 Hongjian Li
Hongjian Li Christy Wing-Sum Tong3
Christy Wing-Sum Tong3 Kenneth Kin-Wah To
Kenneth Kin-Wah To- 1Institute of Future Cities, The Chinese University of Hong Kong, Hong Kong, Hong Kong
- 2Department of Computer Science and Engineering, The Chinese University of Hong Kong, Hong Kong, Hong Kong
- 3Faculty of Medicine, School of Pharmacy, The Chinese University of Hong Kong, Hong Kong, Hong Kong
In advanced lung cancer, epidermal growth factor tyrosine kinase inhibitors (EGFR TKIs) have extraordinary clinical efficacy. However, their usefulness is severely compromised by drug resistance mediated by various mechanisms, the most important of which is the secondary EGFR T790M mutation. The mutation blocks the binding of EGFR TKIs to the receptor kinase, thereby abolishing the therapeutic efficacy. In this study, we used our free and open-source protein-ligand docking software idock to screen worldwide approved small-molecule drugs against EGFR T790M. The computationally selected drug candidates were evaluated in vitro in resistant non-small cell lung cancer (NSCLC) cell lines. The specificity of the drugs toward the mutant EGFR was demonstrated by cell-free kinase inhibition assay. The inhibition of EGFR kinase activity and its downstream signaling pathways in NSCLC cells was shown by immunoblot analysis. The positive hints were revealed to be indacaterol, canagliflozin, and cis-flupenthixol, all of which were shown to induce apoptosis in NSCLC cells harboring the EGFR T790M mutation. Moreover, the combination of indacaterol with gefitinib was also found to produce synergistic anticancer effect in NSCLC cells bearing EGFR T790M. The observed synergistic effect was likely contributed by the enhanced inhibition of EGFR and its downstream signaling molecules.
Introduction
One of the leading causes of cancer-linked deaths worldwide is attributed to lung cancer (1). The prognosis of advanced non-small cell lung cancer (NSCLC), a histologic subtype constituting more than 85% of all lung cancer cases, is very poor. Traditional cytotoxic chemotherapy is the major treatment option for advanced NSCLC, but treatment response is very minimal. Therefore, substantial research efforts have been made to identify novel targets for NSCLC.
As a member of the Erb family of receptor tyrosine kinases, the epidermal growth factor receptor (EGFR) protein plays a key role in the carcinogenesis of lung cancer. Upon stimulation of ligand, EGFR undergoes homo- or heterodimerization, which leads to autophosphorylation and subsequently turns on several downstream intracellular signaling pathways (including Ras/MAPK, PI3K/Akt, and Jak/STAT) to drive cancer growth and proliferation (2). In certain types of NSCLC, the EGFR gene is mutated and becomes constitutively active (i.e., exon 19 deletion or L858R mutation), leading to “addictive” oncogenic signaling (3). EGFR tyrosine kinase inhibitors (TKIs) target this “addictive” signaling and disrupt the downstream pathways to selectively kill cancer cells. However, the clinical efficacy of first-generation EGFR TKIs (gefitinib and erlotinib) is significantly hindered by both primary and acquired resistance (4). Primary resistance is mainly due to the activation of EGFR downstream molecules (KRAS mutation or PTEN loss), thereby bypassing the inhibition of EGFR activation by the drug. On the other hand, acquired resistance is mainly caused by the induction of a secondary EGFR T790M mutation, which reduces the affinity to EGFR TKIs. Aberrant regulation of parallel oncogenic pathways (MET amplification and HER2 mutation) has also been reported. Besides, overexpression of ABC efflux transporters (P-gp and ABCG2) also leads to acquired resistance (5). To this end, most EGFR TKIs are known substrates of these transporters (6, 7). In the clinic, gefitinib and erlotinib are only suggested for the treatment of NSCLC patients if they harbor activating EGFR mutations without the resistance mechanisms. At the effective dose in responsive patients, adverse effects from gefitinb and erlotinib are generally mild, the most frequent of which include rash and diarrhea. Intense research efforts have been made to search for alternative treatment options for patients who have resistance to EGFR TKIs (8, 9).
Numerous second-generation irreversible EGFR TKIs (e.g., neratinib, afatinib) have been developed to bind covalently to Cys-797 of the EGFR ATP-binding domain in order to bypass the most prevalent secondary EGFR T790M mutation (10). Second-generation irreversible EGFR TKIs exhibit potent inhibition of the isolated EGFR kinase in the low-nanomolar range. However, some of them (e.g., neratinib) are not effective against the resistant cells in vivo at a clinically achievable concentration (11, 12). Moreover, neratinib also exhibits severe dose-limiting toxicity of diarrhea due to its inhibition of wild-type EGFR (10). Recently, a few T790M-mutant-selective irreversible third-generation EGFR inhibitors have also been developed (13, 14). They exhibit potent growth inhibition effect on EGFR T790M-bearing NSCLCs in vitro at the low-nanomolar concentration (15). These inhibitors display high-EGFR T790M-mutant selectivity and irreversible binding patterns while sparing the wild-type EGFR activity, thus enhancing tumor selectivity while minimizing adverse effects (16–20). However, resistance to these third-generation EGFR TKI has already been reported, which is due to the emergence of another EGFR mutation C797S and various other mechanisms (21, 22). Most recently, the fourth-generation EGFR TKI (EAI045) has been developed to overcome the concomitant EGFR T790M and C797S mutations mediating resistance to third-generation inhibitor (23).
In this study, we aimed to identify new drug candidates for treating EGFR TKI-resistant lung cancer cells by employing the popular and efficient strategy of drug repurposing, given that new drug development and approval is often protracted and drug attrition rate is notoriously high. In contrast, repurposing drugs, which are already clinically approved with optimized dosing regimens and known side effect profiles, for other indications, has attracted a lot of attention. We hypothesized that clinically approved drugs may be exploited to circumvent EGFR TKI resistance and that they can be identified by in silico methods based on their predicted interaction with the T790M-mutant EGFR receptor.
Indeed, we have been working on in silico drug discovery and devised a couple of software tools and web servers (http://istar.cse.cuhk.edu.hk) for molecular docking, compound scoring and ranking, and molecular visualization. By using these in silico tools to screen a library of FDA-approved drugs, we have recently identified promising drug candidates exhibiting anticancer effect in both in vitro and in vivo assays for the treatment of liver, colon, and bladder cancers (24–26). Inspired by these successful recent cases, in this study we further improved the computational and experimental workflow to search for T790M-mutant EGFR inhibitors, which we believe will likely constitute a novel treatment of EGFR TKI-resistant lung cancer.
Materials and Methods
Structural Data Collection, Ensemble Docking, and Compound Selection
From the Protein Data Bank (PDB) (27), we collected 11 X-ray crystallographic structures of EGFR harboring the resistance-triggering T790M mutation in complex with a ligand (PDB IDs: 3W2O, 3W2P, 3W2Q, 3W2R, 4I22, 4RJ4, 4RJ5, 4RJ6, 4RJ7, 4RJ8, 5HG7). The EGFR structures alone were extracted from their respective complex with the co-crystalized ligand and water molecules detached, and then converted from PDB format to PDBQT format for later use by the docking software. According to the observation that the geometry of the binding site is usually proportional to that of the bound ligand, the cubic search space was placed at the geometrical center of the bound ligand, with the length, width, and height configured to be 30% greater than that of the bound ligand. Then the search space was further stretched by 4 Å in all three dimensions to create sufficient room for the drug molecules to translate and rotate inside and achieve the optimal conformation upon binding.
From the ZINC database (28), we collected the structures of approved drugs worldwide from three catalogs, namely DrugBank-approved, FDA-approved drugs (via DSSTOX), and the NCGC Pharmaceutical Collection (NPC). These compounds, having undergone a filtering and curation procedure, constituted a non-redundant set of 3,167 drugs that have been approved for clinical use by US (FDA), UK (NHS), EU (EMA), Japanese (NHI), and Canadian (HC) authorities. Likewise, the drug compounds downloaded in Mol2 format were also converted to PDBQT format for use by the docking software.
Having retrieved and preprocessed the necessary 3D structures, we then performed the prediction of binding conformations and binding affinities of the 3,167 compounds docking against the 11 T790M-mutant EGFR structures with the free and open-source molecular docking software idock v2.2.1 (29). Software settings were tweaked to make the conformational searching process substantially more exhaustive than the default settings, i.e., grid maps of atomic free energy were calculated with a high density of 0.08 Å for each receptor structure, and 256 conformational searching tasks were executed for each compound to increase the chance of finding the optimal binding pose. In addition, although up to nine putative binding conformations will be generated for each input compound under the default settings, we set to output only the docked conformation with the best idock score (which is always the first outputted conformation among the nine) because such pose had been formerly evaluated to be most likely closest to the crystal pose (30).
Following docking, the compounds were sorted in ascending order of their predicted binding free energy. Meanwhile, the more accurate scoring function RF-Score v3 was executed to rescore all the compounds (30), providing an additional and more reliable estimation of intermolecular binding strength, given the assumption that the compounds were correctly docked. Consequently, the top-scoring compounds would be those predicted to harvest both a low idock score (in terms of binding free energy) and a high RF score (in terms of binding affinity). These top-scoring compounds were visually inspected in the context of the receptor using the convenient web-based visualizer iview (31). Taking commercial availability into consideration, we purchased and consequently validated in vitro some of the potential compounds indicated from the in silico screening results (Table 1).
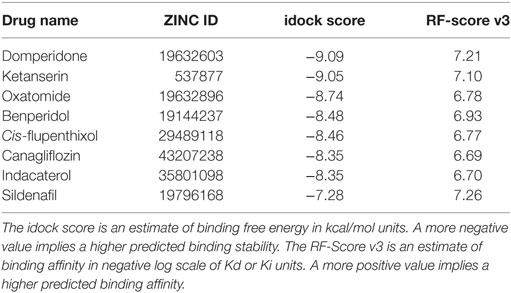
Table 1. The eight drug candidates shortlisted from in silico screening and later purchased and evaluated in vitro.
Cell Culture
Two NSCLC cell lines carrying specific EGFR mutations (HCC827: EGFR sensitizing E746_A750 deletion; H1975: resistance-causing EGFR T790M secondary mutation) were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). A normal human lung epithelial cell line BEAS-2B was also obtained from ATCC to investigate the potential toxic effect of the tested drugs. HCC827, H1975, and BEAS-2B cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum, 100-U/mL streptomycin sulfate, and 100-U/mL penicillin G sulfate, and incubated at 37°C in 5% CO2.
Growth Inhibition Assay
Growth inhibitory effects of various drug candidates on the cell lines were evaluated by the sulforhodamine B assay (32). Cells were seeded into 96-well tissue culture plates in 100 µL at a plating density of 3,000–5,000 cells/well, and allowed to incubate overnight. The cells were then treated with various drug candidates at a range of concentrations and allowed to incubate at 37°C in 5% CO2 for 72 h. Each drug concentration was tested in quadruplicate and controls were tested in replicates of eight.
Evaluation of Drug Combination
Cells growth in 96-well plates were treated with either gefitinib or the repurposed drug alone or their combination in a fixed ratio of each drug in increasing concentrations. The drugs were added either simultaneously (24 h) or the tested drug (24 h) followed by gefitinib (24 h) or vice versa. The ratio of the two drugs in combination was initially determined according to the relative ratio of the IC50 values of the drug alone. For the three positive drug candidates, the range of concentration tested was as follows: indacaterol (10 µM–10 nM, in 1:1 serial dilution), canagliflozin (10 µM–10 nM, in 1:1 serial dilution), and cis-flupenthixol (2 µM–2 nM, in 1:1 serial dilution). Gefitinib was tested in the range of 10 µM–10 nM (1:1 serial dilution). The median-drug effect analysis method was used to evaluate the nature of the drug combination (33). Combination index (CI) was then calculated to assess the outcome of the drug combination at different fraction of cells affected (Fa) as described previously (33).
Cell-Free Biochemical Kinase Inhibition Assay
Inhibition of tyrosine kinase signaling by the drug candidates identified was examined in a cell-free system by assessing the phosphorylation of a poly-EY (4:1 Glu, Tyr) peptide substrate (for EGFR) with recombinant kinases EGFRwt, EGFRL858R, or EGFRL858R+T790M as described previously (34). Inhibition of the recombinant kinases by the drug candidates was evaluated by using the ADP-Glo Kinase assay kit according to the manufacturer’s instruction (Promega, Madison, WI, USA). Briefly, the drug candidates in a range of different concentration (indacaterol: 10 nM–10 µM; canagliflozin: 10 nM–10 µM; cis-flupenthixol: 2 nM–2 µM) were allowed to incubate with 4 ng of the recombinant kinases and 0.2 µg/mL of the poly-EY substrate at room temperature for 60 min. Also, 5 µL of ADP-Glo reagent was then added and incubation continued at room temperature for another 40 min. Afterward, 10 µL of kinase detection reagent was added and the mixture was allowed to incubate at room temperature for 30 min, before the measurement of luminescence by GloMax 20/20 Luminometer (Promega).
Western Blot Analysis
Non-small cell lung cancer (HCC827, H1975) cell lines were treated with the tested drug candidates for the designated time (4 h). The cells were then harvested for Western blot analysis in lysis buffer (0.05-M HEPES pH 7.4, 0.15-M NaCl, 2-mM EDTA, 10% v/v glycerol, 1% v/v Triton X-100) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Whole cell lysates were separated by SDS-PAGE and subjected to immunoblot analysis with the respective antibodies [total EGFR, phosphor-EGFR (Y845), phosphor-ERK1/2 (Thr177/Thr160), ERK1/2, phosphor-Akt, total Akt, and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA)]. Primary antibody incubation was carried out at 4°C overnight in 5% bovine serum albumin/phosphate-buffered saline-Tween 20. Afterward, the membranes were incubated with HRP-conjugated donkey anti-mouse/anti-rabbit secondary antibody at room temperature for 1 h, and developed using the WesternBright Quantum chemiluminescence detection system (Advansta Corporation, Menlo Park, CA, USA). Anti-GAPDH antibody was used as the loading control (Santa Cruz Biotech, Santa Cruz, CA, USA). Digital chemiluminescence images were captured and quantified by using the FluorChem Q Imaging System (Alpha Innotech Corporation, Santa Clara, CA, USA).
Annexin V Apoptosis Assay
Cells were grown on a 60-mm tissue culture dish at a density of about 5.0 × 105 cells/well. They were treated for 48 h with the repurposed drug or its combination with 10-µM gefitinib. At the end of the treatment, both floating and attached cells were collected and washed twice with ice-cold phosphate buffer solution. The extent of apoptosis was determined by using the APC annexin-V apoptosis kit (BD Bioscience, San Jose, CA, USA) according to the manufacturer’s instructions. Cells positive for both annexin V and 7-AAD were considered apoptotic.
Data Analysis and Statistics
All experiments were repeated at least three times. The results were conveyed as mean ± SD. One-way analysis of variance (one-way ANOVA) with Bonferroni’s multiple comparison test was used to compare the differences among various treatment groups. A confidence level of p < 0.05 was considered significant.
Results
Structure-Based Virtual Screening Provided Candidate Inhibitors for Repurposing
A total of 3,167 drugs approved for clinical use by worldwide authorities were individually docked to an ensemble of 11 crystal structures of the T790M-mutant EGFR kinase domain, and ranked in ascending order of their estimated binding free energy. The docking results (predicted binding poses of the compounds) can be openly visualized by accessing http://istar.cse.cuhk.edu.hk/idock/iview/?3W2R-dbap+fda+npc (The PDB ID 3W2R can be changed to 4I22, 4RJ8, or 5HG7 to view the other docking results using the corresponding protein conformation). The top-scoring compounds were manually assessed based on in silico estimations of binding strength, appropriate molecular weight and other drug-like properties, complementary matching of molecular shape, and some sense of intuition from a computational chemist’s experience. Based on commercial availability, eight high-scoring compounds (Table 1) were shortlisted and purchased for subsequent wet-laboratory investigations.
Three Drug Candidates Inhibited Growth of NSCLCs
The anticancer activity of the drug candidates was tested in two NSCLC cell lines harboring EGFR sensitizing mutation (HCC827: E746_A750 deletion) or resistance-causing EGFR secondary mutation (H1975: L858R/T790M). HCC827 is extremely sensitive to erlotinib (first-generation EGFR TKI; IC50 = 6.1 ± 1.8 nM), whereas H1975 is more than 1,000-fold resistant to erlotinib (IC50 = 8.2 ± 2.1 µM) (Figure 1). Among the drug candidates tested, indacaterol, canagliflozin, and cis-flupenthixol were found to exhibit anticancer activity in both HCC827 and H1975 cells (Figure 1). Importantly, indacaterol and canagliflozin were both significantly more potent in inhibiting the growth of H1975 than HCC827 (indacaterol: 14.3 ± 2.2 µM in H1975 versus 41.5 ± 3.9 µM in HCC827; canagliflozin: 25.5 ± 3.1 µM in H1975 versus 45.4 ± 2.9 µM in HCC827). The other five drug candidates tested did not affect cell proliferation of HCC827 and H1975. Representative dose–response curves for two negative drug candidates (silfenafil and ketanserin) are shown in Figure 1. To assess the potential toxic effect of the drug candidates in normal lung tissue, cytotoxicity in a normal lung epithelial cell line BEAS-2B was also evaluated. While erlotinib was found to have an IC50 of around 20 µM, indacaterol, canagliflozin, and cis-flupenthixol only reduced cell viability by less than 20% at their highest concentration tested (indacaterol: 100 µM; canagliflozin: 100 µM; cis-flupenthixol: 20 µM).
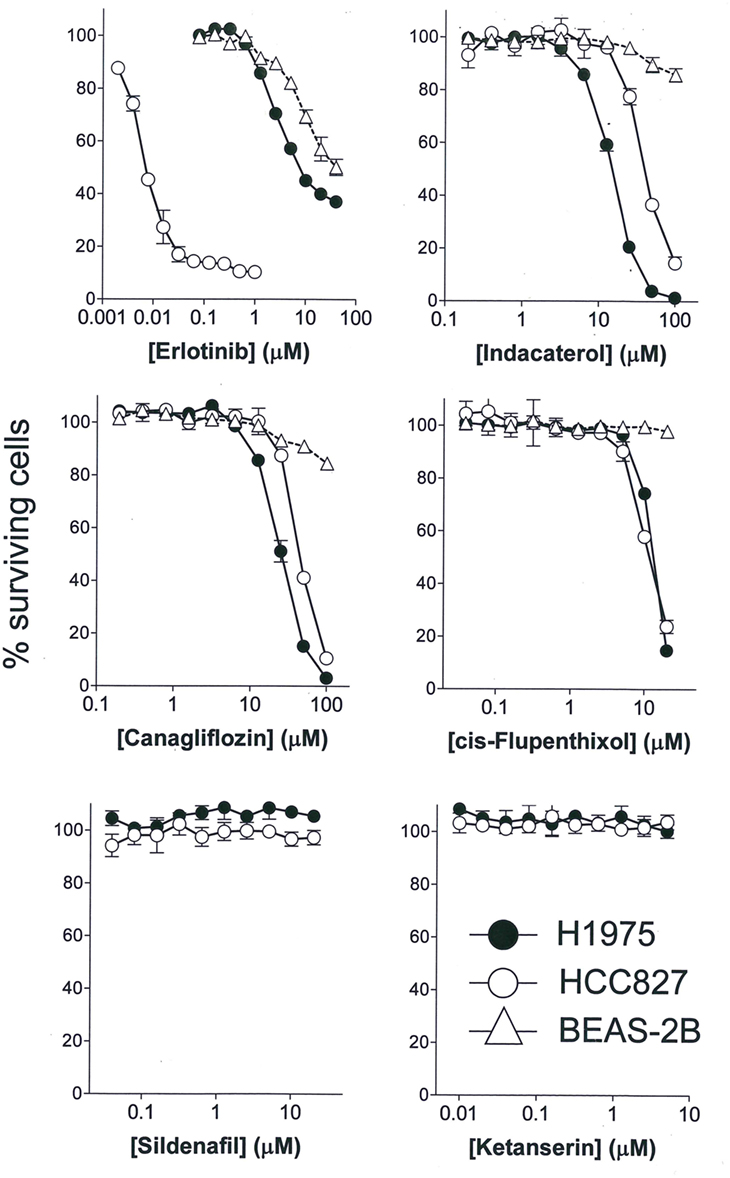
Figure 1. Cytotoxicity of the drugs tested. Cytotoxicity of three positive hits (indacaterol, canagliflozin, and cis-flupenthixol) and two negative drug candidates in the indicated cell lines was determined by the sulforhodamine B assay. A representative first-generation epidermal growth factor tyrosine kinase inhibitor (erlotinib) was also evaluated as control for comparison.
Positive Drug Candidates Inhibited the Kinase Activity of T790M EGFR in Cell-Free Assay
Since the positive drug candidates were identified by idock to interact with T790M EGFR, we hypothesize that their anticancer activity was contributed by their EGFR kinase inhibitory activity. The inhibitory effect of the positive drug candidates on the kinase activity of wild-type, L858R and L858R/T790M EGFR recombinant protein were evaluated in cell-free system. All of the three positive drug candidates were found to inhibit L858R and L858R/T790M EGFR to different extent (Table 2), presumably leading to the anticancer activity. Consistent with the preferential anticancer activity of indacaterol and canagliflozin in H1975 cells (harboring EGFR L858R/T790M resistance-causing mutation) over HCC827 cells (harboring the sensitizing EGFR E746_A750 deletion), the two drug candidates were found to inhibit EGFR L858R/T790M more significantly than EGFR L858R (Table 2). While the adverse effects of EGFR TKIs are known to be due to the inhibition of wild-type EGFR, the IC50 of the three positive hints in wild-type EGFR were at least two times (for cis-flupenthixol) higher than that in EGFR L858R/T790M.
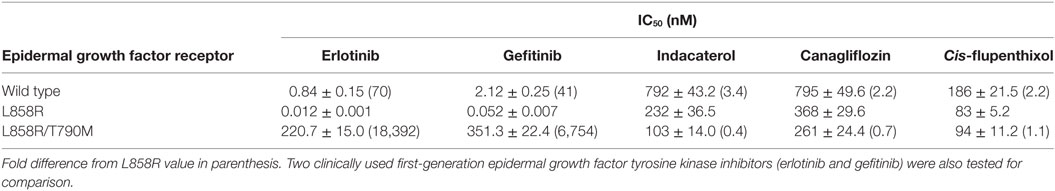
Table 2. Inhibitory effect of tyrosine kinase activity by the tested drug candidates in cell-free in vitro kinase assay.
Positive Hints Inhibited the Autophosphorylation of EGFR and Its Downstream Pathway in NSCLC Cells
The inhibition of EGFR autophosphorylation by the three positive drug candidates were also examined in HCC827 and H1975 cells. Consistent with the data in the cell-free system, both indacaterol and canagliflozin were found to inhibit EGFR phosphorylation more potently in H1975 than in HCC827 cells (Figure 2; Figure S1 in Supplementary Material; albeit more obvious for indacaterol). On the other hand, cis-flupenthixol was found to inhibit EGFR phosphorylation similarly in both H1975 and HCC827 cells (Figure S2 in Supplementary Material). The phosphorylation status of Akt and ERK was chosen as marker to illustrate the inhibition of the EGFR downstream survival signaling pathways. Parallel to the inhibition of EGFR autophosphorylation by indacaterol and canagliflozin, these two positive drug candidates were also found to inhibit the phosphorylation of Akt and ERK more remarkably in H1975 than in HCC827 (Figure 2; Figure S1 in Supplementary Material).
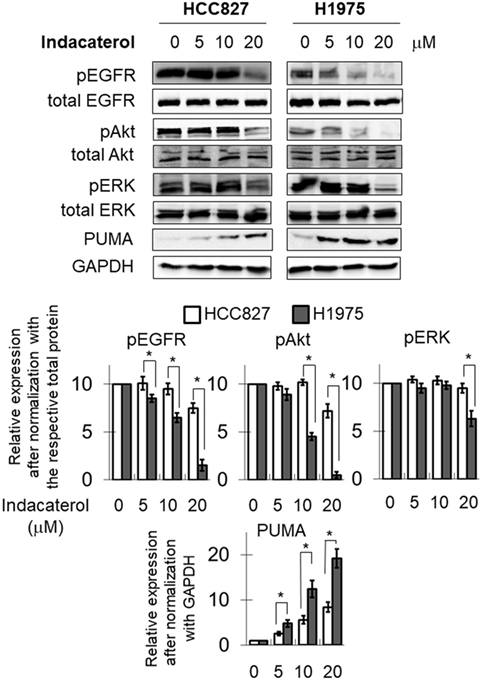
Figure 2. Western blot analysis showing the inhibition of the epidermal growth factor receptor (EGFR)–PI3K–Akt pathway by one of the three positive drug candidates (indacaterol) in both EGFR sensitizing mutation-bearing HCC827 cells and resistance-causing EGFR T790M mutation-bearing H1975 cells. The extent of inhibition of EGFR, Akt, and ERK phosphorylation is summarized after normalization with the respective total protein as bar graphs (lower panel). *p < 0.05, Student’s t-test, compared with the data in HCC827 cells. Cropped blots from different gels are grouped together for clear illustration. The full-length gels are shown in Figure S3 in Supplementary Material.
Positive Hints Induced Apoptosis in Concentration-Dependent Manner
Consistent with the anticancer activity found for indacaterol, canagliflozin and cis-flupenthixol in NSCLC cells, these drugs were also found to induce apoptosis in a concentration-dependent manner in the EGFR TKI-resistant H1975 cells (Figure 3). Since the key apoptotic effector PUMA is required for EGFR TKI inhibition-mediated apoptosis (35), the induction of PUMA by the repurposed drugs were also evaluated by Western blot analysis. Consistent with the more remarkable inhibition of EGFR in H1975 cells than in HCC827 cells by indacaterol and canagliflozin, both drugs were also found to give rise to more pronounced induction of PUMA in H1975 cells than in HCC827 cells (Figure 2; Figure S1 in Supplementary Material).
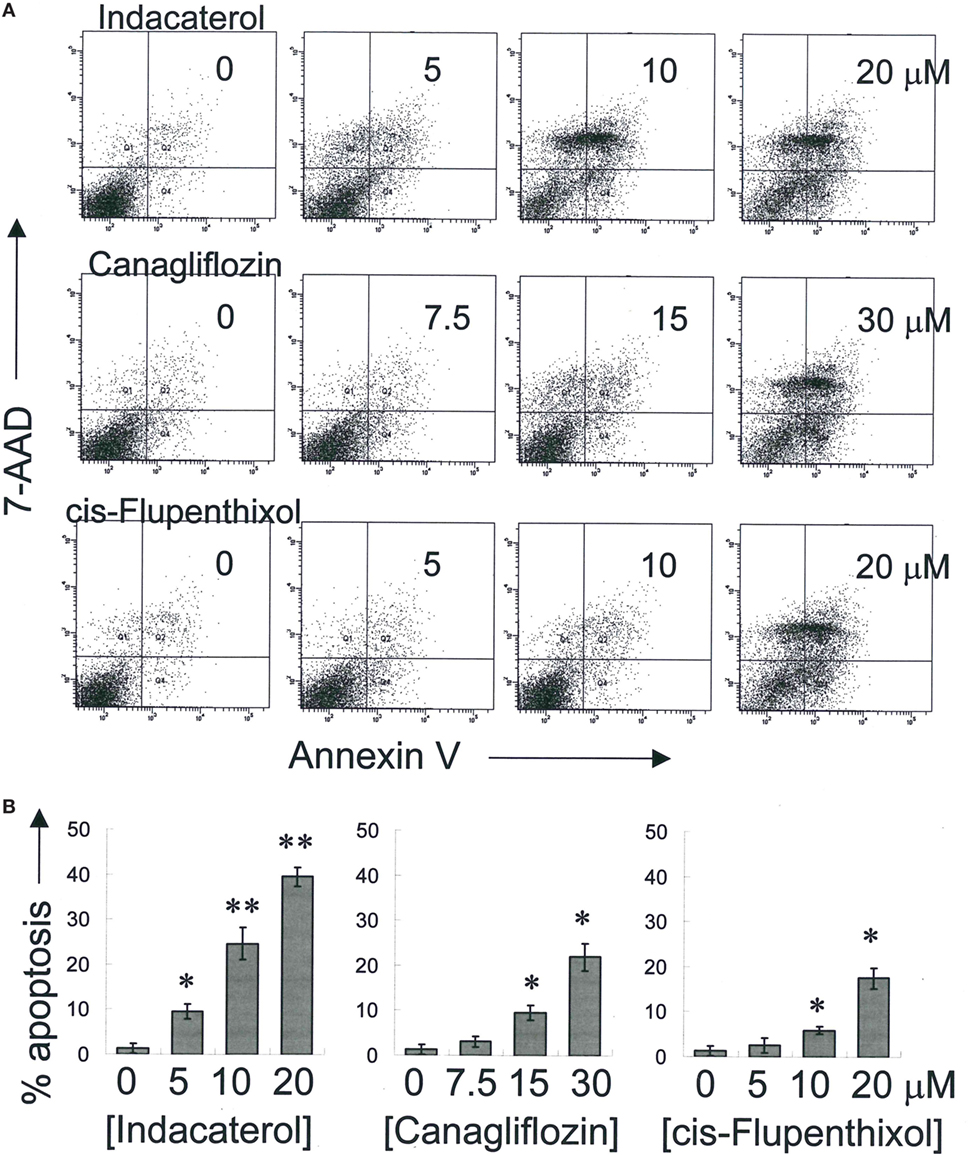
Figure 3. Induction of apoptosis by the three positive drug candidates in epidermal growth factor receptor T790M-bearing H1975 cells in a concentration-dependent manner. (A) Cells were exposed to the drugs at the designated concentration for 48 h before harvest for apoptosis assay. A representative set of data from three independent experiments is shown. (B) Summary of apoptosis assay data in (A) from three independent experiments. Data are presented in histogram as mean ± SD. *p < 0.05, **p < 0.01, Student’s t-test, compared with no treatment control.
Combination of Indacaterol and Gefitinib Synergistic in EGFR T790M-Bearing H1975 Cells
Combinations of gefitinib (a representative first-generation EGFR TKI) with the most potent positive drug candidate (indacaterol) were evaluated in H1975 cells. Equipotent concentration ratio of gefitinib (10 nM–10 µM) and indacaterol (10 nM–10 µM) was used in the combination treatment. The concentrations were chosen so that each drug in the combination contributed equally to the overall anticancer effect. To determine whether the sequence of drug addiction affects the resulting drug combination effect, H1975 cells were treated concomitantly (for 24 h) with gefitinib and indacaterol, or 24 h with indacaterol, followed by drug-free washout and treatment with 24-h gefitinib, or vice versa. The CI was adopted as a measure of combination effect. The effects of the drug combinations are shown in the CI-fraction affected (Fa) plot (Figure 4A). For all drug combination sequence, a CI remarkably <1 was observed over a wide range of growth inhibition levels. At 50% growth inhibition level, a CI of around 0.3 was obtained, indicating a strong synergistic effect.
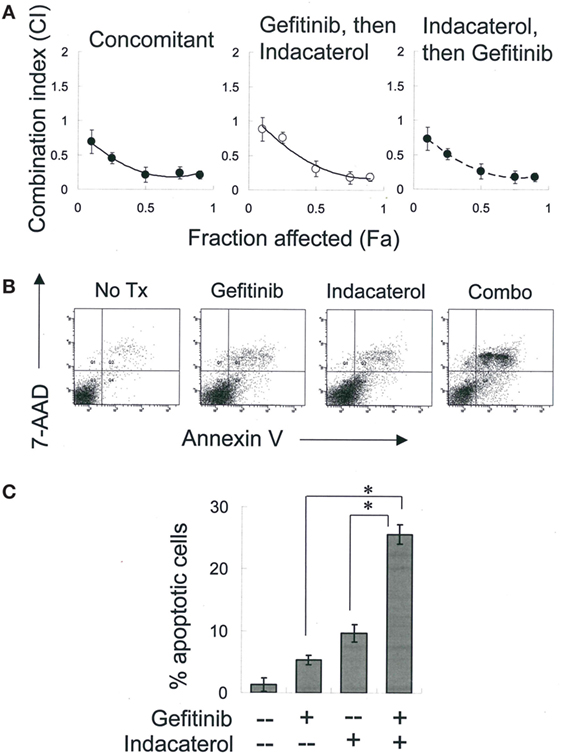
Figure 4. Combination of gefitinib and indacaterol giving rise to synergistic anticancer activity. (A) Combination index (CI) plot showing anticancer activity of combination of gefitinib and indacaterol in different sequence. CI values were plotted as a function of fractional growth inhibition (Fa). CI < 1, =1, and >1 suggest synergism, additivity, and antagonism, respectively. (B) Indacaterol sensitized H1975 cells to gefitinib-mediated apoptosis. Cells were treated with gefitinib alone (4 µM), indacaterol alone (5 µM), or their concomitant combination for 48 h before harvest for apoptosis assay. A representative set of data from three independent experiments is shown. (C) Summary of apoptosis assay data in (B). Data are presented in histogram as mean ± SD. *p < 0.05, Student’s t-test.
H1975 cells were treated with a combination of gefitinib (4 µM) and indacaterol (5 µM) concomitantly for 48 h, after which the extent of apoptosis was measured. While both gefitinib and indacaterol alone at the concentration tested only caused mild apoptosis (<10%), their combination was found to dramatically increase the proportion of apoptotic cells [27.8 ± 2.5% for drug combination versus 6.9 ± 1.2% (gefitinib alone) or 9.7 ± 1.8% (indacaterol alone); p < 0.01] (Figures 4B,C).
The activation of EGFR and its downstream signaling molecules (Akt and ERK) were also evaluated after concomitant combination of gefitinib (10 µM) and indacaterol (10 µM) for 4 h. While both gefitinib and indacaterol were only able to inhibit phosphorylation of EGFR, Akt, and ERK mildly, the drug combination was found to inhibit these signaling molecules more profoundly than the drugs alone (Figure 5).
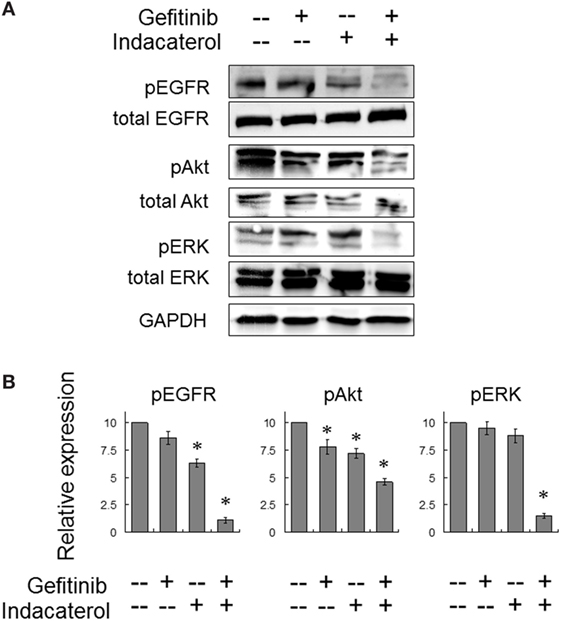
Figure 5. Combination of gefitinib and indacaterol giving rise to more pronounced inhibition of the epidermal growth factor receptor (EGFR)–PI3K–Akt pathway in EGFR T790M-bearing H1975 cells. (A) Cells were treated with gefitinib alone (10 µM), indacaterol alone (10 µM), or their concomitant combination for 4 h before harvest for Western analysis. A representative set of data is shown. Cropped blots from different gels are grouped together for clear illustration. The full-length gels are shown in Figure S6 in Supplementary Material. (B) Summary of the Western blot analysis from three independent experiments. Data are presented in histogram as mean ± SD. *p < 0.05, Student’s t-test, compared with no treatment control.
Putative Binding Conformation of Indacaterol to EGFR L858R/T790M Revealed by Molecular Docking
Figure 6A plots the macromolecular surface of EGFR L858R/T790M as well as the putative binding pose of indacaterol predicted by the docking software. Carbons, oxygens, nitrogens, and sulfurs were, respectively, rendered in gray, red, blue, and yellow. It can be seen that the indacaterol molecule was buried deep down in the binding pocket, with an oxygen atom acting as a hydrogen-bond acceptor and creating a hydrogen bond with the donor atom of the backbone of “gatekeeper” MET790. We hypothesize that the formation of the hydrogen bond might possibly permit the indacaterol molecule to be inserted into the binding cavity and therefore explain its specificity to EGFR T790M expressing cancer cells.
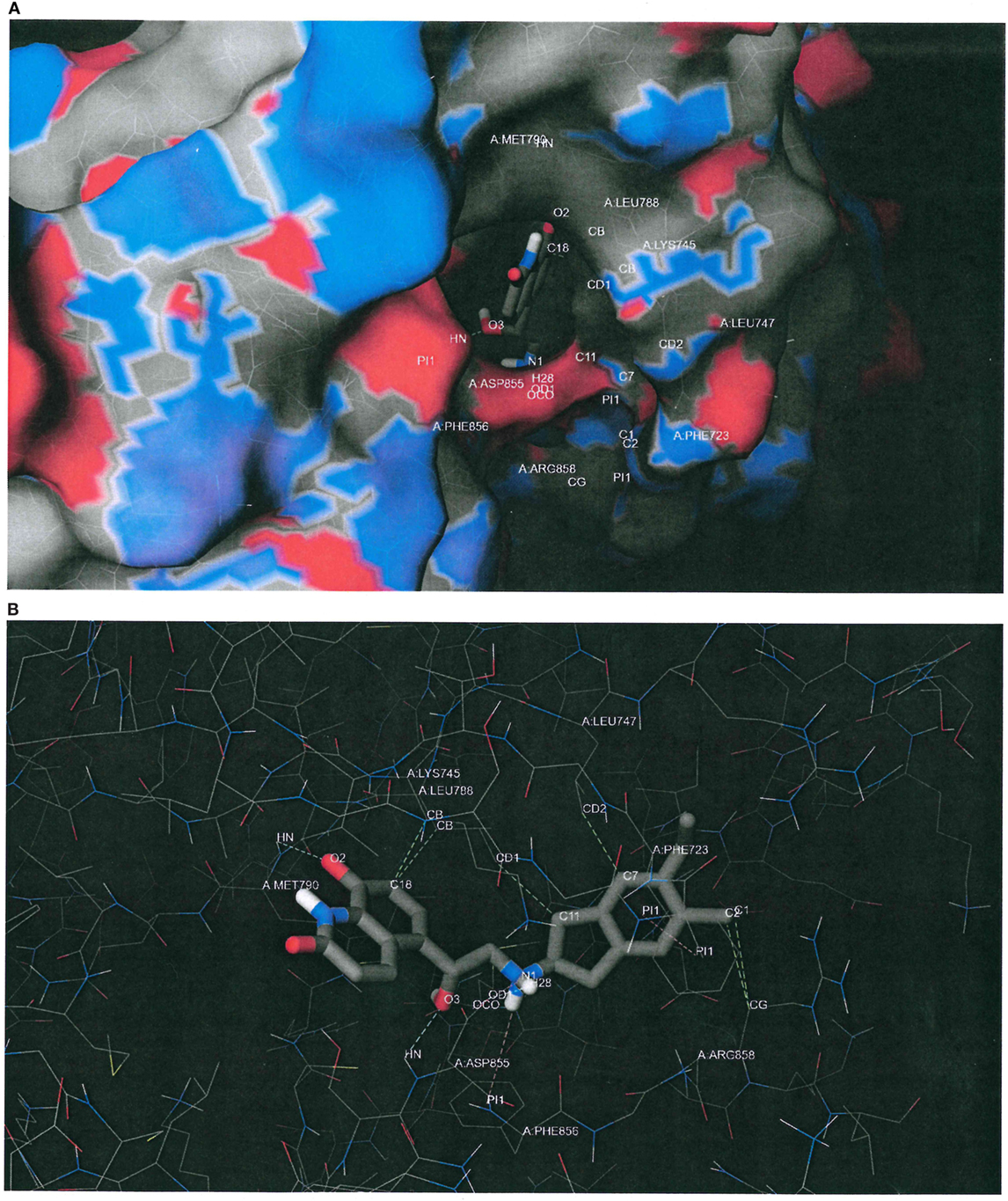
Figure 6. Putative binding conformation of indacaterol to epidermal growth factor receptor (EGFR) L858R/T790M active site (PDB ID 3W2R). (A) The putative binding pose of indacaterol on the macromolecular surface of EGFR L858R/T790M as predicted by the docking software. Carbons, oxygens, nitrogens, and sulfurs were rendered in gray, red, blue, and yellow, respectively. It can be seen that the indacaterol molecule was buried deep down in the binding pocket, with an oxygen atom acting as a hydrogen-bond acceptor and creating a hydrogen bond with the donor atom of the backbone of “gatekeeper” MET790. (B) The same predicted conformation of indacaterol upon binding to EGFR L858R/T790M, but with the macromolecular surface hidden in order to better inspect the putative intermolecular interactions, depicted by dashed lines. In addition to the hydrogen bond with MET790, indacaterol was predicted to establish two hydrogen bonds with ASP855 (cyan color), a salt bridge with ASP855 (purple color), a parallel displaced π stacking with PHE723 (pink color), a cation–π interaction with PHE856 (pink color), and some hydrophobic contacts with LYS745, LEU747, LEU788, and ARG858 (green color).
Figure 6B shows the same predicted conformation of indacaterol upon binding to EGFR L858R/T790M, but with the macromolecular surface hidden in order to better inspect the intermolecular interactions, depicted by dashed lines. In addition to the hydrogen bond with MET790, indacaterol was estimated to establish two hydrogen bonds with ASP855 (cyan color), a salt bridge with ASP855 (purple color), a parallel displaced π stacking with PHE723 (pink color), a cation–π interaction with PHE856 (pink color), and some hydrophobic contacts with LYS745, LEU747, LEU788, and ARG858 (green color).
Discussion
The aim of the study was to identify clinically used drug candidates for repurposing to treat EGFR TKI-resistant NSCLC by in silico docking simulation and to validate the chosen drug candidates in cancer cell models.
Thanks to the rapid growth of number of 3D structures of large biological molecules deposited to the freely available PDB (27), structure-based virtual screening by protein-ligand docking has become a widely used computational method to quickly identify potential small-molecule inhibitors of protein targets of therapeutic interest. From the perspective of macromolecular targets, popular proteins such as kinases usually have more than one experimentally solved 3D structure available. These multiple structures of the same protein provide an opportunity to mine additional knowledge of structural similarity and variability using an approach called ensemble docking, which, in its simplest implementation, estimates the binding affinity of a compound by docking it to a desired binding pocket of multiple protein structures and averaging the individually predicted binding affinities. Hence, the top-scoring compounds would likely possess the capability of binding to diverse conformations of the protein. From the perspective of small-molecule compounds, approved drugs represent an attractive dataset to screen, as they have been clinically proved to be safe for human use, and are often well studied and well annotated. Finding novel therapeutic indications for already approved drugs is commonly referred to as repurposing or repositioning. The rationale is that a drug typically acts on more than one target and may exhibit previously unknown activities due to promiscuous off-target interactions explaining efficacy or side effects. This approach is substantially faster and cheaper with a lower attrition rate than developing new drugs, as all preclinical developments and toxicity tests prior to phase-II clinical trial can be skipped.
Encouragingly, the powerful synergy of drug repositioning combined with in silico ensemble docking has been demonstrated in two recent publications (24, 25), where, by targeting cyclin-dependent kinase 2 (CDK2), two FDA-approved drugs fluspirilene and adapalene have been rediscovered as anticancer agents in vitro and in vivo for the treatment of hepatocellular and colorectal carcinoma, respectively. Inspired by these successful previous cases, in this study we employed the same overall computational workflow with a number of improvements. First, the inclusion of small-molecule drugs from all major regulatory authorities worldwide for in silico screening, instead of limiting to the United States Food and Drug Administration data base, can greatly enhance the chance of obtaining successful hits. Second, we have combined the use of the docking software idock with our recently developed scoring function RF-Score v3 (30). While idock usually gives correct prediction of the binding poses of the drugs, the RF-Score-v3 scoring system is expected to provide an accurate prediction of the binding strength between the drug and the protein target. The additional information on the binding strength can then serve as an alternative classifier to help select a subset of positive hits from docking for consideration. Lastly, the use of a convenient web-based visualizer iview further facilitated the positive hit selection process by allowing complementary shape matching through molecular visualization (31). Therefore, the decision on the positive hits can be made by comprehensively considering different estimates of binding strength, appropriate molecular weight, as well as the binding conformation.
Based on the docking score and commercial availability, eight drug candidates were chosen from the in silico screening for in vitro evaluation in NSCLC-bearing wild-type EGFR, EGFR L858R (sensitizing mutation), or EGFR L858R/T790M (resistance-causing mutation). Three drugs were found to exhibit anticancer activity in the cancer cells and two of them were more potent in NSCLC-harboring EGFR L858R/T790M than those with EGFR L858R. Their higher specificity toward inhibiting EGFR L858R/T790M was further confirmed by cell-free biochemical kinase assay and Western blot analysis in the NSCLC cell models. The positive hits were also showed to inhibit the EGFR–PI3K–Akt signaling pathway and cause apoptosis in EGFR T790M-bearing NSCLC. Since the toxic effect of EGFR TKIs is known to be partially mediated by the inhibition of wild-type EGFR in normal cells, the finding that at T790M EGFR-inhibiting concentration (Table 2) the wild-type EGFR is not much affected suggests a milder adverse effect profile from the repurposing drugs identified.
Of note, one of the identified drug candidates, indacaterol, is a long-acting bronchodilator indicated for the treatment of chronic obstructive pulmonary disease (COPD). COPD and lung cancer are closely linked diseases with high prevalence of comorbidities in patients (36). Therefore, it is not unusual for an NSCLC patient to receive a medication to relieve his/her COPD symptoms. A salient point of identifying indacaterol is that it is administered as an inhalation powder and that it can achieve a locally high concentration in the lung. Although indacaterol has a higher IC50 on L858R/T790M EGFR-bearing NSCLC cells (~14 μM), as compared with a third-generation irreversible mutant-selective EGFR TKI (osimertinib) (~11 nM) (15), the concentration may be achievable for a locally administered drug. Moreover, due to the route of administration, drug–drug interaction is less of a concern. Furthermore, indacaterol may have other advantages compared with the third and fourth generations of EGFR TKIs such as osimertinib, which has only a short history of safe human use. Also, importantly, according to docking prediction, indacaterol does not target EGFR Cys-797, the mutation of which is a known mechanism of resistance for third-generation EGFR TKIs (21). Thus, indacaterol is likely less prone to drug resistance caused by the same mechanism. The drug candidates identified may be repurposed for treating NSCLC resistant to EGFR TKIs. Further research along this line may be able to identify more potent drugs for repurposing. The same in silico screening approach may be used to hunt for other clinically used drugs for repurposing to treat cancer cells resistant to other molecular targeted agents.
To overcome resistance to EGFR TKIs, besides developing new drugs, combination therapy is also a promising strategy. This approach aims at circumventing drug resistance through a so-called bypass signaling mechanism by targeting horizontal or vertical pathways or both. Interestingly, the repurposing drug indacaterol identified also gave rise to pronounced synergistic anticancer activity when used in combination with a model first-generation EGFR specifically in NSCLC cells harboring EGFR T790M TKI gefitinib. From the structural point of view, the anilinoquinazoline core of gefitinib forms a hydrogen bond with MET793 of the EGFR backbone, and its propylmorpholine tail picks up some non-specific interactions. In contrast, the recognition of indacaterol may be explained by different sets of intermolecular forces. According to the docking predictions, the indacaterol molecule was buried in the ATP-binding cavity, making a hydrogen bond to the gatekeeper MET790 backbone and two hydrogen bonds with ASP855, a parallel displaced π stacking with PHE723, a cation–π interaction with PHE856, and some hydrophobic contacts with LYS745, LEU747, LEU788, and ARG858 (Figure 6). We believed that the distinctly different sets of intermolecular forces between EGFR T790M and gefitinib versus indacaterol may allow a synergistic inhibition of the mutant kinase and thus enhanced anticancer activity by the drug combination. The combination of indacaterol and EGFR TKIs may be adopted as a novel means to circumvent resistance to EGFR TKIs. Further mechanistic investigation and animal studies are warranted to fully understand and optimize the beneficial drug combinations.
Ethics Statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Author Contributions
KT, HL, K-SL, M-HW, and YL conceived and designed the study. KT, HL, and CT conducted experiments and analyzed data. KT, HL, and K-SL wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LWD and handling Editor declared their shared affiliation.
Acknowledgments
The provision of graduate studentship to Wing-Sum Tong by the Chinese University of Hong Kong is greatly appreciated.
Funding
This work was supported by the Vice-Chancellor’s One-off Discretionary Fund, Faculty of Social Science Postdoctoral Fellowship Scheme and Institute of Future Cities, The Chinese University of Hong Kong; the Direct Grant from the Chinese University of Hong Kong; the GRF Grant from the Research Grants Council of Hong Kong SAR (project reference 414413); and Lee Hysan Foundation Research from the United College, The Chinese University of Hong Kong (grant number CA11239).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fonc.2017.00288/full#supplementary-material.
Figure S1. Western blot analysis showing the inhibition of the EGFR–PI3K–Akt pathway by one of the three positive drug candidates (canagliflozin) in both EGFR sensitizing mutation-bearing HCC827 cells and resistance-causing EGFR T790M mutation-bearing H1975 cells. The extent of inhibition of EGFR, Akt, and ERK phosphorylation is summarized after normalization with the respective total protein as bar graphs (lower panel). *p < 0.05, Student’s t-test, compared with the data in HCC827 cells. Cropped blots from different gels are grouped together for clear illustration. The full-length gels are shown in Figure S4 in Supplementary Material.
Figure S2. Western blot analysis showing the inhibition of the EGFR–PI3K–Akt pathway by one of the three positive drug candidates (cis-flupenthixol) in both EGFR sensitizing mutation-bearing HCC827 cells and resistance-causing EGFR T790M mutation-bearing H1975 cells. The extent of inhibition of EGFR, Akt, and ERK phosphorylation is summarized after normalization with the respective total protein as bar graphs (lower panel). *p < 0.05, Student’s t-test, compared with the data in HCC827 cells. Cropped blots from different gels are grouped together for clear illustration. The full-length gels are shown in Figure S5 in Supplementary Material.
Figure S3. Full-length gels for the Western blot data shown in Figure 2 (indacaterol).
Figure S4. Full-length gels for the Western blot data shown in Figure S1 in Supplementary Material (canagliflozin).
Figure S5. Full-length gels for the Western blot data shown in Figure S2 in Supplementary Material (cis-flupenthixol).
Figure S6. Full-length gels for the Western blot data shown in Figure 5 (combination of gefitinib and indacaterol).
References
1. Siegel R, Ma J, Zuo Z, Jemal A. Cancer statistics, 2016. CA Cancer J Clin (2016) 66:7–30. doi:10.3322/caac.21332
2. Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol (2001) 2:127–37. doi:10.1038/35052073
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med (2004) 350:2129–39. doi:10.1056/NEJMoa040938
4. Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res (2011) 17:5530–7. doi:10.1158/1078-0432.CCR-10-2571
5. Ozvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist Updat (2005) 8:15–26. doi:10.1016/j.drup.2005.02.002
6. Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (tarceva, OSI-774) antagonizes ATP-binding cassette family B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res (2007) 67:11012–20. doi:10.1158/0008-5472.CAN-07-2686
7. Hegedus C, Truta-Feles K, Antalffy G, Varady G, Nemet K, Ozvegy-Laczka C, et al. Interaction of the EGFR inhibitors gefitinib, vandetanib, pelitinib and neratinib with the ABCG2 multidrug transporter: implications for the emergence and reversal of cancer drug resistance. Biochem Pharmacol (2012) 84:260–7. doi:10.1016/j.bcp.2012.04.010
8. Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol (2015) 16:e447–59. doi:10.1016/S1470-2045(15)00246-6
9. Song Z, Ge Y, Wang C, Huang S, Shu X, Liu K, et al. Challenges and perspectives on the development of small-molecule EGFR inhibitors against T790M-mediated resistance non-small-cell lung cancer. J Med Chem (2016) 59:6580–94. doi:10.1021/acs.jmedchem.5b00840
10. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGFR receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A (2005) 102:7665–70. doi:10.1073/pnas.0502860102
11. Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, et al. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther (2008) 7:874–9. doi:10.1158/1535-7163.MCT-07-2387
12. Sos ML, Rode HB, Heynck S, Peifer M, Fischer F, Kluter S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res (2010) 70:868–74. doi:10.1158/0008-5472.CAN-09-3106
13. Walter AO, Tjin Tham Sjin R, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov (2013) 3:1404–15. doi:10.1158/2159-8290.CD-13-0314
14. Barnes TA, O’Kane GM, Vincent MD, Leighl NB. Third-generation tyrosine kinase inhibitors targeting epidermal growth factor receptor mutations in non-small cell lung cancer. Front Oncol (2017) 7:113. doi:10.3389/fonc.2017.00113
15. Cross DA, Ashton SE, Ghiorqhiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov (2014) 4:1046–61. doi:10.1158/2159-8290.CD-14-0337
16. Finlay MRV, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem (2014) 57:8249–67. doi:10.1021/jm500973a
17. Juchum M, Gunther M, Laufer SA. Fighting cancer drug resistance: opportunities and challenges for mutation-specific EGFR inhibitors. Drug Resist Updat (2015) 20:12–28. doi:10.1016/j.drup.2015.05.002
18. Russo A, Franchina T, Ricciardi GRR, Smiroldo V, Picciotto M, Zanqhi M, et al. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol (2017) 117:38–47. doi:10.1016/j.critrevonc.2017.07.003
19. Passaro A, Guerini-Rocco E, Pochesci A, Vacirca D, Spitaleri G, Catania CM, et al. Targeting EGFR T790M mutation in NSCLC: from biology to evaluation and treatment. Pharmacol Res (2017) 117:406–15. doi:10.1016/j.phrs.2017.01.003
20. Chuang JC, Salahudeen AA, Wakelee HA. Rociletinib, a third generation EGFR tyrosine kinase inhibitor: current data and future directions. Expert Opin Pharmacother (2016) 17:989–93. doi:10.1517/14656566.2016.1162786
21. Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol (2016) 9:59. doi:10.1186/s13045-016-0290-1
22. Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med (2016) 10:383–8. doi:10.1007/s11684-016-0488-1
23. Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett (2017) 38:51–4. doi:10.1016/j.canlet.2016.11.008
24. Shi XN, Li HJ, Yao H, Liu X, Li L, Leung KS, et al. In silico identification and in vitro and in vivo validation of anti-psychotic drug fluspirilene as a potential CDK2 inhibitor and a candidate anti-cancer drug. PLoS One (2015) 10:e0132072. doi:10.1371/journal.pone.0132072
25. Shi XN, Li HJ, Yao H, Liu X, Li L, Kung HF, et al. Adapalene inhibits the activity of cyclin-dependent kinase 2 in colorectal carcinoma. Mol Med Rep (2015) 12:6501–8. doi:10.3892/mmr.2015.4310
26. Ke K, Li H, Yao H, Shi XN, Dong C, Zhu Y, et al. In silico prediction and in vitro and in vivo validation of acaricide fluazuron as a potential inhibitor of FGFR3 and a candidate anticancer drug for bladder carcinoma. Chem Biol Drug Des (2017) 89:505–13. doi:10.1111/cbdd.12872
27. Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Mol Biol (2003) 10:980. doi:10.1038/nsb1203-980
28. Irwin JJ, Shoichet BK. ZINC––a free database of commercially available compounds for virtual screening. J Chem Inf Model (2005) 45:177–82. doi:10.1021/ci049714+
29. Li H, Leung KS, Ballester PJ, Wong MH. istar: a web platform for large scale protein-ligand docking. PLoS One (2014) 9:e85678. doi:10.1371/journal.pone.0085678
30. Li H, Leung KS, Wong MH, Ballester PJ. Improving AutoDock Vina using random forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol Inform (2015) 34:115–26. doi:10.1002/minf.201400132
31. Li H, Leung KS, Nakane T, Wong MH. iview: an interactive WebGL visualizer for protein-ligand complex. BMC Bioinformatics (2014) 15:56. doi:10.1186/1471-2105-15-56
32. Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst (1990) 82:1107–12. doi:10.1093/jnci/82.13.1107
33. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res (2010) 70:440–6. doi:10.1158/0008-5472.CAN-09-1947
34. Wei Y, Poon DC, Fei R, Lam AS, Au-Yeung SC, To KK. A platinum-based hybrid drug design approach to circumvent acquired resistance to molecular targeted tyrosine kinase inhibitors. Sci Rep (2016) 6:25363. doi:10.1038/srep25363
35. Bean GR, Ganesan YT, Dong Y, Takeda S, Liu H, Chan PM, et al. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal (2013) 6:ra20. doi:10.1126/scisignal.2003483
Keywords: epidermal growth factor tyrosine kinase inhibitor resistance, docking, drug repurposing, indacaterol, canagliflozin, cis-flupenthixol
Citation: Li H, Tong CW-S, Leung Y, Wong M-H, To KK-W and Leung K-S (2017) Identification of Clinically Approved Drugs Indacaterol and Canagliflozin for Repurposing to Treat Epidermal Growth Factor Tyrosine Kinase Inhibitor-Resistant Lung Cancer. Front. Oncol. 7:288. doi: 10.3389/fonc.2017.00288
Received: 12 September 2017; Accepted: 13 November 2017;
Published: 29 November 2017
Edited by:
Gautam Sethi, National University of Singapore, SingaporeReviewed by:
Dhiraj Kumar, University of Texas MD Anderson Cancer Center, United StatesLih Wen Deng, National University of Singapore, Singapore
Copyright: © 2017 Li, Tong, Leung, Wong, To and Leung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth Kin-Wah To, JiN4MDAwNUI7KiYjeDAwMDVEO2tlbm5ldGh0byYjeDAwMDQwO2N1aGsuZWR1Lmhr;
Kwong-Sak Leung, JiN4MDAwNUI7KiomI3gwMDA1RDtrc2xldW5nJiN4MDAwNDA7Y3NlLmN1aGsuZWR1Lmhr