 Colleen A. Flanagan
Colleen A. Flanagan Ashmeetha Manilall
Ashmeetha Manilall
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 24 October 2017
Sec. Neuroendocrine Science
Volume 8 - 2017 | https://doi.org/10.3389/fendo.2017.00274
This article is part of the Research Topic Gonadotropin-Releasing Hormone Receptor Signaling and Functions View all 15 articles
Gonadotropin-releasing hormone (GnRH) regulates reproduction. The human GnRH receptor lacks a cytoplasmic carboxy-terminal tail but has amino acid sequence motifs characteristic of rhodopsin-like, class A, G protein-coupled receptors (GPCRs). This review will consider how recent descriptions of X-ray crystallographic structures of GPCRs in inactive and active conformations may contribute to understanding GnRH receptor structure, mechanism of activation and ligand binding. The structures confirmed that ligands bind to variable extracellular surfaces, whereas the seven membrane-spanning α-helices convey the activation signal to the cytoplasmic receptor surface, which binds and activates heterotrimeric G proteins. Forty non-covalent interactions that bridge topologically equivalent residues in different transmembrane (TM) helices are conserved in class A GPCR structures, regardless of activation state. Conformation-independent interhelical contacts account for a conserved receptor protein structure and their importance in the GnRH receptor structure is supported by decreased expression of receptors with mutations of residues in the network. Many of the GnRH receptor mutations associated with congenital hypogonadotropic hypogonadism, including the Glu2.53(90) Lys mutation, involve amino acids that constitute the conserved network. Half of the ~250 intramolecular interactions in GPCRs differ between inactive and active structures. Conformation-specific interhelical contacts depend on amino acids changing partners during activation. Conserved inactive conformation-specific contacts prevent receptor activation by stabilizing proximity of TM helices 3 and 6 and a closed G protein-binding site. Mutations of GnRH receptor residues involved in these interactions, such as Arg3.50(139) of the DRY/S motif or Tyr7.53(323) of the N/DPxxY motif, increase or decrease receptor expression and efficiency of receptor coupling to G protein signaling, consistent with the native residues stabilizing the inactive GnRH receptor structure. Active conformation-specific interhelical contacts stabilize an open G protein-binding site. Progress in defining the GnRH-binding site has recently slowed, with evidence that Tyr6.58(290) contacts Tyr5 of GnRH, whereas other residues affect recognition of Trp3 and Gly10NH2. The surprisingly consistent observations that GnRH receptor mutations that disrupt GnRH binding have less effect on “conformationally constrained” GnRH peptides may now be explained by crystal structures of agonist-bound peptide receptors. Analysis of GPCR structures provides insight into GnRH receptor function.
Gonadotropin-releasing hormone (GnRH) regulates reproduction by binding and activating GnRH receptors on pituitary gonadotrope cells, which synthesize and secrete the gonadotropins, LH, and FSH. The gonadotropins act on the gonads to stimulate gametogenesis, gonadal cell proliferation, and production of the gonadal steroids. GnRH secretion is suppressed during childhood and increases at puberty, when increased production of gonadotropins and gonadal steroids initiate sexual development. Disruption of GnRH receptor function disrupts reproduction and mutations of the GnRH receptor gene disrupt or delay pubertal development, resulting in congenital hypogonadotropic hypogonadism (cHH) (1, 2). This central role in regulation of reproduction has made the GnRH receptor a target for treatment of infertility and of sex steroid-dependent hyperplasias, including uterine fibroids, endometriosis and prostatic cancer, where gonadal steroid production is decreased by administration of GnRH antagonists or high doses of GnRH agonists, which downregulate receptor expression (3–5). Agonist binding to the GnRH receptor activates the Gq/11 family of heterotrimeric G proteins. Activated GTP-bound Gαq/11 subunits activate phospholipase Cβ, which catalyzes production of the second messengers diacylglycerol and inositol trisphosphate, which initiate the cellular signaling pathways that culminate in gonadotropin synthesis and secretion (3, 6, 7). Although the GnRH receptor is also reported to transiently activate Gs proteins in the LβT2 gonadotrope cell line (3, 8, 9) and inhibit cell growth via the inhibitory Gi proteins, no direct GnRH receptor activation of Gαi or Gαs could be shown in a range of cell lines (10–12) and it has been proposed that GnRH-stimulated activation of Gi or Gs proteins may be downstream of activation of the Gq/11 proteins (6, 12). The mammalian (type 1) GnRH receptor does not activate β-arrestin-dependent signaling (3, 6, 7, 13), suggesting that all effects of the GnRH receptor may be mediated by activation of Gq/11 proteins.
The GnRH receptor belongs to the G protein-coupled receptor (GPCR) family, which constitutes the largest family of membrane proteins in the human genome (14, 15). The GPCRs regulate physiological systems ranging from vision and olfaction through neurotransmission and immunology in addition to endocrine systems. Physiological ligands that activate GPCRs range from cations (Ca2+), small molecule neurotransmitters and immune modulators to peptide and protein hormones, cytokines and even light, which changes the 11-cis-retinal prosthetic group of rhodopsin from a covalently bound inverse agonist (an antagonist that actively stabilizes inactive receptor conformations) to an agonist. In spite of their diverse physiological functions and ligands, all GPCRs share a common molecular function, which consists of transducing an extracellular signal across a biological membrane via a change in receptor protein conformation (16–18). This conserved function is supported by a conserved protein structure that consists of an extracellular amino-terminus, a bundle of seven membrane-spanning α-helical segments connected by three intracellular and three extracellular loops and a cytoplasmic carboxy-terminus (16, 19, 20). No crystal structure of the GnRH receptor has yet been reported, but much can be learned about its structure and how it conveys the extracellular GnRH-binding signal to intracellular signaling pathways by studying the structures of related GPCRs that have been crystallized and combining this with biochemical studies. This review will focus on understanding of the structure of the GnRH receptor and ligand binding that has arisen since the last major review (13) with emphasis on the application of recently described GPCR structures and how these may inform mechanisms of GnRH receptor structure, activation and ligand binding.
Based on conserved amino acid sequence features (Table 1), the GnRH receptor is a class A GPCR. Class A is the largest and best-studied class of GPCR proteins and includes rhodopsin, adrenergic and other monoamine neurotransmitter receptors and many peptide and protein-binding receptors. The membrane-spanning segments of GPCRs are most conserved, whereas the loops and termini are more variable (19). To facilitate comparison of amino acid residues of the GnRH receptor with equivalent residues of other class A GPCRs, the Ballesteros and Weinstein numbering system (21) will be used. Residues are numbered relative to the most conserved residue in each transmembrane (TM) segment, which is designated .50, preceded by the TM segment number and followed, where relevant, by the amino acid sequence number in the receptor in parenthesis. For example Asp319 of the human GnRH receptor is designated Asp7.49(319), because it immediately precedes the most conserved residue in TM7, Pro7.50(320). The equivalent residue of the mouse receptor is Asp7.49(318).
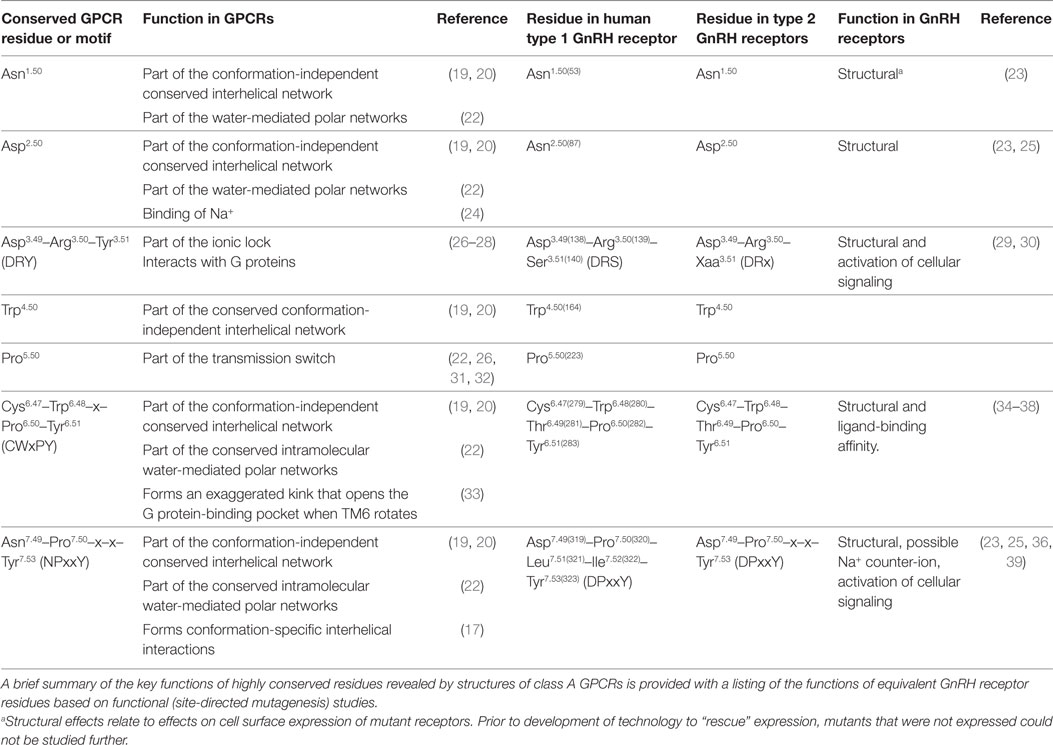
Table 1. Highly conserved amino acid residues and motifs in class A GPCRs and equivalent residues in type 1 and type 2 GnRH receptors.
This review will focus on the mammalian type 1 GnRH receptor, which is characterized by absence of a cytoplasmic carboxy-terminal tail (13, 40) that accounts for the lack of arrestin-dependent desensitization, internalization, and signaling. Many systems of nomenclature have been used for GnRH receptor subtypes, largely because of the unclear relationship between the tailless mammalian receptors and the other GnRH receptors, all of which have carboxy-terminal tails (13, 41, 42). The discovery that some lower vertebrates have tailless GnRH receptors that are structurally and functionally similar to mammalian receptors (43) has now provided some consensus (40, 44–46). All of the tailless GnRH receptors are designated type 1 and all of the tailed GnRH receptors, type 2.
Human GnRH receptors have all of the highly conserved Ballesteros and Weinstein reference residues, except for the acidic Asp2.50 in TM2, which is substituted with uncharged Asn (Table 1; Figure S1 in Supplementary Material). Mutation of Asn2.50(87) to the normal Asp disrupted GnRH receptor expression (23, 25), confirming the functional importance of the substitution. The type 1 GnRH receptors also have variations of the highly conserved amino acid sequence motifs. In TM7 the NPxxY motif (Asn7.49-Pro7.50-x-x-Tyr7.53 where x represents any amino acid) is changed to DPxxY (Asp7.49-Pro7.50-Leu7.51-Ile7.52-Tyr7.53). Mutation of Asp7.49 to Asn reversed the disruption of GnRH receptor expression caused by mutation of Asn2.50 to Asp, suggesting these resides might be close to each other in the three-dimensional structures of class A GPCRs (25). The CWxPY motif in TM6 is preserved as Cys6.47-Trp6.48-Thr6.49-Pro6.50-Tyr6.51, whereas the DRY motif at the cytosolic end of TM3 is DRS (Asp3.49-Arg3.50-Ser3.51) (Table 1; Figure S1 in Supplementary Material).
Type 1 GnRH receptors have a Glu2.53(90) residue in TM2, which has risen to prominence because a cHH-associated Glu2.53(90)Lys mutation disrupts membrane expression of the receptor, but treatment with a pharmacoperone [small-molecule membrane-permeable GnRH receptor antagonists that act as templates for folding of nascent receptor proteins (34, 35, 47)] rescues expression of the mutant receptor, both in vitro and in knock-in transgenic mice (34, 47, 48). In other class A GPCRs the equivalent residue is mostly large and hydrophobic (Leu, Val, or Phe) (49) and is Ile2.53, Val2.53, or Met2.53 in type 2 GnRH receptors (13, 43), suggesting that the carboxyl side chain of Glu2.53(90) may not be required.
The functional importance of the highly conserved Tyr5.58 residue was revealed by crystal structures of active rhodopsin (33, 50). Type 1 GnRH receptors have Asn5.58, but all tailed GnRH receptors have the conserved Tyr5.58 (13, 40, 43). In most class A GPCRs a conserved large aliphatic amino acid, Ile3.40, forms part of a group of conserved residues referred to as the “core triad” (22) or “transmission switch” (26, 31, 32), which changes configuration during receptor activation. GnRH receptors have a small Ala3.40(129) residue, which is also present in type 2 GnRH receptors.
Ligands interact with the variable extracellular half of GPCR molecules. The membrane-spanning domain conveys the ligand-binding signal to the cytosolic surface of the receptor, which interacts with the G protein (20). In order to specifically transduce an agonist signal across a cellular membrane, a GPCR must exist in a silent state that does not activate G proteins. Once agonist binds, the receptor must undergo transition to a state that binds and activates G proteins located on the opposite side of the membrane. Thus, agonist ligands, such as GnRH, can be thought of as allosteric activators of GPCRs, enabling them to catalyze G protein activation (16, 18, 51).
Current theories of receptor activation posit that GPCRs exist in an equilibrium of inactive “R” and activated “R*” conformations, with the equilibrium balanced toward the R conformations in the absence of ligand. The R conformations cannot activate G proteins, are stabilized by binding of inverse agonist (antagonist) ligands and have low affinity for agonist ligands. In contrast, R* conformations bind and activate G proteins, have high affinity for agonist ligands and are stabilized by binding of agonists and/or G proteins (16, 52–54). Thus, agonist binding induces or stabilizes one or more active GPCR conformation(s), which activate G protein signaling. Similarly, G protein binding increases the binding affinity of the receptor for agonist (16, 54, 55). Until recently, the structural correlates of the R and R* conformations were unknown, but a flurry of technical innovations has recently allowed X-ray crystallographic determination of the structures of rhodopsin and then other GPCRs. Initial structures were bound to inverse agonists and thus represent inactive R conformations. These were followed by crystal structures of agonist-bound GPCRs that were partially active, whereas, in most cases, cocrystallization of agonist-bound receptors with a G protein or a G protein mimetic (antibody or truncated G protein) was required to achieve fully active GPCR structures (16, 27, 31, 32, 56). The GPCR crystal structures reveal the differences between the R and R* conformations, although they do not provide dynamic information about the activation process. We will attempt to use information from the structures of class A GPCRs that have been crystallized to understand GnRH receptor structure.
G protein-coupled receptor crystal structures show that the three-dimensional structures of GPCRs are more conserved than the amino acid sequences (15, 19, 20, 26, 57). The convergence of diverse amino acid sequences to a common structure allowed considerable plasticity in the evolutionary development of the diverse GPCR family (17, 19, 57). The crystal structures have defined the relative positions of the known highly conserved amino acids and of the conserved amino acid sequence motifs and shown that the highly conserved Pro5.50, Pro6.50, and Pro7.50 residues in TM5, TM6, and TM7 cause bends in the α-helices that are not classical proline kinks (32, 58, 59).
The exaggerated bend angle around Pro6.50, in the CWxPY motif in TM6, is stabilized by a water molecule that makes hydrogen bonds to the Cys6.47 and Tyr6.51 residues of the CWxPY motif and to a residue in TM7 in GPCR structures (33, 58, 60–62). The importance of this structure in the GnRH receptor is supported by no less than three cHH-associated mutations of the CWxPY motif. The Pro6.50(282)Arg mutation of the GnRH receptor completely disrupts receptor function, which cannot be recovered by pharmacoperone treatment (36), showing that the proline kink is essential for GnRH receptor expression. The Cys6.47(279)Tyr mutant GnRH receptor and a laboratory-produced Cys6.47(279)Ala mutant showed no measurable GnRH binding and severely decreased cellular responses to GnRH stimulation that were recovered after pharmacoperone treatment of cells transfected with the mutant receptor (1, 35, 63, 64). This suggests that the Cys6.47(279) mutations disrupt receptor folding during biosynthesis, but the rescue shows that its effect is less destructive than mutating the Pro6.50(282). A Tyr6.51(283)His GnRH receptor mutation causes cHH and results in no measurable function in vitro (65).
The overall GPCR fold (the relative positions of the seven TM segments) is stabilized by ~200–260 non-covalent intramolecular contacts (hydrogen bonds, van der Waals interactions, etc.) and by a network of hydrogen-bonded water molecules in the interior of the TM domain. Depending on methodology, 24–40 interhelical contacts between topologically equivalent loci (positions) are present in all active and inactive class A GPCR structures (17, 19, 20). These conserved conformation-independent interhelical contacts determine the overall GPCR structure, forming a conserved “scaffold,” on which conformational changes can occur. Some of the conserved interhelical contacts are required for protein folding and insertion into the membrane during biosynthesis. The conserved interhelical contacts involve many of the residues that are highly conserved in class A GPCRs (Table S1 in Supplementary Material) but also involve residues in topologically equivalent loci, where the amino acids are not conserved (19, 20, 57). Most of the conserved conformation-independent interhelical interactions are located toward the central and cytoplasmic side of the TM domain (20) (Figure S2 in Supplementary Material), consistent with the emerging recognition that these are the areas in which GPCR structure is most conserved, whereas the extracellular side of the TM barrel is less conserved, because of the need to accommodate diverse ligands (18).
Gonadotropin-releasing hormone receptor residues topologically equivalent to the residues in conserved conformation-independent interhelical contacts are listed in Table S1 in Supplementary Material, with the predicted interhelical contacts and effects of previously reported mutations of the residues on GnRH receptor expression and function. Many mutations result in undetectable receptor function, consistent with disruption of cell surface GnRH receptor expression or severe misfolding of the receptor protein. The disruption of functional receptor expression confirms the importance of the residues for GnRH receptor structure and indicates that the conserved interhelical contacts constitute part of the GnRH receptor structure, similar to their roles in other GPCRs.
The complete disruption of GnRH receptor expression when Asn2.50(87) in TM2 was substituted with Ala or Asp (Table S1 in Supplementary Material) supports a role for the Asn2.50(87) residue in stabilizing GnRH receptor structure. However, GPCR crystal structures show that Asp2.50 makes conserved contacts with residues in TM1 and TM7, but it is only connected to Asn7.49 via the water-mediated hydrogen bond network (19, 58). Based on the conserved structural scaffold, Asn2.50(87) of the GnRH receptor contacts Asn1.50(53) and Pro7.46(316), whereas Asp7.49(319) does not form any conserved conformation-independent contacts (Table S1 in Supplementary Material).
Many cHH-associated GnRH receptor mutations involve residues that constitute conserved interhelical contacts in the crystallized GPCR structures. These include the Glu2.53(90)Lys, Glu2.53(90)Asp, Ala4.57(171)Thr, Cys6.47(279)Tyr, Tyr6.51(283)His, Tyr6.52(284)Cys, Pro7.50(320)Arg, and Tyr7.53(323)Cys mutations. Most cHH-associated mutant receptors are poorly expressed in vitro (Table S1 in Supplementary Material), consistent with the mutations disrupting the structural scaffold of the receptor. Disruption of interhelical contacts would destabilize receptor protein folding, resulting in fewer correctly folded receptor molecules being transported to the cell membrane or decreased residence time of less stable receptor proteins once they get to the cell membrane. Pharmacoperones act as templates for folding of nascent receptor proteins, stabilizing biosynthesis, and increasing protein expression (34, 35, 47). In most cases, pharmacoperone treatment of cells transfected with cHH-associated mutant GnRH receptors, enhanced mutant receptor expression (Table S1 in Supplementary Material) and the “rescued” receptors showed wild type-like function. This supports a role for the mutated residues in stable folding and cell surface expression of the GnRH receptor (34, 35, 47).
The late Michael Conn’s laboratory and others proposed that the Glu2.53(90) side chain of the GnRH receptor forms a interhelical salt-bridge with Lys3.32(121) in TM3 and that the cHH-associated Glu2.53(90)Lys mutation disrupted receptor biogenesis and folding by breaking this salt bridge (66–69). Although Glu2.53(90) is conserved in type 1 GnRH receptors, type 2 GnRH receptors and other class A GPCRs have large hydrophobic residues at position 2.53 (13, 43, 49, 57). The lack of conservation suggests that the acidic side chain of Glu2.53(90) may not be necessary for type 1 GnRH receptor structure and that the effect of the Glu2.53(90)Lys mutation may result from disruptive effects of introducing Lys, rather than lack of Glu. This is supported by a mutation of Glu2.53(90) to uncharged Gln, which had no effect on receptor function (70) and a conservative Glu2.53(90)Asp, which is associated with cHH (Table S1 in Supplementary Material). A Glu2.53(90)Ala mutation resulted in undetectable GnRH receptor function (68), but we have not found any report of the effect of the Glu2.53(90)Asp mutation, which would formally test the salt bridge hypothesis.
The conserved interhelical contacts in GPCR structures predict that Glu2.53(90) interacts with Ser3.35(124) (Table S1 in Supplementary Material). Interaction of Glu2.53(90) with Ser3.35(124) is supported by an automated (unbiased) structural homology model of the GnRH receptor (71), which shows Glu2.53(90) close to Ser3.35(124), whereas Lys3.32(121) points away, toward Asp2.61(98) (Figure 1). A Ser3.35(124)Asp mutation resulted in undetectable binding, suggesting that the mutation caused receptor instability by introducing close apposition of two carboxyl side chains (37). Thus, it is likely that the cHH-associated Glu2.53(90)Lys mutation disrupts GnRH receptor expression by disrupting the conserved interhelical contact with the 3.35 locus, rather than disrupting a salt-bridge with Lys3.32(121).
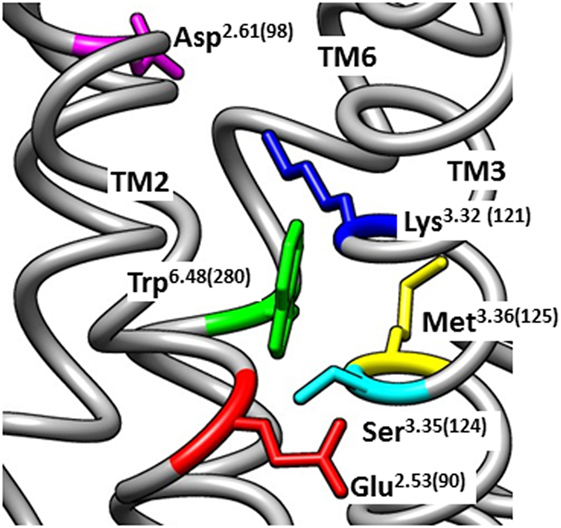
Figure 1. Homology model of the inactive human gonadotropin-releasing hormone (GnRH) receptor. The model was downloaded from the GPCRdb website (www.gpcrdb.org/structure/homology_models) (71) and viewed using the UCSF Chimera software package (72) to show the spatial positioning of Glu2.53(90) (red) relative to the neighboring residues Ser3.35(124) (light blue), Lys3.32(121) (dark blue), Asp2.61(98) (magenta), Met3.36(125) (yellow), and Trp6.48(280) (green). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Another cHH-associated GnRH receptor mutant, Tyr6.51(283)His, also showed undetectable function in vitro (65). Since Tyr6.51 forms a conserved interhelical contact with the residue in the 7.39 locus, the Tyr6.51(283)His mutation may disrupt receptor structure by disrupting an interhelical contact of Tyr6.51(283) with Phe7.39(309) in TM7 of the GnRH receptor (Table S1 in Supplementary Material).
The Trp6.48(280) residue in the CWxPY motif was proposed to directly contact the Trp3 residue of the GnRH peptide (69, 73). Systematic mutagenesis of Trp6.48(280) to Ala, His, Ser, Gln, and Met disrupted GnRH receptor expression, as did mutation of Trp6.48(279) of the rat GnRH receptor to Arg or Ser (74). However, once expression of the mutant receptors was recovered, using a pharmacoperone, the mutant receptors displayed unchanged ligand-binding affinity and signaling (37, 38, 73). This shows that Trp6.48(280) does not directly contact GnRH, but is important for GnRH receptor structure. Trp6.48(280) likely forms conserved interhelical contacts with Met3.36(125) and Ala7.42(312) of the GnRH receptor (Table S1 in Supplementary Material), which would be disrupted by the mutations.
About half of the intramolecular interactions differ between the inactive and active GPCR structures. The GnRH receptor residues equivalent to the residues that form conserved conformation-specific interhelical contacts are listed in Table 2. These conformation-specific interhelical contacts depend on reassignment of the interacting amino acid pairs during the activation process (17, 19) (Figure 2). This section will describe the key features of the inactive GPCR structures and discuss the evidence for similar structural features in the GnRH receptor. The main features of inactive GPCR structures include a “closed” G protein-binding pocket, a water-mediated hydrogen bond network and a hydrophobic barrier separating the water network in the ligand-binding pocket from the G protein-binding pocket (17, 18, 22, 32, 33, 56, 75).
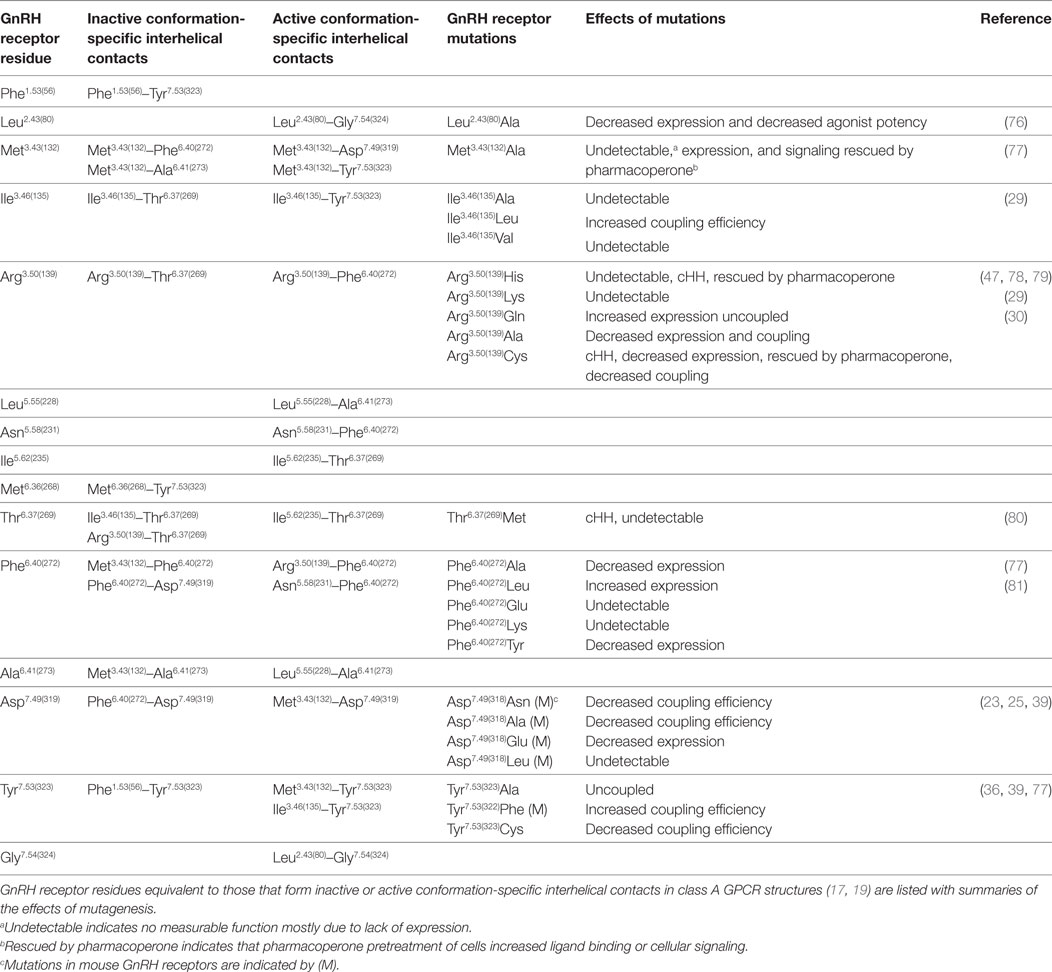
Table 2. GnRH receptor residues potentially involved in conserved conformation-specific interhelical contacts.
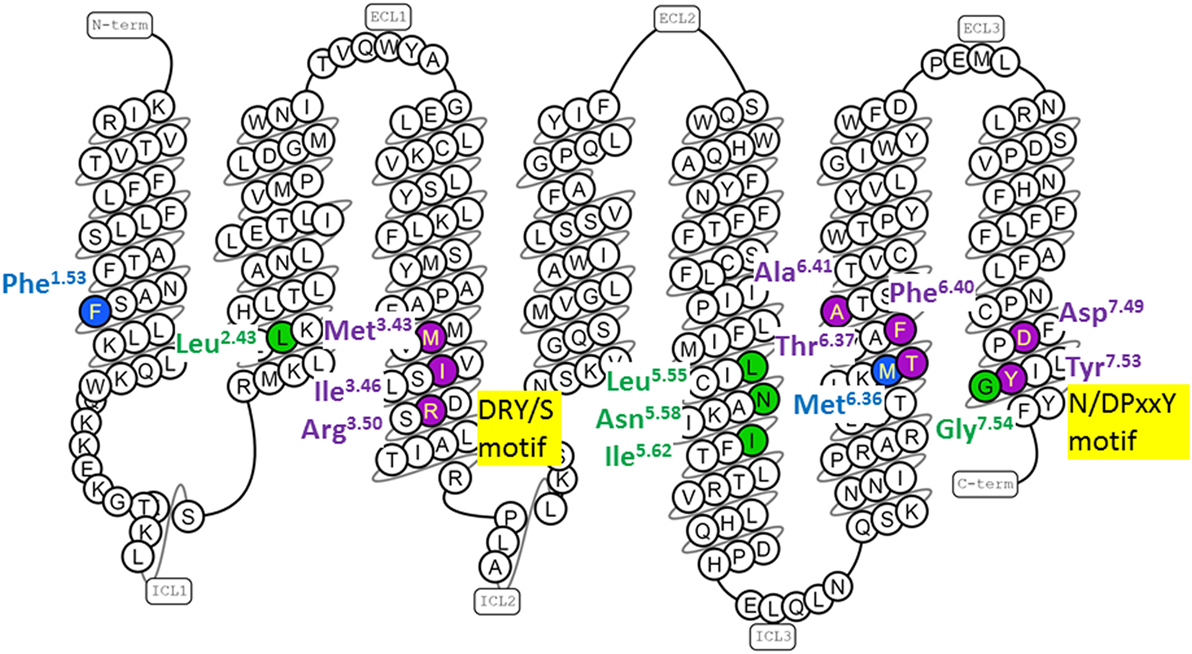
Figure 2. Human gonadotropin-releasing hormone (GnRH) receptor residues topologically equivalent to G protein-coupled receptor (GPCR) residues that form conserved conformation-specific interhelical contacts. The snake diagram of the GnRH receptor amino acid sequence was downloaded from GPCRdb (www.gpcrdb.org/structure/homology_models) (71). GnRH receptor residues topologically equivalent to residues that form conserved conformation-specific contacts (17, 19) only in inactive GPCR structures (blue); only in active GPCR structures (green) and with different partners in inactive and active GPCR structures (purple) are shown.
The inactive rhodopsin structure showed a salt bridge between Arg3.50 of the D/ERY motif at the cytoplasmic end of TM3, and Glu6.30 at the cytosolic end of TM6 (28, 82). This “ionic-lock” interaction stabilizes the inactive GPCR conformation by drawing the cytoplasmic ends of TM3 and TM6 together (26, 83, 84). However, the salt bridge between Arg3.50 and Glu6.30 cannot provide a universal mechanism for stabilizing inactive GPCR conformations, because Glu6.30 is not conserved (83). The GnRH receptor has Arg6.30(262), which clearly cannot form a salt bridge with Arg3.50(139). Nevertheless, mutations show that both Arg residues are important for GnRH receptor structure and function. The cHH-associated Arg6.30(262)Gln mutation decreased ligand binding and cellular signaling (85), which was recovered when cells were treated with pharmacoperone (1, 47), suggesting that the Arg6.30(262) side chain forms an intramolecular interaction that stabilizes folding of the unoccupied GnRH receptor. Since GPCRs are biosynthesized in cellular compartments that are inaccessible to endogenous ligands, receptors are likely synthesized in inactive conformations. Inactive conformation-specific interhelical contacts may thus stabilize receptor biosynthesis, so mutations that disrupt these contacts may also disrupt expression.
Mutation of Arg3.50(139) in the DRY/S motif to other basic amino acids, His or Lys, resulted in no measurable receptor function, consistent with disruption of a structurally important interaction. An Arg3.50(139)Gln mutation increased GnRH receptor expression and decreased activation of cellular signaling (29). This suggests that the Gln side chain mimics an interhelical interaction of Arg3.50(139) that stabilizes receptor folding and stabilizes the inactive receptor conformation. The importance of the Arg3.50(139) side chain for GnRH receptor structure is also supported by cHH-associated Arg3.50(139)His and Arg3.50(139)Cys mutations, which are poorly expressed (30, 78), but rescued by pharmacoperone treatment (Table 2) suggesting that the mutations disrupt biosynthesis of the unoccupied (inactive) GnRH receptor. Since Arg3.50(139) cannot form an ionic lock in the GnRH receptor, it must form a different contact. GPCR structures show a conserved inactive conformation-specific interhelical contact of Arg3.50 with the 6.37 locus (19). Thus, the Arg3.50(139) side chain may stabilize the inactive GnRH receptor conformation by forming an interhelical hydrogen bond with Thr6.37(269) (Table 2), which may be enhanced in the Arg3.50(139)Gln mutant receptor.
Many conserved inactive conformation-specific interhelical contacts involve residues in TM3 and TM6 (19), suggesting that, like the ionic lock, they maintain close proximity of the cytoplasmic ends of TM3 and TM6. Met3.43(132), conserved as a large hydrophobic residue in most GPCRs, is likely to stabilize the inactive GnRH receptor conformation by interacting with Phe6.40(272) and Ala6.41(273) in TM6 (Table 2). Mutation of Met3.43(132) and four mutations of Phe6.40(272) decreased GnRH receptor expression, suggesting that they disrupt receptor biogenesis. In contrast, the Phe6.40(272)Leu mutation increased receptor expression (Table 2; Figure S3 in Supplementary Material) (77, 81), suggesting that the Phe6.40(272)Leu mutation enhances the TM3-TM6 interhelical contact with Met3.43(132) and so enhances expression of the inactive GnRH receptor.
A more recent study, using five pairs of GPCR structures found that only one TM3–TM6 inactive conformation-specific contact is conserved (17). This interaction between the 3.46 and 6.37 loci is the key contact that defines the “closed” conformation of the G protein-binding site and prevents G protein binding (17). In the GnRH receptor, these residues are Ile3.46(135) and Thr6.37(269) (Table 2). Mutation of Ile3.46(135) to Ala and Val both completely ablated GnRH receptor function (29). In the context of the inactive GPCR crystal structures, the Ala and Val side chains are likely too short to “fulfill the distance criteria for contact formation” (17). Substitution of Ile3.46(135) with Leu, which has a large branched aliphatic side chain, partially preserved GnRH receptor expression and increased coupling efficiency (29). Thus, the Ile3.46(135)Leu mutation may destabilize the inactive conformation of the GnRH receptor by weakening the Ile3.46(135)–Thr6.37(269) interhelical contact and favoring formation of the R* active conformation. A cHH-associated Thr6.37(269)Met mutation resulted in no measurable function in several assay systems (80). This result confirms the importance of Thr6.37(269) in GnRH receptor structure, but provides no information about its function in receptor conformation.
The second key inactive conformation-specific interhelical contact orients Tyr7.53 of the NPxxY motif in TM7 toward TM1, where it contacts the residue in the 1.53 locus, keeping the Tyr7.53 side chain away from the interior of the TM domain (17, 19). The corresponding residues of the GnRH receptor are Phe1.53(56) in TM1 and Tyr7.53(323) in the N/DPxxY motif in TM7 (Table 2).
Higher resolution GPCR structures revealed internal water molecules that form a conserved network, with hydrogen bonds connecting the conserved amino acids in different helices, including Asn1.50, Asp2.50, Trp6.48 of the CWxPY motif and residues of the NPxxY motif. The water molecules both stabilize the GPCR structural fold and mediate transition between conformational states, by forming many “low energy switches,” which can be broken and reconfigured during GPCR activation (22, 26, 33, 50, 86). The GnRH receptor likely has a network of intramolecular water molecules between its highly conserved amino acids and mutations that disrupt GnRH receptor expression or function, may do so by disrupting the water-mediated intramolecular network.
The hydrophilic network in inactive GPCRs also includes a sodium ion, which makes a conserved contact with Asp2.50 and stabilizes the inactive receptor conformation. Cations decrease GnRH receptor agonist binding (87). Since type 1 GnRH receptors do not have Asp2.50, the nearby Glu2.53(90) and Asp7.49(319) (of the N/DPxxY motif) residues may provide negative charges that enhance cation binding, as suggested for the PAR1 receptor (22, 24, 88, 89).
All inactive class A GPCR structures have a layer of hydrophobic amino acids on the cytosolic side of Asp2.50 that separates the water molecules in the ligand- and G protein-binding pockets. The barrier stabilizes the inactive GPCR conformation and consists of conserved class A GPCR residues, including some that also form conserved inactive conformation-specific interhelical contacts (26, 33, 75). The partially overlapping “central hydrophobic core,” consisting of the conserved Phe6.44 and conserved hydrophobic residues in the 3.43 and 6.40 loci (90), the “core triad” consisting of Phe6.44, Pro5.50 and the hydrophobic residue at position 3.40 (22) and the “transmission switch,” consisting of the hydrophobic residue at position 3.40, Pro5.50, Leu5.51, and Phe 6.44, which is one helical turn away from Trp6.48 of CWxPY motif, all couple the ligand-binding pocket to the hydrophobic barrier (26, 31, 32). The GnRH receptor has hydrophobic residues in the loci associated with the hydrophobic barrier and the transmission switch, so it is likely that the GnRH receptor has a hydrophobic barrier that stabilizes its inactive conformation.
The diverse GPCR ligands trigger a variety of receptor-specific molecular changes to initiate receptor activation (31, 91). The changes must converge to generate a structurally conserved G protein-binding pocket that can be recognized by- and activate a G protein that interacts with many GPCRs (17, 18). Structural features that define the activated GPCR conformation include rotation of TM6, changed interfaces of TM3, TM5, and TM6, opening of the hydrophobic barrier, movement of the Tyr5.58 (Asn5.58(231) in type 1 GnRH receptors) and Tyr7.53 side chains toward the interior of the TM bundle and opening of a cytoplasmic surface cleft that allows G protein access and binding (20, 31, 32, 56, 90, 91).
In spite of the diversity of ligand-binding surfaces many ligands contact highly conserved residues, particularly Trp6.48 and Tyr6.51 of the CWxPY motif. In the active structure of rhodopsin retinal isomerization induces movement of Trp6.48, which causes rotation of TM6, without changing either the rotamer angle of the Trp6.48 side chain or the hinge angle of the proline kink. The exaggerated bend angle of Pro6.50 amplifies movement of the cytoplasmic end of TM6, which moves outward, away from TM3 (33). Similar rotation of the cytoplasmic end of TM6 was seen in all fully active GPCR structures (22, 27, 56, 92, 93).
Rotation of TM6 also changes its interhelical contacts with TM3 and TM5. The Phe6.44 side chain moves toward TM5, where it rearranges the Leu5.51 and Pro5.50 residues and moves the conserved Leu3.40 of TM3 away from TM5, thus triggering the “transmission switch” (26, 31). Similar activation of the transmission switch via agonist-induced movement of Trp6.48 is seen in the A2A-adenosine and μ-opioid receptors (22, 31). The transmission switch changes the conformation of the proline kink of TM5 and rotates the conserved Tyr5.58 side chain (near the cytoplasmic end) inwards. It also changes the position of TM3, rotating it and moving it slightly toward the extracellular of side the TM domain (90).
Active structures of other GPCRs showed similar outward movement of the cytosolic end of TM6 but showed no agonist contact with Trp6.48 (31, 93–98), so TM6 movement must be achieved via different mechanisms. In the β2-adrenergic receptor, agonist binding at the extracellular end of TM5 causes movement of Pro5.50, which moves Ile3.40 in TM3 and Phe6.44 in TM6, thus opening the core triad, triggering the transmission switch and rotating the cytoplasmic end of TM6 away from the helix bundle (26, 31). A similar opening of the core triad residues occurs in the μ-opioid receptor, except that the agonist binds to two residues (the 3.32 and 3.36 loci) in TM3, resulting in movement of Ile3.40, which activates the core triad (22). Mutation of the Lys3.32(121) residue in TM3 of the GnRH receptor decreased binding of GnRH agonists but not antagonists (99), suggesting that GnRH interaction with Lys3.32(121) initiates activation. Although mutagenesis experiments do not support a role for Trp6.48(280) in ligand binding or activation of the GnRH receptor (38), there is evidence that GnRH contacts Tyr6.58(290) at the extracellular end of TM6 (100) and that Tyr6.51(283) of the CWxPY affects ligand-binding affinity (37), suggesting that these residues may initiate rotation of TM6.
The water-mediated polar network in active GPCR structures differs from that of inactive structures. Agonist-induced movement of the extracellular ends of the TM helices rearranges water molecules at the extracellular side of the TM domain and opens the hydrophobic barrier, which allows formation of a continuous water channel between the ligand-binding pocket and the cytoplasmic surface of the receptor. This changes the conformation of TM7 and causes rotation of Tyr7.53 away from its interaction with TM1 toward the center of the TM bundle (22, 32, 33, 50, 75, 90, 96, 101, 102). The rearranged water molecules link Arg3.50 with Tyr5.58 and Tyr7.53 in a water-mediated interhelical network that can be considered the “open” conformation of the ionic lock that stabilizes the active receptor conformation (22, 28, 96). Movements of TM6 and TM7 collapse the sodium ion-binding pocket, making it too small to accommodate the ion (24, 88, 97) and the cation moves toward the cytoplasm through the open hydrophobic barrier (24). Mutation of the water-associated Arg3.50(139), Asp7.49(318) and Tyr7.53(322) residues decreases GnRH receptor coupling efficiency (23, 29, 30, 39, 77, 103) indicating that they have roles in the active receptor conformation, which may be mediated by the water network.
Rotation of TM6 and opening of the hydrophobic barrier break the inactive conformation-specific interhelical contacts and form new active conformation-specific interhelical contacts. The ionic lock opens and the Arg3.50 side chain moves into the space vacated by TM6 where it orients toward Tyr5.58 of TM5. Arg3.50 and Tyr5.58 form new interhelical contacts with the hydrophobic residue in position 6.40 (19, 22, 28). The GnRH receptor has Asn5.58(231), which is smaller than Tyr. The NTSR1 neurotensin receptor also has Asn5.58 and an “active-like” NTSR1 structure shows a hydrogen bond between Asn5.58(257) and Arg3.50(167) (104), which suggests that Asn5.58(231) stabilizes the open ionic lock in the GnRH receptor.
On activation Met3.43 breaks its inactive conformation-specific interhelical contact with the residues in positions 6.40 and 6.41. The release of the hydrophobic side chain in position 6.41 allows it to contact the hydrophobic residue in position 5.55, forming one of two key active conformation-specific interhelical contacts (17, 19). In the GnRH receptor these residues are Leu5.55(228) and Ala6.41(273) (Table 2). One helical turn closer to the cytoplasmic side of the receptor, the Ile3.46 side chain breaks its inactive conformation-specific contact with the position 6.37 side chain and forms a new interhelical contact with Tyr7.53, forming the second key active conformation-specific interaction (17, 19). The breaking of the TM3-TM6 contacts opens a cleft that allows G protein access and releases the position 6.37 residue to make a conserved interaction with the G protein (17, 27) (Figure S3 in Supplementary Material). In the GnRH receptor these residues are Ile3.46(135), Thr6.37(269) and Tyr7.53(323) (Table 2). Mutation of Ile3.46(135) to Leu increased GnRH receptor coupling efficiency (29), suggesting that the Leu side chain may favor interaction with Tyr7.53(323) over interaction with Thr6.37(269), thus favoring the active conformation. A role for Tyr7.53(323) in stabilizing the active GnRH receptor conformation is supported by the Tyr7.53(323)Ala mutant, which did not activate cellular signaling (39, 77).
Gonadotropin-releasing hormone is a decapeptide with the sequence pGlu1-His2-Trp3-Ser4-Tyr5-Gly6-Leu7-Arg8-Pro9-Gly10NH2. The amino-terminal residues, pGlu1, His2, and Trp3, determine agonist activity, but the carboxy-terminal residues, particularly Arg8, are necessary for high affinity binding to the GnRH receptor (4, 13, 105). Although the GnRH peptide is conformationally flexible, the predominant conformer consists of a β-turn that brings the amino- and carboxy-termini close together. This conformation is stabilized by substituting the achiral Gly6 residue of the GnRH peptide with d-amino acids, which increase receptor-binding affinity, whereas l-amino acids decrease affinity (4, 13, 105). In the absence of a GnRH-receptor crystal structure, alanine-scanning mutagenesis and molecular models have identified potential intermolecular contacts. However, most have not been validated by biochemical studies to distinguish indirect disruption of the GnRH-binding surface (Figure S4 in Supplementary Material). We will discuss potential GnRH-receptor contacts in the context of peptide-bound GPCR structures and recent GnRH receptor mutagenesis studies.
In spite of the diversity of GPCR ligand-binding pockets, Venkatakrishnan et al. identified a consensus ligand-binding pocket consisting of topologically equivalent residues at positions 3.32, 3.33, and 3.36 in TM3, 6.48 and 6.51 in the CWxPY motif, and 7.39 in TM7. These residues include two conserved interhelical contacts, 3.36–6.48 and 6.51–7.39, which couple the ligand-binding pocket to the conserved GPCR structure (20). Structures of peptide-bound GPCRs show that the sections of the peptides that are required for agonist activity, the carboxy-termini of endothelin-1 and apelin and the amino-termini of chemokines, penetrate the TM cores of their receptors and interact with subsets of the consensus ligand-binding residues, whereas other parts of the peptides bind outside of the core (93, 106–109). This suggests that the amino-terminal residues of GnRH may interact with the consensus-binding pocket.
The endothelin-1 peptide penetrates the TM core of the ETB-endothelin receptor and contacts the consensus-binding residues, Trp6.48 and Leu6.51 and Gln3.32 (107). The smaller peptide agonist NTS8–13 binds closer to the extracellular surface of the NTSR1 neurotensin receptor, but may contact the consensus Arg3.32(149) residue (104). Mutation of the equivalent GnRH receptor residue, Lys3.32(121), to Gln or Ala decreased GnRH affinity and signaling, but had minimal effect on binding of a peptide antagonist, which had modified amino-terminal residues. This led to a conclusion that Lys3.32(121) may form a hydrogen bond with the aromatic rings of His2 or Trp3 of GnRH (37, 99). Subsequent models proposed that Lys3.32(121) contacts pGlu1 or His2 (37, 68, 110–113) but, in the absence of further experiments, it remains uncertain whether Lys3.32(121) directly contacts GnRH or initiates receptor activation.
Agonist peptide ligands bound to the NTSR1 neurotensin, US28 viral chemokine and apelin receptors do not penetrate deeply enough to contact Trp6.48 of the CWxPY motif (93, 104, 106) and mutagenesis of Trp6.48 had minimal effects on NTSR1 receptor function (114). Molecular models suggested that Trp6.48(280) of the GnRH receptor interacts with Trp3 of the GnRH peptide (13, 69, 74, 111). However, mutations of Trp6.48(280) had minimal effects on GnRH affinity or cellular signaling (37, 38), indicating that it does not directly contact GnRH. Since TM3 has central roles in ligand binding, the conserved interhelical network and the hydrophobic core, movement of TM3 may provide an alternative molecular pathway to Trp6.48-mediated activation of the transmission switch in the GnRH and NTSR1 receptors (20, 57).
The hydrophobic residue in position 6.51 contacts the ligand in the ETB-endothelin and apelin receptors (106, 107), whereas the conserved Glu7.39 of chemokine receptors is a key determinant of chemokine binding and receptor activation (109). A Tyr6.51(283)Phe mutation in the GnRH receptor and mutations of Phe7.39(309) to Leu or Gln decreased GnRH-binding affinity (37). A recent computational model suggests that Phe7.39(309) may contact Trp3 of GnRH (110). Together, these data are consistent with the mutations disrupting ligand binding by breaking an interhelical contact between residues that may also contact the ligand, but more experiments are needed.
In summary, it is possible that amino-terminal residues of the GnRH peptide contact some of the consensus ligand-binding residues, Lys3.32(121), Tyr6.51(283), and Phe7.39(309), but not Trp6.48(280). It remains uncertain whether GnRH binds to a largely extracellular surface of the receptor like NTS8–13 (104) or penetrates the TM core like the peptide ligands of the apelin and chemokine receptors (106, 109).
A molecular model predicted that Arg1.35(38) in the amino terminus of the GnRH receptor is close to the carboxy-terminal Pro9-Gly10NH2 of GnRH. Mutations of Arg1.35(38) decreased GnRH-binding affinity, but had lesser effects on binding of [Pro9-NHEt]-GnRH, which lacks Gly10NH2 (112). The results support a hydrogen bond contact between Arg1.35(38) and Gly10NH2, but show that both the geometry and charge of the Arg1.35(38) side chain are important for additional inter- or intramolecular interactions. Asn2.65(102) in TM2 has similar functions in distinguishing Gly10NH2 of GnRH (13, 115). These studies suggest that the carboxy-terminus of GnRH may locate close to both Arg1.35(38) and Asn2.65(102) (Figure S4 in Supplementary Material). Systematic mutagenesis of Asp2.61(98) of the GnRH receptor, combined with ligand modification, showed that the Asp2.61(98) side chain determines receptor recognition of His2 of the GnRH peptide, via a hydrogen bond, whereas the charge of the Asp2.61(98) side chain may configure the surface of the ligand-binding pocket by forming an interhelical salt bridge with Lys3.32(121) (13, 116). Thus, residues in the amino-terminus and extracellular ends of TM1 and TM2 of the GnRH receptor appear to affect GnRH binding via direct contacts with amino- and carboxy-terminal residues of the peptide and via intramolecular interactions that affect the shape of the ligand-binding surface.
Molecular models of the GnRH receptor showed contact of Tyr6.58(290), two helical turns toward the extracellular end of TM6 from the CWxPY motif, with the Tyr5 side chain of the GnRH peptide. Systematic mutagenesis showed that both the hydroxyl group and the aromatic ring of the Tyr6.58(290) side chain contribute to high affinity binding of GnRH, but had less effect on binding of [Ala5]-GnRH, consistent with the hydroxyl group of Tyr6.58(290) interacting with the aromatic ring of Tyr5 of the peptide. The receptor mutations also decreased GnRH potency in signaling assays more than they decreased binding affinity, showing that the Tyr6.58(290) side chain has an additional role in coupling agonist binding to receptor activation (100). The Tyr6.58(290)–Tyr5 interaction may initiate movement and rotation of TM6 in the GnRH receptor.
Mutation of His7.36(305) at the extracellular end of TM7 in the mouse GnRH receptor to non-polar amino acids decreased GnRH-binding affinity, suggesting loss of a hydrogen bond interaction. Ligand modification suggested a His7.36(305)-Trp3 hydrogen bond contact. However, mutation of His7.36(305) to polar amino acids had no effect on ligand-binding affinity, making a direct interaction with the ligand unlikely. Molecular modeling showed that His7.36(305) made only intramolecular contacts with the amino terminus of the receptor, whereas Trp3 of GnRH was oriented near the consensus ligand-binding residue, Phe7.39(308). This suggests that His7.36(305) forms an interhelical contact that positions Phe7.39(308) to form π–π contact with Trp3 of the peptide (110). The cHH-associated GnRH receptor mutation, Thr32Ile, which decreases ligand-binding affinity (63), is immediately adjacent to the His7.36(305) interhelical contacts and may disrupt the interhelical contact.
Mammalian GnRH has a basic Arg8 residue, which is important for binding to type 1 GnRH receptors. Mutation of the acidic Asp7.32(302) residue to uncharged Asn decreased binding affinity of GnRH, but had no effect on binding of peptides with uncharged Gln8. This suggests that Asp7.32(302) forms a salt bridge contact with Arg8 of the peptide. However, “conformationally constrained” GnRH peptides, in which the high affinity β-turn was stabilized by a d-amino acid in position 6, retained high affinity binding in the absence of Asp7.32(302) or Arg8 or both. Since the Arg8 side chain also contributes to stabilizing the β-turn in the native GnRH peptide, it was concluded that the interaction of Asp7.32(302) with Arg8 induces the high-affinity peptide conformation (70, 117).
Although it was hypothesized that the Asp7.32(302)-Arg8 interaction induced the high affinity conformation of GnRH on binding to the receptor, mutation of many different GnRH receptor residues causes a similar large decrease in binding affinity of native GnRH, but much smaller decreases in affinity for conformationally constrained GnRH peptides (37, 68, 100, 115). So the ability of constrained peptides to overcome the ligand-binding affinity effects of receptor mutations is not specific to the Asp7.32(302)-Arg8 interaction. In the active GPCR conformations that have increased agonist-binding affinity, a “cap” forms over the extracellular surface of the ligand-binding pocket and increases agonist affinity by hindering dissociation of the ligand and trapping it in the binding pocket (51). In peptide receptors the larger ligand extends beyond the TM-binding pocket, so it cannot be capped. Nevertheless, the extracellular sides of agonist-bound peptide receptors, such as the NTSR1 neurotensin and μ-opioid receptors, move inwards and it has been suggested that this movement “pinches” the peptide ligand, increasing its affinity by hindering its dissociation (51). Comparison of the ETB-endothelin receptor structures with and without endothelin-1 showed that the peptide induces inward movement of the extracellular ends of the TM helices “tightening” the ligand pocket (107). Extrapolating to the GnRH receptor, conformationally constrained peptides may be more compact than GnRH before contacting the receptor and hence enhance narrowing of the ligand-binding pocket via multiple contacts with the ligand-binding pocket. The tightening would overcome mutation-induced loss of individual contacts.
Although only direct determination will confirm the GnRH receptor structure, growing numbers of other GPCR structures provide insight into common features likely to be shared by the GnRH receptor. GPCR structures can be used to hypothesize mechanisms by which agonist binding is coupled to G protein activation, which must be tested by dynamic methods, such as site-directed mutagenesis and functional analysis, regardless of availability of directly determined structures. Identification of the conformation-independent interhelical contact network has provided explanations for decreased expression of many cHH-associated mutant GnRH receptors, whereas the conserved conformation-specific interhelical contacts begin to explain how conserved residues mediate receptor activation. In spite of the diversity of ligand-binding surfaces, recent agonist-bound peptide-binding GPCRs suggest that ligand contacts in TM3 may trigger receptor activation and they may explain the high affinity of conformationally constrained GnRH peptides.
CF conceived the project and wrote the manuscript. AM wrote a preliminary review as part of her MSc dissertation, which was partly used in the current project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by South African National Research Foundation Competitive Program for Rated Researchers Grant no 93464 (CF) and Incentive Funding for Rated Researchers Grant no 81184 (CF).
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fendo.2017.00274/full#supplementary-material.
1. Bedecarrats GY, Kaiser UB. Mutations in the human gonadotropin-releasing hormone receptor: insights into receptor biology and function. Semin Reprod Med (2007) 25(5):368–78. doi:10.1055/s-2007-984743
2. Brioude F, Bouligand J, Trabado S, Francou B, Salenave S, Kamenicky P, et al. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotype-phenotype relationships. Eur J Endocrinol (2010) 162(5):835–51. doi:10.1530/EJE-10-0083
3. Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol (2009) 30(1):10–29. doi:10.1016/j.yfrne.2008.07.001
4. Schally AV, Block NL, Rick FG. Discovery of LHRH and development of LHRH analogs for prostate cancer treatment. Prostate (2017) 77(9):1036–54. doi:10.1002/pros.23360
5. Struthers RS, Nicholls AJ, Grundy J, Chen T, Jimenez R, Yen SS, et al. Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix. J Clin Endocrinol Metab (2009) 94(2):545–51. doi:10.1210/jc.2008-1695
6. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol (2010) 31(3):322–40. doi:10.1016/j.yfrne.2010.04.002
7. Perrett RM, McArdle CA. Molecular mechanisms of gonadotropin-releasing hormone signaling: integrating cyclic nucleotides into the network. Front Endocrinol (2013) 4:180. doi:10.3389/fendo.2013.00180
8. Tsutsumi R, Mistry D, Webster NJ. Signaling responses to pulsatile gonadotropin-releasing hormone in LbetaT2 gonadotrope cells. J Biol Chem (2010) 285(26):20262–72. doi:10.1074/jbc.M110.132662
9. Choi SG, Jia J, Pfeffer RL, Sealfon SC. G proteins and autocrine signaling differentially regulate gonadotropin subunit expression in pituitary gonadotrope. J Biol Chem (2012) 287(25):21550–60. doi:10.1074/jbc.M112.348607
10. Naor Z, Huhtaniemi I. Interactions of the GnRH receptor with heterotrimeric G proteins. Front Neuroendocrinol (2013) 34(2):88–94. doi:10.1016/j.yfrne.2012.11.001
11. White CD, Coetsee M, Morgan K, Flanagan CA, Millar RP, Lu ZL. A crucial role for Galphaq/11, but not Galphai/o or Galphas, in gonadotropin-releasing hormone receptor-mediated cell growth inhibition. Mol Endocrinol (2008) 22(11):2520–30. doi:10.1210/me.2008-0122
12. Grosse R, Schmid A, Schoneberg T, Herrlich A, Muhn P, Schultz G, et al. Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J Biol Chem (2000) 275(13):9193–200. doi:10.1074/jbc.275.13.9193
13. Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev (2004) 25(2):235–75. doi:10.1210/er.2003-0002
14. Munk C, Isberg V, Mordalski S, Harpsoe K, Rataj K, Hauser AS, et al. GPCRdb: the G protein-coupled receptor database – an introduction. Br J Pharmacol (2016) 173(14):2195–207. doi:10.1111/bph.13509
15. Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, et al. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol Sci (2015) 36(1):22–31. doi:10.1016/j.tips.2014.11.001
16. Mahoney JP, Sunahara RK. Mechanistic insights into GPCR-G protein interactions. Curr Opin Struct Biol (2016) 41:247–54. doi:10.1016/j.sbi.2016.11.005
17. Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature (2016) 536(7617):484–7. doi:10.1038/nature19107
18. Tesmer JJ. Hitchhiking on the heptahelical highway: structure and function of 7TM receptor complexes. Nat Rev Mol Cell Biol (2016) 17(7):439–50. doi:10.1038/nrm.2016.36
19. Cvicek V, Goddard WA III, Abrol R. Structure-based sequence alignment of the transmembrane domains of all human GPCRs: phylogenetic, structural and functional implications. PLoS Comput Biol (2016) 12(3):e1004805. doi:10.1371/journal.pcbi.1004805
20. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature (2013) 494(7436):185–94. doi:10.1038/nature11896
21. Ballesteros JA, Weinstein W. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci (1995) 25:366–428. doi:10.1016/S1043-9471(05)80049-7
22. Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, et al. Structural insights into micro-opioid receptor activation. Nature (2015) 524(7565):315–21. doi:10.1038/nature14886
23. Flanagan CA, Zhou W, Chi L, Yuen T, Rodic V, Robertson D, et al. The functional microdomain in transmembrane helices 2 and 7 regulates expression, activation, and coupling pathways of the gonadotropin-releasing hormone receptor. J Biol Chem (1999) 274(41):28880–6. doi:10.1074/jbc.274.41.28880
24. Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium: a key co-factor in class A GPCR signaling. Trends Biochem Sci (2014) 39(5):233–44. doi:10.1016/j.tibs.2014.03.002
25. Zhou W, Flanagan C, Ballesteros JA, Konvicka K, Davidson JS, Weinstein H, et al. A reciprocal mutation supports helix 2 and helix 7 proximity in the gonadotropin-releasing hormone receptor. Mol Pharmacol (1994) 45(2):165–70.
26. Trzaskowski B, Latek D, Yuan S, Ghoshdastider U, Debinski A, Filipek S. Action of molecular switches in GPCRs-theoretical and experimental studies. Curr Med Chem (2012) 19(8):1090–109. doi:10.2174/092986712799320556
27. Carpenter B, Nehme R, Warne T, Leslie AG, Tate CG. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature (2016) 536(7614):104–7. doi:10.1038/nature18966
28. Goncalves JA, South K, Ahuja S, Zaitseva E, Opefi CA, Eilers M, et al. Highly conserved tyrosine stabilizes the active state of rhodopsin. Proc Natl Acad Sci U S A (2010) 107(46):19861–6. doi:10.1073/pnas.1009405107
29. Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, et al. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem (1998) 273(17):10445–53. doi:10.1074/jbc.273.17.10445
30. Topaloglu AK, Lu ZL, Farooqi IS, Mungan NO, Yuksel B, O’Rahilly S, et al. Molecular genetic analysis of normosmic hypogonadotropic hypogonadism in a Turkish population: identification and detailed functional characterization of a novel mutation in the gonadotropin-releasing hormone receptor gene. Neuroendocrinology (2007) 84(5):301–8. doi:10.1159/000098147
31. Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol (2011) 21(4):541–51. doi:10.1016/j.sbi.2011.06.002
32. Deupi X. Relevance of rhodopsin studies for GPCR activation. Biochim Biophys Acta (2014) 1837(5):674–82. doi:10.1016/j.bbabio.2013.09.002
33. Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature (2011) 471(7340):656–60. doi:10.1038/nature09795
34. Conn PM, Leanos-Miranda A, Janovick JA. Protein origami: therapeutic rescue of misfolded geneproducts. Mol Interv (2002) 2(5):308–16. doi:10.1124/mi.2.5.308
35. Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab (2002) 87(7):3255–62. doi:10.1210/jcem.87.7.8582
36. Tello JA, Newton CL, Bouligand J, Guiochon-Mantel A, Millar RP, Young J. Congenital hypogonadotropic hypogonadism due to GnRH receptor mutations in three brothers reveal sites affecting conformation and coupling. PLoS One (2012) 7(6):e38456. doi:10.1371/journal.pone.0038456
37. Betz SF, Reinhart GJ, Lio FM, Chen C, Struthers RS. Overlapping, nonidentical binding sites of different classes of nonpeptide antagonists for the human gonadotropin-releasing hormone receptor. J Med Chem (2006) 49(2):637–47. doi:10.1021/jm0506928
38. Coetsee M, Gallagher R, Millar R, Flanagan C, Lu ZL. Role of Trp280(6.48) in the Gonadotropin-releasing hormone (GnRH) receptor. Abstrct, BioScience2006. Glasgow, UK (2006).
39. Arora KK, Cheng Z, Catt KJ. Dependence of agonist activation on an aromatic moiety in the DPLIY motif of the gonadotropin-releasing hormone receptor. Mol Endocrinol (1996) 10(8):979–86. doi:10.1210/mend.10.8.8843414
40. Roch GJ, Busby ER, Sherwood NM. GnRH receptors and peptides: skating backward. Gen Comp Endocrinol (2014) 209:118–34. doi:10.1016/j.ygcen.2014.07.025
41. Flanagan CA, Chen CC, Coetsee M, Mamputha S, Whitlock KE, Bredenkamp N, et al. Expression, structure, function, and evolution of gonadotropin-releasing hormone (GnRH) receptors GnRH-R1SHS and GnRH-R2PEY in the teleost, Astatotilapia burtoni. Endocrinology (2007) 148(10):5060–71. doi:10.1210/en.2006-1400
42. Kah O, Lethimonier C, Somoza G, Guilgur LG, Vaillant C, Lareyre JJ. GnRH and GnRH receptors in metazoa: a historical, comparative, and evolutive perspective. Gen Comp Endocrinol (2007) 153(1–3):346–64. doi:10.1016/j.ygcen.2007.01.030
43. Sefideh FA, Moon MJ, Yun S, Hong SI, Hwang JI, Seong JY. Local duplication of gonadotropin-releasing hormone (GnRH) receptor before two rounds of whole genome duplication and origin of the mammalian GnRH receptor. PLoS One (2014) 9(2):e87901. doi:10.1371/journal.pone.0087901
44. Chang JP, Pemberton JG. Comparative aspects of GnRH-stimulated signal transduction in the vertebrate pituitary – contributions from teleost model systems. Mol Cell Endocrinol (2017). doi:10.1016/j.mce.2017.06.002
45. Plachetzki DC, Tsai PS, Kavanaugh SI, Sower SA. Ancient origins of metazoan gonadotropin-releasing hormone and their receptors revealed by phylogenomic analyses. Gen Comp Endocrinol (2016) 234:10–9. doi:10.1016/j.ygcen.2016.06.007
46. Williams BL, Akazome Y, Oka Y, Eisthen HL. Dynamic evolution of the GnRH receptor gene family in vertebrates. BMC Evol Biol (2014) 14:215. doi:10.1186/s12862-014-0215-y
47. Leanos-Miranda A, Janovick JA, Conn PM. Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2002) 87(10):4825–8. doi:10.1210/jc.2002-020961
48. Janovick JA, Stewart MD, Jacob D, Martin LD, Deng JM, Stewart CA, et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci U S A (2013) 110(52):21030–5. doi:10.1073/pnas.1315194110
49. Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry (2003) 42(10):2759–67. doi:10.1021/bi027224+
50. Blankenship E, Vahedi-Faridi A, Lodowski DT. The high-resolution structure of activated opsin reveals a conserved solvent network in the transmembrane region essential for activation. Structure (2015) 23(12):2358–64. doi:10.1016/j.str.2015.09.015
51. DeVree BT, Mahoney JP, Velez-Ruiz GA, Rasmussen SG, Kuszak AJ, Edwald E, et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature (2016) 535(7610):182–6. doi:10.1038/nature18324
52. Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem (1993) 268(7):4625–36.
53. Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell (2013) 152(3):532–42. doi:10.1016/j.cell.2013.01.008
54. Kenakin T. Theoretical aspects of GPCR-ligand complex pharmacology. Chem Rev (2017) 117(1):4–20. doi:10.1021/acs.chemrev.5b00561
55. De Lean A, Stadel J, Lefkowitz R. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem (1980) 255(15):7108–17.
56. Carpenter B, Tate CG. Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr Opin Struct Biol (2017) 45:124–32. doi:10.1016/j.sbi.2017.04.010
57. Bortolato A, Dore AS, Hollenstein K, Tehan BG, Mason JS, Marshall FH. Structure of Class B GPCRs: new horizons for drug discovery. Br J Pharmacol (2014) 171(13):3132–45. doi:10.1111/bph.12689
58. Pardo L, Deupi X, Dolker N, Lopez-Rodriguez ML, Campillo M. The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. Chembiochem (2007) 8(1):19–24. doi:10.1002/cbic.200600429
59. Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol (2001) 60(1):1–19. doi:10.1124/mol.60.1.1
60. Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature (2008) 454(7203):486–91. doi:10.1038/nature07101
61. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science (2007) 318(5854):1258–65. doi:10.1126/science.1150577
62. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science (2008) 322(5905):1211–7. doi:10.1126/science.1164772
63. Bedecarrats GY, Linher KD, Janovick JA, Beranova M, Kada F, Seminara SB, et al. Four naturally occurring mutations in the human GnRH receptor affect ligand binding and receptor function. Mol Cell Endocrinol (2003) 205(1–2):51–64. doi:10.1016/S0303-7207(03)00201-6
64. Lu Z-L, Coetsee M, White CD, Millar RP. Structural determinants for ligand-receptor conformational selection in a peptide G protein-coupled receptor. J Biol Chem (2007) 282(24):17921–9. doi:10.1074/jbc.M610413200
65. Beneduzzi D, Trarbach EB, Min L, Jorge AA, Garmes HM, Renk AC, et al. Role of gonadotropin-releasing hormone receptor mutations in patients with a wide spectrum of pubertal delay. Fertil Steril (2014) 102(3):838.e–46.e. doi:10.1016/j.fertnstert.2014.05.044
66. Janovick JA, Patny A, Mosley R, Goulet MT, Altman MD, Rush TS III, et al. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol (2009) 23(2):157–68. doi:10.1210/me.2008-0384
67. Janovick JA, Pogozheva ID, Mosberg HI, Conn PM. Salt bridges overlapping the gonadotropin-releasing hormone receptor agonist binding site reveal a coincidence detector for G protein-coupled receptor activation. J Pharmacol Exp Ther (2011) 338(2):430–42. doi:10.1124/jpet.111.180869
68. Hoffmann SH, ter Laak T, Kuhne R, Reilander H, Beckers T. Residues within transmembrane helices 2 and 5 of the human gonadotropin-releasing hormone receptor contribute to agonist and antagonist binding [In Process Citation]. Mol Endocrinol (2000) 14(7):1099–115. doi:10.1210/mend.14.7.0483
69. Soderhall JA, Polymeropoulos EE, Paulini K, Gunther E, Kuhne R. Antagonist and agonist binding models of the human gonadotropin-releasing hormone receptor. Biochem Biophys Res Commun (2005) 333(2):568–82. doi:10.1016/j.bbrc.2005.05.142
70. Flanagan CA, Becker II, Davidson JS, Wakefield IK, Zhou W, Sealfon SC, et al. Glutamate 301 of the mouse gonadotropin-releasing hormone receptor confers specificity for arginine 8 of mammalian gonadotropin-releasing hormone. J Biol Chem (1994) 269(36):22636–41.
71. Isberg V, Mordalski S, Munk C, Rataj K, Harpsoe K, Hauser AS, et al. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res (2016) 44(D1):D356–64. doi:10.1093/nar/gkv1178
72. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem (2004) 25(13):1605–12. doi:10.1002/jcc.20084
73. Chauvin S, Hibert M, Berault A, Counis R. Critical implication of transmembrane Phe310, possibly in conjunction with Trp279, in the rat gonadotropin-releasing hormone receptor activation. Biochem Pharmacol (2001) 62(3):329–34. doi:10.1016/S0006-2952(01)00675-X
74. Chauvin S, Berault A, Lerrant Y, Hibert M, Counis R. Functional importance of transmembrane helix 6 Trp(279) and exoloop 3 Val(299) of rat gonadotropin-releasing hormone receptor. Mol Pharmacol (2000) 57(3):625–33. doi:10.1124/mol.57.3.625
75. Yuan S, Filipek S, Palczewski K, Vogel H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat Commun (2014) 5:4733. doi:10.1038/ncomms5733
76. Arora KK, Krsmanovic LZ, Mores N, O’Farrell H, Catt KJ. Mediation of cyclic AMP signaling by the first intracellular loop of the gonadotropin-releasing hormone receptor. J Biol Chem (1998) 273(40):25581–6. doi:10.1074/jbc.273.40.25581
77. Lu ZL, Gallagher R, Sellar R, Coetsee M, Millar RP. Mutations remote from the human gonadotropin-releasing hormone (GnRH) receptor-binding sites specifically increase binding affinity for GnRH II but not GnRH I: evidence for ligand-selective, receptor-active conformations. J Biol Chem (2005) 280(33):29796–803. doi:10.1074/jbc.M413520200
78. Costa EM, Bedecarrats GY, Mendonca BB, Arnhold IJ, Kaiser UB, Latronico AC. Two novel mutations in the gonadotropin-releasing hormone receptor gene in Brazilian patients with hypogonadotropic hypogonadism and normal olfaction. J Clin Endocrinol Metab (2001) 86(6):2680–6. doi:10.1210/jcem.86.6.7551
79. Wolczynski S, Laudanski P, Jarzabek K, Mittre H, Lagarde JP, Kottler ML. A case of complete hypogonadotropic hypogonadism with a mutation in the gonadotropin-releasing hormone receptor gene. Fertil Steril (2003) 79(2):442–4. doi:10.1016/S0015-0282(02)04667-8
80. Maione L, Bouligand J, Nettore IC, Flanagan C, Guiochon-Mantel A, Millar RP, et al. Identification et caractérisation moléculaire de deux nouvelles mutations de GNRHR chez deux familles avec retard pubertaire. Ann Endocrinol (Paris) (2015) 76(4):317. doi:10.1016/j.ando.2015.07.083
81. Myburgh DB, Pawson AJ, Davidson JS, Flanagan CA, Millar RP, Hapgood JP. A single amino acid substitution in transmembrane helix VI results in overexpression of the human GnRH receptor. Eur J Endocrinol (1998) 139(4):438–47. doi:10.1530/eje.0.1390438
82. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science (2000) 289(5480):739–45. doi:10.1126/science.289.5480.739
83. Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, et al. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem (2001) 276(31):29171–7. doi:10.1074/jbc.M103747200
84. Vogel R, Mahalingam M, Ludeke S, Huber T, Siebert F, Sakmar TP. Functional role of the “ionic lock” – an interhelical hydrogen-bond network in family A heptahelical receptors. J Mol Biol (2008) 380(4):648–55. doi:10.1016/j.jmb.2008.05.022
85. Bedecarrats GY, Linher KD, Kaiser UB. Two common naturally occurring mutations in the human gonadotropin-releasing hormone (GnRH) receptor have differential effects on gonadotropin gene expression and on GnRH-mediated signal transduction. J Clin Endocrinol Metab (2003) 88(2):834–43. doi:10.1210/jc.2002-020806
86. Angel TE, Chance MR, Palczewski K. Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc Natl Acad Sci U S A (2009) 106(21):8555–60. doi:10.1073/pnas.0903545106
87. Wormald PJ, Eidne KA, Millar RP. Gonadotropin-releasing hormone receptors in human pituitary: ligand structural requirements, molecular size, and cationic effects. J Clin Endocrinol Metab (1985) 61(6):1190–4. doi:10.1210/jcem-61-6-1190
88. Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science (2012) 337(6091):232–6. doi:10.1126/science.1219218
89. Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, et al. Molecular control of delta-opioid receptor signalling. Nature (2014) 506(7487):191–6. doi:10.1038/nature12944
90. Tehan BG, Bortolato A, Blaney FE, Weir MP, Mason JS. Unifying family A GPCR theories of activation. Pharmacol Ther (2014) 143(1):51–60. doi:10.1016/j.pharmthera.2014.02.004
91. Lebon G, Warne T, Tate CG. Agonist-bound structures of G protein-coupled receptors. Curr Opin Struct Biol (2012) 22(4):482–90. doi:10.1016/j.sbi.2012.03.007
92. Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature (2013) 504(7478):101–6. doi:10.1038/nature12735
93. Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, et al. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science (2015) 347(6226):1113–7. doi:10.1126/science.aaa5026
94. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature (2011) 469(7329):175–80. doi:10.1038/nature09648
95. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature (2011) 477(7366):549–55. doi:10.1038/nature10361
96. Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, et al. Adrenaline-activated structure of beta-adrenoceptor stabilized by an engineered nanobody. Nature (2013) 502:575–9. doi:10.1038/nature12572
97. Krumm BE, White JF, Shah P, Grisshammer R. Structural prerequisites for G-protein activation by the neurotensin receptor. Nat Commun (2015) 6:7895. doi:10.1038/ncomms8895
98. Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science (2015) 347(6226):1117–22. doi:10.1126/science.1261064
99. Zhou W, Rodic V, Kitanovic S, Flanagan CA, Chi L, Weinstein H, et al. A locus of the gonadotropin-releasing hormone receptor that differentiates agonist and antagonist binding sites. J Biol Chem (1995) 270(32):18853–7. doi:10.1074/jbc.270.32.18853
100. Coetsee M, Millar RP, Flanagan CA, Lu ZL. Identification of Tyr(290(6.58)) of the human gonadotropin-releasing hormone (GnRH) receptor as a contact residue for both GnRH I and GnRH II: importance for high-affinity binding and receptor activation. Biochemistry (2008) 47(39):10305–13. doi:10.1021/bi800911z
101. Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, et al. Crystal structure of metarhodopsin II. Nature (2011) 471(7340):651–5. doi:10.1038/nature09789
102. Deupi X, Standfuss J, Schertler G. Conserved activation pathways in G-protein-coupled receptors. Biochem Soc Trans (2012) 40(2):383–8. doi:10.1042/BST20120001
103. Arora KK, Cheng Z, Catt KJ. Mutations of the conserved DRS motif in the second intracellular loop of the gonadotropin-releasing hormone receptor affect expression, activation, and internalization. Mol Endocrinol (1997) 11(9):1203–12. doi:10.1210/mend.11.9.9968
104. White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, et al. Structure of the agonist-bound neurotensin receptor. Nature (2012) 490(7421):508–13. doi:10.1038/nature11558
105. Sealfon SC, Weinstein H, Millar RP. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr Rev (1997) 18(2):180–205. doi:10.1210/edrv.18.2.0295
106. Ma Y, Yue Y, Ma Y, Zhang Q, Zhou Q, Song Y, et al. Structural basis for apelin control of the human apelin receptor. Structure (2017) 25(6):858–66.e4. doi:10.1016/j.str.2017.04.008
107. Shihoya W, Nishizawa T, Okuta A, Tani K, Dohmae N, Fujiyoshi Y, et al. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature (2016) 537(7620):363–8. doi:10.1038/nature19319
108. Standfuss J. Structural biology. Viral chemokine mimicry. Science (2015) 347(6226):1071–2. doi:10.1126/science.aaa7998
109. Kufareva I, Gustavsson M, Zheng Y, Stephens BS, Handel TM. What do structures tell us about chemokine receptor function and antagonism? Annu Rev Biophys (2017) 46:175–98. doi:10.1146/annurev-biophys-051013-022942
110. Mayevu NM, Choe H, Abagyan R, Seong JY, Millar RP, Katz AA, et al. Histidine in the conserved peptide receptor activation domain of the gonadotropin releasing hormone receptor couples peptide binding and receptor activation. Mol Cell Endocrinol (2015) 402:95–106. doi:10.1016/j.mce.2015.01.008
111. Hovelmann S, Hoffmann SH, Kuhne R, ter Laak T, Reilander H, Beckers T. Impact of aromatic residues within transmembrane helix 6 of the human gonadotropin-releasing hormone receptor upon agonist and antagonist binding. Biochemistry (2002) 41(4):1129–36. doi:10.1021/bi0113162
112. Stewart AJ, Sellar R, Wilson DJ, Millar RP, Lu ZL. Identification of a novel ligand binding residue Arg38(1.35) in the human gonadotropin-releasing hormone receptor. Mol Pharmacol (2008) 73(1):75–81. doi:10.1124/mol.107.040816
113. Forfar R, Lu ZL. Role of the transmembrane domain 4/extracellular loop 2 junction of the human gonadotropin-releasing hormone receptor in ligand binding and receptor conformational selection. J Biol Chem (2011) 286(40):34617–26. doi:10.1074/jbc.M111.240341
114. Krumm BE, Lee S, Bhattacharya S, Botos I, White CF, Du H, et al. Structure and dynamics of a constitutively active neurotensin receptor. Sci Rep (2016) 6:38564. doi:10.1038/srep38564
115. Davidson JS, McArdle CA, Davies P, Elario R, Flanagan CA, Millar RP. Asn102 of the gonadotropin-releasing hormone receptor is a critical determinant of potency for agonists containing C-terminal glycinamide. J Biol Chem (1996) 271(26):15510–4. doi:10.1074/jbc.271.26.15510
116. Flanagan CA, Rodic R, Konvicka K, Yuen T, Chi L, Rivier JE, et al. Specific interactions of the Asp2.61(98) side chain of the gonadotropin-releasing hormone receptor contribute differentially to ligand interaction and receptor expression. Biochemistry (2000) 39:8133–41. doi:10.1021/bi000085g
Keywords: gonadotropin-releasing hormone receptor, G protein-coupled receptor, receptor structure, receptor activation, ligand binding
Citation: Flanagan CA and Manilall A (2017) Gonadotropin-Releasing Hormone (GnRH) Receptor Structure and GnRH Binding. Front. Endocrinol. 8:274. doi: 10.3389/fendo.2017.00274
Received: 08 August 2017; Accepted: 28 September 2017;
Published: 24 October 2017
Edited by:
Ivana Bjelobaba, University of Belgrade, SerbiaReviewed by:
Zvi Naor, Tel Aviv University, IsraelCopyright: © 2017 Flanagan and Manilall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colleen A. Flanagan, Y29sbGVlbi5mbGFuYWdhbkB3aXRzLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.