 Kyle E. McElroy1,2*†
Kyle E. McElroy1,2*† Laura Bankers2†
Laura Bankers2† Deanna Soper3†Gery Hehman2Jeffrey L. Boore4†John M. Logsdon Jr.2†
Deanna Soper3†Gery Hehman2Jeffrey L. Boore4†John M. Logsdon Jr.2† Maurine Neiman2,5†
Maurine Neiman2,5†- 1Department of Ecology, Evolution, and Organismal Biology, Iowa State University, Ames, IA, United States
- 2Department of Biology, The University of Iowa, Iowa City, IA, United States
- 3Department of Biology, University of Dallas, Dallas, TX, United States
- 4Institute for Systems Biology, Providence St. Joseph Health, Seattle, WA, United States
- 5Department of Gender, Women’s, and Sexuality Studies, The University of Iowa, Iowa City, IA, United States
Why sexual reproduction is so common when asexual reproduction should be much more efficient and less costly remains an open question in evolutionary biology. Comparisons between otherwise similar sexual and asexual taxa allow us to characterize the genetic architecture underlying asexuality, which can, in turn, illuminate how this reproductive mode transition occurred and the mechanisms by which it is maintained or disrupted. Here, we used transcriptome sequencing to compare patterns of ovarian gene expression between actively reproducing obligately sexual and obligately asexual females from multiple lineages of Potamopyrgus antipodarum, a freshwater New Zealand snail characterized by frequent separate transitions to asexuality and coexistence of otherwise similar sexual and asexual lineages. We also used these sequence data to evaluate whether population history accounts for variation in patterns of gene expression. We found that source population was a major source of gene expression variation, and likely more influential than reproductive mode. This outcome for these common garden-raised snails is strikingly similar to earlier results from field-collected snails. While we did not identify a likely set of candidate genes from expression profiles that could plausibly explain how transitions to asexuality occurred, we identified around 1,000 genes with evidence of differential expression between sexual and asexual reproductive modes, and 21 genes that appear to exhibit consistent expression differences between sexuals and asexuals across genetic backgrounds. This second smaller set of genes provides a good starting point for further exploration regarding a potential role in the transition to asexual reproduction. These results mark the first effort to characterize the causes of asexuality in P. antipodarum, demonstrate the apparently high heritability of gene expression patterns in this species, and hint that for P. antipodarum, transitions to asexuality might not necessarily be strongly associated with broad changes in gene expression.
Introduction
Why sexual reproduction is so common despite profound costs has long been considered one of the most important questions in evolutionary biology (Williams, 1975; Maynard-Smith, 1978). A clear explanation for the nearly ubiquitous maintenance of sexual reproduction is still outstanding (Neiman et al., 2018; Otto, 2020). Understanding the proximate causes and consequences of the rare transitions to asexuality that have been observed will provide key information toward an answer to this question (Neiman et al., 2014; Mau et al., 2021). In particular, characterizing the mechanisms underlying the generation of successful asexual lineages from sexual progenitors is a necessary component of understanding the phylogenetic and geographic distribution of asexual lineages and whether and why certain taxa are more likely to transition to successful asexuality.
Two specific traits must emerge for a particular lineage to produce viable asexual descendants: (1) the ability to produce unreduced gametes, and (2) spontaneous development of gametes (i.e., no need for sperm fertilization to trigger development) (reviewed in Neiman et al., 2014). Because successful transitions to asexuality require both of these traits, a simple genetic basis for unreduced gamete production and spontaneous development will facilitate these transitions, with the logical extreme of a single locus controlling both traits. This phenomenon has been observed in several insect systems, e.g., (Sandrock and Vorburger, 2011; Jaquiéry et al., 2014; Mau et al., 2021), including a single gene responsible for thelytokous parthenogenesis in Apis mellifera capensis (Yagound et al., 2020).
Transcriptomic approaches to uncovering the genetic basis of asexuality have been used extensively in plants (reviewed in Schmidt, 2020). Analogous studies in animals are somewhat less common: RNA-seq and microarrays have been used to compare gene expression between parthenogenetic and cyclically parthenogenetic monogonont rotifers (Hanson et al., 2013) and Daphnia (Ye et al., 2021; Xu et al., 2022), and to evaluate sexual vs. asexual oogenesis in reproductively polyphenetic cotton aphids (Gallot et al., 2012; Liu et al., 2014), and reproductive tissues in sexual vs. asexual species of stick insects (Parker et al., 2019), fishes (Ren et al., 2017; Schedina et al., 2018; Bartoš et al., 2019; Lu et al., 2021) and brine shrimp (Huylmans et al., 2021). The candidate genes that emerged from these studies set the stage for future research aimed at characterizing the mechanistic basis of asexual reproduction. For example, several of these animal studies revealed evidence for suppression of meiosis via downregulation of meiotic genes in asexuals (e.g., Gallot et al., 2012; Schedina et al., 2018; Parker et al., 2019; Ye et al., 2021; Xu et al., 2022), despite notable across-taxa variation in the mechanisms underlying asexuality and hybrid vs. non-hybrid origins. Such commonality suggests that RNA-seq should be a useful tool for detecting changes in gene expression related to the production of unreduced games, but nevertheless leaves largely unanswered questions regarding the nature of spontaneous development of unfertilized eggs in asexuals [but see Parker et al. (2019) for possible gene expression changes in asexual female reproductive tract sensory neurons relative to sexual counterparts]. These studies also demonstrate that relatively few genes may be required for transitions from sexual to asexual reproduction, even when multilocus control of asexuality is revealed (e.g., Ye et al., 2021). More broadly, because gene expression is an important determinant of phenotype and evolutionary trajectory (Stajic and Jansen, 2021), characterizing gene expression in asexuals relative to closely related sexuals will provide important insights into the genomic and phenotypic consequences of asexuality.
The New Zealand freshwater snail, Potamopyrgus antipodarum, is a powerful model for the study of sexual reproduction because otherwise similar obligately sexual diploid and obligately asexual triploid and tetraploid individuals frequently coexist within natural populations, enabling direct comparisons between sexual and asexual reproduction (Lively, 1987; Neiman et al., 2011). Several different lines of genetic and genomic evidence (e.g., Phillips and Lambert, 1989; Hauser et al., 1992; Million et al., 2021) indicate that asexual P. antipodarum are apomictic and lack meiosis, though formal cytogenetic analysis still awaits. While the mechanisms underlying ploidy elevation have also not been characterized, phylogenetic (Neiman et al., 2011) and population genetic data (Paczesniak et al., 2013) and the fact that asexual P. antipodarum readily engage in copulation (e.g., (Neiman et al., 2005) point toward the likelihood that ploidy increases might be a consequence of occasional fertilization of unreduced eggs (Neiman et al., 2011). Regardless of the particulars involved, it is evident that reproductive mode is genetically determined in P. antipodarum (Phillips and Lambert, 1989). Both population genetic (Dybdahl and Lively, 1995; Paczesniak et al., 2013) and phylogenetic information (Neiman and Lively, 2004; Neiman et al., 2011; Paczesniak et al., 2013; McElroy et al., 2021) indicate that asexual P. antipodarum have been derived from sexual conspecifics on multiple separate occasions. There is marked population structure in P. antipodarum (Paczesniak et al., 2013), and asexual P. antipodarum are usually more closely related to sympatric sexuals than to asexuals from other lakes (Dybdahl and Lively, 1995; Paczesniak et al., 2013), demonstrating that these transitions to asexuality are frequent and often recent. The implications are that we can treat these different populations of coexisting sexual and asexual P. antipodarum as powerful replicated natural experiments into the maintenance of sex and consequences of asexuality.
Characterizing how asexuality evolves and functions in P. antipodarum will be valuable to the field of reproductive biology because the asexual lineages are somewhat unusual in that they are derived from obligate sexual lineages without hybridization. Most asexual animal and plant lineages have hybrid origins, including nearly all vertebrate unisexual and parthenogenetic taxa (reviewed in Neaves and Baumann, 2011; Fujita et al., 2020). Unlike many other systems in which the genetic mechanisms, or at least strong candidate genes, underlying the transition from sexual to asexual reproduction have been identified, sexual P. antipodarum are not known to harbor some capability for asexual reproduction (e.g., cyclical to obligate asexuality, obligate asexuality in haploid-diploid systems) (Neiman et al., 2014). Indeed, how these transitions from obligate sexual to obligate asexual transitions occur is almost completely unclear, meaning our results will be especially valuable for comparative studies on the genetics of parthenogenesis. Finally, P. antipodarum represents one of the only molluscan species that is polymorphic for sexual vs. asexual reproduction. Two other freshwater species of snail, Melanoides tuberculata (Ben-Ami and Heller, 2005; Dagan et al., 2013) and Campeloma limum (Johnson, 2000) are the only comparable molluscan systems of which we are aware; neither of these species have ever been studied from the perspective of the genetic mechanisms underlying transitions to asexual reproduction. Our focus here on P. antipodarum therefore represents a novel foray into how obligate asexuality is achieved in a mollusk and expands the taxonomic range of natural systems with comparative data for sexual vs. asexual gene expression.
Here, we used RNA-seq to take critical first steps toward characterizing gene expression differences between sexual and asexual P. antipodarum and identifying candidate genes associated with transitions to asexuality. The strategy of sequencing RNA from the ovaries of reproductively active female P. antipodarum was based on the expectation that this tissue would express genes related to ova development, which should be a key process during which sexual and asexual reproduction differ. We also used these RNA-seq data to perform FST-based analyses aimed at evaluating possible connections between genetic and expression divergence. Similar to the expectations for expression, we predicted that candidate genes with functions connected to the causes and consequences of asexuality in P. antipodarum should feature relatively high genetic differentiation between sexual and asexual P. antipodarum.
While P. antipodarum is a compelling natural system to explore parallel vs. unique genetic bases for transitions to asexuality, the availability of snails from our common garden setting that were actively reproducing during the time of RNA isolation prevented us from acquiring the diverse set of sexual and asexual lineages that we would need to formally sort out lineage-specific vs. general expression patterns for asexuality. This limitation imposed by availability of these often difficult-to-culture snails means that this work must be seen as a preliminary rather than definitive evaluation of global patterns of gene expression in sexual vs. asexual P. antipodarum. Because our dataset did include lineages isolated from multiple different lake populations, we were also able to preliminarily estimate the relative influences of population and reproductive mode on global gene expression profiles and consider whether any genes are expressed differently between sexuals vs. asexuals regardless of source population.
Materials and methods
We generated RNA-seq data from three sexual and three asexual P. antipodarum lineages representing three different natural lake populations (see Supplementary Figure 1). Each lineage was descended from a single adult female snail originally collected from the South Island New Zealand lakes Alexandrina (2 sexual lineages and 1 asexual lineage), Sarah (1 asexual lineage), and Selfe (1 sexual lineage and 1 asexual lineage) in 2009–2010. We chose these lineages because each harbored an adequate number of reproductively active females (see below) and, to the extent possible, were from lakes for which we had available both sexual and asexual lineages. Because there is strong evidence that the degree of genetic differentiation in P. antipodarum is correlated with geographical distance among populations (Neiman and Lively, 2004; Paczesniak et al., 2013), sampling from these lakes would also allow for comparisons between geographically distant (Sarah-Alexandrina, separated by ∼255 km, and Selfe-Alexandrina, separated by ∼230 km) and geographically close (Sarah and Selfe, separated by ∼22 km) populations.
Asexual P. antipodarum are polyploid and can be either triploid or tetraploid (sexuals are diploid) (Wallace, 1992; Neiman et al., 2011). Only asexual lineages verified as triploid via flow cytometry (following Krist et al., 2014) were used in this study. The lineages were previously established and ploidy determined in Neiman et al. (2012) and Larkin et al. (2016). All lineages had spent at least 2–3 generations in the laboratory prior to dissection and data collection and all snails used for RNA-seq were born and raised in the same laboratory under standard P. antipodarum rearing conditions (e.g., Zachar and Neiman, 2013) until RNA isolation.
RNA extraction and sequencing
Female P. antipodarum are ovoviviparous (Winterbourn, 1970). We determined whether female snails were reproductively active by either observing embryos through the shell (only possible in light-shelled lineages) or by dissecting the female and opening the brood pouch. Only the ovaries from females that contained developing offspring were extracted. We experienced major challenges in finding enough reproductively active sexual females (a common challenge in laboratory-raised P. antipodarum), which constrained our ability to replicate within and across sexual lineages. All dissections were performed in RNAlater (Thermo Fisher Scientific, Waltham, MA, United States). The dissected ovaries were submerged in 150 μL of RNAlater (Sigma-Aldrich) in a sterile microfuge tube, incubated at 4°C for 24 h to allow the RNAlater to saturate tissue, and then stored at −80°C until RNA extraction. P. antipodarum ovaries are so small (<0.01 mm3 in volume) that adequate RNA isolation for RNA sequencing at the time of data collection in 2014 required pooling of ovarian tissue from at least six females per sample. Accordingly, for each sample other than the Selfe sexuals, ovaries from six females from the same lineage were pooled into a sample for RNA sequencing, with two biological replicates/lineage (12 females total for each lineage). For the Selfe sexuals, the lack of reproductively active females meant that we were only able to sequence one pool of six females.
We homogenized ovary tissue in Trizol reagent (Invitrogen, Carlsbad, CA, United States) using a TissueLyser II (Qiagen, Valencia, CA, United States) with a 5-mm bead and 2 rounds of shaking at 20 cycles/sec. for 2 min. Following the addition of 0.2 volumes of chloroform, we used Phase Lock Gel (5PRIME, Gaithersburg, MD, United States) to achieve phase separation. Next, we isolated total RNA from the aqueous phase with RNA Clean & Concentrator-5 columns (Zymo Research, Irvine, CA, United States) and treated this RNA with Dnase I (Qiagen) to remove contaminating DNA. We then quantified total RNA by fluorometry with Ribogreen (Invitrogen) on a Turner TBS-380 (Promega, Madison, WI, United States), and we assayed RNA quality with an Experion (Bio-Rad, Hercules, CA, United States).
We then used the Illumina TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, United States) to generate each RNA sequencing library from 1 μg of total RNA and sequenced this RNA on an Illumina HiSeq 2000 (2 × 100 bp). This sequencing effort resulted in a mean (±SD) number of reads per run of 28,610,000 (±16,410,000).
Transcriptome assembly and annotation
We used the Fastx-toolkit (Gordon and Hannon, 2010; version 0.0.14) to filter out low-quality reads by retaining only those in which at least 75% of bases had PHRED scores ≥ 20 (mean ± SD of reads following filtering was 27,700,000 ± 15,340,000) and assessed the quality of the reads with FastQC (Andrew, 2010; version 0.10.1). Next, we used Trinity (Grabherr et al., 2011; Hass et al., 2013; version 20131110) for in silico normalization of each read pair (maximum read coverage of 30 and otherwise default settings) to reduce memory use and improve computation ease for de novo assembly. We then pooled the normalized reads from all of the lineages into one assembly using a group pair distance of 350 and otherwise default settings. Following de novo assembly, we used the Trinity plug-in tool TransDecoder (Hass et al., 2013) to identify the most likely coding sequences of the transcriptome and filter out misassembled transcripts. Next, we used CD-HIT-EST (Huang et al., 2010; version 4.6.4) to hierarchically cluster contigs based on 95% identity to reduce redundancy in our transcriptome. Finally, to identify possible contaminant sequences, we used the Blobology (Kumar et al., 2013) pipeline script ‘blast_taxonomy_report.pl’ to assign each transcript to a kingdom and domain based on BLASTX results and removed any non-metazoan transcripts. We annotated the final transcriptome using BLASTX via Blast+ (Camacho et al., 2009; version 2.6.0) against the NCBI non-redundant protein database with an E-value cutoff of 1e-5. Finally, we used Blast2GO (Conesa et al., 2005; version 3) to assign gene ontology (GO) terms, InterPro IDs, and KEGG pathways to each transcript.
Gene expression analysis
We used the final version of the ovary transcriptome as a reference for differential gene expression analysis. We mapped each set of reads to this reference using Tophat2 (Kim et al., 2013; version 2.0.9) and compared gene expression with Cuffdiff 2 (Trapnell et al., 2010, 2012; Kim et al., 2013; version 2.1.1) via two approaches. The first gene expression analysis that we conducted, hereafter “sex-asex,” treated each of the five sexual and six asexual sequence samples as biological replicates for the conditions of “sexual” or “asexual,” with the goal of broadly comparing gene expression between sexual and asexual P. antipodarum. This approach was intended to have sufficient power for detecting changes in gene expression that discriminated between sexuals and asexuals, with the caveat that we might also inflate the likelihood of bias or error (e.g., detecting false positives and negatives) for differential expression by treating samples from the same lineage as independent data points and with uneven representation between original population sources (e.g., two sexual Alexandrina lineages and no sexual Sarah lineages). Furthermore, this analysis does not account for the possible effect lake of origin could have on gene expression differences. Despite its limitations, this analysis was the best we could do with available snails to take initial steps toward our focal question regarding expression differences between sexual and asexual P. antipodarum.
With this logic and goal in mind, we sought to narrow down our candidate gene field by using Cuffdiff 2 to identify the most consistently differentially expressed sequences between sexual and asexual P. antipodarum, here treating all six laboratory lineages (represented by two sequence samples each, except for the single sample from the sexual Selfe) as independent conditions and conducting 15 pairwise gene expression comparisons, again with a false-discovery rate applied via Benjamini-Hochberg correction for multiple tests (q < 0.05). This approach was intended to reduce the possible noise introduced in the sex-asex differential expression comparison and identify sets of genes broadly relevant to differences in reproductive mode and/or specific to distinct transitions to asexuality. We acknowledge that we lose power in this “pairwise” approach relative to the sex-asex comparison because of low or absent replication per lineage, which prevents us from more confidently characterizing population or lineage-specific changes in gene expression and the addressing the possibility of distinct transcriptional signatures of asexuality between different asexual lineages. We therefore relied on these pairwise results primarily for global expression profiles to evaluate the roles of population history and reproductive mode on gene expression and aid our effort to identify candidate genes by highlighting the genes with minimal expression variation outside of the sexual vs. asexual comparison. We similarly used CuffDiff 2 to compare gene expression between the two biological replicates for each of the five lineages for which we had replicated sequence data to assess within-lineage variability. The main caveat that applied to this analysis is that these particular CuffDiff 2 comparisons lack replication (e.g., one sequenced sample from the asexual Alexandrina lineage vs. another sample from the same lineage). Nevertheless, this comparison appears to demonstrate consistent expression between each set of replicates (Supplementary Figure 2) relative to much higher variability across lineages. This result indicates that the lack of replication for the sexual Selfe lineage may not have strongly affected our ability to make comparisons between this lineage and the five lineages for which we did have biological replicates.
To identify transcripts that are particularly likely to be directly associated with sexual vs. asexual reproductive mode, we searched GO annotations to identify 702 genes with putative roles in (sexual) reproduction, oogenesis, embryogenesis, meiosis, germline, ovaries, female-specific functions, mitosis, chromosomes, and cell cycle, hereafter referred to as “focal sequences” for subsequent analyses. This collection of sequences is far from comprehensive and was used as a means to test for potential expression differences that were not captured with the GO-enrichment analysis of the sequences differentially expressed between sexual and asexual snails. For example, a greater proportion of this “focal sequence” set may be differentially expressed between sexuals and asexual than the rest of the transcriptome. To address this possibility, we also collected 702 transcripts for which we used a random number generator to select from the transcriptome as a whole as a control set (both sets of transcripts are listed in Supplementary Tables 5, 6). We used a chi-square test to determine if the “focal” and “control” sets of genes had different proportions of differentially expressed genes (“significant” vs. “non-significant”) in the sex-asex expression analysis results.
Genetic differentiation
We used FST analyses to determine relative levels of genetic differentiation among lineages and to examine within vs. among-lineage differentiation. We also used outlier analyses to identify genes with consistently high differentiation in sexual vs. asexual comparisons. We first filtered the ovary transcriptome to the 11,428 transcripts that were expressed (FPKM > 0) by all six lineages. We then again normalized read counts of each read pair with Trinity’s in silico read normalization to a maximum read coverage of 60 to ease memory demands for downstream analyses and use comparable coverage depths for SNP analysis; we otherwise used default settings. Next, we used Tophat2 (with the same parameters as in the gene expression analyses) to map reads from each lineage to this reduced reference assembly. We then used Picard Tools1 (version 1.99) and elements of the GATK (McKenna et al., 2010; version 3.4) pipeline according to recommendations for using RNA-seq in population genomics (De Wit et al., 2015) and following Bankers et al. (2017) to prepare the BAM files produced by Tophat2 for variant discovery. In short, we used Picard Tools AddOrReplaceReadGroups to tag reads with sample identifiers and MarkDuplicates to identify and remove duplicated reads. We then used the GATK SplitNCigarReads to reassign mapping qualities and then realigned indels with RealignerTargetCreator and IndelRealigner. Next, we used SAMtools (Li et al., 2009; version 0.1.19) to create mpileup files from the processed bam files. From these bam files, we calculated FST for all SNPs using Popoolation2 (Kofler et al., 2011; version 1201) for all pairwise comparisons between lineages and between biological replicates from the same lineage.
To evaluate the effect of lineage and lake of origin on population differentiation we compared mean pairwise FST values from within lineages vs. between lineages and within lakes vs. between lakes with Mann–Whitney U (MWU) tests. We also identified FST outlier SNPs as those SNPs with FST values above the upper 95% confidence interval of the mean FST per SNP for all pairwise comparisons between lineages using GraphPad Prism (version 7.0). We then compared sets of transcripts that contained outlier SNPs across the various pairwise comparisons to identify genes that appear to be consistently differentiated between sexual vs. asexual lineages. We conducted GO enrichment analysis with Blast2GO on sets of transcripts with outlier SNPs in from all pairwise comparisons and identified GO term enrichment for transcripts with outlier FST SNPs in both Alexandrina sexual vs. asexual lineage comparisons and the Selfe sexual vs. asexual lineage comparisons to identify the types of genes, if any, differentiating sexuals and asexuals across multiple populations.
Orthologous sequence identification and phylogenetic analysis
We also used a phylogenetic approach to demonstrate the evolutionary relationships among the six lineages used in this study. First, we assembled transcriptomes for each lineage separately following the same methods described earlier. In brief, we used Trinity to assemble transcripts followed by transdecoder to retain only the coding sequence and CD-HIT-EST to cluster highly similar sequences. Next, we performed reciprocal blastn searches between pairs of transcriptomes with “reciprocalblast_allsteps.py” (Warren et al., 2014) using an E-value cutoff of 1e-20 to identify sequence pairs that were one another’s best hit for each blast search. We then identified sequences from these pairwise comparisons that were reciprocal best blast hits in all 15 comparisons, i.e., 1:1:1:1:1:1 best hits. We considered these 6,757 sequences orthologous and retained only these sequences for further analysis.
We then used MUSCLE (Edgar, 2004; version 3.5) to produce individual alignments for each of the 6,757 orthologous sequence sets. Because we had processed these sequences in TransDecoder, each sequence set was already in frame prior to alignment. Consequently, we kept only alignments in which no gaps were introduced to any sequence during the alignment process for subsequent analysis. Next, we concatenated the 1,225 single-sequence alignments into one file containing 1,135,416 aligned positions for phylogenetic analysis. We then used the 3rd position sites from this alignment within a maximum-likelihood framework and Kimura 2-parameter computed distances to generate an unrooted phylogeny in MEGA (Kumar et al., 2016; version 7) and used 1,000 bootstrap replicates to evaluate phylogeny support.
Results and discussion
We first used Trinity to generate a transcriptome with all our filtered reads, resulting in a transcriptome of 261,666 transcripts. We filtered this transcriptome with TransDecoder, CD-HIT-EST, and Blobology scripts, producing a final reference transcriptome containing 36,754 transcripts (Supplementary Table 1). We annotated our transcriptome by using BLASTX against NCBI’s non-redundant protein database and Blast2GO to assign GO terms to these transcripts. This annotation effort resulted in BLASTX annotations for 23,972 transcripts and GO terms for 21,972 transcripts, for a total of 25,457 annotated transcripts (69.2% of the transcriptome) (see Supplementary Tables 2–4 for Blast2GO results and summaries). We used this annotated transcriptome as a reference to compare gene expression in reproductively active ovaries from sexual vs. asexual P. antipodarum.
Broadly, our analysis of gene expression revealed evidence for lineage, lake of origin, and reproductive mode influences on gene expression in P. antipodarum. Snails derived from the same lake populations tended to have more similar expression profiles than snails from different lakes, regardless of reproductive mode. This observation is consistent with the view that asexuality has arisen from sexual populations independently in each lake (Dybdahl and Lively, 1995; Paczesniak et al., 2013) and that patterns of gene expression are inherited in a phylogenetically consistent manner (Bankers et al., 2017). While our evaluation of differential expression between reproductive modes is preliminary, our results share some notable similarities with other asexual systems, including differential expression relating to metabolic processes and a few genes that appeared to be consistently differentially expressed in sexual vs. asexual lineages involved with chromosome segregation.
Population history seems to be a stronger driver of global gene expression variation in P. antipodarum than reproductive mode
In addition to gene expression analyses, we used these RNA-seq data to evaluate the (1) population genetic differentiation on a per-SNP basis across the transcriptome to determine if lineages from the same lake are more similar genetically than between lakes and (2) phylogenetic history of the sequenced lineages, to address the hypothesis that population history has a strong influence on gene expression, regardless of reproductive mode.
These analyses revealed substantially lower FST values for comparisons within vs. between lineages, with the lowest mean FST/SNP value from an inbred sexual line (0.002723 ± 0.005571; Figure 1A), and the highest mean FST/SNP (∼35× greater than within the inbred sexual line) from the comparison of the asexual Alexandrina lineage and sexual Selfe lineage (0.09754 ± 0.162; Figure 1A). When all within-and between-lineage mean FST/SNP values are rank ordered, a clear pattern of distinctly lower values for within vs. between-lineage differentiation emerges (Supplementary Figure 2; p < 0.025 Bonferroni MWU). We also found that lineages from the same lake tend to be more similar to one another than to lineages from different lakes (Supplementary Figure 2; p < 0.025 Bonferroni MWU). The Alexandrina lineages in particular reveal a clear signal of increasing genetic differentiation across a spectrum of comparisons, with minimum genetic differentiation within lineages, intermediate values of genetic differentiation for Alexandrina sexual lineages, higher (but still intermediate) genetic differentiation for sexual lineages vs. the asexual lineage from Alexandrina, and the highest genetic differentiation for any Alexandria lineage vs. a lineage from a different lake (Figure 1A). We do not see a systematic grouping of the asexual vs. sexual comparisons for mean FST, suggesting that the triploidy of the asexual lineages does not systematically affect genetic differentiation. A good example of the absence of triploid/asexual grouping is provided by the Alexandrina sexual vs. asexual comparisons, which have lower mean FST than Alexandrina sexual vs. Selfe sexual comparisons.
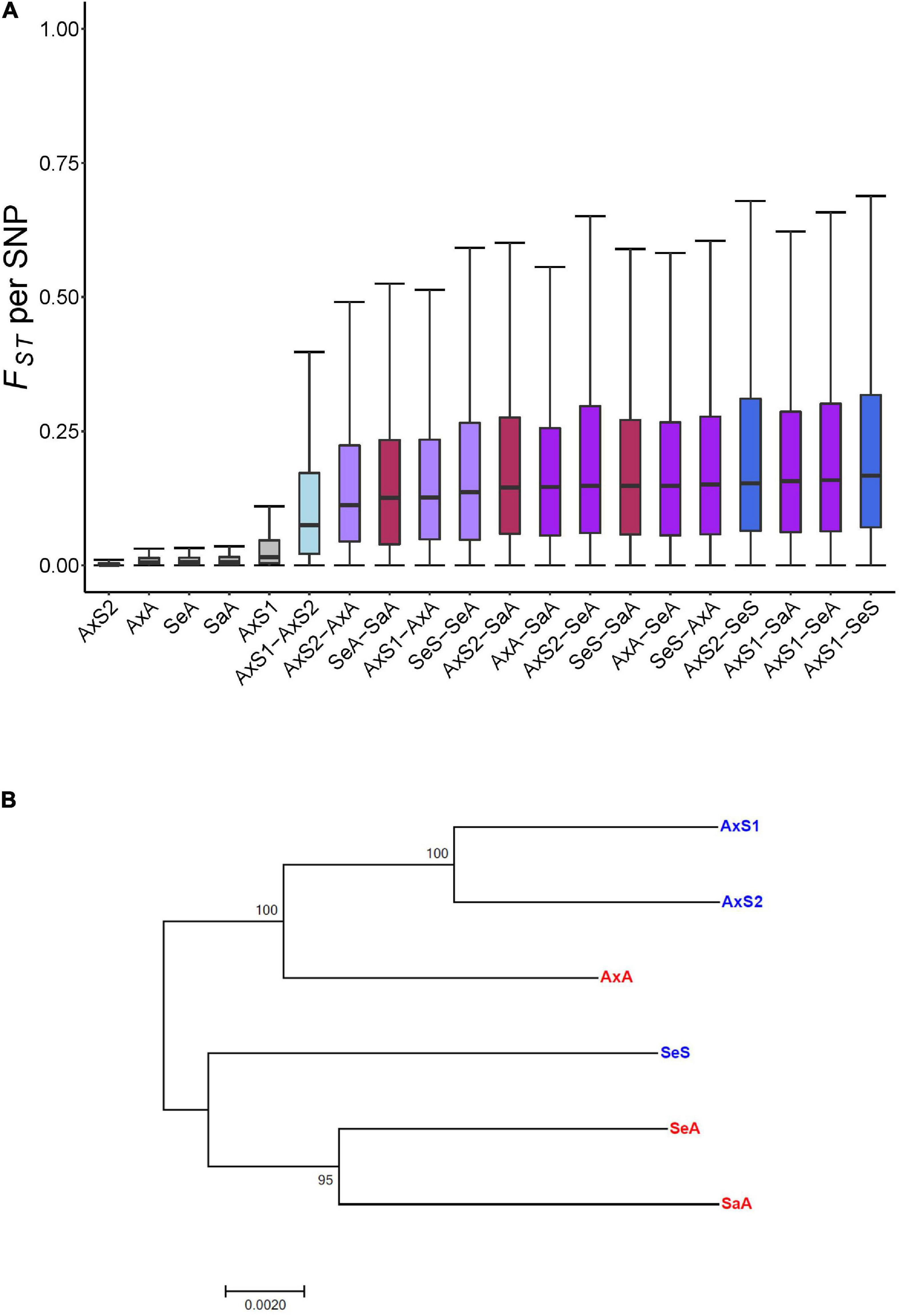
Figure 1. Evolutionary history and relatedness of Potamopyrgus antipodarum lineages in this study. (A) Pairwise comparisons of population differentiation measured by FST per SNP within and between all lineages sequenced. Tukey’s boxplots rank ordered by median FST. Replicates of the same lineage exhibit the lowest amounts of differentiation and across-lake comparisons exhibit the greatest amounts of differentiation. (B) Evolutionary history of the six lineages as inferred by maximum-likelihood method based on the Jukes–Cantor substitution model. Topology with the highest log likelihood (–686934.2673) ratio is shown with 1,000 bootstrap replicate support. 378,472 third codon positions from a concatenation of 1,225 1:1:1:1:1:1 reciprocal best blast hits between separately assembled transcriptomes of each lineage.
Similar results emerged from the phylogenetic approach. We aligned orthologous sequences from lineage-specific transcriptome assemblies and concatenated the gapless alignments to generate a phylogeny for these lineages. We identified 6,757 orthologous sequences that were reciprocal best-blast hits across all six lineages. We aligned these sequences individually and then concatenated the 1,225 gapless sequence alignments into one alignment with 378,472 third codon sites. The resulting phylogeny is very similar to the biogeographic history, with the six lineages grouped into an Alexandrina clade and a Selfe and Sarah clade (Figure 1B). This result is also consistent with our FST analysis: the asexual Selfe and Sarah lineages had the lowest mean FST of any cross-lake comparison (0.0728 vs. 0.0847–0.0975 for others) and was lower than the mean FST from the Selfe sexual vs. the asexual Selfe snails (0.0827). We speculate that the close relationship of the asexual Sarah and Selfe lineages might reflect a recent invasion of Lake Sarah by an asexual lineage from Selfe; Neiman and Lively (2004) found that all 35 sampled snails from Sarah harbored the same recently derived mitochondrial haplotype, which was also found in 19/23 snails collected from Selfe. The very close proximity of these two lakes (22 km as the bird flies) is consistent with this scenario.
The pairwise analysis of gene expression allowed us to perform a variety of comparisons of patterns of gene expression (e.g., sex vs. asex, sex vs. sex, etc.) and identify transcripts consistently differentially expressed between sexual and asexual lineages (FPKM and statistical results from pairwise analysis listed in Supplementary Table 8). Mean proportions of transcripts with differential expression did not differ across sexual vs. sexual or asexual vs. asexual lineage comparisons, sexual vs. asexual lineages, or comparisons of any two lineages (Supplementary Figure 3). With the caveat that these pairwise comparisons necessarily involved some pseudoreplication and will not be able to identify lineage-specific drivers of transitions to asexuality, these results indicate that expression profiles between sexuals and asexuals do not seem to be any more different, in terms of proportion of transcripts differentially expressed, than are any two sexual or asexual lineages. These comparisons also revealed that within-lineage comparisons (i.e., differential expression analysis between biological replicates) were much less likely to reflect differential expression (>14× smaller proportion of transcripts) than any across-lineage comparison (Supplementary Figure 3). While more extensive analyses, including RNA-seq of many individuals from the same lineage raised separately in order to account for culture effects, will be needed to further test the influence of genetic history vs. maternal effects and other shared environmental factors, our results support a scenario where snails from the same lineage tend to express genes at very similar levels to one another relative to snails from other lineages.
A dendrogram-based visualization of overall gene expression profiles revealed that lineages originating from the same lake populations tend to cluster relative to lineages from other lakes (Figure 2A), regardless of reproductive mode. This result suggests that lake of origin is a more important predictor of expression profiles than reproductive mode. We obtained similar results with a multidimensional scaling (MDS) plot (Figure 2B and Supplementary Figure 4), in which the M1 axis groups the three Alexandrina lineages together and splits the sexuals and asexuals on the M2 axis. The M2 axis grouped the gene expression profiles of the Selfe sexual lineage with the Sarah asexual lineage, and the M1 axis separated the Selfe and Sarah lineages from the Alexandrina lineages. Principal component analyses resulted in similar groupings, where PC2 separates these lineages into an Alexandrina group and a Selfe and Sarah group (Figure 2C and Supplementary Figure 5). As noted above, lakes Sarah and Selfe are physically much closer to each other than either lake is to Lake Alexandrina. Altogether, the relationship of these expression profiles is consistent with earlier studies in P. antipodarum suggesting that geographic distance and biogeography are associated with population-level differences in traits (e.g., Verhaegen et al., 2018) including gene expression (e.g., Bankers et al., 2017). Notably, the earlier evidence for population-level clustering of gene expression in P. antipodarum were from field-collected snails (Bankers et al., 2017), while the snails analyzed in this study had been raised in common garden conditions for several generations, suggesting that these field effects persist even under laboratory culture and thus likely represent inherited expression traits. Our results therefore add another layer of understanding of gene regulation in this system.
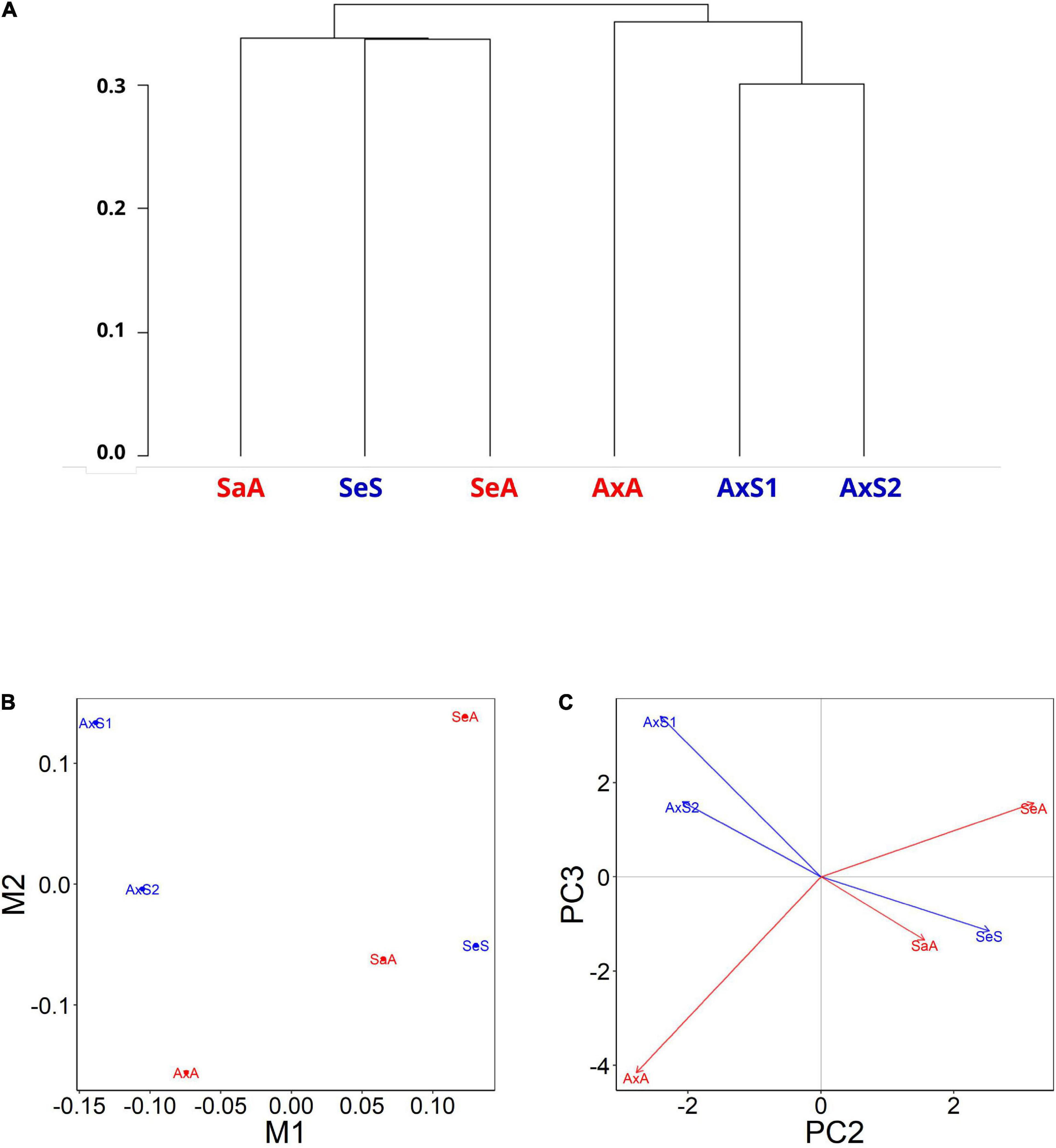
Figure 2. Gene expression profiles are more similar between lineages originating from the same lakes. (A) Dendrogram-based expression patterns from ovaries of three sexual and three asexual lineages of P. antipodarum. (SaA, Sarah, asexual; SeS, Selfe, sexual; SeA, Selfe, asexual; AxA, Alexandrina, asexual; AxS1,2, Alexandrina, sexuals). Generated with the R package CummeRbund. Visualization of multidimensional scaling (MDS) (B) and principal component analysis (PCA) (C) from gene expression values for sexual (blue) and asexual (red) snails. M1 in MDS and PC2 in PCA plots separate the Alexandrina lineages from the Selfe and Sarah. M2 and PC3 both further separate the two sexual Alexandrina lineages from the asexual Alexandrina lineage. MDS and PCA plots generated with R package cummeRbund.
Are gene expression differences related to population genetic differentiation?
We used SNPs with outlier FST values to identify transcripts and gene categories that are significantly differentiated between sexual and asexual P. antipodarum. For this analysis, we used Blast2GO to determine functional enrichment for FST outlier-containing transcripts for all lineage pairwise comparisons. To identify gene classes that might differ according to reproductive mode and to minimize the contribution of lineage effects to this classification effort, we identified GO terms that were overrepresented in sets of transcripts with outlier FST SNPs harbored by both Alexandrina sexual lineages compared to the Alexandrina asexual lineage, while excluding overrepresented terms from the comparison of the two Alexandrina sexual lineages. This collection of 78 GO terms included nine categories related to mitosis, cell cycle, and DNA damage repair and 17 categories related to respiration and mitochondrial functions. The latter group is of interest in light of prior evidence for a higher load of deleterious mutations in the mitochondrial genomes of asexual vs. sexual P. antipodarum (Neiman et al., 2010; Sharbrough et al., 2018). We also conducted GO function enrichment analysis for transcripts with FST outliers for the Selfe sexual vs. asexual lineages and the Selfe sexual lineage vs. the Sarah asexual lineage. This analysis revealed that Selfe sexual vs. asexual transcripts with FST outliers were enriched for 277 GO terms. Similar to the Alexandrina sexual/asexual analysis, while many (e.g., 27/87 cellular component categories) of these overrepresented categories were also associated with mitochondrial function, GO functions related to “single fertilization,” “fertilization,” “zona pellucida receptor complex,” “binding of sperm to zona pellucida,” and “sperm-egg recognition” were also overrepresented for the Selfe sexual/asexual comparison. Finally, 20 GO terms relating to mitosis, cell cycle, and DNA damage repair were overrepresented in the Selfe sexual vs. Sarah asexual FST comparison.
The only GO term categories enriched for transcripts differentially expressed in sexual vs. asexual lineages and transcripts with FST outlier SNPs for sexual vs. asexual comparisons in both groups of lineages (i.e., lineages from Alexandria and ones from Selfe and Sarah) were scavenger receptor activity, cargo receptor activity, and carbohydrate binding. These receptor activities are notable for being part of immune responses and therefore could represent genes that are rapidly evolving in response to pressure from parasites (e.g., Ameline et al., 2021), perhaps not unexpected in this textbook model system for host-parasite coevolution (e.g., Emlen and Zimmer, 2020). Also notable is that P. antipodarum experiences extensive across-population variation in the frequency of parasite infection (e.g., Lively, 1987) as well as population-specific transcriptomic responses to trematode infections (Bankers et al., 2017), meaning that different populations likely experience different selection for immune function.
Chitin-related enzymes were both overrepresented as differentially expressed between sexual and asexual snails in the broad sex-asex comparison and had outlier FST SNPs in comparisons of sexual vs. asexual lineages in lakes Alexandrina and Selfe. “Chitin catabolic process” and “chitinase activity” were also overrepresented GO terms for transcripts with outlier SNPs for the Selfe sexual vs. Sarah asexual FST comparison. Various chitinases are active throughout mollusk development and might play a role in processes from shell development to cell proliferation (Badariotti et al., 2006; Weiss et al., 2006; Schönitzer and Weiss, 2007; Yonezawa et al., 2016), meaning that the differential expression of these genes in sexual and asexual P. antipodarum (and, often, overexpression in the latter) is of particular interest in light of the higher growth rate of asexual snails (Larkin et al., 2016). Both sexual and asexual P. antipodarum are ovoviviparous, featuring internal fertilization followed by internal embryo brooding until birth, so by sampling ovarian tissues of reproductively active snails, we might have also captured expression of early embryos.
The GO terms “chitin binding” and “chitin metabolic process” were also overrepresented in genes differentially expressed between cyclical and obligate parthenogenic monogonont rotifers (Hanson et al., 2013), though this difference between sexual and asexual forms of these rotifers might instead be related to the major differences in egg casing between the sexually produced resting eggs and the eggs produced by asexual females (Hanson et al., 2013). Differences in egg casings in sexual vs. asexual settings may contribute to why chitin binding genes are upregulated in sexual relative to asexual Daphnia similoides (Zhang et al., 2016). It is also possible that the apparently adaptive nature of at least some of the morphological plasticity in shell shape/size in asexual P. antipodarum in the United States and Europe (e.g., Kistner and Dybdahl, 2013; Verhaegen et al., 2018) might also be linked to the expression of chitin-related genes.
We observed SNP FST outliers in two meiosis- related genes, rad51 and msh2, among the three Alexandrina lineages. In particular, we found that for comparisons involving any two Alexandrina lineages, each of these two genes had at least one outlier SNP. For msh2, the two sexuals shared the same outlier SNP when compared with the asexual lineage. This SNP did not appear as an outlier when the two Alexandrina sexual lineages were compared to one another. Neither gene had outlier SNPs for the Selfe sexual vs. asexual lineages or the Selfe sexual vs. Sarah asexual lineage comparisons, and msh2 was not differentially expressed among any of the Selfe lineages. There was no clear pattern or association with reproductive mode for rad51. It is important to note that msh2 and rad51, along with other meiosis-related genes, tended to be expressed at relatively low levels. This low expression likely limits our statistical power to detect significant expression differences in the absence of much deeper coverage than available in our study. While we cannot exclude the possibility that we did not capture enough tissue and/or the correct developmental stage for a rigorous analysis of meiotic gene expression in P. antipodarum, it is relevant to point out that absence of evidence for expression differences in meiosis genes between sexual and asexual lineages have also been reported for obligate vs. cyclical parthenogenetic strains of the monogonont rotifer Brachionus calyciflorus (Hanson et al., 2013) and during meiosis and parthenogenesis in Daphnia pulex (Schurko et al., 2009).
Evidence for little overall difference in sexual vs. asexual gene expression
The sex-asex differential gene expression analysis identified 1,103 transcripts with significantly different (FDR q < 0.05) expression levels between the two reproductive modes. Of these transcripts, 660 (3.39% of the total transcripts tested) were expressed at significantly higher levels by asexual P. antipodarum, and 443 (2.28% of the total transcripts tested) were expressed at significantly higher levels by sexual P. antipodarum (Figure 3A and Supplementary Figure 6). This general pattern of more transcripts with significantly higher expression in asexuals relative to sexuals extended to our pairwise comparisons between sexual and asexual lineages. Here, at lineage-level differential expression comparisons, asexuals overexpressed, on average (±SD), 2293 (±336.5) transcripts and underexpressed, on average, 1897 (±66.4) transcripts relative to sexuals (Figure 3B).
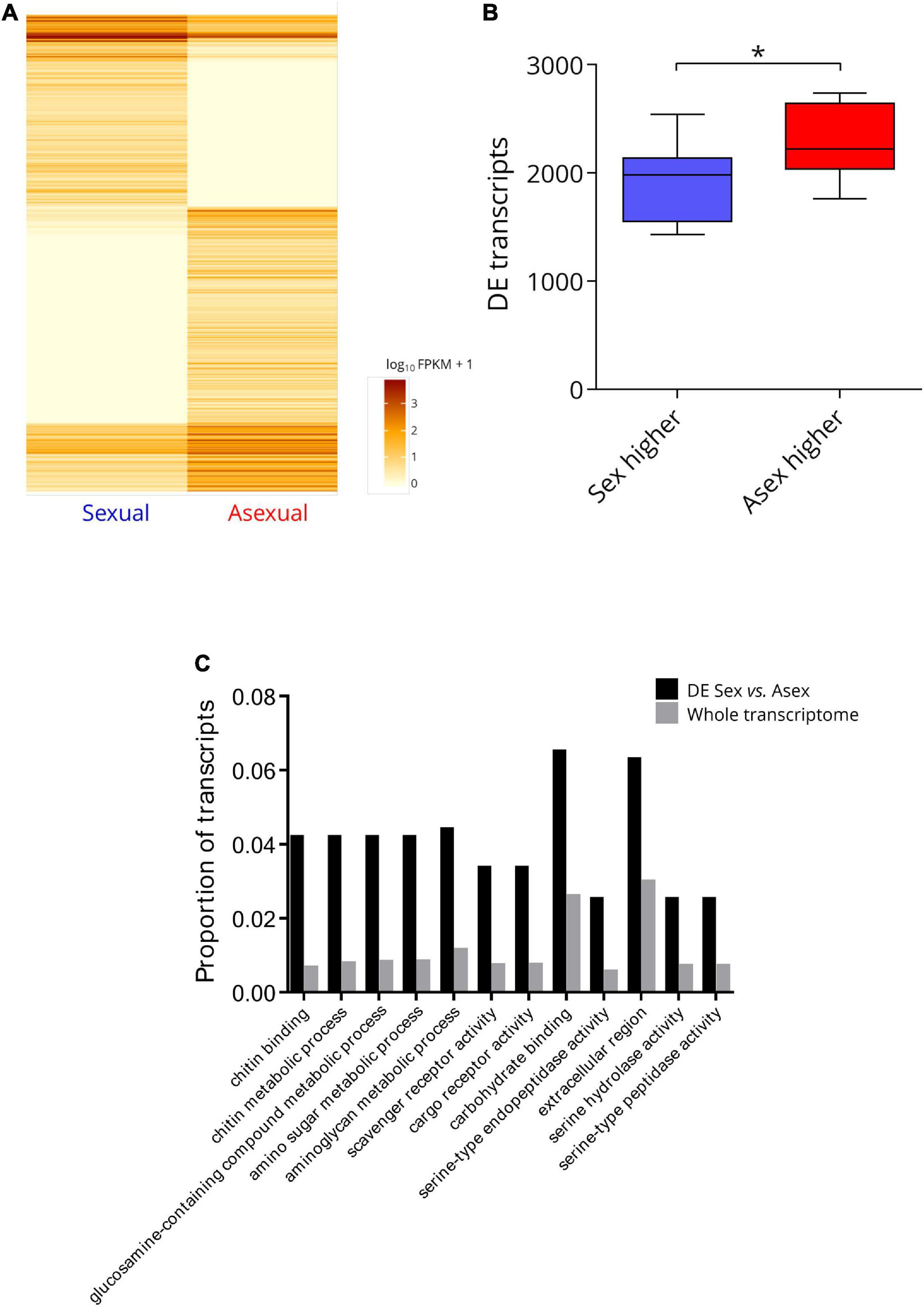
Figure 3. Summary of differentially expression between sexual and asexual P. antipodarum ovaries. (A) Heatmap of significantly differentially expressed transcripts between sexual and asexual P. antipodarum. Darker color indicates higher FPKM values, i.e., higher gene expression. Significance was determined using CuffDiff with an FDR of 5% and Benjamini–Hochberg multiple test correction. “Sexual” represents the data from all three sexual lineages pooled together for this analysis, and “Asexual” represents the data pooled from all three asexual lineages. Heatmap generated with the R package CummeRbund. (B) Asexuals overexpress more transcripts relative to sexuals than vice versa. Differential expression was determined using CuffDiff with an FDR of 5% and Benjamini–Hochberg multiple test correction (*paired t-test p = 0.0014, N = 9 sexual vs. asexual comparisons). (C) GO-term enrichment (Fisher’s Exact test with FDR q < 0.05 in Blast2GO) for transcripts differentially expressed (DE) in the pooled sexual vs. asexual comparison.
We also used the pairwise analysis to assess whether and which transcripts were consistently differentially expressed between sexual and asexual lineages to do the best we could to account for the limitation that our sex-asex comparison could not control for lake or lineage. We narrowed down the pairwise expression results to transcripts only differentially expressed in sexual vs. asexual lineage comparisons, excluding transcripts differentially expressed in any sexual vs. sexual or asexual vs. asexual comparison. This exclusion could inadvertently eliminate results relevant to population or lineage-specific transitions to asexuality, but because we did not generate data sufficient for such characterization, we chose to focus on identifying genes with the strongest evidence for reproductive-mode driven expression differences. This approach enabled us to in part account for the caveats of the sex-asex differential expression analysis (e.g., not structured to consider population history or lineage, which could lead to the appearance of sexual vs. asexual differential expression being driven by lineage-specific overexpression) to determine if common expression differences exist between sexual and asexual P. antipodarum regardless of genetic background. In light of the design limitations with respect to sequencing replication per lineage and the uneven representation of lake populations, we caution that these results are not definitive but instead represent the likeliest set of genes related to reproductive differences that we could identify with the data we generated.
From the pairwise comparisons, we identified 347 sequences only differentially expressed between reproductive modes, and not within reproductive modes. 86 of these sequences (24.8%) were also identified in the sex-asex analysis. Of those 347 sequences, 89 sequences were upregulated by at least two sexual lineages relative to asexuals or at least two asexual lineages relative to sexuals; 60 of those 89 sequences (67.4%) were identified in the sex-asex approach. Of the 23 sequences that were differentially expressed in all sexual vs. asexual pairwise comparisons 21 were identified with the sex-asex approach (91.3%). The increasing degree of overlap between the sex-asex results and specificity of the pairwise results indicate that the pairwise approach was useful in identifying consistently differentially expressed genes in sexual vs. asexual snails. Furthermore, the pairwise results demonstrate that a more thorough consideration for genetic background will need to be taken into account when designing future RNA-seq experiments aimed at uncovering regulatory differences in sexual vs. asexual (and different ploidy levels) gene expression in P. antipodarum.
Similar to Bankers et al. (2017), where almost no transcripts (3/62,862) were consistently differentially expressed between infected and uninfected P. antipodarum across source populations, we see relatively few transcripts consistently differentially expressed in all sexual vs. asexual lineage comparisons (Figure 4). Of those 23 transcripts, 10 were upregulated in sexuals relative to asexuals, and 13 transcripts were upregulated in asexuals relative to sexuals in every comparison (Figure 4, listed in Supplementary Tables 9–14). We applied the SuperExactTest (Wang et al., 2015), which demonstrated that these overlaps in sequences consistently expressed higher in sexuals vs. asexuals and vice versa were larger than expected by chance (10 vs. 0.00180219 in sexuals and 13 vs. 0.0007407894 in asexual; p = 2.031013e-35 and p = 6.984346e-53). Only four of the 10 consistently upregulated transcripts in sexuals were annotated. One of these transcripts was annotated as kinetochore protein spc24, an essential component of the NDC80 complex, which is necessary for chromosome segregation (McCleland et al., 2003). In yeast, the regulation of NDC80 subunits is integral for meiosis I to occur (reviewed in (Chen and Ünal, 2021). Of potential relevance to the P. antipodarum system is that a lineage of baker’s yeast with a unique non-synonymous mutation in NDC80 was also found to have a particularly high incidence of polyploidy (83% vs. 34% of all 144 strained analyzed) (Zhu et al., 2016). Also noteworthy is that one of the 13 transcripts consistently upregulated in asexual relative to sexual lineages is annotated as DNA primase large subunit, which is involved with DNA synthesis and replication. This last result is of interest in light of the polyploid status of asexual P. antipodarum, which have more DNA to replicate during cell division than diploid counterparts. While these examples of genes that are differentially expressed across reproduced modes are of potential relevance to our overarching goals regarding the identification of candidate loci involved in the transition to asexuality, they are not informative enough to construct a model of how asexuality is achieved in P. antipodarum. Characterizing the molecular evolution of these genes across a diverse and representative sample of P. antipodarum sexual and asexual lineages would be a worthwhile next step in evaluating the potential that these candidate loci play a role in the transitions to asexuality.
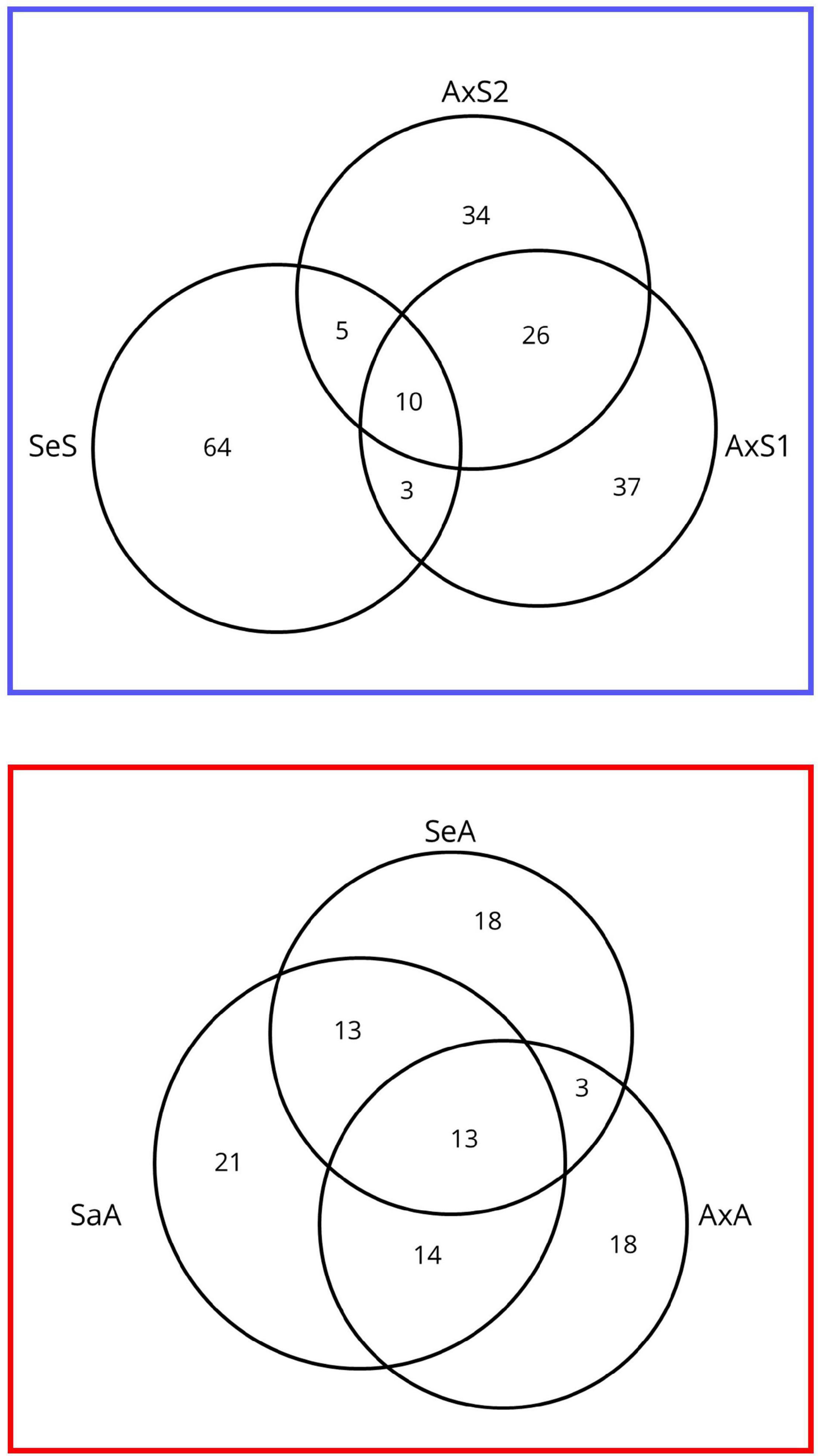
Figure 4. Venn diagram of shared differentially expressed transcripts across lineages from pairwise sexual vs. asexual comparisons of gene expression. (Top) The number of transcripts expressed higher for each sexual lineage relative to asexual lineages in all pairwise comparisons. (Bottom) The number of transcripts expressed higher for each asexual lineage relative to sexual lineages in all pairwise comparisons.
These results are consistent with the apparent lack of systematic differences in gene expression between sexual and asexual reproductively active female P. antipodarum, including our “focal” gene set with GO terms associated with reproduction. We used a chi-square test to evaluate the proportion of differentially expressed genes in the “focal” vs. “control” set of genes from the sex-asex expression analysis. We found that the “focal” genes were less often differentially expressed than the random set of genes (χ2 = 13.3601, p < 0.05). Of the “focal” genes, 0.92% were differentially expressed, compared to the 5.29% in the random set. The random set of genes had a very similar amount of differentially expressed genes detected transcriptome-wide (5.67%). This sampling of genes likely involved with reproduction and cell cycle processes is not exhaustive, but this evidence for relatively few differentially expressed genes from this large set of genes suggests that sexual and asexual P. antipodarum reproduction are not globally different at the level of gene expression. More rigorous evaluation of sexual and asexual gene regulation during reproduction would benefit in identifying genes in P. antipodarum that are upregulated during reproduction or exclusively expressed in ovaries.
Similar results were observed in comparisons of gene expression between sexual and asexual species of Artemia brine shrimp, where only 60 genes were differentially expressed in female gonads (Huylmans et al., 2021). Like brine shrimp, P. antipodarum may experience little reprogramming of reproductive pathways following transitions to asexuality, possibly because asexuality is recently derived in these species (Neiman et al., 2005; Rode et al., 2021) compared to much longer divergence times between sexual and asexual Timema species (Schwander et al., 2011), which have a high degree (∼8% of all genes) demonstrating convergent expression differences between reproductive modes (Parker et al., 2019). Extrapolation from asexual brine shrimp must be done with caution, however, because unlike (to the extent that we can discern) asexual P. antipodarum, asexual brine shrimp reproduce via automixis, in which meiosis occurs. Recent developments regarding the extent of cryptic sex occurring in asexual Artemia also raises the question of whether these lineages can truly be considered to be asexual (Boyer et al., 2021). The likelihood that apparently asexual lineages engage in rare sex is increasing in light of a growing body of evidence for such events, even extending to the bdelloid rotifers (Vakhrusheva et al., 2020; Laine et al., 2022; Paul et al., 2022), (in)famous as the most “ancient” of all asexuals. If long-term asexual persistence relies on occasional sex, then successful asexual lineages could plausibly represent taxa that can still engage in occasional recombination (or recombination-like) processes. To date, there is no evidence for recombinational processes in asexual P. antipodarum, though the occasional production of male offspring by asexual female P. antipodarum (Neiman et al., 2012) hints that this might be possible. On the flip side, that these males produce sperm that have strikingly aberrant morphology compared to sexual counterparts (Jalinsky et al., 2020) suggests that such recombination within asexual lineages might be extremely rare. Similarly, taxa thought to be obligate sexuals that give rise to asexuals may have cryptic asexual reproductive capabilities (i.e., facultative asexuality rather than obligate sexuality) and gradually transition to obligate asexuality, as had been hypothesized in Timema stick insects (Schwander and Crespi, 2009) and more recently supported with genetic evidence (Larose et al., 2022). There is nevertheless no evidence to date for facultative asexuality in P. antipodarum.
Gene expression possibly linked to metabolic and physiological differences in sexual vs. asexual P. antipodarum
Using Blast2GO and Fisher’s Exact test (FDR q < 0.05), we identified 12 GO terms that were significantly overrepresented in the set of transcripts differentially expressed between sexual and asexual lineages (Figure 3C). We did not detect any statistical overrepresentation of GO terms related to reproduction, sex, meiosis/mitosis, chromosome movement, or any specific function we might expect to differ between diploids and triploids in sets of differentially expressed transcripts in sexual vs. asexual comparisons but we did identify 12 GO-terms enriched in differentially expressed genes. Several of these genes were annotated for functions related to metabolism, including the functions of “chitin-metabolic activity,” “glucosamine-containing compound metabolic process,” “amino sugar metabolic process,” and “aminoglycan metabolic process.” Chitin and aminoglycan metabolism were enriched GO terms for DE genes in cyclical vs. obligate parthenogenic rotifers (Hanson et al., 2013). Other RNA-seq studies in asexual vs. sexual animals have also reported metabolic GO terms enriched in sets of differentially expressed genes (Gallot et al., 2012; Liu et al., 2014; Parker et al., 2019). RNA sequencing has revealed evidence for distinct metabolic states, particularly with respect to amino acid metabolism, in sexual vs. asexual stages of flatworms (Sekii et al., 2019). Carter et al. (2012) found evidence for different energy investments between obligate and cyclical asexual aphids that indicate greater investment toward reproduction in obligate asexuals. Reminiscent of these findings, asexual female P. antipodarum grow more rapidly and reach reproductive maturity earlier than sexual females (Larkin et al., 2016). Our results may therefore be indicative of regulatory differences in metabolic processes in sexual vs. asexual P. antipodarum.
A role for polyploidy and future directions
Polyploidy could also contribute to the differential expression observed between sexual and asexual P. antipodarum and enrichment in metabolic processes. Polyploidy and large genome size are also expected to influence standard metabolic rate via increasing cell size (Cavalier-Smith, 1978), as has been observed in polyploid fish (Maciak et al., 2011). It is unclear whether higher-ploidy P. antipodarum actually have larger somatic cells (Neiman et al., 2009), with the exception of sperm cells (Jalinsky et al., 2020). The overwhelming majority of literature on gene expression changes following polyploidization refer to allopolyploids, whereas whether autopolyploidy influences gene expression is not as well studied but is generally not associated with major shifts in gene expression (reviewed in Parisod et al., 2010). Long-term genome reorganization during diploidization following autopolyploidy, however, may be accompanied by significant changes in gene regulation while duplicated sequences diverge (e.g., Gundappa et al., 2022). Despite wide DNA content variation in P. antipodarum asexual polyploids (Neiman et al., 2011; Million et al., 2021), a trend toward DNA reduction in asexuals does not emerge (Million et al., 2021), indicating that polyploidy is maintained. The relatively recent derivation of most asexual lineages of P. antipodarum (Neiman et al., 2005), however, indicates that we might not yet be able to observe major global differences in gene expression between diploids and polyploids. Altogether, whether reproductive mode or ploidy variation is more important to gene regulation-related metabolic and physiological differences in P. antipodarum will need to be carefully evaluated in downstream studies comparing diploid, triploid, and tetraploid snails.
Along these lines, we cannot exclude the possibility that there exist differences in gene expression between sexual and asexual P. antipodarum that went undetected as a function of, for example, relatively small magnitude of differences or as a consequence of our focus on reproductively active females. Other studies have used RNA-seq to compare gene expression in reproductive tissue and other tissues/body regions in sexual and asexual lineages to distinguish the genes that may be most directly related to reproduction (Parker et al., 2019; Huylmans et al., 2021). This approach could also be useful to distinguish between ploidy and reproductive mode driving differences in gene expression. In future efforts to characterize the genetic causes of asexuality in P. antipodarum, improvements in microdissection of ovaries would be particularly useful to more likely capture tissue enriched with active oogenesis, as would characterization of critical time points in reproduction, and particularly, determining where meiosis predominantly occurs (i.e., certain regions of the ovaries).
The discovery of distinguishing features for egg production and development between sexual and asexual P. antipodarum might also provide a powerful means of establishing how to proceed. At this time, we are unaware of any differences in early development or egg formation between sexual and asexual P. antipodarum; profiling gene expression across development may be a useful avenue to explore. While we focused on likely protein-coding genes in this study, it is also possible that non-coding RNAs could play an important role in the differences in sexual and asexual reproduction. Indeed, a central role for non-coding RNAs has been demonstrated in Boechera (Mau et al., 2013), for which the non-coding RNA UPGRADE2 which appears to be responsible for unreduced pollen formation. Finally, we also cannot rule out the possibility that posttranscriptional regulation might be involved with sexual vs. asexual reproduction in P. antipodarum.
Conclusion
Potamopyrgus antipodarum has long been studied as a model system for the maintenance of sexual reproduction. As an emerging model for the genomics of sex, mutation accumulation in mitochondria of asexual relative sexual lineages (Neiman et al., 2010; Sharbrough et al., 2018) and gene copy number increase in asexual relative to sexual lineages (McElroy et al., 2021) have been observed. How the separate and repeated transitions to asexual reproduction for which this system is notable remains uncharacterized. Our results, like several other natural model systems for the maintenance of sex, suggest relatively few consistent gene expression differences between sexuals and asexuals. Among the few differentially expressed sequences are genes related to DNA replication and chromosome movement. The latter is particularly relevant as RNA-seq studies have revealed similar candidates across several systems. Genes differentially expressed between reproductive modes were overrepresented by metabolic gene ontology categories, possibly associated with established and hypothesized physiological differences between sexual and asexual snails. Overall patterns of gene expression, however, were shaped more by lake of origin than reproductive mode, strikingly consistent with the strong population structure in this species (Paczesniak et al., 2013) and previous RNA-seq data from field collected snails (Bankers et al., 2017). In our case, snails had been housed in common-garden conditions for several generations, indicating strong heritability of gene expression. Future efforts to uncover the proximate mechanisms for asexuality in P. antipodarum should sample broadly from its geographic range to better assess whether transitions to asexuality occur via the same or different mechanisms, include tetraploids to comprehensively account for a potential ploidy effect, and investigate gene expression across a variety of tissues to differentiate between genes involved in reproduction and other processes.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRX3560064, SRX3560063, SRX3560062, SRX3560061, SRX3560060, SRX3560059, SRX3560058, SRX3560057, SRX3560056, SRX3560055, SRX3560054, and GGFE00000000.
Author contributions
MN, DS, KM, and LB designed the research. DS, GH, KM, and LB performed the research. KM analyzed the data. KM and MN wrote the manuscript. JB, JL, and MN funded the study. KM, MN, LB, GH, JB, DS, and JL edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was funded by National Science Foundation (NSF MCB grant number 1122176).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Curt Lively, Kayla King, and Dorota Paczesniak for the field collections and Katelyn Larkin for establishing the lineages used. We would also like to thank the Carver College of Medicine’s Flow Cytometry Facility and Roy J. Carver Center for Genomics for their services. We also thank the reviewers, whose thoughtful and rigorous evaluations of our manuscript greatly improved the final version.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.845640/full#supplementary-material
Footnotes
References
Ameline, C., Bourgeois, Y., Vögtli, F., Savola, E., Andras, J., Engelstädter, J., et al. (2021). A two-locus system with strong epistasis underlies rapid parasite-mediated evolution of host resistance. Mol. Biol. Evol. 38, 1512–1528. doi: 10.1093/molbev/msaa311
Andrew, S. (2010). FastQC: A Quality Control Tool For High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed July 6, 2022).
Badariotti, F., Kypriotou, M., Lelong, C., Dubos, M. P., Renard, E., Galera, P., et al. (2006). The phylogenetically conserved molluscan chitinase-like protein 1 (Cg-Clp1), homologue of human HC-gp39, stimulates proliferation and regulates synthesis of extracellular matrix components of mammalian chondrocytes. J. Biol. Chem. 281, 29583–29596. doi: 10.1074/jbc.M605687200
Bankers, L., Fields, P., McElroy, K. E., Boore, J. L., Logsdon, J. M., and Neiman, M. (2017). Genomic evidence for population-specific responses to co-evolving parasites in a New Zealand freshwater snail. Mol. Ecol. 26, 3663–3675. doi: 10.1111/mec.14146
Bartoš, O., Röslein, J., Kotusz, J., Paces, J., Pekárik, L., Petrtýl, M., et al. (2019). The legacy of sexual ancestors in phenotypic variability, gene expression, and homoeolog regulation of asexual hybrids and polyploids. Mol. Biol. Evol. 36, 1902–1920. doi: 10.1093/molbev/msz114
Ben-Ami, F., and Heller, J. (2005). Spatial and temporal patterns of parthenogenesis and parasitism in the freshwater snail Melanoides tuberculata. J. Evol. Biol. 18, 138–146. doi: 10.1111/j.1420-9101.2004.00791.x
Boyer, L., Jabbour-Zahab, R., Mosna, M., Haag, C. R., and Lenormand, T. (2021). Not so clonal asexuals: unraveling the secret sex life of Artemia parthenogenetica. Evol. Lett. 5, 164–174. doi: 10.1002/evl3.216
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Carter, M. J., Simon, J.-C., and Nespolo, R. F. (2012). The effects of reproductive specialization on energy costs and fitness genetic variances in cyclical and obligate parthenogenetic aphids. Ecol. Evol. 2, 1414–1425. doi: 10.1002/ece3.247
Cavalier-Smith, T. (1978). Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J. Cell Sci. 34, 247–278. doi: 10.1242/jcs.34.1.247
Chen, J., and Ünal, E. (2021). Meiotic regulation of the Ndc80 complex composition and function. Curr. Genet. 67, 511–518. doi: 10.1007/s00294-021-01174-3
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dagan, Y., Liljeroos, K., Jokela, J., and Ben-Ami, F. (2013). Clonal diversity driven by parasitism in a freshwater snail. J. Evol. Biol. 26, 2509–2519. doi: 10.1111/jeb.12245
De Wit, P., Pespeni, M. H., and Palumbi, S. R. (2015). SNP genotyping and population genomics from expressed sequences-Current advances and future possibilities. Mol. Ecol. 24, 2310–2323. doi: 10.1111/mec.13165
Dybdahl, M. F., and Lively, C. M. (1995). Diverse, endemic and polyphyletic clones in mixed populations of a freshwater snail (Potamopyrgus antipodarum). J. Evol. Biol. 8, 385–398. doi: 10.1046/j.1420-9101.1995.8030385.x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emlen, D. J., and Zimmer, C. (2020). Evolution: Making Sense of Life, 3rd Edn. New York, NY: W. H. Freeman.
Fujita, M. K., Singhal, S., Brunes, T. O., and Maldonado, J. A. (2020). Evolutionary dynamics and consequences of parthenogenesis in vertebrates. Annu. Rev. Ecol. Evol. Syst. 51, 191–214. doi: 10.1146/annurev-ecolsys-011720-114900
Gallot, A., Shigenobu, S., Hashiyama, T., Jaubert-Possamai, S., and Tagu, D. (2012). Sexual and asexual oogenesis require the expression of unique and shared sets of genes in the insect Acyrthosiphon pisum. BMC Genomics 13:76. doi: 10.1186/1471-2164-13-76
Gordon, A., and Hannon, G. J. (2010). Fastx-toolkit. FASTQ/A Short-Reads Preprocessing Tools (Unpublished). Available online at: http://hannonlab.cshl.edu/fastx_toolkit (accessed July 6, 2022).
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gundappa, M. K., To, T.-H., Grønvold, L., Martin, S. A. M., Lien, S., Geist, J., et al. (2022). Genome-wide reconstruction of rediploidization following autopolyploidization across one hundred million years of salmonid evolution. Mol. Biol. Evol. 39:msab310. doi: 10.1093/molbev/msab310
Hanson, S. J., Stelzer, C. P., Welch, D. B. M., and Logsdon, J. M. (2013). Comparative transcriptome analysis of obligately asexual and cyclically sexual rotifers reveals genes with putative functions in sexual reproduction, dormancy, and asexual egg production. BMC Genomics 14:412. doi: 10.1186/1471-2164-14-412
Hass, B., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., and Bowden, J. (2013). De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hauser, L., Carvalho, G. R., Hughes, R. N., and Carter, R. E. (1992). Clonal structure of the introduced freshwater snail Potamopyrgus antipodarum (Prosobranchia: Hydrobiidae), as revealed by DNA fingerprinting. Proc. R. Soc. London. Ser. B Biol. Sci. 249, 19–25. doi: 10.1098/rspb.1992.0078
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Huylmans, A. K., Macon, A., Hontoria, F., and Vicoso, B. (2021). Transitions to asexuality and evolution of gene expression in Artemia brine shrimp. Proc. R. Soc. B Biol. Sci. 288:20211720. doi: 10.1098/rspb.2021.1720
Jalinsky, J., Logsdon, J. M., and Neiman, M. (2020). Male phenotypes in a female framework: evidence for degeneration in sperm produced by male snails from asexual lineages. J. Evol. Biol. 33, 1050–1059. doi: 10.1111/jeb.13632
Jaquiéry, J., Stoeckel, S., Larose, C., Nouhaud, P., Rispe, C., Mieuzet, L., et al. (2014). Genetic control of contagious asexuality in the Pea Aphid. PLoS Genet. 10:e1004838. doi: 10.1371/journal.pgen.1004838
Johnson, S. G. (2000). Population structure, parasitism, and survivorship of sexual and autodiploid parthenogenetic Campeloma limum. Evolution 54, 167–175. doi: 10.1111/j.0014-3820.2000.tb00017.x
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kistner, E. J., and Dybdahl, M. F. (2013). Adaptive responses and invasion: the role of plasticity and evolution in snail shell morphology. Ecol. Evol. 3, 424–436. doi: 10.1002/ece3.471
Kofler, R., Pandey, R. V., and Schlötterer, C. (2011). PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436. doi: 10.1093/bioinformatics/btr589
Krist, A. C., Kay, A. D., Larkin, K., and Neiman, M. (2014). Response to phosphorus limitation varies among lake populations of the freshwater snail Potamopyrgus antipodarum. PLoS One 9:e85845. doi: 10.1371/journal.pone.0085845
Kumar, S., Jones, M., Koutsovoulos, G., Clarke, M., and Blaxter, M. (2013). Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 4:237. doi: 10.3389/fgene.2013.00237
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Laine, V. N., Sackton, T. B., and Meselson, M. (2022). Genomic signature of sexual reproduction in the bdelloid rotifer Macrotrachella quadricornifera. Genetics 220:iyab221. doi: 10.1093/genetics/iyab221
Larkin, K., Tucci, C., and Neiman, M. (2016). Effects of polyploidy and reproductive mode on life history trait expression. Ecol. Evol. 6, 765–778. doi: 10.1002/ece3.1934
Larose, C., Lavanchy, G., Freitas, S., Parker, D. J., and Schwander, T. (2022). Facultative parthenogenesis in an asexual stick insect: re-evolution of sex or vestigial sexual capacity? bioRxiv [Preprint]. doi: 10.1101/2022.03.25.485836
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, L. J., Zheng, H. Y., Jiang, F., Guo, W., and Zhou, S. T. (2014). Comparative transcriptional analysis of asexual and sexual morphs reveals possible mechanisms in reproductive polyphenism of the cotton aphid. PLoS One 9:e99506. doi: 10.1371/journal.pone.0099506
Lu, Y., Bierbach, D., Ormanns, J., Warren, W. C., Walter, R. B., and Schartl, M. (2021). Fixation of allelic gene expression landscapes and expression bias pattern shape the transcriptome of the clonal Amazon molly. Genome Res. 31, 372–379. doi: 10.1101/gr.268870.120
Maciak, S., Janko, K., Kotusz, J., Choleva, L., Boroń, A., Juchno, D., et al. (2011). Standard metabolic rate (SMR) is inversely related to erythrocyte and genome size in allopolyploid fish of the Cobitis taenia hybrid complex. Funct. Ecol. 25, 1072–1078. doi: 10.1111/j.1365-2435.2011.01870.x
Mau, M., Corral, J. M., Vogel, H., Melzer, M., Fuchs, J., Kuhlmann, M., et al. (2013). The conserved chimeric transcript UPGRADE2 is associated with unreduced pollen formation and is exclusively found in apomictic Boechera species. Plant Physiol. 163, 1640–1659. doi: 10.1104/pp.113.222448
Mau, M., Liiving, T., Fomenko, L., Goertzen, R., Paczesniak, D., Böttner, L., et al. (2021). The spread of infectious asexuality through haploid pollen. New Phytol. 230, 804–820. doi: 10.1111/nph.17174
McCleland, M. L., Gardner, R. D., Kallio, M. J., Daum, J. R., Gorbsky, G. J., Burke, D. J., et al. (2003). The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev. 17, 101–114. doi: 10.1101/gad.1040903
McElroy, K. E., Müller, S., Lamatsch, D. K., Bankers, L., Fields, P. D., Jalinsky, J. R., et al. (2021). Asexuality associated with marked genomic expansion of tandemly repeated rRNA and histone genes. Mol. Biol. Evol. 38, 3581–3592. doi: 10.1093/molbev/msab121
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Million, K. M., Bhattacharya, A., Dinges, Z. M., Montgomery, S., Smith, E., and Lively, C. M. (2021). DNA content variation and SNP diversity within a single population of asexual snails. J. Hered. 112, 58–66. doi: 10.1093/jhered/esaa048
Neaves, W. B., and Baumann, P. (2011). Unisexual reproduction among vertebrates. Trends Genet. 27, 81–88. doi: 10.1016/j.tig.2010.12.002
Neiman, M., and Lively, C. M. (2004). Pleistocene glaciation is implicated in the phylogeographical structure of Potamopyrgus antipodarum, a New Zealand snail. Mol. Ecol. 13, 3085–3098. doi: 10.1111/j.1365-294X.2004.02292.x
Neiman, M., Hehman, G., Miller, J. T., Logsdon, J. M., and Taylor, D. R. (2010). Accelerated mutation accumulation in asexual lineages of a freshwater snail. Mol. Biol. Evol. 27, 954–963. doi: 10.1093/molbev/msp300
Neiman, M., Jokela, J., and Lively, C. M. (2005). Variation in asexual lineage age in Potamopyrgus antipodarum, a New Zealand snail. Evolution 59, 1945–1952. doi: 10.1111/j.0014-3820.2005.tb01064.x
Neiman, M., Larkin, K., Thompson, A. R., and Wilton, P. (2012). Male offspring production by asexual Potamopyrgus antipodarum, a New Zealand snail. Heredity 109, 57–62. doi: 10.1038/hdy.2012.13
Neiman, M., Meirmans, P. G., Schwander, T., and Meirmans, S. (2018). Sex in the wild: how and why field-based studies contribute to solving the problem of sex*. Evolution 72, 1194–1203. doi: 10.1111/evo.13485
Neiman, M., Paczesniak, D., Soper, D. M., Baldwin, A. T., and Hehman, G. (2011). Wide variation in ploidy level and genome size in a new zealand freshwater snail with coexisting sexual and asexual lineages. Evolution 65, 3202–3216. doi: 10.1111/j.1558-5646.2011.01360.x
Neiman, M., Sharbel, T. F., and Schwander, T. (2014). Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. J. Evol. Biol. 27, 1346–1359. doi: 10.1111/jeb.12357
Neiman, M., Theisen, K. M., Mayry, M. E., and Kay, A. D. (2009). Can phosphorus limitation contribute to the maintenance of sex? A test of a key assumption. J. Evol. Biol. 22, 1359–1363. doi: 10.1111/j.1420-9101.2009.01748.x
Otto, S. P. (2020). Selective interference and the evolution of sex. J. Hered. 112, 9–18. doi: 10.1093/jhered/esaa026
Paczesniak, D., Jokela, J., Larkin, K., and Neiman, M. (2013). Discordance between nuclear and mitochondrial genomes in sexual and asexual lineages of the freshwater snail Potamopyrgus antipodarum. Mol. Ecol. 22, 4695–4710. doi: 10.1111/mec.12422
Parisod, C., Holderegger, R., and Brochmann, C. (2010). Evolutionary consequences of autopolyploidy. New Phytol. 186, 5–17. doi: 10.1111/j.1469-8137.2009.03142.x
Parker, D. J., Bast, J., Jalvingh, K., Dumas, Z., Robinson-Rechavi, M., and Schwander, T. (2019). Repeated evolution of asexuality involves convergent gene expression changes. Mol. Biol. Evol. 36, 350–364. doi: 10.1093/molbev/msy217
Paul, S., Jitendra, N., Antoine, H., Alessandro, D., Lyam, B., Emilien, N., et al. (2022). Chromosome-level genome assembly reveals homologous chromosomes and recombination in asexual rotifer Adineta vaga. Sci. Adv. 7:eabg4216. doi: 10.1126/sciadv.abg4216
Phillips, N. R., and Lambert, D. M. (1989). Genetics of Potamopyrgus antipodarum (Gastropoda: Prosobranchia): evidence for reproductive modes. New Zeal. J. Zool. 16, 435–445. doi: 10.1080/03014223.1989.10422911
Ren, L., Tang, C., Li, W., Cui, J., Tan, X., Xiong, Y., et al. (2017). Determination of dosage compensation and comparison of gene expression in a triploid hybrid fish. BMC Genomics 18:38. doi: 10.1186/s12864-016-3424-5
Rode, N. O., Jabbour-Zahab, R., Boyer, L., Flaven, É, Hontoria, F., Van Stappen, G., et al. (2021). The origin of asexual brine shrimps. bioRxiv [Preprint]. doi: 10.1101/2021.06.11.448048
Sandrock, C., and Vorburger, C. (2011). Single-Locus recessive inheritance of asexual reproduction in a parasitoid wasp. Curr. Biol. 21, 433–437. doi: 10.1016/j.cub.2011.01.070
Schedina, I. M., Groth, D., Schlupp, I., and Tiedemann, R. (2018). The gonadal transcriptome of the unisexual Amazon molly Poecilia formosa in comparison to its sexual ancestors, Poecilia mexicana and Poecilia latipinna. BMC Genomics 19:12. doi: 10.1186/s12864-017-4382-2
Schmidt, A. (2020). Controlling Apomixis: shared features and distinct characteristics of gene regulation. Genes 11:329. doi: 10.3390/genes11030329
Schönitzer, V., and Weiss, I. M. (2007). The structure of mollusc larval shells formed in the presence of the chitin synthase inhibitor Nikkomycin Z. BMC Struct. Biol. 7:71. doi: 10.1186/1472-6807-7-71
Schurko, A. M., Logsdon, J. M., and Eads, B. D. (2009). Meiosis genes in Daphnia pulex and the role of parthenogenesis in genome evolution. BMC Evol. Biol. 9:78. doi: 10.1186/1471-2148-9-78
Schwander, T., and Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution 63, 84–103. doi: 10.1111/j.1558-5646.2008.00524.x
Schwander, T., Henry, L., and Crespi, B. J. (2011). Molecular evidence for ancient asexuality in Timena stick insects. Curr. Biol. 21, 1129–1134. doi: 10.1016/j.cub.2011.05.026
Sekii, K., Yorimoto, S., Okamoto, H., Nagao, N., Maezawa, T., Matsui, Y., et al. (2019). Transcriptomic analysis reveals differences in the regulation of amino acid metabolism in asexual and sexual planarians. Sci. Rep. 9:11799. doi: 10.1038/s41598-019-42025-z
Sharbrough, J., Luse, M., Boore, J. L., Logsdon, J. M., and Neiman, M. (2018). Radical amino acid mutations persist longer in the absence of sex. Evolution 72, 808–824. doi: 10.1111/evo.13465
Stajic, D., and Jansen, L. E. T. (2021). Empirical evidence for epigenetic inheritance driving evolutionary adaptation. Philos. Trans. R. Soc. B Biol. Sci. 376:20200121. doi: 10.1098/rstb.2020.0121
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Vakhrusheva, O. A., Mnatsakanova, E. A., Galimov, Y. R., Neretina, T. V., Gerasimov, E. S., Naumenko, S. A., et al. (2020). Genomic signatures of recombination in a natural population of the bdelloid rotifer i. Nat. Commun. 11:6421. doi: 10.1038/s41467-020-19614-y
Verhaegen, G., McElroy, K. E., Bankers, L., Neiman, M., and Haase, M. (2018). Adaptive phenotypic plasticity in a clonal invader. Ecol. Evol. 8, 4465–4483. doi: 10.1002/ece3.4009
Wallace, C. (1992). Parthenogenesis, sex and chromosomes in Potamopyrgus. J. Molluscan Stud. 58, 93–107. doi: 10.1093/mollus/58.2.93
Wang, M., Zhao, Y., and Zhang, B. (2015). Efficient test and visualization of multi-set intersections. Sci. Rep. 5:16923. doi: 10.1038/srep16923
Warren, I. A., Ciborowski, K. L., Casadei, E., Hazlerigg, D. G., Martin, S., Jordan, W. C., et al. (2014). Extensive local gene duplication and functional divergence among paralogs in Atlantic salmon. Genome Biol. Evol. 6, 1790–1805. doi: 10.1093/gbe/evu131
Weiss, I. M., Schönitzer, V., Eichner, N., and Sumper, M. (2006). The chitin synthase involved in marine bivalve mollusk shell formation contains a myosin domain. FEBS Lett. 580, 1846–1852. doi: 10.1016/j.febslet.2006.02.044
Winterbourn, M. J. (1970). The New Zealand species of Potamopyrgus (Gastropoda: Hydrobiidae). Malacologia 10, 283–321.
Xu, S., Huynh, T. V., and Snyman, M. (2022). The transcriptomic signature of obligate parthenogenesis. Heredity 128, 132–138. doi: 10.1038/s41437-022-00498-1
Yagound, B., Dogantzis, K. A., Zayed, A., Lim, J., Broekhuyse, P., Remnant, E. J., et al. (2020). A single gene causes thelytokous parthenogenesis, the defining feature of the cape honeybee Apis mellifera capensis. Curr. Biol. 30, 2248.e6–2259.e6. doi: 10.1016/j.cub.2020.04.033
Ye, Z., Jiang, X., Pfrender, M. E., and Lynch, M. (2021). Genome-Wide allele-specific expression in obligately asexual daphnia pulex and the implications for the genetic basis of asexuality. Genome Biol. Evol. 13:evab243. doi: 10.1093/gbe/evab243
Yonezawa, M., Sakuda, S., Yoshimura, E., and Suzuki, M. (2016). Molecular cloning and functional analysis of chitinases in the fresh water snail, Lymnaea stagnalis. J. Struct. Biol. 196, 107–118. doi: 10.1016/j.jsb.2016.02.021
Zachar, N., and Neiman, M. (2013). Profound effects of population density on fitness-related traits in an invasive freshwater snail. PLoS One 8:e80067. doi: 10.1371/journal.pone.0080067
Zhang, Y.-N., Zhu, X.-Y., Wang, W.-P., Wang, Y., Wang, L., Xu, X.-X., et al. (2016). Reproductive switching analysis of Daphnia similoides between sexual female and parthenogenetic female by transcriptome comparison. Sci. Rep. 6:34241. doi: 10.1038/srep34241
Keywords: asexual (clonal) reproduction, transcriptome (RNA-seq), Mollusca, maintenance of sexual reproduction, polyploidy
Citation: McElroy KE, Bankers L, Soper D, Hehman G, Boore JL, Logsdon JM Jr and Neiman M (2022) Patterns of gene expression in ovaries of sexual vs. asexual lineages of a freshwater snail. Front. Ecol. Evol. 10:845640. doi: 10.3389/fevo.2022.845640
Received: 30 December 2021; Accepted: 27 June 2022;
Published: 15 July 2022.
Edited by:
Astrid Böhne, Centre for Molecular Biodiversity Research, Zoological Research Museum Alexander Koenig, GermanyReviewed by:
Darren J. Parker, University of Lausanne, SwitzerlandChristoph Haag, UMR 5175 Centre d’Ecologie Fonctionnelle et Evolutive (CEFE), France
Karel Janko, Institute of Animal Physiology and Genetics, Academy of Sciences of the Czech Republic (ASCR), Czechia
Copyright © 2022 McElroy, Bankers, Soper, Hehman, Boore, Logsdon and Neiman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyle E. McElroy, kmcelroy@iastate.edu
†ORCID: Kyle E. McElroy, orcid.org/orcid.org/0000-0001-9581-2535; Laura Bankers, orcid.org/0000-0001-6433-9263; Deanna Soper, orcid.org/0000-0002-9083-6375; Jeffrey L. Boore, orcid.org/0000-0001-6751-4549; John M. Logsdon, orcid.org/0000-0002-0031-462X; Maurine Neiman, orcid.org/0000-0002-1543-8115