 Shyra Wilde
Shyra Wilde Anders F. Johnson
Anders F. Johnson Christopher N. LaRock
Christopher N. LaRock- 1Microbiology and Molecular Genetics Program, Graduate Division of Biological and Biomedical Sciences, Laney Graduate School, Emory University, Atlanta, GA, United States
- 2Department of Microbiology and Immunology, Division of Infectious Diseases, Department of Medicine, and Antibiotic Resistance Center, Emory University School of Medicine, Atlanta, GA, United States
Group A Streptococcus is an obligate human pathogen that is a major cause of infectious morbidity and mortality. It has a natural tropism for the oropharynx and skin, where it causes infections with excessive inflammation due to its expression of proinflammatory toxins and other virulence factors. Inflammation directly contributes to the severity of invasive infections, toxic shock syndrome, and the induction of severe post-infection autoimmune disease caused by autoreactive antibodies. This review discusses what is known about how the virulence factors of Group A Streptococcus induce inflammation and how this inflammation can promote disease. Understanding of streptococcal pathogenesis and the role of hyper-immune activation during infection may provide new therapeutic targets to treat the often-fatal outcome of severe disease.
Introduction
Streptococcus pyogenes (group A Streptococcus; GAS) is an exclusively human pathogen (Walker et al., 2014). GAS specifically colonizes the upper respiratory mucosa, primarily the oropharynx, and tonsils and other associated lymphoid tissues as their primary site for carriage, dissemination to other body sites, and transmission between individuals. Humans, particularly children, are commonly transiently colonized without overt symptoms. Infection can be relatively mild (e.g., ‘strep throat’ pharyngitis, impetigo) or serious (including scarlet fever, puerperal sepsis, bacteremia, streptococcal toxic shock syndrome (STSS), necrotizing fasciitis, endocarditis and pneumonia) and result in post-infection immune sequelae (including acute poststreptococcal glomerulonephritis, rheumatic fever, rheumatic heart disease, and Sydenham chorea). GAS is a major health burden and the cause of an estimated 600,000 annual deaths due to these diseases (Ralph and Carapetis, 2012).
The inflammatory symptoms of infection (fever, redness, and swelling) ultimately coordinate antimicrobial processes to act against the pathogen and resolve infection. Successful bacterial pathogens commonly work to avoid immune recognition and delay or prevent their clearance by antimicrobial immune effectors. Strategies include modifying pathogen-associated molecular pattern (PAMP) agonists of Nod-like receptor (NLR) or Toll-like receptor (TLR) pattern recognition receptors (PRRs) to prevent their recognition (or restrict their accessibility), or interfering with signaling downstream of these receptors using toxins and effectors (LaRock and Nizet, 2015). NLR and TLR PRRs detect numerous GAS PAMPs including lipopeptides, lipoteichoic acid, peptidoglycan, CpG-rich DNA, SLO [reviewed in (LaRock and Nizet, 2015)], SIC (Neumann et al., 2021), and the C-type lectin agonist monoglucosyldiacylglycerol (Imai et al., 2018). GAS also encodes additional virulence factors that promote pathogenesis by directly activating immune responses rather than inhibiting them (Table 1). The complex and seemingly paradoxical contributions of these proinflammatory virulence factor mechanisms are the focus of this review. Additional pathogens with tropism for the skin or upper respiratory mucosa such as Staphylococcus aureus and Streptococcus pneumoniae can derive specific benefits from inflammation. We draw parallels with the commonalities in virulence factors and their underlying molecular mechanisms where applicable.
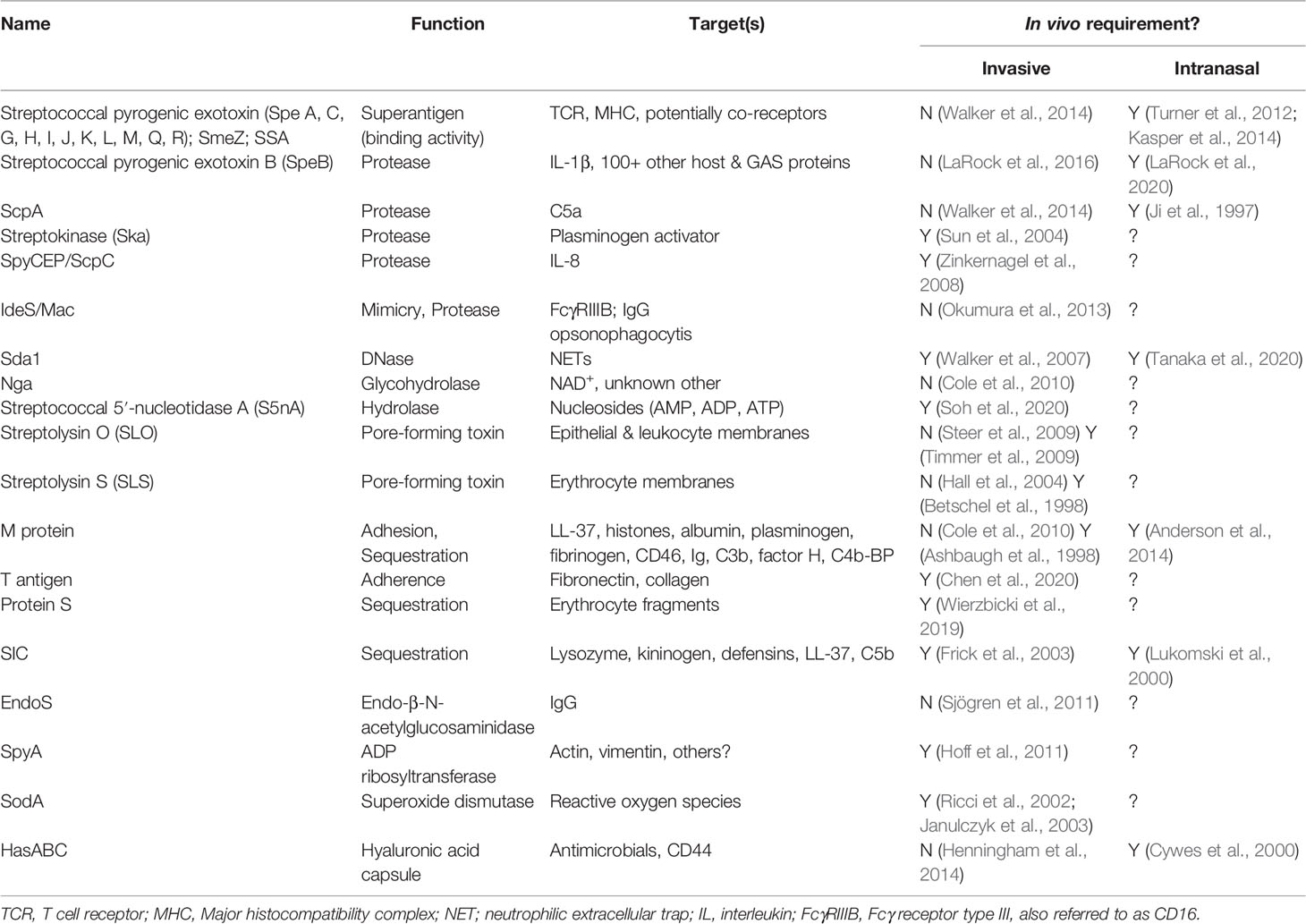
Table 1 Predominant GAS virulence factors, their known mechanisms and targets, and their essentiality in models of invasive (intradermal, subcutaneous, or intravenous) or nasopharyngeal (intranasal) infection.
Roles of Proinflammatory Virulence Factors During Infection and in Disease
Proinflammatory pathways have evolved to initiate and coordinate the immune response against pathogens to protect against infection. Correspondingly, transgenic mice with deficiencies in inflammatory signaling are generally more susceptible to infection, as are humans with immunodeficiencies due to genetic disorders, preexisting conditions, or anti-inflammatory drugs (von Bernuth et al., 2008; Picard et al., 2011; Kidd et al., 2015; LaRock et al., 2016; Rose-John et al., 2017; Wilde et al., 2021). The study of when pathogens instead benefit from inflammation is an emergent field that runs counter to this prevailing paradigm.
During GAS infection of the murine nasopharynx (a model of strep throat), neutrophils, T cells, and the proinflammatory cytokine IL-1β all promote GAS growth (Zeppa et al., 2017; LaRock et al., 2020), despite conventionally having vital antimicrobial roles in immunity and acting to counter GAS invasion at other body sites (LaRock et al., 2016; Pinho-Ribeiro et al., 2018). In antibiotic-treated mice, neutrophils and IL-1β are no longer essential for GAS colonization of the nasopharynx, suggesting this immune axis may be necessary for overcoming interference from antibiotic-sensitive members of the resident microbiota (LaRock et al., 2020). Diverse pathogens including Salmonella enterica serovar Typhimurium (Winter et al., 2010; Arpaia et al., 2011), Helicobacter pylori (Kusters et al., 2006), Pseudomonas aeruginosa (Palomo et al., 2014; Sun et al., 2020), and Candida albicans (Jawhara et al., 2008) also subvert inflammation to their advantage, either to disrupt membrane barrier function, promote dissemination, acquire nutrients, or antagonize competing microbes. For the best-characterized example closely related to GAS, Streptococcus pneumoniae, inflammation broadly promotes growth and transmission, though T cells, neutrophils, and IL-1β each have specifically antagonistic effects on S. pneumoniae growth (Weiser et al., 2018). Thus while several pathogens have a common strategy of subverting inflammation for their benefit, the specific mechanisms and benefits can greatly differ.
The molecular basis underlying the requirement for T cells at the earliest stages of GAS infection is more enigmatic, but later T cells responses are uncoordinated, non-specific, and potentially less effective (Zeppa et al., 2017). Excessive inflammation can promote epitope spreading, whereby increased activation of antigen-presenting cells and T cells leads to broader specificities and increased chance of recognizing self-antigen (Lehmann et al., 1992; Brocke et al., 1993). Recurrent GAS infections drive the generation of autoreactive antibodies that cross-react with heart valve endothelium, lysogangliosides, dopamine receptors, and other human tissues (Cunningham, 2019). These antibodies may give rise to immune sequelae such as acute rheumatic fever (ARF) and rheumatic heart disease (RHD), which account for a majority of annual deaths from GAS (Ralph and Carapetis, 2012); however, the pathogenesis of these conditions remains controversial. A neurological manifestation of these autoreactive antibodies is Sydenham chorea (SC), characterized by an uncontrolled movement of the arms, legs, and facial muscles (Joshi et al., 2015).
The aberrant immune activation GAS has evolved to promote nasopharyngeal infection drives morbidity and mortality when GAS is found in other body sites. Excessive systemic inflammation directly induces death through sepsis, organ failure, disseminated intravascular coagulation, thrombosis, and edema (Cohen et al., 2015). During invasive skin infections like necrotizing fasciitis, inflammation drives localized microvascular thrombosis, tissue hemorrhage, and cell infiltrate, all which further the proinflammatory cycle (Keller et al., 2018). Through these mechanisms, which can limit the perfusion of antibiotics and provide a protective intracellular niche within macrophages, inflammation may also contribute to antibiotic failure (Thulin et al., 2006). In the following sections, we discuss how specific GAS virulence factors that induce inflammation contribute to disease. We focus on those for which inflammation is a specifically beneficial feature for GAS and not only a linked but detrimental consequence ancillary to a secondary function.
SpeB, a Broad-Spectrum Protease
The Streptococcal pyrogenic exotoxin B (SpeB) is a secreted cysteine protease with broad specificity, high turnover, and which can comprise up to 95% of the total secreted protein that can be detected from GAS (Aziz et al., 2004). In tissue, SpeB cleavage of occludin, E-cadherin, and desmoglein can disrupt tight junctions and may enhance tissue invasion and edema (Sumitomo et al., 2013; Sumitomo et al., 2018). SpeB targets several components of the immune system, including cytokines and chemokines, which may inhibit their signaling (Egesten et al., 2009); and cathelicidin/LL-37, defensins, complement proteins, and immunoglobulins, which may enhance GAS resistance to these immune effector molecules (Table 1). Cleavage by SpeB is generally assumed to be exclusively destructive, but SpeB cleavage activates bradykinin (Herwald et al., 1996), the protease-activated receptor PAR-1 (Ender et al., 2013), matrix metalloprotease zymogens (Burns et al., 1996; Tamura et al., 2004), and the proinflammatory cytokine IL-1β (LaRock et al., 2016; LaRock et al., 2020). The pathogenic benefit GAS derives from activating proinflammatory cytokines such as IL-1β requires additional study, but may involve disrupting colonization resistance mediated by the microbiota (LaRock et al., 2020). Conversely, SpeB can cleave and potentially inactivate most GAS virulence factors (Aziz et al., 2004), including those with proinflammatory activities such as SLO, superantigens, and M protein (all discussed in greater detail in following sections). However, this may occur only to a limited degree during infection since both SpeB and the virulence factors it cleaves are essential (Aziz et al., 2004; Cole et al., 2006; Walker et al., 2007; Walker et al., 2014). Nonetheless, SpeB is conserved and promotes GAS survival by intradermal (LaRock et al., 2016), subcutaneous (Ashbaugh et al., 1998; Cole et al., 2006), intraperitoneal (Lukomski et al., 1997; Kuo et al., 1998; Lukomski et al., 1998), and intranasal (Lukomski et al., 1999; Makthal et al., 2018; LaRock et al., 2020) routes in murine experimental infections (Table 1).
Some of the underlying cost-benefit of SpeB expression is clear from mutants recovered from natural and experimental infections. These include mutations in the two-component regulatory system CovRS (CsrRS) that lead to constitutive speB repression, increased expression of other virulence factors that promote tissue invasion, and shifts in cytokine responses (Li et al., 2014; LaRock et al., 2016; Williams et al., 2021). Development of these mutations is variable between GAS serotypes, and more rarely, other mutations are found to disrupt speB expression, such as in the positive regulator ropB (Friaes et al., 2015; Feng et al., 2016). CovRS mutants are widely observed in human invasive infection (Chatellier et al., 2000; Kansal et al., 2000; Olsen et al., 2015) and murine models of invasive infection (Engleberg et al., 2001), and less frequently during human pharyngitis (Sumby et al., 2006; Ikebe et al., 2010; Shea et al., 2011) or murine models of pharyngitis (LaRock et al., 2016, Cole et al., 2010; Fiebig et al., 2015; Horstmann et al., 2018; LaRock et al., 2020). CovRS mutants are attenuated in transmission and colonization (Hollands et al., 2010), but can have pathogenic synergy in combination with non-mutant GAS that increases disease severity in murine models of invasive infection (Horstmann et al., 2018).
Superantigens
Superantigens are secreted virulence factors that act as T cell mitogens and have strong proinflammatory activity (Proft and Fraser, 2016). Superantigens bypass conventional processing by Antigen Presenting Cells (APCs) and directly bind Major Histocompatibility Complex class II (MHC-II) proteins to the variable β-chain of the T cell Receptor (TCR) (Spaulding et al., 2013). The crosslinking allows for activation of up to 30% of human T cells, compared to the 0.01% typical of an antigen (Lappin and Ferguson, 2009). Mucosal associated invariant T (MAIT) cells are the most responsive to superantigens (Shaler et al., 2017) and major contributors to the “cytokine storm” of pathological and proinflammatory IFN-γ, TNF, IL-6, and IL-1β (Figure 1) during STSS (Emgård et al., 2019).
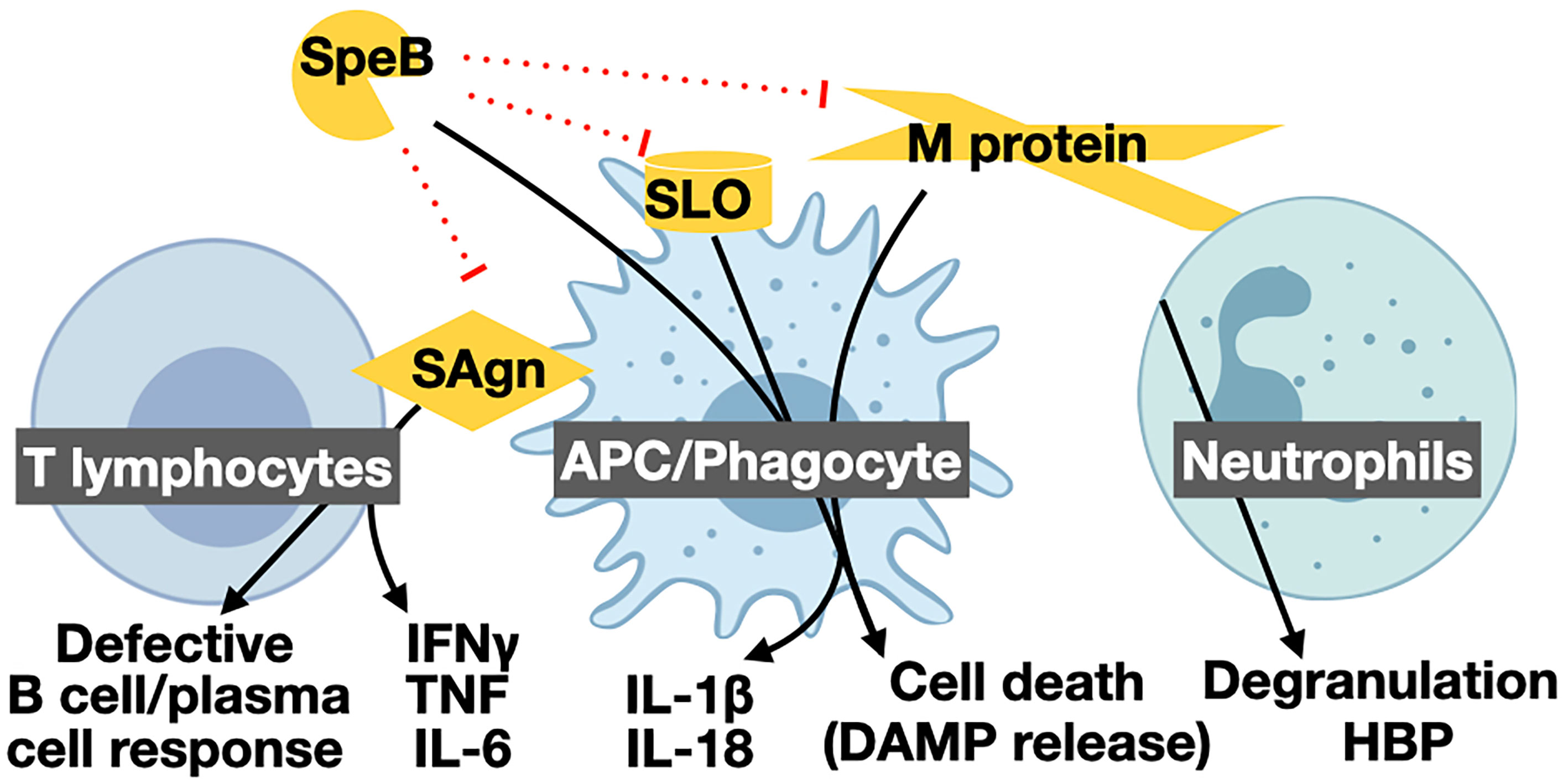
Figure 1 Proinflammatory virulence mechanisms of GAS and their targets. The GAS protease SpeB is directly proinflammatory by activating pro-IL-1β, other host substrates, and inactivating anti-inflammatory GAS effectors. SpeB cleavage of other proinflammatory cytokines, and proinflammatory virulence factors such as superantigens (SAgn), streptolysin O (SLO), and M protein can lead to their inactivation and have anti-inflammatory contributions. Superantigens forcibly bind T lymphocytes and APCs, leading to excessive T cell activation. Activated T cells kill other immune cells and release a “cytokine storm” of IFN-γ, TNF, and IL-6, hallmark of STSS. The pore-forming toxins SLO and streptolysin S (SLS) form large pores in host cells that can lead to the passive release DAMPs, and other cytosolic or organelle-associated proinflammatory compounds, or be detected by the inflammasome to further activate inflammatory cell death by pyroptosis. Proinflammatory effects of SLO can include aiding translocation of the virulence factors Nga. M protein proteolytically released from the GAS surface can similarly form complexes that induce pyroptosis in macrophages or hyper-degranulation by neutrophils. Like other microbes, GAS has numerous TLR agonists that activate proinflammatory regulatory programs [reviewed in (LaRock and Nizet, 2015)]. Created with BioRender.com.
Most GAS encode several superantigens (summarized in Table 1), which are typically encoded on mobile genetic elements like lysogenic phage (Ben Zakour et al., 2015). This wide prevalence suggests there is a selective benefit to express superantigens. The other major superantigen-producing pathogen is Staphylococcus aureus, which can use superantigens to control bacterial number during nasal colonization of mice (Xu et al., 2015). Superantigens and T cells clearly promote GAS growth in murine nasopharyngeal infections (Kasper et al., 2014; Zeppa et al., 2017). While additional work is required to delineate the pathogenic benefit for GAS, superantigens potentially dampen restrictive innate immune responses by inducing T cell anergy or to increase nutrient availability as a result of inflammation (Sriskandan et al., 2007). Recent research suggests this is true in humans, and shows that superantigens can also direct germinal center follicular helper T cells to kill B cells, and patients with recurrent tonsillitis have smaller tonsil germinal centers and reduced antibody responses (Dan et al., 2019). Thus, superantigens may not only promote infection but also induce host susceptibility to subsequent infection.
Pore-Forming Toxins
One of the first virulence factors identified in GAS was streptolysin O (SLO), a toxin that forms pores ~30 μm in diameter in the plasma membrane of target cells (Herbert and Todd, 1941). SLO targets immune cells and keratinocytes for translocation of the co-regulated NAD-glycohydrolase toxin Nga (Sumby et al., 2005), which can promote the intracellular survival of GAS (Bastiat-Sempe et al., 2014). Like many other pore-forming toxins, SLO is TLR4 agonist and can induce proinflammatory cytokine expression (Park et al., 2004), and also activates the NLRP3 inflammasome to induce inflammatory cell death by pyroptosis of macrophages (Figure 1) and the maturation and release of proinflammatory IL-1β (Timmer et al., 2009). This inflammation may be beneficial for GAS during pharyngitis, where IL-1β promotes GAS growth (LaRock et al., 2020), but may also work to eliminate cells that could restrict GAS growth in other circumstances. During interactions with neutrophils, SLO leads to release of azurocidin that induces edema (Nilsson et al., 2006), while in all cells, the released cytosolic contents are detected by PRRs as damage-associated molecular patterns (DAMPs) that induce further inflammation [Reviewed in (LaRock and Nizet, 2015)]. Other recent work indicates that sub-lethal quantities of SLO and NGA can be anti-inflammatory, leading to degradation of pro-IL-1β and suppress IL-1β activation, suggesting there are more complexities to this immunomodulation yet to discover (Hancz et al., 2017; Hancz et al., 2019; Westerlund et al., 2019).
GAS encodes a second pore-forming toxin, streptolysin S (SLS), which resembles bacteriocins in sequence and function (Nizet et al., 2000). SLS lyses a broad array of cells including erythrocytes, platelets, lymphocytes, and keratinocytes (Ofek et al., 1970), and is responsible for β-hemolysis (Table 1). SLS-induced death of keratinocytes can disrupt tissue, promoting lesion formation and dissemination (Sumitomo et al., 2011). SLS also activates the p38 MAPK and NF-kB pathways, broadly inducing of IL-1β, IL-6, and other proinflammatory cytokines (Goldmann et al., 2009) that can be beneficial for GAS at sites where inflammation is beneficial, such as the nasopharynx (LaRock et al., 2020). Lastly, SLS directly activates nociceptor neurons to release the neuropeptide calcitonin gene-related peptide (CGRP), which induces pain and increases necrotizing fasciitis severity (Pinho-Ribeiro et al., 2018).
M Protein
M protein is the most abundant protein on the GAS surface, forming dimeric coil-coils that extend as hair-like fibrils from the cell wall (Smeesters et al., 2010). There are over 250 allelic variants, each with the variable ability for binding different host factors which include fibrinogen, plasminogen, C4b-binding protein, Protein H, IgA, IgG, LL-37, and the histones contained within antimicrobial neutrophil extracellular traps (Table 1). Each of these interactions can promote virulence by both binding host immune effectors and coating the bacterial surface with a barrier of non-immunogenic endogenous proteins. Binding to each of these factors primarily occurs in the variable N-terminal region of M protein; the C-terminus is more conserved and thought to function as a stalk to project these functions distally from the surface (Ghosh, 2018). Few M protein alleles have been comprehensively examined for their host factors they target, but the binding motifs mediating most of these interactions are widespread, with most M alleles predicted to bind several different host factors (Sanderson-Smith et al., 2014).
Surface-anchored M protein is not proinflammatory, however, some alleles of M protein can also be released and gain drastically altered activities (Elliott, 1945; Berge and Bjorck, 1995; Herwald et al., 2004; LaRock et al., 2015; Dohrmann et al., 2017; Valderrama et al., 2017). Both neutrophil proteases (Herwald et al., 2004) and SpeB (Berge and Bjorck, 1995) may be involved in M protein release. Soluble M protein has greater availability as an antigen, a PRR agonist, and can form toxic aggregates with fibrinogen to induce cell death, neutrophil degranulation, and vasculature leakage responses (Herwald et al., 2004; Soehnlein et al., 2008; Macheboeuf et al., 2011; Hertzén et al., 2012; Walker et al., 2014). Some M protein alleles may also have weak T cell mitogen activity (Påhlman et al., 2007). Binding of M protein to platelets leads to their activation and thrombosis (Horváth et al., 2004; Shannon et al., 2007; Palm et al., 2019). Lastly, M protein is a major agonist of TLR2, inducing expression of the numerous proinflammatory molecules regulated by NF-kB (Pahlman et al., 2006), and activates the NLRP3 inflammasome, resulting in proinflammatory cell death by pyroptosis (Valderrama et al., 2017). As with pore-forming toxins, these proinflammatory activities contribute to the pathology and complications of infection, but it is not clear whether they are required for virulence.
Potential for Therapeutics to Manage Infection
There is no GAS vaccine after 100 years of research. Ongoing preclinical and clinical work focus on the immunodominant, but variable, M protein, the conserved group A carbohydrate, and multi-valent vaccines against major GAS virulence factors [reviewed in (Dale et al., 2016)]. Invasive infections have a mortality rate upwards of 20% within seven days of the onset, and treatments include antibiotic therapy and surgical debridement of infected necrotic tissue (Wilkins et al., 2017). GAS remains penicillin sensitive (Vannice et al., 2020), and clindamycin is recommended for severe infections and patients with penicillin allergies (Sriskandan et al., 1997; Mascini et al., 2001; Steer et al., 2012). Macrolide resistance is increasingly prevalent (Silva-Costa et al., 2015; Fyfe et al., 2016; Michos et al., 2016), but tedizolid and linezolid may be used instead (Bryant et al., 2020). Despite the availability of antibiotics to treat infection, antibiotic monotherapy can fail to eradicate GAS during pharyngitis or invasive disease (Kim and Kaplan, 1985; Steer et al., 2012). Antibiotic inefficacy is multifactorial and can be due to bacterial tolerance, reservoirs of protected intracellular bacteria, failure of the drug to reach the infection site due to tissue necrosis and thrombosis, and the rapid progression of disease (Spinaci et al., 2006; Hertzén et al., 2010; Fischetti and Dale, 2016). Below, we will discuss recent developments in therapeutics based on targeting inflammation and proinflammatory virulence factors.
Intravenous immunoglobulin (IVIG) are non-specific antibodies pooled from human donors. As an adjunctive therapy, IVIG may decrease morbidity by not only opsonizing the bacteria, but neutralizing GAS exotoxins (Norrby-Teglund et al., 2005; Low, 2013). In murine models, IVIG treatment in conjunction with penicillin and clindamycin was successful at modulating the systemic inflammatory response as well as increasing bacterial killing (Sriskandan et al., 2006). In human trials, IVIG treatment is associated with a 20-30% increase in survival during STSS (Linnér et al., 2014), but this may have lessor benefit in children (Shah et al., 2009). The use of IVIG for the treatment of GAS disease is an area of active research, but shows promise in safety and efficacy, though may be cost-prohibitive in many regions where the health burden is highest (Williams et al., 2016).
Several FDA-approved drugs have potential for off-label use during GAS disease. The HIV protease inhibitor nelfinavir inhibits streptolysin S, limiting its pro-virulence and proinflammatory activities (Maxson et al., 2015). Excessive inflammation during STSS and other conditions is harmful, and therapeutics targeting inflammation may also have therapeutic benefit. During invasive infections, the opioid-derivative cough suppressant dextromethorphan may prolong survival through its anti-inflammatory activities (Li et al., 2011; Chen et al., 2018; Zhou et al., 2019). Drugs that block nociceptor signaling also promote beneficial immune responses (Pinho-Ribeiro et al., 2018). Lastly, inhibiting protein synthesis can block production of SpeA, the nuclease Sda1, SLO, and other toxins (Herbert et al., 2001; Mascini et al., 2001; Goscinski et al., 2006; Andreoni et al., 2017). Thus, antibiotics like clindamycin may have therapeutic benefits in addition to direct killing of GAS.
Concluding Remarks
As an obligate human pathogen GAS is highly adapt at manipulating human innate and adaptive immune responses. This often serves to resist immune effectors, mask the pathogen, and subvert immune signaling (Table 1), but GAS also induces and thrives off the activation of other immune processes (Figure 1). New insights into the molecular mechanisms involved may come from forward genetic screens based on high-throughput sequencing of transposon mutant libraries, which have already provided new insights into regulatory, metabolic, and antimicrobial resistance mechanisms that contribute to fitness during infection (Le Breton et al., 2015; Zhu et al., 2019). The complicated relationship of GAS with inflammation can underlie infectious risk with some anti-inflammatory therapeutics, like inhibitors of IL-1β, IL-6, or TNF, but may provide opportunities for treatments with other immune-targeted drugs.
Host-directed molecules based on a better understanding of the role of inflammation in GAS pathogenesis hold hope for future therapeutics. Debilitating and difficult-to-treat disease manifestations following GAS infection like rheumatic heart disease, scarlet fever, toxic shock syndrome, and post-streptococcal glomerulonephritis result from aberrant and excessive immune responses during and after infection and can potentially be treated through immunomodulation. Since many GAS deaths are from immune-mediated complications, knowledge of which GAS factors induce inflammation, and which inflammatory pathways drive pathology in immune disease, will be essential for these strategies. Careful dissection of these processes will be essential to develop adjunctive therapies to reduce the significant morbidity and mortality caused by GAS.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
Work in the LaRock lab is supported by NIH/NIAID AI153071. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, E. L., Cole, J. N., Olson, J., Ryba, B., Ghosh, P., Nizet, V., et al. (2014). The Fibrinogen-Binding M1 Protein Reduces Pharyngeal Cell Adherence and Colonization Phenotypes of M1T1 Group A Streptococcus. J. Biol. Chem. 289, 3539–3546. doi: 10.1074/jbc.M113.529537
Andreoni, F., Zürcher, C., Tarnutzer, A., Schilcher, K., Neff, A., Keller, N., et al. (2017). Clindamycin Affects Group A Streptococcus Virulence Factors and Improves Clinical Outcome. J. Infect. Dis. 215, 269–277. doi: 10.1093/infdis/jiw229
Arpaia, N., Godec, J., Lau, L., Sivick, K. E., McLaughlin, L. M., Jones, M. B., et al. (2011). TLR Signaling Is Required for Salmonella Typhimurium Virulence. Cell 144, 675–688. doi: 10.1016/j.cell.2011.01.031
Ashbaugh, C. D., Warren, H. B., Carey, V. J., Wessels, M. R. (1998). Molecular Analysis of the Role of the Group A Streptococcal Cysteine Protease, Hyaluronic Acid Capsule, and M Protein in a Murine Model of Human Invasive Soft-Tissue Infection. J. Clin. Invest. 2 (4), 283–292. doi: 10.1172/JCI3065
Aziz, R. K., Pabst, M. J., Jeng, A., Kansal, R., Low, D. E., Nizet, V., et al. (2004). Invasive M1T1 Group A Streptococcus Undergoes a Phase-Shift In Vivo to Prevent Proteolytic Degradation of Multiple Virulence Factors by SpeB. Mol. Microbiol. 51, 123–134. doi: 10.1046/j.1365-2958.2003.03797.x
Bastiat-Sempe, B., Love, J. F., Lomayesva, N., Wessels, M. R. (2014). Streptolysin O and NAD-Glycohydrolase Prevent Phagolysosome Acidification and Promote Group a Streptococcus Survival in Macrophages. mBio 5, e01690–e01614. doi: 10.1128/mBio.01690-14
Ben Zakour, N. L., Davies, M. R., You, Y., Chen, J. H. K., Forde, B. M., Stanton-Cook, M., et al. (2015). Transfer of Scarlet Fever-Associated Elements Into the Group A Streptococcus M1T1 Clone. Sci. Rep. 5, 15877. doi: 10.1038/srep15877
Berge, A., Bjorck, L. (1995). Streptococcal Cysteine Proteinase Releases Biologically Active Fragments of Streptococcal Surface Proteins. J. Biol. Chem. 270 (17), 9862–9867. doi: 10.1074/jbc.270.17.9862
Betschel, S. D., Borgia, S. M., Barg, N. L., Low, D. E., De Azavedo, J. C. S. (1998). Reduced Virulence of Group A Streptococcal Tn916 Mutants That Do Not Produce Streptolysin S. Infect. Immun. 66, 1671–1679. doi: 10.1128/IAI.66.4.1671-1679.1998
Brocke, S., Gaur, A., Piercy, C., Gautam, A., Gijbels, K., Fathman, C. G., et al. (1993). Induction of Relapsing Paralysis in Experimental Autoimmune Encephalomyelitis by Bacterial Superantigen. Nature 365, 642–644. doi: 10.1038/365642a0
Bryant, A. E., Bayer, C. R., Aldape, M. J., McIndoo, E., Stevens, D. L. (2020). Emerging Erythromycin and Clindamycin Resistance in Group A Streptococcus: Efficacy of Linezolid and Tedizolid in Experimental Necrotizing Infection. J. Glob. Antimicrob. Resist. 22, 601–607. doi: 10.1016/j.jgar.2020.04.032
Burns, E. H., Marciel, A. M., Musser, J. M. (1996). Activation of a 66-Kilodalton Human Endothelial Cell Matrix Metalloprotease by Streptococcus Pyogenes Extracellular Cysteine Protease. Infect. Immun. 64, 4744–4750. doi: 10.1128/iai.64.11.4744-4750.1996
Chatellier, S., Ihendyane, N., Kansal, R. G., Khambaty, F., Basma, H., Norrby-Teglund, A., et al. (2000). Genetic Relatedness and Superantigen Expression in Group A Streptococcus Serotype M1 Isolates From Patients With Severe and Nonsevere Invasive Diseases. Infect. Immun. 68, 3523–3534. doi: 10.1128/IAI.68.6.3523-3534.2000
Chen, C.-L., Cheng, M.-H., Kuo, C.-F., Cheng, Y.-L., Li, M.-H., Chang, C.-P., et al. (2018). Dextromethorphan Attenuates NADPH Oxidase-Regulated Glycogen Synthase Kinase 3β and NF-κb Activation and Reduces Nitric Oxide Production in Group A Streptococcal Infection. Antimicrob. Agents Chemother. 62, e02045-17. doi: 10.1128/AAC.02045-17
Chen, Y.-H., Li, S.-H., Yang, Y.-C., Hsu, S.-H., Nizet, V., Chang, Y.-C. (2020). T4 Pili Promote Colonization and Immune Evasion Phenotypes of Nonencapsulated M4 Streptococcus Pyogenes. mBio 11, e01580-20. doi: 10.1128/mBio.01580-20
Cohen, J., Vincent, J.-L., Adhikari, N. K. J., Machado, F. R., Angus, D. C., Calandra, T., et al. (2015). Sepsis: A Roadmap for Future Research. Lancet Infect. Dis. 15, 581–614. doi: 10.1016/S1473-3099(15)70112-X
Cole, J., McArthur, J. D., McKay, F. C., Sanderson-Smith, M. L., Cork, A. J., Ranson, M., et al. (2006). Trigger for Group A Streptococcal M1T1 Invasive Disease. FASEB J. 20 (10), 1745–1747. doi: 10.1096/fj.06-5804fje
Cole, J., Pence, M. A., von Köckritz-Blickwede, M., Hollands, A., Gallo, R. L., Walker, M. J., et al. (2010). M Protein and Hyaluronic Acid Capsule Are Essential for In Vivo Selection of covRS Mutations Characteristic of Invasive Serotype M1T1 Group A Streptococcus. mBio 1, e00191-10. doi: 10.1128/mBio.00191-10
Cunningham, M. W. (2019). Molecular Mimicry, Autoimmunity and Infection: The Cross-Reactive Antigens of Group A Streptococci and Their Sequelae. Microbiol. Spectr. 7, 10.1128. doi: 10.1128/9781683670131.ch7
Cywes, C., Stamenkovic, I., Wessels, M. R. (2000). CD44 as a Receptor for Colonization of the Pharynx by Group A Streptococcus. J. Clin. Invest. 106, 995–1002. doi: 10.1172/JCI10195
Dale, J. B., Batzloff, M. R., Cleary, P. P., Courtney, H. S., Good, M. F., Grandi, G., et al. (2016). “Current Approaches to Group A Streptococcal Vaccine Development,” in Streptococcus Pyogenes: Basic Biology to Clinical Manifestations. Eds. Ferretti, J. J., Stevens, D. L., Fischetti, V. A. (Oklahoma City, OK: University of Oklahoma Health Sciences Center). Available at: http://www.ncbi.nlm.nih.gov/books/NBK333413/ (Accessed September 15, 2020).
Dan, J. M., Havenar-Daughton, C., Kendric, K., Al-kolla, R., Kaushik, K., Rosales, S. L., et al. (2019). Recurrent Group A Streptococcus Tonsillitis Is an Immunosusceptibility Disease Involving Antibody Deficiency and Aberrant TFH Cells. Sci. Transl. Med. 11, eaau3776. doi: 10.1126/scitranslmed.aau3776
Dohrmann, S., LaRock, C. N., Anderson, E. L., Cole, J. N., Ryali, B., Stewart, C., et al. (2017). Group A Streptococcal M1 Protein Provides Resistance Against the Antimicrobial Activity of Histones. Sci. Rep. 7, 43039. doi: 10.1038/srep43039
Egesten, A., Olin, A. I., Linge, H., Yadav, M., Morgelin, M., Karlsson, A., et al. (2009). SpeB of Streptococcus Pyogenes Differentially Modulates Antibacterial and Receptor Activating Properties of Human Chemokines. PloS One 4, e4769. doi: 10.1371/journal.pone.0004769
Elliott, S. D. (1945). A Proteolytic Enzyme Produced by Group A Streptococci With Special Reference to Its Effect on the Type-Specific M Antigen. J. Exp. Med. 81, 573–592. doi: 10.1084/jem.81.6.573
Emgård, J., Bergsten, H., McCormick, J. K., Barrantes, I., Skrede, S., Sandberg, J. K., et al. (2019). MAIT Cells Are Major Contributors to the Cytokine Response in Group A Streptococcal Toxic Shock Syndrome. Proc. Natl. Acad. Sci. 116 (51), 25923–25931. doi: 10/ggdttg
Ender, M., Andreoni, F., Zinkernagel, A. S., Schuepbach, R. A. (2013). Streptococcal SpeB Cleaved PAR-1 Suppresses ERK Phosphorylation and Blunts Thrombin-Induced Platelet Aggregation. PloS One 8, e81298. doi: 10.1371/journal.pone.0081298
Engleberg, N. C., Heath, A., Miller, A., Rivera, C., DiRita, V. J. (2001). Spontaneous Mutations in the CsrRS Two-Component Regulatory System of Streptococcus Pyogenes Result in Enhanced Virulence in a Murine Model of Skin and Soft Tissue Infection. J. Infect. Dis. 183, 1043–1054. doi: 10.1086/319291
Feng, W., Minor, D., Liu, M., Li, J., Ishaq, S. L., Yeoman, C., et al. (2016). Null Mutations of Group A Streptococcus Orphan Kinase RocA: Selection in Mouse Infection and Comparison With CovS Mutations in Alteration of In Vitro and In Vivo Protease SpeB Expression and Virulence. Infect. Immun. 85 (1), e00790-16. doi: 10.1128/IAI.00790-16
Fiebig, A., Loof, T. G., Babbar, A., Itzek, A., Koehorst, J. J., Schaap, P. J., et al. (2015). Comparative Genomics of Streptococcus Pyogenes M1 Isolates Differing in Virulence and Propensity to Cause Systemic Infection in Mice. Int. J. Med. Microbiol. IJMM. 305 (6), 532–543. doi: 10.1016/j.ijmm.2015.06.002
Fischetti, V. A., Dale, J. B. (2016). One More Disguise in the Stealth Behavior of Streptococcus Pyogenes. mBio 7, e00661-16. doi: 10/gg4q69
Friaes, A., Pato, C., Melo-Cristino, J., Ramirez, M. (2015). Consequences of the Variability of the CovRS and RopB Regulators Among Streptococcus Pyogenes Causing Human Infections. Sci. Rep. 5, 1–12. doi: 10.1038/srep12057
Frick, I.-M., Åkesson, P., Rasmussen, M., Schmidtchen, A., Björck, L. (2003). SIC, a Secreted Protein of Streptococcus Pyogenesthat Inactivates Antibacterial Peptides. J. Biol. Chem. 278, 16561–16566. doi: 10/c28hk9
Fyfe, C., Grossman, T. H., Kerstein, K., Sutcliffe, J. (2016). Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb. Perspect. Med. 6, a025395. doi: 10/gg4q7c
Ghosh, P. (2018). Variation, Indispensability, and Masking in the M Protein. Trends Microbiol. 26, 132–144. doi: 10/gcz3dn
Goldmann, O., Sastalla, I., Wos-Oxley, M., Rohde, M., Medina, E. (2009). Streptococcus Pyogenes Induces Oncosis in Macrophages Through the Activation of an Inflammatory Programmed Cell Death Pathway. Cell. Microbiol. 11, 138–155. doi: 10/dm9jxq
Goscinski, G., Tano, E., Thulin, P., Norrby-Teglund, A., Sjölin, J. (2006). Release of SpeA From Streptococcus Pyogenes After Exposure to Penicillin: Dependency on Dose and Inhibition by Clindamycin. Scand. J. Infect. Dis. 38, 983–987. doi: 10/dfzc4n
Hall, M. A., Stroop, S. D., Hu, M. C., Walls, M. A., Reddish, M. A., Burt, D. S., et al. (2004). Intranasal Immunization With Multivalent Group A Streptococcal Vaccines Protects Mice Against Intranasal Challenge Infections. Infect. Immun. 72, 2507–2512. doi: 10/dqvqn8
Hancz, D., Westerlund, E., Bastiat-Sempe, B., Sharma, O., Valfridsson, C., Meyer, L., et al. (2017). Inhibition of Inflammasome-Dependent Interleukin 1β Production by Streptococcal NAD+-Glycohydrolase: Evidence for Extracellular Activity. mBio 8, e00756–e00717. doi: 10.1128/mBio.00756-17
Hancz, D., Westerlund, E., Valfridsson, C., Aemero, G. M., Bastiat-Sempe, B., Orning, P., et al. (2019). Streptolysin O Induces the Ubiquitination and Degradation of Pro-IL-1β. J. Innate Immun. 11 (6), 457–468. doi: 10.1159/000496403
Henningham, A., Yamaguchi, M., Aziz, R., Kuipers, K., Buffalo, C., Dahesh, S., et al. (2014). Mutual Exclusivity of Hyaluronan and Hyaluronidase in Invasive Group A Streptococcus. J. Biol. Chem. 289 (46), 32303–32315. doi: 10.1074/jbc.M114.602847
Herbert, S., Barry, P., Novick, R. P. (2001). Subinhibitory Clindamycin Differentially Inhibits Transcription of Exoprotein Genes in Staphylococcus Aureus. Infect. Immun. 69, 2996–3003. doi: 10/fcvs56
Herbert, D., Todd, E. W. (1941). Purification and Properties of a Haemolysin Produced by Group A Haemolytic Streptococci (Streptolysin O). Biochem. J. 35, 1124–1139. doi: 10/gg4g6w
Hertzén, E., Johansson, L., Kansal, R., Hecht, A., Dahesh, S., Janos, M., et al. (2012). Intracellular Streptococcus Pyogenes in Human Macrophages Display an Altered Gene Expression Profile. PloS One 7, e35218. doi: 10/f248gs
Hertzén, E., Johansson, L., Wallin, R., Schmidt, H., Kroll, M., Rehn, A. P., et al. (2010). M1 Protein-Dependent Intracellular Trafficking Promotes Persistence and Replication of Streptococcus Pyogenes in Macrophages. J. Innate Immun. 2, 534–545. doi: 10.1159/000317635
Herwald, H., Collin, M., Müller-Esterl, W., Bjorck, L. (1996). Streptococcal Cysteine Proteinase Releases Kinins: A Virulence Mechanism. J. Exp. Med. 184, 665–673. doi: 10.1084/jem.184.2.665
Herwald, H., Cramer, H., Morgelin, M., Russell, W., Sollenberg, U., Norrby-Teglund, A., et al. (2004). M Protein, a Classical Bacterial Virulence Determinant, Forms Complexes With Fibrinogen That Induce Vascular Leakage. Cell. 116 (3), 367–379. doi: 10.1016/S0092-8674(04)00057-1
Hoff, J. S., DeWald, M., Moseley, S. L., Collins, C. M., Voyich, J. M. (2011). SpyA, a C3-Like ADP-Ribosyltransferase, Contributes to Virulence in a Mouse Subcutaneous Model of Streptococcus Pyogenes Infection. Infect. Immun. 79, 2404–2411. doi: 10/c57v5x
Hollands, A., Pence, M. A., Timmer, A. M., Osvath, S. R., Turnbull, L., Whitchurch, C. B., et al. (2010). Genetic Switch to Hypervirulence Reduces Colonization Phenotypes of the Globally Disseminated Group A Streptococcus M1T1 Clone. J. Infect. Dis. 202, 11–19. doi: 10.1086/653124
Horstmann, N., Tran, C. N., Brumlow, C., DebRoy, S., Yao, H., Nogueras Gonzalez, G., et al. (2018). Phosphatase Activity of the Control of Virulence Sensor Kinase CovS Is Critical for the Pathogenesis of Group A Streptococcus. PloS Pathog. 14, e1007354. doi: 10.1371/journal.ppat.1007354
Horváth, A., Olive, C., Karpati, L., Sun, H. K., Good, M., Toth, ,. I. (2004). Toward the Development of a Synthetic Group a Streptococcal Vaccine of High Purity and Broad Protective Coverage. J. Med. Chem. 47, 4100–4104. doi: 10/dwz8z7
Ikebe, T., Ato, M., Matsumura, T., Hasegawa, H., Sata, T., Kobayashi, K., et al. (2010). Highly Frequent Mutations in Negative Regulators of Multiple Virulence Genes in Group A Streptococcal Toxic Shock Syndrome Isolates. PloS Pathog. 6, e1000832. doi: 10/dw7s3q
Imai, T., Matsumura, T., Mayer-Lambertz, S., Wells, C. A., Ishikawa, E., Butcher, S. K., et al. (2018). Lipoteichoic Acid Anchor Triggers Mincle to Drive Protective Immunity Against Invasive Group A Streptococcus Infection. Proc. Natl. Acad. Sci. 18, 201809100. doi: 10.1073/pnas.1809100115
Janulczyk, R., Ricci, S., Björck, L. (2003). MtsABC Is Important for Manganese and Iron Transport, Oxidative Stress Resistance, and Virulence of Streptococcus Pyogenes. Infect. Immun. 71, 2656–2664. doi: 10/fck2nb
Jawhara, S., Thuru, X., Standaert-Vitse, A., Jouault, T., Mordon, S., Sendid, B., et al. (2008). Colonization of Mice by Candida Albicans Is Promoted by Chemically Induced Colitis and Augments Inflammatory Responses Through Galectin-3. J. Infect. Dis. 197, 972–980. doi: 10/cfwx77
Ji, Y., Carlson, B., Kondagunta, A., Cleary, P. P. (1997). Intranasal Immunization With C5a Peptidase Prevents Nasopharyngeal Colonization of Mice by the Group A Streptococcus. Infect. Immun. 65, 2080–2087. doi: 10/gkn7tr
Joshi, A., Shrestha, R. P. B., Shrestha, P. S., Dangol, S., Shrestha, N. C., Poudyal, P., et al. (2015). Sydenham’s Chorea as Presentation of Rheumatic Heart Disease. Kathmandu Univ. Med. J. KUMJ 13, 271–273. doi: 10/gg4g6n
Kansal, R. G., McGeer, A., Low, D. E., Norrby-Teglund, A., Kotb, M. (2000). Inverse Relation Between Disease Severity and Expression of the Streptococcal Cysteine Protease, SpeB, Among Clonal M1T1 Isolates Recovered From Invasive Group A Streptococcal Infection Cases. Infect. Immun. 68, 6362–6369. doi: 10.1128/IAI.68.11.6362-6369.2000
Kasper, K. J., Zeppa, J. J., Wakabayashi, A. T., Xu, S. X., Mazzuca, D. M., Welch, I., et al. (2014). Bacterial Superantigens Promote Acute Nasopharyngeal Infection by Streptococcus Pyogenes in a Human MHC Class II-Dependent Manner. PloS Pathog. 10, e1004155. doi: 10/f57ptc
Keller, N., Andreoni, F., Reiber, C., Luethi-Schaller, H., Schuepbach, R. A., Moch, H., et al. (2018). Human Streptococcal Necrotizing Fasciitis Histopathology Mirrored in a Murine Model. Am. J. Pathol. 188, 1517–1523. doi: 10.1016/j.ajpath.2018.03.009
Kidd, B. A., Wroblewska, A., Boland, M. R., Agudo, J., Merad, M., Tatonetti, N. P., et al. (2015). Mapping the Effects of Drugs on the Immune System. Nat. Biotechnol. 34, 47–54. doi: 10.1038/nbt.3367
Kim, K. S., Kaplan, ,. E. L. (1985). Association of Penicillin Tolerance With Failure to Eradicate Group A Streptococci From Patients With Pharyngitis. J. Pediatr. 107, 681–684. doi: 10/bktcgk
Kuo, C. F., Wu, J. J., Lin, K. Y., Tsai, P. J., Lee, S. C., Jin, Y. T., et al. (1998). Role of Streptococcal Pyrogenic Exotoxin B in the Mouse Model of Group A Streptococcal Infection. Infect. Immun. 66, 3931–3935. doi: 10.1128/IAI.66.8.3931-3935.1998
Kusters, J. G., Vliet, A.H.M., Kuipers, E. J. (2006). Pathogenesis of Helicobacter Pylori Infection. Clin. Microbiol. Rev. 19, 449–490. doi: 10.1128/CMR.00054-05
Lappin, E., Ferguson, A. J. (2009). Gram-Positive Toxic Shock Syndromes. Lancet Infect. Dis. 9, 281–290. doi: 10/bp4rkm
LaRock, C. N., Döhrmann, S., Todd, J., Corriden, R., Olson, J., Johannssen, T., et al. (2015). Group A Streptococcal M1 Protein Sequesters Cathelicidin to Evade Innate Immune Killing. Cell Host Microbe 18, 471–477. doi: 10/f7zgs6
LaRock, C. N., Nizet, V. (2015). Inflammasome/IL-1 β Responses to Streptococcal Pathogens. Front. Immunol. 6, 518. doi: 10.3389/fimmu.2015.00518
LaRock, C. N., Todd, J., LaRock, D. L., Olson, J., O’Donoghue, A. J., Robertson, A. A. B., et al. (2016). IL-1β Is an Innate Immune Sensor of Microbial Proteolysis. Sci. Immunol. 1, eaah3539. doi: 10/gf4ht2
LaRock, D. L., Russell, R., Johnson, A. F., Wilde, S., LaRock, C. N. (2020). Group A Streptococcus Infection of the Nasopharynx Requires Proinflammatory Signaling Through the Interleukin-1 Receptor. Infect. Immun. 88 (10), e00356-20. doi: 10/gg8f4g
Le Breton, Y., Belew, A. T., Valdes, K. M., Islam, E., Curry, P., Tettelin, H., et al. (2015). Essential Genes in the Core Genome of the Human Pathogen Streptococcus Pyogenes. Sci. Rep. 5, 9838. doi: 10.1038/srep09838
Lehmann, P. V., Forsthuber, T., Miller, A., Sercarz, ,. E. E. (1992). Spreading of T-Cell Autoimmunity to Cryptic Determinants of an Autoantigen. Nature 358, 155–157. doi: 10/d94s5w
Li, J., Liu, G., Feng, W., Zhou, Y., Liu, M., Wiley, J. A., et al. (2014). Neutrophils Select Hypervirulent CovRS Mutants of M1T1 Group A Streptococcus During Subcutaneous Infection of Mice. Infect. Immun. 82, 1579–1590. doi: 10/f5vz6m
Li, M.-H., Luo, Y.-H., Lin, C.-F., Chang, Y.-T., Lu, S.-L., Kuo, C.-F., et al. (2011). Dextromethorphan Efficiently Increases Bactericidal Activity, Attenuates Inflammatory Responses, and Prevents Group a Streptococcal Sepsis. Antimicrob. Agents Chemother. 55, 967–973. doi: 10/dfk2vv
Linnér, A., Darenberg, J., Sjölin, J., Henriques-Normark, B., Norrby-Teglund, ,. A. (2014). Clinical Efficacy of Polyspecific Intravenous Immunoglobulin Therapy in Patients With Streptococcal Toxic Shock Syndrome: A Comparative Observational Study. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc Am. 59, 851–857. doi: 10/f24vg3
Low, D. E. (2013). Toxic Shock Syndrome: Major Advances in Pathogenesis, But Not Treatment. Crit. Care Clin. 29, 651–675. doi: 10/ggbr6g
Lukomski, S., Burns, E. H., Wyde, P. R., Podbielski, A., Rurangirwa, J., Moore-Poveda, D. K., et al. (1998). Genetic Inactivation of an Extracellular Cysteine Protease (SpeB) Expressed by Streptococcus Pyogenes Decreases Resistance to Phagocytosis and Dissemination to Organs. Infect. Immun. 66, 771–776. doi: 10.1128/IAI.66.2.771-776.1998
Lukomski, S., Hoe, N. P., Abdi, I., Rurangirwa, J., Kordari, P., Liu, M., et al. (2000). Nonpolar Inactivation of the Hypervariable Streptococcal Inhibitor of Complement Gene (Sic) in Serotype M1 Streptococcus Pyogenes Significantly Decreases Mouse Mucosal Colonization. Infect. Immun. 68, 535–542. doi: 10.1128/IAI.68.2.535-542.2000
Lukomski, S., Montgomery, C. A., Rurangirwa, J., Geske, R. S., Barrish, J. P., Adams, G. J., et al. (1999). Extracellular Cysteine Protease Produced by Streptococcus Pyogenes Participates in the Pathogenesis of Invasive Skin Infection and Dissemination in Mice. Infect. Immun. 67, 1779–1788. doi: 10.1128/IAI.67.4.1779-1788.1999
Lukomski, S., Sreevatsan, S., Amberg, C., Reichardt, W., Woischnik, M., Podbielski, A., et al. (1997). Inactivation of Streptococcus Pyogenes Extracellular Cysteine Protease Significantly Decreases Mouse Lethality of Serotype M3 and M49 Strains. J. Clin. Invest. 99, 2574–2580. doi: 10.1172/JCI119445
Macheboeuf, P., Buffalo, C., Fu, C., Zinkernagel, A. S., Cole, J., Johnson, J., et al. (2011). Streptococcal M1 Protein Constructs a Pathological Host Fibrinogen Network. Nature 472, 64–68. doi: 10.1038/nature09967
Makthal, N., Do, H., VanderWal, A. R., Olsen, R. J., Musser, J. M., Kumaraswami, M. (2018). Signaling by a Conserved Quorum Sensing Pathway Contributes to Growth Ex Vivo and Oropharyngeal Colonization of Human Pathogen Group A Streptococcus. Infect. Immun. 86, e00169-18. doi: 10/gdf7vj
Mascini, E. M., Jansze, M., Schouls, L. M., Verhoef, J., Van Dijk, H. (2001). Penicillin and Clindamycin Differentially Inhibit the Production of Pyrogenic Exotoxins A and B by Group A Streptococci. Int. J. Antimicrob. Agents 18, 395–398. doi: 10/btf34n
Maxson, T., Deane, C. D., Molloy, E. M., Cox, C. L., Markley, A. L., Lee, S. W., et al. (2015). HIV Protease Inhibitors Block Streptolysin S Production. ACS Chem. Biol. 10, 1217–1226. doi: 10/f7crp6
Michos, A., Koutouzi, F. I., Tsakris, A., Chatzichristou, P., Koutouzis, E. I., Daikos, G. L., et al. (2016). Molecular Analysis of Streptococcus Pyogenes Macrolide Resistance of Paediatric Isolates During a 7 Year Perio-13). J. Antimicrob. Chemother. 71, 2113–2117. doi: 10/gg4q7d
Neumann, A., Happonen, L., Karlsson, C., Bahnan, W., Frick, I.-M., Björck, L. (2021). Streptococcal Protein SIC Activates Monocytes and Induces Inflammation. iScience 24, 102339. doi: 10/gknzvn
Nilsson, M., Sørensen, O. E., Mörgelin, M., Weineisen, M., Sjöbring, U., Herwald, H. (2006). Activation of Human Polymorphonuclear Neutrophils by Streptolysin O From Streptococcus Pyogenes Leads to the Release of Proinflammatory Mediators. Thromb. Haemost. 95, 982–990. doi: 10/bwpvbq
Nizet, V., Beall, B., Bast, D. J., Datta, V., Kilburn, L., Low, D. E., et al. (2000). Genetic Locus for Streptolysin S Production by Group A Streptococcus. Infect. Immun. 68, 4245–4254. doi: 10/cf2jw5
Norrby-Teglund, A., Muller, M. P., Mcgeer, A., Gan, B. S., Guru, V., Bohnen, J., et al. (2005). Successful Management of Severe Group A Streptococcal Soft Tissue Infections Using an Aggressive Medical Regimen Including Intravenous Polyspecific Immunoglobulin Together With a Conservative Surgical Approach. Scand. J. Infect. Dis. 37, 166–172. doi: 10/dkm9b4
Ofek, I., Bergner-Rabinowitz, S., Ginsburg, I. (1970). Oxygen-Stable Hemolysins of Group A Streptococci. VII. The Relation of the Leukotoxic Factor to Streptolysin S. J. Infect. Dis. 122, 517–522. doi: 10/c9c3bp
Okumura, C. Y. M., Anderson, E. L., Döhrmann, S., Tran, D. N., Olson, J., von Pawel-Rammingen, U., et al. (2013). IgG Protease Mac/IdeS Is Not Essential for Phagocyte Resistance or Mouse Virulence of M1T1 Group A Streptococcus. mBio 4, e00499-13. doi: 10/f2zn8d
Olsen, R. J., Raghuram, A., Cantu, C., Hartman, M. H., Jimenez, F. E., Lee, S., et al. (2015). The Majority of 9,729 Group A Streptococcus Strains Causing Disease Secrete SpeB Cysteine Protease: Pathogenesis Implications. Infect. Immun. 83, 4750–4758. doi: 10.1128/IAI.00989-15
Pahlman, L., Morgelin, M., Eckert, J., Johansson, L., Russell, W., Riesbeck, K., et al. (2006). Streptococcal M Protein: A Multipotent and Powerful Inducer of Inflammation. J. Immunol. 177, 1221–1228. doi: 10.4049/jimmunol.177.2.1221
Påhlman, L. I., Olin, A. I., Darenberg, J., Mörgelin, M., Kotb, M., Herwald, H., et al. (2007). Soluble M1 Protein of Streptococcus Pyogenes Triggers Potent T Cell Activation. Cell. Microbiol. 0, 070928215112001. doi: 10/crmj8b
Palm, F., Sjöholm, K., Malmström, J., Shannon, O. (2019). Complement Activation Occurs at the Surface of Platelets Activated by Streptococcal M1 Protein and This Results in Phagocytosis of Platelets. J. Immunol. 202, 503–513. doi: 10/ggbzf4
Palomo, J., Marchiol, T., Piotet, J., Fauconnier, L., Robinet, M., Reverchon, F., et al. (2014). Role of IL-1β in Experimental Cystic Fibrosis Upon P. Aeruginosa Infection. PloS One 9, e114884. doi: 10.1371/journal.pone.0114884
Park, J. M., Ng, V. H., Maeda, S., Rest, R. F., Karin, M. (2004). Anthrolysin O and Other Gram-Positive Cytolysins Are Toll-Like Receptor 4 Agonists. J. Exp. Med. 200, 1647–1655. doi: 10.1084/jem.20041215
Picard, C., Casanova, J.-L., Puel, A. (2011). Infectious Diseases in Patients With IRAK-4, MyD88, NEMO, or Iκbα Deficiency. Clin. Microbiol. Rev. 24, 490–497. doi: 10.1128/CMR.00001-11
Pinho-Ribeiro, F. A., Baddal, B., Haarsma, R., O’Seaghdha, M., Yang, N. J., Blake, K. J., et al. (2018). Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173, 1083–1097.e22. doi: 10/gdf4b3
Proft, T., Fraser, J. D. (2016). “Streptococcal Superantigens: Biological Properties and Potential Role in Disease.” Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet]. Eds. Ferretti, J. J., Stevens, D. L., Fischetti, V. A. (Oklahoma City (OK): University of Oklahoma Health Sciences Center) Available at: https://www.ncbi.nlm.nih.gov/books/NBK333435/.
Ralph, A. P., Carapetis, J. R. (2012). Group A Streptococcal Diseases and Their Global Burden. Curr. Top. Microbiol. Immunol. 68, 1–27. doi: 10/gjmhkr
Ricci, S., Janulczyk, R., Björck, L. (2002). The Regulator PerR Is Involved in Oxidative Stress Response and Iron Homeostasis and Is Necessary for Full Virulence of Streptococcus Pyogenes. Infect. Immun. 70, 4968–4976. doi: 10/dv887h
Rose-John, S., Winthrop, K., Calabrese, L. (2017). The Role of IL-6 in Host Defence Against Infections: Immunobiology and Clinical Implications. Nat. Rev. Rheumatol. 13, 399–409. doi: 10/gbhnjn
Sanderson-Smith, M., De Oliveira, D. M. P., Guglielmini, J., McMillan, D. J., Vu, T., Holien, J. K., et al. (2014). A Systematic and Functional Classification of Streptococcus Pyogenes That Serves as a New Tool for Molecular Typing and Vaccine Development. J. Infect. Dis. 210, 1325–1338. doi: 10/f6pdc5
Shah, S. S., Hall, M., Srivastava, R., Subramony, A., Levin, J. E. (2009). Intravenous Immunoglobulin in Children With Streptococcal Toxic Shock Syndrome. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc Am. 49, 1369–1376. doi: 10/ffh29z
Shaler, C. R., Choi, J., Rudak, P. T., Memarnejadian, A., Szabo, P. A., Tun-Abraham, M. E., et al. (2017). MAIT Cells Launch a Rapid, Robust and Distinct Hyperinflammatory Response to Bacterial Superantigens and Quickly Acquire an Anergic Phenotype That Impedes Their Cognate Antimicrobial Function: Defining a Novel Mechanism of Superantigen-Induced Immunopathology and Immunosuppression. PloS Biol. 15, e2001930. doi: 10.1371/journal.pbio.2001930
Shannon, O., Hertzén, E., Norrby-Teglund, A., Mörgelin, M., Sjöbring, U., Björck, L. (2007). Severe Streptococcal Infection Is Associated With M Protein-Induced Platelet Activation and Thrombus Formation. Mol. Microbiol. 65, 1147–1157. doi: 10/b8cs94
Shea, P. R., Beres, S. B., Flores, A. R., Ewbank, A. L., Gonzalez-Lugo, J. H., Martagon-Rosado, A. J., et al. (2011). Distinct Signatures of Diversifying Selection Revealed by Genome Analysis of Respiratory Tract and Invasive Bacterial Populations. Proc. Natl. Acad. Sci. 108, 5039–5044. doi: 10/d6vzz6
Silva-Costa, C., Friães, A., Ramirez, M., Melo-Cristino, J. (2015). Macrolide-Resistant Streptococcus Pyogenes: Prevalence and Treatment Strategies. Expert Rev. Anti Infect. Ther. 13, 615–628. doi: 10/f7f96j
Sjögren, J., Okumura, C. Y., Collin, M., Nizet, V., Hollands, A. (2011). Study of the IgG Endoglycosidase EndoS in Group A Streptococcal Phagocyte Resistance and Virulence. BMC Microbiol. 11, 120. doi: 10/bs5hdh
Smeesters, P., McMillan, D. J., Sriprakash, K. S. (2010). The Streptococcal M Protein: A Highly Versatile Molecule. Trends Microbiol. 18, 275–282. doi: 10.1016/j.tim.2010.02.007
Soehnlein, O., Oehmcke, S., Ma, X., Rothfuchs, A. G., Frithiof, R., van Rooijen, N., et al. (2008). Neutrophil Degranulation Mediates Severe Lung Damage Triggered by Streptococcal M1 Protein. Eur. Respir. J. 32, 405–412. doi: 10/bx4f8c
Soh, K. Y., Loh, J. M. S., Proft, T. (2020). Cell Wall-Anchored 5’-Nucleotidases in Gram-Positive Cocci. Mol. Microbiol. 113, 691–698. doi: 10/ggf35r
Spaulding, A. R., Salgado-Pabón, W., Kohler, P. L., Horswill, A. R., Leung, D. Y. M., Schlievert, P. M. (2013). Staphylococcal and Streptococcal Superantigen Exotoxins. Clin. Microbiol. Rev. 26, 422–447. doi: 10/gdm3tr
Spinaci, C., Magi, G., Varaldo, P. E., Facinelli, B. (2006). Persistence of Erythromycin-Resistant Group a Streptococci in Cultured Respiratory Cells. Pediatr. Infect. Dis. J. 25, 880–883. doi: 10/c6v5nn
Sriskandan, S., Faulkner, L., Hopkins, P. (2007). Streptococcus Pyogenes: Insight Into the Function of the Streptococcal Superantigens. Int. J. Biochem. Cell Biol. 39, 12–19. doi: 10/bdc994
Sriskandan, S., Ferguson, M., Elliot, V., Faulkner, L., Cohen, J. (2006). Human Intravenous Immunoglobulin for Experimental Streptococcal Toxic Shock: Bacterial Clearance and Modulation of Inflammation. J. Antimicrob. Chemother. 58, 117–124. doi: 10/fdtfb2
Sriskandan, S., McKee, A., Hall, L., Cohen, J. (1997). Comparative Effects of Clindamycin and Ampicillin on Superantigenic Activity of Streptococcus Pyogenes. J. Antimicrob. Chemother. 40, 275–277. doi: 10/fpm4fm
Steer, A. C., Lamagni, T., Curtis, N., Carapetis, J. R. (2012). Invasive Group a Streptococcal Disease: Epidemiology, Pathogenesis and Management. Drugs 72, 1213–1227. doi: 10/ggt9pq
Steer, A. C., Law, I., Matatolu, L., Beall, B. W., Carapetis, J. R. (2009). Global Emm Type Distribution of Group A Streptococci: Systematic Review and Implications for Vaccine Development. Lancet Infect. Dis. 9, 611–616. doi: 10/dwpnjk
Sumby, P., Porcella, S. F., Madrigal, A. G., Barbian, K. D., Virtaneva, K., Ricklefs, S. M., et al. (2005). Evolutionary Origin and Emergence of a Highly Successful Clone of Serotype M1 Group A Streptococcus Involved Multiple Horizontal Gene Transfer Events. J. Infect. Dis. 192, 771–782. doi: 10.1086/432514
Sumby, P., Whitney, A. R., Graviss, E. A., DeLeo, F. R., Musser, J. M. (2006). Genome-Wide Analysis of Group a Streptococci Reveals a Mutation That Modulates Global Phenotype and Disease Specificity. PloS Pathog. 2, e5. doi: 10.1371/journal.ppat.0020005
Sumitomo, T., Mori, Y., Nakamura, Y., Honda-Ogawa, M., Nakagawa, S., Yamaguchi, M., et al. (2018). Streptococcal Cysteine Protease-Mediated Cleavage of Desmogleins Is Involved in the Pathogenesis of Cutaneous Infection. Front. Cell. Infect. Microbiol. 8:10. doi: 10.3389/fcimb.2018.00010
Sumitomo, T., Nakata, M., Higashino, M., Jin, Y., Terao, Y., Fujinaga, Y., et al. (2011). Streptolysin S Contributes to Group A Streptococcal Translocation Across an Epithelial Barrier. J. Biol. Chem. 286, 2750–2761. doi: 10/c7thwd
Sumitomo, T., Nakata, M., Higashino, M., Terao, Y., Kawabata, S. (2013). Group A Streptococcal Cysteine Protease Cleaves Epithelial Junctions and Contributes to Bacterial Translocation. J. Biol. Chem. 288, 13317–13324. doi: 10.1074/jbc.M113.459875
Sun, H., Ringdahl, U., Homeister, J., Fay, W. P., Engleberg, N. C., Yang, A. Y., et al. (2004). Plasminogen Is a Critical Host Pathogenicity Factor for Group A Streptococcal Infection. Science. 305 (5688), 1283–1286. doi: 10.1126/science.1101245
Sun, J., LaRock, D. L., Skowronski, E. A., Kimmey, J. M., Olson, J., Jiang, Z., et al. (2020). The Pseudomonas Aeruginosa Protease LasB Directly Activates IL-1β. EBioMedicine 60, 102984. doi: 10/gjmhkt
Tamura, F., Nakagawa, R., Akuta, T., Okamoto, S., Hamada, S., Maeda, H., et al. (2004). Proapoptotic Effect of Proteolytic Activation of Matrix Metalloproteinases by Streptococcus Pyogenes Thiol Proteinase (Streptococcus Pyrogenic Exotoxin B). Infect. Immun. 72, 4836–4847. doi: 10.1128/IAI.72.8.4836-4847.2004
Tanaka, M., Kinoshita-Daitoku, R., Kiga, K., Sanada, T., Zhu, B., Okano, T., et al. (2020). Group A Streptococcus Establishes Pharynx Infection by Degrading the Deoxyribonucleic Acid of Neutrophil Extracellular Traps. Sci. Rep. 10, 3251. doi: 10/ghbj53
Thulin, P., Johansson, L., Low, D. E., Gan, B. S., Kotb, M., McGeer, A., et al. (2006). Viable Group A Streptococci in Macrophages During Acute Soft Tissue Infection. PloS Med. 3, e53. doi: 10.1371/journal.pmed.0030053
Timmer, A. M., Timmer, J. C., Pence, M. A., Hsu, L.-C., Ghochani, M., Frey, T. G., et al. (2009). Streptolysin O Promotes Group A Streptococcus Immune Evasion by Accelerated Macrophage Apoptosis. J. Biol. Chem. 284, 862–871. doi: 10/bztvgs
Turner, C. E., Sommerlad, M., McGregor, K., Davies, F. J., Pichon, B., Chong, D. L. W., et al. (2012). Superantigenic Activity of Emm3 Streptococcus Pyogenes Is Abrogated by a Conserved, Naturally Occurring smeZ Mutation. PloS One 7, e38271. doi: 10/f386hv
Valderrama, J. A., Riestra, A. M., Gao, N. J., LaRock, C. N., Gupta, N., Ali, S. R., et al. (2017). Group A Streptococcal M Protein Activates the NLRP3 Inflammasome. Nat. Microbiol. 27, 264. doi: 10.1038/s41564-017-0005-6
Vannice, K. S., Ricaldi, J., Nanduri, S., Fang, F. C., Lynch, J. B., Bryson-Cahn, C., et al. (2020). Streptococcus Pyogenes Pbp2x Mutation Confers Reduced Susceptibility to β-Lactam Antibiotics. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc Am. 71, 201–204. doi: 10/ggbhkx
von Bernuth, H., Picard, C., Jin, Z., Pankla, R., Xiao, H., Ku, C.-L., et al. (2008). Pyogenic Bacterial Infections in Humans With MyD88 Deficiency. Science 321, 691–696. doi: 10.1126/science.1158298
Walker, M. J., Barnett, T. C., McArthur, J. D., Cole, J. N., Gillen, C. M., Henningham, A., et al. (2014). Disease Manifestations and Pathogenic Mechanisms of Group A Streptococcus. Clin. Microbiol. Rev. 27, 264–301. doi: 10/f5x97b
Walker, M. J., Hollands, A., Sanderson-Smith, M. L., Cole, J., Kirk, J. K., Henningham, A., et al. (2007). DNase Sda1 Provides Selection Pressure for a Switch to Invasive Group A Streptococcal Infection. Nat. Med. 13, 981–985. doi: 10.1038/nm1612
Weiser, J. N., Ferreira, D. M., Paton, ,. J. C. (2018). Streptococcus Pneumoniae: Transmission, Colonization and Invasion. Nat. Rev. Microbiol. 16, 355–367. doi: 10/gc8z9z
Westerlund, E., Valfridsson, C., Yi, D. X., Persson, J. J. (2019). The Secreted Virulence Factor NADase of Group A Streptococcus Inhibits P2X7 Receptor-Mediated Release of IL-1β. Front. Immunol. 10, 1385. doi: 10.3389/fimmu.2019.01385
Wierzbicki, I. H., Campeau, A., Dehaini, D., Holay, M., Wei, X., Greene, T., et al. (2019). Group A Streptococcal S Protein Utilizes Red Blood Cells as Immune Camouflage and Is a Critical Determinant for Immune Evasion. Cell Rep. 29, 2979–2989.e15. doi: 10/ggdxf5
Wilde, S., Olivares, K. L., Nizet, V., Hoffman, H. M., Radhakrishna, S., LaRock, C. N. (2021). Opportunistic Invasive Infection by Group A Streptococcus During Anti–Interleukin-6 Immunotherapy. J. Infect. Dis. 223, 1260–1264. doi: 10/gg8f36
Wilkins, A. L., Steer, A. C., Smeesters, P. R., Curtis, N. (2017). Toxic Shock Syndrome – The Seven Rs of Management and Treatment. J. Infect. 74, S147–S152. doi: 10/gg4g6z
Williams, J. G., Ly, D., Geraghty, N. J., McArthur, J. D., Vyas, H. K. N., Gorman, J., et al. (2021). Streptococcus Pyogenes M1T1 Variants Activate Caspase-1 and Induce an Inflammatory Neutrophil Phenotype. Front. Cell. Infect. Microbiol. 10, 596023. doi: 10.1101/2020.03.02.972240
Williams, K. A., Swedo, S. E., Farmer, C. A., Grantz, H., Grant, P. J., D’Souza, P., et al. (2016). Randomized, Controlled Trial of Intravenous Immunoglobulin for Pediatric Autoimmune Neuropsychiatric Disorders Associated With Streptococcal Infections. J. Am. Acad. Child Adolesc. Psychiatry 55, 860–867.e2. doi: 10/f9qmcg
Winter, S. E., Thiennimitr, P., Winter, M. G., Butler, B. P., Huseby, D. L., Crawford, R. W., et al. (2010). Gut Inflammation Provides a Respiratory Electron Acceptor for Salmonella. Nature 467, 426–429. doi: 10.1038/nature09415
Xu, S. X., Kasper, K. J., Zeppa, J. J., McCormick, J. K. (2015). Superantigens Modulate Bacterial Density During Staphylococcus Aureus Nasal Colonization. Toxins 7, 1821–1836. doi: 10/gg4q68
Zeppa, J. J., Kasper, K. J., Mohorovic, I., Mazzuca, D. M., Haeryfar, S. M. M., McCormick, J. K. (2017). AndNasopharyngeal Infection by Streptococcus Pyogenes Requires Superantigen-Responsive Vβ-Specific T Cells. Proc. Natl. Acad. Sci. 114, 10226–10231. doi: 10/gbx4qm
Zhou, R., Chen, S.-H., Li, G., Chen, H.-L., Liu, Y., Wu, H.-M., et al. (2019). Ultralow Doses of Dextromethorphan Protect Mice From Endotoxin-Induced Sepsis-Like Hepatotoxicity. Chem. Biol. Interact. 303, 50–56. doi: 10/gg4g69
Zhu, L., Olsen, R. J., Beres, S. B., Eraso, J. M., Saavedra, M. O., Kubiak, S. L., et al. (2019). Gene Fitness Landscape of Group A Streptococcus During Necrotizing Myositis. J. Clin. Invest. 129, 887–901. doi: 10/gf4r9r
Keywords: Group A Streptococcus, Streptococcus pyogenes, inflammation, virulence factors, pathogenesis, toxins
Citation: Wilde S, Johnson AF and LaRock CN (2021) Playing With Fire: Proinflammatory Virulence Mechanisms of Group A Streptococcus. Front. Cell. Infect. Microbiol. 11:704099. doi: 10.3389/fcimb.2021.704099
Received: 01 May 2021; Accepted: 23 June 2021;
Published: 06 July 2021.
Edited by:
Charles T. Spencer, The University of Texas at El Paso, United StatesReviewed by:
Timothy Barnett, University of Western Australia, AustraliaMartina Sanderson-Smith, University of Wollongong, Australia
Copyright © 2021 Wilde, Johnson and LaRock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher N. LaRock, Y2xhcm9ja0BlbW9yeS5lZHU=