al. MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget (2016) 7:84508–19. doi:10.18632/oncotarget.13022
72. Tahiri A, Leivonen SK, Luders T, Steinfeld I, Ragle Aure M, Geisler J, et al. Deregulation of cancer-related miRNAs is a common event in both benign and malignant human breast tumors. Carcinogenesis (2014) 35:76–85. doi:10.1093/carcin/bgt333
73. Riccioni R, Lulli V, Castelli G, Biffoni M, Tiberio R, Pelosi E, et al. miR-21 is overexpressed in NPM1-mutant acute myeloid leukemias. Leuk Res (2015) 39:221–8. doi:10.1016/j.leukres.2014.11.001
74. Li Y, Vecchiarelli-Federico LM, Li YJ, Egan SE, Spaner D, Hough MR, et al. The miR-17-92 cluster expands multipotent hematopoietic progenitors whereas imbalanced expression of its individual oncogenic miRNAs promotes leukemia in mice. Blood (2012) 119:4486–98. doi:10.1182/blood-2011-09-378687
75. Ren P, Gong F, Zhang Y, Jiang J, Zhang H. MicroRNA-92a promotes growth, metastasis, and chemoresistance in non-small cell lung cancer cells by targeting PTEN. Tumour Biol (2016) 37:3215–25. doi:10.1007/s13277-015-4150-3
76. Joyce CE, Novina CD. miR-155 in acute myeloid leukemia: not merely a prognostic marker? J Clin Oncol (2013) 31:2219–21. doi:10.1200/JCO.2012.48.3180
77. Sun Y, Bai Y, Zhang F, Wang Y, Guo Y, Guo L. miR-126 inhibits non-small cell lung cancer cells proliferation by targeting EGFL7. Biochem Biophys Res Commun (2010) 391:1483–9. doi:10.1016/j.bbrc.2009.12.098
78. Pekarsky Y, Balatti V, Croce CM. BCL2 and miR-15/16: from gene discovery to treatment. Cell Death Differ (2017) 25(1):21–6. doi:10.1038/cdd.2017.159
79. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis (2000) 21:485–95. doi:10.1093/carcin/21.3.485
80. Houston A, O’Connell J. The Fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol (2004) 4:321–6. doi:10.1016/j.coph.2004.03.008
81. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi:10.1038/nrc1478
82. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal (2013) 11:97. doi:10.1186/1478-811X-11-97
83. Chiariello M, Marinissen MJ, Gutkind JS. Regulation of c-myc expression by PDGF through Rho GTPases. Nat Cell Biol (2001) 3:580–6. doi:10.1038/35078555
84. Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, et al. MYC deregulation in primary human cancers. Genes (2017) 8:E151. doi:10.3390/genes8060151
85. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
86. Bien E, Balcerska A. Serum soluble interleukin 2 receptor alpha in human cancer of adults and children: a review. Biomarkers (2008) 13:1–26. doi:10.1080/13547500701674063
87. Picard C, Belot A. Does type-I interferon drive systemic autoimmunity? Autoimmun Rev (2017) 16:897–902. doi:10.1016/j.autrev.2017.07.001
88. Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front Immunol (2013) 4:266. doi:10.3389/fimmu.2013.00266
89. Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol (2017) 38:542–57. doi:10.1016/j.it.2017.05.005
90. Atanackovic D, Arfsten J, Cao Y, Gnjatic S, Schnieders F, Bartels K, et al. Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood (2007) 109:1103–12. doi:10.1182/blood-2006-04-014480
91. Ciechomska M, van Laar JM, O’Reilly S. Emerging role of epigenetics in systemic sclerosis pathogenesis. Genes Immun (2014) 15:433–9. doi:10.1038/gene.2014.44
92. Zhou B, Zuo XX, Li YS, Gao SM, Dai XD, Zhu HL, et al. Integration of microRNA and mRNA expression profiles in the skin of systemic sclerosis patients. Sci Rep (2017) 7:42899. doi:10.1038/srep42899
93. Pfeffer SR, Yang CH, Pfeffer LM. The role of miR-21 in Cancer. Drug Dev Res (2015) 76:270–7. doi:10.1002/ddr.21257
94. Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther (2017) 19:144. doi:10.1186/s13075-017-1331-z
95. Christmann RB, Wooten A, Sampaio-Barros P, Borges CL, Carvalho CR, Kairalla RA, et al. miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther (2016) 18:155. doi:10.1186/s13075-016-1054-6
96. Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ (2010) 17:215–20. doi:10.1038/cdd.2009.69
Keywords: systemic sclerosis, cancer, gene expression, miRNA, gene module, protein–protein interaction network
Citation: Dolcino M, Pelosi A, Fiore PF, Patuzzo G, Tinazzi E, Lunardi C and Puccetti A (2018) Gene Profiling in Patients with Systemic Sclerosis Reveals the Presence of Oncogenic Gene Signatures. Front. Immunol. 9:449. doi: 10.3389/fimmu.2018.00449
Received: 08 November 2017; Accepted: 20 February 2018;
Published: 06 March 2018
Edited by:
Raffaele De Palma, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyReviewed by:
Serena Vettori, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyCopyright: © 2018 Dolcino, Pelosi, Fiore, Patuzzo, Tinazzi, Lunardi and Puccetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Lunardi, Y2xhdWRpby5sdW5hcmRpQHVuaXZyLml0
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.