The Mitochondrial Genome–on Selective Constraints and Signatures at the Organism, Cell, and Single Mitochondrion Levels
 Noam Shtolz
Noam Shtolz Dan Mishmar
Dan Mishmar- Department of Life Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
Natural selection acts on the phenotype. Therefore, many mistakenly expect to observe its signatures only in the organism, while overlooking its impact on tissues, cells and subcellular compartments. This is particularly crucial in the case of the mitochondrial genome (mtDNA), which, unlike the nucleus, resides in multiple cellular copies that may vary in sequence (heteroplasmy) and quantity among tissues. Since the mitochondrion is a hub for cellular metabolism, ATP production, and additional activities such as nucleotide biosynthesis and apoptosis, mitochondrial dysfunction leads to both tissue-specific and systemic disorders. Therefore, strong selective pressures act to maintain mitochondrial function via removal of deleterious mutations via purifying (negative) selection. In parallel, selection also acts on the mitochondrion to allow adaptation of cells and organisms to new environments and physiological conditions (positive selection). Nevertheless, unlike the nuclear genetic information, the mitochondrial genetic system incorporates closely interacting bi-genomic factors (i.e., encoded by the nuclear and mitochondrial genomes). This is further complicated by the order of magnitude higher mutation rate of the vertebrate mtDNA as compared to the nuclear genome. Such mutation rate difference generates a generous mtDNA mutational landscape for selection to act, but also requires tight mito-nuclear co-evolution to maintain mitochondrial activities. In this essay we will consider the unique mitochondrial signatures of natural selection at the organism, tissue, cell, and single mitochondrion levels.
Introduction
All cells require ATP, as it is the most common cellular currency to do work. Although glycolysis provides the means to produce ATP when glucose is available, an order of magnitude and more efficient energy-production system emerged ~2.5 billion years ago in eukaryotes through endosymbiosis between the ancestor of the mitochondria and the progenitor of eukaryotic cells (Sagan, 1967). Ever since, genetic material migrated from the genome of the former free-living alpha proteo-bacterium to the host nucleus (the formation of which will not be discussed here). This apparent lateral gene transfer created interdependence between the host and tenant, not only due to the cellular reliance on mitochondrial ATP production, but also due to the involvement of mitochondria in many other major activities, such as nucleotide biosynthesis, the generation of Iron-Sulfur protein clusters, apoptosis etc. Consequently, mitochondria cannot be grown outside of the eukaryotic cells, and the vast majority of eukaryotic cells cannot survive without their mitochondria.
Such relocation of genetic material from the cytoplasm to the cell nucleus required adaptation of the former mitochondrial DNA (mtDNA)-encoded genes to the nuclear genetic code and translation machinery, assimilation of the “new” genetic immigrants into the nuclear mode of gene regulation, which respond to chromatin remodeling, and finally required the acquisition of the protein properties that allow their re-import into the mitochondria to maintain their function. This became further complicated by the emergence of metazoans, which required differential energy expenditure per tissues and cell types (Lane and Martin, 2010). Hence, strong selective constraints should have been inflicted to preserve the activity (via negative selection) of the factors which generate such energy. In parallel, positive selection likely also acted to enable adaptation of the energy metabolism system to a variety of environments, and possibly allows each cell type to incorporate its specific activities within the tissues of the organism. In the current essay, we will discuss evidence supporting selective signatures that marked the mitochondrial genetic system after its emergence. We will demonstrate such signatures at three different levels—the organism, the cell and the individual mitochondrion. As much literature studied and discussed the organism level, we will put more emphasis on the cellular and mitochondrial levels.
Selection Acts on the Phenotype—the Various Levels of Mitochondrial Phenotypic Expression
In 1983 the neutral theory of molecular evolution was put forward by Motoo Kimura, who argued that most population genetic variants result from mutations which propagate via genetic drift and not selection, and therefore, different alleles generally do not affect the individual's fitness (Kimura, 1983). This theory is frequently misinterpreted, as it does not imply that organisms are not adapted to their environments, and it does not state that natural selection is negligible in shaping genetic variation and genomes. Although geneticists and their next generation of scientists, genomicists, are keen to identify the signatures of selection readily written as an epitaph in the genetic material, selection acts first on the phenotype and (indirectly) on the genotype. Indeed, although many of the traits of a given individual, in any given metazoan species, are mostly the result of changes in the inherited matter, many traits may vary in their appearance due to interactions between two or more genetic factors (i.e., epistasis), and due to variable interactions with environmental conditions. While considering the mitochondria, this scheme of interaction is further complicated by the involvement of factors encoded by both the nuclear and the mitochondrial genomes (G × G) (Levin et al., 2014). Therefore, when also considering the environment, mitochondrial-encoded traits are influenced by interactions between two genomes and a variety of environments and physiological conditions (G × G × E) (Zhu et al., 2014). This scheme experienced another level of complexity, due to the existence of multiple mitochondria per cell, the mtDNAs of which may vary in sequence (heteroplasmy). Specifically, heteroplasmy patterns and their phenotypic consequences notably differs between dividing (mitotic) and post-mitotic tissues (Kowald and Kirkwood, 2013; Filograna et al., 2019). Therefore, mitochondrial phenotypic implications should be considered not only at the level of the organism which has been widely discussed in the past (Meiklejohn et al., 2007; Levin et al., 2014), but also at the level of the cell, and even in the single mitochondrion (Figure 1). At first, analysis of a single mitochondrion was limited by the technological resolution of mutational detection (Reiner et al., 2010). However, a recent study investigated mtDNA sequence variation within 118 isolated mitochondria from mouse neurons and astrocytes (Morris et al., 2017). Whole mtDNA sequencing of each of the isolated mitochondria revealed an average of 3.9 (± 5.71 SD) single heteroplasmic nucleotide variants (SNVs) in the tested mitochondrial population per nucleotide position. Hence, despite the fact that all samples originate from the same mouse strain (C57BL/6N), a notable repertoire of mtDNA heteroplasmic SNVs was discovered. While analyzing this mutational repertoire, these researchers plotted the most frequent non-reference alleles against log2 appearance counts. Unexpectedly, they observed a distribution that did not match the symmetric U-shaped distribution, which is the expected distribution of alleles, under the assumption of neutrality (Birky et al., 1983). In addition to the distribution of non-reference alleles, the authors identified three specific SNVs with high potential impact. They found a negative correlation between the predicted impact of the specific mutations and their degree of variability among the tested samples. These pieces of evidence suggest that selection likely shaped the mutational distribution pattern at the single mitochondrion level and was responsible for the divergence from a random pattern.
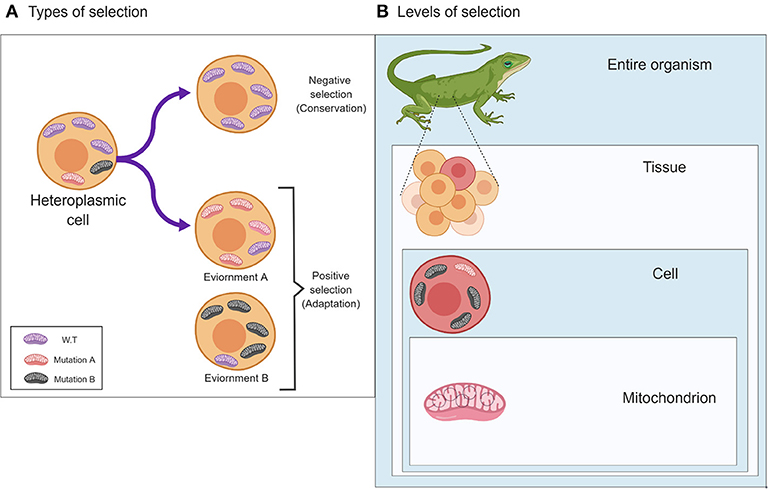
Figure 1. The two main types of selection, Negative and Positive, and their effects on patterns of mitochondrial genetic variability. (A) Different types of selection (negative or positive) affect the pattern of mitochondrial mutations in response to different environments (lower panels) or will undergo a selective sweep (negative selection to maintain function (upper panel). (B) The different levels in which selection may act on the mitochondria, i.e., (down-up) the single mitochondrion, cells and the entire organism. The figure was designed using ©BioRender—www.biorender.com.
While considering intercellular patterns of heteroplasmy, under the assumption of neutrality, with genetic drift being the primary force that shapes genetic variation over-time, it is likely that certain cells will develop a heterogenous mitochondrial population, without any phenotypic implications. Such a scenario predicts stochastic accumulation of mutations during each replication cycle, and random assortment of mtDNA molecules between daughter cells. To test such prediction, heteroplasmy patterns were studied in colonies derived from single cells of two human cell lines—MDA-MB-157 (breast cancer) and U20S (osteosarcoma) (Jayaprakash et al., 2015). Jayaprakash et al. found stably maintained heteroplasmy in daughter cells over multiple passages. This finding slightly departs from the expected random mitochondrial segregation, which predicts diverse levels of heteroplasmy regardless of the nature of the identified mtDNA mutations. Although these findings reveal constant levels of heteroplasmy regardless of the mtDNA position in which the mutations occurred, one cannot easily explain the results either by random forces, or by selective constraints. However, a recent study of heteroplasmy in single cells identified consistent cell-lineage-specific segregation of heteroplasmic mtDNA mutations during differentiation of hematopoietic cells, which did not comply with random segregation of mutations during cell division (Ludwig et al., 2019). The growing availability of single cell genomics data holds much promise in future investigation of the forces that govern patterns of mtDNA heteroplasmy during cell division and differentiation (Ludwig et al., 2019).
Deep sequencing of many human individuals (Li et al., 2010; Payne et al., 2013; Ye et al., 2014), family members (Goto et al., 2011) and identical twins (Avital et al., 2012) attest for the common phenomenon of heteroplasmy, its inheritance and accumulation during the life of the individual. Notably, the distribution of these mutations across the mtDNA was not random, with much higher incidence of heteroplasmy in non-coding mtDNA sequences than expected by chance (Avital et al., 2012). Thus, it can be inferred that heteroplasmic mutations are likely subjected to selective constraints. We previously found that such non-random mutational distribution throughout the mtDNA occurred regardless of the heteroplasmy levels in two cell populations (blood and skeletal muscle) in identical twins, thus attesting for the impact of selection, not only at the level by which phenotypic consequences are expected for the organism, but also at the cellular level (Avital et al., 2012). Nevertheless, while investigating very low-level heteroplasmic mutations, it has been argued that known mtDNA disease-causing mutations could be present in healthy individuals in the population, if their heteroplasmy is maintained at low levels (Ye et al., 2014). A growing body of evidence suggests that since the mitochondria within cells are interconnected in a network, pathological mutations could survive due to inter-mitochondrial functional complementation, and exchange of nutrients (Schon and Gilkerson, 2010). As mitochondria go through cycles of fission and fusion, which build such network (fusion), or disconnect it (fission), dysfunctional mitochondria could be removed by mitophagy (Twig and Shirihai, 2011). When the latter is compromised in model organisms, the level of deleterious mutations elevates to a degree that may have phenotypic consequences (Valenci et al., 2015). Accordingly, Ferree et al. (2013) showed that knockout of mitophagy factors led to an accumulation of partly dysfunctional, aged mitochondria. Hence, mitochondria within single mammalian cells are frequently not uniform in function (Aryaman et al., 2018), and hence will be under differential selective constraints between cell types, tissues, and cells in the same tissue. Taken together, functional differences in mitochondrial activities are observed not only at the organism level, but also between and within cells.
Selection and Drift Shape the Mitochondrial Population During Female Germline Formation
Variation in heteroplasmy patterns between tissues could either result from somatic accumulation of mutations during the lifetime of an individual, or could be inherited. Therefore, one has to consider patterns of heteroplasmy already in the ovum, and specifically during the emergence of the maternal germ line. Indeed, much discussion revolved around the nature of mitochondrial bottleneck in the primordial maternal germ line in mouse and humans (Cree et al., 2008; Cao et al., 2009; Rebolledo-Jaramillo et al., 2014). A ~1000-fold reduction in mtDNA content was estimated during the development of the human female germ cell (Freyer et al., 2012; Floros et al., 2018), followed by intense replication as cells migrate to form the gonad. Although neutral effects (genetic drift) are widely thought to govern this process, one has to take into account that the female germline formation requires OXPHOS activity (Ginsburg et al., 1990). Therefore, it is logical in addition to neutrality that two types of selection come into play: firstly, positive selection, which prefers highly replicating variants, and secondly negative selection acts to remove variants with reduced ATP production (Wei et al., 2019). A relatively straightforward way to study the signatures of selection in the mtDNA of the germline is by comparing heteroplasmy patterns between mothers and offspring. This approach has recently been taken by Wei et al. by analyzing 1,526 human mother-offspring pairs (Wei et al., 2019). Firstly, higher levels of heteroplasmy across the entire mtDNA were observed in the mothers as compared to their offspring. Secondly, they identified, from the distribution of heteroplasmic mutations across the mtDNA sequence of the offspring, that these mutations were more frequent in the first or second codon positions as compared to the third position. Furthermore, while considering the entire coding mtDNA sequence, evolutionary conserved positions tended to retain heteroplasmy, in contrast to the tendency toward homoplasmy in positions with lesser conservation. Third, homoplasmic mutations were seldom found in evolutionary conserved positions. Fourth, most of the heteroplasmic mutations were characterized as de novo (found only in the offspring) or lost (found only in the mother), which again was best interpreted as the result of negative selection in the maternal germline. Finally, analysis of the mtDNA control region (D-loop) revealed reduced frequency of heteroplasmic positions within regions of regulatory importance for replication and transcription. Taken together, these findings support the action of negative selection during maternal germ line formation in the human mtDNA sequence. Although the mitochondrial population in the maternal germline determines the initial mitochondrial repertoire in the zygote, such population is still prone to the effects of evolutionary forces during differentiation, and in the lifetime of the organism.
Natural Selection Acts on the Mitochondria at the Whole Organism Level
The potential phenotypic impact, and hence the signatures of natural selection, on mitochondrial changes is mostly considered at the organism level (Stewart et al., 2008; Castellana et al., 2011). Probably the best example for such is disease-causing mutations, as thoroughly reviewed previously (Dowling, 2014), and hence will be discussed here only briefly. In 1988, the research group led by Douglas C Wallace was the first to have discovered a clear association between high levels of heteroplasmy of an mtDNA mutation and the tendency to develop a disease—Leber's hereditary optic neuropathy (LHON) (Wallace et al., 1988). In the same year, Ian Holt and others showed that a high level heteroplasmic mtDNA deletion led to mitochondrial myopathy (Holt et al., 1988). Ever since these discoveries, the association between certain threshold levels of heteroplasmy (~85% for point mutations) with expression of disease phenotypes was found in multiple mitochondrial disorders (Stewart and Chinnery, 2015; Wallace, 2018). As mitochondrial diseases are relatively rare (Craven et al., 2017), the most logical explanation is that negative selection acts to remove such mutations from the population both in the mtDNA, and in mitochondrial genes encoded by the nucleus, although, in the latter, mutations could be retained in the population due to recessive modes of inheritance, and/or due to partial penetrance. Supportive evidence for such negative selection, while considering the mtDNA, came from large-scale deep sequence analysis of multiple individuals (mentioned above), which revealed that, although deleterious mutations are relatively prevalent in the human population, they only appear at very low heteroplasmy levels (Payne et al., 2013; Ye et al., 2014). Negative selection is also exemplified by the strong mutational bias in population genetics mtDNA variants (Gu et al., 2019). The differential accumulation pattern of mtDNA deleterious mutations in Drosophila males as compared to females, termed the “mother's curse”, also supports the impact of natural selection on mtDNA sequences at the organism level (Innocenti et al., 2011), since in general, the mtDNA is maternally inherited. The signature of negative selection is also evident at the very early stages of embryo development, i.e., during oogenesis, as discussed above (De Fanti et al., 2017). Although bottleneck of mitochondrial transmission has been suggested to occur during the development of female germ cells in mice (Cree et al., 2008; Floros et al., 2018) and humans (Rebolledo-Jaramillo et al., 2014), divergence between mtDNA mutational patterns between tissues support the impact of natural selection (Latorre-Pellicer et al., 2016). Finally, certain inherited mtDNA mutations associate with altered tendency to develop age-related disorders (Marom et al., 2017), i.e., diseases whose onset is during post-reproductive age; similarly, large mtDNA deletions tend to accumulate preferentially in aging humans, again during post-reproductive ages (Arnheim and Cortopassi, 1992; Simonetti et al., 1992), hence with little effect on fitness. These pieces of evidence clearly attest for the impact of negative selection on mitochondrial function at the organism level.
Unlike negative selection, adaptive selection (i.e., positive selection) is less obvious to observe. The first evidence for signatures of positive selection in the human mtDNA has been demonstrated by the analysis of multiple whole mitochondrial genomes from individuals representing all major global populations (Mishmar et al., 2003; Ruiz-Pesini et al., 2004). These analyses correlated the pattern of ancient mtDNA mutations with global geographic distribution of mtDNA haplotypes, and argued for differential advantage of mtDNA haplogroups to survive in different climatic conditions (Mishmar et al., 2003). These predictions gained recent experimental support from the identification of sharp differentiation in the geographic distribution of Drosophila mtDNA haplotypes between cold and warm latitudinal regions in Australia: the haplotype that predominated low (subtropical) latitudes displayed greater resilience to heat than to cold stresses, as compared to haplotypes predominating higher (temperate) latitudes (Camus et al., 2017; Lajbner et al., 2018). Nevertheless, as mitochondrial function depends on interactions between mtDNA and nuclear DNA-encoded factors, one still awaits testing the impact of each of the tested mtDNA haplotypes in different nuclear genetic backgrounds.
Mito-Nuclear Interactions: Corresponding Signatures of Selection in Both the mtDNA and the nDNA
As mentioned above, one unique characteristic of the mitochondrial genomic system is the G x G interaction, or in other words, the interaction between factors encoded by the nuclear and mitochondrial genomes. The possible, and actual, impact of compatible vs. incompatible mito-nuclear genotypes were investigated in cell culture harboring different combinations of mtDNAs and nDNAs (cybrids) (Suissa et al., 2009; Gomez-Duran et al., 2010; Ji et al., 2012; Kenney et al., 2014; Crawford et al., 2018), and from repeated backcrossing of model organisms for the sake of generating animals with differential combinations of mito-nuclear genotypes (conplastic animals) originating from different strains of model organisms (Dingley et al., 2014; Latorre-Pellicer et al., 2016). Additionally, population genetics studies from species and different population isolates from the same species in the copepod Tigriopus californicus (Burton et al., 2006; Ellison and Burton, 2006), in reptiles (chameleons) (Bar-Yaacov et al., 2015) and in birds (sparrows) (Trier et al., 2014), revealed hybrid incompatibility, which constitutes an important step toward speciation. These pieces of evidence provide strong support for the importance of genetic compatibility between the nuclear and mitochondrial genomes, that when interfered with can either lead to diseases (Gershoni et al., 2014) or lead to the creation of reproductive barriers in both invertebrates and in vertebrates (Gershoni et al., 2009; Trier et al., 2014; Telschow et al., 2019; Tobler et al., 2019). The underlying mechanism of mito-nuclear genetic compatibility has previously been thoroughly discussed at the protein-protein, RNA-protein and protein-mtDNA levels at the whole organism level (Bar-Yaacov et al., 2012; Levin et al., 2014; Hill, 2019; Hill et al., 2019). However, although mito-nuclear interactions occur at the inter-cellular and intracellular levels, they are currently more technically challenging to identify. One good example that might reflect possible intra-cellular mito-nuclear incompatibilities is the establishment of heteroplasmic animals harboring a mixed population of mitochondria from two different strains (Sharpley et al., 2012). Although the phenotypes of such heteroplasmic mice could result from defective interactions between mitochondria with different haplotypes in the cells, they could also result from differential interactions of the two mtDNA genotypes with products coming from the nuclear genome, as reflected in a similar study (Latorre-Pellicer et al., 2016). Finally, it has been shown in yeast that the mixture of mtDNA mulecules from two different strains have slowly “drifted” into a situation where the original mtDNA had outcompeted the donor mtDNA to reconstruct the original mtDNA-nDNA genotype combination (Lee et al., 2008). Taken together, genotype compatibility between the mitochondrial and nuclear genomes are likely important for life. Nevertheless, one has to consider the fact that human offspring that were born from fertilized eggs in which the nuclei were transferred to an enucleated donor ovum (i.e., the mitochondrial replacement therapy) had no apparent phenotypes (Hyslop et al., 2016). Although mito-nuclear incompatibility has been shown in mitochondrial replacement experiments in multiple species (Dobler et al., 2018), some have questioned its wide impact on mitochondrial activity (Eyre-Walker, 2017). Interestingly, a recent analysis of human admixed populations revealed that mtDNA copy number decreases with increasing discordance between nuclear and mtDNA ancestry (Zaidi and Makova, 2019). However, since the lack of phenotype in human offspring that were born from mitochondrial replacement therapy was only demonstrated for generation F1, and as backcrossing is not possible in humans, the long-term impact of mito-nuclear incompatibility in humans still awaits a longitudinal study in the years to come.
Identifying the Signatures of Selection—Assessing the Functional Potential of Changes in DNA Sequences
How can one predict the effects of selection by mere sequence analysis? As protein-coding genes served as the focus of many studies, and since the genetic code and mutations in protein-coding genes are well-investigated, many bioinformatics computational tools were developed to assess the potential functional implications in protein-coding genes. The logic underlying such analyses stem from the notion that functional potential of changes in protein-coding sequences range from missense mutations, which alter the amino-acid composition of the translation product, to non-sense mutations and splice variants (both exon inclusion or skipping). This not only changes the amino acid composition, but also alters the length of the resultant protein. The latter may alter protein domains, and hence may have the potential to fundamentally affect the activity of a given protein, and even its subcellular localization. In contrast, it is believed that by-and-large, lesser functional impact is caused by sequence alterations in codon positions that do not change the amino acid sequence (Stern et al., 2007). Nevertheless, such so-called synonymous mutations change the choice of tRNAs during the translation process, which is directly influenced by differences in the abundance of different tRNAs that recognize different codons of the same amino acid (Levin et al., 2013). Previously, we have shown that, in the human mtDNA, there is preference for codons recognized by mtDNA-encoded tRNAs, which are more prevalent in the mitochondria as compared to imported tRNAs (Levin et al., 2013). Notably, as some mtDNA protein-coding genes harbor codons which are not recognized by the mtDNA-encoded tRNAs, nuclear-DNA encoded tRNA come into play and are imported into the mitochondria; such import depends on the varying tRNA gene contents of the mitochondrial genomes of different organisms, such as yeast, plants and mammals [reviewed in: (Rubio and Hopper, 2011; Schneider, 2011)]. For example, , which is not encoded by both mouse and human mtDNAs, was identified in-vivo within the mitochondria of cells from both organisms, where it was incorporated into the translation machinery (Rubio et al., 2008). It was suggested that the import of such nuclear tRNA into the mitochondria occurs via an ATP-dependent mechanism, which is likely distinct from the general mitochondrial protein import machinery. Can the usage of a rare tRNA affect protein function? Synonymous mutations in the nuclear DNA-encoded Multidrug Resistance 1(MDR1) gene affected protein folding (Kimchi-Sarfaty et al., 2007). Specifically, the authors argued that the slow incorporation of a rare tRNAGly led to slight hindrance of translation, which in turn affected protein folding, and its subsequent functionality. tRNA sequences themselves are under selective constraints, yet it was previously argued that tRNA mutations in the mtDNA that cause diseases in humans can appear as polymorphic variants in other species (i.e. recurrent mutations), thus supporting the putative impact of epistatic interactions acting as functional compensation (Kern and Kondrashov, 2004; Breen et al., 2012; Levin et al., 2013; Levin and Mishmar, 2017). Accordingly, experiments in Drosophila suggested that, in some cases, tRNA synthetase undergoes coordinated changes alongside their corresponding tRNAs to maintain function (Meiklejohn et al., 2013; Holmbeck et al., 2015). The availability of ribosome profiling as a highly quantitative approach to assess rates of translation, and its recent adaptation to the mtDNA (Rooijers et al., 2013; Gao et al., 2017), serves as a promising future experimental approach to compare rates of mtDNA translation between samples differing in synonymous mtDNA mutations.
As mutations in protein-coding genes are easier to interpret, most computational tools that screen for the signatures of natural selection calculate the ratios of missense (non-synonymous) to synonymous mutations (Ka/Ks) in genes of interest. Another measurable parameter that may reflect signatures of selection is whether the mutations occurred in evolutionary conserved sequence positions, and physico-chemical properties of amino acids. Tools (Bank et al., 2014), such as SELECTON (Stern et al., 2007), PAML (Yang, 1997), and PANTHER (Thomas et al., 2003), are designed to perform such calculations. Sequence conservation is also a useful parameter to assess the functional impact of mutations in RNA genes, i.e., those transcripts that are not ultimately translated, including tRNAs, rRNAs and the various types of non-coding RNAs (e.g., long, small and microRNAs). Human mtDNA encodes for 2 rRNAs, 22 tRNAs, and few relatively recently discovered long and microRNAs (Mercer et al., 2011; Rackham et al., 2011). As such transcripts overlap the coding regions of protein-coding genes, it is difficult to distinguish the potential functional impact of mutations in such elements. Despite all of the above, although it is possible to assess the potential impact of mutations that alter codons but not the amino acid code (the so-called silent mutations) by assessing codon bias index (Levin et al., 2013), only minor phenotypic effects have been demonstrated regarding such in the mitochondria, leading some researchers to argue for lack of selective signatures in most mtDNA mutations (Stoeckle and Thaler, 2018). Nevertheless, phenotypic impact of a mutation is not only limited to the subsequent gene function—an apparently silent mutation could alter a previously un-noticed regulatory element (Blumberg et al., 2018). Indeed, it has previously been demonstrated that certain coding sequences could also act as regulatory elements both in the nucleus (Birnbaum et al., 2012) and in the mitochondria (Blumberg et al., 2014). However, unlike the functional potential of mutations in genes, which could be assessed using the above-mentioned parameters, the functionality of mutations in mtDNA regulatory elements is much harder to predict, mainly since the “language and grammar” of these mtDNA regulatory elements are yet to be deciphered. In the subsequent sub-section, we will discuss efforts to assess the impact of mtDNA sequence variation on mtDNA regulation.
The Impact of mtDNA Mutations on Regulation of Gene Expression and mtDNA Copy Number
As mentioned above, although changes in a given sequence will not affect the amino acid sequence, or the activity of an RNA gene, a regulatory element which might reside within the same sequence could suffer from the same mutation and lead to altered transcription (Blumberg et al., 2014, 2017, 2018), replication, and possibly recombination or repair. Sequencing the human mtDNA in multiple tissues (Samuels et al., 2013) revealed that certain tissues (kidney, liver and skeletal muscle), shared the same recurrent heteroplasmic mutations, all in regulatory mtDNA regions, which were undetectable in other tissues in the same individuals, supporting non-random mutational segregation. Previously, ourselves and others showed that changes in mtDNA regulatory elements affect mtDNA regulation in vitro (Asari et al., 2007; Suissa et al., 2009) and in cytoplasmic hybrids (cybrids) (Gomez-Duran et al., 2010; Kenney et al., 2014). RNA-seq analysis of multiple lymphoblastoid cell lines demonstrated an association of ancient mtDNA SNPs and linked sets of mutations (haplogroups) with altered mtDNA gene expression (Cohen et al., 2016). In the latter study, DNA SNPs in the nucleus marked nuclear regulators of mtDNA gene expression as candidate modulators of such association. Similarly, a recent study used expression SNPs (eSNP) association in the nuclear genome to identify nuclear regulators of mtDNA gene expression, while using mtDNA genetic backgrounds as co-variants (Ali et al., 2019). These association studies with gene expression suggest that co-adaptation of the nuclear and mitochondrial genomes should also be extended to co-regulation of the two genomes. Indeed, analysis of ~8500 RNA-seq experiments from multiple human individuals sampled from 48 different human body sites demonstrated mito-nuclear co-expression and co-regulation of gene expression (Barshad et al., 2018). Hence, compatibility of the two genomes should be extended beyond the direct interactions of proteins within the OXPHOS system to a regulatory cross talk. In summary, as a growing set of functional genomics tools are being adapted to investigate mitochondrial regulation (mtDNA gene expression, replication, translation and even repair), one will be able to directly assess the impact of mtDNA SNPs on mtDNA regulation in cells and organisms in the near future.
Summary and Conclusions
In the current essay, we argued that selection is a significant force that shaped, and still shapes, the landscape of mtDNA variation at the organismal, cellular, and single mitochondrial levels. To support this argument, we brought ample evidence from a variety of experimental systems, and demonstrated newly-generated evidence from tissues and cells, enabled by the adaptation of next generation sequencing techniques to investigate functional genomics of the mitochondrion. At the organism level, it is worth mentioning the multiple disease-association studies that identified association of mtDNA genetic variants with altered susceptibility to develop genetic disorders, which strongly attest for the phenotypic impact of mtDNA mutations and hence their “visibility” to selective constraints (Marom et al., 2017). The fact that such association studies reveal positive association in certain populations, but not in others, supports the likelihood of an epistatic impact on nuclear genotypes as modifying factors, which compensate for the phenotypic impact of mtDNA variants, thus supporting the importance of mito-nuclear genotype compatibility. The three levels of functional impact on the mitochondria, e.g., the organism, cell, and single mitochondrion, set forth another dimension for considering mitochondrial phenotypes and assessment of the functionality of mitochondrial mutations. The availability of single cell functional genomics data, and consideration of the population of RNA transcripts per cell suggests, that the logic presented in the current essay may also imply to discussion of phenotypes caused by changes in nuclear genes in other, non-mitochondrial, systems.
Author Contributions
DM and NS wrote the paper. DM conceived the idea and supervised the analyses and literature.
Funding
The study was funded by grants from the Israeli Science Foundation (372/17) and by the Life Sciences division of the US army (LS67993), awarded to DM. Both agents were not involved and did not influence the writing of the current manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by research grants from the Israel Science Foundation (ISF grant 372/17) and by the US army life sciences division grant (LS67993) awarded to DM.
References
Ali, A. T., Boehme, L., Carbajosa, G., Seitan, V. C., Small, K. S., and Hodgkinson, A. (2019). Nuclear genetic regulation of the human mitochondrial transcriptome. Elife 8:41927. doi: 10.7554/eLife.41927
Arnheim, N., and Cortopassi, G. (1992). Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat. Res. 275, 157–167. doi: 10.1016/0921-8734(92)90020-P
Aryaman, J., Johnston, I. G., and Jones, N. S. (2018). Mitochondrial Heterogeneity. Front. Genet. 9:718. doi: 10.3389/fgene.2018.00718
Asari, M., Tan, Y., Watanabe, S., Shimizu, K., and Shiono, H. (2007). Effect of length variations at nucleotide positions 303-315 in human mitochondrial DNA on transcription termination. Biochem. Biophys. Res. Commun. 361, 641–644. doi: 10.1016/j.bbrc.2007.07.055
Avital, G., Buchshtav, M., Zhidkov, I., Tuval Feder, J., Dadon, S., Rubin, E., et al. (2012). Mitochondrial DNA heteroplasmy in diabetes and normal adults: role of acquired and inherited mutational patterns in twins. Hum. Mol. Genet. 21, 4214–4224. doi: 10.1093/hmg/dds245
Bank, C., Ewing, G. B., Ferrer-Admettla, A., Foll, M., and Jensen, J. D. (2014). Thinking too positive? Revisiting current methods of population genetic selection inference. Trends Genet. 30, 540–546. doi: 10.1016/j.tig.2014.09.010
Barshad, G., Blumberg, A., Cohen, T., and Mishmar, D. (2018). Human primitive brain displays negative mitochondrial-nuclear expression correlation of respiratory genes. Genome Res. 28, 952–967. doi: 10.1101/gr.226324.117
Bar-Yaacov, D., Blumberg, A., and Mishmar, D. (2012). Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim. Biophys. Acta 1819, 1107–1111. doi: 10.1016/j.bbagrm.2011.10.008
Bar-Yaacov, D., Hadjivasiliou, Z., Levin, L., Barshad, G., Zarivach, R., Bouskila, A., et al. (2015). Mitochondrial Involvement in Vertebrate Speciation? The case of mito-nuclear genetic divergence in Chameleons. Genome Biol. Evol. 7, 3322–3336. doi: 10.1093/gbe/evv226
Birky, C. W., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527.
Birnbaum, R. Y., Clowney, E. J., Agamy, O., Kim, M. J., Zhao, J., Yamanaka, T., et al. (2012). Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 22, 1059–1068. doi: 10.1101/gr.133546.111
Blumberg, A., Danko, C. G., Kundaje, A., and Mishmar, D. (2018). A common pattern of DNase I footprinting throughout the human mtDNA unveils clues for a chromatin-like organization. Genome Res. 28, 1158–1168. doi: 10.1101/gr.230409.117
Blumberg, A., Rice, E. J., Kundaje, A., Danko, C. G., and Mishmar, D. (2017). Initiation of mtDNA transcription is followed by pausing, and diverges across human cell types and during evolution. Genome Res. 27, 362–373. doi: 10.1101/gr.209924.116
Blumberg, A., Sailaja, B. S., Kundaje, A., Levin, L., Dadon, S., Shmorak, S., et al. (2014). Transcription factors bind negatively-selected sites within human mtDNA genes. Genome Biol. Evol. 6, 2634–2646. doi: 10.1093/gbe/evu210
Breen, M. S., Kemena, C., Vlasov, P. K., Notredame, C., and Kondrashov, F. A. (2012). Epistasis as the primary factor in molecular evolution. Nature 490, 535–538. doi: 10.1038/nature11510
Burton, R. S., Ellison, C. K., and Harrison, J. S. (2006). The sorry state of F2 hybrids: consequences of rapid mitochondrial DNA evolution in allopatric populations. Am. Nat. 168(Suppl. 6), S14–24. doi: 10.1086/509046
Camus, M. F., Wolff, J. N., Sgro, C. M., and Dowling, D. K. (2017). Experimental support that natural selection has shaped the latitudinal distribution of mitochondrial haplotypes in australian Drosophila melanogaster. Mol. Biol. Evol. 34, 2600–2612. doi: 10.1093/molbev/msx184
Cao, L., Shitara, H., Sugimoto, M., Hayashi, J., Abe, K., and Yonekawa, H. (2009). New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet. 5:e1000756. doi: 10.1371/journal.pgen.1000756
Castellana, S., Vicario, S., and Saccone, C. (2011). Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein coding genes. Genome Biol. Evol. 3, 1067–1079. doi: 10.1093/gbe/evr040
Cohen, T., Levin, L., and Mishmar, D. (2016). Ancient out-of-africa mitochondrial DNA variants associate with distinct mitochondrial gene expression patterns. PLoS Genet. 12:e1006407. doi: 10.1371/journal.pgen.1006407
Craven, L., Alston, C. L., Taylor, R. W., and Turnbull, D. M. (2017). Recent advances in mitochondrial disease. Annu. Rev. Genomics Hum. Genet. 18, 257–275. doi: 10.1146/annurev-genom-091416-035426
Crawford, N., Prendergast, D., Oehlert, J. W., Shaw, G. M., Stevenson, D. K., Rappaport, N., et al. (2018). Divergent patterns of mitochondrial and nuclear ancestry are associated with the risk for preterm birth. J. Pediatr. 194, 40–46 e4. doi: 10.1016/j.jpeds.2017.10.052
Cree, L. M., Samuels, D. C., de Sousa Lopes, S. C., Rajasimha, H. K., Wonnapinij, P., Mann, J. R., et al. (2008). A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 40, 249–254. doi: 10.1038/ng.2007.63
De Fanti, S., Vicario, S., Lang, M., Simone, D., Magli, C., Luiselli, D., et al. (2017). Intra-individual purifying selection on mitochondrial DNA variants during human oogenesis. Hum. Reprod. 32, 1100–1107. doi: 10.1093/humrep/dex051
Dingley, S. D., Polyak, E., Ostrovsky, J., Srinivasan, S., Lee, I., Rosenfeld, A. B., et al. (2014). Mitochondrial DNA variant in COX1 subunit significantly alters energy metabolism of geographically divergent wild isolates in Caenorhabditis elegans. J. Mol. Biol. 426, 2199–2216. doi: 10.1016/j.jmb.2014.02.009
Dobler, R., Dowling, D. K., Morrow, E. H., and Reinhardt, K. (2018). A systematic review and meta-analysis reveals pervasive effects of germline mitochondrial replacement on components of health. Hum. Reprod. Update 24, 519–534. doi: 10.1093/humupd/dmy018
Dowling, D. K. (2014). Evolutionary perspectives on the links between mitochondrial genotype and disease phenotype. Biochim. Biophys. Acta 1840, 1393–1403. doi: 10.1016/j.bbagen.2013.11.013
Ellison, C. K., and Burton, R. S. (2006). Disruption of mitochondrial function in interpopulation hybrids of Tigriopus californicus. Evol. Int. J. Org. Evol. 60, 1382–1391. doi: 10.1111/j.0014-3820.2006.tb01217.x
Eyre-Walker, A. (2017). Mitochondrial replacement therapy: are mito-nuclear interactions likely to be a problem? Genetics 205, 1365–1372. doi: 10.1534/genetics.116.196436
Ferree, A. W., Trudeau, K., Zik, E., Benador, I. Y., Twig, G., Gottlieb, R. A., et al. (2013). MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy 9, 1887–1896. doi: 10.4161/auto.26503
Filograna, R., Koolmeister, C., Upadhyay, M., Pajak, A., Clemente, P., Wibom, R., et al. (2019). Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 5:eaav9824. doi: 10.1126/sciadv.aav9824
Floros, V. I., Pyle, A., Dietmann, S., Wei, W., Tang, W. C. W., Irie, N., et al. (2018). Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 20, 144–151. doi: 10.1038/s41556-017-0017-8
Freyer, C., Cree, L. M., Mourier, A., Stewart, J. B., Koolmeister, C., Milenkovic, D., et al. (2012). Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat. Genet. 44, 1282–1285. doi: 10.1038/ng.2427
Gao, F., Wesolowska, M., Agami, R., Rooijers, K., Loayza-Puch, F., Lawless, C., et al. (2017). Using mitoribosomal profiling to investigate human mitochondrial translation. Wellcome Open Res. 2:116. doi: 10.12688/wellcomeopenres.13119.1
Gershoni, M., Levin, L., Ovadia, O., Toiw, Y., Shani, N., Dadon, S., et al. (2014). Disrupting mitochondrial-nuclear co-evolution affects OXPHOS complex I integrity and impacts human health. Genome Biol. Evol. 6, 2665–2680. doi: 10.1093/gbe/evu208
Gershoni, M., Templeton, A. R., and Mishmar, D. (2009). Mitochondrial bioenergetics as a major motive force of speciation. Bioessays 31, 642–650. doi: 10.1002/bies.200800139
Ginsburg, M., Snow, M. H., and McLaren, A. (1990). Primordial germ cells in the mouse embryo during gastrulation. Development 110, 521–528.
Gomez-Duran, A., Pacheu-Grau, D., Lopez-Gallardo, E., Diez-Sanchez, C., Montoya, J., Lopez-Perez, M. J., et al. (2010). Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 19, 3343–3353. doi: 10.1093/hmg/ddq246
Goto, H., Dickins, B., Afgan, E., Paul, I. M., Taylor, J., Makova, K. D., et al. (2011). Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol. 12:R59. doi: 10.1186/gb-2011-12-6-r59
Gu, X., Kang, X., and Liu, J. (2019). Mutation signatures in germline mitochondrial genome provide insights into human mitochondrial evolution and disease. Hum. Genet. 138, 613–624. doi: 10.1007/s00439-019-02009-5
Hill, G. E. (2019). Reconciling the mitonuclear compatibility species concept with rampant mitochondrial introgression. Integr. Comp. Biol. doi: 10.1093/icb/icz019
Hill, G. E., Havird, J. C., Sloan, D. B., Burton, R. S., Greening, C., and Dowling, D. K. (2019). Assessing the fitness consequences of mitonuclear interactions in natural populations. Biol. Rev. Camb. Philos. Soc. 94, 1089–1104. doi: 10.1111/brv.12493
Holmbeck, M. A., Donner, J. R., Villa-Cuesta, E., and Rand, D. M. (2015). A Drosophila model for mito-nuclear diseases generated by an incompatible interaction between tRNA and tRNA synthetase. Dis. Model Mech. 8, 843–854. doi: 10.1242/dmm.019323
Holt, I. J., Harding, A. E., and Morgan-Hughes, J. A. (1988). Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719. doi: 10.1038/331717a0
Hyslop, L. A., Blakeley, P., Craven, L., Richardson, J., Fogarty, N. M., Fragouli, E., et al. (2016). Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 534, 383–386. doi: 10.1038/nature18303
Innocenti, P., Morrow, E. H., and Dowling, D. K. (2011). Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 332, 845–848. doi: 10.1126/science.1201157
Jayaprakash, A. D., Benson, E. K., Gone, S., Liang, R., Shim, J., Lambertini, L., et al. (2015). Stable heteroplasmy at the single-cell level is facilitated by intercellular exchange of mtDNA. Nucleic Acids Res. 43, 2177–2187. doi: 10.1093/nar/gkv052
Ji, F., Sharpley, M. S., Derbeneva, O., Alves, L. S., Qian, P., Wang, Y., et al. (2012). Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc. Natl. Acad. Sci. U.S.A. 109, 7391–7396. doi: 10.1073/pnas.1202484109
Kenney, M. C., Chwa, M., Atilano, S. R., Falatoonzadeh, P., Ramirez, C., Malik, D., et al. (2014). Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim. Biophys. Acta 1842, 208–219. doi: 10.1016/j.bbadis.2013.10.016
Kern, A. D., and Kondrashov, F. A. (2004). Mechanisms and convergence of compensatory evolution in mammalian mitochondrial tRNAs. Nat. Genet. 36, 1207–1212. doi: 10.1038/ng1451
Kimchi-Sarfaty, C., Oh, J. M., Kim, I. W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528. doi: 10.1126/science.1135308
Kimura, M. (1983). The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press.
Kowald, A., and Kirkwood, T. B. (2013). Mitochondrial mutations and aging: random drift is insufficient to explain the accumulation of mitochondrial deletion mutants in short-lived animals. Aging Cell 12, 728–731. doi: 10.1111/acel.12098
Lajbner, Z., Pnini, R., Camus, M. F., Miller, J., and Dowling, D. K. (2018). Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci. Rep. 8:9500. doi: 10.1038/s41598-018-27805-3
Lane, N., and Martin, W. (2010). The energetics of genome complexity. Nature 467, 929–934. doi: 10.1038/nature09486
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. V., Sanchez-Cabo, F., Torroja, C., Acin-Perez, R., et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. doi: 10.1038/nature18618
Lee, H. Y., Chou, J. Y., Cheong, L., Chang, N. H., Yang, S. Y., and Leu, J. Y. (2008). Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 135, 1065–1073. doi: 10.1016/j.cell.2008.10.047
Levin, L., Blumberg, A., Barshad, G., and Mishmar, D. (2014). Mito-nuclear co-evolution: the positive and negative sides of functional ancient mutations. Front. Genet. 5:448. doi: 10.3389/fgene.2014.00448
Levin, L., and Mishmar, D. (2017). The genomic landscape of evolutionary convergence in mammals, birds and reptiles. Nat. Ecol. Evol. 1:0041. doi: 10.1038/s41559-016-0041
Levin, L., Zhidkov, I., Gurman, Y., Hawlena, H., and Mishmar, D. (2013). Functional recurrent mutations in the human mitochondrial phylogeny - dual roles in evolution and disease. Genome Biol. Evol. 5, 876–890. doi: 10.1093/gbe/evt058
Li, M., Schonberg, A., Schaefer, M., Schroeder, R., Nasidze, I., and Stoneking, M. (2010). Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 87, 237–249. doi: 10.1016/j.ajhg.2010.07.014
Ludwig, L. S., Lareau, C. A., Ulirsch, J. C., Christian, E., Muus, C., Li, L. H., et al. (2019). Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339 e22. doi: 10.1016/j.cell.2019.01.022
Marom, S., Friger, M., and Mishmar, D. (2017). MtDNA meta-analysis reveals both phenotype specificity and allele heterogeneity: a model for differential association. Sci. Rep. 7:43449. doi: 10.1038/srep43449
Meiklejohn, C. D., Holmbeck, M. A., Siddiq, M. A., Abt, D. N., Rand, D. M., and Montooth, K. L. (2013). An Incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9:e1003238. doi: 10.1371/journal.pgen.1003238
Meiklejohn, C. D., Montooth, K. L., and Rand, D. M. (2007). Positive and negative selection on the mitochondrial genome. Trends Genet. 23, 259–263. doi: 10.1016/j.tig.2007.03.008
Mercer, T. R., Neph, S., Dinger, M. E., Crawford, J., Smith, M. A., Shearwood, A. M., et al. (2011). The human mitochondrial transcriptome. Cell 146, 645–658. doi: 10.1016/j.cell.2011.06.051
Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark, A. G., Hosseini, S., et al. (2003). Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U.S.A. 100, 171–176. doi: 10.1073/pnas.0136972100
Morris, J., Na, Y. J., Zhu, H., Lee, J. H., Giang, H., Ulyanova, A. V., et al. (2017). Pervasive within-mitochondrion single-nucleotide variant heteroplasmy as revealed by single-mitochondrion sequencing. Cell Rep. 21, 2706–2713. doi: 10.1016/j.celrep.2017.11.031
Payne, B. A., Wilson, I. J., Yu-Wai-Man, P., Coxhead, J., Deehan, D., Horvath, R., et al. (2013). Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390. doi: 10.1093/hmg/dds435
Rackham, O., Shearwood, A. M., Mercer, T. R., Davies, S. M., Mattick, J. S., and Filipovska, A. (2011). Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 17, 2085–2093. doi: 10.1261/rna.029405.111
Rebolledo-Jaramillo, B., Su, M. S., Stoler, N., McElhoe, J. A., Dickins, B., Blankenberg, D., et al. (2014). Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc Natl Acad Sci U.S.A. 111, 15474–15479. doi: 10.1073/pnas.1409328111
Reiner, J. E., Kishore, R. B., Levin, B. C., Albanetti, T., Boire, N., Knipe, A., et al. (2010). Detection of heteroplasmic mitochondrial DNA in single mitochondria. PLoS ONE. 5:e14359. doi: 10.1371/journal.pone.0014359
Rooijers, K., Loayza-Puch, F., Nijtmans, L. G., and Agami, R. (2013). Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat. Commun. 4:2886. doi: 10.1038/ncomms3886
Rubio, M. A., and Hopper, A. K. (2011). Transfer RNA travels from the cytoplasm to organelles. Wiley Interdiscip. Rev. RNA 2, 802–817. doi: 10.1002/wrna.93
Rubio, M. A., Rinehart, J. J., Krett, B., Duvezin-Caubet, S., Reichert, A. S., Soll, D., et al. (2008). Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc. Natl. Acad. Sci. U.S.A. 105, 9186–9191. doi: 10.1073/pnas.0804283105
Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V., and Wallace, D. C. (2004). Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226. doi: 10.1126/science.1088434
Sagan, L. (1967). On the origin of mitosing cells. J. Theor. Biol. 14, 255–274. doi: 10.1016/0022-5193(67)90079-3
Samuels, D. C., Li, C., Li, B., Song, Z., Torstenson, E., Boyd Clay, H., et al. (2013). Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 9:e1003929. doi: 10.1371/journal.pgen.1003929
Schneider, A. (2011). Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu. Rev. Biochem. 80, 1033–1053. doi: 10.1146/annurev-biochem-060109-092838
Schon, E. A., and Gilkerson, R. W. (2010). Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim. Biophys. Acta 1800, 245–249. doi: 10.1016/j.bbagen.2009.07.007
Sharpley, M. S., Marciniak, C., Eckel-Mahan, K., McManus, M., Crimi, M., Waymire, K., et al. (2012). Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. doi: 10.1016/j.cell.2012.09.004
Simonetti, S., Chen, X., DiMauro, S., and Schon, E. A. (1992). Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim. Biophys. Acta 1180, 113–122. doi: 10.1016/0925-4439(92)90059-V
Stern, A., Doron-Faigenboim, A., Erez, E., Martz, E., Bacharach, E., and Pupko, T. (2007). Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 35, W506–W511. doi: 10.1093/nar/gkm382
Stewart, J. B., and Chinnery, P. F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. doi: 10.1038/nrg3966
Stewart, J. B., Freyer, C., Elson, J. L., Wredenberg, A., Cansu, Z., Trifunovic, A., et al. (2008). Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6:e10. doi: 10.1371/journal.pbio.0060010
Stoeckle, M. Y., and Thaler, D. S. (2018). Why should mitochondria define species? BioRxiv doi: 10.1101/276717
Suissa, S., Wang, Z., Poole, J., Wittkopp, S., Feder, J., Shutt, T. E., et al. (2009). Ancient mtDNA genetic variants modulate mtDNA transcription and replication. PLoS Genet. 5:e1000474. doi: 10.1371/journal.pgen.1000474
Telschow, A., Gadau, J., Werren, J. H., and Kobayashi, Y. (2019). Genetic incompatibilities between mitochondria and nuclear genes: effect on gene flow and speciation. Front. Genet. 10:62. doi: 10.3389/fgene.2019.00062
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. doi: 10.1101/gr.772403
Tobler, M., Barts, N., and Greenway, R. (2019). Mitochondria and the origin of species: bridging genetic and ecological perspectives on speciation processes. Integr. Comp. Biol. doi: 10.1093/icb/icz025
Trier, C. N., Hermansen, J. S., Saetre, G. P., and Bailey, R. I. (2014). Evidence for mito-nuclear and sex-linked reproductive barriers between the Hybrid Italian sparrow and its parent species. PLoS Genet. 10:e1004075. doi: 10.1371/journal.pgen.1004075
Twig, G., and Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxidants Redox Signal. 14, 1939–1951. doi: 10.1089/ars.2010.3779
Valenci, I., Yonai, L., Bar-Yaacov, D., Mishmar, D., and Ben-Zvi, A. (2015). Parkin modulates heteroplasmy of truncated mtDNA in Caenorhabditis elegans. Mitochondrion 20, 64–70. doi: 10.1016/j.mito.2014.11.001
Wallace, D. C. (2018). Mitochondrial genetic medicine. Nat. Genet. 50, 1642–1649. doi: 10.1038/s41588-018-0264-z
Wallace, D. C., Singh, G., Lott, M. T., Hodge, J. A., Schurr, T. G., Lezza, A. M., et al. (1988). Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242, 1427–1430. doi: 10.1126/science.3201231
Wei, W., Tuna, S., Keogh, M. J., Smith, K. R., Aitman, T. J., Beales, P. L., et al. (2019). Germline selection shapes human mitochondrial DNA diversity. Science 364. doi: 10.1126/science.aau6520
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. CABIOS 13, 555–556. doi: 10.1093/bioinformatics/13.5.555
Ye, K., Lu, J., Ma, F., Keinan, A., and Gu, Z. (2014). Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. U.S.A. 111, 10654–10659. doi: 10.1073/pnas.1403521111
Zaidi, A. A., and Makova, K. D. (2019). Investigating mitonuclear interactions in human admixed populations. Nat. Ecol. Evol. 3, 213–222. doi: 10.1038/s41559-018-0766-1
Keywords: mitochondria, mtDNA, selection, single cell, single mitochondrion, evolution
Citation: Shtolz N and Mishmar D (2019) The Mitochondrial Genome–on Selective Constraints and Signatures at the Organism, Cell, and Single Mitochondrion Levels. Front. Ecol. Evol. 7:342. doi: 10.3389/fevo.2019.00342
Received: 28 May 2019; Accepted: 26 August 2019;
Published: 18 September 2019.
Edited by:
David S. Thaler, Biozentrum, Universität Basel, SwitzerlandReviewed by:
Geoffrey E. Hill, Auburn University, United StatesMark Stoeckle, The Rockefeller University, United States
Copyright © 2019 Shtolz and Mishmar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Mishmar, dmishmar@bgu.ac.il